
Abstract
Cellular senescence and its senescence-associated secretory phenotype (SASP) represent a pivotal role in the development of skeletal diseases. Targeted elimination or rejuvenation of senescent cells has shown potential as a therapeutic strategy to reverse age-related skeletal senescence and promote bone regeneration. Meanwhile, other age-related mechanisms, involving altered cellular functions, impaired intercellular crosstalk, disturbed tissue microenvironment, and decreased regenerative capacity, synergistically contribute to the pathogenesis. In this review, we outline the cellular senescence and other age-related mechanisms in developing skeletal diseases, including osteoporosis, intervertebral disc degeneration, osteoarthritis, rheumatoid arthritis, bone tumors and ankylosing spondylitis, with the aim of comprehensively understanding their detrimental effects on the aged skeleton and screening the potential targets for anti-aging therapy within the skeletal system.

Similar content being viewed by others
Introduction
The skeleton, as a metabolically active and continuously remodeling tissue, serves to protect internal organs and support body exercise.1 It also has active endocrine functions that closely regulate the health of the body. As a reservoir of various types of cells, the collaborative efforts of different cells in the skeletal system promote the establishment of bone homeostasis.2 However, cellular senescence and other age-related mechanisms synergistically lead to impaired bone cell function, facilitating the onset and progression of bone diseases, such as osteoporosis (OP), intervertebral disc degeneration (IVDD), and osteoarthritis (OA). Cellular senescence is a particular cellular state characterized by irreversible arrest of the cell cycle and the emergence of a distinctive senescence-associated secretory phenotype (SASP), which is one of the key mechanisms in the development and progression of skeletal diseases.3 In addition, other age-related mechanisms occur in the skeletal system that synergistically contribute to the development of bone diseases. For example, the impaired intercellular crosstalk leads to an abnormal accumulation of senescence phenotypes in the bone marrow,4 the generation of a disturbed microenvironment promotes senescence in the skeletal system.5
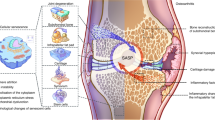
The elderly population has a high prevalence of age-related bone diseases.6 Particularly in elderly individuals (aged 65 and above), age-related bone diseases represent the leading cause of disability worldwide.7 The associated pain and limited mobility lead to a decrease in quality of life, posing a significant healthcare burden to the government.8 Prospects for the development of drugs targeting skeletal aging diseases are currently not optimistic, largely owing to a lack of understanding of the cellular senescence and other age-related mechanisms that lead to bone dysfunction during aging.9 In this review, we summarize the current understanding of cellular senescence and other age-related mechanisms in the pathogenesis of bone diseases, highlighting the diversity of the mechanisms involved in cellular senescence within different skeletal aging microenvironments. Moreover, we provide an overview of therapeutic approaches involving selective elimination or the reversion of cellular senescence (Fig. 1).

Cellular senescence and other age-related mechanisms in skeletal diseases. The synergistic action of various cells maintains bone homeostasis. Cellular senescence and other age-related mechanisms lead to impaired function of bone cells, contributing to the onset and progression of bone disorders, including osteoporosis (OP), intervertebral disc degeneration (IVDD), osteoarthritis (OA), bone tumors, and ankylosing spondylitis (AS)
Cellular senescence and other age-related mechanisms in osteoporosis
As a complex and dynamic organ, bone is maintained by the coordination of multiple cell types, including bone marrow mesenchymal stem cells (BMSCs), osteoblasts (OBs), osteoclasts (OCs), osteocytes (OCYs), adipocytes (ADs), immune cells, etc.10 As rich cellular reservoirs, the various bone cells in the bone marrow undergo a cellular change and cause bone aging as the lifespan increases, ultimately leading to the loss of bone integrity and the development of OP.11,12 The cellular senescence and other age-related mechanisms cause OP through various mechanisms involving different cell types.
Bone marrow mesenchymal stem cells
Recruiting BMSCs to sites of bone destruction and differentiating them toward osteogenic lineage is an approach for maintaining bone homeostasis.13 BMSCs obtained from elderly individuals exhibit replicative senescence and decreased osteogenic potential.14 Advancing age induces changes in the differentiation potential of BMSCs, with a tendency toward adipogenic differentiation, leading to a lack of new bone supply and the accumulation of ADs in the bone marrow.15
Single-cell RNA sequencing (scRNA-seq) analysis has recently been applied to reveal changes in the characteristics of BMSCs during aging. BMSCs labeled with fibroblast growth factor receptor 3 (Fgfr3)-creER exhibit strong colony-forming ability at a young age, but this ability is lost when these cells are aged.16 In addition, leptin receptor (LepR)-cre-labeled BMSCs presented an increase in the proportion of colony-forming cells with advancing age.16 These changes may reduce the osteogenic differentiation of BMSCs during aging and contribute to the development of OP. Senescence triggers the differentiation of C-X-C motif chemokine ligand 12+ (CXCL12+) LepR+ BMSCs toward a lipogenic cell lineage, which provides a rationale for ADs accumulation in aged bone marrow.17,18 Recent work has provided an in-depth analysis of human BMSCs (hBMSCs) via scRNA-seq, which classified these cells into 10 distinct subpopulations. In particular, functional enrichment analysis of a specific sub-population expressing P62 revealed a series of biological processes, including cellular redox homeostasis, the endoplasmic reticulum stress response, and the inhibitory regulation of cell growth, that may be involved in BMSC aging.19 scRNA-seq also revealed a notable decline in the paired related homeobox 1Cre+OsterixCre− (Prx1Cre+OsxCre−) sub-population of BMSCs in Prx1-Cre; Zmpste24f/f (Z24f/f) mice that exhibited premature aging.20 This sub-population has been observed to undergo activation of apoptotic signaling pathways and a reduction in mechanosensation, both of which are strongly linked to age-related bone loss. hBMSCs with premature aging syndrome were generated by knocking out Werner syndrome protein, and they exhibited reduced self-renewal capacity and increased senescence-associated-β-galactosidase (SA-β-gal) activity. A genome-wide CRISPR activation screen utilizing hBMSCs in a senescence model demonstrated that the deletion of sry-box transcription factor 5 (SOX5) resulted in a reduction in the enhancer activity of high mobility group box 2 (HMGB2) and a decrease in the levels of H3K27ac and H3K4me1, thereby facilitating cellular senescence.21 Data support the hypothesis that the in vitro and in vivo inhibition of transcription factor E Box-binding (TFEB) nuclear translocation-mediated autophagy results in the induction of a senescent state in BMSCs, thereby contributing to the development of aging-associated bone loss.22 Recent studies have proposed that epigenetic changes are also significant causes of aging.23,24,25 The expression of the DNA methyltransferases DMNT-1 and DNMT-3B, which increase the expression of p16 and p21, is significantly reduced in senescent BMSCs. In senescent BMSCs, a significant increase in the active chromatin marker H3K4me1 is found in hypomethylated regions, which underscores the role of histone methylation in the process of age-related cellular senescence.26
Extracellular vesicles (EVs) secreted by skeletal myocytes are taken up by BMSCs through the blood circulation. A significant increase in the expression of miRNA-34a-5p has been observed in EVs derived from both senescent human myotubular cells and mouse skeletal muscle cells, which induces senescence in BMSCs.4 A study further demonstrated that lactate dehydrogenase in EVs secreted by young skeletal muscle cells promotes osteogenesis by activating the glycolytic pathway in BMSCs,27 thus establishing the foundation for bone loss therapies for the elderly based on skeletal muscle cell-BMSCs crosstalk. Endothelial cells (ECs), immune cells, and BMSCs are closely related to the OP microenvironment. Vascular ecological niche analyses in older populations and mice demonstrated increased secretion of miRNA-31-5p in the senescent ECs, which inhibits BMSCs’ osteogenesis by suppressing wingless-related integration site (WNT) signaling.28 Macrophages (MACs) in the OP microenvironment release oxidatively damaged mitochondria. The transfer of mitochondria from MACs to BMSCs triggers a burst of reactive oxygen species (ROS), leading to restricted osteogenic differentiation of BMSCs.29 Notably, senescent BMSCs show increased SASP secretion, which promotes the aged bone marrow microenvironment.30 Senescent BMSCs also facilitate OCs maturation and myeloid skewing of hematopoietic stem and progenitor cells via the SASP.5 These findings provide important insights into the functional changes in BMSCs during aging and may provide new targets and approaches to promote bone regeneration.
Osteoblasts
OBs are intermediate cells derived from BMSCs’ osteogenic differentiation, and their senescence is closely associated with abnormal bone metabolism and OP occurrence.31,32 Senescent OBs may help form a malignant microenvironment, as young mice injected with OBs from aged mice exhibit severe bone loss.33,34 Notably, after OBs from young mice were directly injected into the bone marrow cavity of aged mice through bone marrow transplantation, the aged mice presented increased histological trabecular bone and bone mineral density.35 The reversal of OB senescence has potential in the treatment of OP.
One recent study revealed that CD24+ OBs exhibit significant growth arrest, SA-β-gal positivity, and osteogenesis restriction, as shown by time-of-flight mass cytometry.36 Investigators have demonstrated that the elimination of CD24+ OBs in the aged mice suggests the concept that senescent cell elimination may improve aspects of skeletal aging. The production of lymphoid progenitor cells in the periphery was found to be supported by a specific type of osteoblastic progenitor cell in the bone marrow, namely osteolectin-positive (Osteolectin+) cells. Following reduced exercise due to aging, Osteolectin+ cells exhibit less proliferation, decreased bone formation, and reduced lymphocytes, which ultimately lead to poor bone quality and diminished immune function.37 This population of cells provides a novel therapeutic target for intervention in bone aging. The senescence of OBs is regulated by multiple pathways. Mechanistic target of rapamycin complex 1 (mTORC1) was demonstrated to induce membrane potential depolarization by modulating the sodium channel Scn1a, thereby accelerating pre-OBs senescence by increasing Ca²+ influx and activating downstream NFAT/ATF3/p53 signaling.38 It has been shown that methyltransferase-like 3 (METTL3) deficiency in senile OP promotes OB senescence by decreasing Hspa1a mRNA stability in a YTH N6-methyladenosine RNA-binding protein 2 (YTHDF2)-dependent manner.39 In the past decade, the “oxidative stress theory of aging” has attracted widespread attention. The excessive accumulation of ROS in the bone marrow microenvironment induces OB malfunction by activating the forkhead box O (FOXO) pathway and blocking Wnt signal transduction.40,41 Moreover, senescent OBs exhibit reduced responsiveness to insulin-like growth factor (IGF)-I, thereby inhibiting osteogenic proliferation and differentiation.42
Direct cell-to-cell contact between OBs and OCs is achieved through the interaction of the Ephrin B2 (EFNB2)-EPHB4, FAS-FAS ligand (FASL), and Neuropilin-1 (NRP1)-Semaphorin 3A (SEMA3A) signaling pathways, which in turn regulate cell proliferation and differentiation.43 In the bone microenvironment, OBs secrete a variety of key factors, including macrophage colony-stimulating factor (M-CSF), receptor activator of nuclear factor-κB ligand (RANKL), and WNT5A, which promote OCs formation.43 An RNA sequencing data suggests that schnurri-3 (SHN3) secreted by OBs inhibits H-type blood vessel formation through negative regulation of the transcription and expression of the angiogenic factor SLIT3, consequently contributing to the development of OP.44 The present work reveals the OBs-ECs crosstalk and its important role in bone regeneration. Substantial evidence suggests that the OBs-immune cell crosstalk also plays a central role in osteogenesis. The CD39-CD93-adenosine receptor (AdoR) signaling pathway, which is reduced by Treg cells, directly inhibits OBs’ osteogenesis.45 Future studies need to further explore the potential cellular and molecular mechanisms linking OBs senescence and OP development, hopefully providing new insights into the development of targeted therapeutic strategies and new perspectives for clinical treatment.
Osteoclasts
Aged bone shows enhanced bone resorption mediated by hematopoietic-derived OCs under osteoporotic conditions. In aged animals, OCs are overactivated, resulting in a greater capacity for bone removal than young bones do, which ultimately leads to negative bone homeostasis in the aged skeletal system.46,47,48 The development of OCs is dependent on crosstalk with OBs, OCYs, BMSCs, and even ADs. Reports indicate that increased secretion of RANKL and M-CSF, as well as decreased secretion of osteoprotegerin (OPG), in OBs, OCYs, and BMSCs in aging individuals promote OCs maturation and activation.49,50,51,52 Therefore, it seems that aging promotes increased OCs abundance and activation by regulating the RANKL/OPG axis, which leads to accelerated bone loss. In addition, bone marrow ADs have been shown to act on OCs activation through RANKL secretion.53,54 Unfortunately, the mechanism of ADs-OCs crosstalk remains to be elucidated in elderly individuals, and its role in bone aging requires further investigation.
Recent findings have confirmed a novel form of communication between OCs and BMSCs. Analysis of OCs-derived EVs has revealed an enrichment of thrombin cleavage-secreted phosphoprotein 1 (SPP1), and SPP1 deficiency inhibits osteogenic differentiation of BMSCs by reducing TGFβ1/SMAD3 signaling.55 Furthermore, OCs have been shown to influence OBs’ behavior by secreting different soluble factors. On the one hand, RANKL-stimulated OCs secrete more sphingosine 1 phosphate (S1P), which binds to the S1P receptor on the surface of OBs, leading to increased OBs proliferation and bone formation.56 Collagen triple helix repeat containing 1 (CTHRC1) released by OCs also induced OBs differentiation by targeting stromal cells, and knockdown of CTHRC1 expression in OBs led to a significant decrease in bone formation.57 On the other hand, OCs-derived semaphorin 4D (SEMA4D) binds to plexin-b1 (PLXNB1) on the surface of OBs, inhibiting OBs differentiation and accelerating bone loss.58 Studies have identified angiopoietin (ANG) secreted by OCs as a key factor in preventing senescence of neighboring ECs.59 ANG inhibits ECs’ senescence through plexin B2 (PLXNB2)-mediated ribosomal RNA transcription. OCs-ECs crosstalk provides a new molecular basis for studies of bone and vascular regeneration. Notably, changes in the bone matrix also contribute to changes in OCs activity. The presence of type I collagen β-isomerization of C-telopeptide in the bone matrix is considered to induce OC activation, and its content is three times greater in aged bones than in young bones.60 These findings suggest that changes in the aged bone matrix enhance the ability of OCs to resorb. Interestingly, in vitro experiments did not reveal an increase in or activation of OCs when these cells were co-cultured with the aged bone matrix, possibly because of the lack of a bone marrow microenvironment.61 OCs also promote OBs-mediated bone formation through the secretion of transforming growth factor (TGF)-β and IGF-1 secreted in the bone matrix.43 Crosstalk between OCs and other cells contributes to the regulation of OC activity and thus affects bone resorption. Hence, manipulating the crosstalk between various cells and OCs in the aging bone marrow is expected to be a potential therapeutic target for future treatment of OP.
Osteocytes
OCYs account for more than 90% of the cells in the skeletal system, and their dendritic structures provide the basis for communication with other cells.62 OCYs maintain bone homeostasis by responding to mechanical loads and hormonal stimuli in the bone marrow.63 Research has demonstrated that reduced activity and decreased mechanical loading in elderly individuals lead to severe decreases in bone density.64 Numerous evidence have demonstrated that senescent OCYs accelerate bone loss by secreting SASP factors.65,66 Farr et al. identified more than 20 SASP genes that are significantly expressed in senescent OCYs from both humans (aged 72–87 years) and mice (aged 24 months).11 Additionally, the connection between autophagy and OCYs senescence has been elucidated. Atg7 and Map1lc3a, which are highly expressed mitochondrial autophagy genes in OCYs, are expressed at decreased levels in aged animals.11 Reduced mitochondrial autophagy promotes the accumulation of damaged mitochondria and ROS in OCYs, further contributing to OCYs’ senescence.67
Compared with healthy OCYs, senescent OCYs exhibit impaired mechanical signaling and disruption of perilacunar/canalicular remodeling (PLR).10 OCYs actively remodel their surroundings by reabsorbing and redepositing the surrounding bone matrix via PLR.68,69 The presence of PLR is confirmed by “OCYs osteolysis”, which manifests as enlarged lumina and rough borders around OCYs.70 Increasing evidence of reduced luminal volume and hypermineralized lacunae in older human and mouse bones strongly suggests that PLR is impaired in senescent OCYs.63,71 Furthermore, RNA-seq data from OCYs-rich cortical bone indicate that the expression levels of bone resorption-related genes cathepsin K (Ctsk) and tartrate-resistant acid phosphatase (Acp5) are significantly lower in senescent OCYs than in young OCYs.72 These findings suggest that OCs markers in OCYs are progressively lost during aging, which leads to the impaired PLR function of OCYs and the inhibited activation of OCs. In addition, senescent OCYs also activate OCs by secreting M-CSF and RANKL and inhibit the function of OBs by secreting sclerostin.73 Young OCYs-derived EVs and senescent OCYs-derived EVs (S-EVs) were obtained from primary OCYs of 2-month-old and 16-month-old mice, respectively. Proteomic analysis showed that S-EVs lack promyosin-1 (TPM1), which promotes osteogenic differentiation of BMSCs.74 In-depth exploration of aging-induced cellular senescence in OCYs provides potential strategies for the treatment of senile OP.
Adipocytes
Substantial research confirms the regulatory role of bone marrow ADs in skeletal health, as their accumulation leads to bone loss.75 This phenomenon may be attributed to changes in the fate of BMSCs differentiation during aging. Although age-dependent accumulation of ADs in the bone marrow resulting in OP has been observed in elderly individuals, exploration of signaling pathways is still in the early stage.76
Studies have revealed that dipeptidyl peptidase-4 (DPP4) derived from ADs in aged bone marrow has a significant osteogenic inhibitory effect. Conversely, a significant increase in bone mass was observed upon targeted removal of DPP4.17 Moreover, bone marrow ADs produce more receptor activators for RANKL in response to parathyroid hormone deficiency or glucocorticoid intervention, leading to abnormal activation of OCs and bone resorption.77,78 Notably, a recent study explored the mechanisms of senescent ADs in the bone marrow and revealed that bone marrow ADs transform into senescent ADs after glucocorticoid administration. The secretion of SASP from senescent ADs further promotes secondary senescence of ECs and OBs in the skeleton. Mechanistically, the formation of a positive feedback loop of 15d-PGJ2-PPARγ-INK signaling in the skeletal system ultimately leads to bone degeneration.75 In addition, the expression level of bone marrow macrophage-derived proliferating cell nuclear antigen clamp-associated factor (PCLAF) was found to be significantly elevated in aged mice compared with young mice. PCLAF binds to the ADGRL2 receptor on bone marrow ADs, promoting bone marrow AD senescence and disrupting bone homeostasis.79 These studies have sparked strong interest in senescent ADs in the development of OP and indicate that clearing mediators derived from senescent ADs may be an effective approach to alleviate OP.
Immune cells
In addition to the negative impact of senescent bone cells in the skeleton, immune cells in elderly individuals also participate in the regulation of bone homeostasis. Elderly individuals exhibit chronic inflammation and changes in the immune cell spectrum.80 Previous studies have revealed that various immune cells establish communication with bone cells through direct contact or paracrine pathways.
MACs and neutrophils (NEs) accumulate and secrete large amounts of grancalcin (GCA) protein in the bone marrow during aging, and GCA binds to Plexin-B2, a cell surface receptor for BMSCs in the bone marrow, and inhibits the downstream FAK-SRC-YAP signaling pathway, which inhibits the osteogenic differentiation of BMSCs.81 Injection of GCA antibody into aged mice resulted in an increase in the number of OBs and OCs on the surface of the bone trabeculae and a significant decrease in the number and area of ADs. These findings suggest that the GCA antibody significantly improves the skeletal health of aged mice. Furthermore, inflammatory factors from M1 MACs and anti-inflammatory factors secreted by M2 MACs play important roles in the fate of bone cells.82 Non-polarized MACs can also promote the differentiation of BMSCs into the OBs lineage via oncostatin M (OSM).83 TGFβ1+CCR5+ NEs were reported to be the major source of TGFβ1 in mouse bone marrow.84 With age, increased TGFβ1+CCR5+ NEs in the bone marrow induce TNF receptor-associated factor 3 (TRAF3) degradation in mesenchymal progenitor cells via TGFβ1, which promotes NF-κB-mediated CCL5 expression, ultimately leading to bone loss. NEs infiltrating the aging bone marrow also induce OCs formation through the secretion of RANKL and IL-8.85 T lymphocytes are closely related to T lymphocyte subsets and cytokines in regulating bone regeneration. T help 1 (Th1) cells obtained from aged animal models show increased production of tumor necrosis factor-α (TNF-α), which promotes OBs apoptosis and mediates B cell secretion of RANKL, leading to OCs activation and severe bone loss.86 Interleukin 17 (IL-17) secreted by Th17 cells upregulates RANKL expression on the surface of pre-OCs, BMSCs, and OBs, thereby promoting OCs maturation and inhibiting OBs differentiation.87 T regulatory (Treg) cells promote osteogenic differentiation of pre-OBs and BMSCs directly through the CD39-CD93-AdoR signaling pathway.45 Additionally, Treg cells inhibit OCs’ function by secreting cytotoxic T-lymphocyte-associated protein 4 (CTLA4) and activating the CD80/CD86 receptors on pre-OCs.88 These data indicate that T lymphocytes and their derived mediators constantly influence bone balance. Clinical data also revealed that B lymphocytes in the bone marrow of elderly individuals are either normal or reduced, accompanied by increased secretion of G-CSF and RANKL.89,90 This is inextricably linked to the increased formation and activation of OCs. In summary, the crosstalk between immune cells and bone cells directly or indirectly influences the fate of bone cells. Exploring the molecular mechanisms between immune cells and bone cells is beneficial for alleviating the progression of OP.
Endothelial cells
Evidence suggests that angiogenesis and osteogenesis are commonly coupled in the mammalian skeletal system. However, reduced angiogenesis and osteogenesis are observed in aged organisms and ultimately lead to the development of OP.59,91,92 ECs are highly heterogeneous in different tissues and acquire specific morphological, molecular, and functional properties in the local microenvironment.93,94
In recent years, researchers have reported that in mouse bones, capillaries can be differentiated into H-type (CD31high/Emcnhigh) and L-type (CD31low/Emcnlow) ECs, which are categorized on the basis of their shape, molecular properties, and function.91 In particular, H-type ECs, located at the end of the skeletal arterial network, may play a central role in the specialized metabolic organization of the environment. H-type ECs enjoy preferential access to oxygen and nutrients, which may have an important impact on the senescence and regeneration of other cell types. Studies have also shown that H-type ECs provide essential signals to surrounding OBs through the HIF and Notch signaling pathways. H-type ECs are expressed at significantly lower levels in bone with age, which may provide a plausible explanation for OP.91 Moreover, the inhibition of epiphyseal ECs’ senescence ameliorates impaired bone angiogenesis and the decoupling of the osteogenic process through the ANG/PLXNB2 signaling axis.59 Decreased production of pre-OCs in the epiphysis results in a significant decrease in ANG secretion, which leads to impaired rRNA transcription in ECs, ultimately causing aging-related cellular senescence. Recent studies have demonstrated that senescent ECs-derived miRNA-31-5p promotes BMSC lipogenic differentiation by regulating the WNT pathway.28 Conversely, young ECs-exo upregulate zinc finger and BTB domain containing 16 (ZBTB16) expression to promote BMSCs osteogenic differentiation.95 These findings confirmed that senescent ECs and bone cells mediate angiogenesis and osteogenesis in the aged skeleton.
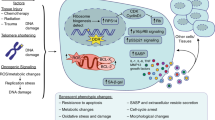
The crosstalk between cells in the aging bone marrow microenvironment collectively determines the onset and progression of OP. Under aging conditions, BMSCs tend to undergo adipogenic differentiation, leading to reduced bone formation and increased bone marrow fat deposition. In the aging microenvironment, skeletal muscle cells, ECs, and MACs synergistically promote the senescence and adipogenic differentiation of BMSCs. The interaction between OBs and OCs is the core process of bone remodeling. Senescent OBs enhance the activation of OCs, thereby exacerbating bone loss. Additionally, OCs are excessively activated in the aging bone marrow environment because of factors secreted from OBs, OCYs, and ADs, resulting in a negative bone balance. The role of immune cells in the aging bone marrow cannot be overlooked. Senescent MACs, NEs, and B cells mediate the formation of OCs, whereas T cells reverse the adipogenic differentiation of BMSCs, influencing the balance between osteogenesis and adipogenesis. ECs exhibit dysfunction during aging, and a reduction in H-type vessels affects OB differentiation. Senescent ECs promote the adipogenic differentiation of BMSCs. In-depth research into the mechanisms of these cellular interactions will contribute to the development of precise intervention strategies for senile OP. The crosstalk between advancing age-related cellular senescence and OP pathogenesis is shown in Fig. 2 and Table 1.

Cellular senescence and other age-related mechanisms in the pathogenesis of OP. The senescence of different bone cells and OP is interwoven into a complex network of interactions. Senescent cells co-secreted the SASP to accelerate inflammatory microenvironment formation and bone loss. Fgfr3-creER, Lepr-cre, CXCL12+LepR+, P62+, and Prx1Cre+OsxCre− labeled BMSCs were shown to be involved in the biological process of BMSCs senescence. Moreover, senescent BMSCs also showed increased expression of P62 and H3K4me1 and decreased expression of SOX5, TFEB, DMNT-1, and DNMT-3B. In addition, the senescence of BMSCs is induced by miRNA-34a-5p secreted by senescent skeletal muscle cells and by senescent ECs through the secretion of miRNA-31-5p. The OBs were differentiated osteoblastically from BMSCs. CD24+ and Osteolectin+ OBs exhibited a distinct senescent phenotype and osteogenic restriction. Increased mTORC1 and FOXO and decreased METTL3 and IGF-I in senescent OBs induced malfunction in OBs. Senescent OBs promoted OCs activation through massive secretion of M-CSF, RANKL, and WNT5A. SHN3 secreted by OBs promoted OP development through the inhibition of H-type angiogenesis. Treg cell-mediated inhibition of the CD39-CD93-AdoR signaling pathway directly suppresses osteogenesis in OBs. Overactivation of OCs is dependent on RANKL secreted by senescent BMSCs, OBs, OCYs, and ADs. In addition, the deficiency of OCs-derived SPP1 inhibited the osteogenic differentiation of BMSCs. OCs-secreted ANG is a key factor preventing the senescence of neighboring ECs. Increased C-telopeptide in the aged bone matrix is thought to induce OC activation. The expression of Atg7 and Map1lc3a in OCYs is reduced in aged animals. Senescent OCYs-derived TPM1 was significantly reduced, thereby inhibiting the osteogenic differentiation of BMSCs. Senescent ADs-derived DPP4 also significantly inhibited osteogenesis. In addition, PCLAF, derived from bone marrow MACs in aged mice, binds to ADGRL2 receptors on ADs and promotes the senescence of ADs. Senescent immune cells secrete increased GCA, TGFβ1+CCR5+, and decreased IL-17, OSM, leading to bone loss. Elevated levels of immune cell-derived TNF-α, IL-8, and RANKL markedly promoted the activation of OCs. Senescent ECs exhibited decreased HIF/Notch signaling pathway and exo secretion, as well as increased miRNA-31-5p, accelerating bone loss. Decreased Cystatin-A secretion by aged skin keratinocytes leads to restricted OB differentiation and enhanced OC differentiation. FOXO3 in skeletal muscle cells plays a key role in counteracting aging-related cellular senescence. EP4 in neuronal cells regulates bone homeostasis and promotes bone regeneration by sensing prostaglandin E2 secreted by OBs. These aging-related cellular senescence leads to changes in bone cell function and fate, which ultimately contribute to the onset and development of OP. The red frames represent upregulated genes, and the blue frames represent down-regulated genes
Cellular senescence and other age-related mechanisms in osteoarthritis
OA is a disease that causes chronic pain in elderly individuals and accelerates disability worldwide.96 As a degenerative disease, aging-related cellular senescence has been confirmed to promote the progression of OA.97 Research data indicate that the incidence of OA increases with age.98 There is a significant increase in SA-β-Gal-positive cells, including chondrocytes, mesenchymal cells (MSCs), synovial cells (SYCs), ADs, and muscle cells, in vertebrate OA tissue, indicating that age-related significant cellular senescence occurs in OA.99,100 Senescent cells within joints continue to secrete SASP factors, further damaging the intra-articular structure.101 As research on OA continues to expand, the connection between senescent cells and OA has become increasingly clear. The various cellular senescence and other age-related mechanisms within osteoarticular joints in OA are reviewed below.
Chondrocytes
In the early stages of arthritis, previously quiescent chondrocytes begin to proliferate, thus repairing damaged cartilage and preventing further tissue degradation. Unfortunately, newborn chondrocytes in this microenvironment are prone to entering a state of aging-related cellular senescence, leading to tissue degeneration and the onset of OA.97,102 Over time, chondrocytes in OA exhibit a more pronounced senescence phenotype and secrete SASP factors, such as matrix metalloproteinase (MMP)-13 and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)-5.103 Research has confirmed that the number of senescent chondrocytes is directly proportional to the severity of OA.104 Symptoms of OA develop after intra-articular injection of senescent chondrocytes into the joint cavity of healthy mice, suggesting that senescent chondrocytes are strongly associated with the development of OA.105 These data provide a rationale for senescent chondrocytes as drivers of OA.
Current studies reveal the crosstalk between OA and senescent chondrocytes. CircRREB1 is highly expressed in senescent chondrocytes and is involved in chondrocyte senescence and age-associated OA by regulating fatty acid synthase-related lipid metabolism.106 Research has shown that PDZ domain containing 1 (PDZK1) belongs to the Na+/H+ exchange regulatory factor family.107 Mechanical overload leads to a reduction in PDZK1, which has been observed in the articular cartilage of OA patients, elderly mice, and OA mice. Deficiency of PDZK1 is a key factor in the progression of OA, where biomechanical induction leads to mitochondrial dysfunction and chondrocyte aging.107 Additionally, SIRT6 was found to be significantly down-regulated in the cartilage tissues of OA patients, which promotes chondrocyte senescence by up-regulating senescence markers via the SIRT6-STAT5 signaling pathway.108 One study revealed that ADAM19 is a key senescence-related gene in cartilage and that lowering ADAM19 expression significantly reduces the senescent phenotype of chondrocytes.109 The expression of senescence markers is significantly increased in chondrocytes from OA patients, and further mechanistic studies revealed that myosin light chain 3 (MYL3) deficiency in chondrocytes promotes clathrin-mediated endocytosis and Notch signaling, leading to chondrocyte senescence and OA.110
Senescent chondrocytes also support the formation of an intra-articular inflammatory microenvironment through the secretion of SASP, which mediates crosstalk between senescent chondrocytes and neighboring cells and ultimately promotes neighboring cell senescence.111,112 In addition to the SASP, senescent chondrocytes secrete additional EVs to trigger senescence in their neighboring cells.113 These data confirmed that the production of EVs in senescent chondrocytes is positively correlated with the number of senescent cells. Mechanistically, EVs affect neighboring cell senescence by regulating the expression of miR-34a, miR-24, and miR-150.113 Recent crosstalk between extracellular matrix (ECM) and senescent chondrocytes has also been revealed. ECM sclerosis downregulates histone deacetylase 3 (HDAC3), which contributes to Parkin acetylation, which in turn activates mitochondrial autophagy, leading to a senescent phenotype in chondrocytes and accelerated OA symptoms.114 Further explorations are needed to reverse chondrocyte senescence to maintain the integrity of the articular cartilage.
Mesenchymal cells
The MSCs that primarily influence joint structure are mainly subchondral bone marrow MSCs, whose depletion and abnormal repair accelerate pathological changes in OA.115 Dysfunction of RNA-binding proteins is closely associated with aging-related cellular senescence and OA. An exploration of the interaction of the RNA-binding protein PUM1 with the 3ʹ-UTR of Toll-like receptor 4 (TLR4) and its effect on NF-κB activity in MSCs revealed that the PUM1 protein alleviated MSCs senescence and OA progression by inhibiting TLR4-mediated NF-κB activation.116 Moreover, RNA modification is involved in the regulation of aging-related cellular senescence. Elevated m6A levels and reduced expression of the m6A demethylase ALKBH5 were detected in senescent MSCs. Downregulation of ALKBH5 expression promoted MSC senescence and OA by enhancing m6A modification to increase the stability of CYP1B1 mRNA and induce mitochondrial dysfunction.117 A study also demonstrated that MSC senescence and OA injury could be reversed by rescuing the levels of the heterochromatin-associated protein HP1α and the nuclear lamina protein LAP2, promoting cell division and inhibiting aging-associated inflammation.118
Crosstalk between chondrocytes and subchondral MSCs is also closely linked to the development of OA. By co-culturing senescent subchondral bone marrow MSCs with chondrocytes from OA patients in vitro, chondrocytes were found to still exhibit a catabolic phenotype.119 The same conclusion was also confirmed in vivo. After intra-articular injection of senescent subchondral bone marrow MSCs into the joints of young male mice, histological analysis revealed a significant increase in articular cartilage decomposition, and micro-CT analysis revealed a significant reduction in subchondral bone volume. These data suggest that senescent subchondral bone marrow MSCs destroy cartilage and joints.119 Cao et al. noted that senescent chondrocytes restrict the multipotency of subchondral bone marrow MSCs, whereas MSCs accelerate the apoptosis of senescent chondrocytes, which leads to OA via crosstalk.120 Furthermore, the immunomodulatory properties of MSCs have been demonstrated to significantly influence the progression of OA. The synthesis and catabolism of metabolic factors are regulated by cytokines secreted by MSCs, which promote the secretion of anti-inflammatory factors by SYCs.121
The aged skeletal stem cells in the cartilage have been reported to potentially promote OA. Murphy et al. reported a significant decrease in the number and secretory function of skeletal stem cells in the cartilage of elderly animals compared with young animals, indicating that aged skeletal stem cells contribute to cartilage pathology in OA.122 Interestingly, a special type of skeletal stem cell, Gremlin 1+ cells, in joint cartilage potentially promotes OA formation.123,124 Gremlin 1+ cells have been demonstrated to obtain the capacity to differentiate into OBs, chondrocytes, and reticular BMSCs. A gradual loss of Gremlin 1+ cells was observed in the joints of the aged mice, which resulted in the progression of OA. This study confirmed that the absence of Gremlin 1+ cells may lead to OA progression and that OA progression can be reversed by developing therapeutic regimens that target Gremlin 1+ cells.124 Overall, altering the differentiation potential and secretion behavior of aged subchondral bone marrow MSCs and skeletal stem cells may offer a promising avenue for OA treatment.
Synovial cells
SYCs primarily include synovial fibroblasts and synovial MACs, which are responsible for lubricating joints and maintaining microenvironment homeostasis.125 While the role of the synovium in OA has gradually been recognized, the relationship between senescent SYCs and OA remains unclear.
Studies have indicated that during arthritis inflammation, synovial tissue from mice stained with SA-β-gal presents visibly senescent cells, although the specific cell types involved are not clearly identified.99 Research has revealed a significant presence of P16-expressing MACs around aged tissues, suggesting that the infiltration of senescent MACs may contribute to pathological changes in joint tissues.126 However, the impact of the secretory function of senescent MACs in OA remains unclear. It has been suggested that synovial MACs deplete the cell surface molecule CD38 by secreting nicotinamide adenine dinucleotide (NAD), thereby accelerating senescence.127 Concurrently, the SASP derived from senescent cells also induces aging in synovial MACs. Additionally, synovial fibroblasts in elderly OA exhibit decreased synovial fluid secretion and increased levels of pro-inflammatory mediators, leading to cartilage degradation and joint damage.128 Synovial fibroblasts in aged OA patients also accelerate cartilage deterioration by activating the PI3K and ERK pathways through the secretion of adipokines.129 Consequently, while the relationship between senescent synoviocytes and age-related OA remains to be elucidated, crosstalk between these cells and bone cells appears to influence the progression of age-related OA.
Subchondral bone cells
In addition to the pivotal role of chondrocytes in the development of OA, the impact of subchondral bone cells has also garnered widespread attention in recent years. Compared with that in young joints, the microenvironment of the subchondral bone in elderly patients with OA significantly changes. Subchondral senescent pre-OCs with a unique secretory phenotype have been identified in the early stages of OA.130 At the molecular level, senescent pre-OCs enhance the osteogenic differentiation of BMSCs by activating the cyclooxygenase 2 (COX2)-prostaglandin E2 (PGE2) pathway.130 The bone resorption activity of subchondral OCs leads to a sharp increase in the activity of TGF-β1 in the subchondral bone cells of OA patients.131 Increased TGF-β1 activates the Smad2/3 signaling pathway, attracting bone progenitor cells to the area of bone remodeling and promoting the formation of abnormal bone tissue.131 Furthermore, subchondral OCs in natural senescence-induced OA ubiquitinated HIF-1α in chondrocytes by inducing H-type vasculature to elevate the oxygen content, leading to chondrocyte senescence.132 The knockdown of lymphocyte cytoplasmic protein 1 (Lcp1) maintains the hypoxic state of the subchondral bone microenvironment by disrupting angiogenesis and delaying pathological changes in OA.132 Moreover, senescent OCs in the subchondral bone microenvironment also promote the formation of sensory nerves by secreting Netrin-1.133 Additionally, RANKL and MMP-9 derived from H-type vessels induce the migration and maturation of OCs.134 With aging, the ratio of RANKL to OPG in subchondral OCYs increases, thereby promoting the activation of OCs.135 Various factors are released into subchondral OBs, such as PGE2, interleukin-6 (IL-6), matrix metallopeptidase 9, vascular endothelial growth factor (VEGF), and RANKL, further promoting the differentiation of OCs and the erosion of subchondral bone.133 Importantly, after the use of the INK-ATTAC “suicide” transgene, “senolytic” compounds, or JAK inhibitors to remove or inhibit SASP secretion from aged osteogenic lineage cells, elderly mice exhibited improved bone microstructure and reduced OA.12 These mechanisms act on subchondral bone cells to exacerbate the pathological process of OA. Inhibiting the senescence of subchondral bone cells may be an effective treatment for OA.
Other cells
Individuals with a body mass index exceeding 28 have a high incidence of OA, as increased mechanical loading is a key factor driving OA occurrence.100 Recent studies have shown that senescent ADs in joints may be a significant source of inflammatory mediators in OA.136 Similar to other senescent cells, senescent ADs secrete SASP factors that accelerate the aging of other cells and the progression of OA.75 Additionally, OA patients commonly experience muscle function impairments. Studies indicate that the regeneration of senescent musculoskeletal cells is inhibited by aging satellite cells. In 24-month-old mice, satellite cells enter an aging state by unblocking p16. Senescent satellite cells significantly inhibited the regeneration of musculoskeletal cells, suggesting that senescent satellite cells and muscle cells may also be involved in the development of OA.137
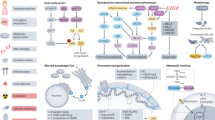
The onset of OA is driven by complex crosstalk among multiple cell types. In the early stages of OA, chondrocytes proliferate to repair damage, but with the advancement of age, they secrete SASP factors that degrade the cartilage matrix and induce inflammation. The depletion of MSCs accelerates OA progression. Senescent MSCs inhibit chondrocyte proliferation while promoting their senescence, creating a vicious cycle. Senescent SYCs reduce the secretion of synovial fluid and release pro-inflammatory factors that exacerbate inflammation. Subchondral bone cells promote OC activity through factors such as RANKL. Abnormal H-type vessels accelerate chondrocyte senescence. Overactivated OCs enhance bone resorption and secrete factors that promote osteogenesis in BMSCs and remodeling of subchondral bone, further aggravating OA pathological progression. ADs secrete SASP to induce senescence in neighboring cells, and the senescence of satellite cells inhibits muscle regeneration, affecting joint stability. Intervention strategies targeting intercellular crosstalk may provide new directions for OA treatment. Molecules or signals involved in cellular senescence and other age-related mechanisms during OA are shown in Fig. 3 and Table 2.

Cellular senescence and other age-related mechanisms in the knee joint lead to OA progression. Senescent chondrocytes promote the expression of the senescent phenotype by up-regulating SASP, CircRREB1, and ADAM19 and down-regulating PDZK1, SIRT6, and MYL3. Meanwhile, senescent chondrocytes also promote senescence in neighboring cells by secreting SASP and EVs (containing miR-34a, miR-24, and miR-150). In addition, ECM sclerosis downregulates HDAC3 and activates mitochondrial autophagy, leading to a chondrocyte senescence phenotype and accelerated OA symptoms. Depletion and aberrant repair of MSCs accelerate pathological changes in OA. Senescent MSCs presented decreased expression of PUM1, ALKBH5, and HP1α/LAP2. Senescent chondrocytes limit the pluripotency of MSCs, while MSCs accelerate the apoptosis of senescent chondrocytes. The absence of Gremlin 1+ cells may contribute to the progression of OA. Senescent SYCs accelerate senescence and cartilage damage by increasing NAD, SASP, PI3K/ERK, and TNF-α levels. Changes in subchondral bone cells with senescence in OA also promote OA progression. OCs accelerate intra-articular senescence and OA pathology by regulating COX2/PGE2, TGF-β1, HIF-1α, Lcp1, and Netrin-1. ECs, subchondral OCYs, and subchondral OBs activate OCs via RANKL and accelerate subchondral bone erosion. OBs release PGE2, IL-6, MMP-9, and VEGF to further promote OCs differentiation. Senescent ADs secrete SASP factors, accelerating the senescence of other cells and the progression of OA. Senescent satellite cells significantly inhibit musculoskeletal cell regeneration by unblocking p16. All of these factors contribute to the malignant crosstalk of senescence in OA and accelerate its progression. The red frames represent upregulated genes, and the blue frames represent down-regulated genes
Cellular senescence and other age-related mechanisms in intervertebral disc degeneration
The intervertebral disc (IVD) is composed of the inner nucleus pulposus (NP) and the outer annulus fibrosus (AF). The incorporation of the upper and lower vertebral endplates creates a relatively enclosed microenvironment within the IVD.138 Increased expression of aging-associated cell phenotypes has been detected in IVD within elderly vertebrates.139,140,141 The accumulation of aging-related cellular senescence in the IVD provides a new research direction for IVD development.142,143,144,145,146 Initially, aging prevents the proliferation and formation of new IVD cells. Subsequently, aging-related cellular senescence to the senescent state results in increased SASP secretion, which accelerates inflammation and degradation of the disc matrix.147,148 In conclusion, aging promotes the transition of the IVD microenvironment to an inflammatory microenvironment by activating multiple pathways. Precisely exploring the mechanisms underlying cellular senescence and other age-related mechanisms holds promise for reversing IVD.
Nucleus pulposus cells
As the main effector cells, nucleus pulposus cells (NPCs) are responsible for ECM synthesis and regulate homeostasis within the IVD.149 NPCs accelerate the production of tissue growth factors such as connective tissue growth factor (CTGF/CCN2) by secreting TGF-β. Moreover, CTGF/CCN2 promotes the proliferation of NPCs.150
As age increases, NPCs exhibit reduced CTGF/CCN2 and increased SASP secretion, leading to increased matrix metalloproteinases (MMP3 and MMP7), which results in ECM breakdown.151 Studies have shown that abnormal mechanical stress is a primary driving factor for NPCs aging. Piezo1, a mechanosensitive ion channel, links mechanical stimuli to signal transduction.152 Increased Piezo1 was observed in NPCs from excessive stress or elderly individuals, leading to mitochondrial dysfunction and inflammatory infiltration and triggering senescence in NPCs.153 Furthermore, accumulated ROS in aged IVD accelerates NPCs aging through ROS-mediated biological signaling.154 Interestingly, senescent NPCs are also associated with the epigenetic regulation of non-coding RNAs. Upregulation of miR-182-5p and downregulation of circERCC2 are found in the NPCs of elderly individuals. circERCC2 regulates NPCs aging through the miR-182-5p/SIRT1 cascade.155 The Keap1/Nrf2/Wnt5a axis was identified as a novel signaling pathway that protects NPCs from senescence.156 In addition, aberrant genomic DNA damage promoted inflammatory senescence in NPCs through activation of the cyclic GMP-AMP synthase/IFN gene-stimulating factor (cGAS/STING) axis but not melanoma absent 2 (AIM2) inflammasome assembly.157 ROS accumulation and mitochondrial dysfunction were observed in degenerating NP tissues from rats and young adults, which caused the senescence of NPCs. Senescent NPCs promote disc degeneration through the p53‒p21 signaling axis, as confirmed by a series of experiments, including RNA-seq.142 Sustained oxidative stress in the IVD also leads to impaired antioxidant systems in NP cells. Transcriptome sequencing revealed that the antioxidant glutaredoxin3 (GLRX3) is significantly reduced in senescent NPCs and that dysregulation of GLRX3 disrupts redox homeostasis and contributes to NPCs’ senescence and disc degeneration.158 Notably, senescence of NPCs has been shown to be closely related to autophagy. NLR family member X1 (NLRX1) deficiency in NPCs exhibits mitochondrial rupture and excessive mitochondrial autophagy, which in turn leads to cellular senescence and disc degeneration.159 Another study confirmed that the inflammatory factor TNF-mediated senescence of NPCs involves chaperone-mediated autophagy (CMA) inhibition.160 Sequencing analyses revealed that CMA deficiency led to the accumulation of phospholipase C gamma 1 (PLCG1), which triggered calcium overload-induced cellular senescence in NPCs. These data indicate that senescent NPCs expedite the process of IVDD, providing new therapeutic approaches for rescuing aging NPCs and combating IVDD.
Annulus fibrosus cells
Research has revealed significant heterogeneity in external and internal AF cells (AFCs).138 External AFCs are spindle-shaped and rich in fibroblasts and type I collagen. The internal AFCs are more rounded and enriched in chondrocytes and type II collagen. Healthy AFCs exhibit a sensitive stress response and strong tensile properties that effectively limit the prominence of NP.161 Sonic hedgehog hormone (SHH) from NPCs has been shown to promote the survival and normal differentiation of AFCs.162 With age, the SHH pathway is gradually inhibited in NPCs.163,164 The absence of SHH in NPCs has been demonstrated to result in AFCs losing their polarity and characteristic hierarchical structure.162 This change may greatly reduce the tensile strength of the AFCs and the matrix degradation in the AF, which ultimately fails to prevent NP protrusion.
Recent studies have found that activation of the Keap1/Nrf2/Wnt5a signaling axis inhibits senescence and extracellular matrix breakdown in AFCs, thereby effectively delaying natural aging-induced disc degeneration.156 Furthermore, studies have confirmed that sites of AF injury in IVDD exhibit an abnormal accumulation of ROS and a significantly reduced number of AFCs, thus limiting AF regeneration.165 Observations of human AF tissue have shown that the AF/chondral endplate border is adherent to CD133+ stem cells that are committed to either the chondrogenic or adipogenic spectrum.166 A significant loss of the stem cell pluripotency markers Notch1, Jagged1, C-KIT, and CD166 was found in the AF of aged rabbits, which may contribute to disc aging.167 Inhibiting the aging of AFCs may provide reliable information for IVD regeneration.
Cartilaginous endplate cells
Cartilage endplate cells (CEPCs), located between the IVD and vertebrae, not only serve a transitional function but also support the healthy growth of IVD cells.168 Similar to cartilage cells, CEPCs are responsible for ECM synthesis. These cells restrict the entry of NPCs into the vertebral body and alleviate the hydrostatic pressure generated by mechanical loads.169
Senescence of CEPCs has also been observed in aged IVDs. Decreased proteoglycan secretion and increased type X collagen secretion in CEPCs of aged individuals lead to ECM calcification.170,171 It has been shown that the permeability of the endplate is attributed to the presence of a microvascular network within the endplate. Various nutrients of different sizes and charges maintain the health of IVD cells through the cartilage endplate.172,173 However, calcified CEPCs exhibit reduced permeability, limiting the absorption of nutrients and oxygen and slowing the clearance of harmful metabolites to disrupt IVD homeostasis.138 Cao et al. reported that the number of p16+ TRAP+ OCs significantly increased in the endplates of aged mice, while the endplates degenerated. These OCs induce sensory nerves to innervate the porotic endplates by mediating the secretion of Netrin-1 and nerve growth factor (NGF), which suggests that the senescent OCs in the endplates of aged mice are closely related to degenerative changes in the endplates and the development of low back pain.174 Ni et al. reported that Trap+ OCs secrete the neurotransmitter Netrin-1, which mediates the gradual growth of sensory nerves into aged endplate tissue. Furthermore, they also revealed that significantly elevated levels of PGE2 in aged endplate tissue stimulated EP4 receptors on sensory nerves and activated the downstream PKA/CREB signaling pathway, which mediated the inward flow of Na ions, thereby leading to pain signaling.175 Meanwhile, Chen et al. revealed a new mechanism of OCs in endplate remodeling. The abnormal stress induced by lumbar spine instability activates Hippo signaling, which initiates cartilage endplate remodeling through activation of the OCs differentiation gene CCL3. Inhibition of Hippo signaling activation or resupply of YAP in CEPCs effectively prevents their transformation into cheese-like endplates and subsequent lumbar degeneration.146 Furthermore, Ni et al. discovered that senescent MACs accumulated in sclerotic endplates in an aged male mouse model. Senescent-like MACs affect immunovascular communication by secreting the pro-angiogenic cytokine IL-10, which leads to endplate sclerosis by affecting signal transduction and activator of transcription 3 (STAT3) signaling pathways.176 Measures to rescue senescent CEPCs are indispensable for the treatment of IVDD.
IVDD is driven by a complex interplay of NPCs, AFCs, CEPCs, OCs, and immune cells. NPCs maintain ECM production, but as they age, their SASP increases and promotes matrix degradation. Moreover, mechanical stress, ROS accumulation, and non-coding RNA dysregulation accelerate the aging of NPCs and trigger IVDD. The AFCs depend on SHH secreted by NPCs to maintain and lose polarity with decreasing SHH, which reduces AF strength. In addition, excessive accumulation of ROS and loss of stem cell markers reduced the regenerative ability of AFCs, exacerbating IVD degeneration. Senescence of CEPCs leads to calcification of the ECM, reducing endplate permeability and disrupting IVD homeostasis. Senescent OCs promote endplate remodeling and secrete Netrin-1 and PGE2, which induce nerve invasion and cause pain. MACs accumulate and secrete IL-10 to promote endplate sclerosis and exacerbate IVD degeneration. In conclusion, IVDD is driven by multiple cellular crosstalk, and interventions targeting key cellular interactions may become a new therapeutic strategy in the future. The mechanisms linking cellular senescence and other age-related changes to IVDD are shown in Table 3 and Fig. 4.

Cellular senescence and other age-related mechanisms in IVD accelerate IVDD. With age, NPCs lead to ECM breakdown through decreased CTGF/CCN2 and increased SASP secretion. An increase in Piezo1 and a decrease in circERCC2 were also observed in senescent NPCs. Blockade of the Keap1/Nrf2/Wnt5a axis or activation of the cGAS/STING axis promoted the senescence of NPCs. Furthermore, ROS accumulation, p53/p21 activation, GLRX3 dysregulation, a lack of NLRX1, and TNF infiltration also contributed to the senescence of NPCs. Decreased SHH secretion in senescent NPCs leads to loss of polarity and the characteristic hierarchical structure of AFCs. ROS accumulation and deletion of Keap1/Nrf2/Wnt5a, CD133+, Notch1, Jagged1, C-KIT, and CD166 trigger senescence in AFCs. Decreased proteoglycan secretion and increased X-type collagen secretion in CEPCs led to calcification of the ECM. Calcified CEPCs limit nutrient and oxygen uptake, thereby disrupting IVD homeostasis. Senescent OCs induce pain via Netrin-1 by inducing sensory nerves into the aging endplate. Abnormal stress activates Hippo signaling to initiate cartilage endplate remodeling. Senescent macrophages induce endplate sclerosis by secreting IL-10. These factors ultimately contribute to the cellular changes associated with aging and the development of IVDD. The red frames represent upregulated genes, and the blue frames represent down-regulated genes
Cellular senescence and other age-related mechanisms in other bone disorders
Cellular senescence and other age-related mechanisms in rheumatoid arthritis
Rheumatoid arthritis (RA) is characterized by chronic inflammatory infiltration that severely impairs joint function.177 Studies indicate that aging promotes the onset and progression of RA. Clinical data confirm that the incidence of RA in elderly individuals is ~14 times greater than that in younger individuals and that later onset corresponds to more severe loss of joint function.178,179 Premature immune senescence, including expansion of late-differentiated T cell proliferation, increased SASP secretion, and shortened telomeres, has been reported in RA patients.180 Late-differentiated T cells are defined as CD4+ and CD8+CD28− T cells, which are detected in both elderly and RA patients.181,182 Importantly, the number of late-differentiated T cells is proportional to the severity of RA.183 Additionally, senescent synovial fibroblasts in RA exhibit increased SASP secretion, further exacerbating RA.184 Recent studies have also shown that telomere shortening in senescent cells promotes the extra-articular manifestations of RA.185 Telomere shortening also triggers enhanced autoantigen reactivity, indicating that cellular senescence is one of the risk factors for RA.186 In summary, the senescence of immune cells and synovial fibroblasts exacerbates the symptoms of RA, and rejuvenating senescent cells may hold significant value in RA.
Cellular senescence and other age-related mechanisms in bone tumors
Growing data have supported the contradictory dual role of senescent cells in bone tumors.187,188 On the one hand, dysfunction of senescent cells promotes tumorigenesis. Studies have revealed the abnormal accumulation of senescent immune cells in the osteosarcoma of elderly patients.189 Senescent immune cells exhibit weak antigen presentation, reduced antibody production, and low cytotoxic activity, thereby promoting tumor cell evasion.190,191 Senescent immune cells fail to proliferate and exhibit a weakened response to antigen stimulation in tumors.192 Moreover, the accumulation of senescent immune cells in osteosarcoma leads to increased secretion of SASP components. An increased SASP in the tumor microenvironment is linked to chronic inflammation, angiogenesis, mitosis, migration, and invasion, thereby supporting tumor cell proliferation and migration.193,194 Known SASP factors, such as IL-6, IL-8, and CCL5, are key mediators driving tumor cell proliferation.195,196 VEGF and CXCL5 have been shown to promote angiogenesis in tumors.197,198 The MMP contributes to tumor migration by degrading the ECM.199 Additionally, senescent immune cells exhibit attenuated bone tumor cell attack due to poor memory function.200,201
On the other hand, cell cycle arrest in senescent cells limits tumor development. The senescent cells induced by p16 and p21 exhibit irreversible growth arrest, directly inhibiting tumor cell proliferation. Several components of the senescent cell-derived SASP restrict tumor cell growth. IGF binding protein 7 and GROα in the SASP have been found to promote tumor cell growth arrest.202,203 Plasminogen activator inhibitor 1 also enhances replicative senescence in tumor cells.204 Thus, the induction of tumor cell senescence has a significant effect on anti-bone tumor therapy. Indeed, the accumulation of senescent cells has been observed in tumor specimens following the administration of chemotherapy, radiotherapy, or targeted therapy.205,206,207 This is closely related to the severe DNA damage triggered by anti-tumor therapies. Notably, the threshold for the therapeutic induction of tumor cell senescence requires further definition to reduce the occurrence of this process in non-tumor cells.187 Further studies are needed to document the behavior of various senescent cells in bone tumors to gain a more comprehensive understanding of their dual role in patients with bone tumors.
Cellular senescence and other age-related mechanisms in ankylosing spondylitis
Currently, there is a lack of research revealing the relationship between aging-induced cellular senescence and ankylosing spondylitis (AS). However, some clues can be obtained from the clinical manifestations of elderly AS patients. Elderly individuals always exhibit reduced function of various types of cells combined with specific lesions of AS, increasing the susceptibility of elderly AS patients to severe complications. The current observation that elderly AS patients exhibit severe OP, rapid sarcopenia, and an increased incidence of cardiovascular events indicates that aging-related cellular senescence is involved in the aggravation of AS.208 It is well known that pathological new bone formation accelerates bone fusion in AS. Recent studies have indicated that chondrocytes promote ectopic new bone formation in AS through the BMP6/pSmad1/5 pathway, suggesting that inducing chondrocyte senescence in AS may help alleviate its progression.209 Future research on the role of cellular senescence and other age-related mechanisms in the occurrence and development of AS is worthy of attention.
Cellular senescence and other age-related mechanisms in “bone‒organ” disorders
In addition to cells within the bone marrow, cells in other organs play pivotal roles in aging-related bone disease. Studies have shown that premature skin aging leads to bone loss in mice.210 Cystatin-A, which is secreted by skin keratinocytes, binds to receptors for activated C-kinase 1 in OBs and OCs progenitor cells, resulting in enhanced OBs differentiation and restricted OCs differentiation. This study connects skin aging with age-related bone loss and confirms that improving the level of Cystatin-A in the skin could serve as a potential topical treatment for age-related OP.210 The same research group also generated a single-cell nuclear transcriptome atlas of skeletal muscle, revealing the critical role of FOXO3 in counteracting skeletal muscle aging.211 These findings provide a theoretical basis for further development of diagnostic and intervention strategies for skeletal muscle aging. Senescent skeletal muscle cells secrete fewer myogenic factors, which impairs the maintenance of skeletal homeostasis.211 Neuronal cells have also been proven to regulate bone homeostasis.212 PGE2 receptor 4 in neuronal cells regulates bone homeostasis and promotes regeneration by sensing PGE2 secreted by OBs. Aging-induced hypofunction of neuronal cells may affect bone regeneration through reduced receptor expression. Moreover, the abnormal elevation of OB-derived sclerostin associated with aging dysregulates the Wnt/β-catenin pathway in the brain and increases the production of β-amyloid through the β-catenin-BACE1 signaling pathway, thereby accelerating cognitive impairment.213 This study reveals the mechanism of “bone‒brain” interactions and provides a new strategy for the treatment of Alzheimer’s disease. This study demonstrated that the expression of Sirtuin 2 (SIRT2) was increased in the hepatocytes of aged individuals. Liver-specific SIRT2 deletion upregulated the transfer of hepatic leucine α-2-glycoprotein 1 (LRG1) to bone marrow mononuclear cells, which inhibited the nuclear translocation of NF-κB p65 and the activation of OCs, ultimately slowing the progression of OP in elderly individuals.214 The discovery of the “bone‒liver” axis provides a new perspective for the treatment of OP.
Aging is usually accompanied by disruption of gut barrier integrity, changes in the composition of the gut microbiota, and increased susceptibility to a variety of aging-related diseases.215 An oral hydrogel microsphere system has been developed to achieve targeted delivery of BMSCs by adhering to intestinal epithelial cells.216 Following 8 weeks of continuous oral treatment, a significant increase in bone density and trabecular number and a decrease in trabecular spacing were observed in aged mice.216 RNA sequencing revealed that the system effectively reversed mitochondrial senescence in BMSCs by activating the AMPK-SIRT1 pathway and promoted regeneration of aged bone tissue.216 Furthermore, the probiotic strain Lactobacillus plantarum TWK10 has been found to attenuate aging-related bone loss by modulating intestinal dysbiosis and increasing total short-chain fatty acid levels.217 It has been reported that gut microbial tryptophan metabolites promote osteogenesis and inhibit OCs activation by enhancing the production of IL-10 by M2 MACs in the intestinal lamina propria into the bone marrow.218 Collectively, these data support the inextricable link between the “bone-gut” axis and suggest that the “bone-gut” axis holds promise for the treatment of OP in the elderly. Owing to the inadequacy of existing experimental techniques, the mechanism of the complex crosstalk between bone and various organs remains obscure. It is believed that advanced technologies, such as bone microarrays, organoids, and assemblies, will be able to elucidate the bone‒organ communication mechanism in a more systematic way in the future, thus contributing to the treatment of anti-bone aging diseases. The cellular senescence and other age-related mechanisms in “bone‒organ” disorders are shown in Table 4.
Perspective and translation of the cellular senescence and other age-related mechanisms in bone diseases
The commonalities and differences in cellular senescence and other age-related mechanisms are important in understanding the role of cells in bone disease and in developing targeted therapeutic strategies. Cells in different skeletal diseases exhibit common features related to aging, such as the secretion of SASP and oxidative stress. However, there are significant differences in cellular senescence in different diseases, mainly in the specific changes of cell functions and different mechanisms of disease progression. For example, the senescence of MSCs manifests as a decrease in osteogenic differentiation capacity in OA, leading to bone loss. A decrease in the number of MSCs in OA weakens the cartilage repair capacity. However, a decrease in osteogenic capacity may reduce the ossification and alleviate the progression of OA. Senescent OBs lead to reduced bone formation, and their promotion of OCs activity in OP exacerbates bone loss, while in OA, it accelerates cartilage degeneration and inflammation. OCs enhance bone resorption, leading to OP. While in OA, their senescence contributes to the osteogenic differentiation of MSCs, exacerbating joint damage. Moreover, OCs induce H-type angiogenesis, which contributes to the alleviation of bone loss in OP, but elevated oxygen levels exacerbate the process of chondrocyte senescence in OA. In IVDD, OCs exacerbate pain through innervation. Senescent synoviocytes have been shown to lead to synovial fluid reduction and inflammation, which is manifested by synovial fibroblast dysfunction in OA, accelerating cartilage degeneration, whereas in RA, it is manifested by synovial MACs infiltration, promoting chronic inflammation and joint destruction. Currently, four principal strategies have been identified for targeting age-related cellular senescence, including drug administration, immune clearance, genetic intervention, and vaccine treatment. The translation and application of these strategies indicate directions for anti-cellular senescence in the future. Potential therapeutics for disorders that target age-related cellular senescence are shown in Fig. 5 and Table 5.

Modes of action for anti-cellular senescence. a Selective elimination of senescent cells by inhibiting the increase in BCL-2 and promoting senescent cell apoptosis. b CAR-T cells were cultured in vitro to target senescent cell-specific antigens to effectively eliminate senescent cells in vivo. c Genetic intervention rejuvenates senescent cells. d Vaccine treatment to prevent aging increased cellular senescence
Drug administration
Targeted killing of senescent cells through drugs has been shown to be effective.219 Researchers have observed a significant reduction in the proportion of senescent cells by simultaneously using dasatinib (D) and quercetin (Q). Aged mice treated with D and Q presented prolonged lifespan, enhanced osteogenic differentiation, and reduced aging-related bone senescence.219 The survival of senescent cells is closely related to the antiapoptotic proteins BCL-2 and BCL-XL. ABT263, an excellent specific inhibitor of BCL-2 and BCL-X, rapidly reduces the number of senescent cells when it is administered to aged mice, in which the levels of aging markers in bone cells are also significantly reduced.220 Fisetin (F) and cardiac glycosides are also used to reduce the number of senescent cells. F clears senescent cells from tissues, restores tissue homeostasis, and reduces aging-related pathologies.221 Cardiac glycosides cause extensive death of senescent cells by creating a low-pH microenvironment.222 In addition to directly killing cells, inhibiting SASP secretion from senescent cells is also an effective strategy for managing bone aging.101,223 In an aged mouse model, SASP components were successfully blocked by TNF-α inhibitors and an IL-1 receptor antagonist, which rescued bone aging.224,225 Drugs that target cellular senescence have shown promising anti-aging effects in animal models. However, more compelling is the generation of good data from anti-aging clinical trials. Physical function is being monitored in patients with age-related bone, lung, kidney and brain diseases after treatment with D + Q, F, or D + Q + F (NCT04313634, NCT04476953, NCT02848131, and NCT04785300).9,226 At least 20 clinical trials targeting age-related cellular changes are underway or planned.227
Cell therapy
The off-target effects of drugs against aging-related cellular senescence lead to reduced efficacy.228 Cell therapy, including immune clearance and stem cell transplantation, provides better results in slowing the aging process. Immune clearance involves the removal of senescent cells by targeting specific antigens on the surface membrane of senescent cells, including the urokinase-type plasminogen activator receptor (uPAR), DPP4, and CD9 receptors.227 In vitro culture of uPAR-specific chimeric antigen receptor (CAR) T cells has been shown to be effective in reducing the number of senescent cells in vivo.229 Natural killer (NK) cells have been shown to target senescent cells through perforin-mediated granule cytotoxicity.230 MACs eliminate senescent cells via direct contact with senescent cells during limb regeneration in salamanders, although the exact mechanism is not fully understood.231 CAR-T cells, CAR-NK cells, or CAR-MACs may lead to successful removal of senescent cells with enhanced immune cell-specific targeting. The strong differentiation capacity of stem cells offers tantalizing possibilities for anti-cellular senescence. Reduced amyloid deposition and improved cognitive performance have been observed in Alzheimer’s disease mice after transplantation of neural stem cells from young mice.232 Transplantation of muscle stem cells has been applied in trials of myotonic dystrophy and traumatic muscle injury.233,234 Cell therapy promises to yield unexpected results in future anti-aging research.
Genetic intervention
Genetic intervention rejuvenates senescent cells, restoring their youthful capacity for repair. A study revealed that the lack of YAP and chromobox (CBX) 4 in mice led to significant bone aging. After the expression of YAP and CBX4 was upregulated through adeno-associated virus (AAV) gene therapy, the mice showed with reduced signs of bone aging.235,236 Severe OP was observed after lysine-specific demethylase 4B (KDM4B) was knocked out in mouse BMSCs, but overexpressing KDM4B in BMSCs may have tremendous potential in OP treatment.18 Non-coding RNAs are closely involved in the aging process. miR-204 reduces the secretion of proteoglycans, accelerating the progression of OA.237 Local injection of lentiviruses containing anti-miR-204 sequences may effectively rescue OA. In addition, the overexpression of miR-290 and miR-302 reverses aging by inhibiting p21.238
Vaccine treatment
Recently developed vaccines against senescent cells offer promising strategies for reversing age-related cellular senescence by targeting specific antigens and inducing immune responses.239 Senescence is associated with the accumulation of a number of molecular markers in senescent tissues that are absent or under-expressed in healthy tissues. Studies have revealed significantly increased levels of NGF in OA, IL-1β in type 2 diabetes mellitus (T2D), and TAU in AD, making it a potential target for anti-aging vaccines.240,241,242,243 In one study, recombinant NGF covalently linked to cucumber mosaic virus-derived tetanus toxoid epitopes on mosaic virus-derived virus-like particles (VLPs) significantly reversed OA-associated pain after inoculation into mice.240 A vaccine (NCT00924105) containing full-length recombinant IL-1β-coupled VLPs is currently being observed for its efficacy in patients with T2D, as demonstrated by ClinicalTrials.gov.241 Two TAU-targeted AD vaccines, AADvac1 (NCT02579252) and ACI-35 (NCT04445831), have been identified in currently conducted clinical trials, and they demonstrate unexpected translational promise.242,243 Despite the encouraging results observed in the current studies, further validation, including robust human clinical trials for safety, efficacy, and long-term outcomes, is needed to ascertain the anti-aging potential of these vaccines.
Conclusion and perspectives
Cellular senescence and other age-related mechanisms has been shown to be strongly associated with the onset and progression of bone disease. The present study suggests that aging-induced cellular senescence involves collective phenotypes composed of a complex network of effector programs. Crosstalk between senescent bone cells involves changes in intrinsic senescence mechanisms, local factors, and signaling pathways. A full understanding of the commonalities and differences exhibited by senescent cells in skeletal diseases provides significant clues for potential interventions for age-related cellular senescence. The targeting of senescent changes in these cells to rescue skeletal disease has been attempted, but many key issues must be addressed before the clinical translation of these therapies can be realized. For example, while the SASP is recognized as a key driver of bone aging, the exact mechanisms by which it accelerates skeletal degeneration are not fully understood. Further investigation of the intricate crosstalk between various bone cells induced by aging is needed to reveal its impact on skeletal pathology. The identification of appropriate biomarkers is crucial for distinguishing between normal, physiologically senescent, and pathologically senescent cells, which is essential for the effective treatment of aging-related diseases.
To facilitate advancements in the field, a more comprehensive comparative analysis of aging-related cellular senescence in different skeletal diseases is needed to identify both common mechanisms and those changes that are specific to individual diseases. This will provide a theoretical basis for the design of universal and customized treatments. From a clinical perspective, current therapeutic strategies, including drug administration, cell therapy, genetic intervention, and vaccine treatment, require further refinement. Future studies should integrate multiple therapeutic modalities to comprehensively regulate intercellular and intracellular crosstalk networks, with an emphasis on improving the stability, efficacy and safety of interventions. In addition, given the heterogeneity of the individual aging process, a precision medicine approach should be prioritized to tailor anti-aging therapies to specific molecular and cellular aging profiles. By addressing these challenges, future research could bridge the gap between basic research and clinical applications, ultimately providing more effective and personalized interventions for aging-related diseases.
Cellular senescence and other age-related mechanisms in skeletal diseases not only contribute to the development of a variety of skeletal diseases, but also tend to affect organ dysfunction throughout the body, resulting in a deleterious cascade effect. This review also provides important insights into the field of systemic age-related diseases. Understanding the mechanisms associated with cellular senescence in bone tissue may provide new perspectives for treating age-related dysfunction in other organs. In addition, therapeutic strategies targeting senescent cells have great potential for improving the quality of life of elderly individuals and thereby promoting healthy aging. From a societal perspective, age-related diseases generate significant healthcare costs and long-term care needs. Effective interventions to delay the onset of age-related diseases can reduce hospitalization rates, decrease the need for surgical interventions, reduce the reliance on assisted care, and ultimately benefit both the individual and the healthcare system. Future research that integrates skeletal aging with systemic aging diseases may pave the way for comprehensive anti-aging therapies that improve longevity and overall health.
References
Wan, M., Gray-Gaillard, E. F. & Elisseeff, J. H. Cellular senescence in musculoskeletal homeostasis, diseases, and regeneration. Bone Res. 9, 41 (2021).
Chandra, A. & Rajawat, J. Skeletal aging and osteoporosis: mechanisms and therapeutics. Int. J. Mol. Sci. 22, 3553 (2021).
Ogrodnik, M. et al. Guidelines for minimal information on cellular senescence experimentation in vivo. Cell 187, 4150–4175 (2024).
Fulzele, S. et al. Muscle-derived miR-34a increases with age in circulating extracellular vesicles and induces senescence of bone marrow stem cells. Aging 11, 1791–1803 (2019).
Ambrosi, T. H. et al. Aged skeletal stem cells generate an inflammatory degenerative niche. Nature 597, 256–262 (2021).
Risbud, M. V. & Shapiro, I. M. Role of cytokines in intervertebral disc degeneration: pain and disc content. Nat. Rev. Rheumatol. 10, 44–56 (2014).
Patel, J. Economic implications of osteoporotic fractures in postmenopausal women. Am. J. Manag. Care 26, S311–S318 (2020).
Ma, V. Y., Chan, L. & Carruthers, K. J. Incidence, prevalence, costs, and impact on disability of common conditions requiring rehabilitation in the United States: stroke, spinal cord injury, traumatic brain injury, multiple sclerosis, osteoarthritis, rheumatoid arthritis, limb loss, and back pain. Arch. Phys. Med. Rehabil. 95, 986–995.e1 (2014).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: an expanding universe. Cell 186, 243–278 (2023).
Cui, J., Shibata, Y., Zhu, T., Zhou, J. & Zhang, J. Osteocytes in bone aging: advances, challenges, and future perspectives. Ageing Res. Rev. 77, 101608 (2022).
Farr, J. N. et al. Identification of senescent cells in the bone microenvironment. J. Bone Miner. Res. 31, 1920–1929 (2016).
Farr, J. N. et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 23, 1072–1079 (2017).
Wang, T., Huang, S. & He, C. Senescent cells: a therapeutic target for osteoporosis. Cell Prolif. 55, e13323 (2022).
Qadir, A. et al. Senile osteoporosis: the involvement of differentiation and senescence of bone marrow stromal cells. Int. J. Mol. Sci. 21, 349 (2020).
Corrado, A., Cici, D., Rotondo, C., Maruotti, N. & Cantatore, F. P. Molecular basis of bone aging. Int. J. Mol. Sci. 21, 3679 (2020).
Matsushita, Y. et al. Bone marrow endosteal stem cells dictate active osteogenesis and aggressive tumorigenesis. Nat. Commun. 14, 2383 (2023).
Ambrosi, T. H. et al. Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell 20, 771–784.e6 (2017).
Deng, P. et al. Loss of KDM4B exacerbates bone-fat imbalance and mesenchymal stromal cell exhaustion in skeletal aging. Cell Stem Cell 28, 1057–1073.e7 (2021).
Xie, Z. et al. Single-cell RNA sequencing analysis of human bone-marrow-derived mesenchymal stem cells and functional subpopulation identification. Exp. Mol. Med. 54, 483–492 (2022).
Yang, R. et al. Premature aging of skeletal stem/progenitor cells rather than osteoblasts causes bone loss with decreased mechanosensation. Bone Res. 11, 35 (2023).
Jing, Y. et al. Genome-wide CRISPR activation screening in senescent cells reveals SOX5 as a driver and therapeutic target of rejuvenation. Cell Stem Cell 30, 1452–1471.e10 (2023).
Luo, Z. et al. Rejuvenation of BMSCs senescence by pharmacological enhancement of TFEB-mediated autophagy alleviates aged-related bone loss and extends lifespan in middle aged mice. Bone Res. 12, 45 (2024).
Musri, M. M., Corominola, H., Casamitjana, R., Gomis, R. & Párrizas, M. Histone H3 lysine 4 dimethylation signals the transcriptional competence of the adiponectin promoter in preadipocytes. J. Biol. Chem. 281, 17180–17188 (2006).
Hamam, D. et al. microRNA-320/RUNX2 axis regulates adipocytic differentiation of human mesenchymal (skeletal) stem cells. Cell Death Dis. 5, e1499 (2014).
Bowers, R. R., Kim, J. W., Otto, T. C. & Lane, M. D. Stable stem cell commitment to the adipocyte lineage by inhibition of DNA methylation: role of the BMP-4 gene. Proc. Natl. Acad. Sci. USA 103, 13022–13027 (2006).
Fernández, A. F. et al. H3K4me1 marks DNA regions hypomethylated during aging in human stem and differentiated cells. Genome Res. 25, 27–40 (2015).
Ma, S. et al. Skeletal muscle-derived extracellular vesicles transport glycolytic enzymes to mediate muscle-to-bone crosstalk. Cell Metab. 35, 2028–2043.e7 (2023).
Fleischhacker, V. et al. Aged-vascular niche hinders osteogenesis of mesenchymal stem cells through paracrine repression of Wnt-axis. Aging Cell 23, e14139 (2024).
Cai, W. et al. Mitochondrial transfer regulates cell fate through metabolic remodeling in osteoporosis. Adv. Sci. 10, e2204871 (2023).
Fang, C. L., Liu, B. & Wan, M. Bone-SASP” in skeletal aging. Calcif. Tissue Int. 113, 68–82 (2023).
Manolagas, S. C. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 21, 115–137 (2000).
Kassem, M. & Marie, P. J. Senescence-associated intrinsic mechanisms of osteoblast dysfunctions. Aging Cell 10, 191–197 (2011).
Ueda, Y. et al. Induction of senile osteoporosis in normal mice by intra-bone marrow-bone marrow transplantation from osteoporosis-prone mice. Stem Cells 25, 1356–1363 (2007).
Li, K. et al. Targeting ROS-induced osteoblast senescence and RANKL production by Prussian blue nanozyme based gene editing platform to reverse osteoporosis. Nano Today 50, 101839 (2023).
Takada, K. et al. Treatment of senile osteoporosis in SAMP6 mice by intra-bone marrow injection of allogeneic bone marrow cells. Stem Cells 24, 399–405 (2006).
Doolittle, M. L. et al. Multiparametric senescent cell phenotyping reveals targets of senolytic therapy in the aged murine skeleton. Nat. Commun. 14, 4587 (2023).
Shen, B. et al. A mechanosensitive peri-arteriolar niche for osteogenesis and lymphopoiesis. Nature 591, 438–444 (2021).
Chen, A. et al. mTORC1 induces plasma membrane depolarization and promotes preosteoblast senescence by regulating the sodium channel Scn1a. Bone Res. 10, 25 (2022).
Wang, Y. et al. METTL3-mediated m6A modification increases Hspa1a stability to inhibit osteoblast aging. Cell Death Discov. 10, 155 (2024).
Rached, M. T. et al. FoxO1 expression in osteoblasts regulates glucose homeostasis through regulation of osteocalcin in mice. J. Clin. Invest. 120, 357–368 (2010).
Ambrogini, E. et al. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab. 11, 136–146 (2010).
Cao, J. J. et al. Aging impairs IGF-I receptor activation and induces skeletal resistance to IGF-I. J. Bone Miner. Res. 22, 1271–1279 (2007).
Kim, J. M., Lin, C., Stavre, Z., Greenblatt, M. B. & Shim, J. H. Osteoblast-osteoclast communication and bone homeostasis. Cells 9, 2073 (2020).
Xu, R. et al. Targeting skeletal endothelium to ameliorate bone loss. Nat. Med. 24, 823–833 (2018).
Lei, H., Schmidt-Bleek, K., Dienelt, A., Reinke, P. & Volk, H. D. Regulatory T cell-mediated anti-inflammatory effects promote successful tissue repair in both indirect and direct manners. Front. Pharmacol. 6, 184 (2015).
Pietschmann, P. et al. Bone structure and metabolism in a rodent model of male senile osteoporosis. Exp. Gerontol. 42, 1099–1108 (2007).
Lin, W. et al. Breaking osteoclast-acid vicious cycle to rescue osteoporosis via an acid responsive organic framework-based neutralizing and gene editing platform. Small 20, e2307595 (2024).
Almeida, M. et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 282, 27285–27297 (2007).
Makhluf, H. A., Mueller, S. M., Mizuno, S. & Glowacki, J. Age-related decline in osteoprotegerin expression by human bone marrow cells cultured in three-dimensional collagen sponges. Biochem. Biophys. Res. Commun. 268, 669–672 (2000).
Cao, J. J. et al. Aging increases stromal/osteoblastic cell-induced osteoclastogenesis and alters the osteoclast precursor pool in the mouse. J. Bone Miner. Res. 20, 1659–1668 (2005).
Chung, P. L. et al. Effect of age on regulation of human osteoclast differentiation. J. Cell. Biochem. 115, 1412–1419 (2014).
Cao, J., Venton, L., Sakata, T. & Halloran, B. P. Expression of RANKL and OPG correlates with age-related bone loss in male C57BL/6 mice. J. Bone Miner. Res. 18, 270–277 (2003).
Yu, W. et al. Bone marrow adipogenic lineage precursors promote osteoclastogenesis in bone remodeling and pathologic bone loss. J. Clin. Invest. 131, e140214 (2021).
Hariri, H., Kose, O., Bezdjian, A., Daniel, S. J. & St-Arnaud, R. USP53 regulates bone homeostasis by controlling RANKL expression in osteoblasts and bone marrow adipocytes. J. Bone Miner. Res. 38, 578–596 (2023).
Faqeer, A. et al. Cleaved SPP1-rich extracellular vesicles from osteoclasts promote bone regeneration via TGFβ1/SMAD3 signaling. Biomaterials 303, 122367 (2023).
Ryu, J. et al. Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. EMBO J. 25, 5840–5851 (2006).
Takeshita, S. et al. Osteoclast-secreted CTHRC1 in the coupling of bone resorption to formation. J. Clin. Invest. 123, 3914–3924 (2013).
Negishi-Koga, T. et al. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat. Med. 17, 1473–1480 (2011).
Liu, X. et al. Osteoclasts protect bone blood vessels against senescence through the angiogenin/plexin-B2 axis. Nat. Commun. 12, 1832 (2021).
Henriksen, K. et al. Osteoclasts prefer aged bone. Osteoporos. Int. 18, 751–759 (2007).
Groessner-Schreiber, B., Krukowski, M., Lyons, C. & Osdoby, P. Osteoclast recruitment in response to human bone matrix is age related. Mech. Ageing Dev. 62, 143–154 (1992).
Ikpegbu, E. et al. FGF-2 promotes osteocyte differentiation through increased E11/podoplanin expression. J. Cell. Physiol. 233, 5334–5347 (2018).
Zhang, C. et al. Ageing characteristics of bone indicated by transcriptomic and exosomal proteomic analysis of cortical bone cells. J. Orthop. Surg. Res. 14, 129 (2019).
Gaudio, A. et al. Increased sclerostin serum levels associated with bone formation and resorption markers in patients with immobilization-induced bone loss. J. Clin. Endocrinol. Metab. 95, 2248–2253 (2010).
Piemontese, M. et al. Old age causes de novo intracortical bone remodeling and porosity in mice. JCI Insight 2, e93771 (2017).
Farr, J. N., Kaur, J., Doolittle, M. L. & Khosla, S. Osteocyte Cellular Senescence. Curr. Osteoporos. Rep. 18, 559–567 (2020).
Dalle Pezze, P. et al. Dynamic modelling of pathways to cellular senescence reveals strategies for targeted interventions. PLoS Comput. Biol. 10, e1003728 (2014).
Dole, N. S. et al. Osteocyte-intrinsic TGF-β signaling regulates bone quality through perilacunar/canalicular remodeling. Cell Rep. 21, 2585–2596 (2017).
Schurman, C. A., Verbruggen, S. W. & Alliston, T. Disrupted osteocyte connectivity and pericellular fluid flow in bone with aging and defective TGF-β signaling. Proc. Natl. Acad. Sci. USA 118, e2023999118 (2021).
Yee, C. S., Schurman, C. A., White, C. R. & Alliston, T. Investigating osteocytic perilacunar/canalicular remodeling. Curr. Osteoporos. Rep. 17, 157–168 (2019).
Jilka, R. L. & O’Brien, C. A. The role of osteocytes in age-related bone loss. Curr. Osteoporos. Rep. 14, 16–25 (2016).
Youlten, S. E. et al. Osteocyte transcriptome mapping identifies a molecular landscape controlling skeletal homeostasis and susceptibility to skeletal disease. Nat. Commun. 12, 2444 (2021).
Nakashima, T. et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 17, 1231–1234 (2011).
Wang, Z. X. et al. Young osteocyte-derived extracellular vesicles facilitate osteogenesis by transferring tropomyosin-1. J. Nanobiotechnol. 22, 208 (2024).
Liu, X. et al. Oxylipin-PPARγ-initiated adipocyte senescence propagates secondary senescence in the bone marrow. Cell Metab. 35, 667–684.e6 (2023).
Justesen, J. et al. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology 2, 165–171 (2001).
Hozumi, A. et al. Bone marrow adipocytes support dexamethasone-induced osteoclast differentiation. Biochem. Biophys. Res. Commun. 382, 780–784 (2009).
Fan, Y. et al. Parathyroid hormone directs bone marrow mesenchymal cell fate. Cell Metab. 25, 661–672 (2017).
Xie, L. et al. PCLAF induces bone marrow adipocyte senescence and contributes to skeletal aging. Bone Res. 12, 38 (2024).
Fischer, V. & Haffner-Luntzer, M. Interaction between bone and immune cells: Implications for postmenopausal osteoporosis. Semin. Cell Dev. Biol. 123, 14–21 (2022).
Li, C. J. et al. Senescent immune cells release grancalcin to promote skeletal aging. Cell Metab. 33, 1957–1973.e6 (2021).
Chen, K. et al. Communications between bone marrow macrophages and bone cells in bone remodeling. Front. Cell Dev. Biol. 8, 598263 (2020).
Sims, N. A. & Quinn, J. M. Osteoimmunology: oncostatin M as a pleiotropic regulator of bone formation and resorption in health and disease. Bonekey Rep. 3, 527 (2014).
Li, J. et al. TGFβ1+CCR5+ neutrophil subset increases in bone marrow and causes age-related osteoporosis in male mice. Nat. Commun. 14, 159 (2023).
Hu, X., Sun, Y., Xu, W., Lin, T. & Zeng, H. Expression of RANKL by peripheral neutrophils and its association with bone mineral density in COPD. Respirology 22, 126–132 (2017).
Zhang, W., Dang, K., Huai, Y. & Qian, A. Osteoimmunology: the regulatory roles of T lymphocytes in osteoporosis. Front. Endocrinol. 11, 465 (2020).
Adamopoulos, I. E. et al. Interleukin-17A upregulates receptor activator of NF-kappaB on osteoclast precursors. Arthritis Res. Ther. 12, R29 (2010).
Axmann, R. et al. CTLA-4 directly inhibits osteoclast formation. Ann. Rheum. Dis. 67, 1603–1609 (2008).
Nordqvist, J., Bernardi, A., Islander, U. & Carlsten, H. Effects of a tissue-selective estrogen complex on B lymphopoiesis and B cell function. Immunobiology 222, 918–923 (2017).
Weitzmann, M. N. The role of inflammatory cytokines, the RANKL/OPG axis, and the immunoskeletal interface in physiological bone turnover and osteoporosis. Scientifica 2013, 125705 (2013).
Kusumbe, A. P., Ramasamy, S. K. & Adams, R. H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 507, 323–328 (2014).
Yang, P. et al. Preservation of type H vessels and osteoblasts by enhanced preosteoclast platelet-derived growth factor type BB attenuates glucocorticoid-induced osteoporosis in growing mice. Bone 114, 1–13 (2018).
Ribatti, D., Nico, B., Vacca, A., Roncali, L. & Dammacco, F. Endothelial cell heterogeneity and organ specificity. J. Hematother. Stem Cell Res. 11, 81–90 (2002).
Garlanda, C. & Dejana, E. Heterogeneity of endothelial cells. Specific markers. Arterioscler. Thromb. Vasc. Biol. 17, 1193–1202 (1997).
Liu, L. et al. Endothelial cell-derived exosomes trigger a positive feedback loop in osteogenesis-angiogenesis coupling via up-regulating zinc finger and BTB domain containing 16 in bone marrow mesenchymal stem cell. J. Nanobiotechnol. 22, 721 (2024).
Cleveland, R. J. & Callahan, L. F. Can osteoarthritis predict mortality? North Carol. Med. J. 78, 322–325 (2017).
Loeser, R. F., Collins, J. A. & Diekman, B. O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 12, 412–420 (2016).
Prieto-Alhambra, D. et al. Incidence and risk factors for clinically diagnosed knee, hip and hand osteoarthritis: influences of age, gender and osteoarthritis affecting other joints. Ann. Rheum. Dis. 73, 1659–1664 (2014).
Jeon, O. H. et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 23, 775–781 (2017).
Jeon, O. H., David, N., Campisi, J. & Elisseeff, J. H. Senescent cells and osteoarthritis: a painful connection. J. Clin. Invest. 128, 1229–1237 (2018).
Childs, B. G., Durik, M., Baker, D. J. & van Deursen, J. M. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat. Med. 21, 1424–1435 (2015).
He, S. & Sharpless, N. E. Senescence in health and disease. Cell 169, 1000–1011 (2017).
Kapoor, M., Martel-Pelletier, J., Lajeunesse, D., Pelletier, J. P. & Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 7, 33–42 (2011).
Gao, S. G. et al. Correlation between senescence-associated beta-galactosidase expression in articular cartilage and disease severity of patients with knee osteoarthritis. Int. J. Rheum. Dis. 19, 226–232 (2016).
Xu, M. et al. Transplanted senescent cells induce an osteoarthritis-like condition in mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 72, 780–785 (2017).
Gong, Z. et al. CircRREB1 mediates lipid metabolism related senescent phenotypes in chondrocytes through FASN post-translational modifications. Nat. Commun. 14, 5242 (2023).
Shao, Y. et al. PDZK1 protects against mechanical overload-induced chondrocyte senescence and osteoarthritis by targeting mitochondrial function. Bone Res. 12, 41 (2024).
Ji, M. L. et al. Sirt6 attenuates chondrocyte senescence and osteoarthritis progression. Nat. Commun. 13, 7658 (2022).
Wang, J. et al. Rejuvenating hyaline cartilage with senescence-targeting Si-ADAM19 delivery for osteoarthritis therapy. Adv. Sci. 12, e2414419 (2025).
Cao, H. et al. MYL3 protects chondrocytes from senescence by inhibiting clathrin-mediated endocytosis and activating of Notch signaling. Nat. Commun. 14, 6190 (2023).
Xie, J., Huang, Z., Yu, X., Zhou, L. & Pei, F. Clinical implications of macrophage dysfunction in the development of osteoarthritis of the knee. Cytokine Growth Factor Rev. 46, 36–44 (2019).
Ferrucci, L. & Fabbri, E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 15, 505–522 (2018).
Jeon, O. H. et al. Senescence cell-associated extracellular vesicles serve as osteoarthritis disease and therapeutic markers. JCI Insight 4, e125019 (2019).
Fu, B. et al. Matrix stiffening promotes chondrocyte senescence and the osteoarthritis development through downregulating HDAC3. Bone Res. 12, 32 (2024).
Čamernik, K. et al. Increased exhaustion of the subchondral bone-derived mesenchymal stem/stromal cells in primary versus dysplastic osteoarthritis. Stem Cell Rev. Rep. 16, 742–754 (2020).
Yoon, D. S. et al. TLR4 downregulation by the RNA-binding protein PUM1 alleviates cellular aging and osteoarthritis. Cell Death Differ. 29, 1364–1378 (2022).
Ye, G. et al. ALKBH5 facilitates CYP1B1 mRNA degradation via m6A demethylation to alleviate MSC senescence and osteoarthritis progression. Exp. Mol. Med. 55, 1743–1756 (2023).
Lei, J. et al. Exosomes from antler stem cells alleviate mesenchymal stem cell senescence and osteoarthritis. Protein Cell 13, 220–226 (2022).
Malaise, O. et al. Mesenchymal stem cell senescence alleviates their intrinsic and seno-suppressive paracrine properties contributing to osteoarthritis development. Aging 11, 9128–9146 (2019).
Cao, X. et al. Intraarticular senescent chondrocytes impair the cartilage regeneration capacity of mesenchymal stem cells. Stem Cell Res. Ther. 10, 86 (2019).
Cavallo, C. et al. Small extracellular vesicles from adipose derived stromal cells significantly attenuate in vitro the NF-κB dependent inflammatory/catabolic environment of osteoarthritis. Sci. Rep. 11, 1053 (2021).
Murphy, M. P. et al. Articular cartilage regeneration by activated skeletal stem cells. Nat. Med. 26, 1583–1592 (2020).
Worthley, D. L. et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 160, 269–284 (2015).
Ng, J. Q. et al. Loss of Grem1-lineage chondrogenic progenitor cells causes osteoarthritis. Nat. Commun. 14, 6909 (2023).
Xie, J. et al. Cellular senescence in knee osteoarthritis: molecular mechanisms and therapeutic implications. Ageing Res. Rev. 70, 101413 (2021).
Hall, B. M. et al. Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging 8, 1294–1315 (2016).
Covarrubias, A. J. et al. Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages. Nat. Metab. 2, 1265–1283 (2020).
Liu-Bryan, R. & Terkeltaub, R. Emerging regulators of the inflammatory process in osteoarthritis. Nat. Rev. Rheumatol. 11, 35–44 (2015).
Chang, T. K. et al. Apelin enhances IL-1β expression in human synovial fibroblasts by inhibiting miR-144-3p through the PI3K and ERK pathways. Aging 12, 9224–9239 (2020).
Su, W. et al. Senescent preosteoclast secretome promotes metabolic syndrome associated osteoarthritis through cyclooxygenase 2. eLife 11, e79773 (2022).
Zhen, G. & Cao, X. Targeting TGFβ signaling in subchondral bone and articular cartilage homeostasis. Trends Pharmacol. Sci. 35, 227–236 (2014).
Zhang, H. et al. Maintaining hypoxia environment of subchondral bone alleviates osteoarthritis progression. Sci. Adv. 9, eabo7868 (2023).
Hu, W., Chen, Y., Dou, C. & Dong, S. Microenvironment in subchondral bone: predominant regulator for the treatment of osteoarthritis. Ann. Rheum. Dis. 80, 413–422 (2021).
Romeo, S. G. et al. Endothelial proteolytic activity and interaction with non-resorbing osteoclasts mediate bone elongation. Nat. Cell Biol. 21, 430–441 (2019).
Cabahug-Zuckerman, P. et al. Osteocyte apoptosis caused by hindlimb unloading is required to trigger osteocyte RANKL production and subsequent resorption of cortical and trabecular bone in mice femurs. J. Bone Miner. Res. 31, 1356–1365 (2016).
Ushiyama, T., Chano, T., Inoue, K. & Matsusue, Y. Cytokine production in the infrapatellar fat pad: another source of cytokines in knee synovial fluids. Ann. Rheum. Dis. 62, 108–112 (2003).
Sousa-Victor, P. et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506, 316–321 (2014).
Roberts, S. et al. Ageing in the musculoskeletal system. Acta Orthop. 87, 15–25 (2016).
Vo, N. et al. Accelerated aging of intervertebral discs in a mouse model of progeria. J. Orthop. Res. 28, 1600–1607 (2010).
Le Maitre, C. L., Freemont, A. J. & Hoyland, J. A. Accelerated cellular senescence in degenerate intervertebral discs: a possible role in the pathogenesis of intervertebral disc degeneration. Arthritis Res. Ther. 9, R45 (2007).
Gruber, H. E., Ingram, J. A., Norton, H. J. & Hanley, E. N. Jr. Senescence in cells of the aging and degenerating intervertebral disc: immunolocalization of senescence-associated beta-galactosidase in human and sand rat discs. Spine 32, 321–327 (2007).
Shi, Y. et al. Rescuing nucleus pulposus cells from senescence via dual-functional greigite nanozyme to alleviate intervertebral disc degeneration. Adv. Sci. 10, e2300988 (2023).
Shi, Y. et al. Rescuing nucleus pulposus cells from ROS toxic microenvironment via mitochondria-targeted carbon dot-supported prussian blue to alleviate intervertebral disc degeneration. Adv. Health. Mater. 13, e2303206 (2024).
Bu, W. et al. Rescue of nucleus pulposus cells from an oxidative stress microenvironment via glutathione-derived carbon dots to alleviate intervertebral disc degeneration. J. Nanobiotechnol. 22, 412 (2024).
Wu, S. et al. N-acetylcysteine-derived carbon dots for free radical scavenging in intervertebral disc degeneration. Adv. Health. Mater. 12, e2300533 (2023).
Li, H. et al. Lumbar instability remodels cartilage endplate to induce intervertebral disc degeneration by recruiting osteoclasts via Hippo-CCL3 signaling. Bone Res. 12, 34 (2024).
Hiyama, A. et al. Enhancement of intervertebral disc cell senescence by WNT/β-catenin signaling-induced matrix metalloproteinase expression. Arthritis Rheum. 62, 3036–3047 (2010).
Markova, D. Z. et al. An organ culture system to model early degenerative changes of the intervertebral disc II: profiling global gene expression changes. Arthritis Res. Ther. 15, R121 (2013).
Erwin, W. M., Islam, D., Inman, R. D., Fehlings, M. G. & Tsui, F. W. Notochordal cells protect nucleus pulposus cells from degradation and apoptosis: implications for the mechanisms of intervertebral disc degeneration. Arthritis Res. Ther. 13, R215 (2011).
Tran, C. M. et al. Regulation of CCN2/connective tissue growth factor expression in the nucleus pulposus of the intervertebral disc: role of Smad and activator protein 1 signaling. Arthritis Rheum. 62, 1983–1992 (2010).
Freund, A., Orjalo, A. V., Desprez, P. Y. & Campisi, J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol. Med. 16, 238–246 (2010).
Guo, S. et al. Role of the mechanosensitive piezo1 channel in intervertebral disc degeneration. Clin. Physiol. Funct. imaging 43, 59–70 (2023).
Shi, S. et al. Excessive mechanical stress-induced intervertebral disc degeneration is related to Piezo1 overexpression triggering the imbalance of autophagy/apoptosis in human nucleus pulpous. Arthritis Res. Ther. 24, 119 (2022).
Cheng, F. et al. The role of oxidative stress in intervertebral disc cellular senescence. Front. Endocrinol. 13, 1038171 (2022).
Xie, L. et al. CircERCC2 ameliorated intervertebral disc degeneration by regulating mitophagy and apoptosis through miR-182-5p/SIRT1 axis. Cell Death Dis. 10, 751 (2019).
Xue, Q. et al. Nrf2 activation by pyrroloquinoline quinone inhibits natural aging-related intervertebral disk degeneration in mice. Aging Cell 23, e14202. (2024).
Zhang, W. et al. Disassembly of the TRIM56-ATR complex promotes cytoDNA/cGAS/STING axis-dependent intervertebral disc inflammatory degeneration. J. Clin. Invest. 134, e165140 (2024).
Liu, C. et al. A redox homeostasis modulatory hydrogel with GLRX3+ extracellular vesicles attenuates disc degeneration by suppressing nucleus pulposus cell senescence. ACS Nano 17, 13441–13460 (2023).
Song, Y. et al. The NLRX1-SLC39A7 complex orchestrates mitochondrial dynamics and mitophagy to rejuvenate intervertebral disc by modulating mitochondrial Zn2+ trafficking. Autophagy 20, 809–829 (2024).
Cheng, Z. et al. Impaired degradation of PLCG1 by chaperone-mediated autophagy promotes cellular senescence and intervertebral disc degeneration. Autophagy 21, 352–373 (2025).
Molladavoodi, S., McMorran, J. & Gregory, D. Mechanobiology of annulus fibrosus and nucleus pulposus cells in intervertebral discs. Cell Tissue Res. 379, 429–444 (2020).
Dahia, C. L., Mahoney, E. & Wylie, C. Shh signaling from the nucleus pulposus is required for the postnatal growth and differentiation of the mouse intervertebral disc. PLoS One 7, e35944 (2012).
Veras, M. A., McCann, M. R., Tenn, N. A. & Séguin, C. A. Transcriptional profiling of the murine intervertebral disc and age-associated changes in the nucleus pulposus. Connect. Tissue Res. 61, 63–81 (2020).
Zhuang, Y. et al. The sonic hedgehog pathway suppresses oxidative stress and senescence in nucleus pulposus cells to alleviate intervertebral disc degeneration via GPX4. Biochim. Biophys. Acta Mol. Basis Dis. 1870, 166961 (2024).
Han, F. et al. Targeting endogenous reactive oxygen species removal and regulating regenerative microenvironment at annulus fibrosus defects promote tissue repair. ACS Nano 17, 7645–7661 (2023).
Risbud, M. V. et al. Evidence for skeletal progenitor cells in the degenerate human intervertebral disc. Spine 32, 2537–2544 (2007).
Yasen, M. et al. Changes of number of cells expressing proliferation and progenitor cell markers with age in rabbit intervertebral discs. Acta Biochim. Biophys. Sin. 45, 368–376 (2013).
Moore, R. J. The vertebral end-plate: what do we know? Eur. Spine J. 9, 92–96 (2000).
Broberg, K. B. On the mechanical behaviour of intervertebral discs. Spine 8, 151–165 (1983).
Paietta, R. C., Burger, E. L. & Ferguson, V. L. Mineralization and collagen orientation throughout aging at the vertebral endplate in the human lumbar spine. J. Struct. Biol. 184, 310–320 (2013).
Aigner, T., Gresk-otter, K. R., Fairbank, J. C., von der Mark, K. & Urban, J. P. Variation with age in the pattern of type X collagen expression in normal and scoliotic human intervertebral discs. Calcif. Tissue Int. 63, 263–268 (1998).
Benneker, L. M., Heini, P. F., Alini, M., Anderson, S. E. & Ito, K. 2004 Young Investigator Award Winner: vertebral endplate marrow contact channel occlusions and intervertebral disc degeneration. Spine 30, 167–173 (2005).
Hee, H. T., Chuah, Y. J., Tan, B. H., Setiobudi, T. & Wong, H. K. Vascularization and morphological changes of the endplate after axial compression and distraction of the intervertebral disc. Spine 36, 505–511 (2011).
Pan, D. et al. Senescence of endplate osteoclasts induces sensory innervation and spinal pain. eLife 12, RP92889 (2024).
Ni, S. et al. Sensory innervation in porous endplates by Netrin-1 from osteoclasts mediates PGE2-induced spinal hypersensitivity in mice. Nat. Commun. 10, 5643 (2019).
Fan, Y. et al. Senescent-like macrophages mediate angiogenesis for endplate sclerosis via IL-10 secretion in male mice. Nat. Commun. 15, 2939 (2024).
Smolen, J. S., Aletaha, D. & McInnes, I. B. Rheumatoid arthritis. Lancet 388, 2023–2038 (2016).
Shah, L. et al. Do menopause and aging affect the onset and progression of rheumatoid arthritis and systemic lupus erythematosus? Cureus 12, e10944 (2020).
Doran, M. F., Pond, G. R., Crowson, C. S., O’Fallon, W. M. & Gabriel, S. E. Trends in incidence and mortality in rheumatoid arthritis in Rochester, Minnesota, over a forty-year period. Arthritis Rheum. 46, 625–631 (2002).
Bauer, M. E. Accelerated immunosenescence in rheumatoid arthritis: impact on clinical progression. Immun. Ageing 17, 6 (2020).
Petersen, L. E. et al. Premature immunosenescence is associated with memory dysfunction in rheumatoid arthritis. Neuroimmunomodulation 22, 130–137 (2015).
Effros, R. B. The role of CD8 T cell replicative senescence in human aging. Discov. Med. 5, 293–297 (2005).
Martens, P. B., Goronzy, J. J., Schaid, D. & Weyand, C. M. Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum. 40, 1106–1114 (1997).
Goronzy, J. J. et al. Prognostic markers of radiographic progression in early rheumatoid arthritis. Arthritis Rheum. 50, 43–54 (2004).
Ormseth, M. J. et al. Telomere length and coronary atherosclerosis in rheumatoid arthritis. J. Rheumatol. 43, 1469–1474 (2016).
Prelog, M. Aging of the immune system: a risk factor for autoimmunity? Autoimmun. Rev. 5, 136–139 (2006).
Schmitt, C. A., Wang, B. & Demaria, M. Senescence and cancer—role and therapeutic opportunities. Nat. Rev. Clin. Oncol. 19, 619–636 (2022).
Li, K. et al. Thermo-sensitive hydrogel-mediated locally sequential release of doxorubicin and palbociclib for chemo-immunotherapy of osteosarcoma. Mater. Des. 224, 111365 (2022).
Lian, J., Yue, Y., Yu, W. & Zhang, Y. Immunosenescence: a key player in cancer development. J. Hematol. Oncol. 13, 151 (2020).
Inagaki, Y. et al. Dendritic and mast cell involvement in the inflammatory response to primary malignant bone tumours. Clin. Sarcoma Res. 6, 13 (2016).
Bischof, J. et al. Immune cells and immunosenescence. Folia Biol. 65, 53–63 (2019).
Zhang, C. et al. Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of osteosarcoma. Aging 12, 3486–3501 (2020).
Acosta, J. C. et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018 (2008).
Wang, B., Kohli, J. & Demaria, M. Senescent cells in cancer therapy: friends or foes? Trends Cancer 6, 838–857 (2020).
Aldinucci, D. & Colombatti, A. The inflammatory chemokine CCL5 and cancer progression. Mediat. Inflamm. 2014, 292376 (2014).
Ancrile, B., Lim, K. H. & Counter, C. M. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 21, 1714–1719 (2007).
Chen, C. et al. CXCL5 induces tumor angiogenesis via enhancing the expression of FOXD1 mediated by the AKT/NF-κB pathway in colorectal cancer. Cell Death Dis. 10, 178 (2019).
Coppé, J. P., Kauser, K., Campisi, J. & Beauséjour, C. M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 281, 29568–29574 (2006).
Kessenbrock, K., Plaks, V. & Werb, Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67 (2010).
Haynes, L. The effect of aging on cognate function and development of immune memory. Curr. Opin. Immunol. 17, 476–479 (2005).
Fan, L., Ru, J., Liu, T. & Ma, C. Identification of a novel prognostic gene signature from the immune cell infiltration landscape of osteosarcoma. Front. Cell Dev. Biol. 9, 718624 (2021).
Yang, G. et al. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc. Natl. Acad. Sci. USA 103, 16472–16477 (2006).
Wajapeyee, N., Serra, R. W., Zhu, X., Mahalingam, M. & Green, M. R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 132, 363–374 (2008).
Kortlever, R. M., Higgins, P. J. & Bernards, R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 8, 877–884 (2006).
Schoetz, U. et al. Early senescence and production of senescence-associated cytokines are major determinants of radioresistance in head-and-neck squamous cell carcinoma. Cell Death Dis. 12, 1162 (2021).
Sanoff, H. K. et al. Effect of cytotoxic chemotherapy on markers of molecular age in patients with breast cancer. J. Natl. Cancer Inst. 106, dju057 (2014).
Perez, M. et al. Efficacy of bortezomib in sarcomas with high levels of MAP17 (PDZK1IP1). Oncotarget 7, 67033–67046 (2016).
Kiltz, U., Baraliakos, X., Buehring, B. & Braun, J. [Becoming older with axial spondyloarthritis]. Z. Rheumatol. 77, 363–368 (2018).
Shao, F. et al. Targeting chondrocytes for arresting bony fusion in ankylosing spondylitis. Nat. Commun. 12, 6540 (2021).
Liang, W. et al. Skin chronological aging drives age-related bone loss via secretion of cystatin-A. Nat. Aging 2, 906–922 (2022).
Jing, Y. et al. Single-nucleus profiling unveils a geroprotective role of the FOXO3 in primate skeletal muscle aging. Protein Cell 14, 497–512 (2023).
Chen, H. et al. Prostaglandin E2 mediates sensory nerve regulation of bone homeostasis. Nat. Commun. 10, 181 (2019).
Shi, T. et al. Osteocyte-derived sclerostin impairs cognitive function during ageing and Alzheimer’s disease progression. Nat. Metab. 6, 531–549 (2024).
Lin, L. et al. SIRT2 regulates extracellular vesicle-mediated liver-bone communication. Nat. Metab. 5, 821–841 (2023).
Hu, H., Huang, Y., Liu, F. Z., Wang, Q. & Yao, Y. Z. How aging affects bone health via the intestinal micro-environment. Biocell 48, 353–362 (2024).
Qu, X. et al. Regulating mitochondrial aging via targeting the gut-bone axis in BMSCs with oral hydrogel microspheres to inhibit bone loss. Small 21, e2409936 (2025).
Lee, C. C. et al. Lactobacillus plantarum TWK10 attenuates aging-associated muscle weakness, bone loss, and cognitive impairment by modulating the gut microbiome in mice. Front. Nutr. 8, 708096 (2021).
Chen, C. et al. Microbial tryptophan metabolites ameliorate ovariectomy-induced bone loss by repairing intestinal AhR-mediated gut-bone signaling pathway. Adv. Sci. 11, e2404545 (2024).
Zhu, Y. et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 (2015).
Chang, J. et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78–83 (2016).
Yousefzadeh, M. J. et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28 (2018).
Triana-Martínez, F. et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 10, 4731 (2019).
Sun, S. et al. A single-cell transcriptomic atlas of exercise-induced anti-inflammatory and geroprotective effects across the body. Innovation 4, 100380 (2023).
Croft, M. & Siegel, R. M. Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat. Rev. Rheumatol. 13, 217–233 (2017).
Witkin, S. S., Gerber, S. & Ledger, W. J. Influence of interleukin-1 receptor antagonist gene polymorphism on disease. Clin. Infect. Dis. 34, 204–209 (2002).
Wissler Gerdes, E. O., Misra, A., Netto, J. M. E., Tchkonia, T. & Kirkland, J. L. Strategies for late phase preclinical and early clinical trials of senolytics. Mech. Ageing Dev. 200, 111591 (2021).
Zhang, L. et al. Cellular senescence: a key therapeutic target in aging and diseases. J. Clin. Invest. 132, e158450 (2022).
Partridge, L., Fuentealba, M. & Kennedy, B. K. The quest to slow ageing through drug discovery. Nat. Rev. Drug Discov. 19, 513–532 (2020).
Amor, C. et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020).
Sagiv, A. & Krizhanovsky, V. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology 14, 617–628 (2013).
Yun, M. H., Davaapil, H. & Brockes, J. P. Recurrent turnover of senescent cells during regeneration of a complex structure. eLife 4, e05505 (2015).
Kim, J. A. et al. Neural stem cell transplantation at critical period improves learning and memory through restoring synaptic impairment in Alzheimer’s disease mouse model. Cell Death Dis. 6, e1789 (2015).
Mihaly, E., Altamirano, D. E., Tuffaha, S. & Grayson, W. Engineering skeletal muscle: building complexity to achieve functionality. Semin. Cell Dev. Biol. 119, 61–69 (2021).
Sun, C., Serra, C., Lee, G. & Wagner, K. R. Stem cell-based therapies for Duchenne muscular dystrophy. Exp. Neurol. 323, 113086 (2020).
Ren, X. et al. Maintenance of nucleolar homeostasis by CBX4 alleviates senescence and osteoarthritis. Cell Rep. 26, 3643–3656.e7 (2019).
Fu, L. et al. Up-regulation of FOXD1 by YAP alleviates senescence and osteoarthritis. PLoS Biol. 17, e3000201 (2019).
Kang, D. et al. Stress-activated miR-204 governs senescent phenotypes of chondrocytes to promote osteoarthritis development. Sci. Transl. Med. 11, eaar6659 (2019).
Borgdorff, V. et al. Multiple microRNAs rescue from Ras-induced senescence by inhibiting p21(Waf1/Cip1). Oncogene 29, 2262–2271 (2010).
Wu, R., Sun, F., Zhang, W., Ren, J. & Liu, G. H. Targeting aging and age-related diseases with vaccines. Nat. Aging 4, 464–482 (2024).
von Loga, I. S. et al. Active immunisation targeting nerve growth factor attenuates chronic pain behaviour in murine osteoarthritis. Ann. Rheum. Dis. 78, 672–675 (2019).
Cavelti-Weder, C. et al. Development of an interleukin-1β vaccine in patients with type 2 diabetes. Mol. Ther. 24, 1003–1012 (2016).
Theunis, C. et al. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau.P301L mice that model tauopathy. PLoS One 8, e72301 (2013).
Novak, P. et al. FUNDAMANT: an interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimer’s Res. Ther. 10, 108 (2018).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (82172468, 82372436), Outstanding Youth Fund of Jiangsu Province (BK2024047), China Postdoctoral Science Foundation (2023T160553), and Postgraduate Research & Practice Innovation Program of Jiangsu Province (SJCX22-1819).
Author information
Authors and Affiliations
Contributions
K.L. wrote the original draft and contributed to drawing the illustrations. S.H.H. and H.C. contributed to conceptualization and funding acquisition, review and editing. All authors have reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, K., Hu, S. & Chen, H. Cellular senescence and other age-related mechanisms in skeletal diseases. Bone Res 13, 68 (2025). https://doi.org/10.1038/s41413-025-00448-7
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41413-025-00448-7
