
Abstract
Background
Metastatic prostate cancer (PCa) has much lower survival and ultimately develops castration resistance, which expects novel targets and therapeutic approaches. As a result of iron-dependent lipid peroxidation, ferroptosis triggers programmed cell death and has been associated with castration-resistant prostate cancer (CRPC).
Subjects
To better understand how ferroptosis can be used to treat CRPC, we reviewed the following: First, ferroptosis mechanisms and characteristics. We then pay attention to ferroptosis effects on CRPC, and the relationship between ferroptosis and CRPC treatment. Finally, we’d like to figure out if ferroptosis could predict the prognosis of CRPC thus screening early for populations that may benefit from appropriate therapies.
Results
The review demonstrated that ferroptosis regulators like PI3K/AKT/mTOR, DECR1 et al., have a significant role in the development of CRPC and that several inducers of ferroptosis, such as erastin, BSO, RSL3, and FIN56, have already demonstrated their effects in that area. What’s more, ferroptosis is crucial for radiation-induced anticancer effects by inducing lipid peroxidation and regulating p53, AMPK, and others. Additionally, it has been discovered that certain GPX4 and SLC7A11 inhibitors can increase radiosensitivity, which brings new combination strategies. Finally, among the genes associated with ferroptosis, which may be excellent predictors of prostate cancer prognosis, several risk models have been developed and shown promising predictive capabilities.
Conclusions
Ferroptosis can serve as a potential therapeutic target for CRPC, and could be a new strategy for combination therapy. Moreover, ferroptosis-related genes may be great indicators of PCa prognosis. Further research on ferroptosis in CRPC therapy can benefit from the frameworks provided by this review.

Similar content being viewed by others


Introduction
The GLOBOCAN 2020 database reports that among male cancers, PCa ranks second in incidence and fifth in fatalities worldwide [1]. Patients’ chances of surviving PCa are proportional to the stage of the disease when it is first diagnosed. According to statistics from the US Surveillance, Epidemiology and End Results Database 2014–2020, patients with clinically localized PCa had a 5-year survival rate of nearly 100% after having standard treatment, which includes radical prostatectomy and radiation. On the other hand, patients with metastatic PCa had a much worse 5-year survival rate of 36% [2]. This is because metastatic PCa eventually develops castration resistance, and androgen deprivation therapy (ADT) has a temporal limit [3]. Until 2004, the only effective medication for improving survival was docetaxel with prednisone. Yet, the enhancement in overall survival remains somewhat restricted [4]. CRPC is still incurable, even though several treatment options have been suggested, such as androgen signaling or synthesis inhibitors (enzalutamide and abiraterone), chemotherapy based on taxanes (docetaxel-DTAX and cabazitaxel), radionuclide therapy that targets the bones (radium-223), and immunotherapy (sipuleucel-T) [5]. Hence, finding ways to enhance the survival rate of individuals with metastatic PCa is a pressing concern. Consequently, more and more research is anticipated to discover novel targets and therapeutic approaches.
One type of programmed cell death that arises from lipid peroxidation dependent on iron was first proposed by Dixon SJ in 2012. It is known as ferroptosis. In terms of both morphology and mechanism, this process differs from other forms of programmed cell death, such as necrotic apoptosis, autophagy, and apoptosis. In most cases, ferroptosis cannot be suppressed by inhibitors of other forms of programmed cell death [6]. From a morphological standpoint, ferroptosis does not exhibit any hallmarks of autophagy, such as autophagic formation, or apoptotic characteristics, such as chromatin condensation and apoptosome formation. Ferroptosis cells, on the other hand, usually have smaller mitochondria, denser mitochondrial membranes, and fewer mitochondria overall [7]. According to the scientific community, ferroptosis is a type of abnormal cell death that is not apoptotic and is linked to lipid peroxidation and an imbalance in the antioxidant system, which in turn triggers an imbalance in cellular iron metabolism. Apoptosis and this process are comparable, but they are not the same [8]. The pathophysiological processes involved in the migration and mesenchymal transformation of urological malignancies, including the PCa oncogenic pathway and CRPC treatment, are thought to be significantly impacted by ferroptosis, according to recent research [9]. Radiotherapy can enhance the prognosis for low metastatic burden patients with metastatic PCa and serve as a palliative measure. Research has recognized ferroptosis as a crucial component of the anticancer effect induced by radiotherapy [10,11,12]. However, the mechanism and specific role of ferroptosis in the irradiation of PCa cells remain unclear. It is important to investigate how ferroptosis inducers can impact patients with CRPC by potentially providing direct anti-tumor effects and enhancing tumor reduction when combined with radiotherapy.
In the following sections, we first review the current understanding of ferroptosis characteristics and mechanisms. We then discuss its impact on CRPC, including the application of ferroptosis inducers in CRPC. Subsequently, we expound on the relationship between ferroptosis and CRPC radiotherapy, which reviews the mechanisms of radiotherapy regulating ferroptosis and the ability of ferroptosis inducers to sensitize radiotherapy. Finally, we summarize ferroptosis-related genes that could be great indicators of PCa prognosis. For the future of CRPC treatment, we are excited to propose novel therapeutic targets and combination methods, as well as ideas for increasing the survival rate of CRPC patients.
Overview of ferroptosis
Mechanism of ferroptosis
Dysfunction iron metabolism and oxidation of lipids
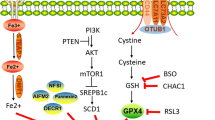
Many biochemical and metabolic activities rely on Fe2+, including energy metabolism, oxygen transport, and the cellular mitochondrial synthesis of iron-sulfur proteins. Additionally, it has a multifaceted function in oxidative stress as a cofactor [8]. Abnormalities in intracellular iron metabolism result in excess iron undergoing a Fenton reaction with hydrogen peroxide (a result of mitochondrial superoxide dismutase reacting on mitochondrial superoxide radicals). In this way, the polyunsaturated fatty acids (PUFA) undergo peroxidation. In the presence of acyl-CoA synthetase long-chain family member 4 (ACSL4), PUFA can be converted into polyunsaturated fatty acid coenzyme A (PUFA-CoA). Lysophospholipid acyltransferase 3 (LPCAT3) can further esterify, and PUFA-CoA can be integrated into membrane phospholipids [13].
The oxidation of lipids generates additional oxidized lipids and other toxic byproducts when exposed to oxygen. These harmful substances can accumulate and endanger the stability of biological membranes, ultimately resulting in cell death [7].
Imbalance in the antioxidant system
The unique ability of glutathione peroxidase 4 (GPX4), an intracellular lipid peroxidase, to reduce lipid peroxide derivatives to hydroxyl derivatives in the presence of Fe2+ makes it unique among lipid peroxidases [14]. Toxic accumulations such as lipids require glutathione (GSH) for their recycling process, which in turn relies on NADPH for recycling [15]. The Xc transporter protein system is essential for cysteine entry into cells, where it is converted into glutathione, an essential antioxidant for cells. To maintain glutathione production, a series of events leading to the exchange of extracellular cystine for intracellular glutamate occurs, and the antiporter for amino acids is comprised of solute carrier family 7 member 11 (SLC7A11) and SLC3A2 [13]. Peroxidation of lipids cellular with free iron accessible, generation of hydrogen peroxide, and polyunsaturated fatty acids can be accelerated by depriving cells of cysteine, inhibiting the Xc transporter protein system, reducing NADPH, inhibiting SLC7A11, or inhibiting GPX4 [6]. Uncontrolled lipid peroxidation, as described through cascade amplification, ultimately results in ferroptosis [15]. However, other therapeutic targets can protect cells from ferroptosis, such as iron chelation, hydrogen or lipid peroxide scavenging, and the suppression of polyunsaturated fatty acid production [15]. The mechanism of ferroptosis and several of its inducers can be seen in Fig. 1.

Schematic diagram of ferroptosis and its inducers mechanism.
Ferroptosis characteristics
When compared to other types of programmed cell death, including necrotic apoptosis, autophagy, and apoptosis, ferroptosis cells exhibit several unique morphological and biochemical features. These characteristics include changes in the cellular ultrastructure, such as an unbroken nucleus, an increase in mitochondrial density, a decrease in mitochondrial volume and cristae, and an interruption of the outer mitochondrial membrane [6, 16]. Lipid peroxidation, elevated ferric ions, the buildup of intracellular reactive oxygen species (ROS), reduction of GSH, decreased GPX4 activity/levels, and decreased SLC7A11 production are biochemical indicators of this process [16]. Iron response element-binding protein 2 (IREB2), citrate synthase (CS), ATP synthase F0 complex subunit C3 (ATP5G3), acyl-CoA synthetase family member 2 (ACSF2), tetratricopeptide repeat domain 35 (TTC35), and ribosomal protein L8 (RPL8) are the six genes that, at the molecular level, are upregulated during ferroptosis [6].
Ferroptosis can serve as a potential new therapeutic target for CRPC
Negative regulators of ferroptosis promote CRPC progression
Activation of PI3K/AKT/mTOR leads to the development of CRPC by inhibiting ferroptosis
The PI3K/AKT pathway is engaged in 70% of advanced PCa, and PTEN loss happens often with human PCa development [17]. The androgen receptor (AR) transcription factor activity is regulated by PTEN loss, which in turn inhibits androgen-responsive gene expressions. By reducing the expression of the androgen-responsive gene Fkbp5 and blocking PHLPP-mediated AKT inhibition, conditional AR deletion in the epithelium increases the proliferation of PTEN null cancer cells. Possible significant implications for PCa pathogenesis and therapy were highlighted by Mulholland et al., who found PI3K and AR pathway crosstalk as a mechanism of CRPC formation [18].
As a downstream player of the PI3K/AKT signaling cascade, mTOR regulates biological processes in tumors. High levels of cholesteryl ester have been associated with an increased risk of PCa in multiple investigations [19]. Yue et al. demonstrated that stimulation of the PI3K/AKT/mTOR pathway can generate the buildup of cholesteryl ester, which supports PCa progression [20]. Yi et al. found that PCa cells could activate downstream sterol regulatory element-binding protein 1 (SREBP1) and its transcriptional target stearoyl-CoA desaturase-1 (SCD1) through the PI3K/AKT/mTORC1 pathway to mediate monounsaturated fatty acids production. What’s more, mTORC1 can mediate nuclear factor erythroid 2-related factor 2 (NRF2) signaling, acting as an important regulator of cellular redox homeostasis. Finally prevents lipids from being peroxidized, thus preventing ferroptosis in cancer cells [21]. To sum up, mTORC1 is an antioxidant and a negative regulator of ferroptosis; it also inhibits ferroptosis in PCa cells by interfering with lipid metabolism.
In a nutshell, increased cell proliferation and castration resistance are all outcomes of PTEN loss or activation of the PI3K/AKT/mTOR pathway.
Overexpression of DECR1 inhibits CRPC cells from ferroptosis
The rate-limiting enzyme in PUFA oxidation, 2,4-Dienoyl-CoA reductase 1 (DECR1), is shown to be elevated in prostate tumor samples and castration-resistant mice models [22]. Decreased DECR1 expression significantly inhibits PCa cell growth through PUFA buildup, heightened lipid peroxidation vulnerability, and ferroptosis induction, according to recent studies [23]. It is intriguing that temporarily repressing the DECR1 gene improved the inhibitory impact of enzalutamide on PCa cells. On the flip side, temporary overexpression of the DECR1 gene boosted cancer cell proliferation. Additionally, CRPC cells become more susceptible to ferroptosis when DECR1 is downregulated which raises GPX4 expression [22]. In addition, it is highly important for prognosis prediction that increased DECR1 expression is associated with shorter disease-free survival in PCa patients [23].
PANX2 promotes PCa progression via suppressing ferroptosis
Pannexin 2 (PANX2) is a protein that forms a cell-specific channel and is mostly found in postnatal neural precursor cells and mature neurons. A study discovered that PANX2 expression levels were notably increased in PCa cells and showed a favorable correlation with the Gleason score in PCa [24]. In this study, disrupting PANX2 expression notably raised Fe2+ levels in cancer cells. PCa cells can upregulate iron metabolism via PANX2, enabling them to evade ferroptosis, promoting the progression of PCa. On the flip side, PCa cells are unable to proliferate, migrate, or invade when PANX2 is silenced. The study found that PANX2 may promote the growth and progression of PCa cells by controlling NRF2, a significant controller of intracellular oxidative balance and effectively prevents ferroptosis by collaborating with the antioxidant transcription factor STAT3 to enhance SLC7A11 expression [25]. PANX2 functions as a suppressor of ferroptosis by specifically affecting iron metabolism and promoting the progression of PCa through the up-regulation of SLC7A11 levels.
Up-regulation of SLC7A11 may lead to docetaxel-resisitance
The inhibition of ferroptosis and the acceleration of PCa progression may result from up-regulating the levels of SLC7A11. Zhang et al. found that long non-coding RNA OIP5-AS1 expression was greatly elevated in PC3 and DU145 cells upon chronic Cd exposure, which targets and enhances the miR-128-3p/SLC7A11 signaling, and finally promotes cell growth and suppresses ferroptosis. As a result, PCa cells migrate and form colonies [26]. In addition, Jiang X and colleagues [27] observed that lncRNA-PCAT1 competes for miR-25-3p, which increases the stability of c-Myc through physical interactions. This transcriptionally activates SLC7A11 expression, leading to an increase in docetaxel resistance, which is recommended to be used for CRPC. Furthermore, lncRNA-PCAT1 knockdown promotes ferroptosis, effectively impairing docetaxel resistance.
HSPB1 may inhibit erastin-induced ferroptosis of PCa
As a member of heat shock protein family B, HSPB1 can degrade unfolded or misfolded proteins, which it uses to protect cells from death [28]. The upregulation of HSPB1, which has been linked to extremely aggressive cancer and bad clinical results, suggests that it is an important regulator of PCa cell activity [29]. Sun et al. showed that erastin-induced ferroptosis was greatly amplified in PCa cell lines when HSPB1 was knocked down, but inhibited when HSPB1 was overexpressed. Phosphorylation of HSPB1 by protein kinase C could prevent cancer cell ferroptosis by blocking cytoskeletal-mediated iron absorption and lipid ROS generation [30]. To sum up, HSPB1 suppresses erastin-induced PCa ferroptosis and acts as a negative regulator of ferroptosis.
Enhancing the expression of negative regulators of ferroptosis may be linked to PCa invasion and progression, as well as the development and progression of CRPC. Conversely, it may enhance the sensitivity of PCa cells to ferroptosis, thus impeding disease advancement. Ferroptosis inducers show significant potential as a novel target for treating CRPC sufferers.
Positive regulators of ferroptosis inhibit CRPC progression
Inducing ferroptosis by critical down-regulation of SLC7A11 and GPX4 expression slows the growth of PCa. As an illustration, research shows that the amount of glutathione-specific γ-glutamylcyclotransferase 1 (CHAC1) is related to the viability of PCa cells and the levels of GSH [31]. The ferroptosis activator induced a significant up-regulation in the expression level of CHAC1, which led to lower GPX4 levels and higher intracellular lipid peroxides. The process progressed to ferroptosis, which rendered the PCa cells more sensitive to docetaxel and reduced their viability. CHAC1 is indicated to be a promoter of ferroptosis. The correlation between the expression level of CHAC1 and endoplasmic reticulum stress is known, but the connection between CHAC1, endoplasmic reticulum stress, and ferroptosis has not been extensively researched. Investigating this relationship could provide more insight into the mechanism of ferroptosis.
Positive regulators of ferroptosis increase the sensitivity of PCa cells to docetaxel, indicating that triggering ferroptosis could be a promising approach for treating CRPC.
Ferroptosis Inducers (FIN) and CRPC
Metastatic PCa progresses to castration resistance, endocrine therapy is not very effective, and docetaxel plus prednisone provides minimal improvement in overall survival [4]. Hence, there is a pressing requirement for innovative CRPC treatment medications. The analysis of the aforementioned data indicates that ferroptosis inducers could be a promising therapy approach.
Class I FIN: Inhibits Xc transport system [32], depletes GSH
The Xc transporter impacts cell viability by promoting nutrient absorption at the plasma membrane. Its inhibitors decrease cystine uptake and hinder protein folding. Accumulation of misfolded proteins results in endoplasmic reticulum stress, which triggers ferroptosis [33, 34]. For instance, Erastin reduces glutathione (GSH) levels by interrupting Xc function and, in addition, obstructs mitochondrial voltage-dependent anion channels and fosters ferroptosis in RAS-positive cells (so named because of the first identification in rat sarcoma virus). According to constitutive relationship studies, erastin’s effectiveness is conditional on the quinazolinone moiety’s integrity, the piperazine linker’s stiffness, and the chlorine atom’s existence [34]. In addition, Ghoochani et al. discovered that AR-expressing cells showed enhanced anticancer activity of enzalutamide and abiraterone when treated with erastin [35]. Furthermore, Yang et al. discovered an additional erastin mechanism for the treatment of CRPC. Expression of GPX4, AR and its splice variants, and their transcriptional activity were all downregulated in erastin-exposed cells. Together, ferroptosis inhibitors (ferrostatin-1 and liproxstatin-1) and these results (e.g., increased ROS and reduced GSH) reverted the effects of decreased proliferation, cell cycle arrest, and induction of Ferroptosis. Erastin slowed the progression of 22Rv1 xenograft tumors when administered alone, and when combined with DTAX, it enhanced the anticancer drug’s growth inhibitory effect [36].
Furthermore, in addition to using iron or Buthionine Sulphoximine (BSO) alone, combining AR antagonists with it is another approach that is successful in CRPC. Qin and colleagues administered an isothiocyanate-modified AR antagonist to PCa cells with BSO. The drug combination caused notable lipid peroxidation, both AR/AR-V7 were downregulated and further ferroptosis was induced as a consequence of the combination’s synergistic action. Agents that antagonize iron (such as antioxidants and iron chelators) were able to suppress this cellular response. Combining BSO-mediated inhibition of GSH-defense mechanisms with enhanced availability of modified-AR antagonists to AR likely resulted in lower AR levels [37].
But erastin’s pharmacological efficacy is constrained by its poor bioavailability and solubility, whereas the application of its two derivatives, piperazine erastin and imidazole ketone erastin (IKE), may improve its pharmacological activity [38, 39]. The mechanism of action of sulfasalazine is akin to that of Erastin but with significantly lower potency [40]. Also, by blocking the Xc transporter system, the kinase inhibitor sorafenib causes ferroptosis. This drug is now used to treat thyroid, hepatocellular, and renal cell carcinomas. The molecular mechanism behind this effect, however, is not fully understood. As noted by Dixon et al. [34], therefore, sorafenib may cause ferroptosis by blocking the kinases that control Xc activity or by blocking components that are structurally unconnected to the kinases that they target. However, additional research is needed to determine if these medications are effective in treating CRPC.
Class II FIN: Direct inhibition of GPX4 activity via covalent binding
To prevent ferroptosis, GPX4 expression levels must remain normal. GSH plays a key role in this process by transforming dangerous lipid ROS into safe lipid alcohols [41]. RSL3 inhibits GPX4 explicitly, with the maximum activity observed in (1S,3 R)-RSL3, one of four diastereoisomers. Chemical proteomics studies have found that the drug’s activity is primarily attributed to the chloroacetamide portion. This compound inhibits GPX4 through covalent alkylation of selenocysteine and could potentially demonstrate a preference for cells expressing HRAS because of its affinity for HRAS pathway proteins [38, 41].
Restraining enzalutamide exposure over time decreased cell growth, increased tolerance to several drugs, and accumulated lipids, according to Tousignant and colleagues. The cells become resistant to ADT but sensitive to ferroptosis induced by RSL3 because of an increase in GPX4 dependence, which in turn causes an increase in PUFA levels, increased membrane fluidity, and lipid peroxidation [42]. Evidence suggests that RSL3 might work against CRPC.
Class III FIN: Depletion of coenzyme Q and degradation of GPX4
FIN56, a derivative of CIL56, induces GPX4 degradation in response to activation of acetyl-CoA carboxylase (ACC) and squalene synthase (SQS) through a mechanism of action different from cysteine asparaginase activation, relying on oxime and piperidine residues [43, 44]. The antitumor activity in vitro and in a xenograft mouse model in vivo was further enhanced when DECR1-directed siRNA, which induced selective DECR1 silencing, was combined with FIN56 [23]. This suggests that inhibitors of negative regulators of ferroptosis and inducers of ferroptosis may work together to treat CRPC.
Class IV FIN: Oxidation of iron and polyunsaturated fatty acids [45]
A 1,2-dioxolane ring is present in FINO2, making it a potent inducer of ferroptosis. Investigations into the composition of FINO2 have identified (-)-FINO2 as the most active enantiomer, with its drug activity attributed to polar residues near the peroxide ring. The drug induces ferroptosis by directly oxidizing intracellular iron pools and inactivating GPX4 [46].
Ferroptocide (FPT) is an active small molecule natural product modifier capable of inducing ferroptosis by covalently modifying the active site of thioredoxin (TNX), thereby interfering with the cellular antioxidant system and altering lipid peroxidation and redox status. According to constitutive relationship assays, the presence of the electrophilic alpha-chloroester group is necessary for FPT to have biological action [47]. Artemisinins (ART) are a derivative of sesquiterpene lactones with antimalarial and antitumor activities [48]. Recent research has indicated that the anticancer mechanisms of these derivatives may relate to ferroptosis, suggesting that artemisinins are becoming increasingly applicable in broader fields. One example is the ability of artesunate to display important ferroptosis-inducing activity in anti-apoptotic KRAS-transformed cancer cells [49], while dihydroartemisinin promotes ferritin autophagic degradation, leading to ferritin down-regulation and ferroptosis [50]. Furthermore, we anticipate that future studies will explore if these drugs have value in the treatment of CRPC. The ferroptosis inducers that have been shown to affect CRPC are listed in Table 1.
Ferroptosis could be a new strategy for CRPC combination therapy
Multiple forms of DNA damage, including base damage, single-strand breaks, and double-strand breaks, can be directly caused by ionizing radiation [51], Additionally, it can radioactively break down water molecules within cells and trigger oxidative enzymes to release highly reactive oxygen and hydrogen peroxide radicals, among other ROS, which can impact proteins, lipids, and nucleic acids in a dose-dependent manner [52]. Tumor cells are subjected to detrimental biological processes such as cellular senescence, apoptosis, and cell cycle arrest as a result of both direct and indirect influences, which aid in the eradication of cancer cells. Additional investigation into the functions and processes of alternative types of programmed cell death in radiation treatment is necessary [53].
Radiotherapy (RT) plays an important role in the treatment of PCa, it is an increasingly common treatment for localized PCa, especially in males aged 65 and up [54]. In cases of CRPC, particularly in individuals with metastatic disease, the National Comprehensive Cancer Network (NCCN) recommends radiotherapy as a local treatment for painful bone metastases [55]. New evidence suggests that ferroptosis is essential for radiation-induced anticancer effects [11, 12], which could be a potential target in improving radiosensitivity, identifying possible indicators of RT effectiveness, and investigating the role of RT in conjunction with other medicinal approaches.
Ferroptosis may be a possible indicator of RT effectiveness
Radiotherapy can induce lipid peroxidation to promote ferroptosis
Ionizing radiation activates ferroptosis through three parallel pathways that result in characteristic lipid peroxidation. Fatty acid radicals (PUFA-) are formed in the first pathway when ionizing radiation removes electrons from polyunsaturated fatty acids, leading to the production of reactive oxygen species. Lipid peroxyl radicals (PUFA-OO-) are quickly produced when unstable carbon-centered radicals react with molecular oxygen. These radicals then undergo the Fenton reaction and can remove hydrogen from other molecules, resulting in lipid peroxide (PUFA-OOH) [56]. Furthermore, ionizing radiation enhances ACSL4 expression, an enzyme critical for lipid metabolism and the synthesis of phospholipids containing polyunsaturated fatty acids (PUFA-PLs), a peroxidation-prone lipid type. Inhibition of ferroptosis and ionizing radiation-induced lipid peroxidation is prevented by ACSL4 deficiency [57]. Evidence suggests that ionizing radiation can enhance PUFA-PL production via ACSL4 expression. This, in turn, can cause PUFA-PL peroxidation (PUFA-PL-OOH) and ferroptosis when coupled with ROS generated by ionizing radiation [11]. However, it is still not clear how ionizing radiation triggers ACSL4 expression. It is unclear whether transcription factors or chromatin-modifying enzymes influence ACSL4 expression, which in turn controls radiation reaction and ferroptosis. For example, p53 [58] and BRCA1-associated protein-1 (BAP1) [59, 60] are potential candidates. Furthermore, radiation can induce ferroptosis by depleting glutathione (GSH) and suppressing GPX4-mediated defenses against ferroptosis. Finally, SLC7A11 expression can be increased as an adaptive response to prevent ferroptosis or downregulated in an ATM-dependent way to enhance ferroptosis [61].
Radiotherapy can induce the activation of p53 to regulate ferroptosis
Furthermore, various studies have suggested that radiotherapy-induced ferroptosis may have additional molecular mechanisms. For instance, research has shown that p53 promotes ferroptosis through multiple pathways. According to research by Wang Y and colleagues, p53 can either interact with ubiquitin-specific protease 7 or directly bind to the p53 response element in the SLC7A11 promoter region, both of which decrease the SLC7A7 level [62]. This reduces the level of mono-ubiquitylation of H11B in the regulatory region of the SLC2A7 gene, this triggers ferroptosis as a defense mechanism against oxidative damage. Ou et al. discovered that in response to ROS stress, ferroptosis is promoted by inducing the production of spermidine/spermine N1-acetyltransferase 1 (SAT1), which overrides ALOX15 [63]. Chu et al. discovered that ferroptosis is enhanced in an ALOX12-dependent way through p53-mediated regulation of SLC7A11 [64]. However, p53 can prevent ferroptosis in two ways: first, by transcriptionally inhibiting dipeptidyl peptidase-4 (DPP4) activity [65]; second, by increasing p21 to preserve GSH levels during stress from metabolism [66]. Given p53’s environment-dependent role in ferroptosis regulation, additional research is required to ascertain if radiotherapy-induced activation of p53 enhances or hinders ferroptosis.
Radiotherapy can activate AMPK to regulate ferroptosis
Ferroptosis and AMPK are both closely related to cellular metabolism, which indicates that AMPK may be a potential signaling node for radiotherapy-induced ferroptosis. Evidence suggests that radiotherapy can activate AMPK [67, 68], environment also plays a role in how its activation regulates ferroptosis. Inhibition of the Xc transport system by AMPK-phosphorylated beclin-1, according to studies, promotes ferroptosis [69]. Energy stress-induced AMPK activation, on the other hand, inhibits ferroptosis by reducing PUFA-PL production [70, 71]. So, to find out how AMPK causes radiotherapy-induced ferroptosis and what role it plays, additional research is needed.
Radiotherapy can induce the expression of MDM2 to promote ferroptosis
Expression of the mouse double minute 2 homolog (MDM2) gene is induced by radiation exposure in a way that is reliant on either ATM or p53 [72, 73]. Venkatesh and colleagues [74] have shown that MDM2 is involved in radiotherapy-induced ferroptosis because it promotes ferroptosis by controlling lipid metabolism and the production of ferroptosis suppressor protein 1 (FSP1).
Radiotherapy can inhibit the GCH1-BH4 signaling to promote ferroptosis
Furthermore, the GCH1(guanosine triphosphate cyclohydrolase 1)-BH4(tetrahydrobiopterin) signaling axis serves as a GPX4-independent system that inhibits ferroptosis [75]. BH4, which is an effective antioxidant that reduces lipid peroxidation and ferroptosis by capturing free radicals in the membranes of cells, encourages the regeneration of CoQH2 and α-tocopherol [75, 76]. Dihydrofolate reductase (DHFR) converts boron dihydride (BH2) into BH4. Cells become far more vulnerable to ferroptosis when DHFR is rendered inactive [75]. Ionizing radiation reduces BH4 levels and bioavailability in vivo. The activation of GCH1 feedback-regulatory proteins (GFRP) could be the reason behind this, as radiation amplifies the inhibition of GCH1 activation that is mediated by GFRP [77, 78]. These findings lend credence to the idea that radiation treatment has the potential to regulate ferroptosis by blocking the GCH1-BH4 signaling.
Taken together, radiotherapy may regulate ferroptosis and thus exert antitumor effects through a variety of mechanisms (seen in Fig. 2), suggesting that all of these molecules may serve as markers for predicting the effectiveness of radiotherapy for CRPC. Furthermore, induction of ferroptosis by genetic or pharmacological approaches has also proved important in treating some types of cancer [45, 79]. Inducing ferroptosis may enhance the elimination of cancer cells with radiotherapy when combined with ADT, chemotherapy, immunotherapy, or targeted therapy. Hence, additional research is needed to determine the function of RT in combination with other pharmaceutical methods and to see if this combination strategy heightens normal tissue toxicity.

Radiotherapy-associated ferroptosis regulators and signaling pathways.
Ferroptosis could be a potential target in improving radiosensitivity
Radiotherapy is beneficial in relieving pain in patients with limited metastatic disease in the vertebra or paravertebral region [80]. The overall response rate for pain was approximately 58%, indicating a need for improvement. A randomized controlled trial comparing ADT to ADT combined with external beam radiation therapy for primary bone metastatic PCa patients did not demonstrate a meaningful difference in overall survival (OS) [81]. Further research is required to validate these results. Several studies have demonstrated that radiation can enhance progression-free survival (PFS) [82] and OS [83] in patients with oligometastatic progression. However, the enhancement remains constrained. The results suggest that enhancing the sensitivity and efficacy of radiation is a crucial issue in treating metastatic CRPC.
The use of ferroptosis inducers may increase radiation sensitivity, according to several recent studies. For instance, in vitro studies have demonstrated that non-small cell lung cancer (NSCLC) cells can be rendered more radiosensitive by certain FINs, including class I FINs (which target SLC7A11, erastin, and salazosulfapyridine), class II FINs (which target GPX4, RSL3, and ML162), and class III FINs (which deplete coenzyme Q and degrade GPX4, such as FIN56) [11].
SLC7A11 inhibitors may safely improve radiosensitivity
When administered to xenografts derived from cell lines (CDX) and patients (PDX), salazosulfapyridine significantly reduced the radiosensitivity of ovarian and KEAP1 mutant lung cancers [10, 11]. Another study showed that radiotherapy in combination with IKE or sorafenib in both CDX and PDX is effective for tumor suppression [12]. These results indicate that in vivo, the use of class I FINs in conjunction with radiation is well-tolerated, and could be a promising treatment option for cancer patients. Furthermore, SLC7A11 inhibitors are quite safe, as SLC7A11-deficient animals did not show any noticeable phenotype [84, 85].
GPX4 inhibitors may have chance to improve radiosensitivity
Buthionine Sulphoximine (BSO) has been shown to inhibit GSH synthesis, which sensitizes cancer cells to radiotherapy [86, 87]. One possible way to make cancer cells more sensitive to radiation is to suppress GPX4, which is essential for the survival of drug-resistant cancer cells [88]. In vitro, RSL-3, ML162, and FIN-56 exhibit strong radiosensitizing properties [10,11,12], but suboptimal pharmacokinetics make them unsuitable for in vivo treatment [88]. Cancer therapy medicines with FDA approval, such as withaferin A and altretamine, both inhibit GPX4. These drugs show promise as anticancer agents in animal studies and offer another way to target GPX4 in living organisms [89, 90]. A key point to keep in mind is that GPX4 knockout animals die during embryonic development, which brings up questions regarding the safety of GPX4 inhibitors as in vivo treatments [41, 91]. But GPX4 inhibitors seem to work better on clear cell tumors than normal cells [92], perhaps there is a chance to target GPX4 in certain malignancies, which requires further investigation.
These findings offer potential strategies to improve the effectiveness of radiation therapy in treating metastatic CRPC, including increasing the overall response rate for pain or OS in bone metastatic patients and enhancing PFS and OS in oligometastatic progression. It should be noted that ferroptosis may occur alongside typical radiation-induced tissue damage, such as lung injury [93, 94]. Therefore, finding out if radiation and FIN can operate together ideally is of the utmost importance. Additionally, the function of ferroptosis in X-ray radiotherapy—photons with non-linear energy transfer—has been the subject of recent studies. Future studies should focus on ferroptosis and its possible role in proton therapy and other high-linear-energy-transfer radiations such as carbon ion radiotherapy. Table 2 displays ferroptosis inducers that can act as radiosensitizers in vitro or in vivo.
Ferroptosis-related genes may be great indicators of PCa prognosis
Several diseases have been identified as ferroptosis-associated genes, which could potentially serve as biomarkers for disease diagnosis (such as acute myocardial infarction [95], smoking-associated chronic obstructive pulmonary disease [96], and Alzheimer’s disease [97]) or to predict tumor prognosis (such as leukemia [98], prostate cancer [99], hepatocellular carcinoma [100], papillary cell carcinoma of the kidney [101], and colorectal carcinoma [102]).
For example, a multifunctional enzyme known as aldo-keto reductase family 1 member C3 (AKR1C3) plays a key role in the metabolism of steroidal compounds in living organisms. It is also known as type 5 17β-hydroxysteroid dehydrogenase or prostaglandin F2α synthase and is one of the four isoforms of enzymes in subfamily 1C (AKR1C) of the AKR superfamily, which includes AKR1C1, AKR1C2, AKR1C3, and AKR1C4 [103].
It was discovered that AKR1C3 expression levels are present in a range of hormone-dependent, hormone-independent, metabolic, solid, and hematological tumors. There were more cases of cancer than of normal cells. These cases included a variety of cancers, including esophageal cancer [104], prostate cancer [105], endometrial carcinoma [106], breast carcinoma [107], tumors of the central nervous system [108], leukemia [109], and renal cell carcinoma, gastric carcinoma, pancreatic carcinoma, cervical carcinoma, colon carcinoma, rectal carcinoma, non-small-cell lung carcinoma, hepatocellular carcinoma, bladder carcinoma. The up-regulation of this gene is strongly linked to other cancer-related outcomes, like tumor cell differentiation [110], invasion [111], proliferation [112], metastasis [113], poor patient prognosis, and lower survival [114]. It also leads to tumor cell resistance to radiotherapy [111, 115, 116] and chemotherapy [117]/immunotherapy [118], resulting in poor patient prognosis and lower survival. Localized and progressed prostate adenocarcinomas are characterized by an upregulation of AKR1C3 [119]. When it is overexpressed, PCa is more likely to form and spread aggressively [114]. In addition, it can cause the buildup of prostaglandin F2α, which can accelerate the development of PCa cells and increase their resistance to radiation. Nevertheless, tumor cells regain their radiation sensitivity when AKR1C3 is inhibited. AKR1C3 is involved in every step to androgens of PCa progression and has been linked to CRPC [120].
Furthermore, Liu et al. examined seven genes related to ferroptosis: AKR1C3, ALOXE3, ATP5MC3, CARS1, MT1G, PTGS2, and TFRC to create a risk model for PCa. This model was proven to accurately predict prognosis when verified in the MSKCC dataset [99]. These results point to the possibility that genes associated with ferroptosis could be useful as biological indicators for prognosis and as therapeutic targets. And by utilizing the UCSC XENA database, a ferroptosis-related gene prognostic index (FGPI) was developed by Shi et al. as a research tool [121]. To determine whether PCa patients who had radical radiation would survive without metastases, researchers screened two genes, ACSL3 and ACTC1, using LASSO and Cox regression models. Biochemical recurrence (BCR) and metastasis in PCa patients were successfully predicted by the FGPI, which is based on ACSL3 and ACTC1.
Ferroptosis-related genes like AKR1C3 may serve as valuable prognostic indicators for PCa and are strongly associated with CRPC, making them possible biomarkers for PCa therapy. Several risk models of Pca have been developed and demonstrated strong predictive capability.
Conclusions
Recent studies show that ferroptosis regulators like PI3K/AKT/mTOR, DECR1, PANX2, SLC7A11, HSPB1, CHAC1 plays an important role in CRPC development, which implies that ferroptosis inducers could be a promising therapy approach. Class I FIN (erastin and BSO), Class II FIN (RSL3), and Class III (FIN56) have already shown effect in CRPC. In addition, radiotherapy serves as a significant local treatment for painful bone metastases of CRPC and oligometastatic progression, but the enhancement remains constrained. Ferroptosis is essential for radiation-induced anticancer effects by inducing lipid peroxidation and regulating p53, AMPK, MDM2, and GCH1-BH4 signaling. Several SLC7A11 inhibitors and GPX4 inhibitors have been found to have the capability of improving radiosensitivity. Moreover, several risk models of PCa based on ferroptosis-related genes have been developed and demonstrated great predictive capability. In conclusion, ferroptosis shows hopeful potential in the treatment of CRPC, but further clinical studies are required to evaluate whether ferroptosis inducers in combination with ADT produce a survival benefit in CRPC patients compared with ADT alone and whether the genes associated with ferroptosis can achieve risk stratification of patients to select the targeted therapeutic modalities, or to guide the application of ferroptosis inducers based on predictors of ferroptosis that are sensitive to therapeutic modalities or not.
Data availability
The data presented in this study are available in this manuscript.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca Cancer J Clin. 2021;71:209–49.
The Surveillance, Epidemiology, and End Results (SEER) Program. 2024. Available from: https://seer.cancer.gov/canques/survival.html.
Loblaw DA, Virgo KS, Nam R, Somerfield MR, Ben-Josef E, Mendelson DS, et al. Initial hormonal management of androgen-sensitive metastatic, recurrent, or progressive prostate cancer: 2006 update of an American Society of Clinical Oncology practice guideline. J Clin Oncol. 2007;25:1596–605.
Petrylak DP, Tangen CM, Hussain MH, Lara PN Jr., Jones JA, Taplin ME, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–20.
Nuhn P, De Bono JS, Fizazi K, Freedland SJ, Grilli M, Kantoff PW, et al. Update on Systemic Prostate Cancer Therapies: Management of Metastatic Castration-resistant Prostate Cancer in the Era of Precision Oncology. Eur Urol. 2019;75:88–99.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell. 2012;149:1060–72.
Stockwell BR, Angeli JPF, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171:273–85.
Tang DL, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–25.
Zhou X, Zou L, Liao H, Luo J, Yang T, Wu J, et al. Abrogation of HnRNP L enhances anti-PD-1 therapy efficacy via diminishing PD-L1 and promoting CD8+ T cell-mediated ferroptosis in castration-resistant prostate cancer. Acta Pharmaceutica Sin B. 2022;12:692–707.
Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019;9:1673–85.
Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30:146–62.
Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, et al. Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem Biol. 2020;15:469–84.
Schoeler M, Caesar R. Dietary lipids, gut microbiota and lipid metabolism. Rev Endocr Metab Dis. 2019;20:461–72.
Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic Biol Med. 2020;152:175–85.
Green DR. The Coming Decade of Cell Death Research: Five Riddles. Cell. 2019;177:1094–107.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–79.
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22.
Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792–804.
Peck B, Schulze A. Cholesteryl esters: fueling the fury of prostate cancer. Cell Metab. 2014;19:350–2.
Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014;19:393–406.
Yi J, Zhu J, Wu J, Thompson CB, Jiang X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci USA. 2020;117:31189–97.
Blomme A, Ford CA, Mui E, Patel R, Ntala C, Jamieson LE, et al. 2,4-dienoyl-CoA reductase regulates lipid homeostasis in treatment-resistant prostate cancer. Nat Commun. 2020;11:2508.
Nassar ZD, Mah CY, Dehairs J, Burvenich IJ, Irani S, Centenera MM, et al. Human DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis. Elife. 2020;9:e54166.
Liao D, Yang G, Yang Y, Tang X, Huang H, Shao J, et al. Identification of Pannexin 2 as a Novel Marker Correlating with Ferroptosis and Malignant Phenotypes of Prostate Cancer Cells. Onco Targets Ther. 2020;13:4411–21.
Qiang Z, Dong H, Xia Y, Chai D, Hu R, Jiang H. Nrf2 and STAT3 Alleviates Ferroptosis-Mediated IIR-ALI by Regulating SLC7A11. Oxid Med Cell Longev. 2020;2020:5146982.
Zhang Y, Guo S, Wang S, Li X, Hou D, Li H, et al. LncRNA OIP5-AS1 inhibits ferroptosis in prostate cancer with long-term cadmium exposure through miR-128-3p/SLC7A11 signaling. Ecotoxicol Environ Safety. 2021;220:112376.
Jiang X, Guo S, Xu M, Ma B, Liu R, Xu Y, et al. TFAP2C-Mediated lncRNA PCAT1 Inhibits Ferroptosis in Docetaxel-Resistant Prostate Cancer Through c-Myc/miR-25-3p/SLC7A11 Signaling. Front Oncol. 2022;12:862015.
Okuno M, Adachi S, Kozawa O, Shimizu M, Yasuda I. The Clinical Significance of Phosphorylated Heat Shock Protein 27 (HSPB1) in Pancreatic Cancer. Int J Mol Sci. 2016;17:137.
Vasiljevic N, Ahmad AS, Beesley C, Thorat MA, Fisher G, Berney DM, et al. Association between DNA methylation of HSPB1 and death in low Gleason score prostate cancer. Prostate Cancer Prostatic Dis. 2013;16:35–40.
Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. 2015;34:5617–25.
He S, Zhang M, Ye Y, Zhuang J, Ma X, Song Y, et al. ChaC glutathione specific γ‑glutamylcyclotransferase 1 inhibits cell viability and increases the sensitivity of prostate cancer cells to docetaxel by inducing endoplasmic reticulum stress and ferroptosis. Exper Therapeutic Med. 2021;22:997.
Zaffaroni N, Beretta GL. Ferroptosis Inducers for Prostate Cancer Therapy. Curr Medicinal Chem. 2022;29:4185–201.
Lu B, Chen XB, Ying MD, He QJ, Cao J, Yang B. The Role of Ferroptosis in Cancer Development and Treatment Response. Front Pharmacol. 2018;8:992.
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e02523.
Ghoochani A, Hsu EC, Aslan M, Rice MA, Nguyen HM, Brooks JD, et al. Ferroptosis Inducers Are a Novel Therapeutic Approach for Advanced Prostate Cancer. Cancer Res. 2021;81:1583–94.
Yang Y, Liu T, Hu C, Xia H, Liu W, Chen J, et al. Ferroptosis inducer erastin downregulates androgen receptor and its splice variants in castration‑resistant prostate cancer. Oncol Rep. 2021;45:25.
Qin Z, Ou S, Xu L, Sorensen K, Zhang Y, Hu DP, et al. Design and synthesis of isothiocyanate-containing hybrid androgen receptor (AR) antagonist to downregulate AR and induce ferroptosis in GSH-Deficient prostate cancer cells. Chem Biol Drug Des. 2021;97:1059–78.
Yang WanS, SriRamaratnam R, Welsch MatthewE, Shimada K, Skouta R, Viswanathan VasanthiS, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell. 2014;156:317–31.
Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol. 2019;26:623–33.e9.
Sehm T, Fan Z, Ghoochani A, Rauh M, Engelhorn T, Minakaki G, et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget. 2016;7:36021–33.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–91.
Tousignant KD, Rockstroh A, Poad BLJ, Talebi A, Young RSE, Taherian Fard A, et al. Therapy-induced lipid uptake and remodeling underpin ferroptosis hypersensitivity in prostate cancer. Cancer Metab. 2020;8:11.
Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497–503.
Cotto-Rios XM, Gavathiotis E. Unraveling cell death mysteries. Nat Chem Biol. 2016;12:470–1.
Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell. 2019;35:830–49.
Abrams RP, Carroll WL, Woerpel KA. Five-Membered Ring Peroxide Selectively Initiates Ferroptosis in Cancer Cells. Acs Chem Biol. 2016;11:1305–12.
Llabani E, Hicklin RW, Lee HY, Motika SE, Crawford LA, Weerapana E, et al. Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nat Chem. 2019;11:521–32.
Efferth T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin Cancer Biol. 2017;46:65–83.
Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2:517–32.
Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. 2019;131:356–69.
Baidoo KE, Yong K, Brechbiel MW. Molecular Pathways: Targeted α-Particle Radiation Therapy. Clin Cancer Res. 2013;19:530–7.
Reisz JA, Bansal N, Qian J, Zhao WL, Furdui CM. Effects of Ionizing Radiation on Biological Molecules-Mechanisms of Damage and Emerging Methods of Detection. Antioxid Redox Sign. 2014;21:260–92.
Adjemian S, Oltean T, Martens S, Wiernicki B, Goossens V, Vanden Berghe T, et al. Ionizing radiation results in a mixture of cellular outcomes including mitotic catastrophe, senescence, methuosis, and iron-dependent cell death. Cell Death Dis. 2020;11:1003.
Miller KD, Nogueira L, Devasia T, Mariotto AB, Yabroff KR, Jemal A, et al. Cancer treatment and survivorship statistics, 2022. Ca Cancer J Clin. 2022;72:409–36.
Schaeffer EM, Srinivas S, Adra N, An Y, Bitting R, Chapin B, et al. Prostate Cancer, Version 3.2024. J Natl Compr Canc Netw. 2024;22:140–50.
Azzam EI, Jay-Gerin JP, Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012;327:48–60.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingo IdI, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–8.
Jiang L, Kon N, Li T, Wang S-J, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Zhang YL, Zhuang L, Gan BY. BAP1 suppresses tumor development by inducing ferroptosis upon SLC7A11 repression. Mol Cell Oncol. 2019;6:1536845.
Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20:1181–92.
Lei G, Mao C, Yan Y, Zhuang L, Gan B. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell. 2021;12:836–57.
Wang Y, Yang L, Zhang X, Cui W, Liu Y, Sun QR, et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019;20:e47563.
Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci USA. 2016;113:E6806–E12.
Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21:579–91.
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017;20:1692–704.
Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018;22:569–75.
Li L, Rezvan A, Salerno JC, Husain A, Kwon K, Jo H, et al. GTP cyclohydrolase I phosphorylation and interaction with GTP cyclohydrolase feedback regulatory protein provide novel regulation of endothelial tetrahydrobiopterin and nitric oxide. Circ Res. 2010;106:328–36.
Sanli T, Steinberg GR, Singh G, Tsakiridis T. AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol Ther. 2014;15:156–69.
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc– Activity. Curr Biol. 2018;28:2388–99.e5.
Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22:225–34.
Li C, Dong X, Du W, Shi X, Chen K, Zhang W, et al. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transd Targeted Therapy. 2020;5:187.
Chen CY, Oliner JD, Zhan Q, Fornace AJ, Vogelstein B, Kastan MB. Interactions between p53 and MDM2 in a mammalian cell cycle checkpoint pathway. Proc Natl Acad Sci. 1994;91:2684–8.
Maya R, Balass M, Kim S-T, Shkedy D, Leal J-FM, Shifman O, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15:1067–77.
Venkatesh D, O’Brien NA, Zandkarimi F, Tong DR, Stokes ME, Dunn DE, et al. MDM2 and MDMX promote ferroptosis by PPARα-mediated lipid remodeling. Genes Dev. 2020;34:526–43.
Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. 2020;16:1351–60.
Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci. 2019;6:41–53.
Pathak R, Pawar SA, Fu Q, Gupta PK, Berbée M, Garg S, et al. Characterization of TransgenicGfrpKnock-In Mice: Implications for Tetrahydrobiopterin in Modulation of Normal Tissue Radiation Responses. Antioxid Redox Sign. 2014;20:1436–46.
Cheema AK, Pathak R, Zandkarimi F, Kaur P, Alkhalil L, Singh R, et al. Liver Metabolomics Reveals Increased Oxidative Stress and Fibrogenic Potential in Gfrp Transgenic Mice in Response to Ionizing Radiation. J Proteome Res. 2014;13:3065–74.
Liang C, Zhang XL, Yang MS, Dong XC. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv Mater. 2019;31:e1904197.
Chow E, Harris K, Fan G, Tsao M, Sze WM. Palliative radiotherapy trials for bone metastases: a systematic review. J Clin Oncol. 2007;25:1423–36.
Boeve LMS, Hulshof M, Vis AN, Zwinderman AH, Twisk JWR, Witjes WPJ, et al. Effect on Survival of Androgen Deprivation Therapy Alone Compared to Androgen Deprivation Therapy Combined with Concurrent Radiation Therapy to the Prostate in Patients with Primary Bone Metastatic Prostate Cancer in a Prospective Randomised Clinical Trial: Data from the HORRAD Trial. Eur Urol. 2019;75:410–8.
Phillips R, Shi WY, Deek M, Radwan N, Lim SJ, Antonarakis ES, et al. Outcomes of Observation vs Stereotactic Ablative Radiation for Oligometastatic Prostate Cancer: The ORIOLE Phase 2 Randomized Clinical Trial. JAMA Oncol. 2020;6:650–9.
Palma DA, Olson R, Harrow S, Gaede S, Louie AV, Haasbeek C, et al. Stereotactic Ablative Radiotherapy for the Comprehensive Treatment of Oligometastatic Cancers: Long-Term Results of the SABR-COMET Phase II Randomized Trial. J Clin Oncol. 2020;38:2830–8.
Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, et al. Redox Imbalance in Cystine/Glutamate Transporter-deficient Mice. J Biol Chem. 2005;280:37423–9.
McCullagh EA, Featherstone DE. Behavioral characterization of system xc- mutant mice. Behavioural Brain Res. 2014;265:1–11.
Bump EA, Brown JM. Role of glutathione in the radiation response of mammalian cells invitro and in vivo. Pharmacol Therapeutics. 1990;47:117–36.
Zeng L, Ding S, Cao Y, Li C, Zhao B, Ma Z, et al. A MOF-Based Potent Ferroptosis Inducer for Enhanced Radiotherapy of Triple Negative Breast Cancer. ACS Nano. 2023;17:13195–210.
Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247–50.
Woo JH, Shimoni Y, Yang WS, Subramaniam P, Iyer A, Nicoletti P, et al. Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell. 2015;162:441–51.
Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY, et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Investig. 2018;128:3341–55.
Yoo S-E, Chen L, Na R, Liu Y, Rios C, Van Remmen H, et al. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic Biol Med. 2012;52:1820–7.
Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10:1617.
Li X, Duan L, Yuan S, Zhuang X, Qiao T, He J. Ferroptosis inhibitor alleviates Radiation-induced lung fibrosis (RILF) via down-regulation of TGF-β1. J Inflammation. 2019;16:11.
Li X, Zhuang X, Qiao T. Role of ferroptosis in the process of acute radiation-induced lung injury in mice. Biochemical Biophysical Res Commun. 2019;519:240–5.
Liang J, Cao Y, He M, Li W, Huang G, Ma T, et al. AKR1C3 and Its Transcription Factor HOXB4 Are Promising Diagnostic Biomarkers for Acute Myocardial Infarction. Front Cardiovasc Med. 2021;8:694238.
Lin Z, Xu Y, Guan L, Qin L, Ding J, Zhang Q, et al. Seven ferroptosis-specific expressed genes are considered as potential biomarkers for the diagnosis and treatment of cigarette smoke-induced chronic obstructive pulmonary disease. Ann Transl Med. 2022;10:331.
Wang B, Fu C, Wei Y, Xu B, Yang R, Li C, et al. Ferroptosis-related biomarkers for Alzheimer’s disease: Identification by bioinformatic analysis in hippocampus. Front Cell Neurosci. 2022;16:1023947.
Pan B, Li Y, Xu Z, Miao Y, Yin H, Kong Y, et al. Identifying a novel ferroptosis-related prognostic score for predicting prognosis in chronic lymphocytic leukemia. Front Immunol. 2022;13:962000.
Liu H, Gao L, Xie T, Li J, Zhai T-s, Xu Y. Identification and Validation of a Prognostic Signature for Prostate Cancer Based on Ferroptosis-Related Genes. Front Oncol. 2021;11:623313.
Dai T, Li J, Lu X, Ye L, Yu H, Zhang L, et al. Prognostic Role and Potential Mechanisms of the Ferroptosis-Related Metabolic Gene Signature in Hepatocellular Carcinoma. Pharmacogenomics Personalized Med. 2021;ume 14:927–45.
Yin H, Lin M, Liang S, Wei M, Huang C, Qin F, et al. Ferroptosis-related gene signature predicts prognosis in kidney renal papillary cell carcinoma. Front Oncol. 2022;12:988867.
Miao Y-D, Kou Z-Y, Wang J-T, Mi D-H. Prognostic implications of ferroptosis-associated gene signature in colon adenocarcinoma. World J Clin Cases. 2021;9:8671–93.
Penning TM. The aldo-keto reductases (AKRs): Overview. Chem Biol Interact. 2015;234:236–46.
Guise CP, Abbattista MR, Singleton RS, Holford SD, Connolly J, Dachs GU, et al. The Bioreductive Prodrug PR-104A Is Activated under Aerobic Conditions by Human Aldo-Keto Reductase 1C3. Cancer Res. 2010;70:1573–84.
Adeniji AO, Chen M, Penning TM. AKR1C3 as a target in castrate resistant prostate cancer. J Steroid Biochem. 2013;137:136–49.
Ito K, Utsunomiya H, Yaegashi N, Sasano H. Biological roles of estrogen and progesterone in human endometrial carcinoma - New developments in potential endocrine therapy for endometrial cancer. Endocr J. 2007;54:667–79.
Yoda T, Kikuchi K, Miki Y, Onodera Y, Hata S, Takagi K, et al. 11β-Prostaglandin F2α, a bioactive metabolite catalyzed by AKR1C3, stimulates prostaglandin F receptor and induces slug expression in breast cancer. Mol Cell Endocrinol. 2015;413:236–47.
Park AL, Lin HK, Yang Q, Sing CW, Fan M, Mapstone TB, et al. Differential expression of type 2 3 alpha/type 5 17 beta-hydroxysteroid dehydrogenase (AKR1C3) in tumors of the central nervous system. Int J Clin Exp Patho. 2010;3:743–54.
Evans K, Duan JX, Pritchard T, Jones CD, McDermott L, Gu ZH, et al. OBI-3424, a Novel AKR1C3-Activated Prodrug, Exhibits Potent Efficacy against Preclinical Models of T-ALL. Clin Cancer Res. 2019;25:4493–503.
Desmond JC, Mountford JC, Drayson MT, Walker EA, Hewison M, Ride JP, et al. The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Res. 2003;63:505–12.
Sun SQ, Gu XB, Gao XS, Li Y, Yu HL, Xiong W, et al. Overexpression of AKR1C3 significantly enhances human prostate cancer cells resistance to radiation. Oncotarget. 2016;7:48050–8.
Ciccarelli C, Vulcano F, Milazzo L, Gravina GL, Marampon F, Macioce G, et al. Key role of MEK/ERK pathway in sustaining tumorigenicity and in vitro radioresistance of embryonal rhabdomyosarcoma stem-like cell population. Mol Cancer. 2016;15:16.
Zhao JG, Zhang MN, Liu JD, Liu ZH, Shen PF, Nie L, et al. AKR1C3 expression in primary lesion rebiopsy at the time of metastatic castration-resistant prostate cancer is strongly associated with poor efficacy of abiraterone as a first-line therapy. Prostate. 2019;79:1553–62.
Powell K, Semaan L, Conley-LaComb MK, Asangani I, Wu YM, Ginsburg KB, et al. ERG/AKR1C3/AR Constitutes a Feed-Forward Loop for AR Signaling in Prostate Cancer Cells. Clin Cancer Res. 2015;21:2569–79.
Xiong W, Zhao J, Yu HL, Li XY, Sun SQ, Li Y, et al. Elevated Expression of AKR1C3 Increases Resistance of Cancer Cells to Ionizing Radiation via Modulation of Oxidative Stress. Plos One. 2014;9:e111911.
Xie L, Yu J, Guo W, Wei L, Liu Y, Wang X, et al. Aldo-keto reductase 1C3 may be a new radioresistance marker in non-small-cell lung cancer. Cancer Gene Ther. 2013;20:260–6.
Penning TM, Jonnalagadda S, Trippier PC, Rizner TL. Aldo-Keto Reductases and Cancer Drug Resistance. Pharm Rev. 2021;73:1150–71.
Ascierto ML, McMiller TL, Berger AE, Danilova L, Anders RA, Netto GJ, et al. The Intratumoral Balance between Metabolic and Immunologic Gene Expression Is Associated with Anti-PD-1 Response in Patients with Renal Cell Carcinoma. Cancer Immunol Res. 2016;4:726–33.
Nakamura Y, Suzuki T, Nakabayashi M, Endoh M, Sakamoto K, Mikami Y, et al. In situ androgen producing enzymes in human prostate cancer. Endocr-Relat Cancer. 2005;12:101–7.
Penning TM. AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase): Roles in malignancy and endocrine disorders. Mol Cell Endocrinol. 2019;489:82–91.
Shi X, Feng D, Han P, Wei W. Ferroptosis-related ACSL3 and ACTC1 predict metastasis-free survival for prostate cancer patients undergoing radical radiotherapy. Asian J Surg. 2023;46:2489–90.
Acknowledgements
The graphical abstract and all figures created with BioRender.com. Color should be used for all figures in print.
Funding
This work was funded by the National Natural Science Foundation of China (82271771) and the Beijing Natural Science Foundation (7244422).
Author information
Authors and Affiliations
Contributions
HC wrote most of the manuscript with assistance from other co-authors. HC generated all figures. FL supervised and revised the manuscript. All authors commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, H., Lyu, F. & Gao, X. Advances in ferroptosis for castration-resistant prostate cancer treatment: novel drug targets and combination therapy strategies. Prostate Cancer Prostatic Dis 29, 36–46 (2026). https://doi.org/10.1038/s41391-024-00933-w
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41391-024-00933-w
This article is cited by
-
Multi-omics genetic study revealing ferroptosis regulator CTSB driving prostate cancer progression by modulating the immune microenvironment
Naunyn-Schmiedeberg's Archives of Pharmacology (2026)
-
Fucoidan-decorated metal-zoledronic acid nanocomplexes suppress tumor metastasis by inducing ferroptotic cell death and enhancing cancer immunotherapy
Journal of Nanobiotechnology (2025)
-
The role of the NcRNA/ferroptosis axis in lung cancer: molecular mechanisms and potential therapeutic targets
Apoptosis (2025)
