
Abstract
We investigated the correlation between increased gene copy number of gamma aminobutyric acid type A (GABAA) receptor α5-containing subunits and electrophysiological and behavioral phenotypes in a mouse model of Dup15q syndrome (15q dup) and tested the hypothesis that selectively inhibiting the activity of GABAA-α5 receptors may have therapeutic effects. Dup15q syndrome is a rare neurodevelopmental disorder caused by copy number gains of the 15q11.2-q13.1 chromosomal region, which includes UBE3A and a cluster of three genes (GABRA5, GABRB3, and GABRG3) encoding GABAA receptor subunits, all of which are critical for neural development and function. Most affected children display hypotonia, motor delays, intellectual disability, and epilepsy, as well as a characteristic electroencephalography (EEG) beta-band phenotype. There is no disease-modifying therapy available. Autoradiography showed increased density of GABAA-α5 receptors in the brains of 15q dup mice, while electrophysiology revealed enhanced GABAergic transmission in hippocampal slices from these mice. A GABAA-α5 negative allosteric modulator, RO4938581, decreased inhibitory synaptic charge transfer in 15q dup hippocampal slices. The behavioral analyses confirmed inflexibility in learning and abnormal social behaviors in 15q dup mice, and both phenotypes were normalized following chronic treatment with RO4938581. EEG recordings showed increased beta-power in 15q dup mice – which resembled the spectral signature of subjects with Dup15q – and was partially normalized following RO4938581 treatment. Our results suggest that excessive expression and function of the GABAA-α5 receptor subtype plays a key role in the pathophysiology of Dup15q and GABAA-α5 NAMs may represent a potential precision medicine therapeutic option.
Similar content being viewed by others
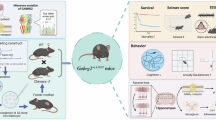


Introduction
Variation in DNA copy number is a well-described cause of human genetic disorder [1]. Dup15q syndrome is a rare genetic neurodevelopmental disorder caused by copy number gains of the 15q11.2-q13.1 region on chromosome 15. These duplications range from 4 to 12 Mb and occur de novo in most cases, either through the generation of supernumerary isodicentric chromosomes or as interstitial duplications, and, in rare cases, as interstitial triplications [2, 3]. The syndrome is characterized by a distinctive but heterogeneous neurodevelopmental phenotype, ranging from mild to very severe symptomatology, and a disease-characteristic electroencephalography (EEG) signature [4, 5]. The most common features are hypotonia and motor impairments, as well as a global developmental delay with cognitive delay, which manifests as intellectual disability, language impairment, autism, seizures, and behavioral challenges [2, 3]. Dup15q syndrome is a life-long and debilitating disorder precluding independent functioning and creating a significant burden for affected individuals and their caregivers. With no approved disease-modifying therapy available, there is a high, unmet medical need for people with this syndrome. Since their life expectancy is in the range of 40 years, they receive supportive care focusing on physical and behavioral therapies and seizures management.
The 15q11.2-q13.3 region overlaps the Prader-Willi syndrome/Angelman syndrome (PWS/AS) region between breakpoints 2 and 3 or 1 and 3 (BP2-BP3 or BP1-BP3). This region includes imprinted genes whose activity is dependent on their parental origin. Therefore, individuals exhibit different symptoms according to the parental origin of the duplication. The 15q11.2-13.1 region includes more than 10 characterized genes and non-coding regions, several of which have been associated with neurodevelopmental conditions, including UBE3A, CYFIP1, and a cluster of GABR genes which are non-imprinted, namely GABRA5, GABRB3, and GABRG3 (Supplementary Fig. 1A) [2, 6, 7]. The individual contributions and importance of the different genes for the pathophysiology of Dup15q syndrome are unknown but it is plausible to assume that, given their role in neurodevelopment and function, the three GABR genes have an important contribution. The GABRA5 and GABRB3 genes have been linked to epileptic encephalopathies and some studies have found association between genetic variations of these genes, as well as GABRG3, and autism spectrum disorders (ASD) [8,9,10,11] although these findings are not universally confirmed across all populations or studies. They encode for the α5, β3, and γ3 subunits of gamma aminobutyric acid type A (GABAA) receptor, which can co-assemble to form the pentameric GABAA receptor α5β3γ3 and α5β3γ2 subtypes [12, 13]. Collectively, these receptors are termed GABAA α5 subunit-containing receptors (hereafter GABAA-α5 receptors). The abundance of GABAA receptors containing the α5β2/3γ3 subunit composition is very low in rat hippocampus in contrast to that of receptors containing α5β2/3γ2 subunits which constitute approximately 25% of the total rat hippocampal GABAA receptors [12]. In addition, it was shown that GABAA receptors containing the α5 subunit in both rat and human hippocampus exhibit α5β3γ2 subtype characteristics [13]. There is also evidence that both γ2 and γ3 subunits can coexist within the same pentamer [12, 14].
In line with the excess dose of GABR genes, clinical evidence suggests that excess GABAA receptor expression and function contribute to Dup15q syndrome. Specifically, increased GABRB3, GABRA5, and GABRG3 mRNA expression was detected in dental pulp stem cell-derived neurons from subjects with Dup15q syndrome [15]. Moreover, Germain et al. reported increased GABRB3 expression (the only GABAA receptor subunit assessed) in induced pluripotent stem cell−derived neurons from subjects with Dup15q syndrome [16]. Individuals with Dup15q syndrome, both in maternal and paternal duplications, have a distinct EEG phenotype characterized by excessive beta-band oscillations [4, 5] that closely resembles the EEG signature of GABAA receptor–enhancing drugs (e.g., benzodiazepines) [17], thereby providing strong evidence for excess GABAA receptor activity in this syndrome. In line with this observation, the 15q11.2-q13.1 deletion Angelman syndrome is characterized by reduced EEG beta-power compared to genotypes that only affect imprinted genes [18]. Lastly, children with Dup15q syndrome have atypical responses to GABAergic drugs, particularly benzodiazepines, suggesting abnormal GABAA receptor function [19, 20].
Considering the above and the fact that the GABAergic system plays pivotal roles in brain development and function and is commonly disturbed in many neurodevelopmental disorders, including ASD [21], GABAA-α5 receptors may represent a desirable candidate target for therapeutic intervention in Dup15q syndrome. The expression of GABAA-α5 receptors starts very early in development in both rodents and humans. In the adult brain, these receptors comprise about 5% of all GABAA receptors and are preferentially localized in limbic structures such as the hippocampus, prefrontal cortex, anterior cingulate cortex, insular cortex, nucleus accumbens, and amygdala in typical developing individuals [22, 23]. At the subcellular level, GABAA-α5 receptors are localized mainly extrasynaptically, in close proximity to N-methyl-D-aspartate (NMDA) receptors at distal dendrites of pyramidal cells, where they mediate tonic inhibition [24]. Hence, GABAA-α5 receptors are well-positioned to regulate neuronal excitability and consequently excitatory/inhibitory circuit balance in the brain. Both genetic and pharmacological studies have demonstrated that GABAA-α5 receptors play an important modulatory role in learning and memory processes [25]. Therefore, GABAA-α5 negative allosteric modulation may oppose the functional consequences of the 15q GABR cluster duplication, thus directly addressing part of the pathomechanism underlying Dup15q phenotypes.
In this study we investigated the correlation between increased gene copy number of GABAA-α5 receptors and electrophysiological and behavioral phenotypes of a mouse model of Dup15q syndrome (15q dup). This engineered mouse model carries an interstitial paternal duplication of murine chromosome 7 corresponding to the region between common breakpoints (BP2-BP3) in human chromosome 15q11-13 [26] and displays abnormal behaviors that resemble aspects of the human condition [26, 27]. We also tested the hypothesis that selectively inhibiting the activity of GABAA-α5 receptors with a negative allosteric modulator, RO4938581 [28] in the 15q dup mice, may have therapeutic effects that might translate in Dup15q syndrome.
Materials and methods
Materials
RO4938581, diazepam, [3H]flumazenil, [3H]L-655,708, and [3H]RO0154513 were synthesized at F.Hoffmann-La Roche AG, Switzerland.
Animals
For all experiments, we used male patDp/+ (15q dup) mice and wild type (WT) littermates: autoradiography (n = 7 mice per group for [3H]L-655,708, n = 3 mice per group for [3H]flumazenil and [3H]RO0154513); electrophysiology (WT: n = 16; 15q dup = 20); EEG (WT: n = 8; 15q dup = 8); behavior (WT: n = 27; 15q dup = 34). The experiments were performed in male mice because we aimed to gather comparable data regarding the phenotypes of the mice, as those published previously [26]. Frozen embryos were used to generate the mouse line at Charles River Laboratories (France). The animals were then maintained on a C57BL/6J genetic background at F. Hoffman-La Roche (Basel, Switzerland). Mice were housed in groups of three animals per cage in standard laboratory conditions (temperature 22 ± 2 °C, 12-h light/dark cycles) and had access to food and water ad libitum. Genotyping was performed using the following primers: forward primer: 5′−ATA TGT ACT TTT GCA TAT AGT ATA C−3′, reverse primer: 5′–AGA GGA GGG CCT TAC TAA TTA CTT A−3′. All experimental procedures were approved by the City of Basel Cantonal Animal Protection Committee (AUP #2359) based on adherence to federal and local regulations on animal maintenance and testing.
In vitro autoradiography
Receptor autoradiography was performed on sagittal sections of fresh-frozen brains from WT and 15q dup mice, by using established GABAA receptor radioligands. Tissue sections (10-μm thick) were prepared from fresh-frozen mouse brains by using a cryostat (Leica CM3050). Sections were collected on microscope glass slides (HistoBond, Paul Marienfeld GmbH).
[3H]L-655,708 [29] was used to assess the expression of GABAA-α5 receptors. Brain sections were pre-incubated for 10 min in Ringer’s solution (146.5 mM NaCl, 2.7 mM KCl, 1.2 mM CaCl2, 0.85 mM MgCl2, 1.2 mM Na2HPO4, 0.27 mM NaH2PO4 pH 7.4), and incubated for 1 h at room temperature in Ringer’s solution containing 2 nM [3H]L655,708 (53 Ci/mmol). Sections were rinsed twice for 2 min in ice-cold Ringer’s solution, dipped three times into ice-cold H2O, and air-dried at 4 °C before being exposed to tritium-sensitive imaging plates (BAS-IP TR2025, Fujifilm) by a tritium microscale at RT for 5 days. The imaging plates were scanned by a high-resolution phosphor imager (Fuji BAS-5000, Bucher Biotec AG), and the binding intensity for selected brain areas was quantified using an MCID M2 image analysis software (version 7, InterFocus Imaging GmbH). For each section, non-specific binding (NSB) of [3H]L-655,708 was taken from the cerebellum and subtracted from the total binding (TB) in the brain area of interest to yield the specific binding (SB) according to the following formula: SB = TB − NSB. The SB of the radioligand was quantified and averaged from 3−4 adjacent sections of each mouse brain and expressed in fmol of bound radioligand per mg of protein.
To analyze the expression of GABAA receptor subtypes containing the α1, α2, α3, and α5 subunits, [3H]flumazenil (84 Ci/mmol) was used at 2 nM. For quantification of GABAA receptor subtypes containing the α4 and α6 subunits, [3H]RO0154513 (47 Ci/mmol) was used at 10 nM in the presence of 10 µM flunitrazepam. The conditions for incubation, washing, and exposure were the same as for [3H]L655,708.
Electrophysiology: Paired-pulse inhibition recordings
Paired-pulse inhibition (PPI) experiments were performed by using 2-month-old mice. Animals were deeply anesthetized in a 2.5% isoflurane/96.5% oxygen mixture and sacrificed. All the slicing and recording solutions were saturated with 95% O2 and 5% CO2.
The hippocampi were dissected, and 400-μm slices were transversely cut by a tissue chopper (Sorvall). Slices were maintained in a submerged chamber and perfused at RT with a solution containing (in mM): NaCl 124, KCl 5, MgSO4 2, CaCl2 2, KH2PO4 1.25, NaHCO3 25, and D-glucose 11, pH 7.4. Population spikes (PS) were recorded from the CA1. A glass micropipette (1−3 MΩ) containing 2 M NaCl was positioned in the stratum pyramidale and insulated bipolar platin/iridium electrodes were positioned in Schaffer collaterals for orthodromic stimulation (0.1 ms, 60−90 μA). The stimulus intensity that induced a PS near to the maximum amplitude was used as the test stimulus. PS were amplified by a Cyberamp 380 amplifier (Molecular Devices), filtered at 2.4 KHz, and digitized at 20 KHz via a Digidata 1322 acquisition board (Molecular Devices) for subsequent storage on a computer (Compaq Deskpro EN). The paired-pulse protocol used throughout this study consisted of two stimuli timely spaced by 20 ms, applied at Schaffer collaterals, and resulted in two PS. Data are normalized to the amplitude of the first PS.
Behavioral experiments
All behavioral experiments were performed during the light phase. The experimenters were blind to genotype and pharmacological treatment throughout the entire behavioral assessment. Treatment groups were assigned randomly. To minimize potential confounders regarding time of day for behavioral testing, animals were split into two cohorts (balanced for treatment groups) and tested during the same period. To decrease the chances of behavioral responses being altered by previous test history, procedures with more intensive handling were performed last, according to the following order: open field, direct social interaction, ultrasonic vocalization, olfactory habituation/dishabituation, Morris water maze (Supplementary Fig. 6A).
Pharmacological treatment
RO4938581 was synthesized at F. Hoffmann-La Roche Ltd. and prepared in a vehicle containing 5% PEG 400 and 20% HP-beta-cyclodextrin v/v water. RO4938581 (10 mg/kg) was administered per os (p.o.) by oral gavage (volume of 10 ml/kg) once per day for 3 weeks prior to the behavioral experiments and continued throughout the period of the behavioral experiments after the completion of testing each day.
Open field test
Locomotor activity was recorded for 60 min in large Plexiglas chambers (40 × 40 cm) equipped with an automated VersaMax tracking system (AccuScan Instruments). The brightest area, in the middle of the test chamber, was 20 Lux. The total distance covered in the entire open field was recorded using the VersaMax tracking system.
Direct social interaction test
Two mice of identical genotype (and identical treatment) that were originally housed in different cages were placed into a clear Perspex box together (18.5 × 38.5 cm) and allowed to explore freely for 5 min. Social behavior was monitored by a camera on the ceiling. Global interaction (i.e., the total time spent with any contact) was manually scored using the Noldus EthoVision XT system (Noldus, Netherlands).
Ultrasonic vocalization
Test male animals received female sexual experience for 10 min approximately 1 week before starting the experiment. On the test day, mice were first habituated to the test chamber (40 × 40 cm) for 5 min. Each subject was monitored in the presence of 100 μl distilled water and then urine from female mice in estrus, presented at a specific location on the paper floor covering, for a 5 min trial each. Vocalizations and animal movements were recorded using Avisoft USGH (Avisoft Bioacoustics) and EthoVision tracking system (Noldus).
Olfactory habituation/dishabituation test
The ability to discriminate non-social and social odors was measured using modifications of the olfactory habituation/dishabituation task, as previously described [30,31,32]. Mice were individually tested for time spent sniffing cotton tipped swabs suspended from the cage lid. The olfactory cues were designed to measure familiar and unfamiliar odors, with and without social valence. Sequences of two or three identical swabs were used to assess habituation to the same odor. Switching to a different odor on the swab was used to assess dishabituation (i.e., recognition that an odor is new). Swabs were dipped in distilled water, almond extract (1:100 dilution; McCormick), banana flavoring (1:100 dilution; McCormick), or wiped in a zig-zag pattern across the bottom surface of two different plastic cages (social1 and social2) that contained three stimulus mice each. The order of swab presentation was as follows: water, water, almond, almond, banana, banana, social1, social1, social1, social2, social2, and social2. Each swab was presented for a 2-min period immediately following the last swab presentation. Each test session was conducted in a clean mouse cage containing fresh litter. Before starting the experiment, animals were habituated to a neutral swab for 30 min.
Morris water maze test
The water maze was a large pool (1 m in diameter, 40-cm high) filled with water that was made opaque by using a white artificial opacifier (E308, Bronx Chemicals), and was surrounded by extramaze visual cues. The maze was arbitrarily divided into four quadrants: NE, NW, SE, and SW; a colorless Perspex circular platform (10 cm in diameter) was positioned at the center of one of these quadrants, 1 cm below the water level. A computer tracking system (HVS Image Ltd.) was used to analyze the mouse trajectories and measure escape latency. The maximum duration of each trial was 60 seconds. If a mouse found the platform during the trial, it was left on the platform for 10 seconds. If the mouse did not find the platform by the end of the trial, it was guided towards it and then allowed to remain on the platform for 10 seconds. After each trial the animal was towel dried and returned to its home cage, which was placed under a heat lamp. All the different start positions (north, south, east, and west) were applied to each trial, and a 15-min inter-trial interval was applied. Six trials per day were performed for 2 successive days for the visible platform task. A hidden platform task was also performed either by maintaining the platform at the original location (four trials per day for 7 successive days; acquisition trials) or by changing the platform to a new location rotated by 180 degrees relative to the original platform location (four trials for 4 successive days; reversal trials). On the days following the last acquisition trial and the last reversal trial of the hidden platform test (i.e., on the 10th and 15th day of the experiment), a probe test and a reversal probe test were performed, respectively. During this 60-second probe trial, the platform was removed from the tank.
Electroencephalography
This experiment investigated EEG in male 15q dup mice and WT controls. At ~P150, the mice received acute drug treatment intraperitoneally (i.p.) in a randomized cross-over design with RO4938581 (two doses: 3 and 10 mg/kg), diazepam (1 mg/kg), and a vehicle control (0.3% Tween80 in 0.9% NaCl); with 2–3 days washout between doses. Dosing was at the start of the dark cycle (active period) followed by EEG recordings for 6 h. Additional EEG recordings before and after the acute experiment are not reported here.
Surgical procedures
Eight male 15q dup mice and eight male WT controls (11–13 weeks old) were implanted for chronic tethered recordings of EEG and electromyography (EMG) using aseptic techniques. Under isoflurane anesthesia (1−4%), the fur on the top of the head was shaved off, and the skin was disinfected with Betadine and sterile water. A dorsal midline incision on top of the head was made, and the skull was cleaned. The EEG/EMG headmount (Pinnacle Technologies) was affixed to the skull by using cyanoacrylate and four stainless steel screws (2× frontal, 0.10″; and 2× parietal, 0.12″), which also functioned as EEG electrodes. The front edge of the headmount was placed 3.0 mm anterior to the bregma, aligned centrally along the sagittal suture. EEG electrodes were positioned 1.5 mm lateral to the sagittal suture, 2.0 mm anterior of the bregma, and −4.0 mm posterior to the bregma. The threads of the EEG electrodes were coated with silver epoxy (8226, Pinnacle Technologies) to ensure a solid connection with the headmount. EMG electrodes were inserted and sutured into the trapezius muscle. Electrical conductivity between the screws and the headmount was verified using a digital multimeter before anchoring the implant to the skull by dental acrylic (Lang Dental Manufacturing Co). Mice were administered post-operative hydration (0.9% NaCl), analgesics (0.05−0.1 mg/kg buprenorphine, and 5−10 mg/kg meloxicam), thermal support, and soft chow. Notably, 15q dup mice took much longer to arouse from the isoflurane anesthesia than did the WT mice (30−45 vs. 1−5 min). An opioid analgesic (buprenorphine) was administered in doses from the low end of the dose range. During the 14-day post-surgery recovery period and for the remainder of the study, mice were individually housed in cages (280 × 175 × 130 mm) with raised vertical sides to prevent escape during EEG/EMG recordings.
Data collection
EEG/EMG data were collected using iox2 (Version 2.8.0.11, EMKA Technologies) installed on a PC (Windows 7, OS). Electrophysiological signals were collected from the mice by using a 6-channel cable (NMUF6/30-4046U, Cooner Wire) and double-sided 6-pin connectors (8271, Pinnacle Technologies) connected to swivel commutators (8204, Pinnacle Technologies) mounted above the cage, so that mice could move freely. Mice were habituated to the recording cable for 2−3 days before data collection. EEG signals were derived from a frontal-parietal electrode configuration. Additional custom-made cables (546-441/6, Plastics One) connected each commutator to an amplifier system (M15 bipolar amplifier consisting of eight 15A54 modules; Grass Instruments). EEG and EMG signals were amplified 10,000×, routed to iox2 interface boxes (EMKA Technologies), and sampled at 500 Hz.
For two of the 15q dup mice, the EEG power gradually decreased over the course of the study. This is not a physiological effect and was probably due to a gradual increase in impedance. Therefore, these mice could not be used for spectral power comparisons between the different dosing days.
Drug administration occurred using a repeated measures, counter-balanced design. Treatment was administered by experimenters that were blind to the genotype and dose conditions.
Sleep/Wake scoring
EEG recordings were scored in 10-second epochs by expert scorers (with >95% inter-scorer reliability) for waking, rapid eye movement sleep, and non-rapid eye movement sleep. Scorers were blind to genotype and dose conditions. Only the quantitative analyses of EEGs in the wake state from the acute drug challenge experiments are presented here.
Power spectral estimates
Power spectral estimates were derived from the whole 6 h continuous EEG recording of the vehicle condition to investigate the 15q dup phenotype compared to WT mice and from 10 min to 3 h post dose to investigate drug effects (i.e., time-range where drug effects are expected given the half-lives).
Power spectral estimates were derived for logarithmically scaled frequencies by a logarithmic frequency smoothing by using Morlet Wavelets [33] with a spectral smoothing of 1/3 octave (f/σf = 8.7). This frequency transformation accounts for the logarithmic nature of electrophysiological data [34]. Center frequencies were spaced logarithmically according to the exponentiation of base 2, with exponents ranging from 1 (2 Hz) to 7.5 (181.0 Hz) in steps of 1/12. Spectral estimates were derived in successive 3/4-overlapping temporal windows of clean data.
The two doses of RO4938581 were averaged for analysis. Power values were then scaled and log-transformed to have units of 10*log10(μV2/log2 (Hz)). For final illustration, values were back-transformed to μV2/log2 (Hz) or presented as percent change. To allow comparison of RO4938581-related power changes with the 15q dup phenotype, we scaled the drug-related change at baseline in 15q dup mice to the vehicle change in WT mice, by the following equation:
Statistical analysis
Sample sizes were determined based on historical data for each experiment within each laboratory.
Autoradiographical data
Quantitative analysis of autoradiographical data show the relative expression of different GABAA receptor subtypes in the cortex, hippocampus, and striatum of 15q dup and WT mice. Each value is presented as a relative value to the expression level in WT animals. Results are expressed as mean ± standard error of the mean. Statistical differences were analyzed by a two-way ANOVA Sidak’s multiple comparisons test.
Electrophysiology data
Statistical comparisons of data obtained from electrophysiological experiments were performed using GraphPad Prism 7. Shapiro-Wilk test was used to determine data distribution. For normally distributed data, differences between two groups were examined by using two-tailed paired and unpaired Student’s t-test. Differences among more than two groups were assessed using ordinary and repeated-measures analysis of variance (ANOVA) and non-parametric methods with post-hoc tests, as appropriate.
Behavioral data
Numerical behavioral data are expressed as mean ± standard error of the mean (SEM). Statistical analysis was performed using R Statistical Software version 2.14.1 (Foundation for Statistical Computing, Vienna, Austria). The water maze acquisition data were analyzed using either a three-way repeated-measures ANOVA (Genotype, Treatment, Days or Quadrants). All other behavioral data were analyzed by two-way ANOVA or unpaired t-test. Ryan’s post-hoc tests were used to compare groups if the analysis indicated a statistically significant (p < 0.05) main effect or a significant interaction.
Electroencephalography data
Student’s t-tests were used for single comparisons of log-transformed spectral power and for deriving confidence intervals for each frequency. To account for multiple testing across frequencies, we used a randomization procedure that involved a min operation. More specifically, for every 10,000 randomizations of the group assignments (WT, 15q dup), we derived the smallest p-value across all frequencies to generate a null-hypothesis distribution. This approach accounts for multiple comparisons across all frequencies, as well as for positive and negative changes (two-tailed) in a data-adaptive manner. All analyses were performed in MATLAB using custom scripts.
Results
GABAA-α5 receptors are overexpressed in the brain of 15q dup mice
We first confirmed by RTqPCR that the expression levels of the non-imprinted GABR genes in the hippocampus of 15q dup adult male mice were higher (approximately 1.5-fold) than the respective levels in WT animals (Supplementary Fig. 1B). The protein level of the GABAA-α5 subunit was also increased in brain slices and in neuronal cultures from the 15q dup mouse brain as indicated by immunofluorescent staining, with the levels being 50% higher than in WT tissue and cells (Supplementary Fig. 1C–G).
We further validated the increased density of GABAA-α5 subunit-containing receptors in the brain (cortex, hippocampus and striatum) of 15q dup mice by in vitro autoradiography. The binding of [3H]L-655708, which selectively binds at the interphase of α5 and γ2 as well as α5 and γ3 subunits of the GABAA receptor complex, increased by about 1.5-fold in the 15q dup mouse brain compared to the WT mice (Fig. 1A, B). We also showed that the GABAA-α5 receptor is likely the only GABAA benzodiazepine sensitive receptor subtype with increased expression in the brains of 15q dup mice and that there are no apparent changes in the expression of GABAA α4γ2 and α6γ2 containing receptors. There was only a trend toward increased [3H]flumazenil binding in the 15q dup brains compared to WT brains. [3H]flumazenil labels benzodiazepine sensitive receptors (α1, α2, α3, α5 together with γ2 or γ3) whereas [3H]RO0154513 in the presence of 10 µM flunitrazepam allowed quantification of GABAA receptor subtypes containing the α4 and α6 together with the γ2 subunits.

A Representative in vitro autoradiographical images of sagittal brain sections prepared from wild type (WT) and 15q dup mice and incubated with 0.1 nM [3H]L-655,708 (α5; top), 2 nM [3H]flumazenil (α1,α2,α3,α5; middle), and 10 nM [3H]RO0154513 in the presence of 10 μM flunitrazepam (α4,α6; bottom). B Bar graphs showing the relative expression of different GABAA γ2 receptor subtypes in the cortex, hippocampus, and striatum of 15q dup and WT mice. Each value is presented as a relative value to the WT expression level. Results are expressed as individual points (circles) and mean ± standard error of the mean (n = 7 mice per group for [3H]L-655,708, n = 3 mice per group for [3H]flumazenil and [3H]RO0154513). Statistics: ***p < 0.001, 15q dup vs. WT.
15q dup mice exhibit enhanced GABAergic synaptic transmission in CA1 pyramidal neurons
There were no differences between 15q dup and WT on spontaneous excitatory synaptic currents (sEPSCs) (Supplementary Fig. 2). Analysis of the measured spontaneous inhibitory synaptic current (sIPSC) parameters revealed no differences, except for sIPSC frequency which was significantly higher in 15q dup mice than in WT CA1 hippocampal neurons (Supplementary Fig. 3).
RO4938581 had no effect on sIPSC frequency (Supplementary Fig. 3D), but slightly reduced the mean sIPSC area in 15q dup neurons compared to the respective area in WT neurons (Supplementary Fig. 3E), indicating a decrease in inhibitory synaptic charge transfer.
Paired-pulse inhibition (PPI) in brain slices containing the CA1 region was more pronounced in 15q dup mice than in WT littermates (Fig. 2). Application of RO4938581 significantly increased the PPI ratio in both 15q dup and WT due to an increased amplitude of the second PS that was almost identical to that of the first PS in both genotypes (Fig. 2).

A Superimposed representative traces of population spikes recorded in the CA1 region of hippocampal brains slices of WT and 15q dup mice. The paired-pulse protocol consisted of two stimuli timely spaced by 20 ms applied to Schaffer collaterals. Dark traces were obtained in the presence of vehicle and light traces in the presence of RO4938581 at 0.3 μM. B Paired-pulse inhibition was first measured in the absence (vehicle) and then in the presence of 0.3 μM RO4938581 and expressed as the ratio of the amplitude of population spike recorded after the first of two stimuli (as represented in A; 15q dup n = 4 mice, 8 slices; WT n = 3 mice, 7 slices). Results are expressed as individual points (circles) and mean ± standard error of the mean. *** p = 0.0007 effect of RO4938581 in 15q dup slices; ** p = 0.0031 effect of RO4938581 in WT slices.
There were no differences in LTP between 15q dup and WT mice (Supplementary Fig. 4).
15q dup mice exhibit abnormal cognitive and social behaviors that are mitigated by RO4938581 treatment
We examined the behavior of 15q dup mice using a battery of assays designed to 1) test the reproducibility of previously reported phenotypes, and 2) explore novel endpoints that are designed to model the symptomatology of individuals with Dup15q syndrome. Compared to WT mice, 15q dup mice showed reduced exploration, a trend for increased social interaction, reduced USV calls, reduced preference for social odors, and impaired reversal learning in the water maze (Supplementary Fig. 5).
We repeated the behavioral test battery in a new cohort of mice after chronic administration of RO4938581 at 10 mg/kg p.o. There was a significant reduction in activity in 15q dup mice (p = 0.006), which was not affected by drug treatment (Supplementary Fig. 6, B, C). There were no significant phenotypic or drug treatment effects in the adult ultrasonic vocalization test (Supplementary Fig. 6D), or the olfactory habituation/dishabituation test (Supplementary Fig. 6E).
In the Morris water maze, 15q dup exhibited normal acquisition and memory for the platform location during the visual and hidden phases, but an increased latency when the hidden platform location was changed (reversal phase; Fig. 3A). Administration of RO4938581 significantly reduced the escape latency of 15q dup mice during the reversal learning phase as compared to WT controls, particularly on Day 2 and Day 3 (Fig. 3A). It did not impact the time spent in each quadrant of the maze during the probe tests following acquisition (Supplementary Fig. 6F) and reversal learning (Supplementary Fig. 6G). In the direct social interaction test, the abnormal interaction of 15q dup mice was normalized by the chronic administration of RO4938581 (Fig. 3B). At the end of the behavioral experiments, the mean plasma concentration of RO4938581 at 60 min post-administration was 253 ± 229.3 ng/mL (Supplementary Table 1). The receptor occupancy calculated for this average plasma concentration is about 40–60%.

A Morris water maze: mean escape latency (s) each day during the visible and hidden platform acquisition phases and reversal phase (hidden platform in new position) of WT and 15q dup mice following chronic treatment with vehicle or RO4938581 (10 mg/kg p.o.). B Direct social interaction test: Total time touching or investigating their partner for WT and 15q dup mice following chronic treatment with vehicle or RO4938581 (10 mg/kg p.o.). Data are presented as individual points (circles) and mean ± standard error of the mean (n = 8 WT vehicle; n = 9 WT RO4938581; n = 12 15q dup vehicle; n = 12 15q dup RO4938581). Statistics for Morris water maze: *p < 0.05, **p < 0.01, WT-vehicle vs. 15q dup-vehicle; #p < 0.05, ###p < 0.001, 15q dup-vehicle vs. 15q dup-RO4938581 Statistics for direct social interaction test: §p = 0.051 WT-vehicle vs. 15q dup-vehicle.
15q dup mice exhibit increased beta-band activity that is partially normalized by RO4938581 treatment
We examined EEG power spectra (vehicle data) in 15q dup and WT mice and found that 15q dup mice exhibited increased power ( ~ 50%) in the beta frequency range (Fig. 4A, B), which resembled the spectral signature of acute benzodiazepine treatment in WT mice (Fig. 4C). The 15q dup EEG phenotype was partially normalized by acute administration of RO4938581 (low and high doses averaged; 9.2% power decrease at minimum; Fig. 4D). Both doses of RO4938581 revealed a decrease in signal power in the investigated frequency range, which was significant for the low dose (3 mg/kg; cluster permutation test: ~20.1–26.9 Hz, p < 0.05; −11.8% power decrease at minimum, −16.8–6.6, 95% confidence interval, Supplementary Fig. 7A) but not for the high dose (10 mg/kg: cluster permutation test: p > 0.05; −6.8% power decrease at minimum, −12.1–1.2, 95% confidence interval; Supplementary Fig. 7B).

A Power spectrum derived from 6-h continuous EEG recordings after vehicle dosing in wild type (WT; n = 8) and 15q dup (n = 6) mice during the awake state. B Ratio of spectrally-resolved power between 15q dup and WT mice shown in A. Black bar indicates significantly elevated power in the beta frequency range in 15q dup mice (~20−30 Hz; p = 0.017, cluster permutation test). C Changes in EEG power spectrum in response to diazepam (DZP) in WT mice. Diazepam increased the power in the beta to low gamma frequency range (~16−45 Hz; p = 0.007, cluster permutation test) and decreased it in the high gamma frequency range (~68−128 Hz, p = 0.015). D Changes in EEG power spectrum in response to RO4938581 (low and high doses combined) in 15q dup mice (left) and WT mice (right) in the frequency range of 20−30 Hz, i.e., focused on the frequency range of the 15q dup phenotype shown in (A). A significant decrease in power was observed in 15q dup mice after RO4938581 administration (~22.6−25.4 Hz; p = 0.030, cluster randomization test) but not in WT mice. CI = confidence interval. See Supplementary Fig. 7 for separate analyses for low and high doses of RO4938581 in 15q dup mice.
Discussion
Immunohistochemistry and [3H]L-655,708 autoradiography confirmed the higher density of GABAA-α5 receptors in 15q dup mouse cortex, hippocampus, and striatum in line with the increased expression of the α5, β3 and γ3 subunit mRNA, all methods showing about 1.5-fold increase of the signal compared to WT mice samples. Both the [3H]L-655,708 and [3H]flumazenil autoradiography data indicated that the GABAA-α5 receptor subtype is likely the only benzodiazepine-sensitive GABAA receptor subtype with increased expression in the brains of 15q dup mice. Only a trend toward increased [3H]flumazenil binding was observed in the 15q dup brains compared to WT brains and this fit the estimated 5% increase in the total pool of benzodiazepine sensitive subtypes. This number can be estimated by considering the relative abundance of the α5 containing receptors and the corresponding Ki values of the radioligand i.e. in the hippocampus, about 20–25% of total GABAA receptors belong to the α5γ2 subtype, and Flumazenil has about 3-fold higher affinity for the α1, α2 and α3γ2 containing receptors compared to the α5γ2 subtype. Therefore the 50% increase in GABAA α5 receptors determined by [3H]L-655,708 binding corresponds to about 5% increase in the [3H]flumazenil binding.
Our findings also indicate no significant changes in the expression levels of GABAA receptors containing the α4γ2 and α6γ2 subunits. However, we cannot rule out the possibility that increased β3 mRNA expression may affect the assembly of receptor complexes containing β3 alongside other subunits, such as δ or ε subunit. Additionally, our data do not provide evidence for compensatory changes in β2 or β1 subunit expression in response to the increased β3 levels.
Consistent with the enhanced GABAA receptor expression in both Dup15q individuals [15, 16] and 15q dup mice, and the known effect of GABAA-α5 receptors in mediating inhibitory neuronal activity [24], we found enhanced hippocampal GABAergic transmission in ex-vivo electrophysiological experiments performed using hippocampal slices prepared from 15q dup mice. There was an increase in sIPSC frequency in CA1 pyramidal neurons of 15q dup mice, while sIPSC current amplitude was unaltered. The presence of more inhibitory synapses in 15q dup CA1 pyramidal neurons may explain these results. In agreement, NL3R451C mice, a well-characterized animal model relevant for ASD that also exhibits increased sIPSC frequency, were reported to have more cortical and hippocampal VGAT-positive puncta compared to WT littermates [35]. Moreover, Isshiki et al. detected enhanced inhibitory synapse turnover on the dendritic shaft of 15q dup mice as shown by the inhibitory postsynaptic marker gephyrin [36, 37]. Alternatively, this could be a consequence of increased excitability of interneurons projecting to CA1 pyramidal neurons. In contrast, an opposite phenotype, i.e., reduced mIPSC frequency, has been observed in the somatosensory cortex of 15q dup mice, which points to circuit-specific effects.
Acute treatment of hippocampal slices with RO4938581 had no direct effect on the sIPSC frequency or amplitude, and this is in agreement with previous reports that GABAA-α5 receptors do not mediate fast kinetic synaptic events i.e. fast sIPSCs in CA1 [38, 39]. However, RO4938581 dampened the inhibitory tone in 15q dup mice, thus counterbalancing the increased frequency by reducing the mean sIPSC area of 15q dup pyramidal neurons. The PPI results indicate that GABAA-α5 receptors in hippocampal neurons impact inhibitory feedback in the tri-synaptic hippocampal circuit, and that RO4938581 normalizes feedback/feedforward inhibition in 15q dup slices by increasing the excitability of CA1 pyramidal neurons. We selected the hippocampus for these studies because the GABAA-α5 receptor is prominently expressed in this region and it is known to play a key role in memory, navigation, and cognition [40]. Interestingly, LTP was not affected which suggests that, under the current experimental conditions, moderately altered inhibition in 15q dup mice does not impact this form of synaptic plasticity.
We assessed only male mice in behavior tests so that we could compare our data with a previous study which was performed in male 15q dup mice [26]. Our behavioral analysis confirmed that male 15q dup mice exhibit abnormal social behavior and memory-related deficits that suggest cognitive inflexibility, and further revealed novel behavioral phenotypes such as reduced ultrasonic vocalizations in adult male mice and reduced preference for social versus non-social odors. However, the latter findings were not reproduced in the chronic treatment study, which may have been a consequence of chronic daily treatment and handling for approximately 4 weeks prior to testing. Only the social interaction test and reversal learning in the Morris water maze showed impairments during the chronic treatment study.
15q dup mice showed increased interaction compared to WT mice in the social interaction test which is inconsistent with a previous study [41] that showed a reduction in social interaction compared to WT mice. Our study used a small arena which was identical to the housing and so was likely to be familiar to the mice, whereas the Tsurugizawa study used a large unfamiliar arena. Since 15q dup mice have been shown to have an anxiety-like phenotype [27], the difference between the two studies may be due to increased anxiety in response to the novel, large arena leading to a reduction in social interaction.
Chronic treatment with RO4938581 normalized social interaction behaviors, affirming that altered GABAergic signaling contributes to this phenotype. Numerous studies have shown that cortico-limbic and striatal structures are involved in social behavior [42, 43]. Hence, it is possible that RO4938581 normalized the abnormal social behavior of 15q dup mice by re-establishing the circuit’s inhibitory/excitatory balance that was disrupted by the increased density of GABAA-α5 receptors in these regions. Gabrb3−/− mice, a mouse model relevant for ASD [44], also exhibit significant deficits in activities related to social behavior, including sociability and responses to exposure to unknown mice [45]. This is intriguing given that β3 subunits co-assemble with α5 subunits [13, 46], and that GABRB3 is included in the 15q11-13 cluster of GABR genes and overexpressed in the hippocampus of 15q dup mice according to our RT-qPCR results. Moreover, multiple genetic studies link mutations in the GABRB3 and GABRA5 genes to other neurodevelopmental disorders, including ASD and epilepsy [47,48,49,50].
Regarding spatial learning, the performance of 15q dup mice was normal during the visual platform task and acquisition phase of the place task of the Morris water maze. This is consistent with the comparable LTP at the Schaffer collateral-CA1 pathway (in vitro) between 15q dup and WT littermates, since synaptic plasticity at hippocampal excitatory synapses has been proposed as the cellular mechanism underlying spatial learning and memory [51, 52]. However, in our current study, the mice may have also used a non-spatial strategy whilst learning to find the platform location, since there was no preference for the platform position during the probe trial.
Consistent with a previous study [26], 15q dup mice showed impaired learning in the reversal phase of the water maze, suggesting that they do not respond as quickly to a platform location change as WT mice. This may be comparable to the inflexibility in daily routine that characterizes individuals with autism [53]. This phenotype (i.e., normal acquisition phase and impaired reversal learning) has also been observed in mice with a conditional knockout of the α5 subunit in dentate gyrus granule cells [54]. Thus, both a reduction of α5 subunit expression and an increase of expression result in reversal learning deficits. This suggests that the normal levels of GABAA α5 subunit-containing receptors are required for reversal learning and perhaps more generally for cognitive flexibility.
In reversal learning paradigms, mice initially persevere towards the original platform position, until they learn to switch their strategy to find and learn the position of the new platform, as shown in the current study by the reduction in latency from day 1 to day 2/3 by WT-vehicle mice. RO4938581 treated 15q dup mice adapted as easily as WT littermates to the reversal task. Despite an increase in latency on day 4, the key variable for reversal learning is the comparison between day 1 to day 2/3, particularly since the control group (WT-vehicle) reaches a plateau at this point. Therefore, this study indicates that GABAA-α5 receptors contribute to cognitive inflexibility in 15q dup mice.
A similar phenotype (i.e., normal acquisition phase and impaired reversal learning), together with increased hippocampal inhibition rescued by α5 negative allosteric modulation, was also reported by Hausrat et al [55]. The authors claimed that depletion of radixin, an actin-binding protein, increases synaptic GABAA-α5 receptor cluster sizes after redistribution from the extrasynaptic space, which was enough to impair reversal learning. Consistent with these findings, a more recent study showed that changes of GABAA-α5 receptor expression and distribution contributes to sevoflurane-induced partial long-term learning and memory impairment in young mice [56]. In 15q dup mice, the excessive expression of GABAA-α5 receptors due to the chromosomal duplication might similarly cause an abnormal distribution to the synaptic and extrasynaptic compartments which contribute to the observed phenotypes.
Individuals with Dup15q syndrome have a characteristic EEG signature, excessive beta-band oscillations [4, 5], that is distinct from both typically-developing controls and individuals with idiopathic ASD, and closely resembles the EEG pattern observed in response to treatment with benzodiazepines both in humans [17] and rats [57], and has an opposite pattern to the 15q11.2-q13.1 deletion Angelman syndrome [18]. We observed an increase of beta-power in 15q dup mice compared to WT controls, which was similar to the characteristic EEG signature seen in individuals with Dup15q syndrome [4, 5]. In line with human data, 15q dup mice EEG phenotype also closely resembles the increase in beta-power observed in response to treatment with benzodiazepines in WT mice in the current study. Treatment with RO4938581 partially restored the EEG phenotype in 15q dup mice. Given the low number of animals (n = 6) and the large overlap of confidence intervals a conclusion cannot be drawn on dose-dependence (reason for reporting the pooled analysis in the main text). In contrast to typically developing humans, where the GABAA α5-NAM, basmisanil, decreases beta-power [57], there was no effect observed in WT mice with RO4938581. This may relate to the limited sample size or different circuit dynamics and receptor expression in mice compared to humans. Unfortunately, plasma samples were not collected for the EEG study and so RO4938581 receptor occupancy could not be determined.
There are some limitations to our results and interpretation which need to be considered. Firstly, electrophysiology was only undertaken in the hippocampal region and in acute experiments and can therefore not be directly linked to observed behavioral changes and effects shown in vivo with chronic RO4938581 treatment. Secondly, only one dose of RO4938581 was assessed in the behavioral experiments. However, this dose was selected based on our experience with this compound across many behavioral paradigms and based on predicted receptor occupancy above 40%. Since we observed behavioral effects at this dose, we included a lower dose in the subsequent EEG experiment to explore a dose range. Thirdly, the EEG experiment was performed in a small group of animals, acute administration only, and plasma levels were not determined. Notably, the positive pharmacological control, i.e. diazepam, induced the expected increase in beta-power in WT mice, while RO4938581 induced the opposite effect in 15q dup mice, i.e. a decrease in beta-power, suggesting that plasma levels were in the pharmacologically relevant range in the EEG experiment.
Despite these limitations, we think that these results support clinical testing of GABAA-α5 NAMs in Dup15q syndrome. RO4938581 has been well characterized in rodents and was selected for this study because it has the most appropriate pharmacokinetic properties for in vivo assessment in mice, but not for clinical testing in humans. We have selected basmisanil [57] for testing in humans since it has an ideal profile to explore the potential clinical benefits of GABAA-α5 receptor negative modulation. Basmisanil was not tested in the 15q dup mice given its less favorable pharmacokinetic profile in rodents, in contrast to that in humans. Importantly, both compounds display similar potency, functional selectivity and intrinsic efficacy (negative allosteric modulation) [28, 57]. Basmisanil has an EEG signature in healthy volunteers which is opposite to the increase in beta-band power observed in Dup15q syndrome [57] and the 15q dup mouse model as reported here. Therefore, beta-band EEG power was selected as a translational biomarker to quantify GABAA-α5 receptor related pathophysiology and the effects of basmisanil treatment in individuals with Dup15q syndrome. A clinical trial aimed at testing the efficacy of basmisanil in this indication (NCT05307679) has unfortunately been prematurely terminated by the Sponsor for reasons independent of efficacy and safety of the compound in development. Learnings from this work will be published in another manuscript. Basmisanil has been exclusively licensed to Newleos Therapeutics for further development.
Data availability
EEG data from 15q dup mice are available from the corresponding author on reasonable request. All other data are available in the main text or the Supplementary Materials.
References
Takumi T, Tamada K. CNV biology in neurodevelopmental disorders. Curr Opin Neurobiol. 2018;48:183–92.
Finucane BM, Lusk L, Arkilo D, et al. 15q duplication syndrome and related disorders. In: Adam MP AH, Pagon RA, et al (ed). GeneReviews. Seattle: University of Washington; 2016.
Kalsner L, Chamberlain SJ. Prader-Willi, Angelman, and 15q11-q13 duplication syndromes. Pediatr Clin North Am. 2015;62:587–606.
Frohlich J, Reiter LT, Saravanapandian V, DiStefano C, Huberty S, Hyde C, et al. Mechanisms underlying the EEG biomarker in Dup15q syndrome. Mol Autism. 2019;10:29.
Urraca N, Cleary J, Brewer V, Pivnick EK, McVicar K, Thibert RL, et al. The interstitial duplication 15q11.2-q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Res. 2013;6:268–79.
Smith SE, Zhou YD, Zhang G, Jin Z, Stoppel DC, Anderson MP. Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci Transl Med. 2011;3:103ra197.
Vatsa N, Jana NR. UBE3A and its link with Autism. Front Mol Neurosci. 2018;11:448.
Boonsimma P, Suwannachote S, Phokaew C, Ittiwut C, Suphapeetiporn K, Shotelersuk V. A case of GABRA5-related developmental and epileptic encephalopathy with response to a combination of antiepileptic drugs and a GABAering agent. Brain Dev. 2020;42:546–50.
Wang L, Li J, Shuang M, Lu T, Wang Z, Zhang T, et al. Association study and mutation sequencing of genes on chromosome 15q11-q13 identified GABRG3 as a susceptibility gene for autism in Chinese Han population. Transl Psychiatry. 2018;8:152.
Ali ZA, Yasseen AA, McAllister KA, Al-Dujailli A, Al-Karaqully AJ, Jumaah AS. SNP-PCR genotyping links alterations in the GABAA receptor (GABRG3: rs208129) and RELN (rs73670) genes to autism spectrum disorder among peadiatric Iraqi Arabs. Mol Biol Rep. 2022;49:6019–28.
Coskunpinar EM, Tur S, Cevher Binici N, Yazan Songur C. Association of GABRG3, GABRB3, HTR2A gene variants with autism spectrum disorder. Gene. 2023;870:147399.
Benke D, Honer M, Michel C, Mohler H. GABAA receptor subtypes differentiated by their gamma-subunit variants: prevalence, pharmacology and subunit architecture. Neuropharmacology. 1996;35:1413–23.
Sur C, Quirk K, Dewar D, Atack J, McKernan R. Rat and human hippocampal alpha5 subunit-containing gamma-aminobutyric AcidA receptors have alpha5 beta3 gamma2 pharmacological characteristics. Mol Pharmacol. 1998;54:928–33.
Quirk K, Gillard NP, Ragan CI, Whiting PJ, McKernan RM. gamma-Aminobutyric acid type A receptors in the rat brain can contain both gamma 2 and gamma 3 subunits, but gamma 1 does not exist in combination with another gamma subunit. Mol Pharmacol. 1994;45:1061–70.
Urraca N, Hope K, Victor AK, Belgard TG, Memon R, Goorha S, et al. Significant transcriptional changes in 15q duplication but not Angelman syndrome deletion stem cell-derived neurons. Mol Autism. 2018;9:6.
Germain ND, Chen PF, Plocik AM, Glatt-Deeley H, Brown J, Fink JJ, et al. Gene expression analysis of human induced pluripotent stem cell-derived neurons carrying copy number variants of chromosome 15q11-q13.1. Mol Autism. 2014;5:44.
Greenblatt DJ, Ehrenberg BL, Gunderman J, Locniskar A, Scavone JM, Harmatz JS, et al. Pharmacokinetic and electroencephalographic study of intravenous diazepam, midazolam, and placebo. Clin Pharmacol Ther. 1989;45:356–65.
Frohlich J, Miller MT, Bird LM, Garces P, Purtell H, Hoener MC, et al. Electrophysiological phenotype in Angelman syndrome differs between genotypes. Biol Psychiatry. 2019;85:752–9.
Conant KD, Finucane B, Cleary N, Martin A, Muss C, Delany M, et al. A survey of seizures and current treatments in 15q duplication syndrome. Epilepsia. 2014;55:396–402.
Di Rocco A, Loggini A, Di Rocco M, Di Rocco P, Rossi RP, Gimelli G, et al. Paradoxical worsening of seizure activity with pregabalin in an adult with isodicentric 15 (IDIC-15) syndrome involving duplications of the GABRB3, GABRA5 and GABRG3 genes. BMC Neurol. 2013;13:43.
Tang X, Jaenisch R, Sur M. The role of GABAergic signalling in neurodevelopmental disorders. Nat Rev Neurosci. 2021;22:290–307.
Myers JF, Comley RA, Gunn RN. Quantification of [(11)C]Ro15-4513 GABA(A)α5 specific binding and regional selectivity in humans. J Cereb Blood Flow Metab. 2017;37:2137–48.
Sequeira A, Shen K, Gottlieb A, Limon A. Human brain transcriptome analysis finds region- and subject-specific expression signatures of GABA(A)R subunits. Commun Biol. 2019;2:153.
Schulz JM, Knoflach F, Hernandez MC, Bischofberger J. Dendrite-targeting interneurons control synaptic NMDA-receptor activation via nonlinear α5-GABA(A) receptors. Nat Commun. 2018;9:3576.
Jacob TC. Neurobiology and therapeutic potential of α5-GABA type A receptors. Front Mol Neurosci. 2019;12:179.
Nakatani J, Tamada K, Hatanaka F, Ise S, Ohta H, Inoue K, et al. Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell. 2009;137:1235–46.
Tamada K, Tomonaga S, Hatanaka F, Nakai N, Takao K, Miyakawa T, et al. Decreased exploratory activity in a mouse model of 15q duplication syndrome; implications for disturbance of serotonin signaling. PLoS One. 2010;5:e15126.
Ballard TM, Knoflach F, Prinssen E, Borroni E, Vivian JA, Basile J, et al. RO4938581, a novel cognitive enhancer acting at GABAA alpha5 subunit-containing receptors. Psychopharmacology. 2009;202:207–23.
Quirk K, Blurton P, Fletcher S, Leeson P, Tang F, Mellilo D, et al. 3H]L-655,708, a novel ligand selective for the benzodiazepine site of GABAA receptors which contain the alpha 5 subunit. Neuropharmacology. 1996;35:1331–5.
Crawley JN. Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol. 2007;17:448–59.
Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11:490–502.
Yang M, Crawley JN. Simple behavioral assessment of mouse olfaction. Curr Protoc Neurosci. 2009. https://doi.org/10.1002/0471142301.ns0824s48.
Tallon-Baudry C, Bertrand O, Delpuech C, Permier J. Oscillatory gamma-band (30-70 Hz) activity induced by a visual search task in humans. J Neurosci. 1997;17:722–34.
Buzsáki G, Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926–9.
Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–76.
Isshiki M, Tanaka S, Kuriu T, Tabuchi K, Takumi T, Okabe S. Enhanced synapse remodelling as a common phenotype in mouse models of autism. Nat Commun. 2014;5:4742.
Nakai N, Nagano M, Saitow F, Watanabe Y, Kawamura Y, Kawamoto A, et al. Serotonin rebalances cortical tuning and behavior linked to autism symptoms in 15q11-13 CNV mice. Sci Adv. 2017;3:e1603001.
Prenosil GA, Schneider Gasser EM, Rudolph U, Keist R, Fritschy JM, Vogt KE. Specific subtypes of GABAA receptors mediate phasic and tonic forms of inhibition in hippocampal pyramidal neurons. J Neurophysiol. 2006;96:846–57.
Zarnowska ED, Keist R, Rudolph U, Pearce RA. GABAA receptor alpha5 subunits contribute to GABAA,slow synaptic inhibition in mouse hippocampus. J Neurophysiol. 2009;101:1179–91.
Lisman J, Buzsáki G, Eichenbaum H, Nadel L, Ranganath C, Redish AD. Viewpoints: how the hippocampus contributes to memory, navigation and cognition. Nat Neurosci. 2017;20:1434–47.
Tsurugizawa T, Tamada K, Ono N, Karakawa S, Kodama Y, Debacker C, et al. Awake functional MRI detects neural circuit dysfunction in a mouse model of autism. Sci Adv. 2020;6:eaav4520.
Báez-Mendoza R, Schultz W. The role of the striatum in social behavior. Front Neurosci. 2013;7:233.
Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–8.
Noroozi R, Taheri M, Ghafouri-Fard S, Bidel Z, Omrani MD, Moghaddam AS, et al. Meta-analysis of GABRB3 gene polymorphisms and susceptibility to Autism spectrum disorder. J Mol Neurosci. 2018;65:432–7.
DeLorey TM, Sahbaie P, Hashemi E, Homanics GE, Clark JD. Gabrb3 gene deficient mice exhibit impaired social and exploratory behaviors, deficits in non-selective attention and hypoplasia of cerebellar vermal lobules: a potential model of autism spectrum disorder. Behav Brain Res. 2008;187:207–20.
Lüddens H, Seeburg PH, Korpi ER. Impact of beta and gamma variants on ligand-binding properties of gamma-aminobutyric acid type A receptors. Mol Pharmacol. 1994;45:810–4.
Menzikov SA, Morozov SG, Kubatiev AA. Intricacies of GABA(A) receptor function: the critical role of the β3 subunit in norm and pathology. Int J Mol Sci. 2021;22:1457.
Butler KM, Moody OA, Schuler E, Coryell J, Alexander JJ, Jenkins A, et al. De novo variants in GABRA2 and GABRA5 alter receptor function and contribute to early-onset epilepsy. Brain. 2018;141:2392–405.
Hernandez CC, XiangWei W, Hu N, Shen D, Shen W, Lagrange AH, et al. Altered inhibitory synapses in de novo GABRA5 and GABRA1 mutations associated with early onset epileptic encephalopathies. Brain. 2019;142:1938–54.
Absalom NL, Liao VWY, Kothur K, Indurthi DC, Bennetts B, Troedson C, et al. Gain-of-function GABRB3 variants identified in vigabatrin-hypersensitive epileptic encephalopathies. Brain Commun. 2020;2:fcaa162.
Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711.
Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–38.
Sanders J, Johnson KA, Garavan H, Gill M, Gallagher L. A review of neuropsychological and neuroimaging research in autistic spectrum disorders: attention, inhibition and cognitive flexibility. Res Autism Spectr Disord. 2008;2:1–16.
Engin E, Zarnowska ED, Benke D, Tsvetkov E, Sigal M, Keist R, et al. Tonic inhibitory control of dentate gyrus granule cells by alpha5-Containing GABAA receptors reduces memory interference. J Neurosci. 2015;35:13698–712.
Hausrat TJ, Muhia M, Gerrow K, Thomas P, Hirdes W, Tsukita S, et al. Radixin regulates synaptic GABAA receptor density and is essential for reversal learning and short-term memory. Nat Commun. 2015;6:6872.
Wang S, Wang S, Wang Z, Dong J, Zhang M, Wang Y, et al. The changing of alpha5-GABAA receptors expression and distribution participate in sevoflurane-induced learning and memory impairment in young mice. CNS Neurosci Ther. 2024;30:e14716.
Hipp JF, Knoflach F, Comley R, Ballard TM, Honer M, Trube G, et al. Basmisanil, a highly selective GABA(A)-α5 negative allosteric modulator: preclinical pharmacology and demonstration of functional target engagement in man. Sci Rep. 2021;11:7700.
Acknowledgements
We thank Grégoire Friz, Maria Karg and Judith Lengyel for molecular biology support; Marie-Claire Pflimlin and Michael Weber for electrophysiology support; Svenja Moes, Patricia Glaentzlin and Jennifer Beck for autoradiography support; Vanessa De Barros Urbano and Julie Lambotte for mouse breeding support; Theresa Ballard, Marie Haman, Roger Wyler and Audrey Genet for behavior pharmacology support; Jeremiah Palmerston, Diana Lee and Jackie DeRose for support with the EEG experiments; Andrew Thomas and Henner Knust for compound supply; Fethallah Benmansour for image analysis support; and Thomas Thelly for formulation support; Theresa M. Ballard of Galwyn (UK) Ltd. for medical writing and editorial support.
Author information
Authors and Affiliations
Contributions
TT, MS and MCH conceptualized and supervised the study. RN, FN, JH, MH, FK, RG, MS, LO, AV, LT, FF and SRM designed and performed the experiments. RN, JH, MH, FK, FF and MS analyzed the results. TT and KT provided the 15q dup mice. RN, FN, MH, JH, MS and MCH wrote the original draft. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Ethics approval
All experimental procedures were approved and performed in accordance with the City of Basel Cantonal Animal Protection Committee (AUP #2359) based on adherence to federal and local regulations on animal maintenance and testing.
Competing interests
This study was funded by F. Hoffmann-La Roche Ltd. At the time of the study, Ryoko Nakagawa, Francesca Nani, Joerg Hipp, Michael Honer, Frederic Knoflach, Rodolfo Gasser, Laurence Ozmen, Fatiha Fjeldskaar, Livia Takacs, Audrey Vautheny, Michael Saxe, Maria-Clemencia Hernandez were employees of F. Hoffmann-La Roche Ltd. Switzerland. All employees (former and current) may be eligible for stock and stock options. Kota Tamada, Toru Takumi and Stephen R. Morairty have no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nakagawa, R., Nani, F., Hipp, J.F. et al. RO4938581, a GABAA-α5 negative allosteric modulator rescued behavioral and EEG phenotypes of a mouse model of Dup15q syndrome. Mol Psychiatry 31, 1351–1360 (2026). https://doi.org/10.1038/s41380-025-03247-y
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41380-025-03247-y
