
Abstract
Nutrient competition between tumor and immune cells is a hallmark of the glioblastoma (GBM) microenvironment, yet the mechanisms underlying amino acid metabolic reprogramming and immune evasion remain incompletely understood. Here, we demonstrate that GBM cells outcompete NK cells for branched-chain amino acid (BCAA), leading to BCAA depletion, suppression of NK and CD8+ T cell cytotoxicity, and immune escape. Mechanistically, we identify a positive feedback circuit involving PSMD14, BCKDK, and IGF2BP3 that stabilizes BCKDK post-translationally and promotes SLC7A5/SLC7A8-mediated BCAA uptake by GBM cells. PSMD14 directly interacts with and deubiquitinates BCKDK, antagonizing TRIM21-mediated proteasomal degradation. This metabolic remodeling disrupts NK cell signaling and function, as BCAA deprivation impairs PI3K/Akt and cGAS–STING pathways and disrupts mitochondrial integrity. Preclinical models reveal that pharmacologic inhibition of PSMD14 by O-phenanthroline (OPA) or PSMD14 knockdown restores immune cell infiltration, enhances CAR-NK cytotoxicity, and synergizes with immunotherapy to suppress GBM growth. Clinical analysis further establishes that elevated PSMD14 and BCKDK expression in GBM correlates with decreased CD8+ T and NK cell infiltration and poorer patient survival. These findings highlight the PSMD14–BCKDK axis as a central regulator of tumor metabolic adaptation and immune suppression, and support PSMD14 inhibition—alone or in combination with CAR-NK therapy—as a promising strategy for precision immunometabolic intervention in GBM.
Similar content being viewed by others


Introduction
Natural killer (NK) cells are being utilized well as a prospective contender in tumor immunotherapy [1]. Their distinctive physiology provides two significant benefits: autonomy from antigen presentation by Major Histocompatibility Complex (MHC) molecules and safety for allogeneic applications, making them highly suitable for off-the-shelf utilization [2]. Subsequent to the success of chimeric antigen receptor (CAR)-T cells, the technology has also been utilized for NK cells [3]. No dose-limiting toxicities were seen, and no patient experienced cytokine release syndrome or immune effector cell-associated neurotoxicity syndrome [4]. Moreover, the extent of CD8+ T cell infiltration in recurrent tumor tissue before CAR-NK cell administration exhibited a good correlation with progression-free survival [5]. Glioblastoma (GBM) constitutes about 50% of all primary central nervous system (CNS) neoplasms and is the predominant primary malignant brain tumor in adults, exhibiting an annual occurrence of about three cases per 100,000 individuals [6]. GBM utilizes many strategies to evade NK cell cytotoxicity, leading to insufficient migration and survival of NK cells within solid tumors [7, 8]. Notwithstanding their inherent cytotoxic potential, NK cells are frequently impeded by the tumor microenvironment (TME), such as indoleamine 2,3-dioxygenase-induced tryptophan depletion and kynurenine generation, which leads to CD8+ T cell apoptosis and NK cell suppression, while promoting the recruitment of Treg cells [9]. Clinical studies indicate that two criteria are essential for the adoptive transfer of NK cells to exert therapeutically significant benefits in individuals with solid tumors: (a) intratumoral accumulation [10]. and (b) sustained activation [11]. Thus, combinatorial techniques designed to facilitate effective tumor infiltration and safeguard NK cells from the metabolic and immunological conditions of the TME are essential to realize the therapeutic promise of adoptively transferred NK cells for human solid tumors [12].
Cancer cells exhibit heightened metabolic activity and excel in food absorption as well as the secretion of TME-specific metabolites. They establish an acidic, hypoxic, and nutrient-deficient TME that hinders the adaptation of cytotoxic immune cells to the metabolically adverse conditions [13]. For example, tumor cells often maintain intracellular pools of branched-chain amino acid (BCAA), which leads to increased mTOR signaling, hence promoting tumorigenesis and aggressiveness [14]. Moreover, obstructing the absorption of BCAA by tumors may augment macrophage infiltration and phagocytosis within the TME, whereas the BCAA accumulation enhances the effector role and anti-tumor immunity of CD8+ T cells by reprogramming glucose metabolism [15, 16]. BCAA, comprising leucine, isoleucine, and valine, are vital amino acids for immune cells that can be integrated into proteins or oxidized into branched-chain ketoacid (BCKA) for biosynthesis and subsequent oxidative phosphorylation. Furthermore, exposure to tumor-derived BCKA diminished the phagocytic activity of macrophages [17]. Additionally, cancer cells release BCKA into the extracellular environment, which can influence specific macrophage polarization by modifying pro-inflammatory and anti-inflammatory phenotypic markers [18]. However, the impact of tumor-associated BCAA metabolism and its metabolite BCKA on NK cells is still ambiguous.
The modification of BCAA metabolism is linked to metabolic diseases. Many malignancies were included, such as pancreatic cancer, colorectal cancer, and GBM [19,20,21,22]. BCAA comprise about 35% of the essential amino acids in circulation and are critical nitrogen and carbon sources for cancer cell growth. Given that the majority of research on GBM has concentrated on systemic or cancer cell autonomous BCAA metabolism, comprehending the cancer-immune ecology necessitates an understanding of the interplay between cancer-associated alterations in the immune system and the reprogramming of their BCAA metabolism [21, 22]. Neither systemic in vivo BCAA metabolism nor the metabolism of BCAA in malignant cells alone is adequate to elucidate the immune function. The challenge of comprehending BCAA metabolism inside the tumor environment is intensified by nutritional deficiency, exchange processes, and metabolite transfer between cancer and immunological cells [23].
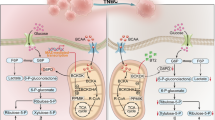
This study presents evidence that the concentration of BCAA in the TME of GBM is markedly decreased. The reduction in BCAA impedes the activation of immunological effector cells, including NK and CD8+ T cells. Furthermore, we recognize the protein PSMD14 as an essential regulator that sustains elevated levels of BCKDK (branched-chain α-keto acid dehydrogenase kinase, a pivotal enzyme in BCAA metabolism) in GBM [24]. PSMD14 functions by deubiquitinating and stabilizing BCKDK, hence inhibiting its degradation facilitated by the E3 ligase TRIM21. The metabolism of BCAA is impaired, resulting in its increased absorption and immune evasion in GBM. Furthermore, the inhibition of PSMD14 diminishes BCKDK protein levels, restores immunological tolerance in GBM, and increases the vulnerability of GBM tumors to CAR-NK therapy. These findings underscore the critical function of PSMD14 in the post-translational control of BCKDK and its capacity to inhibit anti-tumor immunity. Thus, PSMD14 presents itself as a viable target for immunotherapeutic approaches in GBM. It is anticipated to serve as a possible objective aimed at enhancing the efficacy of CAR-NK cell-based treatments for GBM.
Results
Tumor cells outcompete NK cells for BCAA, with BCKDK as a key regulator
Nutrient competition between tumor and immune cells is a hallmark of the TME, but the role of amino acid metabolism in GBM immune evasion remains unclear. Clinical data revealed reduced plasma levels of BCAA in GBM patients, correlating with poor prognosis (Fig. 1A–C). Using in vivo and in vitro models, we found that BCAA deprivation impaired NK cell survival and function, with isoleucine, leucine, and valine loss markedly reducing NK cell cytotoxicity and cytokine production (Fig. S1A–H).

A Workflow for investigating the influence of BCAA deficiency on NK cell activity in GBM patients, mouse models, and in vitro cultures. B Plasma BCAA concentrations in GBM patients (n = 15) and healthy controls (n = 14). All data are mean ± SEM. P values were calculated using unpaired two-tailed Student’s t test. C Correlation between plasma BCAA levels and patient survival. D Rescue of NK cell IFN-γ and TNF-α production by BCAA supplementation following treatment with conditioned media from GBM cells. TEM presenting the immunological synapse (E) and mitochondrial morphology (F) of NK cells cultured in BCAA-sufficient and -deficient conditions, scale bars: 2 μm (overview), 500 nm (boxed area). G BCAA levels in CSF (n = 15 vs. 15) and tumor tissues (n = 12 vs. 5) from GBM patients and healthy controls. All data are mean ± SEM. P values were calculated using an unpaired two-tailed Student’s t test. H BCAA concentrations in tumor, peritumoral, and normal brain tissues from orthotopic GBM-bearing mice at different time points (n = 3 mice per group). I WB analysis of PI3K/Akt and cGAS-STING pathway activity in NK cells under BCAA-sufficient and -deficient conditions. J Flow cytometric analysis of NK cell infiltration in GBM tissues. K, L WB analysis of BCKDK protein level in GBM cell lines. M, N BCKDK protein levels in GBM tissues (n = 10) and normal-appearing brain tissue beyond the tumor (NTB) (n = 10).
Exposure of NK cells to GBM-conditioned media suppressed effector function and viability, which could be rescued by BCAA supplementation (Figs. 1D and S1I, J). Transmission electron microscopy (TEM) showed that BCAA deficiency also altered NK cell membrane topology and mitochondrial integrity, leading to impaired immune synapse formation and enhanced mitophagy (Figs. 1E, F and S1M). In addition, NK cells cultured in BCAA-rich conditions formed tight junctions and synapses with GBM cells, whereas BCAA-depleted NK cells failed to do so (Fig. S1L). Metabolomic profiling revealed elevated BCAA levels in tumor tissues but reduced levels in cerebrospinal fluid (CSF) and adjacent brain regions of both human and mouse GBM (Fig. 1G, H). Intratumoral BCAA abundance negatively correlated with NK and CD8⁺ T cell infiltration (Figs. 1J and S1K, N), and BCAA deprivation suppressed PI3K/Akt and cGAS–STING signaling in NK cells (Fig. 1I). Given reports that tumor-derived branched-chain ketoacids (BCKAs) can modulate immune cells, we treated NK cells with increasing BCKA concentrations and assessed effector cytokines by flow cytometry (Fig. S1O–R). Low-dose BCKAs modestly increased granzyme B, perforin, and TNF-α, whereas higher BCKA levels progressively suppressed these markers and significantly reduced IFN-γ. Notably, BCKA-induced impairment was consistently weaker than that caused by BCAA deprivation, supporting BCAA depletion as the dominant driver of NK dysfunction in our models.
To probe how BCAA deprivation dampens PI3K/Akt and cGAS–STING signaling, we performed untargeted metabolomics in NK cells cultured under BCAA-negative versus BCAA-positive conditions (n = 5 per group; 1,554 metabolites detected) (Table S1 and Fig. S2A–F). PCA showed robust group separation, indicating global metabolic reprogramming (Fig. S2A). Differential analysis (VIP/FC/P-value) identified 577 altered metabolites (389 increased, 188 decreased) (Fig. S2B). BCAA deprivation led to accumulation of purine catabolites (e.g., xanthine and uric acid) alongside depletion of nucleosides/nucleotides and related metabolites, consistent with suppressed nucleotide metabolism (Fig. S2C). In parallel, multiple organic acids and derivatives (e.g., proline, citrate, and cysteine) were reduced, indicating compromised metabolic fitness and biosynthetic support (Fig. S2D). Pathway-level analysis further highlighted coordinated downregulation of nucleotide metabolism and amino acid biosynthesis programs, with enrichment of nutrient-sensing pathways linked to mTOR/FoxO (Fig. S2F and Supplementary Data 1). Collectively, these metabolomic features align with impaired nutrient/anabolic signaling (PI3K/Akt/mTOR axis) and weakened innate immune activation capacity, providing mechanistic support for the reduced PI3K/Akt and cGAS–STING outputs observed under BCAA deprivation.
Bioinformatic analysis of TCGA data showed higher expression of BCKDK, a key BCAA metabolic enzyme, in GBM with low immune cell infiltration (Fig. S2G, H). Western blot (WB) analysis revealed that BCKDK was upregulated in GBM cell lines and patient tumors, while its mRNA levels remained unchanged (Fig. 1K–N and Fig. S3A, B). Immunohistochemistry (IHC) and multiplex immunofluorescence (IF) confirmed a negative association between BCKDK expression and immune cell infiltration, potentially linked to BCAA levels in the TME. (Fig. S3C–K), and BCKDK knockdown reduced intracellular BCAA levels in GBM cells (Fig. S3L). These findings suggest that BCKDK protein abundance, rather than transcriptional activity, regulates BCAA availability and immune exclusion in GBM, prompting further investigation into its post-translational control.
BCKDK phosphorylates IGF2BP3 to mediate the expression of SLC7A5/SLC7A8
To illustrate the regulatory function of BCKDK in tumor BCAA metabolism, we either knocked down or pharmacologically inhibited BCKDK and examined transcriptional changes in key genes within the BCAA metabolic pathway. Notably, the mRNA levels of the BCAA transporters SLC7A5 and SLC7A8, located at the GBM cell membrane, were significantly reduced following BCKDK suppression (Fig. 2A, B). Consistently, both IF and WB analyses confirmed a marked decrease in SLC7A5/SLC7A8 protein expression upon BCKDK inhibition (Figs. 2C, D and S4A–C). Previous studies have reported that the RNA-binding protein IGF2BP3 stabilizes SLC7A5 mRNA [25]. Based on this, we hypothesized that BCKDK, IGF2BP3, and SLC7A5/SLC7A8 mRNAs may assemble into a functional protein-RNA ternary complex that facilitates BCAA uptake in GBM. Mass spectrometry (MS) analysis identified IGF2BP3 as a potential BCKDK-interacting partner (Table S2), and reciprocal co-immunoprecipitation (co-IP) assays validated their physical association (Fig. S4D).

RT-PCR analysis showing reduced SLC7A5 and SLC7A8 mRNA levels after BCKDK inhibition (A) or knockdown (B) in GBM cells. All data are mean ± SEM. P values were calculated using an unpaired two-tailed Student’s t test. C, D IF showing decreased SLC7A5/SLC7A8 protein levels following BCKDK inhibition or knockdown in A172 cells, scale bars: 100 μm. E, F IP and WB showing BCKDK-dependent threonine phosphorylation of IGF2BP3. G, H WB and RT-qPCR showing reduced SLC7A5/SLC7A8 expression following IGF2BP3 knockdown in A172 and U251 cells. I A172 cells were transfected stably with scramble shRNA and IGF2BP3 shRNA, and were treated with actinomycin D (20 μg/mL) for the indicated time, and SLC7A5 and SLC7A8 mRNA levels were analyzed.
Given the intrinsic kinase activity of BCKDK, we treated GBM cells with the selective inhibitor BT2 and observed a time-dependent decrease in threonine phosphorylation after 8 h, while total IGF2BP3 protein levels remained unchanged (Fig. S4E). IP of IGF2BP3, followed by phospho-threonine detection, further confirmed a BCKDK-dependent increase in IGF2BP3 phosphorylation (Fig. 2E, F). Consistent with this regulatory relationship, knockdown of IGF2BP3 in both A172 and U251 cells led to marked downregulation of SLC7A5 and SLC7A8 expression (Fig. 2G, H). To validate the existence of a BCKDK-IGF2BP3-mRNA complex, RNA immunoprecipitation (RIP) was performed. Knockdown of either BCKDK or IGF2BP3 reduced the mRNA binding capacity of IGF2BP3 (Fig. S4F, G). To determine whether IGF2BP3 regulates SLC7A5/SLC7A8 expression by influencing mRNA stability, we assessed mRNA half-lives in actinomycin D-treated GBM cells. IGF2BP3 knockdown significantly decreased the stability of both transcripts in A172 and U251 cells (Fig. 2I and Fig. S4H).
To identify the BCKDK-responsive phospho-site on IGF2BP3, we expressed Flag-IGF2BP3 in HEK293T cells, treated cells with BT2 or DMSO, and performed LC-MS/MS on immunoprecipitated IGF2BP3. Phosphorylation at Thr457 was markedly reduced by BT2, nominating Thr457 as a major BCKDK-sensitive threonine residue (Table S3). We then generated phospho-deficient T457A and phospho-mimetic T457D IGF2BP3 mutants and expressed them in PSMD14-silenced A172 cells. Compared with WT IGF2BP3, T457A failed to sustain SLC7A5/SLC7A8 protein levels, whereas T457D largely maintained their expression under reduced BCKDK activity (Fig. S5A). Consistently, mRNA stability assays showed that the WT-mediated stabilization of SLC7A5/SLC7A8 transcripts was significantly attenuated by T457A but preserved by T457D, indicating that Thr457 phosphorylation is required for BCKDK-enhanced IGF2BP3-dependent mRNA stabilization (Fig. S5B, C).
In summary, BCKDK promotes BCAA uptake in GBM cells by forming a ternary complex with IGF2BP3 and SLC7A5/SLC7A8 mRNAs, which enhances mRNA stability and represents a pivotal mechanism of post-transcriptional control in tumor metabolic adaptation.
PSMD14 interacts with and stabilizes the BCKDK protein
To identify deubiquitinating enzymes (DUBs) regulating BCKDK, we performed a DUB siRNA library screen followed by WB validation, and identified five DUBs (PSMD14, USP11, USP19, USP2, USP20) whose knockdown significantly reduced BCKDK protein levels (>40% knockdown efficiency) (Figs. 3A–C and S6A, B). In addition, IP assays demonstrated that only PSMD14 directly interacts with BCKDK (Fig. 3F, G). MS analysis in A172 and U251 cells identified BCKDK-binding proteins, with pathway enrichment highlighting “negative regulation of innate immune response” and “ubiquitin ligase inhibitor activity” (Table S2, 4). Subsequent cross-referencing of siRNA and IP/MS results pinpointed PSMD14 as a specific BCKDK-interacting DUB. PSMD14 showed a high expression in GBM cell lines and tissues, and BCKDK protein levels positively correlated with PSMD14 expression (Figs. 3D, E and S6C). The interaction between PSMD14 and BCKDK was validated by endogenous co-IP and GST pull-down assays (Fig. S6I–K). Exogenous co-IP in HEK293T cells further confirmed reciprocal binding between Flag-BCKDK and Myc-PSMD14 (Fig. S6L). WB analysis revealed that BCKDK was also upregulated in GBM patient tumors (Fig. 3H, I). Further molecular mapping revealed that the N-terminal D1 region (residues 1–103) of BCKDK interacts with the N-terminal MPN domain (residues 1–166) of PSMD14, which is associated with the ubiquitin-proteasome system (Fig. 3J–L).

A Identification of BCKDK-regulating DUBs by intersecting DUB siRNA screen with BCKDK IP/MS interactome. B, C PSMD14, USP11, USP19, USP2, and USP20 reduce BCKDK levels upon siRNA knockdown. All data are mean ± SEM. P values were calculated using one-way ANOVA for Dunnett’s multiple-comparisons test. D Western blot analysis of PSMD14 protein level in GBM cell lines. E BCKDK expression correlates with PSMD14 protein levels in GBM cell lines. F, G Co-IP in HEK293T cells confirms PSMD14–BCKDK interaction. H, I PSMD14 protein levels in 10 paired GBM vs. adjacent normal tissues. J Design of PSMD14 and BCKDK truncations. K, L BCKDK N-terminal region binds the MPN domain of PSMD14. M, N PSMD14 knockdown reduces, and overexpression increases, BCKDK levels in A172 and U251 cells. O, P PSMD14 modulates extracellular BCAA levels in A172 and U251 cells. All data are mean ± SEM. P values were calculated using one-way ANOVA for Dunnett’s multiple-comparisons test. Q, R CHX chase assay shows PSMD14 knockdown accelerates BCKDK degradation in U251 cells. S, T OPA (10 µM) treatment reduces BCKDK protein levels over time.
To evaluate the impact of PSMD14 on BCKDK stability, we observed that PSMD14 overexpression elevated BCKDK protein levels, while shRNA-mediated knockdown reduced BCKDK protein levels in GBM cells (Figs. 3M, N and S6F), with no effect on BCKDK mRNA expression (Fig. S6D, E). Both intracellular and extracellular BCAA levels were regulated by PSMD14 in A172 and U251 cells (Figs. 3O, P and S6G, H). Overexpression of wild-type PSMD14 extended the half-life of BCKDK (Fig. S6M), whereas PSMD14 knockdown (Fig. 3Q, R) or OPA inhibition (Fig. 3S, T) reduced BCKDK half-life and protein levels. IF staining revealed strong co-localization of endogenous PSMD14 and BCKDK in A172 and U251 cells (Fig. S6N, O).
To further elucidate the molecular mechanisms by which PSMD14 regulates BCAA metabolic reprogramming, WB analysis demonstrated that PSMD14 regulates SLC7A5 and SLC7A8 expression in A172 and U251 cells (Fig. S7A). Notably, PSMD14 expression was significantly reduced in IGF2BP3-knockdown cells (Fig. S7B, C), implicating a PSMD14–BCKDK–IGF2BP3 positive feedback circuit that facilitates GBM-driven BCAA acquisition and aggravates the development of an immunosuppressive TME. RNA pull-down assays further showed that a PSMD14-specific probe, but not a control, enriched IGF2BP3 protein in U251 cells (Fig. S7D, E). Consistently, anti-IGF2BP3 IP demonstrated a significant decrease in threonine phosphorylation upon PSMD14 knockdown or pharmacological inhibition, which was rescued by BCKDK overexpression (Fig. S7F, G). To directly map the PSMD14 mRNA–binding region within IGF2BP3, we generated a series of Flag- IGF2BP3 truncation constructs spanning its major RNA-binding modules (two RRM motifs and four KH domains) (Fig. S7H). RIP-qPCR in A172 cells showed that both the N- and C-terminal fragments of IGF2BP3 were capable of associating with PSMD14 mRNA, with the N-terminal truncation exhibiting the strongest enrichment (Fig. S7I). Additionally, knockdown of BCKDK or IGF2BP3 in U251 cells diminished the association of PSMD14 mRNA with IGF2BP3 (Fig. S7J), and actinomycin D chase assays confirmed a shortened half-life of PSMD14 mRNA in IGF2BP3-knockdown cells (Fig. S7K, L). Collectively, these results support a positive feedback circuit in which PSMD14 stabilizes BCKDK, BCKDK promotes IGF2BP3 phosphorylation, and activated IGF2BP3 further stabilizes PSMD14 in GBM cells.
PSMD14 deubiquitinates and overexpresses BCKDK
BCKDK proteasomal degradation was reported in neurodevelopmental disorders [26]. prompting us to investigate whether PSMD14 modulates this process in GBM cells. WB analysis illustrated that BCKDK protein levels were markedly elevated in a time-dependent manner following MG132 treatment (Fig. S8A, B), and the proteasome inhibitor MG132 fully rescued BCKDK from cycloheximide (CHX)-induced degradation (Fig. S8C), confirming that BCKDK is degraded via the proteasome pathway in GBM. Notably, the exogenous expression of PSMD14 suppressed BCKDK ubiquitination in HEK293T cells (Fig. 4A), whereas either OPA or PSMD14 knockdown significantly increased BCKDK ubiquitination in A172 and U251 cells (Figs. 4B, C and S8D, E). These findings demonstrate that PSMD14 antagonizes BCKDK ubiquitination and preserves its stability in GBM cells.

A HEK293T cells co-expressing Flag-BCKDK, HA-ubiquitin, and Myc-PSMD14 were treated with MG132 (10 μM, 6 h); BCKDK polyubiquitination assessed by Flag-IP. B, C U251 cells treated with OPA (10 μM, 24 h) or stably expressing PSMD14 shRNA were exposed to MG132 and subjected to BCKDK-IP to assess ubiquitination. D HEK293T cells co-expressing Flag-BCKDK, Myc-PSMD14, and wild-type or mutant HA-ubiquitin were analyzed for BCKDK ubiquitination after MG132 treatment. E PSMD14 catalytic mutants (H113Q, H115Q, C120S) reduce deubiquitination of BCKDK in HEK293T cells. F Ubiquitination of BCKDK K48R and K184R mutants assessed in HEK293FT cells co-expressing PSMD14 and HA-ubiquitin. G TRIM21 wild-type and LD mutant co-expression in HEK293T cells analyzed for effect on BCKDK ubiquitination. H TRIM21 knockdown in U251 cells decreases BCKDK ubiquitination following MG132 treatment.
We next characterized the specific polyubiquitin linkages of BCKDK regulated by PSMD14. HEK293T cells were co-transfected with Flag-BCKDK, Myc-PSMD14, and various HA-tagged ubiquitin constructs (WT, K11, K27, K29, K33, K48, and K63). IP followed by WB revealed that exogenous PSMD14 overexpression markedly reduced WT and K48-linked BCKDK ubiquitination (Fig. 4D). Given that K48-linked polyubiquitination primarily mediates proteasomal degradation, we further showed that PSMD14 knockdown or OPA treatment increased endogenous K48-linked BCKDK ubiquitination in U251 cells (Fig. S8F). Notably, PSMD14 mutants lacking JAMM domain catalytic residues (H113Q, H115Q, C120S) retained BCKDK binding but failed to decrease BCKDK ubiquitination or enhance its stability [27, 28]. highlighting the requirement for PSMD14 deubiquitinase activity in BCKDK regulation (Fig. 4E). Additionally, consistent with reports that BCKDK is ubiquitinated at K48 and K184 [29]. PSMD14 could not reduce ubiquitination of BCKDK K48R and K184R mutants (Fig. 4F). To further substantiate the PSMD14 dependence of OPA-associated phenotypes, we additionally employed Capzimin (CZM), a recently developed PSMD14-selective small-molecule inhibitor, as an orthogonal pharmacological comparator [30,31,32]. In both A172 and U251 cells, PSMD14 knockdown or treatment with either OPA or CZM comparably increased K48-linked ubiquitination of BCKDK, consistent with compromised PSMD14 deubiquitinase activity. Notably, in PSMD14-silenced cells, neither OPA nor CZM further enhanced BCKDK K48-linked ubiquitination, indicating that both inhibitors act through PSMD14 (Fig. S9A, B). Given the extensive validation and widespread use of OPA as a PSMD14 inhibitor, we therefore selected OPA as the primary pharmacological tool for subsequent experiments.
MS considered TRIM21 as an E3 ubiquitin ligase strongly interacting with BCKDK (Table S2). This interaction was validated by silver staining and both exogenous and endogenous co-IP (Fig. S8G–I). Overexpression of TRIM21 in A172 and U251 cells reduced BCKDK protein levels without altering BCKDK mRNA (Fig. S8J). Inhibition experiments showed that the reversion of TRIM21-mediated BCKDK degradation was conducted by the proteasome inhibitor MG132, but not by the lysosomal inhibitor CQ (chloroquine) (Fig. S8K), indicating that TRIM21 targets BCKDK for proteasomal degradation. TRIM21 overexpression markedly increased BCKDK ubiquitination, whereas the TRIM21 LD (ligase-dead) mutant had no effect (Fig. 4G). Conversely, TRIM21 knockdown abolished BCKDK ubiquitination following MG132 treatment (Figs. 4H and S8L). Importantly, PSMD14 overexpression rescued BCKDK protein levels in TRIM21-overexpressing cells, while catalytically inactive PSMD14 mutants failed to do so (Fig. S8M). These results demonstrate that PSMD14 protects BCKDK from TRIM21-mediated ubiquitination and proteasomal degradation.
PSMD14 triggers BCKDK-mediated immune suppression in vitro
PSMD14 regulates BCKDK post-translationally; thus, we hypothesized that dysregulated PSMD14 expression could modulate BCAA metabolism and thereby affect immunosuppression. To assess whether PSMD14/BCKDK-dependent BCAA metabolism contributes to NK cell inhibition, we purified NK cells from murine spleens and performed co-culture proliferation assays (Fig. S10A). Supernatants from PSMD14-knockdown tumor cells significantly enhanced NK cell proliferation, whereas BCKDK overexpression reversed this effect. Supplementation with BCAA robustly restored NK cell proliferation, regardless of PSMD14 or BCKDK status (Fig. S10B). Furthermore, dose-response analyses revealed that NK cells exhibited greater sensitivity to BCAA deprivation than A172 and U251 cells, as demonstrated by their respective EC50 values (Fig. S10C–E).
To investigate the effects of the PSMD14/BCKDK axis on CAR-NK cytotoxicity, we generated EGFRvIII. CAR-GFP-NK92 cells with high transduction efficiency for immune killing assays (Fig. S10F). Direct-contact co-culture assays demonstrated that CAR-NK cells exhibited increased cytotoxicity against PSMD14-knockdown A172 and U251 cells across multiple E/T (effector-to-target) ratios (Figs. 5A, B and S10G). This enhancement was rescued by BCKDK overexpression in PSMD14-knockdown A172 and U251 cells (Fig. 5D, E). To further assess the influence of TME BCAA availability, we compared immune killing efficiency in co-cultures with or without BCAA supplementation. BCAA depletion impaired CAR-NK cytotoxicity, which was restored by exogenous BCAA addition (Figs. 5C, F and S11A–D). Furthermore, supernatant from PSMD14-knockdown A172 and U251 cells reduced CAR-NK apoptosis (Fig. 5H).

A, B CAR-NK cytotoxicity against PSMD14-knockdown A172 and U251 cells at different E:T ratios. All data are mean ± SEM. P values were calculated using two-way ANOVA for Dunnett’s multiple-comparisons test. C Assessment of the effects of BCAA supplementation or deprivation on the viability of A172 and U251 cells. All data are mean ± SEM. P values were calculated using two-way ANOVA for Dunnett’s multiple-comparisons test. D, E Effect of BCKDK overexpression on CAR-NK cytotoxicity in PSMD14-knockdown GBM cells. All data are mean ± SEM. P values were calculated using one-way ANOVA for Dunnett’s multiple-comparisons test. F BCAA supplementation in co-culture and its impact on CAR-NK cytotoxicity. All data are mean ± SEM. P values were calculated using two-way ANOVA for Dunnett’s multiple-comparisons test. G Differential fluorescent labeling (CFSE/CTV) of knockdown and control A172 cells, followed by co-culture with CAR-NK cells to evaluate target-specific cytotoxicity. H Assessment of CAR-NK apoptosis following treatment with supernatants from PSMD14-knockdown A172 and U251 cells. All data are mean ± SEM. P values were calculated using two-way ANOVA for Dunnett’s multiple-comparisons test. I Quantification of the killing efficiency of CAR-NK cells toward CFSE/CTV-labeled knockdown and control tumor cells. All data are mean ± SEM. P values were calculated using unpaired two-tailed Student’s t test. J, K Crystal violet staining analysis of PSMD14-knockdown A172 and U251 cells’ sensitivity to CAR-NK killing at varying E:T ratios. L Transwell migration assay measuring CAR-NK infiltration toward PSMD14-knockdown U251 cells, scale bars: 100 μm.
To determine whether the effect of PSMD14 on CAR-NK cytotoxicity was mediated by altered BCAA metabolism or changes in tumor immunogenicity, we differentially labeled PSMD14-knockdown, shPSMD14/BCKDK (rescue), and control U251 cells with CFSE or CTV and co-cultured them with CAR-NK cells at a 1:1 ratio. No significant variation was detected in the relative susceptibility of knockdown versus control tumor cells to CAR-NK-mediated killing within the same microenvironment, with overall cytotoxicity increasing as the proportion of PSMD14 knockdown U251 cells increased, and this effect was restored by BCKDK overexpression (Figs. 5G–I and S11E, F). Crystal violet staining further demonstrated that immune killing efficiency was enhanced with higher CAR-NK cell ratios, and PSMD14-knockdown tumor cells exhibited increased sensitivity to CAR-NK-mediated lysis (Fig. 5J, K); this effect was reversed by BCKDK overexpression (Fig. S11H, I). In vitro transwell assays revealed that PSMD14-knockdown GBM cells significantly promoted CAR-NK migration and infiltration, peaking at day 2 (Figs. 5L and S11G), while BCKDK overexpression attenuated this effect (Fig. S11J, K).
To assess the potential off-target cytotoxicity of PSMD14 inhibition on normal CNS cells, we treated primary GBM cultures (IL60-11 and 140-55) and normal brain-derived cells, including normal human astrocytes (NHA), microglia (HMC3), and oligodendrocyte precursor-like cells (MO3.13), with OPA at the same concentrations used in tumor cell assays. Flow-cytometry-based apoptosis profiling following OPA or CAR-NK challenge showed that both interventions induced markedly higher apoptosis in GBM cells than in normal CNS cells (Fig. S12A–E). Consistently, dose-escalation OPA treatment further revealed robust, concentration-dependent activation of canonical apoptotic signaling in GBM cells by immunoblotting, whereas activation of these pathways remained minimal in normal CNS cells under identical conditions (Fig. S12F–J).
Collectively, these findings support the translational feasibility of PSMD14 targeting by demonstrating preferential vulnerability of GBM cells with limited toxicity in normal CNS cells. Mechanistically, PSMD14 deubiquitinates and stabilizes BCKDK, thereby remodeling tumor BCAA metabolism and elevating BCAA availability in the TME, which suppresses NK cell activity and facilitates immune evasion.
PSMD14 promotes BCKDK-mediated immune suppression in vivo
To demonstrate the function of the PSMD14/BCKDK axis in reprogramming BCAA metabolism and mediating immunosuppression in vivo, we established an orthotopic model in C57BL/6 mice using luc-GL261 cells stably expressing PSMD14 shRNA, BCKDK open reading frame (ORF), or PSMD14 shRNA plus BCKDK ORF. Compared to other groups, PSMD14 knockdown significantly reduced tumor size and improved survival outcomes (Figs. 6A, B and S13A). PSMD14 knockdown markedly decreased BCAA levels within GL261 tumors, whereas BCKDK overexpression restored BCAA content in PSMD14-deficient tumors. Moreover, GL261 tumors with BCKDK overexpression in the scramble group displayed further elevated BCAA levels, as assessed by HPLC-MS/MS (Fig. S13B) (Table S5). Multiplex IF analysis showed that PSMD14 knockdown markedly increased TNF-α+ NK cell infiltration and reduced TIM3+ NK cell infiltration in GL261 tumors, while BCKDK overexpression attenuated the infiltration of functionally active NK cells in PSMD14-deficient tumors (Fig. 6C). Tumors were harvested and processed for flow cytometric analysis to evaluate NK cell infiltration. The amount of NK cells among tumor-infiltrating immune cells, as well as the frequencies of CD107+, PD-1+, TNF-α+, IFN-γ+, perforin+, and granzyme B+ subsets among NK cells, were determined by flow cytometry. PSMD14 knockdown led to a significant increase in overall NK cell infiltration and higher percentages of CD107+, TNF-α+, IFN-γ+, perforin+, and granzyme B+ NK cells, while reducing the amount of PD-1+ NK cells. In contrast, BCKDK overexpression reversed the immunostimulatory effects of PSMD14 knockdown and further exacerbated immune suppression in scramble tumors (Fig. S13C–I). IHC analysis confirmed that PSMD14/BCKDK expression patterns in vivo were consistent with in vitro findings: PSMD14 knockdown significantly enhanced NK cell infiltration and inhibited tumor proliferation, whereas BCKDK overexpression almost completely rescued the loss of NK cell infiltration and restored tumor proliferation in PSMD14-deficient tumors. TUNEL staining further demonstrated a significant increase in the apoptotic index in PSMD14-knockdown tumors (Fig. S13J–P).

A, B Tumor growth and survival analysis of C57 mice orthotopically implanted with GL261 cells stably expressing PSMD14 shRNA, BCKDK ORF, or PSMD14 shRNA plus BCKDK ORF (n = 6 mice per group). C Multiplex IF analysis of TNF-α⁺ and TIM3⁺ NK cell infiltration in GL261 tumors from different experimental groups, 50 μm (overview), 25 μm (boxed area). D Schematic of experimental workflow for scRNA-seq of dissociated scramble and shPSMD14 GL261 tumors. E UMAP visualization of scRNA-seq showing clustering by canonical markers. F Quantitative assessment of NK, CD8⁺ T, and DC populations within tumor-infiltrating immune cells. G Functional enrichment analysis highlighting changes in NK cell-mediated cytotoxicity in shPSMD14 GL261 tumors.
To further investigate the role of PSMD14 in modulating the tumor immune microenvironment, we dissociated scramble and PSMD14-knockdown GL261 tumors and conducted single-cell RNA sequencing (scRNA-seq) (Fig. 6D). Dimensionality reduction and clustering based on canonical marker genes (Figs. 6E and S14A) revealed a marked elevation in the proportions of intratumoral NK, CD8+ T, and dendritic cells (DCs) in shPSMD14 GL261 tumors (Fig. 6F). Functional enrichment analysis of cell subsets demonstrated that PSMD14 knockdown significantly influenced NK cell-mediated cytotoxicity within the NK cell compartment (Fig. 6G), and also affected regulating the T cell stimulation and immune cell differentiation within other immune subpopulations (Fig. S14B, C). CellChat analysis further indicated that intercellular communication was significantly enhanced in shPSMD14 GL261 tumors (Fig. S14D), with NK cells acting as key “Senders” orchestrating intercellular signaling, most notably in communication between NK and CD8+ T cells (Fig. S14E).
To assess the in vivo effects of the PSMD14 inhibitor OPA on tumor BCAA metabolic reprogramming and the immune microenvironment, we established an orthotopic luc-CT2A model in C57 mice. Compared to controls, OPA treatment resulted in significantly reduced tumor burden and improved survival (Fig. S15A–C). IHC demonstrated markedly decreased expression of PSMD14, BCKDK, and Ki-67, along with increased infiltration of NK1.1+ cells in OPA-treated tumors; TUNEL staining further indicated an elevated apoptosis index (Fig. S15D, E). HPLC-MS/MS analysis revealed that OPA administration significantly lowered BCAA levels in CT2A tumor tissues (Fig. S15F) (Table S5). Flow cytometric analysis showed that OPA treatment significantly increased NK cell tumor infiltration and enhanced the percentages of activated NK cells, while reducing PD-1+ NK cells within the tumor (Fig. S15G–I). Multiplex IF further confirmed that OPA treatment increased TNF-α+ NK cell infiltration and decreased TIM3+ NK cell infiltration in CT2A tumors compared to the control group (Fig. S15J).
Previous studies have established the existence of an NK-cDC1(conventional DC subset 1)-CD8⁺ T cell immune circuit mediating synergistic anti-tumor responses within the TME [33, 34]. Our single-cell transcriptomic analysis further demonstrated that PSMD14 knockdown markedly enhanced intercellular communication between intratumoral NK and CD8⁺ T cells. To disrupt the bridge facilitating NK and CD8⁺ T cell interactions in vivo, we utilized Batf3-deficient (Batf3–/–) mice, characterized by defective cDC1 development and function [35]. and established orthotopic luc-GL261 tumors with OPA treatment (Fig. S16A). In vivo imaging revealed that OPA-mediated tumor suppression was substantially attenuated in Batf3–/– mice compared to wild-type controls (Fig. S16B, D). Flow cytometric analysis of tumor tissues indicated that OPA significantly increased both NK and CD8⁺ T cell infiltration in wild-type mice, whereas only NK cell infiltration was elevated in Batf3–/– mice (Fig. S16C). Survival analysis further showed that the therapeutic efficacy of OPA was markedly diminished in Batf3–/– mice compared to wild-type animals (Fig. S16E). Given that tumor-driven BCAA depletion suppresses NK cell infiltration and immune activation, we next investigated whether in vivo BCAA supplementation, alone or combined with PD-1 blockade, could enhance anti-tumor efficacy in GL261 tumor–bearing mice (Fig. S16F). Tumor size and survival were comparable between the high and normal BCAA diet groups. However, adding anti–PD-1 antibody in the high BCAA group significantly reduced tumor burden and improved survival outcomes (Fig. S16G–J). IHC analysis confirmed a noticeable elevation in NK and CD8⁺ T cell infiltration following PD-1 immunotherapy in the high BCAA diet group (Fig. S16H). Collectively, these findings illustrate that while an increased BCAA diet alone does not inhibit tumor growth, such an immune-supportive microenvironment may facilitate the recruitment of cDC1s by restored NK cells, thereby promoting CD8⁺ T cell cytotoxicity during immunotherapy.
Inhibition of PSMD14 activity increases CAR-NK sensitivity
Our studies have indicated that PSMD14 has a critical function in regulating tumor BCAA metabolism and modulating the tumor immune microenvironment via influencing NK cell activity, indicating its potential as a therapeutic target for endogenous immune regulation. To investigate the potential synergistic effects of combining the PSMD14-specific inhibitor OPA with CAR-NK cells therapy, we established an orthotopic luc-IL60-11 patient-derived xenograft (PDX) GBM model. This model included groups treated with OPA alone, CAR-NK cells alone, and their combination, with therapeutic efficacy dynamically monitored by bioluminescent imaging (BLI) and magnetic resonance imaging (MRI) (Fig. 7A). While OPA monotherapy exhibited limited efficacy, both CAR-NK cell and combination therapies produced significant tumor growth inhibition, with the combined treatment displaying superior synergistic anti-tumor effects (Figs. 7B and S17A), as in vivo DiR fluorescent tracing demonstrated significantly increased CAR-NK cell persistence and tumor-infiltrating immunological function in the combination group (Figs. 7C and S17B). Combined therapy significantly prolonged overall survival, delayed weight loss, and improved the quality of life in treated mice compared to all other groups (Fig. S17C, D). MRI confirmed marked tumor regression and necrotic cavity formation following combination treatment (Fig. 7D, E). TUNEL staining indicated extensive tumor cell apoptosis (Fig. 7F, G), while WB analysis showed activation of the Bax/Bcl-2 apoptotic pathway in tumors from the combination group (Fig. 7H). On day 21 post-treatment, ELISA analyses of serum and tumor tissue demonstrated increased levels of pro-inflammatory cytokines (TNF-α, IFN-γ, IL-2, IL-6, IL-17A) and reduced anti-inflammatory cytokines (IL-4, IL-10) with combined therapy, highlighting an enhanced anti-tumor immune response (Fig. S17E, F).

A Schematic of the treatment protocol and experimental groups in the IL60-11-luc orthotopic PDX model (n = 5 mice per group). B Bioluminescence imaging (BLI) assessment of intracranial tumor growth following treatment with OPA, EGFRvIII-CAR NK cells, or their combination. C In vivo DiR fluorescent tracking of CAR-NK cell persistence and tumor infiltration. D, E MRI analysis of tumor burden and necrosis formation across groups. All data are mean ± SEM. P values were calculated using two-way ANOVA for Dunnett’s multiple-comparisons test. F, G TUNEL staining of tumor sections from each treatment group to assess apoptotic activity, 50 μm (overview), 25 μm (boxed area). All data are mean ± SEM. P values were calculated using two-way ANOVA for Dunnett’s multiple-comparisons test. H WB analysis of Bax/Bcl-2 pathway activation following therapy.
To further evaluate the influence of BCAA metabolism on the effectiveness of CAR-NK immunotherapy, we established an orthotopic luc-140-55 PDX model and administered either standard or high BCAA diets during PBS or CAR-NK cell treatment (Fig. S18A). High dietary BCAA intake markedly enhanced CAR-NK cell persistence in TME, promoted tumor-infiltrating immune activity, and significantly suppressed tumor growth (Fig. S18B–D). To evaluate therapeutic safety, TUNEL staining was performed on liver and spleen tissues after treatment, revealing no significant differences in apoptosis among groups, thus indicating an absence of detectable hepatic or splenic toxicity with OPA, CAR-NK therapy, or their combination (Fig. S18E, F).
PSMD14 and BCKDK clinical relevance in immune infiltration
To evaluate the relationship between PSMD14, BCKDK, and antitumour immune responses in GBM, we conducted multiplex IF staining using anti-CD8 and anti-CD56 antibodies on clinical GBM samples. Tumors with high PSMD14 expression displayed elevated levels of BCKDK, along with a marked reduction in the infiltration of CD8+ T and CD56+ NK cells (Fig. 8A–C). To further validate the link between PSMD14 and BCKDK expression in clinical samples, we conducted IHC in a cohort of 114 GBM specimens, including 41 with low and 73 with high PSMD14 expression (Figs. 8D and S19A, B). A total of 82.19% of the PSMD14-high samples illustrated elevated BCKDK levels compared to 14.63% of the PSMD14-low samples (Fig. S19B). Consistently, high PSMD14 expression was significantly associated with MGMT status (P = 0.017; χ² test) and with elevated BCKDK expression (P < 0.001; χ² test) (Table S6). In addition, multivariable Cox regression analyses identified PSMD14 and BCKDK levels as independent prognostic factors for 4-year overall survival in patients with GBM (Table S7).

A Representative multiplex IF images of two GBM cases showing the strategy for tumor-infiltrating lymphocyte (TIL) identification based on CD56 and CD8 expression; each marker is displayed in a distinct color, scale bars: 50 μm (overview), 25 μm (boxed area). Quantification of CD8⁺ T cells (B) and CD56⁺ NK cells (C) in GBM tissues with high (n = 73) or low (n = 41) PSMD14 expression, assessed by multiplex IF staining. All data are mean ± SEM. P values were calculated using unpaired two-tailed Student’s t test. D Representative IHC images of PSMD14 and BCKDK in human GBM samples, 200 μm (overview), 25 μm (boxed area). Kaplan–Meier survival analysis of GBM patients (n = 114), stratified by PSMD14 (E) or BCKDK (F) expression (log-rank test, P < 0.0001). G Schematic illustration of the PSMD14/BCKDK/IGF2BP3 positive feedback circuit, which stabilizes SLC7A5/SLC7A8 mRNAs to promote GBM-driven BCAA acquisition, suppress NK cell activation, and exacerbate the formation of an immunosuppressive TME.
Transcriptomic profiling from TCGA revealed significantly higher PSMD14 mRNA levels in GBM compared with low-grade glioma (LGG) and non-tumour brain (NTB) tissues (Fig. S19C). The CGGA analysis further illustrated a positive link between PSMD14 expression and glioma WHO grade (Fig. S19D). Kaplan-Meier analysis demonstrated that elevated expression of PSMD14 or BCKDK was linked to significantly worse overall survival (Fig. 8E, F), consistent with findings from the TCGA cohort (Fig. S19E, F). Moreover, multivariate survival analysis illustrated an inverse survival correlation with PSMD14 expression even when accounting for IDH mutation, 1p/19q codeletion, MGMT promoter methylation, as well as patient age and sex (Fig. S19G–P).
Collectively, these data indicate that the PSMD14–BCKDK axis is negatively associated with immune cell infiltration in GBM, supporting the therapeutic potential of PSMD14 in enhancing CAR-NK immunotherapy responses (Fig. 8G).
Discussion
Metabolic cross-talk within the GBM TME critically influences immune surveillance. Our study demonstrated that GBM promoted immune evasion by competitively depleting BCAA, thereby suppressing NK and CD8+ T cell function. This is mediated by a PSMD14–BCKDK–IGF2BP3 positive feedback loop, which stabilizes SLC7A5/SLC7A8 mRNAs and enhances tumor BCAA uptake. These findings highlight the significance of TME intercellular metabolic competition in tumor immune escape and suggest that additional immune effector populations, such as NK-cDC1-CD8+ T cell circuit, may also play important roles in anti-tumor immunity [33, 36].
We identified PSMD14 as a critical post-translational regulator that stabilizes BCKDK in GBM by preventing TRIM21-mediated ubiquitination and degradation. This stabilization reprograms BCAA metabolism, leading to BCAA depletion and suppressed NK cell activity in the TME, in part through inhibition of PI3K/Akt and cGAS-STING pathways. Given BCKDK’s established role as a therapeutic target in cancer and BCAA metabolism [24]. targeting PSMD14 or BCKDK may enhance anti-tumor immunity, improve CAR-T/CAR-NK efficacy, and potentially synergize with PD-1 blockade [16, 37]. Furthermore, a prospective study revealed that supplementation with BCAA significantly reduces the risk of hepatocarcinogenesis. Nevertheless, whether similar benefits can be observed in patients with GBM remains to be elucidated [38]. Clinically, elevated PSMD14 correlates with increased immunosuppression in GBM. Importantly, multivariable Cox regression analyses of our clinical cohort further identified PSMD14 and BCKDK expression levels as independent prognostic factors for 4-year overall survival in GBM, underscoring their clinical relevance. Together, these findings elucidate a novel post-translational regulatory mechanism of BCKDK and support direct BCKDK inhibition as a promising strategy for boosting immunotherapy in solid tumors.
BCKDK is a key enzyme that phosphorylates and inhibits the BCKDH complex, controlling the rate-limiting step of BCAA catabolism. Beyond this metabolic role, BCKDK also activates MEK/ERK and MAPK signaling to promote cancer cell growth and regulates homologous recombination repair, contributing to resistance against DNA damage-based therapies [24, 39, 40]. Both genetic and pharmacologic inhibition of BCKDK in triple-negative breast cancer models reduces intracellular and secreted BCKAs, which can strongly suppress tumor immunity [41, 42]. Whether decreasing BCKAs reverses NK cell suppression remains to be determined. Thus, BCKDK is an attractive therapeutic target, but its post-translational regulation is poorly understood.
Numerous factors—including growth factors, cytokines, antioxidants, chaperones, protein kinases, transcriptional regulators, and histone modifiers—are central to metabolic disease progression [43,44,45]. Enzymes that mediate post-translational modifications, such as phosphorylation [46]. oxidation [47]. O-GlcNAcylation [48] and ubiquitination [49]. They are particularly important in metabolic dysregulation. Ubiquitination, a reversible modification regulated by ubiquitin ligases and DUBs, targets proteins for proteasomal degradation [50]. Our study shows that high BCKDK protein levels, which do not correlate with mRNA abundance, are linked to reduced immune infiltration in GBM, highlighting post-translational control. MS identified PSMD14—a DUB associated with the proteasome—as a novel BCKDK interactor. PSMD14 stabilizes BCKDK by removing K48-linked ubiquitin chains and counteracting TRIM21-mediated ubiquitination and degradation [51]. These findings uncover a new mechanism by which PSMD14 regulates BCKDK abundance and function in GBM cells. The 26S proteasome, composed of a 20S core and 19S regulatory particles, processes polyubiquitinated substrates, with PSMD family DUBs in the 19S subunit determining substrate fate [52, 53]. PSMD14, a JAMM–domain DUB in the 19S regulatory particle (also known as POH1/RPN11), promotes tumor progression by deubiquitinating and stabilizing oncogenic substrates[54, 55]. Elevated PSMD14 expression is observed in various cancers, including breast, ovarian, and esophageal tumors, and multiple myeloma, and is associated with tumor development [31, 32, 56, 57]. These insights suggest that targeting PSMD14 could regulate BCKDK, remodel tumor BCAA metabolism, and modulate the immune microenvironment. Notably, OPA is a specific PSMD14 inhibitor [54, 58]. In preclinical models, OPA alleviated immunosuppression, improved immunotherapy efficacy, and reduced BCKDK-mediated BCAA metabolism and immune escape. In addition, we identified Thr457 as a major BCKDK-sensitive phosphorylation site within the KH domain of IGF2BP3. Although both N- and C-terminal fragments of IGF2BP3 were able to associate with PSMD14 mRNA, phosphorylation at Thr457 may modulate RNA binding by altering IGF2BP3 conformation, inter-domain coordination, or cofactor interactions, thereby fine-tuning IGF2BP3–PSMD14 mRNA association [59].
In summary, our study identifies a PSMD14–BCKDK–IGF2BP3 positive feedback axis that enables GBM cells to competitively deplete BCAA, suppress NK cell-mediated immunity, and establish an immunosuppressive microenvironment. Thus, PSMD14 is a promising therapeutic target—especially in BCKDK-high tumors—and its inhibition may be leveraged alone or combined with CAR-NK therapy in GBM. Further clinical studies are needed to advance these findings toward precision medicine for GBM.
Methods and materials
Tumor models and treatments
All animal research received approval from the Institutional Animal Care and Use Committee of Huazhong University of Science and Technology ([2023] IACUC Number: 3807). Mice were euthanized upon exhibiting severe neurological deficits or significant weight loss (up to 20%). NOD/SCID/IL-2rg (NSG), Batf3 knockout (KO), and C57BL/6J mice were acquired from Shulaibao (Wuhan) Biotechnology Co., Ltd. Tumor proliferation was assessed with bioluminescence imaging (Bruker Corporation, Billerica, MA). The weight of the mouse and its survival duration were meticulously documented. More details are included in the Supplementary Materials.
Patient information and tissue specimens
Clinical data and 114 GBM tissue samples were acquired from the Department of Neurosurgery at Wuhan Union Hospital, along with 20 comparable non-neoplastic brain tissues (NBT). There was a complete absence of anti-cancer treatment among the patients who underwent surgery. All surgical specimens were pathologically confirmed as glioblastoma, and, in accordance with the latest WHO classification and clinical guidelines, were classified as IDH-wildtype [60]. Following their removal, all specimens were rapidly frozen in liquid nitrogen. In compliance with the Declaration of Helsinki (for humans) and the U.S. Public Health Service Policy on Human Care and Use of Laboratory Animals (2015 reprint; for mice), the institutional review boards of Union Hospital, Tongji Medical College, Huazhong University of Science and Technology provided their approval for all experiments and the use of human tissues (Approval No. UHCT240826). All adults were asked to sign a document indicating their understanding and agreement. For minors, informed consent was obtained from a parent or legal guardian.
Reagents
Amino acids, including l-isoleucine, l-leucine, l-lysine, l-methionine, l-phenylalanine, l-threonine, l-tryptophan, l-valine, l-histidine, l-arginine, l-cystine, and l-tyrosine, as well as MEM non-essential amino acids (100×) containing l-alanine, l-aspartic acid, l-asparagine, l-glutamic acid, l-glycine, l-proline, and l-serine, were obtained from Sigma. L-Glutamine (100×), 2-mercaptoethanol, and dialyzed fetal bovine serum (FBS) were purchased from GIBCO. Standard MEM-α medium and a modified version devoid of amino acids were acquired from Procell.
Cell culture
Human GBM cell lines (U251, A172, U87, LN229, U118, KNS89, and T98G), the normal astrocyte cell line (NHA), microglia (HMC3), oligodendrocyte precursor-like cells (MO3.13), murine GBM cell lines (GL261, CT2A), and human embryonic kidney cells HEK293T were purchased from the American Type Culture Collection (ATCC). Human NK cell line NK92 was purchased from Leibniz Institute DSMZ (Brunswick, Germany) and cultured in MEMα complete medium containing the following components: 0.2 mM inositol, 0.1 mM β-mercaptoethanol, 0.02 mM folic acid, 200 U/mL recombinant IL-2 (Peprotech), 12.5% horse serum (Sigma), 12.5% FBS (Gibco), 100 U/mL penicillin, and 0.1 mg/mL streptomycin (Gibco). Cell line identity was recently confirmed by short tandem repeat (STR) profiling, and all cultures were routinely tested for mycoplasma contamination with negative results.
To pre-activate and expand isolated resting NK cells, the medium was supplemented with recombinant human IL-2. Half of the IL-2-containing medium was replaced with fresh medium three times a week. The cell density was maintained between 1–3 × 107 cells/mL, and fresh complete medium was added to dilute the cell culture when necessary. Resting NK cells were used for cytotoxicity assays after 3 days of pre-activation and were utilized within two weeks of activation (from Day 3 to 14). Unless otherwise specified, all cells were cultured in DMEM supplemented with 10% FBS (Sigma), 100 U/mL penicillin, and 0.1 mg/mL streptomycin (Gibco). All cell lines were authenticated by STR profiling and regularly tested for mycoplasma contamination.
Transient transfection, lentiviral infection, and shRNA knockdown
Genomeditech (Shanghai, China) generated the lentiviral KO plasmids for PSMD14, BCKDK, TRIM21, and IGF2BP3, together with the lentiviral overexpression plasmid for BCKDK. Table S8 enumerates the sequences of the shRNA employed. The human BCKDK (NM_005881.4) and its truncated cDNA were amplified by PCR and subsequently cloned into the pECMV-3xFLAG-C expression vector. The commercially available NK92-GFP-EGFRvIII·CAR (CD28·CD3ζ) (VGC-0011-017S) was acquired from Vigen Biotechnology (Zhenjiang, China). Transient transfection was conducted utilizing Lipofectamine 3000 (Thermo Fisher Scientific, USA) in accordance with the manufacturer’s instructions. Tumor cells were subsequently exposed to the viral supernatant for 24 h and then subjected to selection with 2 μg/mL puromycin (Santa Cruz Biotechnology) for an additional 48 h.
The detection assays of BCAA-associated metabolites
The BCAA levels of GBM patients’ tumor tissues, plasma, CSF, and glioma cell lines, along with their respective culture supernatants, were measured using the BCAA assay kit (Sigma) according to the manufacturer’s instructions. For tissue or cell samples, 10 mg of tissue or 2 × 106 cells were rapidly homogenized in cold BCAA assay buffer. The homogenate was centrifuged at 13,000 × g for 10 min at 4 °C to remove insoluble material. Plasma and culture supernatants were directly assayed. The absorbance was measured at 450 nm, and the BCAA content in samples was determined from a standard curve.
Approximately 20 mg of mouse tumor tissue samples were homogenized with 500 μL of pre-cooled methanol-water (2:1, v/v) utilizing a tissuelyser at 45 Hz for 90 s. The aforementioned sample was combined with 500 μL of pre-chilled methanol-water (2:1, v/v) and subsequently homogenized, a process that was done twice. The combination underwent centrifugation at 12,000 rpm for 10 min at 4 °C, and the supernatant was subsequently filtered using a 0.22 µm membrane filter before UPLC-MS/MS analysis. All samples were evaluated utilizing an ultra-high-performance liquid chromatography system coupled with tandem MS (1290-6470 UPLC-MS/MS, Agilent, USA). Peak determination and peak area integration were conducted using MassHunter Workstation software (Agilent, Version B.08.00). Standard curves were obtained from plots of the peak area versus the known concentration of the standards (three amino analyte standards were purchased from Sigma-Aldrich, USA). The concentration of the metabolites in samples was determined by the equations of linear regression obtained from the calibration.
TEM
NK cells subjected to BCAA deprivation alone or co-cultured with U251 tumor cells were preserved in a 2.5% glutaraldehyde solution at 4 °C for 12 h. The samples were subsequently post-fixed with 2% osmium tetroxide. Subsequent to washing, the samples were subjected to staining with 1% aqueous uranyl acetate and dehydrated through a graded series of ethanol (50%, 70%, 90%, 95%, and 100%). The desiccated samples were infiltrated and encapsulated in Eponate 12 resin. Ultrathin pieces were affixed to copper grids and stained with lead citrate and uranyl acetate. Sections were examined using a TEM (Tecnai G2 Spirit; 120 kV; FEI). To measure the quantity of membrane synapses and the degree of mitochondrial autophagy in each slice (representing a single cell), TEM imaging of individual NK cells was conducted. Furthermore, TEM imaging of NK cells co-cultured with U251 cells was performed to examine the interaction between the two cell types.
In vitro cytotoxicity assays
A172 and U251 glioma cells, exhibiting exponential growth, were trypsinized and subsequently tagged with CFSE (Abcam) following the manufacturer’s methodology. Labeled cells were resuspended in pre-warmed media and plated in flat-bottom 6-well plates (1 × 105 cells/well, 2 mL) for flow cytometric analysis or in 12-well plates (0.5 × 105 cells/well, 1 mL) for crystal violet staining-based cytotoxicity experiments. To achieve uniform cell distribution, agitation was reduced before attachment, and cells were incubated in a low-vibration incubator for 3 h. Subsequent to initial attachment, the media were carefully substituted with new medium containing the specified chemicals, with or without NK cells at designated effector-to-target ratios. Plates were subsequently incubated under low-vibration conditions for the specified duration.
For flow cytometric analysis, supernatants were harvested, and adhering cells were dissociated using trypsin and subsequently mixed with the supernatant. Annexin V binding buffer (100 mM HEPES, 40 mM KCl, 1.4 M NaCl, 7.5 mM MgCl2, 25 mM CaCl2, pH 7.4) with Annexin V was introduced, and samples were incubated at ambient temperature for 15 min prior to flow cytometric analysis.
In crystal violet experiments, non-adherent cells were removed after 24 h. Following an additional 3–5 days, adherent tumor cells were fixed and stained with 0.1% crystal violet in 50% methanol for 1 h. Images were obtained, and cytotoxicity was assessed by evaluating the stained region.
IHC and IF
Tumor tissues were collected from mouse models, fixed in 4% polyoxymethylene overnight, and paraffin-embedded. IHC was conducted on 114 paraffin-embedded GBM samples using rabbit anti-PSMD14 and anti-BCKDK antibodies, following the manufacturer’s instructions. Slides were scanned at 20× magnification using a Vectra Polaris system (Akoya Biosciences). Staining was independently evaluated by two pathologists and scored as strong (+3), moderate (+2), weak (+1), or negative (0). High expression was defined as a score of +3 or +2; low expression as +1 or 0.
Prior to conducting cellular IF, the cells were prepared by seeding them onto glass coverslips, fixing them with 4% paraformaldehyde, permeabilizing them with 0.25% Triton X-100 for 10 min, and finally, blocking them with 2% BSA in PBS. A combination of primary and fluorescent secondary antibodies was used to incubate the samples. A confocal microscope from Zeiss, the LSM 880, was used to obtain imagery. In terms of tissue, antigen retrieval was performed on 4 μm paraffin slices in citrate buffer (microwave, 95 °C, 30 min). The steps for double IF were as follows: samples were pre-incubated with two primary antibodies at 4 °C for an overnight period, rinsed, and then a 1-h incubation was conducted in the dark at 37 °C with secondary antibodies. DAPI was used as a counterstain.
RNA extraction and RT‑qPCR
Total RNA was isolated from tissues or cell lines utilizing Trizol reagent (Thermo Fisher, USA) in accordance with the manufacturer’s guidelines and evaluated using Nanodrop. cDNA was produced from 1 µg of total RNA via the Evo M-MLV RT kit (AG11728, Accurate Biotechnology, Hunan, China). The following RT-qPCR was conducted utilizing the SYBR Green PreMix Taq HS qPCR kit (AG11728, Accurate Biotechnology, Hunan, China). All reactions were conducted using a Bio-Rad real-time quantitative PCR equipment (Bio-Rad, USA). The RT-qPCR findings were standardized to GAPDH as the reference gene, and relative levels were detected via the 2(-ΔΔCT) technique. Table S9 enumerates the primer sequences utilized in the PCR reactions.
WB and immunoprecipitation (IP) assays
The cell lysis was conducted via RIPA lysis buffer (P0013B, Beyotime) enriched with a protease and phosphatase inhibitor cocktail (HY-K0013, MedChemExpress). Equal quantities of total protein from each sample were exposed to SDS-PAGE and subsequently transferred to a polyvinylidene fluoride (PVDF) membrane (IPVH00010, Merck Millipore). Then, the membrane was blocked with 5% skim milk and treated overnight with the primary antibody at 4 °C. Detection was conducted with a chemiluminescence imaging system (Bio-Rad) featuring enhanced chemiluminescence (ECL, New Cell & Molecular Biotechnology, China). The supplementary table contains details regarding the antibody information and dilution ratios.
Cell lysates for IP tests were produced from specified cells utilizing a lysis solution composed of 150 mM NaCl, 10 mM HEPES (pH 7.4), and 1% NP-40. An overnight incubation of the lysates was conducted at 4 °C with anti-PSMD14 Rabbit antibody, or anti-BCKDK Rabbit antibody, or anti-TRIM21 Rabbit antibody, or anti-IGF2BP3 Rabbit antibody, and Protein A/G-conjugated agarose or Flag, Myc affinity agarose (Sigma-Aldrich). The beads with affinity-bound proteins were rinsed six times with IP wash buffer (150 mM NaCl, 10 mM HEPES, pH 7.4, 0.1% NP-40), and proteins were eluted with 1 M glycine (pH 3.0). The eluates were mixed with sample buffer, denatured, and analyzed by WB. Specific 3xFlag-BCKDK bands were subjected to MS analysis. GST fusion proteins were prepared following standard protocols. For in vitro binding assays, BCKDK-GST fusion proteins were bound to GSH-Sepharose (Sigma-Aldrich) and incubated with cell lysates. After washing, bound proteins were separated by SDS-PAGE and visualized by Coomassie blue staining. Table S10 provides data on the antibodies.
Bacterial expression and in vitro pull-down assay
The bacteria harboring pET23a-GST-PSMD14 and pET23a-His-BCKDK constructions were cultivated at 37 °C, achieving an optical density at 600 nm (A600 nm) between 1.2 and 1.4. The GST-PSMD14 protein purification was conducted via Glutathione Sepharose 4B (GE Healthcare Bio-Sciences AB, SE-75184, Uppsala, Sweden), whereas the His-BCKDK protein was isolated via Ni-NTA Agarose (Genscript, L00250). The purified proteins underwent three washes with cold wash buffer to eliminate unattached proteins. Subsequently, loading buffer was incorporated, and the samples were subjected to heating at 95 °C for 10 min. The protein samples were analysis via Coomassie Brilliant Blue staining and WB.
MS analysis
A172 and U251 cells were transfected with 3xFlag-BCKDK or Vector to determine the binding proteins of BCKDK. Lysates were subjected to IP using Flag beads. Beads with affinity-bound proteins were subjected to six washes with wash buffer (150 mM NaCl, 10 mM HEPES, pH 7.4, 0.1% NP-40), then elution was conducted with 1 M glycine (pH 3.0). The elutes underwent MS. Data regarding the peptides and quantities of BCKDK-binding proteins examined using IP/MS experiments, together with a comprehensive list of the enriched pathways of BCKDK-interacting proteins, are included in Tables S2 and 4.
RNA pull‑down and RIP assays
Probes specific to SLC7A5, SLC7A8, and PSMD14, along with negative control probes, were produced by GeneCreate Biological Engineering in Wuhan, China. RNA pull-down tests were conducted utilizing the Pierce Magnetic RNA-Protein Pull-Down Kit (Thermo Fisher Scientific) in accordance with the manufacturer’s guidelines. Proteins immunoprecipitated from the beads were examined by WB.
RIP trials were performed as per the instructions of the Magna RIP RNA-Binding Protein IP Kit (Millipore, Burlington, MA, USA). Cells were lysed using RIP lysis buffer and subsequently treated with antibodies targeting IGF2BP3 or rabbit IgG. Co-precipitated RNAs were isolated for sequencing or cDNA synthesis and utilized as templates for the measurement of gene expression via qRT-PCR using specified primers. Total RNA (Input) or antibody (Rabbit IgG) served as references.
Actinomycin D assay
For the mRNA stability assay, A172 and U251 cells were seeded into 6-well plates and treated with si-PSMD14 or si-IGF2BP3 and then treated with actinomycin D (20 μg/mL, M4881, AbMole BioScience) or vehicle for 0, 1, 2, 4, 6, and 9 h. The total RNA was extracted by Trizol Reagent and analyzed by RT-qPCR as described above. The results were normalized to the values measured at 0 h.
Single-cell dissociation and sequencing
The tissues were excised surgically and preserved in MACS Tissue Storage Solution (Miltenyi Biotec) until processing. The tissue specimens were treated as outlined below. Initially, samples were rinsed with PBS, then sliced into small fragments (about 1 mm³) on ice and subjected to enzymatic digestion using 1500 U/mL collagenase I (Diamond) and 100 U/mL DNase I (Diamond) for 45 min at 37 °C, with agitation. Post-digestion, materials were filtered using a 70 µm cell strainer and centrifuged at 3000 × g for 5 min. After being rinsed with PBS containing 0.04% BSA, the cell pellets were resuspended in PBS containing 0.04% BSA and passed through a 30 μm cell strainer again. In order to assess cell viability using the Countstar Fluorescence Cell Analyzer, dissociated individual cells were further stained with AO/PI. The MACS dead cell removal kit (Miltenyi Biotec) was used to enrich the single-cell solution further.
10X Genomics’ Chromium Controller Instrument and Chromium Single Cell 3’ V3.1 Reagent Kits (10X Genomics, Pleasanton, CA) were used for constructing the scRNA-Seq libraries. Cells were concentrated to 1000 cells/μL, and roughly 10,000 cells were introduced into each channel to produce single-cell Gel Bead-In-Emulsions (GEMs), yielding anticipated mRNA barcoding of 5000 single cells per sample. Subsequent to the RT process, GEMs were disrupted, and barcoded cDNA was isolated and amplified. The amplified barcoded cDNA was fragmented, A-tailed, ligated using adaptors, and subjected to index PCR amplification. The resultant libraries were measured with the Qubit High Sensitivity DNA assay (Thermo Fisher Scientific), and their size distribution was assessed using a High Sensitivity DNA chip on a Qsep100 (Bioptic). All libraries were sequenced via the NovaSeq 6000 (Illumina, San Diego, CA) in a 150 bp paired-end configuration.
Single-cell isolation and flow cytometry assay
A cocktail of antibodies for surface and intracellular staining was created in advance of the experiment. Afterwards, the Mouse Tumor Dissociation Kit (#130-096-730, Miltenyi Biotec, Germany) was used to separate tumor tissues into single-cell suspensions in accordance with the manufacturer’s protocol. Before being centrifuged at 600 × g for 6 min at 4 °C, the cells were passed through a 200-mesh strainer and resuspended in RPMI-1640. After 1 min of incubation at room temperature, red blood cells were lysed using RBC lysis buffer. The mononuclear cells were grown with 1 mg/mL of GolgiPlugTM for 4 h or tested for NK and T cell infiltration before their stimulation capacity was evaluated using flow cytometry.
Up to 2 × 106 cells per sample were blocked for 10 min, incubated with surface antibody cocktails for 30 min at room temperature, and stained with Dead Cell dye (2.5 mM, 4 °C, 15 min) for viability assessment. After washing with PBS (1% FBS), surface staining was performed at 4 °C for 1 h. For intracellular staining, cells were fixed, permeabilized, and incubated with intracellular antibody cocktails at 4 °C for 1 h, followed by washing and resuspension in 500 μL PBS. Samples were analyzed using a CytoFLEX LX flow cytometer (Beckman Coulter) with CytExpert software. Cell population frequencies were quantified with FlowJo (Tree Star). Table S11 presents the flow cytometry staining panels.
Bioinformatics analysis
Clinical and transcriptomic data for glioma samples were acquired from TCGA (https://xenabrowser.net/heatmap/) and CGGA (CGGA693 and CGGA325; http://www.cgga.org.cn). Normal brain transcript data were retrieved from the GTEx database (https://gtexportal.org).
Statistical analysis
Analyses were performed using SAS 9.4, GraphPad Prism (v8.0), and R (v3.3.1). Data are presented as mean ± SD or mean ± SEM, as specified in the figure legends, with exact n and the definition of biological versus technical replicates indicated for each experiment. Normality and homogeneity of variance were assessed prior to parametric testing. For two-group comparisons, a two-tailed Student’s t test was used when assumptions were met; Welch’s t-test was applied when variances were unequal, and nonparametric tests (Mann–Whitney U test) were used when normality could not be assumed. For comparisons involving more than two groups, one-way or two-way ANOVA with appropriate post hoc multiple-comparison procedures was used for parametric data; otherwise, Kruskal–Wallis tests were applied where appropriate. Linear mixed-effects models were used to account for repeated measures from the same donor. Survival was analyzed using the Kaplan–Meier method with two-sided log-rank tests. Multiple comparisons were controlled using Holm’s method when applicable. A two-sided P < 0.05 was considered statistically significant.
Data availability
All data associated with this study are present in the paper or the Supplementary Materials.
References
Poorva P, Mast J, Cao B, Shah MV, Pollok KE, Shen J. Killing the killers: natural killer cell therapy targeting glioma stem cells in high-grade glioma. Mol Ther. 2025;33:2462–2478.
Kyrysyuk O, Wucherpfennig KW. Designing cancer immunotherapies that engage T cells and NK cells. Annu Rev Immunol. 2023;41:17–38.
Peng L, Sferruzza G, Yang L, Zhou L, Chen S. CAR-T and CAR-NK as cellular cancer immunotherapy for solid tumors. Cell Mol Immunol. 2024;21:1089–1108.
Kong R, Liu B, Wang H, Lu T, Zhou X. CAR-NK cell therapy: latest updates from the 2024 ASH annual meeting. J Hematol Oncol. 2025;18:22.
Burger MC, Forster MT, Romanski A, Straßheimer F, Macas J, Zeiner PS, et al. Intracranial injection of natural killer cells engineered with a HER2-targeted chimeric antigen receptor in patients with recurrent glioblastoma. Neuro Oncol. 2023;25:2058–2071.
Liu Y, Zhou F, Ali H, Lathia JD, Chen P. Immunotherapy for glioblastoma: current state, challenges, and future perspectives. Cell Mol Immunol. 2024;21:1354–1375.
Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer. 2020;20:12–25.
Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15:422–442.
O’Brien KL, Finlay DK. Immunometabolism and natural killer cell responses. Nat Rev Immunol. 2019;19:282–290.
Zhang S, Liu W, Hu B, Wang P, Lv X, Chen S, et al. Prognostic significance of tumor-infiltrating natural killer cells in solid tumors: a systematic review and meta-analysis. Front Immunol. 2020;11:1242.
Page A, Chuvin N, Valladeau-Guilemond J, Depil S. Development of NK cell-based cancer immunotherapies through receptor engineering. Cell Mol Immunol. 2024;21:315–331.
Hosseinalizadeh H, Wang LS, Mirzaei H, Amoozgar Z, Tian L, Yu J. Emerging combined CAR-NK cell therapies in cancer treatment: finding a dancing partner. Mol Ther. 2025;33:2406–2425.
Tyagi A, Wu SY, Watabe K. Metabolism in the progression and metastasis of brain tumors. Cancer Lett. 2022;539:215713.
You M, Xie Z, Zhang N, Zhang Y, Xiao D, Liu S, et al. Signaling pathways in cancer metabolism: mechanisms and therapeutic targets. Signal Transduct Target Ther. 2023;8:196.
Wang Z, Li B, Li S, Lin W, Wang Z, Wang S, et al. Metabolic control of CD47 expression through LAT2-mediated amino acid uptake promotes tumor immune evasion. Nat Commun. 2022;13:6308.
Yao CC, Sun RM, Yang Y, Zhou HY, Meng ZW, Chi R, et al. Accumulation of branched-chain amino acids reprograms glucose metabolism in CD8(+) T cells with enhanced effector function and anti-tumor response. Cell Rep. 2023;42:112186.
Silva LS, Poschet G, Nonnenmacher Y, Becker HM, Sapcariu S, Gaupel AC, et al. Branched-chain ketoacids secreted by glioblastoma cells via MCT1 modulate macrophage phenotype. EMBO Rep. 2017;18:2172–2185.
Cai Z, Li W, Brenner M, Bahiraii S, Heiss EH, Weckwerth W. Branched-chain ketoacids derived from cancer cells modulate macrophage polarization and metabolic reprogramming. Front Immunol. 2022;13:966158.
Halbrook CJ, Lyssiotis CA. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell. 2017;31:5–19.
Budhathoki S, Iwasaki M, Yamaji T, Yamamoto H, Kato Y, Tsugane S. Association of plasma concentrations of branched-chain amino acids with risk of colorectal adenoma in a large Japanese population. Ann Oncol. 2017;28:818–823.
Tönjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM, et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med. 2013;19:901–908.
Boskovic P, Wilke N, Man KH, Lichter P, Francois L, Radlwimmer B. Branched-chain amino acid transaminase 1 regulates glioblastoma cell plasticity and contributes to immunosuppression. Neuro Oncol. 2024;26:251–265.
Lemos H, Huang L, Prendergast GC, Mellor AL. Immune control by amino acid catabolism during tumorigenesis and therapy. Nat Rev Cancer. 2019;19:162–175.
East MP, Laitinen T, Asquith CRM. BCKDK: an emerging kinase target for metabolic diseases and cancer. Nat Rev Drug Discov. 2021;20:498.
Ma Q, Yang F, Huang B, Pan X, Li W, Yu T, et al. CircARID1A binds to IGF2BP3 in gastric cancer and promotes cancer proliferation by forming a circARID1A-IGF2BP3-SLC7A5 RNA-protein ternary complex. J Exp Clin Cancer Res. 2022;41:251.
Cheon S, Kaur K, Nijem N, Tuncay IO, Kumar P, Dean M, et al. The ubiquitin ligase UBE3B, disrupted in intellectual disability and absent speech, regulates metabolic pathways by targeting BCKDK. Proc Natl Acad Sci USA. 2019;116:3662–3667.
Spataro V, Buetti-Dinh A. POH1/Rpn11/PSMD14: a journey from basic research in fission yeast to a prognostic marker and a druggable target in cancer cells. Br J Cancer. 2022;127:788–799.
Pathare GR, Nagy I, Śledź P, Anderson DJ, Zhou HJ, Pardon E, et al. Crystal structure of the proteasomal deubiquitylation module Rpn8-Rpn11. Proc Natl Acad Sci USA. 2014;111:2984–2989.
Beltrao P, Albanèse V, Kenner LR, Swaney DL, Burlingame A, Villén J, et al. Systematic functional prioritization of protein posttranslational modifications. Cell. 2012;150:413–425.
Zhou B, Luo Y, Bi H, Zhang N, Ma M, Dong Z, et al. Amelioration of nonalcoholic fatty liver disease by inhibiting the deubiquitylating enzyme RPN11. Cell Metab. 2024;36:2228–2244.e2227.
Lv J, Zhang S, Wu H, Lu J, Lu Y, Wang F, et al. Deubiquitinase PSMD14 enhances hepatocellular carcinoma growth and metastasis by stabilizing GRB2. Cancer Lett. 2020;469:22–34.
He L, Yu C, Qin S, Zheng E, Liu X, Liu Y, et al. The proteasome component PSMD14 drives myelomagenesis through a histone deubiquitinase activity. Mol Cell. 2023;83:4000–4016.e4006.
Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172:1022–1037.e1014.
Meiser P, Knolle MA, Hirschberger A, de Almeida GP, Bayerl F, Lacher S, et al. A distinct stimulatory cDC1 subpopulation amplifies CD8(+) T cell responses in tumors for protective anti-cancer immunity. Cancer Cell. 2023;41:1498–1515.e1410.
Hernández-García E, Cueto FJ, Cook ECL, Redondo-Urzainqui A, Charro-Zanca S, Robles-Vera I, et al. Conventional type 1 dendritic cells protect against age-related adipose tissue dysfunction and obesity. Cell Mol Immunol. 2022;19:260–275.
Wang M, Zhou Z, Wang X, Zhang C, Jiang X. Natural killer cell awakening: unleash cancer-immunity cycle against glioblastoma. Cell Death Dis. 2022;13:588.
Yang Q, Zhu X, Huang P, Li C, Han L, Han Y, et al. BCKDK modification enhances the anticancer efficacy of CAR-T cells by reprogramming branched-chain amino acid metabolism. Mol Ther. 2024;32:3128–3144.
Kawaguchi T, Shiraishi K, Ito T, Suzuki K, Koreeda C, Ohtake T, et al. Branched-chain amino acids prevent hepatocarcinogenesis and prolong the survival of patients with cirrhosis. Clin Gastroenterol Hepatol. 2014;12:1012–1018.e1011.
Xue P, Zeng F, Duan Q, Xiao J, Liu L, Yuan P, et al. BCKDK of BCAA Catabolism cross-talks with the MAPK pathway promotes tumorigenesis of colorectal cancer. EBioMedicine. 2017;20:50–60.
Liu, H, Feng, J, Pan, T, Zhang, P, Ye, L, Jiang, Z, et al. Nuclear-localized bckdk facilitates homologous recombination repair to support breast cancer progression and therapy resistance. Adv Sci. 2025;12:e2416590.
Biswas D, Slade L, Duffley L, Mueller N, Dao KT, Mercer A, et al. Inhibiting BCKDK in triple-negative breast cancer suppresses protein translation, impairs mitochondrial function, and potentiates doxorubicin cytotoxicity. Cell Death Discov. 2021;7:241.
Zhang B, Peng H, Zhou M, Bao L, Wang C, Cai F, et al. Targeting BCAT1 combined with α-ketoglutarate triggers metabolic synthetic lethality in glioblastoma. Cancer Res. 2022;82:2388–2402.
Xourafa G, Korbmacher M, Roden M. Inter-organ crosstalk during development and progression of type 2 diabetes mellitus. Nat Rev Endocrinol. 2024;20:27–49.
Yao W, Hu X, Wang X. Crossing epigenetic frontiers: the intersection of novel histone modifications and diseases. Signal Transduct Target Ther. 2024;9:232.
Liu YQ, Yang Q, He GW. Post-translational acylation of proteins in cardiac hypertrophy. Nat Rev Cardiol. 2025;22:944–960.
Choi E, Duan C, Bai XC. Regulation and function of insulin and insulin-like growth factor receptor signalling. Nat Rev Mol Cell Biol. 2025;26:558–580.
Masenga SK, Kabwe LS, Chakulya M, Kirabo A. Mechanisms of oxidative stress in metabolic syndrome. Int J Mol Sci. 2023;24:7898.
Chatham JC, Zhang J, Wende AR. Role of O-Linked N-acetylglucosamine protein modification in cellular (patho)physiology. Physiol Rev. 2021;101:427–493.
Kitamura, H. Ubiquitin-specific proteases (USPs) and metabolic disorders. Int J Mol Sci. 2023;24:3219.
Atta H, Kassem DH, Kamal MM, Hamdy NM. Targeting the ubiquitin proteasome system in cancer stem cells. Trends Cell Biol. 2025;35:97–101.
Li JY, Zhao Y, Gong S, Wang MM, Liu X, He QM, et al. TRIM21 inhibits irradiation-induced mitochondrial DNA release and impairs antitumour immunity in nasopharyngeal carcinoma tumour models. Nat Commun. 2023;14:865.
Trulsson F, Akimov V, Robu M, van Overbeek N, Berrocal DAP, Shah RG, et al. Deubiquitinating enzymes and the proteasome regulate preferential sets of ubiquitin substrates. Nat Commun. 2022;13:2736.
Bashore C, Prakash S, Johnson MC, Conrad RJ, Kekessie IA, Scales SJ, et al. Targeted degradation via direct 26S proteasome recruitment. Nat Chem Biol. 2023;19:55–63.
Arkinson C, Dong KC, Gee CL, Martin A. Mechanisms and regulation of substrate degradation by the 26S proteasome. Nat Rev Mol Cell Biol. 2025;26:104–122.
Sun T, Liu Z, Bi F, Yang Q. Deubiquitinase PSMD14 promotes ovarian cancer progression by decreasing enzymatic activity of PKM2. Mol Oncol. 2021;15:3639–3658.
Yu Y, Hu J, Wang W, Lei H, Xi Z, Zhang P, et al. Targeting PSMD14 combined with arachidonic acid induces synthetic lethality via FADS1 m(6)A modification in triple-negative breast cancer. Sci Adv. 2025;11:eadr3173.
Jing C, Li X, Zhou M, Zhang S, Lai Q, Liu D, et al. The PSMD14 inhibitor Thiolutin as a novel therapeutic approach for esophageal squamous cell carcinoma through facilitating SNAIL degradation. Theranostics. 2021;11:5847–5862.
Song Y, Li S, Ray A, Das DS, Qi J, Samur MK, et al. Blockade of deubiquitylating enzyme Rpn11 triggers apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Oncogene. 2017;36:5631–5638.
Schneider T, Hung LH, Aziz M, Wilmen A, Thaum S, Wagner J, et al. Combinatorial recognition of clustered RNA elements by the multidomain RNA-binding protein IMP3. Nat Commun. 2019;10:2266.
Zakharova G, Efimov V, Raevskiy M, Rumiantsev P, Gudkov A, Belogurova-Ovchinnikova O, et al. Reclassification of TCGA diffuse glioma profiles linked to transcriptomic, epigenetic, genomic and clinical data, according to the 2021 WHO CNS tumor classification. Int J Mol Sci. 2022;24:157.
Funding
This work was supported by the Open Foundation of Hubei Key Laboratory of Biological Targeted Therapy (202414 to SY), Youth Fund of the National Natural Science Foundation of China (82203140 to MW) and Fund of the National Natural Science Foundation of China (82273210 to XJ).
Author information
Authors and Affiliations
Contributions
SJY and MJW conceived and designed the study. SJY drafted the manuscript. SJY, CJ, XDL, QHC, and JH performed the in vitro and in vivo experiments. CJ and XDL performed IHC staining of human and mouse tumor samples and contributed to data interpretation. SJY, XDL, ZHG, and ZZ performed single-cell isolation and flow cytometry assays. SJY and MJW performed scRNA-seq experiments (including sample preparation, library construction, and sequencing). SJY, MJW, and CJ analyzed the data and prepared figures. XW provided critical technical advice and contributed to manuscript revision. WX, JJL, and XBJ supervised the project, provided resources, and critically revised the manuscript. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, S., Wang, M., Jiang, C. et al. Targeting the PSMD14-BCKDK pathway overcomes immune suppression and enhances CAR-NK infiltration in glioblastoma. Cell Death Differ (2026). https://doi.org/10.1038/s41418-026-01725-6
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41418-026-01725-6
