
Abstract
Perineural invasion (PNI), a prominent pathological feature of pancreatic ductal adenocarcinoma (PDAC), is closely associated with poor prognosis. Clarifying its mechanism is therefore critical for developing new therapies. Recent research has focused on the crosstalk between tumors and Schwann cells (SCs), particularly the role of SC dedifferentiation in facilitating PNI. In this study, by integrating RNA-seq, spatial transcriptomics, and single-cell analysis of clinical samples, we identified significant enrichment of dedifferentiated SCs and upregulation of key markers (p75NTR, SOX2, and c-Jun) in PNI regions. Moreover, PTGES was more highly expressed in the central region of the PNI than in the other regions of the PNI. Coculture experiments revealed that PANC-1 and BxPC-3 cells enhanced SC dedifferentiation, and this process facilitated pancreatic cancer cell malignancy. Furthermore, PTGES upregulation in the coculture system mediated prostaglandin E2 (PGE2) synthesis. Functional experiments revealed that PGE₂ drove morphological alterations in SCs—characterized by bipolar stretching—and elevated the expression of dedifferentiation markers, including p75NTR, c-Jun, SOX2, and GDNF. In the 3D coculture model, treatment with a PTGES inhibitor (CAY10526), siPTGES or PTGES-KO impaired the directional migration and neurite outgrowth of SCs toward PDAC cells. Mechanistically, PGE₂-stimulated SCs secrete elevated levels of LIF and ADAMTS-1, factors that promote extracellular matrix degradation and neural remodeling to facilitate tumor invasion. In summary, we delineate a novel paracrine axis in which PDAC-derived PGE₂ drives SC dedifferentiation and the production of proinvasive factors (LIF and ADAMTS-1), collectively establishing a microenvironment conducive to PNI. Our findings suggest that the PTGES-SC axis is a promising therapeutic target for inhibiting PNI in PDAC.
Similar content being viewed by others


Introduction
Pancreatic ductal adenocarcinoma (PDAC), accounting for 90% of all pancreatic cancers,1 with a 5-year overall survival (OS) rate of only 10%, is characterized by rapid progression and poor prognosis.2 Perineural invasion (PNI), defined as the infiltration and encasement of the perineurium by cancer cells, is a hallmark pathological feature of PDAC. PNI occurs in 70–100% of PDAC patients,3,4,5,6 and its presence predicts an increased incidence of local recurrence, metastasis and poor OS.7 Despite its significance, PNI is diagnosed postoperatively by histology, with no molecular or cytological biomarkers available for prediction or intervention. Thus, investigating the underlying mechanisms of PNI and identifying reliable predictive targets and biomarkers for PNI are crucial for the clinical prediction and therapeutic management of this pathological process. Although recent studies implicate bidirectional signaling in the tumor-nerve microenvironment—wherein tumors promote axonal regeneration8 and nerves enhance the neurophilic phenotype of cancer cells9—the initiating cues for how nerves grow toward the tumor orientation and how tumors overcome spatial distance to invade toward nerves are still unclear.
PNI is characterized by tumor cell infiltration of nerves, in which the abnormal growth of axons and dynamic tumor invasion and metastasis along nerves represent a key step. This process involves intricate molecular regulatory mechanisms and intercellular communication and crosstalk. As the most predominant glial cells in the peripheral nervous system, Schwann Cells (SCs) have attracted increasing attention for their critical roles in the occurrence and development of PNI. Early in pancreatic tumorigenesis, nerve hypertrophy and an expansion of activated SCs occur, which exhibit tropism for tumors and initiate nerve‒tumor interactions.8 Similar to their crucial role in repairing injured nerves, SCs in pancreatic cancer undergo dedifferentiation into a progenitor-like, “repair” state, characterized by the upregulation of specific markers, including the transcription factors SOX2 and c-Jun and various neurotrophic factors. This “repair” phenotype entails increased secretion of neurotrophic factors, increased proliferative and migratory capacity, and an elongated morphology that can organize into Büngner’s band-like structures. Consequently, these reprogrammed SCs exhibit tropism for cancer cells and migrate preinvasively.10,11 Critically, by integrating these capabilities, they actively construct a microenvironmental bridge between tumors and nerves: their structural features and neurotrophic secretion guide cancer cell invasion, whereas their secreted chemokines12 and inflammatory mediators13 stimulate tumor growth and neural affinity, thereby orchestrating PNI. Given this pivotal role, SC dedifferentiation constitutes a compelling therapeutic target. However, the origin of this proinvasive reprogramming—the tumor-derived signal that launches it—remains unidentified.
In this study, we identified the mutual crosstalk process involved in the induction of SC dedifferentiation by cancer cells and the reinforcement of the malignant phenotype of cancer cells by SCs through an SC-cancer cell coculture system. In combination with data from the TCGA database, PNI tissues from pancreatic cancer patients were subjected to combined RNA-seq, spatial transcriptome, and single-cell histology strategies to characterize the spatial patterns of cellular and gene expression; the prostaglandin synthetase PTGES was identified as the focal gene in tumors, and its expression level was positively correlated with the PNI status of patients with PDAC. PTGES is a key synthetase of prostaglandin E2 (PGE2), and its level in SCs and cancer cell coculture systems is elevated, suggesting that tumor-derived PGE2 may promote SC dedifferentiation and mediate the PNI process in pancreatic cancer. PGE2 is a biologically active lipid that signals to bind to the G protein-coupled receptors EP1-EP4 and is critical for tumor development and neurodevelopment. Increased secretion of PGE2 in pancreatic cancer affects peritumoral cells,14 and increased intratumoral PGE2 secretion is positively correlated with low survival in pancreatic cancer patients. PGE2 has been shown to favor axonal growth,15,16 and SC-derived PGE2 promotes sensory neuron excitability and maturation.17
While SC-derived PGE₂ is known to influence neuronal function, the specific role of PGE₂ signaling in regulating the proinvasive dedifferentiation of SCs within the PNI microenvironment remains completely unexplored. PGE₂, a key pro-inflammatory prostaglandin overproduced in the PDAC tumor microenvironment due to aberrant upregulation of cyclooxygenase-2 (COX-2) and prostaglandin E synthase (PTGES) in cancer and stromal cells, is well recognized for its pleiotropic pro-tumorigenic effects, including promoting tumor cell proliferation, angiogenesis, and immune suppression. However, its paracrine regulatory role in modulating the phenotypic plasticity of SCs—an essential stromal component of the perineural niche—has been largely overlooked in the context of PDAC-associated PNI, leaving a critical gap in our understanding of PGE₂-driven tumor-stroma crosstalk. We therefore hypothesize that pancreatic cancer-secreted PGE₂ activates EP receptor signaling on SCs, driving their dedifferentiation into a progenitor-like, “repair” state, which in turn orchestrates the PNI program. This study is designed to test the aforementioned hypothesis and delineate the PGE2-mediated axis underlying tumor-SC crosstalk, with the dual goals of uncovering novel mechanistic insights and rational therapeutic strategies to target PNI in PDAC, and translating these mechanistic discoveries into clinically actionable interventions that alleviate local tumor invasion and distant metastasis, thereby enhancing survival outcomes for patients with PDAC.
Results
SCs are enriched in the PNI-positive region
We analyzed samples from 149 PDAC patients in the TCGA-PAAD cohort. The classification of PNI presence (PNI+) or absence (PNI-) was based on the available surgical pathological reports and hematoxylin and eosin (H&E)-stained images from TCGA, Kaplan‒Meier (KM) analysis of PNI− and PNI+ cases revealed no significant associations between PNI status and OS (p = 0.56). We subsequently performed quantitative scoring of the PNI on the basis of H&E-stained images following the PNI scoring system and divided the samples into two subgroups accordingly: the high PNI group (PNI score ≥ 6, n = 48) and the low PNI plus PNI- group (PNI score < 6 and without PNI, n = 101) (Supplementary Fig. 1a, b). Analysis of baseline characteristics revealed that there were no statistically significant differences in the distributions of sex, age, pathological T/N stage, or treatment modality between the two groups (p > 0.05). However, PNI subgrouping was significantly correlated with distant metastasis status (p = 0.008) and molecular subtype (p = 0.011) (Supplementary Table 1). Specifically, the low PNI plus PNI- group was predominantly composed of the classical-PDAC subtype (56.4% in the low PNI plus PNI- group vs. 35.4% in the high PNI group), whereas the high PNI group had a greater proportion of the quasimesenchymal-pancreatic-ductal adenocarcinoma (QM-PDAC) subtype (54.2% in the high PNI group vs. 41.6% in the low PNI plus PNI- group) (Supplementary Table1). KM analysis revealed that high PNI was associated with shorter OS (Supplementary Fig. 1c, p = 0.011). In the classical-PDAC subgroup, the OS of the high PNI subgroup was significantly shorter than that of the low PNI plus PNI− subgroup (Supplementary Fig. 1d, p = 0.014), whereas no such difference was observed in the QM-PDAC subgroup (Supplementary Fig. 1e, p = 0.8). Stratified analysis by treatment modality revealed no significant difference in survival between the high PNI group and the low PNI plus PNI− group in the “no treatment” group (not receiving treatment) (Supplementary Fig. 1f, p = 0.3) or the “two treatment” group (receiving radiotherapy and chemotherapy) (Supplementary Fig. 1g, p = 0.82). In contrast, the “one treatment” group (receiving single radiotherapy or chemotherapy) in the high PNI group had significantly shorter OS (Supplementary Fig. 1h, p = 0.00015).
To elucidate the molecular mechanisms governing the PNI microenvironment in pancreatic cancer, we conducted spatial transcriptomic sequencing on four PNI + PDAC tissue samples and integrated seven independent single-cell RNA-sequencing (scRNA-seq) datasets obtained from public repositories (Fig. 1a). On the basis of H&E staining, we delineated the following PNI and no-PNI (NPNI) regions: the PNI region (PNI) (17 spots), PNI near region (PNI-near) (70 spots), PNI mid region (PNI-mid) (253 spots), PNI away region (PNI-away) (358 spots), NPNI region (NPNI) (9 spots), NPNI near region (NPNI-near) (27 spots), NPNI mid region (NPNI-mid) (100 spots), and the NPNI away region (NPNI-away) (151 spots) (Fig. 1b). Gene set variation analysis (GSVA) of these regions revealed a gradient of enriched biological functions. Notably, SC-associated pathways, such as “axon guidance”, showed progressively stronger enrichment from the PNI-away region toward the PNI region, suggesting that dynamic alterations in SCs within the tumor microenvironment are critical for PNI (Fig. 1c).

SCs are enriched in the PNI-positive region. a Schematic diagram of the workflow; some of the graphical elements were created wtith BioGDP. b Division of perineural invasion regions (PNI-region: PNI, PNI-near, PNI-mid, PNI-away) and nonperineural invasion regions (NPNI: NPNI, NPNI-near, NPNI-mid, NPNI-away) in representative spatial transcriptomic images of PDAC PNI samples, with corresponding representative HE-stained images of spot points (right) (n = 4). N: Nerve, T: Tumor. Scale bar: 500 μm. c GSVA of the PNI region and the NPNI region. d Correlations between different cell types and the expression levels of marker genes. e Cell type distribution scatter plot based on the UMAP dimensionality reduction algorithm; f UMAP plot of SC subsets in pancreatic cancer tissues. g Feature score of nomyelinating SCs at the single-cell level in each of the two SC subsets (p = 0.025). h Representative images of RCTD single-cell mapping results based on PNI regions and NPNI regions (n = 4); i Stacked bar charts of different PNI regions and NPNI regions, showing the proportions of various cell types in different samples. j, k Representative mIHC images of human normal pancreatic tissues, PDAC-PNI- tissues, and PDAC-PNI+ tissues stained with PGP9.5, SOX2, c-Jun, and MUC1 antibodies. Scale bar: 100 μm (left), 50 μm (right). The ratios of SOX2+ and c-Jun+ positive cells in normal pancreatic tissues, PDAC-PNI- tissues, and PDAC-PNI+ tissues (n = 5). All the results are presented as the means ± SDs. Each data point in the bar graphs represents an individual sample. Statistical significance was determined by a two-tailed unpaired t test (Fig. 1g) and one-way analysis of variance followed by Tukey’s HSD post hoc test (Fig. 1j). ****p < 0.0001
Single-cell analysis of 14,723 cells from seven PDAC patients was performed, identifying 11 major cell clusters: fibroblasts (n = 5359, COL1A1, COL1A2, and COL3A1), epithelial cells (n = 5349, EPCAM, KRT18, and KRT19), endothelial cells (n = 1196, CDH5, PECAM1, and VWF), myeloid cells (n = 1089, CD14, CD68, and AIF1), smooth muscle cells (n = 487, ACTA2, TAGLN, and SNCG), plasma cells (n = 461, CD38, XBP1, and JCHAIN), endocrine cells (n = 245, INS,GCG and SST), T cells (n = 325, CD3E, CD3D, and CD3G), acinar cells (n = 223, CPA1, SPINK1, and PRSS1), mast cells (n = 142, TPSB2, and SNCB1, and CPA3), and Schwann cells (n = 92, CRYAB, CDH19, and SOX10) (Fig. 1d, e). Uniform manifold approximation and projection (UMAP) visualization revealed two distinct SC subpopulations. Subcluster 1 (red), which showed significantly greater enrichment for a nomyelinating_SC gene signature (p = 0.026), was defined as “Nomyelinating_SC”, in contrast to “Myelinating_SC” (subcluster 2, blue) (Fig. 1f, g). High expression of canonical markers for each SC state was validated by violin plots: Nomyelinating_SC: NGFRAP1, JUND, L1CAM (Supplementary Fig. 1i–k); Myelinating_SC: ERBB3, PLP1, PMP22 (Supplementary Fig. 1l–n). Spatial mapping of these subpopulations via RCTD was subsequently performed (Fig. 1h). Quantification of the mapping revealed a specific spatial distribution: myelinating SCs were distributed in both the PNI and the NPNI regions, whereas Nomyelinating_SCs were exclusively confined to the PNI regions (Fig. 1i). This distinct spatial restriction suggests that the Nomyelinating_SC subpopulation constitutes the principal SC type associated with PNI.
We performed multiplex immunohistochemistry (mIHC) using markers for pancreatic cancer (MUC1) and neurons (PGP9.5) and putative markers for dedifferentiated SCs (p75NTR, SOX2, and c-Jun). In normal pancreatic tissues and PNI tissues, the expression of SOX2 and c-Jun is low. In contrast, within PNI+ tissues, tumor-encircled nerve fibers presented high expression of both SOX2 and c-Jun (Fig. 1j, k). Immunohistochemical staining of SCs in clinical samples of PDAC with a marker for SC dedifferentiation, nerve growth factor receptor (p75NTR), revealed that the percentage of p75 NTR-positive cells was significantly greater in PNI+ tissues than in PNI- tissues (p = 0.0152) (Supplementary Fig. 1o, p). The density of SCs in the pancreas may be correlated with the occurrence of PNI. In summary, dedifferentiated SCs are associated with the pathological state of PNI + PDAC.
Pancreatic cancer-SC crosstalk in the PNI microenvironment
As mentioned previously, SC dedifferentiation is closely correlated with the PNI. To further elucidate the molecular crosstalk between SCs and pancreatic cancer cells, we performed RNA sequencing on SCs following in vitro coculture with pancreatic cancer cells. Pathway enrichment analysis via a Venn diagram revealed that the DEGs in SCs were significantly enriched in pathways related to “Motor protein”, “Axon regeneration” and “Cell cycle”, among others (Fig. 2a, b; Supplementary Dataset 1). These findings demonstrate that SCs transition into an activated, proliferative phenotype following coculture with pancreatic cancer cells.

Pancreatic cancer cells promote SC dedifferentiation. a Schematic diagram of the coculture system of PDAC cells and SCs. b Venn diagram of upregulated pathways in RSC96 (+PANC-1) cells and RSC96 (+BxPC-3) cells compared with RSC96 cells (n = 3). c, e Transwell assays were used to evaluate the effects of PANC-1 and BxPC-3 cells on the migration of sNF96.2 cells (n = 6). Scale bar: 100 μm. d Microscopy images showing the morphological changes in sNF96.2 cells induced by PANC-1 and BxPC-3 cells. Scale bars: 50 μm (upper panels) and 20 μm (lower panels). f–j Western blotting was performed to detect the protein expression levels of dedifferentiation markers (p75NTR, c-Jun, SOX2, N-cadherin, and GDNF) in sNF96.2 cells after coculture with PANC-1 or BxPC-3 cells (n = 3). k, m Transwell assays were used to assess the effects of PANC-1 and BxPC-3 cells on the migration of RSC96 cells (n = 6). Scale bar: 100 μm. l Microscopy images showing the morphological changes in RSC96 cells induced by PANC-1 and BxPC-3 cells. Scale bars: 50 μm (upper panels) and 20 μm (lower panels). n–r Western blotting was conducted to determine the protein expression levels of dedifferentiation markers (p75NTR, c-Jun, SOX2, N-cadherin, and GDNF) in RSC96 cells after coculture with PANC-1 or BxPC-3 cells (n = 3). Relative expression levels were quantified as p75NTR/GAPDH, c-Jun/GAPDH and GDNF/GAPDH; since the molecular weight of SOX2 (~35 kDa) is close to that of GAPDH (~36 kDa), SOX2/β-actin was used for relative expression quantification. All the results are presented as the means ± SDs. Each data point in the bar graphs represents an individual sample. Statistical significance was determined by one-way analysis of variance followed by Tukey’s HSD post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
To verify this hypothesis, we performed a noncontact coculture assay using pancreatic cancer cell lines (PANC-1 and BxPC-3 cells) and SC lines (RSC96 and sNF96.2 cells). The results revealed that coculture with pancreatic cancer cells significantly increased the migratory capacity of RSC96 and sNF96.2 cells (Fig. 2c, e, k, m). In parallel, Supplementary Dataset 1 and experiments revealed that under stimulation by pancreatic cancer cells, RSC96 and sNF96.2 cells exhibited morphological changes: the cells shifted from a morphology with short, sparse protrusions to one characterized by long, slender processes, accompanied by an increased number of protrusions, more complex branching patterns, and even the formation of mesh-like or bundle-like structures via protrusion entanglement (Fig. 2d, l). Consistent with these changes, N-cadherin, a marker associated with migration, was markedly upregulated in RSC96 and sNF96.2 cells. Concurrently, the expression levels of multiple dedifferentiation markers, including p75NTR, c-Jun, SOX2 and GDNF, were also significantly elevated (Fig. 2f–j, n–r). Collectively, these findings demonstrate that pancreatic cancer cells not only promote the migration and morphological remodeling of SCs but also induce the upregulation of core genes implicated in SC dedifferentiation.
We also assessed the reciprocal effect of SCs on pancreatic cancer cells via a contact-independent coculture system. The results demonstrated that SCs markedly enhanced the migratory, proliferative, and invasive capacities of both PANC-1 and BxPC-3 cells. To further elucidate the molecular underpinnings of this crosstalk in the PNI, we performed spatial transcriptomic gene enrichment analysis by comparing mRNA expression levels in spots derived from the PNI-near region and the NPNI region. The analysis revealed that perineural cancer cells were functionally enriched in pathways related to cell migration and cell cycle progression (Fig. 3a). Functional assays validated these transcriptional signatures. Transwell migration and invasion assays revealed that PANC-1 and BxPC-3 cells exhibited significantly increased migratory and invasive abilities when exposed to conditioned factors from RSC96 or sNF96.2 cells (Fig. 3b, d, e). Moreover, 5-ethynyl-2’-deoxyuridine (EdU) incorporation assays confirmed that the proliferative activity of PANC-1 and BxPC-3 cells was markedly elevated following coculture with SCs (Fig. 3c, f). Furthermore, Western blot analysis provided mechanistic evidence that RSC96 and sNF96.2 cells induced epithelial‒mesenchymal transition (EMT) in PANC-1 and BxPC-3 cells. Specifically, we detected upregulated protein expression of the mesenchymal markers N-cadherin, vimentin, and SNAI1, along with downregulated expression of the epithelial marker E-cadherin, in these pancreatic cancer cells (Fig. 3g–v).

SCs induce the malignant progression of pancreatic cancer cells. a Differential gene enrichment analysis of the spatial transcriptome in the PNI-near and NPNI-near regions. b, d, e Transwell assays were used to evaluate the effects of RSC96 or sNF96.2 cells on the migration and invasion of PANC-1 or BxPC-3 cells (n = 6). Scale bar: 100 μm. c, f An EdU assay was performed to assess the effects of RSC96 or sNF96.2 cells on the proliferation of PANC-1 or BxPC-3 cells (n = 6). Scale bar: 50 μm. g‒n Western blot analysis was conducted to detect the protein expression levels of EMT markers (N-cadherin, E-cadherin, vimentin, and SNAI1) in PANC-1 and BxPC-3 cells after coculture with RSC96 cells (n = 3). o-v Western blot analysis was conducted to detect the protein expression levels of EMT markers (N-cadherin, E-cadherin, vimentin, and SNAI1) in PANC-1 and BxPC-3 cells after coculture with sNF96.2 cells (n = 3). Relative expression levels were quantified as N-cadherin/GAPDH, vimentin/GAPDH and E-cadherin/GAPDH; since the molecular weight of SNAI1 ( ~ 29 kDa) is close to that of GAPDH ( ~ 36 kDa), SNAI1/β-actin was used for relative expression quantification. All the results are presented as the means ± SDs. Each data point in the bar graphs represents an individual sample. Statistical significance was determined by one-way analysis of variance followed by Tukey’s HSD post hoc test (for Fig. 3d-f) and a two-tailed unpaired t test (for Fig. 3k-n, s-v). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Elevation of PTGES/PGE2 in the coculture system is correlated with PNI
To characterize the paracrine signaling events underlying PNI, pancreatic cancer cells were collected after 24 h of coculture with RSC96 cells and subjected to RNA-seq analysis. For a more comprehensive investigation, we integrated RNA-seq datasets from two other independent sources: 9 clinical PDAC samples with confirmed PNI and transcriptomic data of 28 PNI+ versus 28 PNI- samples obtained from the GEO database (GSE102238). We subsequently identified overlapping genes across these three datasets via Venn diagram analysis (Supplementary Dataset 2). Within the intersection set, six protein-coding genes—NUF2, NEK2, MELK, C1GALT1, ADGRF1, and PTGES—were significantly upregulated in PNI+ samples (|log2FC | ≥1, p < 0.05; Fig. 4a). Among these genes, NUF2, NEK2, and MELK are primarily involved in cell proliferation processes, including cell cycle progression and mitosis, suggesting their potential role in driving rapid tumor cell division. However, the pathogenesis of PNI involves not only cancer cell proliferation but also intricate and dynamic paracrine crosstalk between tumor cells and the neural microenvironment. Notably, while C1GALT1 and ADGRF1 expression levels were comparable between the PNI and NPNI regions according to spatial transcriptomic analyses, PTGES exhibited robust expression in tumor areas and was specifically enriched in the PNI regions, with negligible expression in the NPNI regions (Fig. 4b, c). This distinct expression pattern further corroborated the association between PTGES and the PNI. On this basis, PTGES (prostaglandin E synthase gene) was designated as the key candidate gene for the present study. PGE2 plays an important role in SC-mediated neurogenesis, axon repair and neural regeneration. In tumor research, the focus is mainly on the regulation of immune cells by tumor-derived PGE2, but the role of PGE2 in SC reprogramming and the mechanism by which it promotes PNI are still unclear. mIHC analysis revealed no expression of MUC1 or PTGES in normal tissues. Colocalization of PTGES with MUC1, but not with PGP9.5, was observed in PNI- and PNI+ tissues. Conversely, p75NTR colocalized with PGP9.5 but not with MUC1, confirming the expression specificity of p75NTR and PTGES (Fig. 4d). Immunohistochemical (IHC) analysis further revealed a lower PTGES H-score in normal tissues and PDAC-PNI- tissues than in PDAC-PNI+ tissues (Fig. 4e, l). The western blot results further confirmed that PTGES was overexpressed in PDAC tissue and that the interaction between PANC-1 and BxPC-3 cells and SCs increased the expression level of prostaglandin synthase PTGES (Fig. 4f-k). Consistent with these findings, PGE2 levels in the coculture system, as measured by enzyme-linked immunosorbent assay (ELISA), were significantly elevated, and this effect was attenuated by either the PTGES inhibitor (CAY10526) or siPTGES knockdown (Fig. 4m, Supplementary Fig. 2). In summary, high PTGES/PGE2 expression is associated with the PNI of PDAC. Inhibition of PTGES in pancreatic cancer cells can reduce the content of PGE2 in the coculture system.

Elevation of PTGES/PGE2 in the coculture system is correlated with PNI. a Venn diagram showing the intersection of multiomics data identifying PTGES as a key gene in PNI. b Expression of PTGES genes via spatial transcriptome analysis. Scale bar: 500 μm (n = 4). c Violin plot showing the expression of PTGES in various PNI regions and NPNI regions via spatial transcriptomics (n = 4). d Representative mIHC images of human normal pancreatic tissues, PDAC-PNI- tissues, and PDAC-PNI+ tissues stained with PGP9.5, p75NTR and MUC1 antibodies (n = 3). Scale bar: 100 μm (left), 50 μm (right). e IHC staining of PTGES in human normal pancreatic tissues, PDAC-PNI- tissues, and PDAC-PNI+ tissues (n = 9), N: nerve, T: tumor. Scale bar: 100 μm. l H-scores of PTGES. f, i Western blotting was performed to detect the protein expression level of PTGES in human normal pancreatic tissues and PDAC tissues (n = 3), N: normal tissue, T: tumor tissue. g, j Western blotting was used to determine the protein expression level of PTGES in PANC-1 and BxPC-3 cells after coculture with RSC96 cells (n = 3). h, k Western blotting was conducted to measure the protein expression level of PTGES in PANC-1 and BxPC-3 cells after coculture with sNF96.2 cells (n = 3). m ELISA was used to detect the concentration of PGE2 in the culture medium (n = 3), and some of the graphical elements were created wtith Figdraw. All the results are presented as the means ± SDs. Each data point in the graphs represents an individual sample. Statistical significance was determined via two-tailed unpaired t tests (for Fig. 4i–k), one-way analysis of variance followed by Tukey’s HSD post hoc test (for Fig. 4m) and the Kruskal‒Wallis H test followed by Dunn’s multiple comparisons test (for Fig. 4l). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Pancreatic cancer-derived PGE2 induces SC activation
Cellular IF further confirmed the colocalization of the membrane marker Dil with the PGE2 receptors PTGER1, PTGER2, and PTGER4 in both RSC96 and sNF96.2 cells (Fig. 5a, Supplementary Fig. 3a). To explore the potential role of PTGES/PGE2 in inducing SC dedifferentiation, SCs were treated with a series of PGE2 concentrations. After 24 h of incubation, phalloidin staining combined with confocal laser scanning microscopy was performed to evaluate the morphological changes in the cells. These results demonstrated that PGE2 treatment triggered distinct morphological alterations in SCs. The originally elongated RSC96 and sNF96.2 cells underwent phenotypic transition, exhibiting typical spindle-shaped and bipolar features associated with the dedifferentiated state (Fig. 5b). Western blot analysis revealed that the protein expression levels of p75NTR, c-Jun, and SOX2 in RSC96 and sNF96.2 cells were increased in a PGE2 concentration-dependent manner (Fig. 5c, e–g; Supplementary Fig. 3c, e–g). Furthermore, cellular IF assays revealed that the relative fluorescence intensity of p75NTR, c-Jun, and SOX2 was significantly increased in both cell lines following PGE2 treatment (Fig. 5d, h–j; Supplementary Fig. 3d, h–j). Notably, pretreatment of pancreatic cancer cells with CAY10526 attenuated their capacity to induce SC migration in the coculture system (Supplementary Fig. 3b). In addition, the administration of CAY10526 or siPTGES to pancreatic cancer cells resulted in decreased expression levels of p75NTR, SOX2, and c-Jun in cocultured SCs, and this suppressive effect was abolished by supplementation with exogenous PGE2 (Fig. 5k, o–q, l, r–t; Supplementary Fig. 4a‒l). 3D cell migration assays demonstrated that RSC96 and sNF96.2 cells exhibited enhanced migratory ability, accompanied by the acquisition of an elongated morphological phenotype. Importantly, this promigratory effect was not elicited by nonmalignant HPNE cells. Conversely, treatment with CAY10526 or siPTGES markedly impaired the directional migration of SCs toward pancreatic cancer cells (Fig. 5m, Supplementary Fig. 4m). Coculture of dorsal root ganglion (DRG) explants with pancreatic cancer cells revealed that treatment with CAY10526 or siPTGES significantly inhibited DRG axon outgrowth, with the outgrowth index markedly decreased (p < 0.01) (Fig. 5n, u–v; Supplementary Fig. 4n, o). To clarify the specificity of the PTGES/PGE2 regulatory pathway, this study selected the pancreatic cancer cell line BxPC-3, which has high PTGES expression, as the research subject. Using gene editing technology, we constructed a PTGES-knockout BxPC-3 cell line (BxPC-3 PTGES-KO) and repeated the aforementioned experimental procedures. The experimental results were consistent with those obtained previously (Supplementary Fig. 5a–r). Collectively, these findings suggest that pancreatic cancer-derived PGE2 is critical for promoting SC activation and migration and that blocking PTGES function in pancreatic cancer cells attenuates the dedifferentiation capacity of SCs.

Pancreatic cancer-derived PGE2 induces SC activation. a Representative IF images of DiI and PTGER1/2/4 in RSC96 cells. Scale bars: 20 μm (left), 5 μm (right). b Representative F-actin IF images of RSC96 and sNF96.2 cells after treatment with different doses of PGE2 (0 nM, 0.1 nM, 1 nM, or 10 nM). Scale bar: 50 μm. c, e–g Protein expression levels of p75NTR, c-Jun, and SOX2 in RSC96 cells treated with different doses of PGE2 (0 nM, 0.1 nM, 1 nM, or 10 nM). Relative expression levels were quantified as p75NTR/GAPDH and c-Jun/GAPDH; since the molecular weight of SOX2 (~35 kDa) is close to that of GAPDH (~36 kDa), the relative expression of SOX2/β-actin was quantified (n = 3). d, h–j Representative IF images of p75NTR, c-Jun, and SOX2 in RSC96 cells after PGE2 intervention. Scale bar: 100 μm. Statistics of the relative fluorescence intensity between the control and treatment groups are shown (n = 3). k, o-q Protein expression levels of p75NTR, c-Jun, and SOX2 in RSC96 cells (RSC96-PANC-1 coculture system) following CAY10526 treatment and PGE2 supplementation (n = 3). l, r–t Protein expression levels of p75NTR, c-Jun, and SOX2 in RSC96 cells (RSC96-BxPC-3 coculture system) following CAY10526 treatment and PGE2 supplementation (n = 3). m Effects of CAY10526 treatment and PGE2 supplementation on 3D cocultures of RSC96 cells with HPNE, PANC-1 or BxPC-3 cells (n = 3).Scale bar: 200 μm. +B/P/H: +BxPC-3/PANC-1/HPNE, + DMSO:+BxPC-3/PANC-1/HPNE + DMSO, + CAY:+BxPC-3/PANC-1/HPNE + DMSO + CAY10526, + CAY + PGE2:+BxPC-3/PANC-1/HPNE + DMSO + CAY10526 + PGE2. n, u, v effects of CAY10526 treatment and PGE2 supplementation on PANC-1/BxPC-3-DRG cocultures, along with quantification of the DRG growth index (n = 3). Scale bar: 500 μm. +B/P: +BxPC-3/PANC-1, +DMSO: +BxPC-3/PANC-1 + DMSO, +CAY: +BxPC-3/PANC-1 + DMSO + CAY10526, +CAY+. PGE2: +BxPC-3/PANC-1 + DMSO + CAY10526 + PGE2. All the results are presented as the means ± SDs. Each data point in the bar graphs represents an individual sample. Statistical significance was determined by one-way analysis of variance followed by Tukey’s HSD post hoc test (for e–g, o–t, and u, v) and a two-tailed unpaired t test (for Fig. 5h-j). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Dedifferentiated SCs secrete LIF and ADAMTS-1 to promote PNI
To reveal the mechanism by which PGE2 promotes PNI, RNA-seq analysis was performed after treating RSC96 cells with PGE2 (10 nM) for 24 h. After coculture with pancreatic cancer cells, the transcriptome of RSC96 cells was differentially expressed. The Venn diagram results revealed that the leukemia inhibitory factor LIF (|log2FC | =1.12, p < 0.001) and the disintegrin and metalloproteinase with thrombospondin motif 1 ADAMTS-1 (|log2FC | =1.67, p < 0.001) were significantly upregulated in the treated group (Fig. 6a, Supplementary Dataset 2). RT‒qPCR revealed that RSC96 and sNF96.2 cell LIF mRNA could be upregulated by pancreatic cancer cells and PGE2 (Fig. 6b–e). Western blot analysis confirmed that the protein expression of ADAMTS-1 in SCs was increased by pancreatic cancer cells and PGE2 treatment (Fig. 6f–i, l–o). Pretreatment with CAY10526 or siPTGES can reduce the ADAMTS-1 protein expression level in RSC96 and sNF96.2 cells, and this effect can be reversed by PGE2 (Fig. 6j, k, p, q; Supplementary Fig. 6a–d, f–i). Treatment with CAY10526 or siPTGES knockdown reduced LIF mRNA expression in sNF96.2 cells, and this effect was reversed by PGE2 (Supplementary Fig. 6e, j). Treatment with a LIF-neutralizing antibody (AB-449-NA) reduced the axon outgrowth index in the coculture system (Fig. 6r, u) and reduced the number of pancreatic cancer cells invading nerves (Fig. 6 s, t, v, w; Supplementary Fig. 6k, l, m). Using PTGES-KO BxPC-3 cells, we confirmed that the induction of ADAMTS-1 and LIF in SCs required functional PTGES in cancer cells (Supplementary Fig. 5s–w). In animal experiments, pancreatic cancer cells preconditioned by coculture with SCs formed heavier tumors and resulted in worse sciatic nerve function scores. Treatments with either CAY10526 or AB-449-NA significantly reduced tumor weight and improved sciatic nerve scores (Supplementary Fig. 6n–q). Transmission electron microscopy (TEM) revealed that tumor neural invasion caused damage to the myelin sheath, which manifested as severe demyelination (Supplementary Fig. 6r). The administration of either CAY10526 or AB-449-NA ameliorated this demyelination. In summary, PGE2 induced the secretion of the trophic factor LIF and the degradation of the extracellular matrix by SCs to facilitate neural invasion (Fig. 6x).

PGE2 promotes the secretion of LIF and ADAMTS-1 from SCs to drive PNI development. a After RSC96 cells were treated with PGE2, PANC-1, or BxPC-3, RNA-seq differential gene intersection analysis was performed to identify commonly upregulated genes; LIF and ADAMTS-1 were found to be associated with PNI (n = 3). b, c RT‒qPCR was conducted to assess the effects of treatment and pancreatic cancer cell coculture on LIF mRNA expression in RSC96 and sNF96.2 cells (n = 3). d, e RT‒qPCR analysis was used to determine the effect of PGE2 (0 nM, 0.1 nM, 1 nM, or 10 nM) treatment on LIF mRNA expression in RSC96 and sNF96.2 cells (n = 3). f, g Protein expression levels of ADAMTS-1 in RSC96 cells cocultured with PANC-1 or BxPC-3 cells (n = 3). h, i Protein expression levels of ADAMTS-1 in RSC96 cells treated with PGE2 (0 nM, 0.1 nM, 1 nM, or 10 nM) (n = 3). l, m Protein expression levels of ADAMTS-1 in sNF96.2 cells cocultured with PANC-1 or BxPC-3 cells (n = 3). n, o Protein expression levels of ADAMTS-1 in sNF96.2 cells treated with PGE2 (0 nM, 0.1 nM, 1 nM, or 10 nM) (n = 3). j, k, p, q Protein expression levels of ADAMTS-1 in sNF96.2 cells (cocultured with BxPC-3/PANC-1) following CAY10526 treatment and PGE2 supplementation (n = 3). r, u Effect of a LIF neutralizing antibody on DRG axon growth (n = 3). Scale bar: 200 μm. s, t, v, w Effect of LIF neutralizing antibody on PNI in the PANC-1/BxPC-3-RSC96 coculture system (n = 3). Scale bar=100 μm. x Schematic diagram illustrating the mechanism: pancreatic cancer-derived PTGES/PGE2 promotes SC dedifferentiation and the secretion of LIF and ADAMTS-1, thereby inducing PNI. This image was created with Adobe Illustrator. All the results are presented as the means ± SDs. Each data point in the bar graphs represents an individual sample. All statistical significance was determined by one-way analysis of variance followed by Tukey’s HSD post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Discussion
Numerous studies have demonstrated that the dynamic crosstalk between neural cells and tumor cells represents a critical mechanism underlying PNI. However, the sequential mechanisms underlying how tumor cells and nerve cells overcome spatial barriers and mutually attract one another remain largely elusive. Both the findings of our study and those reported by Guo et al.18 demonstrated that there was no significant association between OS and PNI status when the PNI was simply dichotomized as PNI+ or PNI-. However, previous studies have indicated that the PNI is closely correlated with poor prognosis and reduced survival rates in PDAC patients.5,6 We noted that the inconsistency between the conclusions of our study and those of previous reports might be attributed primarily to differences in the inclusion criteria and definitions of PNI. Nozzoli,19 Schiavo20 and other researchers have proposed that a graded assessment of the PNI may be more conducive to accurate prognostic risk stratification than a simple binary determination of the presence or absence of the PNI. By stratifying patients into high PNI and low PNI plus PNI- groups, we further investigated the impacts of different PNI classification methods on prognosis. The results revealed a poorer prognosis in the high PNI group than in the low PNI group, confirming that this grading strategy reflects prognostic risk stratification. We also identified a clear link between PNI degree and PDAC molecular subtypes: QM-PDAC predominated in the high PNI group, whereas classical-PDAC dominated in the low PNI plus PNI− group. The aggressive phenotypes of QM-PDAC (e.g., epithelial‒mesenchymal transition, high proliferation) enhance neurotropism and severe PNI, whereas classical-PDAC is indolent with reduced nerve invasion. Survival analysis stratified by subtype highlighted the heterogeneous prognostic value of the PNI: the inherent malignancy of QM-PDAC masks the additional prognostic impact of the PNI, given its robust invasiveness. In contrast, for the indolent classical-PDAC subtype, the PNI acts as a key variable accelerating survival deterioration. Monotherapy (chemotherapy or radiotherapy alone) resulted in significantly shortened OS in the high PNI group, as it failed to eliminate nerve-spread minimal residual lesions. Conversely, combination therapy mitigates PNI-related adverse effects via synergistic modalities. These findings highlight the need for PNI-guided treatment: combination therapy for high PNI classical-PDAC patients should be prioritized to improve survival, and monotherapy should be selected on the basis of tolerance for low PNI plus PNI- patients to avoid overtreatment side effects. In summary, refining PNI grading and clarifying its neural involvement mechanisms are critical for optimizing surgical strategies and developing novel therapies, thereby increasing diagnostic accuracy. However, more precise machine learning models are needed for occult PNI detection to increase sensitivity, enable early intervention, and improve patient prognosis.
Analysis of spatially resolved transcriptomes from PNI and NPNI regions in PDAC revealed that SCs localized in PNI areas exhibit a state of increased biological activity, underscoring their critical role in the PNI process. This finding aligns with the findings of Demir et al.,11 who proposed that SCs accumulate at preinvasive neoplastic foci—indicating their early involvement in tumor microenvironment establishment—and that during PDAC progression, SCs possess a strong affinity for tumor cells, thereby initiating nerve‒tumor cell crosstalk.8 Notably, in the tumor microenvironment, SCs undergo dedifferentiation under the influence of tumor cells, transforming into unmyelinated SCs with reparative properties; this phenotypic shift mirrors the response of SCs to nerve injury.8
Single-cell spatial transcriptome profiling confirmed that Nomyelinating_SC with dedifferentiated features presented higher expression in PNI+ regions than in PNI- regions. Consistent with our findings, Wong et al. reported that PDAC-derived extracellular vesicles induce SC migration and p75NTR expression, validating p75NTR as a robust marker for the dedifferentiated state—a state characterized by high c-Jun and SOX2 expression.21,22 mIHC revealed minimal SC infiltration in normal and PNI− tissues but extensive infiltration in PNI+ tissues. p75NTR staining further revealed expanded dedifferentiated SC populations in PNI+ versus PNI − PDAC patients, indicating that these cells are enriched and correlate with the PNI+ subtype. RNA-seq of PDAC-SC cocultures revealed that tumor cell exposure enriched SCs in cell cycle-related pathways, suggesting that it modulated SC proliferation and migration. Functional assays confirmed that PDAC-cocultured SCs exhibited enhanced migration, which was concurrent with upregulated N-cadherin, dedifferentiation markers (p75NTR, c-Jun, and SOX2) and GDNF. Conversely, PDAC cells treated with RSC96 or sNF96.2 displayed invasive phenotypic switching, with significantly increased proliferation, migration and invasion, indicating paracrine crosstalk between SCs and PDAC cells. Collectively, tumors drive the dedifferentiation of SCs; in turn, dedifferentiated SCs not only promote axonogenesis in adjacent nerves but also facilitate the acquisition of a malignant phenotype by tumor cells. This cascade of dynamic bidirectional interactions may synergistically promote the progression of PNI. While the mechanisms underlying SC-mediated modulation of tumor behavior have been extensively investigated, how SCs sense tumor-derived cues and initiate the dedifferentiation program remains largely elusive.
To identify mediators of pancreatic cancer cell-induced SC dedifferentiation, we performed a multiomics coanalysis of sequenced samples of SC-cultured tumors with clinical neural invasion, and in the standard set of genes, we detected high expression of the gene encoding the prostaglandin microsomal enzyme PTGES, which is involved in the production of PGE2. PTGES converts PGH2 to PGE2. PGE2 and its high-affinity receptors are widely distributed in the nervous system,23 peripheral tissues, and immune cells.24,25 According to a report by Villagrana et al. in Signal Transduction and Targeted Therapy, elevated PGE2 in the tumor-neural niche stimulates immunosuppressive MDSCs, thereby blocking immune cell maturation and maintaining immunosuppression as an evasion mechanism.26,27 These cells further exacerbate tumor neurogenesis via neurotrophic factor secretion, forming a feed-forward loop that promotes tumor growth and immune escape. PTGES overexpression and PGE₂-EP axis activation drive pathological processes in most solid tumors, such as colorectal cancer,28 breast cancer,14 and pancreatic cancer,29 with PGE₂ enhancing tumor cell proliferation, differentiation and invasion via autocrine/paracrine signaling. However, the roles of PGE₂ in PDAC PNI and SC function remain unclear. Spatial transcriptomics and PTGES immunohistochemistry revealed high PTGES expression in PNI+ regions but low PTGES expression in noninvaded perineural areas, indicating that PTGES facilitates tumor PNI.
Relevant studies have demonstrated that cardiovascular cell-derived PGE₂ upregulates neuronal sodium channel (NaV) expression and enhances neuronal excitability, which is essential for sensory neuron development.17 It also mediates DRG neuron neurogenesis and synaptogenesis via the EP4/PKA/Cav3.2 pathway.30 PGE2 also stimulates sensory neurons to secrete calcitonin-related gene peptide (CGRP).31 A recent study revealed that the density of CGRP+ neurons is increased in PDAC. The secretion of CGRP is thought to be an essential factor in the promotion of important neuropeptides for PNI in PDAC,32 which can drive tumor progression through the RAMP1 receptor.33 In this study, we suggest that tumor PTGES/PGE2 produces a novel paracrine effect through which the activation of SCs initiates PNI. Interference of RSC96 and sNF96.2 with exogenous PGE2 induced morphological changes in SCs, stimulated the expression of indexes associated with the dedifferentiation of SCs, and significantly increased the proliferation and migration of RSC96 and sNF96.2 cells, confirming the effect of PGE2 on SC activation promotion, which was reversed by the use of CAY10526 or siPTGES. 3D coculture of tumor cells and SCs revealed that migration of tumor cells preceded the cancer cells' migration to the periphery. CAY10526 or siPTGES attenuated the specific migration of SCs to pancreatic cancer cells, which is consistent with the findings of Deborde34 and Demir,11 which disproves the inherent belief that PNI is caused by the attack of nerves by cancer cells first. In summary, we demonstrated that SC dedifferentiation is associated with tumor PGE2 and that tumor-derived PGE2 reprograms SCs to act as an initiating signal for PNI.
From a mechanistic perspective, PGE2 signaling that induces PNI enhances the alteration of the SC dedifferentiation phenotype of pancreatic cancer cells, which is a dedifferentiated phenotype that favors PNI. Our data show that PGE2 increases the expression of the SC leukemia inhibitory factor LIF and the ADAM metallopeptidase of platelet reactive protein type 1 motif 1 (ADAMTS-1). LIF is a pleiotropic cytokine that regulates cell differentiation, self-renewal, and survival. In the nervous system, LIF exerts neurotrophic effects and promotes neuronal development and survival.35 Within the PDAC tumor microenvironment, elevated LIF levels have been shown to induce intratumoral nerve growth and facilitate tumor-associated neural remodeling. Furthermore, microenvironmental LIF binds to the LIF receptor (LIFR) on SCs, activating the JAK/STAT3/AKT signaling pathway, which in turn stimulates SC proliferation and migration.36 In addition to its effects on the nervous system, LIF promotes the growth and metastasis of various tumor types.37,38 SC-derived LIF serves as a critical intermediate messenger that mediates crosstalk between nerves and tumor cells. ADAMTS-1, a member of the metalloproteinase family, disrupts the perineurial matrix barrier, thereby providing a physical route for cancer cell infiltration along nerve fibers. Our findings confirmed that PGE2 induces ADAMTS-1 expression in SCs. ADAMTS-1 enhances cancer cell invasion through multiple mechanisms, including ECM degradation,39 cleavage of cell adhesion molecules,40 acceleration of tumor proliferation, inhibition of apoptosis, and alteration of angiogenic processes.41 In vivo studies revealed that treatment with either CAY10526 or AB-449-NA alleviated PNI-induced hindlimb paralysis and demyelination. Historically, PGE2 inhibition has targeted cyclooxygenase-2 (COX-2; e.g., celecoxib); however, its clinical application is limited by significant risks of myocardial infarction and gastrointestinal toxicity.42 In contrast, our data suggest that targeting PTGES in the prostaglandin synthesis pathway may represent a more favorable therapeutic strategy. Additionally, circulating LIF is emerging as a potential novel biomarker for pancreatic cancer, and LIF blockade has been shown to significantly attenuate tumor progression and enhance the chemotherapy response.43
In this study, activated SCs presented upregulated ADAMTS-1 expression. PGE₂ promotes nerve growth, tumor progression and ECM catabolism by upregulating LIF and ADAMTS-1, with these factors synergistically accelerating PNI. We hypothesize that PGE₂ induces SC dedifferentiation and the secretion of LIF/ADAMTS-1 to drive PNI; however, the molecular mechanisms underlying PGE₂-mediated SC dedifferentiation and LIF/ADAMTS-1 upregulation remain elusive and require further investigation. While LIF is preliminarily linked to PDAC progression and PNI, how LIF/LIFR mediates PDAC cell neurotropism remains unclear. Subsequent studies will clarify the regulatory effects of PGE₂ on LIF/ADAMTS-1 via transcription factors and histone modifications and define the role of LIF/LIFR in neurophilic switching in PDAC.
In summary, PDAC-SC crosstalk in the tumor-neural microenvironment drives PNI: PDAC-derived PGE₂ induces SC dedifferentiation, and the resulting LIF/ADAMTS-1 secretion promotes nerve growth and ECM degradation to facilitate tumor neuroinvasion. Thus, targeting the PTGES/PGE₂ or LIF microenvironment may modulate SC activity, reduce intratumoral nerve density, shorten tumor–neuron spatial proximity, and improve PDAC prognosis, suggesting a promising therapeutic strategy against PNI.
Materials and methods
Public databases for clinical testing data
H&E-stained slides from 149 patients with PDAC were retrieved from TCGA database (https://portal.gdc.cancer.gov/). PNI annotations for 23 cases were extracted from surgical pathology reports available on cBioPortal (https://www.cbioportal.org/), PNI status for the remaining 126 cases was independently evaluated by two board-certified pathologists through review of the aforementioned H&E-stained slides. For each image, corresponding clinical data, patient survival information, and raw genomic data were also obtained from the TCGA database. All diagnostic images were publicly available for research purposes and did not require institutional review board approval, as the data were publicly available. Single-cell sequencing data were obtained from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), including the GSE231535 (GSM728739, GSM7289740) and GSE154778 (GSM4679532, GSM4679533, GSM4679534, GSM4679535, GSM4679536) datasets, covering 7 PDAC single-cell samples. RNA-seq analysis data for public database PNI samples were obtained from 28 pairs of PNI+ tumor tissues and their matched paracancerous tissues in the GEO database GSE102238 dataset.
Criteria for evaluating the pathology of perineural infiltrates
The PNI scoring criteria include the following features: (a) nerve diameter; (b) number of distinct neural structures involved per tumor; (c) intratumoral vs. extratumoral nerve involvement (i.e., located within the malignant tumor or separated from the primary lesion by at least 1 mm); (d) localization pattern: focal (tumor cells encircle ≤ 50% of the nerve sheath), circumferential (tumor cells encircle ≥50% of the nerve sheath), or intraneural (if different nerves show varying degrees of involvement, the most severe grade is considered); and (e) presence or absence of mitotic figures within the perineural invasion area. Three of the five features (a, b, d) were assigned a three-tier score (0, 1, or 2), whereas the remaining two (c, e) were given a binary score of 0 or 1. Finally, these five histopathological features of the PNI were combined to generate a cumulative PNI score ranging from 0 to 8. Patients were divided into two groups on the basis of the median score: high PNI (PNI score ≥ 6), low PNI plus PNI- (PNI score < 6 and without PNI). The detailed scoring guidelines are provided in Supplementary Table 2.
Differential expression analysis and pathway enrichment analysis
The gene expression dataset GSE102238, comprising 28 pancreatic cancer samples with PNI, was downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). The preprocessed expression matrix was obtained and analyzed via the edgeR package. Differentially expressed genes (DEGs) were screened with a significance threshold defined as p ≤ 0.05 and |log2FC | ≥ 1.
Single-cell analysis
Single-cell RNA sequencing raw data (FASTQ format) were downloaded from the GEO database. The raw reads were processed via Cell Ranger v9.0.1 for quality control, alignment to the reference genome, and quantification on the basis of cellular barcodes and unique molecular identifiers (UMIs), generating quality metrics, including the number of high-quality cells, median genes per cell, and sequencing saturation. Subsequent quality control was performed via Seurat v4.0.0. The cells were filtered on the basis of the following criteria: number of detected genes > 200, UMI counts > 1000, log₁₀(genes per UMI) > 0.7, mitochondrial UMI ratio < 5%, and erythrocyte gene ratio < 5%. Potential doublets were removed via DoubletFinder v2.0.2. Highly variable genes (HVGs) were identified via the FindVariableFeatures function in Seurat. Dimensionality reduction was performed via principal component analysis (PCA) on the HVGs, followed by two-dimensional visualization via UMAP. Marker genes for each cluster were identified via the FindAllMarkers function, which detects genes that are differentially upregulated in each cluster compared with all others. Visualization of marker genes was achieved via ViolinPlot and FeaturePlot.
We extracted SCs from the scRNA-seq data and performed clustering analysis as described above to identify subsets of SCs. The gene signature for the Nomyelinating-SC subsets was obtained from OnClass, as previously described by Sun et al. (genes: COL1A2, DCN, IGFBP6, FBLN1, C1S, C1R, PLAC9, PCOLCE, COL6A2, IGFBP5, MGP, AEBP1, COL6A1, FSTL1, FBLN2, CRISPLD2, ABI3BP, FBN1, TIMP3, COL6A3, EBF1, LHFPL6, CALD1, and EFEMP1). Signature scores were calculated for each cell via the AddModuleScore function in Seurat. ViolinPlots were used to visualize the module scores of each cluster.
Kaplan‒Meier Survival Analysis and Molecular Subtype Classification
Survival outcomes were analyzed via the Kaplan‒Meier method. OS was defined as the time from initial diagnosis to death from any cause. Kaplan‒Meier curves were generated via the “Survival” program package and stratified into high PNI and low PNI plus PNI- groups. Log-rank tests, χ² statistics, and two-sided P values were used to assess differences between groups. The median OS with 95% confidence intervals (CIs) was calculated. To perform molecular subtyping of PDAC patients, we employed the single-sample gene set enrichment analysis (ssGSEA) algorithm. The analysis was based on previously established gene signatures from the research of Zhang Zemin’s team. The “classical-PDAC” subtype signatures included GATA6, FOXA2, TMEM45B, SDR16C5, GPRC5A, AGR2, EMTP, FXYD3, ST6GALNAC1, CEACAM5, CEACAM6, TFF1, TFF3, CAPN8, FOXQ1, ELF3, ERBB3, TSPAN8, TOX3, LGALS4, PLS1, GPX2, ATP10B, and MUC13. The “QM-PDAC” subtype signature included ZEB1, VIM, CDH2, AIM2, FAM26F, GPM6B, S100A2, KRT14, CAV1, LOX, SLC2A3, TWIST1, PAPPA, NT5E, CKS2, HMMR, SLC5A3, PMAIP1, PHLDA1, SLC16A1, FERMT1, HK2, and AHNAK2. Using ssGSEA of the expression profile data of the entire patient cohort, we calculated an enrichment score for each patient corresponding to each of the two subtypes. Patients were then assigned to one of two groups on the basis of their enrichment scores: the subtype with the higher enrichment score was defined as the patient’s molecular subtype. To investigate the impact of different treatments on the prognosis of patients with specific PDAC molecular subtypes, we performed treatment-stratified survival analyses within each subtype (classical-PDAC or QM-PDAC).
Patient and tissue samples and immunohistochemistry
Clinical specimens of pancreatic cancer were obtained from the Department of Pathology of the First Affiliated Hospital of Fujian Medical University with the approval of the Ethics Committee of the First Affiliated Hospital of Fujian Medical University. The ethical approval numbers for human samples were 2015084-3, [2021]008 and [2025]473. The ethical approval number for the animal experiments was IACUC FJMU 2025-Y-1980.
The tumor samples were incubated with specific primary antibodies against p75NTR (1:200, Abways, CY5411) and PTGES (1:200, Invitrogen, PA5--60916). Horseradish peroxidase-labeled goat anti-rabbit secondary antibody (ABclonal, AS014) was used. Finally, immunoreactivity was observed with DAB (UElandy, D6076S), and the samples were counterstained with hematoxylin.
The positive cell ratio was calculated to quantify p75NTR expression. Quantitative analysis of the immunohistochemical sections was performed via the image analysis software ImageJ. Five nonoverlapping fields of view (100× magnification) were randomly selected, and the percentage of the positively stained area relative to the total cellular area in each field was calculated. The average value of these percentages was used as the final data for the sample. PTGES expression was quantitatively analyzed via the H-score method. Two pathologists unaware of the sample grouping information independently evaluated the IHC sections. The selection of representative ROI regions included the normal pancreatic tissue region, the PDAC-PNI− region (containing both neural and tumor tissues simultaneously), and the PDAC-PNI+ region (the PNI area where nerves are surrounded by tumor cells). For each tissue section, 5 nonoverlapping high-power fields (100× magnification) were randomly selected. Staining intensity (intensity): 0 = no staining; 1 = low positive (light brown); 2 = moderate positive (yellowish brown); 3 = high positive (dark brown/blackish brown). Positive cell percentage (percentage, P): The percentage of positive cells corresponding to each staining intensity relative to the total number of counted cells in the ROI (range: 0–100%), denoted as P1 (percentage of weakly positive cells), P2 (percentage of moderately positive cells), and P3 (percentage of strongly positive cells). P0 (percentage of negative cells) was excluded from subsequent calculations. The H-score formula was as follows: H-score = (P1 × 1) + (P2 × 2) + (P3 × 3), with a value range of 0 ≤ H-score ≤ 300.
Cell culture and reagents
The human pancreatic cancer cell lines PANC-1 and BxPC-3 were purchased from Starfish Biotechnology Co., Ltd. The human normal pancreatic cell line HPNE was purchased from Wuhan Ponosi Life Technology Co., Ltd. The rat SC line RSC96 was purchased from Wuhan PhD Bioengineering Co. The human SC line sNF96.2 was purchased from Xiamen Yimo Biotechnology Co., Ltd. BxPC-3 cells were cultured in RPMI-1640 medium (Gibco, C11875500BT). HPNE, PANC-1, RSC96 and sNF96.2 cells were cultured in DMEM (Gibco, C11995500BT). All media were supplemented with 10% fetal bovine serum (VivaCell, C04002--500), 100 U/mL penicillin, and 100 μg/mL streptomycin (Servicebio, G4003--100ML). The cells were cultured in a humidified incubator with 5% CO₂ at 37 °C. PGE2 (Selleck, S3003) was used at concentrations of 0 nM, 0.1 nM, 1 nM, and 10 nM in subsequent experiments.
Cell migration and invasion
Cell invasion and migration assays were performed in 24-well plates with 8.0 μm pore size transparent Transwell chambers (LABSELECT, 14341). To assess the invasive ability of SCs against pancreatic cancer cells, 1×10^5 cancer cells were added to 0.2 mL of FBS-free medium in the upper chamber precoated with matrix adhesive (MCE, HYK6002). In contrast, RSC96 cells were placed at the bottom, and 0.6 mL of medium supplemented with 10% FBS was added. After incubation at 37 °C for 24 h, the cells were fixed with 4% paraformaldehyde at room temperature for 30 min, stained with crystal violet staining solution (Servicebio, G1014-50ML) for 30 min, and finally photographed with a microscope (Nikon ECLIPSE Ts2R). To assess the ability of SCs to promote the migration of pancreatic cancer cells, cancer cells were inoculated in the upper chamber without stromal gel, the lower chamber was inoculated with RSC96 cells, and the above operation was repeated. To assess the ability of pancreatic cancer cells to migrate into SCs, RSC96 cells were inoculated in the upper chamber without matrix gel, cancer cells were inoculated in the lower chamber, and the above operation was repeated.
Collection of Cell-conditioned Media
RSC96 and sNF96.2 cells were cocultured with cancer cells until they reached 70%–80% confluence in complete medium. The cells were rinsed twice with sterile phosphate-buffered saline (PBS, Servicebio, G4202-500ML) and then incubated in serum-free medium for 24 h. The culture supernatant was harvested as conditioned medium and centrifuged at 1,200 rpm and 4 °C for 10 min to pellet the cellular debris. The supernatant was subsequently filtered through a 0.22 μm nylon membrane filter (Merck Millipore, SLGV033) to remove residual floating cells and debris. The resulting conditioned medium was aliquoted and stored at −80 °C for subsequent experiments.
Induction and coculture of dedifferentiated SCs
A total of 5×105 RSC96 and sNF96.2 cells were cultured in a 4.0 μm lower chamber overnight, and then 1×105 PANC-1 cells or BxPC-3 cells were inoculated into the upper chamber of a 6-well plate (LABSELECT, 14111). The lower chamber medium was replaced with cancer cell-conditioned medium to allow for cytokine crossover in the absence of cell-to-cell contact at 37 °C and 5% CO2. Subsequent operations, such as RSC96 and sNF96.2 cell protein and RNA extraction, were performed.
Western blot
After the cells were collected, total protein was extracted from the cell lysate (Epizyme, PC101). A BCA protein concentration assay kit (Boster, AR1189) was used to determine the total protein concentration, which was separated via 10% SDS‒PAGE (Epizyme, PG112) or 15% SDS‒PAGE (Epizyme, PG114) and transferred to a PDVF membrane. The membranes were blocked with 5% skim milk and then incubated with anti-E-cadherin rabbit polyclonal antibody (1:2000, Proteintech, 20874-1-AP), N-cadherin rabbit polyclonal antibody (1:2000, Abways, CY5015), c-Jun rabbit recombinant antibody (1:2000, Selleck, F0168), SNAI1 rabbit polyclonal antibody (1:1000, Proteintech,13099-1-AP), vimentin rabbit monoclonal antibody (1:2000, Diagbio, db12686), GDNF rabbit monoclonal antibody (1:2000, Abways, CY6856), p75NTR rabbit recombinant antibody (1:2000, ABclonal, A11169), PTGES rabbit polyclonal antibody (1:2000, Invitrogen, PA5-60916), ADAMTS-1 rabbit polyclonal antibody (1:2000, Immunoway, YT0112), Sox2 rabbit monoclonal antibody (1:2000, CST, 23064 T), β-actin mouse monoclonal antibody (1:2000, Servicebio, GB12001-100), and GAPDH mouse monoclonal antibody (1:2000, Servicebio, GB12001-100) specific primary antibodies were incubated in a shaker at 4 °C overnight.
RT‒qPCR
Total RNA was extracted from cells via an RNA extraction kit (R711-01, Vazyme), and first-strand cDNA was synthesized via a reverse transcription kit (Accurate Biology, AG11728). Relative mRNA expression was detected via a fluorescence quantitative premixing kit (Accurate Biology, AG11718) on a QuantStudio 1 Plus real-time fluorescence quantitative PCR instrument for the detection of relative mRNA expression, according to the QuantStudio 1 Plus software used to obtain the Ct value of the target gene RT‒qPCR product amplified from the sample for relative quantification, and the results are expressed as 2-△△Ct values. Ct = target gene Ct value - internal reference gene Ct value, △△Ct=experimental group △Ct - control group △Ct. The primer sequences are shown in Supplementary Table 3.
RNA-seq sequencing analysis
To characterize the cellular properties of pancreatic cancer cell–SC coculture systems and elucidate the molecular alterations in SC-derived proteins induced by PGE₂, we performed transcriptome sequencing. Total RNA was isolated from pancreatic cancer cells subjected to coculture, RSC96 SCs, and PGE2-treated RSC96 cells via TRIzol reagent (Vazyme, Cat. No. R401). RNA-seq analyses were jointly conducted by Weiyu Zhihe Technology & Biotechnology Co., Ltd. (Guangzhou, China) and General Bio Co., Ltd. (Anhui, China) on the Illumina NovaSeq 6000 high-throughput sequencing platform. All the experiments were designed with three biological replicates to ensure the reliability and reproducibility of the results. Raw sequencing reads in FASTQ format were first subjected to quality control and filtering via Cutadapt software (v1.9.1) to remove adapter sequences and low-quality bases. The parameters were set as follows: Phred quality score cutoff of 20 for read trimming, maximum adapter mismatch rate of 0.1, minimum length of retained valid reads of 75 bp, and maximum allowable proportion of ambiguous nucleotides (N) of 0.1. After quality control, gene-level read counts were quantified via HTSeq software (v0.6.1). Specifically, only reads uniquely mapped to exon regions were counted, while multimapped reads and those overlapping with multiple genes were excluded from subsequent analyses. DEG analysis was implemented via DESeq2 software (v1.34.0), which employs a negative binomial distribution model for statistical analysis of read count data. For the identified DEGs, Gene Ontology (GO) enrichment analysis was performed via GOSeq software (v1.34.1), with the Wallenius approximation method applied to correct for biases caused by variations in gene length. Finally, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was conducted by mapping DEGs to the KEGG database, and a hypergeometric test was used to identify significantly enriched signaling pathways.
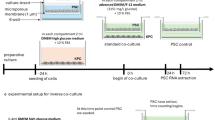
Experimental process and analysis method for the spatial transcriptome
Spatial transcriptomics analysis of human pancreatic cancer tissue samples was performed via the 10x Genomics Visium platform by Ouyi Biomedical Technology Co., Ltd. (Shanghai, China). The tissue sections were hybridized to spatial barcode probes, and total RNA was extracted for library preparation. Sequencing was performed on the NovaSeq 6000 platform, and normalization, feature detection, and spatial cell type annotation were performed via Cell Ranger and Seurat. Raw sequencing data and bright-field microscopy images from the Visium platform were processed via Space Ranger (version 2.0.1) to identify tissue capture areas, align reads to the reference genome (human: GRCh38), demultiplex reads according to spatial barcodes, and generate quality metrics, including total spot count, reads per spot, genes detected, and UMI counts. Subsequent quality control and normalization were performed via Seurat (version 4.1.0). The sctransform function was applied for normalization and variance stabilization, with highly variable features detected and data stored in the SCT assay. The top 3000 highly variable genes (HVGs) were identified via the Find VariableFeatures function. Principal component analysis (PCA) was performed on the basis of HVG expression profiles, followed by batch effect correction via the RunHarmony function from the Harmony package (version 1.0). The results were visualized in two-dimensional space via UMAP. The marker genes for each spatial cluster were identified via the FindAllMarkers function (test.use = “bimod”) to detect genes that were differentially upregulated in each cluster compared with all other clusters. The identified markers were visualized via the VlnPlot and FeaturePlot functions. DEGs between specific comparisons were identified via the FindMarkers function (test.use = “presto”) with thresholds of adjusted p value < 0.05 and absolute |log2FC | ≥ 1. GO and KEGG pathway enrichment analyses of significant DEGs were performed via hypergeometric tests.
On the basis of the spatial location of the tumor and the nerve, regions were manually annotated via Loupe Browser 8 software. The center of the nerve was defined as the PNI-center region, and the tumor region 100 μm away from the nerve (the distance between the centers of the two adjacent spot points was 100 μm) was defined as the PNI-near region (PNI-near). The areas at 200 μm and beyond were designated the PNI-mid region (PNI-mid) and the PNI-away region (PNI-away), respectively. NPNIs were also divided into NPNI-center, NPNI-near, NPNI-mid, and NPNI-away as described above, and these spots contained the spatial location of perineural tumors and gene expression information.
RCTD (version 1.1.0), a robust cell type deconvolution method, was employed to decompose cell type mixtures via cell type profiles derived from single-cell RNA-seq data while correcting for cross-platform sequencing differences. During execution, the create. The RCTD function was run with default parameters, requiring a minimum of one cell per cell type and at least one unique molecular identifier (UMI) per spot. The run. The RCTD function was executed with doublet_mode = FALSE to infer the cellular composition within each spot. Gene set background files were first downloaded and organized from the KEGG database (https://www.kegg.jp/) and the GO database (https://geneontology.org/) via the GSEABase package (version 1.44.0). Pathway activity scores for individual cells were then calculated via the GSVA package (version 1.30.0). Finally, the LIMMA package (version 3.38.3) was used to compute signaling differences between defined groups.
Peptide staining
EdU proliferation assays were performed using an EdU Imaging Kit (APExBIO, K2243). Nuclei were labeled with DAPI (Servicebio, G1012-10ML) and visualized at an excitation wavelength of 594 nm. Photographs were taken using a Leica inverted fluorescence microscope (Leica, DMi8). The ratio of the number of EdU-positive cells to the total number of DAPI-positive cells was calculated as a measure of the extent of EdU adulteration.
Cytoskeletal F-actin was stained with 488-labeled ghost pen cyclic peptide (1:200, UElandy, YP0059S), and the nuclei were labeled with DAPI by incubation for 20 min at 37 °C in the dark according to the manufacturer’s instructions. Images were captured via confocal microscopy (Olympus FV-3000).
3D Coculture Migration Experiments
3D migration assays were performed with HPNE cells, PANC-1 cells, BxPC-3 cells and RSC96 cells. Each cell line was resuspended at 2 × 106 cells/mL in a cold ECM gel (Mogengel Bio, 082733), and droplets of 2 × 10^4 cells/10 μL were placed in 12-well tissue culture plates 1 mm apart from each other and connected by ECM bridges. ECM droplets containing RSC96 cells were used as blanks, and cocultures of HPNE cells and RSC96 cells were used as the control group. After ECM gel hardening at 37 °C for 15 min, DMEM supplemented with 1% FBS was added to each well, and the cancer cells were treated with or without 25 μM PTGES inhibitor (MCE, CAY10526), siPTGES or PTGES-KO. The migration of 3D cultures was monitored at different time points via an inverted microscope (Nikon ECLIPSE Ts2R).
Extraction and coculture of DRG explants
Six-week-old Sprague‒Dawley rats were euthanized by cervical dislocation, and their abdominal skin was sterilized with 75% alcohol. Surgery was performed via a ventral approach: the abdomen was incised on a sterile operating table to expose the abdominal cavity, and intra-abdominal organs were removed en bloc. The sternum was incised to expose the thoracic cavity, and the spinal cord was exposed after the thoracic organs were removed. DRGs, which appear as small nodular structures on each nerve, were extracted one by one under a microscope, placed in PBS containing two antibodies, and then transferred to DMEM containing two antibodies for later use.
An appropriate amount of melted Matrigel was mixed with an equal volume of DMEM in an EP tube. All pipette tips and EP tubes in contact with Matrigel were precooled. Individual DRGs were placed on sterile circular coverslips, and 20 μL of Matrigel was added dropwise onto each DRG and carefully spread over the entire coverslip. All operations were performed on ice. Coverslips were placed in 12-well cell culture plates, and 5 × 104 cancer cells were added 1 mm around each DRG. The plates were incubated in a cell culture incubator for 30 min to allow for Matrigel solidification. Subsequently, 1 mL of 10% FBS-containing medium was added to each well and incubated overnight at 37 °C in an incubator.
ELISA
The secretion of PGE2 in the cell culture supernatant was measured via a human PGE2 ELISA kit (JONLNBIO, JL11266) according to the manufacturer’s protocol. Briefly, serum-free conditioned medium (CM) was collected from PANC-1 and BxPC-3 cells following 24 h of coculture with either RSC96 or sNF96.2 cells. A volume of 100 μL of standard or sample was added to each well, followed by incubation at room temperature for 2.5 h. After the samples were washed, 100 μL of the prepared biotinylated antibody was added to each well, and the samples were incubated at room temperature for 1 h. Following another wash step, 100 μL of prepared streptavidin solution was added to each well and incubated at room temperature for 45 min. After further washing, 100 μl of TMB one-step substrate reagent was added to each well, and the mixture was incubated at room temperature for 30 min. Then, 50 μL of stop solution was added to each well. Finally, the absorbance was measured at 450 nm via a microplate reader.
Mouse Model of Sciatic Nerve Invasion
Nude mice (approximately 4‒6 weeks old) were purchased from Fujian ZaiJi Biotechnology Co., Ltd. All the animal experiments were approved by the Institutional Animal Care and Use Committee (Approval No. IACUC FJMU 2025-Y-1980.) This study was conducted in strict accordance with the Guide for the Care and Use of Laboratory Animals. The mice were anesthetized, and the right lower limb was sterilized. A 3–4 mm incision was made along the femoral shaft approximately 1–2 mm below the femoral head. Muscles were gently separated via blunt dissection along their fibers to locate and expose the sciatic nerve. Control mice (n = 5) received perisciatic injections of 1 million BxPC-3 cells (1:1 mixed with Matrigel), whereas the experimental group (n = 5) received injections of 1 million BxPC-3 cells that had been preconditioned by coculture with SCs. The incision was then sutured, and tumor formation was monitored dynamically. Once the tumors became palpable (approximately 1-week post-injection), drug interventions were initiated. The mice received intratumoral injections of either 200 μM CAY10526 (10 μL, MCE, HY-118119) or LIF neutralizing antibody (10 μL, R&D Systems, AB-449-NA) every three days for three weeks. Sciatic nerve function was assessed weekly via a hindlimb motor scoring system (4: normal extension response, 1: complete paralysis). At the endpoint, the mice were euthanized by cervical dislocation. Tumors were harvested, and the tumor nerve junction was carefully dissected and fixed in 2.5% glutaraldehyde (Servicebio, G1102) for transmission electron microscopy (TEM) analysis.
Perineural invasion assay
DRG cells were seeded in growth factor-reduced Matrigel. Pancreatic cancer cells were labeled with 25 μM fluorescent CellTracker Red CMTPX (GLPBIO, GC26327) at 37 °C for 1 h and then cocultured with DRG explants at a density of (1–2) × 105 cells per drop for 5 days. At the experimental endpoint, the culture medium was removed, and the samples were washed three times with PBS. Fluorescence images were acquired via a fluorescence microscope. PC cells invading the nerves were defined as fluorescent cancer cells associated with neurites extending into the Matrigel. The extent of PNI by PDAC cells was quantified by measuring the area of fluorescent cancer cell invasion into the Matrigel in tumor–nerve cocultures via ImageJ software.
siRNA Interference and establishment of the PTGES frameshift-mutated BxPC-3 cell line
Small interfering RNA (siRNA) targeting PTGES was purchased from General Bio (Anhui, China) Corp. Ltd. Transient transfection was performed via LipoZG RNAi (Zolgene) according to the manufacturer’s instructions. The knockdown efficiency was validated by Western blot analysis.
The PTGES frameshift-mutated BxPC-3 cell line was generated by General Bio Co., Ltd. (Anhui, China). The endonuclease activity of the CRISPR/Cas9 system allows it to cleave the genomic DNA at the target locus of the PTGES gene, resulting in double-strand breaks (DSBs). When cells repair broken DNA via the nonhomologous end joining (NHEJ) pathway, random base insertions or deletions (indels) are introduced, which further lead to frameshift mutations in the PTGES gene and ultimately cause loss of gene function. Cas9 protein and gRNA were preassembled into ribonucleoprotein (RNP) complexes, which were then delivered into BxPC-3 cells via electroporation. At 48–72 h post-transfection, single-cell sorting was performed, and the obtained single-cell clones were cultured and expanded in 96-well plates. The genomic DNA of the cells was extracted, and PCR amplification was performed via specific primers flanking the target exons. The amplified products were subjected to sequencing validation. The verified homozygous knockout clones were subjected to expansion culture and quality control prior to being used in subsequent experiments. The sequence information of all gRNAs used in this study is provided in Supplementary Table 4.
Cellular Immunofluorescence
Sterile cell climbing slides were placed in culture plates and seeded with RSC96 or sNF96.2 cells. After cell adhesion, PGE2 was added to the treatment groups, while the control groups received no treatment. After 24 h, the slides were retrieved and fixed with precooled 4% paraformaldehyde at room temperature for 15 min. Permeabilization was then performed with 0.5% Triton X-100 at room temperature for 10 min to increase membrane permeability. Subsequently, blocking was carried out with 5% BSA in PBS at room temperature for 1 h to prevent nonspecific binding. Following blocking, the slides were incubated overnight at 4 °C in a humidified chamber with primary antibodies against SOX2 (1:200, Diagbio, db16528), c-Jun (1:200, Selleck, F0168), and P75NTR (1:200, ABclonal, A11169). The next day, after thorough washing with PBST, the slides were incubated with the corresponding Alexa Fluor 488-conjugated secondary antibodies (Servicebio, GB25303) at room temperature for 1 h in the dark. Finally, the slides were mounted with anti-fade mounting medium containing DAPI (Servicebio, G1407) and imaged under a fluorescence microscope.
Multiple immunofluorescence
For mIHC staining, formalin-fixed sections were stained with antibodies against PGP9.5 (1:200, Abways, CY6722), MUC1 (1:200, BOSTER, BM4548), SOX2 (1:200, Diagbio, db16528), c-Jun (1:200, Selleck, F0168), and p75NTR (1:200, ABclonal, A11169) according to the manufacturer’s instructions for the multicolor immunofluorescence kit (Zolgene, PR-004507) and imaged with a KFbio Pathology Imaging System (KF-FL-005).
Transmission electron microscopy
For transmission electron microscopy (TEM) analysis, the following procedures were performed. First, targeted fresh tissues were selected to minimize mechanical damage (e.g., pulling, contusion, extrusion), and fresh tissue blocks were quickly cut and harvested via a sharp blade within 1–3 min (tissue block size ≤1 mm³). Prior to sampling, Petri dishes containing TEM fixative were prepared in advance; small tissue blocks were removed from the animal body, immediately placed into Petri dishes, cut into 1 mm³ pieces in the fixative, transferred to an EP tube with fresh TEM fixative, fixed at 4 °C for preservation and transportation, and subsequently washed with 0.1 M phosphate buffer (PB, pH 7.4) 3 times (15 min each). Next, the tissues were postfixed with 1% OsO4 in 0.1 M PB (pH 7.4) for 2 h at room temperature in the dark; after removing OsO4, the tissues were rinsed with 0.1 M PB (pH 7.4) 3 times (15 min each). Dehydration was conducted at room temperature as follows: 30% ethanol (20 min), 50% ethanol (20 min), 70% ethanol (20 min), 80% ethanol (20 min), 95% ethanol (20 min), two changes of 100% ethanol (20 min each), and finally two changes of acetone (15 min each). For resin penetration and embedding, the tissues were treated with acetone:EMB 812 (1:1) at 37 °C for 2–4 h, acetone:EMB 812 (1:2) at 37 °C overnight, and pure EMB812 at 37 °C for 5–8 h. Then, pure EMB812 was poured into the embedding models, the tissues were inserted, and the models were kept in a 37 °C oven overnight. Polymerization was carried out by moving the embedding models with resin and samples into a 60 °C oven for more than 48 h, after which the resin blocks were removed from the models and set aside at room temperature for later use. For positioning, the resin blocks were sectioned into 1.5 μm semithin slices via a semithin microtome, stained with toluidine blue, and positioned under a light microscope. Ultrathin sections (60–80 nm) were cut from the resin blocks via an ultramicrotome, and the tissues were fished onto 150-mesh copper grids with formvar film. The staining steps included staining with 2% uranyl acetate saturated alcohol solution in the dark for 8 min, rinsing with 70% ethanol 3 times and then ultrapure water 3 times, and staining with 2.6% lead citrate (avoiding CO₂) for 8 min and rinsing with ultrapure water 3 times. After drying with filter paper, the copper grids were placed on a grid board and dried overnight at room temperature. Finally, the copper grids were observed via transmission electron microscopy (TEM), and images were captured.
Statistical analysis
The IHC-positive area was calculated via ImageJ (Fiji). The data were processed via GraphPad Prism 9.0 software and are presented as the means ± SDs. Two-tailed unpaired Student’s t test was used for comparisons between two independent groups; one-way analysis of variance followed by Tukey’s HSD post hoc test was used for comparisons among three or more groups with equal variances; the Kruskal‒Wallis H test followed by Dunn’s multiple comparisons test was used for nonparametric comparisons among three or more groups; and the Mann‒Whitney U test was used for nonparametric comparisons between two independent groups. Differences were considered statistically significant when p < 0.05.
Data availability
All data supporting the findings of this study are readily available from the corresponding author upon reasonable request. The H&E-stained slides images from 149 PDAC patients and corresponding clinical data and patient survival information were openly obtained from the TCGA database (https://portal.gdc.cancer.gov/), while partial pathological reports on PNI were obtained from the cBioPortal database (https://www.cbioportal.org/). The RNA-seq and spatial transcriptomic data generated in this study have been deposited in the Genome Sequence Archive (GSA) database (https://ngdc.cncb.ac.cn/gsa/) under accession numbers CRA032340 (RSC96 RNA-seq)(https://ngdc.cncb.ac.cn/gsa/search?searchTerm=CRA032340), HRA014324 (PANC-1 RNA-seq) (https://ngdc.cncb.ac.cn/gsa-human/browse/HRA014324) and HRA014327 (pancreatic cancer spatial transcriptomics) (https://ngdc.cncb.ac.cn/gsa-human/browse/HRA014327).
References
Park, W., Chawla, A. & O’Reilly, E. M. Pancreatic cancer: a review. JAMA 326, 851–862 (2021).
De Dosso, S. et al. Treatment landscape of metastatic pancreatic cancer. Cancer Treat. Rev. 96, 102180 (2021).
Liebl, F. et al. The impact of neural invasion severity in gastrointestinal malignancies: a clinicopathological study. Ann. Surg. 260, 900–907 (2014). 907-908.
Hirai, I. et al. Perineural invasion in pancreatic cancer. Pancreas 24, 15–25 (2002).
Schorn, S. et al. The influence of neural invasion on survival and tumor recurrence in pancreatic ductal adenocarcinoma—a systematic review and meta-analysis. Surg. Oncol. 26, 105–115 (2017).
Mitsunaga, S. et al. Detail histologic analysis of nerve plexus invasion in invasive ductal carcinoma of the pancreas and its prognostic impact. Am. J. Surg. Pathol. 31, 1636–1644 (2007).
Ferdoushi, A. et al. Tumor innervation and clinical outcome in pancreatic cancer. Sci. Rep. 11, 7390 (2021).
Demir, I. E., Friess, H. & Ceyhan, G. O. Neural plasticity in pancreatitis and pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 12, 649–659 (2015).
Li, F. et al. Glutamate from nerve cells promotes perineural invasion in pancreatic cancer by regulating tumor glycolysis through HK2 mRNA-m6A modification. Pharmacol. Res. 187, 106555 (2023).
Weitz, J. et al. Pancreatic ductal adenocarcinoma induces neural injury that promotes a transcriptomic and functional repair signature by peripheral neuroglia. Oncogene 42, 2536–2546 (2023).
Demir, I. E. et al. Investigation of Schwann cells at neoplastic cell sites before the onset of cancer invasion. J. Natl. Cancer Inst. 106, dju184 (2014).
Huang, T. et al. Schwann cell-derived CCL2 promotes the perineural invasion of cervical cancer. Front. Oncol. 10, 19 (2020).
Zhang, B. et al. Tumour-associated macrophages and Schwann cells promote perineural invasion via paracrine loop in pancreatic ductal adenocarcinoma. Br. J. Cancer 130, 542–554 (2024).
Santiso, A., Heinemann, A. & Kargl, J. Prostaglandin E2 in the tumor microenvironment, a convoluted affair mediated by EP receptors 2 and 4. Pharmacol. Rev. 76, 388–413 (2024).
Kissoondoyal, A. & Crawford, D. A. Prostaglandin E2 increases neurite length and the formation of axonal loops, and regulates cone turning in differentiating NE4C cells via PKA. Cell. Mol. Neurobiol. 42, 1385–1397 (2022).
Hiruma, H. et al. Prostaglandin E(2) enhances axonal transport and neuritogenesis in cultured mouse dorsal root ganglion neurons. Neuroscience 100, 885–891 (2000).
Kantarci, H. et al. Schwann cell-secreted PGE(2) promotes sensory neuron excitability during development. Cell 187, 4690–4712.e30 (2024).
Guo, J. A. et al. Pan-cancer transcriptomic predictors of perineural invasion improve occult histopathologic detection. Clin. Cancer Res. 27, 2807–2815 (2021).
Nozzoli, F. et al. Perineural invasion score system and clinical outcomes in resected pancreatic cancer patients. Pancreatology 24, 553–561 (2024).
Schiavo Lena, M. et al. Quantification of perineural invasion in pancreatic ductal adenocarcinoma: proposal of a severity score system. Virchows Arch. 483, 225–235 (2023).
Wong, F. C. et al. Extracellular vesicles from pancreatic cancer and its tumour microenvironment promote increased Schwann cell migration. Br. J. Cancer 132, 326–339 (2025).
Kim, Y. H. et al. p75 and neural cell adhesion molecule 1 can identify pathologic Schwann cells in peripheral neuropathies. Ann. Clin. Transl. Neurol. 6, 1292–1301 (2019).
Kanda, H. et al. Localization of prostaglandin E2 synthases and E-prostanoid receptors in the spinal cord in a rat model of neuropathic pain. Brain Res. 1750, 147153 (2021).
Knudsen, N. H. & Manguso, R. T. Tumor-derived PGE2 gives NK cells a headache. Immunity 53, 1131–1132 (2020).
Cuenca-Escalona, J. et al. Distinct roles of PGE2 signaling via EP2 and EP4 in circulating pDCs: implications for immune modulation in the tumor microenvironment. J. Leukoc. Biol. 117, qiaf034 (2025).
Cervantes-Villagrana, R. D., Albores-García, D., Cervantes-Villagrana, A. R. & García-Acevez, S. J. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct. Target. Ther. 5, 99 (2020).
Sinha, P., Clements, V. K., Fulton, A. M. & Ostrand-Rosenberg, S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res 67, 4507–4513 (2007).
Mizuno, R., Kawada, K. & Sakai, Y. Prostaglandin E2/EP signaling in the tumor microenvironment of colorectal cancer. Int. J. Mol. Sci. 20, 6254 (2019).
Gubbala, V. B. et al. Eicosanoids in the pancreatic tumor microenvironment - a multicellular, multifaceted progression. Gastro Hep Adv. 1, 682–697 (2022).
Maeda, T. et al. Opioid modulation of T-type Ca(2+) channel-dependent neuritogenesis/neurite outgrowth through the prostaglandin E(2)/EP(4) receptor/protein kinase A pathway in mouse dorsal root ganglion neurons. Biochem. Biophys. Res. Commun. 639, 142–149 (2023).
Wu, M. et al. Innervation of nociceptor neurons in the spleen promotes germinal center responses and humoral immunity. Cell 187, 2935–2951.e19 (2024).
Wang, K. et al. Nociceptor neurons promote PDAC progression and cancer pain by interaction with cancer-associated fibroblasts and suppression of natural killer cells. Cell Res. 35, 362–380 (2025).
Zhi, X. et al. Nociceptive neurons promote gastric tumour progression via a CGRP-RAMP1 axis. Nature 640, 802–810 (2025).
Deborde, S. et al. Schwann cells induce cancer cell dispersion and invasion. J. Clin. Invest. 126, 1538–1554 (2016).
Chen, Q., Liu, Q., Zhang, Y., Li, S. & Yi, S. Leukemia inhibitory factor regulates Schwann cell proliferation and migration and affects peripheral nerve regeneration. Cell Death Dis. 12, 417 (2021).
Bressy, C. et al. LIF drives neural remodeling in pancreatic cancer and offers a new candidate biomarker. Cancer Res. 78, 909–921 (2018).
Zhang, F. et al. TGF-β-driven LIF expression influences neutrophil extracellular traps (NETs) and contributes to peritoneal metastasis in gastric cancer. Cell Death Dis. 15, 218 (2024).
Yin, S. et al. Solamargine induces hepatocellular carcinoma cell apoptosis and autophagy via inhibiting LIF/miR-192-5p/CYR61/Akt signaling pathways and eliciting immunostimulatory tumor microenvironment. J. Hematol. Oncol. 15, 32 (2022).
Tang, B. L. ADAMTS: a novel family of extracellular matrix proteases. Int. J. Biochem. Cell. Biol. 33, 33–44 (2001).
Chien, M. H. et al. Cyclic increase in the ADAMTS1–L1CAM–EGFR axis promotes the EMT and cervical lymph node metastasis of oral squamous cell carcinoma. Cell Death Dis. 15, 82 (2024).
Tan Ide, A., Ricciardelli, C. & Russell, D. L. The metalloproteinase ADAMTS1: a comprehensive review of its role in tumorigenic and metastatic pathways. Int. J. Cancer 133, 2263–2276 (2013).
Trelle, S. et al. Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ 342, c7086 (2011).
Shi, Y. et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 569, 131–135 (2019).
Funding
This work was supported by the National Natural Science Foundation of China (No. 82172953 and 82090053), the National Key Clinical Specialty Construction Project Fund (No. 2022YBL-ZD-02) and the National Natural Science Foundation of Fujian (2022Y2004, 2023Y9052, 2025J01109, and 2024Y9158). These funding sources played key supportive roles in sample collection, molecular analysis of patient samples, and bioinformatics analysis.
Author information
Authors and Affiliations
Contributions
J.H.D., Q.C.L., L.W., S.G.W., and F.G. contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Q.C.L., Z.B.Z, S.Z.Y., Y.Q.Z., L.W., J.T., G.B.P., Y.T.Z., S.G.W., and F.G. The first draft of the manuscript was written by L.W., Q.M.W., F.G. and Q.C.L. All the authors commented on previous versions of the manuscript. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
All the techniques used in this study met the requirements of the Medical Ethics Committee of the First Affiliated Hospital of Fujian Medical University. The ethical approval numbers for the human samples were 2015 084-3, [2021]008 and [2025]473. The ethical approval for the animal experiments was IACUC FJMU 2025-Y-1980.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, L., Liu, Q., Zhang, Z. et al. Prostaglandin E2-driven dedifferentiation of Schwann cells leads to perineural invasion in pancreatic ductal adenocarcinoma. Sig Transduct Target Ther 11, 122 (2026). https://doi.org/10.1038/s41392-026-02648-x
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41392-026-02648-x
