
Abstract
The transition of embryonic stem cells (ESCs) from a pluripotent state to lineage commitment is governed by complex regulatory mechanisms, including chromatin remodeling, as well as transcriptional and post-transcriptional processes. Recent studies have emphasized the interplay between these mechanisms, revealing intricate, multilayered regulatory networks that require further elucidation. In this study, we reveal a new connection between the RNA-binding protein LIN28A and the epigenetic regulation of ESC differentiation. LIN28A is upregulated during the early stages of neural commitment and undergoes a shift in subcellular localization from the nucleus to the cytoplasm upon differentiation. Generation and analysis of Lin28a knockout (KO) ESCs revealed that, although these cells can self-renew, they exhibit a pronounced defect in differentiating into neural precursors. However, mesodermal and endodermal differentiation proceeds normally in Lin28a KO cells, suggesting a neuronal-specific function for LIN28A. Proteomic analyses revealed a dynamic, context-dependent LIN28A interactome, with distinct sets of putative interacting partners in ESCs compared to those in differentiating cells. Among the ESC-specific putative interactors, we validated an RNA-dependent association of LIN28A with components of Polycomb Repressive Complex 2 (PRC2), a key chromatin-modifying complex that deposits the repressive histone modification H3K27me3. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) demonstrated that loss of LIN28A results in persistent PRC2 occupancy at the promoters of developmental genes, accompanied by partial uncoupling between PRC2 binding and H3K27me3 deposition. Lin28a KO causes differentiation defects that are not rescued by pharmacological inhibition of PRC2 enzymatic activity, suggesting that LIN28A regulates PRC2 chromatin dynamics independently of H3K27me3 deposition. Furthermore, we identified an interaction between LIN28A and the long non-coding RNA Neat1, which may serve as a scaffold facilitating PRC2 eviction from chromatin. Taken together, our findings reveal a previously unrecognized role for LIN28A in regulating PRC2-mediated chromatin dynamics and underscore its importance in epigenetic control of neuronal differentiation.

Similar content being viewed by others


Introduction
Embryonic stem cells (ESCs), derived from the inner cell mass of the blastocyst, have the unique ability to self-renew and differentiate into all cell types comprising the three germ layers during development [1]. These properties have traditionally been attributed to transcription factors such as octamer-binding transcription factor 4 (OCT4), sex-determining region Y-box 2 (SOX2), and the homeodomain-containing protein NANOG, as well as chromatin-related factors [1]. However, post-transcriptional regulation by RNA-binding proteins (RBPs) has recently emerged as an additional key mechanism controlling ESC pluripotency and early differentiation [2,3,4]. Among RBPs, LIN28A stands out for its crucial role in regulating developmental timing, cellular proliferation, differentiation, stem cell pluripotency, and metabolism [5,6,7,8,9,10,11,12,13]. Initially, LIN28A was identified as a negative regulator of the processing of precursor let-7 microRNAs (pre-let-7) into mature miRNAs [2]. Subsequent studies revealed that LIN28A binds to thousands of RNA transcripts, thereby influencing their stability and translation efficiency [5,6,7,8,9].
Mouse Lin28a is highly expressed during embryogenesis and absent in most adult tissues [10]. In vitro, Lin28a is present in the naïve state of ESCs, which resembles the pre-implantation embryo, and its levels increase in the primed state, which mimics the post-implantation epiblast [6]. The loss of both Lin28a and its paralog Lin28b traps pluripotent stem cells (PSCs) in a more naïve state due to their function in the translation of metabolic enzymes [11]. Our group has demonstrated that, during the transition of ESCs into epiblast-like cells (EpiLCs), LIN28A limits the translation of the architectural factor Hmga2 while enhancing that of the epigenetic regulator Dnmt3a, facilitating the proper exit of ESCs from the naïve state [6, 7]. LIN28A also plays an important role in establishing pluripotency [11, 12]. Remarkably, LIN28A is the only known RBP that, in combination with the Yamanaka factors (OCT4, SOX2, KLF4, and c-MYC), enhances the efficiency of reprogramming from somatic cells into induced pluripotent stem cells (iPSCs) [12]. LIN28A achieves this by repressing the translation of mRNAs encoding proteins involved in oxidative phosphorylation, thereby conferring to somatic cells the metabolic characteristics of PSCs [11].
We and others have observed that the effects of LIN28A depend not only on the RNAs it binds but also on its interactions with specific protein partners [5, 7,8,9, 13]. Therefore, in this study, we analyzed the interactome of LIN28A in undifferentiated ESCs and ESC-derived neural precursors. We found limited overlap among LIN28A’s putative interactors, suggesting different protein functions in undifferentiated and differentiated cells. Among the ESC-specific LIN28A interactors, we found subunits of the Polycomb repressive complex 2 (PRC2), a chromatin repressor complex responsible for trimethylating histone H3 at lysine 27 (H3K27me3) [14]. The targeting of PRC2 to chromatin and the regulation of its activity are very complex processes that are still under investigation. Numerous studies have implicated RNA in the recruitment/eviction of PRCs from chromatin to regulate gene expression during development and differentiation [15,16,17,18]. Here, we show that LIN28A binds to PRC2 in an RNA-dependent manner, and its absence leads to persistent PRC2 chromatin occupancy and repression of developmental genes. Additionally, LIN28A associates with the long non-coding RNA Neat1, potentially facilitating PRC2 eviction from chromatin, thereby uncovering a novel mechanism by which LIN28A modulates chromatin dynamics during neural differentiation.
Results
Lin28a relocalizes during ESC differentiation
Mouse Lin28a was previously described by us and other groups as being expressed in naïve ESCs, with increased expression during the transition into EpiLCs (Fig. 1A) [6, 19]. However, it remains unclear whether Lin28a expression is also regulated during the formation of the three germ layers. To investigate this, we examined the expression profile of Lin28a during ESC differentiation into the ectoderm, mesoderm, and endoderm lineages. To model neuronal differentiation, we employed the previously described serum-free embryoid body (SFEB) method, which is based on the aggregation of ESCs in a chemically defined, serum-free medium without adding specific inducing factors [6, 20,21,22]. The SFEB method predominantly promotes neuroectodermal differentiation leading to the generation of post-mitotic neurons through an intermediate population of Sox1-positive neural precursors (Supplementary Fig. 1A, B), with minimal mesodermal or endodermal contribution (Supplementary Fig. 1C), differently from the classical embryoid bodies (EBs) [23]. After 4 days of differentiation, only a small fraction of cells retains expression of pluripotency markers (Supplementary Fig. 1A). Under these conditions, we observed that LIN28A expression peaks on day four of differentiation (Fig. 1A and Supplementary Fig. 1D), coinciding with the stage at which SFEBs are largely composed of Sox1-positive neural precursors (Supplementary Fig. 1A). LIN28A levels then rapidly decline as differentiation progresses into neurons (12 days of differentiation) (Fig. 1A and Supplementary Fig. 1D). In contrast, during mesodermal differentiation using the hanging drop method to generate EBs (Fig. 1B and Supplementary Fig. 1E) and during endodermal differentiation (Fig. 1C and Supplementary Fig. 1F), LIN28A expression progressively decreases (Supplementary Fig. 1G). A recent study demonstrated that LIN28A exhibits dynamic temporal and spatial expression during murine embryo development [24]. Therefore, we explored the possibility that the subcellular distribution of LIN28A also changes between undifferentiated and differentiated ESCs. Immunofluorescence analysis of undifferentiated cells showed that LIN28A is predominantly localized in the nucleus, with a detectable cytoplasmic signal present as well. The nuclear localization displayed an apparent nucleolar pattern (Fig. 1D and Supplementary Fig. 1H), consistent with prior reports [8, 24]. However, this pattern reverted upon differentiation, with LIN28A predominantly localized to the cytoplasm in SFEBs (Fig. 1D and Supplementary Fig. 1H). To validate these observations, we performed cell fractionation and confirmed that LIN28A protein levels in the nucleus and cytoplasm were inversely correlated as ESCs were induced to differentiate (Fig. 1E). Together, these findings suggest a possible role for LIN28A in neural lineage formation and highlight its dynamic regulation during differentiation.

Expression profile of LIN28A in mouse ESCs induced to differentiate into EpiLCs (2 days of differentiation), neural precursors (SFEBs, 4 days of differentiation), neurons (neur. diff., 12 days of differentiation) (A), and mesodermal (4 and 12 days of differentiation) (B) and endodermal (4 and 8 days of differentiation) (C) lineages. The quantification of three independent differentiation experiments is reported in Supplementary Fig. 1D, G. The graphs under the blots represent the relative mRNA expression (using real-time RT-PCR) of the following specific markers: Oct4 for ESCs (also used as a marker for EpilCs), Sox1 for neural precursors, Map2 for neurons, Mlc2v and Foxa2 for mesodermal and endodermal derivatives, respectively. Data are expressed as the mean ± SD of three biological replicates (n = 3), relative to the expression levels in ESCs (Oct4), SFEBs (Sox1), 12 days of neural differentiation (Map2), 12 days of mesodermal differentiation (Mlc2v), or 8 days of endodermal differentiation (Foxa2). Asterisks represent statistical significance based on the Student’s t test (*p ≤ 0.05, **p ≤ 0.005). D Localization of endogenous LIN28A by immunofluorescence in undifferentiated ESCs and sectioned SFEBs. Oct4 and Sox1 were used as markers of undifferentiated ESCs and neural precursors, respectively. Scale bars: 50 μm. DAPI was used to counterstain the nuclei. Magnified images of cropped areas of LIN28A staining are shown. E Cell fractionation followed by western blot was performed to evaluate the distribution of LIN28A in cell compartments of undifferentiated ESCs and SFEBs. The graph shows the mean of LIN28A levels ± SD relative to the quantity measured in the total lysate and normalized against GAPDH for the cytoplasmic fraction (CF) and against histone H3 for the total nuclear fraction (NF). n = 5 biological replicates, *p ≤ 0.05 (Student’s t test).
Lin28a KO impairs neuronal differentiation
LIN28A has been reported to significantly improve the establishment of a pluripotent state during somatic cell reprogramming [12]. However, its expression and localization change dynamically during ESC differentiation, suggesting a role in the establishment of the neuronal lineage. To investigate this possibility, we generated Lin28a KO ESCs using CRISPR-Cas9 technology with two different single guide RNAs (sgRNAs) (Supplementary Fig. 2A). Western blot and immunofluorescence analyses confirmed LIN28A protein depletion in both KO ESCs and SFEBs (Supplementary Fig. 2B, C) and viability assay (Supplementary Fig. 2D) showed comparable behaviors between wild type (WT) and Lin28a KO undifferentiated and differentiated cells. Next, we analyzed whether LIN28A depletion affects ESC maintenance or differentiation potential. To evaluate the impact of LIN28A loss on ESC maintenance, we examined the ability of undifferentiated ESCs to form alkaline phosphatase (AP)-positive colonies and observed no differences between WT and Lin28a KO cells (Supplementary Fig. 3A). Additionally, the percentage of undifferentiated ESCs expressing pluripotency markers was comparable between WT and KO cells (Supplementary Fig. 3B). Finally, qPCR and western blot analyses over multiple passages revealed similar levels of pluripotency factors in WT and KO cells (Supplementary Fig. 3C, D). Altogether, these results indicate that Lin28a depletion does not significantly impact the maintenance of undifferentiated ESCs. However, when we induced differentiation into SFEBs, Lin28a KO cells displayed impaired neural differentiation, as revealed by qPCR and western blot analyses showing a failure to upregulate neural differentiation markers (Sox1 and Pax6) and persistent expression of pluripotency markers (Oct4 and Nanog) (Fig. 2A, B). Consistently, immunostaining showed that less than half of the Lin28a KO SFEB cells were Sox1-positive (~30%) compared to more than 80% of the WT cells. Conversely, a higher proportion ( ~ 60%) of cells within KO SFEBs retained expression of Oct4 and Nanog (Fig. 2C). Overall, these data indicate that Lin28a KO ESCs are unable to properly leave the undifferentiated state to develop into neural precursors.

A The expression levels of pluripotency (Oct4 and Nanog) and neural differentiation (Sox1 and Pax6) markers were evaluated by qPCR in WT and Lin28a KO SFEBs. The bars represent the mean ± SD relative to the expression levels in WT SFEBs. n ≥ 3 biological replicates, *p ≤ 0.05, **p ≤ 0.005 (Student’s t test). B Western blot analysis of pluripotency and differentiation markers in WT and Lin28a KO undifferentiated ESCs and SFEBs. The graph shows the relative intensity ± SD of the indicated proteins normalized for GAPDH in WT and Lin28a KO SFEBs. Quantification of OCT4 and NANOG in undifferentiated ESCs is reported in Supplementary Fig. 3D. n = 4 biological replicates, *p ≤ 0.05, **p ≤ 0.005 (Student’s t test). C Immunofluorescence analysis of pluripotency (Oct4 and Nanog) and neural differentiation (Sox1) markers in sectioned SFEBs obtained from WT and Lin28a KO ESCs. DAPI was used to counterstain the nuclei. Scale bars: 50 μm. The graph shows the number of positive cells ± SD counting >400/cells across 30 fields from three independent biological replicates. Two-tailed, unpaired Student’s t test: ***p ≤ 0.0001. D WT and Lin28a KO cells were transfected with a vector encoding a FLAG-tagged form of LIN28A or an empty vector (Mock) and differentiation into neural precursors was evaluated by western blot analysis using antibodies against the pluripotency (Oct4 and Nanog) and the neural (Sox1) markers. The antibody against LIN28A shows the presence of both the endogenous protein (white arrow) and the FLAG-tagged form (black arrow). The graphs show the mean of the relative quantitation ± SD from three independent biological replicates. *p ≤ 0.05; **p ≤ 0.0001 (Student’s t test).
To assess whether Lin28b, the paralog of Lin28a, contributes to the observed phenotype in Lin28a KO cells during neuronal differentiation, we first compared the expression profiles of Lin28a and b in ESCs and SFEBs and we found that the expression of Lin28b parallels that of Lin28a (Supplementary Fig. 3E). Next, we analyzed whether Lin28b expression changes in response to Lin28a loss. We found that Lin28b levels remained unchanged in Lin28a KO ESCs and SFEBs compared to wild-type controls (Supplementary Fig. 3F). These findings suggest that Lin28b does not compensate for the loss of Lin28a and is unlikely to contribute to the Lin28a KO phenotype.
To confirm that the observed phenotype is specifically due to Lin28a, we transfected a FLAG-tagged Lin28a construct in both WT and KO cells and induced differentiation. We found that the expression of exogenous Lin28a leads to downregulation of the pluripotency markers (Oct4 and Nanog) and upregulation of the neural differentiation marker Sox1 in KO SFEBs (Fig. 2D and Supplementary Fig. 4A). To assess this effect at the cellular level, we performed an immunofluorescence analysis on SFEBs and found that approximately 60% of cells in Lin28a KO SFEBs became Sox1-positive upon the ectopic expression of Lin28a (Supplementary Fig. 4B). These rescue experiments confirmed that the impaired differentiation phenotype is a specific consequence of Lin28a deficiency.
To evaluate whether the differentiation defect in Lin28a KO cells is restricted to the neuroectodermal lineage or reflects a broader impairment, we analyzed the ability of KO cells to differentiate into mesodermal and endodermal lineages. These analyses demonstrated that Lin28a KO ESCs differentiated into both lineages to the same extent as WT cells (Supplementary Fig. 4C, D).
These findings, together with the prominent expression of Lin28a in neural precursors, demonstrated that this protein is required for proper neural lineage development, while being dispensable for mesendodermal lineage formation.
LIN28A interacts with PRC2 in undifferentiated ESCs
To elucidate the mechanistic role of LIN28A in the regulation of ESC fate, we analyzed its interactome in both undifferentiated and differentiated cells. For this purpose, we expressed FLAG-tagged LIN28A in ESCs and induced their differentiation into SFEBs. Then, we affinity-purified lysates from both ESCs and SFEBs using anti-FLAG beads and then employed a gel-based proteomic approach to identify LIN28A protein interactors. To obtain a high-confidence list of putative LIN28A interactors in ESCs, we subtracted all proteins identified in the corresponding mock control immunoprecipitation from those detected in LIN28A-FLAG sample. We applied the same approach to the SFEB samples (Supplementary Fig. 5A). We identified 816 and 364 proteins as LIN28A putative interactors in ESCs and SFEBs, respectively (Dataset 1). The reliability of this proteomic analysis is supported by the minimal presence of potential nonspecific targets, as assessed using the CRAPome database [25] (Supplementary Table 1). Comparing our combined dataset with those of previously reported LIN28A interactomes from other stem cell types (mouse EpiLCs), their derivatives (neural precursors derived from human iPSCs), and public repositories such as BioGRID and IntAct [7, 13, 26, 27] (Supplementary Table 1) revealed that 68% of the putative LIN28A interactors identified in our study were unique. This finding indicates that LIN28A engages in context-dependent interactomes. Gene ontology (GO) analysis of these putative LIN28A-binding partners showed that, in agreement with previously published results, the most enriched biological processes were related to RNA processing, RNA metabolism and translation (Fig. 3A) [5,6,7, 9, 11, 28, 29]. On the other hand, GO terms in cellular component analysis showed that putative LIN28A interactors in ESCs are mainly localized in the nucleus, particularly in the nucleolus and nucleoplasm (Fig. 3B). Conversely, LIN28A interactors in SFEBs were primarily localized to the cytoplasm, cytosol, and mitochondria (Fig. 3B), consistent with LIN28A’s subcellular distribution during differentiation (Fig. 1D, E). Moreover, a comparison of LIN28A interactomes across the two states revealed that 666 were specific for ESCs and 227 were unique to SFEBs, while only 135 were shared (Fig. 3C). Interestingly, this indicates that LIN28A forms distinct molecular complexes in undifferentiated versus differentiated states, most likely exerting different functions therein. Next, we constructed a protein–protein interaction (PPI) network using STRING database to further interpret the proteomics results. The putative LIN28A interactors common to both ESCs and SFEBs formed a small cluster associated with mitochondrial gene expression and translation, suggesting a previously unrecognized role for LIN28A in mitochondrial translation (Supplementary Fig. 5B). In contrast, STRING analysis of SFEB-specific interactors did not reveal an extensive interaction network (Supplementary Fig. 5C). Conversely, the ESC-specific interactome formed a large, complex network with distinct subclusters related to chromatin organization and remodeling, comprising 88 and 28 entries, respectively (Supplementary Fig. 5D and Fig. 3D). Notably, this subset of LIN28A putative interactors included the core PRC2 components EED (embryonic ectoderm development) and EZH2 (enhancer of zeste 2), as well as the PRC2 accessory proteins AEBP2 (adipocyte enhancer-binding protein 2) and JARID2 (Jumonji and AT-rich interaction domain 2). PRC2 is well known for its central role in ESC differentiation, regulating developmental genes through epigenetic modifications and chromatin organization [30, 31]. Given that Lin28a KO ESCs display pronounced differentiation defects, we hypothesized that a potential interaction between LIN28A and PRC2 could clarify the role of LIN28A in ESC differentiation. To validate the putative interaction between LIN28A and PRC2, we performed co-immunoprecipitation (co-IP) assays using lysates from ESCs ectopically expressing FLAG-tagged LIN28A and analyzed the immunoprecipitates by western blot to detect PRC2 subunits. Consistent with the proteomic data, the PRC2 components EZH2 and JARID2 co-immunopurified with FLAG-LIN28A (Fig. 3E). Furthermore, although the PRC2 subunit SUZ12 (suppressor of zeste 12) was initially filtered out due to the cutoff threshold applied in the proteomic analysis, it was successfully validated in the co-IP assay (Fig. 3E). The presence of JARID2 and AEBP2 suggests that LIN28A is associated with the PRC2.2 variant, thus we investigated whether LIN28A interacts with PRC2.1 using the same co-IP experiments. However, LIN28A immunoprecipitates did not contain the Polycomb-like protein MTF2 (Fig. 3E), which functions as PRC2.1 counterpart to JARID2 in recruiting PRC2 to chromatin [32]. This finding is consistent with our proteomic analysis. To further support the interaction between LIN28A and PRC2, we performed immunoprecipitation using EZH2 antibody in WT ESCs and found that endogenous LIN28A co-immunoprecipitates with EZH2 (Supplementary Fig. 5E). Additionally, a proximity ligation assay (PLA) revealed in situ interactions between LIN28A and the same PRC2 components (Fig. 3F, G and Supplementary Fig. 5F). Indeed, prominently higher levels of PLA-positive spots were observed between LIN28A and PRC proteins compared to P-Smad1/5, which was used as a negative control since it was not detected through proteomic experiments (Fig. 3F, G and Supplementary Fig. 5F).

A Metascape-based enrichment analysis of the biological processes of LIN28A interactors identified in ESCs or SFEBs. B FunRich-based enrichment analysis of the cellular components of the LIN28A interactors identified in ESCs or SFEBs. C Venn diagram showing the large number of unique LIN28A interactors identified in ESCs and SFEBs, as well as the common proteins. D STRING analysis of proteins related to chromatin remodeling from LIN28A-interacting components uniquely identified in ESCs (medium-confidence, strength 0.53, False discovery rate 1.30 e-10). E Western blot analysis of LIN28A immunoprecipitates to assess its interaction with PRC2 core subunits (EZH2 and SUZ12) and accessory subunits (JARID2 for PRC2.2 and MTF2 for PRC2.1). Immunoprecipitation was performed using anti-FLAG beads in ESCs transfected with the LIN28A-FLAG expressing vector or the empty (Mock) vector. F, G Proximity ligation assay (PLA) to assess the interaction of LIN28A with PRC2. ESCs expressing LIN28A-FLAG were analyzed by PLA coupling the anti-FLAG antibody with the following antibodies: anti-P-SMAD1/5 (as negative control), anti-EZH2, anti-SUZ12 and anti-JARID2. Quantification of positive spots/cell (from >480 cells) from three independent (n = 3) biological replicates is shown in the graph (F). In the corresponding images, PLA positive spots are visible in green (G). Controls of single antibodies are shown in Supplementary Fig. 5F. Scale bars: 10 µm. Bars represent SD. Wilcoxon test: ***p ≤ 0.0001.
Given that the main function of PRC2 is to deposit trimethylation on lysine 27 of histone H3 (H3K27me3) [31], and with the aim of understanding whether LIN28A influences PRC2 activity, we evaluated this epigenetic mark in WT and Lin28a KO cells. We found an accumulation of H3K27me3 in undifferentiated KO ESCs compared to WT cells, a pattern that persisted in differentiated cells (Fig. 4A). Despite comparable EZH2 expression levels in WT and KO cells (Supplementary Fig. 6A, B), we hypothesized that the increase in H3K27me3 deposition observed in Lin28a KO cells might be due to changes in protein activity or distribution on chromatin. ESCs are characterized by a high presence of bivalent promoters decorated with both the activating mark H3K4me3 and the repressive mark H3K27me3 [33]. To investigate whether the H3K4me3 mark parallels the accumulation of the H3K27me3 counterpart in Lin28a KO ESCs, we performed western blot analysis, but no significant differences in H3K4me3 levels were observed between WT and KO cells (Fig. 4B). Next, since trimethylation of H3K27 follows ubiquitination of H2AK119, a process catalyzed by Polycomb Repressive Complex 1 (PRC1) [31], we analyzed whether the increase in H3K27me3 observed in Lin28 KO cells was in turn associated with PRC1 hyperactivity. We observed no changes in H2AK119ub mark between Lin28a KO and WT ESCs (Fig. 4B), further suggesting that Lin28a depletion specifically affects PRC2 activity. To determine whether Lin28a KO ESCs are also characterized by the alteration of other epigenetic marks, we analyzed other histone modifications. As shown in Fig. 4B, none of the other analyzed epigenetic marks were altered in the absence of LIN28A. To determine whether re-expression of LIN28A can restore normal H3K27me3 levels, we transfected exogenous LIN28A-FLAG into WT and Lin28a KO cells and assessed H3K27me3 levels in undifferentiated and differentiated ESCs. As shown in Fig. 4C, re-expression of LIN28A had no apparent effect on H3K27me3 levels in undifferentiated KO cells. However, in SFEBs, exogenous LIN28A reduced H3K27me3 deposition to levels comparable with those in WT cells. Taken together, these results indicate that the absence of Lin28a alters the level of the PRC2 specific mark H3K27me3, rather than causing a general change in the epigenetic state. This points to PRC2 as a likely culprit in the inability of Lin28a KO cells to properly differentiate. PRC2 is well known for its role in establishing cell-type-specific gene expression programs that enable differentiation. Therefore, since Lin28a KO cells are unable to differentiate properly and show high H3K27me3 levels, we asked whether the inhibition of PRC2 enzymatic activity might rescue the Lin28a KO cell phenotype. To explore this hypothesis, we treated WT and KO differentiating cells with GSK126, a highly selective inhibitor of EZH2 methyltransferase. Analysis of pluripotency and differentiation markers revealed that the reduction in H3K27me3 deposition induced by GSK126 led to only modest downregulation of the pluripotency marker Oct4, while the activation of the neuronal marker Sox1 remained impaired (Fig. 4D). Immunofluorescence confirmed the Western blot results and revealed that the partial downregulation of Oct4 in Lin28a KO SFEBs treated with GSK126 corresponds mainly to reduced Oct4 expression in a subset of cells (38% ± 15% of cells showing a faint Oct4 signal in 60% of the analyzed SFEBs), rather than a substantial decrease in the overall number of undifferentiated cells (Fig. 4E). These findings indicate that reduced H3K27me3 deposition alone is insufficient to rescue the differentiation defects of Lin28a KO cells, implying that the differentiation block in Lin28a KO ESCs involves mechanisms beyond PRC2 enzymatic activity.

A Western blot analysis and relative quantification of the H3K27me3 histone mark in WT and Lin28a KO cells in both the undifferentiated and differentiated states. Histone H3 and GAPDH were used as loading control. The use of an antibody against LIN28A confirmed the absence of protein in Lin28a KO cells. The graph shows the mean of the relative quantification ± SD of from six independent biological replicates (n = 6). **p < 0.005 (Student’s t test). B Western blot analysis and relative quantification of different histone marks in WT and Lin28a KO cells. Histone H3 and GAPDH were used as loading controls. The graph shows the mean of the relative quantification ± SD for six biological replicates of H3K4me3 and three biological replicates of H2AK119ub, H3K9me3 and H3K27ac. Student’s t test: ns (not significant). C Western blot analysis and relative quantification of H3K27me3 levels in WT and KO ESCs and SFEBs upon ectopic expression of LIN28A. WT and Lin28a KO cells were transfected with the vector encoding a FLAG-tagged form of LIN28A or the empty vector (Mock) and then differentiated into SFEBs. The LIN28A antibody detects both the endogenous protein (white arrow) and the FLAG-tagged form (black arrow). GAPDH and histone H3 were used as loading controls. The graphs report the mean of the relative quantitation ± SD of four independent (n = 4) biological replicates. *p ≤ 0.05; **p ≤ 0.005 (Student’s t test). D Western blot analysis and relative quantification of pluripotency (OCT4) and differentiation (SOX1) markers in WT and Lin28a KO SFEBs treated with the PRC2 inhibitor, GSK126 during ESCs differentiation toward the neural lineage through SFEB formation (4 days of differentiation). GAPDH and histone H3 were used as loading controls. n = 3 biological replicates. Mean ± SD is reported in the graph. *p < 0.05 (Student’s t test). E Immunofluorescence analysis of whole WT and Lin28a KO SFEBs treated or untreated (NT) with the PRC2 inhibitor GSK126. WT and Lin28a KO ESCs were induced to differentiate into SFEBs for 4 days in the presence or absence of GSK126. Staining for the pluripotency marker OCT4 and the neural differentiation marker SOX1 is shown. Images were generated using the Maximum Projection function (see “Methods”). The graph shows the number of positive cells (mean ± SD) quantified from 50 SFEBs across three independent biological replicates. Statistical significance was assessed using a two-tailed, unpaired Student’s t-test: *p ≤ 0.05. ***p ≤ 0.0001.
LIN28A can modulate PRC2 binding to chromatin during differentiation
Changes in PRC2 enzymatic activity alone do not fully explain the impaired differentiation of Lin28a KO ESCs into neural precursors. Therefore, we hypothesized that LIN28A modulates the previously described dynamic balance between PRC2 chromatin occupancy and its RNA interactions [15, 17, 34, 35]. To test this hypothesis, we evaluated the chromatin- associated fraction of PRC2 in WT and Lin28a KO ESCs and differentiating SFEBs. We found that the fraction of EZH2 associated with chromatin is higher in Lin28a KO cells compared to WT (Fig. 5A, B). This suggests that LIN28A, possibly through its RNA-binding capacity, could compete with EZH2 and facilitate its eviction from chromatin during differentiation. Previous studies have reported that PRC2 is recruited to and displaced from Polycomb target genes during ESC differentiation [36]. Based on this, we hypothesized that LIN28A might play a role in this dynamic during ESC differentiation. To investigate this hypothesis, we performed chromatin immunoprecipitation for EZH2, followed by deep sequencing (ChIP-seq), in undifferentiated and differentiating cells (2 days of SFEB differentiation). This allowed us to evaluate the association and distribution of the PRC2 catalytic subunit to specific genomic loci in the presence or absence of LIN28A. We identified 4859 ChIP-seq peaks in WT ESCs and 2,516 in Lin28a KO ESCs (Fig. 5C). In differentiating cells, PRC2 occupancy increased in Lin28a KO cells compared to their WT (6338 versus 4495 EZH2 peaks) (Fig. 5C). The majority (≥75%) of EZH2-binding peaks localized to gene promoters (within 1000 bp from transcriptional start site, TSS and transcriptional end site, TES), with EZH2 signal enriched at TSSs under all analyzed conditions (Fig. 5D). Meta-gene profiling revealed increased EZH2 enrichment in KO SFEBs compared to their WT counterpart, whereas undifferentiated ESC KO and WT cells displayed similar profiles (Supplementary Fig. 6C). Next, we associated EZH2 peaks with the nearest gene whose TSS lies within ± 1000 bp (Dataset 2). To validate our ChIP-seq data, we compared the EZH2 target genes identified in WT ESCs and SFEBs with those in previously published datasets [37] in ESCs and neural progenitor cells (NPCs), which are comparable to SFEBs. We found that nearly all of our target genes were also present in the Kundu et al. [37] dataset (>98% for ESCs/ESCs and > 70% for SFEBs/NPCs) (Supplementary Fig. 6D). Next, we examined the overlap of EZH2 target genes between WT and Lin28 KO cells in both undifferentiated and differentiated states. We identified 1,866 EZH2-bound genes common to all four conditions (Fig. 5E). Intensity plots revealed increased EZH2 enrichment at these shared targets specifically in Lin28a KO SFEBs (Fig. 5F). In ESCs, nearly all EZH2-bound genes in KO cells were also bound in WT cells. However, approximately one-third of the EZH2 targets identified in WT ESCs were absent in KO ESCs (Fig. 5E). Conversely, in differentiating cells approximately 30% of EZH2-bound genes (1214 out of 4224) were unique to KO cells (Fig. 5E). When examining dynamic changes in EZH2 occupancy during differentiation, we observed that 83% of EZH2 targets in WT ESCs were retained in SFEBs, while 17% (555/3225) lost EZH2 binding, and 226 new targets were acquired (Fig. 5E). By contrast, Lin28a KO cells retained nearly all EZH2 targets during differentiation (1959/1981; 99%), lost only 1% (22/1981), and gained 2084 new EZH2-bound promoters upon SFEB differentiation. (Fig. 5E). Strikingly, 90% (1146/1260) of EZH2 targets unique to WT ESCs became bound by EZH2 in KO SFEBs (Supplementary Fig. 6E), indicating massive recruitment to chromatin. These newly acquired targets were strongly enriched for genes involved in developmental processes (Supplementary Fig. 6F), consistent with a wave of transcriptional repression in KO SFEBs that correlates with impaired differentiation. Moreover, GO enrichment analysis of the 1214 EZH2 targets found in Lin28a KO SFEBs but not in WT SFEBs revealed significant enrichment of genes connected to developmental processes including the development of the nervous system and neurogenesis (Fig. 5G). Many of these developmental genes, which are normally transcriptionally activated in differentiating SFEBs, remained repressed in KO cells, coinciding with persistent EZH2 occupancy at their promoters. These observations are supported by the behavior of developmental genes that are also known PRC2 targets [17, 38] and fall within the subset common to all conditions, i.e., occupied by Ezh2, that resulted always repressed (Supplementary Fig. 6G, H). When we analyzed the expression profiles of genes involved in neuronal differentiation that acquired (Cbx2 and Tead3), maintained (Sall2) or are enriched (Pax6) for Ezh2 occupancy in Lin28a KO cells, we found significant transcriptional repression compared to WT (Fig. 5H, I), consistent with the impairment of neuronal differentiation described above.

A Western blot analysis of EZH2 in WT and Lin28a KO ESCs after cell fractionation. The total nuclear fraction is indicated as “nucleus” (both chromatin and non-chromatin bound) and chromatin fraction. n = 4 biological replicates. B Western blot analysis of EZH2 in the total nuclear and chromatin fractions of WT and Lin28a KO cells after 2 days of SFEB differentiation. n = 5 biological replicates. In both (A, B) GAPDH and H3 were used as loading control. Emerin was used as a nuclear marker. The graphs represent the levels of EZH2 and H3K27me3 normalized to H3 signal (mean ± SD) in the relative fractions. Student’s t test: *p < 0.05, ns not significant. C Bar-plot showing the number of peaks detected in each cell line tested by analyzing ChIP-seq NGS experiments with MACS2 software. For each condition, only peaks detected in at least 2 out of the 3 biological replicates were reported. The analysis detected 2516 peaks in Lin28a KO ESCs, 4859 in WT ESCs, 6338 peaks in Lin28a KO SFEBs, and 4495 peaks in WT SFEBs. D Bar-plot reporting the percentage of peak distribution at promoters, genic or intergenic regions. (Promoter: peaks within ±1000 bp from the TSS; genic: peaks outside the promoter region but within a gene, intergenic outside annotated genes). E Venn diagrams showing the overlap of the genes mapped to detected peaks (±1000 bp) for EZH2 in all analyzed conditions (complete list in Dataset 2). F Heatmap showing EZH2 ChIP-seq signals (RPKM) at 1866 common target genes among the WT and Lin28a KO ESCs and SFEBs. G Diagram showing the GO terms for the 1214 genes enriched for EZH2 peaks specifically in Lin28a KO SFEBs compared to their WT counterparts (Benjamini Hochberg corrected p-value ≤0.05). The top 30 significantly enriched biological processes and the relative p-value are reported. H EZH2 ChIP-seq tracks at the promoters of differentiation genes. The plots highlight the binding fingerprint of EZH2 at the promoters of selected genes (Cbx2, Tead3, Sall2 and Pax6), as well as the pluripotency gene Nanog (as control). The intensity of the peaks is reported as reads per million. I Expression profile of differentiation genes on which EZH2 occupancy was enriched at promoters specifically in Lin28a KO SFEBs compared to the WT counterpart. The graphs show the expression levels (mean ± SD) at three time-points: undifferentiated ESCs (t0), 2 and 4 days after the induction of differentiation through SFEBs. Nanog was used as control. n = 3 biological replicates, *p ≤ 0.05, **p ≤ 0.01 (Student’s t test). J Bar plot showing the number of peaks detected in each cell line tested by analyzing ChIP-seq NGS experiments with MACS2 software. For each condition, only peaks detected in at least 2 out of the 3 biological replicates analyzed were reported. The analysis detected 113,921 peaks in Lin28a KO ESCs, 115,789 in WT ESCs, 143,043 peaks in Lin28a KO SFEBs, and 142,907 peaks in WT SFEBs. K Venn diagrams showing the overlap of genes mapped to the detected peaks (±1000 bp) in H3K27me3 ChIP-seq (complete list in Dataset 3) and EZH2 analyses in WT and Lin28a KO ESCs and SFEBs.
To distinguish EZH2 chromatin occupancy from its enzymatic activity, we performed ChIP-seq for H3K27me3 in WT and Lin28a KO ESCs, as well as in differentiating SFEBs (2 days). WT and KO cells exhibited comparable numbers of global H3K27me3 peaks in both undifferentiated and differentiated states (Fig. 5J). The higher overall number of H3K27me3 peaks relative to EZH2 binding sites is consistent with previous findings showing that PRC2 occupies a limited number of loci, while H3K27me3 can spread more broadly and can be deposited independently of stable PRC2 binding [39, 40]. Mapping H3K27me3 peaks to gene promoters revealed substantial overlap with published ESC and NPC datasets [37] (>76% for ESCs/ESCs and > 56% for SFEBs/NPCs) (Supplementary Fig. 7A). Next, we analyzed the overlap of H3K27me3-marked promoters between WT and KO cells. In ESCs, 920 promoters were unique to KO, 1,623 were unique to WT cells, and 3,500 were shared (Supplementary Fig. 7B). While this pattern mirrored EZH2 occupancy trends (Fig. 5E), the differences were less pronounced at the level of H3K27me3. Notably, a disproportionately larger number of promoters were marked only in KO ESCs by H3K27me3 (920), compared to the corresponding subset marked by EZH2 (16), suggesting differential dynamics between binding and enzymatic activity. In SFEBs, a different trend was observed, more in line with EZH2 data: 1,479 promoters were marked only in KO cells, 1144 only in WT cells, and 5150 were shared (Supplementary Fig. 7B). Analysis of H3K27me3 dynamics during differentiation revealed similar proportions of promoters that lost and retained the mark (16% in WT vs. 14% in KO and 84% in WT vs. 86% in KO, respectively) (Supplementary Fig. 7B). However, Lin28a KO SFEBs acquired a significantly higher number of new H3K27me3-marked promoters upon differentiation (2816/6629; 42%) compared to WT cells (1970/6294; 31%), consistent with the global accumulation of H3K27me3 observed by western blot (Fig. 4A) and the increased EZH2 occupancy (Fig. 5E). Then, we intersected the targets of EZH2 with the promoters marked by K27me3 (Fig. 5K) and we found that a subset of EZH2 targets lacked H3K27me3 across all conditions. This effect was slightly more pronounced in KO cells: 46% of EZH2 targets in KO ESCs and 31% in KO SFEBs lacked H3K27me3, compared to 37% and 26%, respectively, in WT cells. Importantly, among the 2084 promoters to which EZH2 was specifically recruited in KO SFEBs, 60% (1259) were also marked by H3K27me3 (Supplementary Fig. 7C), indicating that local PRC2 enzymatic activity accompanied EZH2 binding at many of these sites.
However, a substantial proportion of EZH2-bound promoters in KO SFEBs remained unmarked by H3K27me3. Specifically, 567 out of 1214 EZH2 targets unique to KO SFEBs compared to the WT counterpart lacked the modification (Fig. 5K). Notably, 92% of these unmarked targets fell into the category of promoters bound exclusively by EZH2 in KO SFEBs. This category includes key developmental regulators, such as Sall2, Tead3, and Cbx2 (Supplementary Fig. 7D), that were transcriptionally repressed in KO SFEBs (Fig. 5I). These observations support the idea that EZH2 occupancy in ESCs is dysregulated and can be responsible for the phenotype of KO cells in a manner that is not necessarily constrained to K27me3 deposition.
Taken together, our results demonstrate that in Lin28a KO cells, EZH2 fails to undergo proper eviction and is aberrantly recruited to many developmental gene promoters during differentiation. This disrupts the normal recruitment–eviction balance and contributes to gene repression. The incomplete correspondence between EZH2 binding and H3K27me3 deposition, along with the inability of PRC2 enzymatic inhibition to fully rescue the differentiation phenotype (Fig. 4D, E), supports a model in which LIN28A is required to regulate PRC2 chromatin dynamics at developmental gene loci rather than solely modulating its enzymatic activity.
LIN28A interaction with PRC2 is RNA-dependent
The association of PRC2 with mRNAs and/or non-coding RNAs (ncRNAs) is critical to regulate its recruitment to specific gene loci [14, 17]. To investigate whether LIN28A regulates PRC2 recruitment to specific gene loci through its RNA-binding capacity, we treated lysates of cells expressing LIN28A-FLAG with RNase and analyzed the interaction between LIN28A and PRC2 subunits by co-immunoprecipitation. We found that RNase treatment disrupted LIN28A’s ability to efficiently capture PRC2 components (EZH2, SUZ12, and JARID2), thus indicating that the physical association between LIN28A and PRC2 is RNA-dependent (Fig. 6A). Similar results were obtained in Lin28a KO ESCs expressing exogenous LIN28A-FLAG (Supplementary Fig. 7E). To further support the finding that the LIN28A-PRC2 interaction is RNA-dependent, we performed an endogenous co-IP in WT ESCs using an anti-EZH2 antibody. We found that LIN28A immunoprecipitates with the PRC2 complex and this interaction is severely impaired by RNA degradation, confirming its RNA dependence (Fig. 6B).

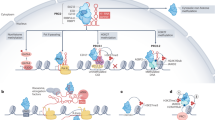
A Western blot analysis of LIN28A immunoprecipitates with and without RNase treatment. Immunoprecipitation was performed using anti-FLAG beads in ESCs transfected with a LIN28A-Flag expressing vector or an empty vector. B Western blot analysis after immunoprecipitation using an anti-EZH2 antibody in WT and KO ESCs. Lysates were treated or not with RNase before immunoprecipitation, as indicated. Black arrow indicates the unspecific band of IgG light chains. C RNA immunoprecipitation followed by qPCR analysis to evaluate the binding of ncRNA Neat1 to LIN28A. The 3’UTR of Lin28b was used as negative control. The graph shows the mean of the relative levels ±SD of four biological replicates, *p < 0.05 (Student’s t test). D Proximity ligation assay (PLA) to assess the interaction between LIN28A and the PRC2 subunit EED upon silencing of the ncRNA Neat1. ESCs were analyzed by PLA coupling the anti-LIN28A with anti-EED antibodies. Quantification of positive PLA spots/nucleus (for >300 cells) ± SD from three independent biological replicates is shown in the graph. Two-tailed, unpaired Student’s t test: ****p ≤ 0.00001. E qPCR analysis showing the mean of the relative levels ± SD of Neat1 RNA upon silencing with a control or a specific siRNA for the experiments reported in (D). n = 3 biological replicates, *p ≤ 0.05 (Student’s t test). F The proposed model for LIN28A involvement in epigenetic regulation. When differentiation is induced EZH2 is redistributed in WT cells allowing for the timely activation of developmental genes. By contrast, Lin28a KO ESCs exhibit restricted EZH2 occupancy under pluripotent conditions, likely due to a disrupted eviction/recruitment dynamic. During differentiation, this imbalance leads to persistent EZH2 binding and widespread recruitment to developmental gene promoters, resulting in their repression and ultimately impairing proper cell differentiation. Adapted from BioRender.com.
Recent studies have identified the long non-coding RNA nuclear paraspeckle assembly transcript 1 (Neat1) as a key regulator of PRC2 activity [17]. Although Neat1 has been shown to be enriched in the nucleus and to be essential for nuclear paraspeckle formation [41, 42], but undifferentiated PSCs do not form paraspeckles [43], suggesting that Neat1 may have additional roles in these cells. Recently, Huang and colleagues demonstrated that Neat1 is required for the activation of developmental genes to enable proper ESC differentiation. They also found that this ncRNA is essential for PRC2 eviction from chromatin and activation of lineage-specific genes, potentially through interactions with yet unidentified RBPs [17]. Notably, CLIP-seq analysis Neat1 indicated an association between Neat1 and LIN28A in mESCs [5]. Thus, we hypothesized that LIN28A could be one of the RBPs interacting with Neat1 to remove EZH2 from chromatin. First, we analyzed LIN28A binding to Neat1 through RNA immunoprecipitation and found that LIN28A efficiently binds to Neat1 RNA in ESCs (Fig. 6C). Next, we investigated whether the interaction between LIN28A and PRC2 could be mediated by Neat1. Using the PLA approach, we analyzed the spatial proximity of LIN28A and the PRC2 subunit EED upon Neat1 silencing in ESCs (Fig. 6D and Supplementary Fig. 7F). As shown in Fig. 6D, quantification of PLA-positive spots per nucleus revealed a significant reduction in LIN28A-EED proximity in cells in which Neat1 was silenced (Fig. 6E). Consistent with these results, co-immunoprecipitation experiments demonstrated that the interaction between LIN28A and PRC2 is markedly impaired by Neat1 silencing (Supplementary Fig. 7G). Although the involvement of other molecules (RNAs and/or proteins) cannot be ruled out, these results suggest that Neat1 serves as a scaffold facilitating the formation of the PRC2/LIN28A complex.
Discussion
Beyond its role in miRNA biogenesis, the RNA-binding protein LIN28A has already been described as a key regulator of translation in ESCs [5, 9]. During the transition from ESCs into EpiLCs, LIN28A regulates the translation of two important epigenetic regulators: the DNA methyltransferase DNMT3A and the architectural factor HMGA2 [6, 7]. Here, we uncover a previously unrecognized role of LIN28A in epigenetic regulation of ESC differentiation.
Generation of Lin28a KO ESCs revealed that, while LIN28A is dispensable for ESC self-renewal, it is essential for proper differentiation into neuronal precursors, particularly in the early stages of this process. Protein localization and interactomics experiments in both undifferentiated and differentiated ESCs revealed that LIN28A undergoes changes in cellular localization and molecular interactions following differentiation. Our proteomic data align with LIN28A’s established roles in ribosome biogenesis, rRNA processing, nucleolar remodeling, and translation [5,6,7,8,9, 24, 44]. We highlight that only a small subset of putative interactor proteins is shared between undifferentiated and differentiated ESCs. Notably, over 13% of the interactors specific to undifferentiated ESCs (88 proteins) are associated with chromatin organization.
Zeng and colleagues proposed that LIN28A promotes the recruitment of TET1 to specific promoters to remove the 5-methylcytosine (5mC) mark, thereby influencing the epigenetic landscape of ESCs [45]. Our proteomic data confirmed TET1 as an ESC-specific partner of LIN28A (Dataset 1), further supporting the aforementioned function. In addition, among other epigenetic regulators detected as putative LIN28A partners, we validated the interaction of LIN28A with the core and accessory subunits of PRC2 in undifferentiated ESCs. PRC2 deposits H3K27me3 on histone H3, an epigenetic modification essential for fine-tuning transcriptional programs during development [14, 46]. Along with PRC1, PRC2 plays a central role in silencing lineage-specific genes in pluripotent cells, repressing pluripotency genes, and regulating key developmental genes as cells commit to specific lineages [14, 36, 47]. In ESCs and pre-implantation embryos, PRC2 is enriched at bivalent genes, which are chromatin domains marked by both H3K4me3 and H3K27me3 [33]. These domains include lineage-specific genes whose premature activation is prevented by PRC2 in early development and in undifferentiated ESCs [48]. Upon differentiation, this bivalent state must resolve toward activation or repression to support lineage-specific transcriptional programs [33]. Activation requires the eviction of PRC2 from nucleosomes, selectively removing H3K27me3 from lineage-specific genes [32]. Numerous studies support the idea that this selective eviction is mediated in cis by nascent mRNA that regulates its own production by removing PRC2 from the promoter, thereby further promoting gene transcription, and in trans by the interactions with some RBPs and ncRNAs [15, 17, 18, 49,50,51,52]. While the direct binding of PRC2 to RNA is still debated [53,54,55,56,57], evidence suggests that PRC2 activity may indirectly rely on RNA via protein-protein interactions with RBPs [17, 58, 59].
Our findings show that the absence of LIN28A impairs the proper distribution of PRC2 on chromatin, accompanied by increased H3K27me3 levels despite unchanged EZH2 protein levels, during neuronal differentiation. We propose that LIN28A modulates PRC2-chromatin association and its eviction to regulate epigenetic programs during neural differentiation. Without LIN28A, PRC2 remains bound to chromatin and represses neural differentiation genes while maintaining pluripotency gene expression (Fig. 6F). We have shown that, in Lin28a KO differentiating cells EZH2 occupancy is enriched, widespread and persistent on the promoters of developmental genes (Fig. 5C–I) and that EZH2 enzymatic inhibition does not rescue the differentiation phenotype of Lin28a KO (Fig. 4D, E). This finding aligns with previous reports showing that EZH2 catalytic inhibition alone is insufficient to fully displace PRC2 from chromatin [60]. Accordingly, in the absence of LIN28A, EZH2 may remain bound to developmental gene promoters even in the presence of its enzymatic inhibitor, thereby preventing the activation of genes essential for proper differentiation.
Of note, we observed that ectopic LIN28A expression does not restore normal H3K27me3 levels in undifferentiated KO cells. However, LIN28A re-expression is able to re-establish proper H3K27me3 levels during SFEB differentiation, a stage at which PRC2 binding and H3K27 methylation are rearranged to permit the activation of lineage-specific genes [61]. These observations suggest that, in Lin28a KO ESCs, EZH2 dynamics are impaired and do not respond to the reintroduction of LIN28A. We speculate that upon differentiation induction additional factors (yet to be identified) may be expressed or activated which, in combination with ectopic LIN28A, can at least partially restore PRC2 function.
Our ChIP-seq analysis revealed that EZH2 occupancy is more restricted in undifferentiated KO ESCs compared to WT cells, despite no apparent impact on ESC phenotype. Upon induction of differentiation, EZH2 remains abnormally bound to chromatin in KO cells and is massively recruited to new target promoters, indicating a failure in the normal eviction–recruitment dynamics and resulting in impaired activation of developmental genes. Importantly, the comparison of ChIP-seq data for EZH2 and H3K27me3 reveals that EZH2 occupancy does not tightly correlate with H3K27me3 deposition, consistent with previous reports [39, 40]. Although EZH2 binding is reduced in Lin28a KO ESCs, we did not observe major global differences in H3K27me3-marked regions between WT and KO cells. This apparent discrepancy can be explained by the multifaceted role of PRC2 in chromatin organization. In ESCs, PRC2 not only promotes H3K27me3 deposition [34, 62, 63] but also mediates long-range chromatin interactions [30, 64], facilitating the recruitment of additional Polycomb complexes [65]. Beyond its local effects, PcG-mediated chromatin compaction and higher-order chromatin architecture significantly contribute to the silencing of developmental genes in ESCs [37, 64, 66]. Notably, Cruz-Molina and colleagues demonstrated that PRC2-dependent regulatory topology establishes a permissive chromatin environment for neural gene activation during ESC differentiation [67]. Moreover, recent studies have linked PRC2 to nuclear lamins, indicating that chromatin boundaries may restrict H3K27me3 spreading [68, 69]. These findings align with our data: in Lin28a KO ESCs, we observe persistent and spatially restricted EZH2 occupancy, which may reflect aberrant long-range interactions and altered H3K27me3 distribution. This is supported by our finding that only a subset of H3K27me3-marked promoters is shared between WT and KO ESCs (920 are unique to KO, 1623 are unique to WT, and 3500 are shared). Upon differentiation into SFEBs, Lin28a KO cells display a substantial increase in EZH2-bound promoters: specifically, 1,214 promoters that are not occupied by EZH2 in WT SFEBs, many of these are associated with developmental genes. Notably, 47% of these newly bound promoters lack detectable H3K27me3, suggesting a functional decoupling between EZH2 binding and its catalytic activity. Therefore, the changes observed in Lin28a KO cells may also reflect altered chromatin conformation and boundary establishment, consistent with the structural roles of EZH2 that are independent of its methyltransferase activity. Investigating changes in 3D chromatin architecture in Lin28a KO cells will be an important avenue for future research.
Our data support the eviction model of PRC2 regulation [70], potentially reconciling various perspectives on PRC2/RNA interactions [15, 16, 18, 34, 49,50,51,52,53,54,55,56,57, 70, 71]. Specifically, we propose that LIN28A binds to PRC2 in an RNA-dependent manner, facilitating its eviction from chromatin during differentiation. The ncRNA Neat1 appears to mediate this interaction at least partially. These findings are consistent with the model in which PRC2’s interactions with RNA and chromatin are mutually antagonistic [15, 16, 52] and with the idea that RNA degradation does not displace PRC2 from chromatin [56, 60]. RNA binding has been shown to displace PRC2 from chromatin [15], a process that can be facilitated by RNA-binding proteins such as LIN28A [59]. A recent study shows that, during ESC-to-EpiLC transition, Neat1 absence disrupts PRC2 chromatin balance, impairing developmental gene repression and differentiation [17]. LIN28A may use Neat1 as an RNA scaffold to interact with PRC2, facilitating PRC2 chromatin eviction and contributing to the proper activation of differentiation genes. Further investigations are required to determine whether LIN28A influences the availability of nascent mRNA at PRC2 target genes.
Our data suggest that LIN28A plays a specific role in ectodermal lineage differentiation. Indeed, Lin28a KO cells do not exhibit evident defects in mesodermal and endodermal differentiation. Furthermore, Lin28a levels increase during neural differentiation but decrease during mesendodermal lineage commitment. Notably, emerging evidence shows that PRC2 recruitment is developmentally regulated and guided by interactions with accessory proteins that are dynamically expressed in a cell type–specific manner [14]. We propose that LIN28A contributes to conferring specificity to PRC2 in neuronal differentiation. The Lin28a KO phenotype may reflect the combined impact of disrupted epigenetic and translational regulation, consistent with the predominantly cytoplasmic localization of LIN28A in ESCs.
In conclusion, our findings uncover a previously unrecognized RNA-dependent functional link between LIN28A and PRC2 in modulating the epigenetic landscape during ESC differentiation.
Materials and methods
Cell culture, differentiation, treatments, and transfection
Mouse ESCs (E14Tg2a, BayGenomics) were grown on gelatin-coated plates in the following ESC medium: Glasgow minimum essential medium (Sigma-Aldrich) supplemented with 2 mM glutamine, 1 mM sodium pyruvate, 1 × non-essential amino acids (all from Thermo Fisher Scientific), 0.1 mM β-mercaptoethanol (Sigma-Aldrich), 10% fetal bovine serum (Hyclone Laboratories), and 103 U/mL leukemia inhibitory factor (LIF, EMD Millipore).
The generation of EpiLCs [6] was induced by plating ESCs on fibronectin coated plates in EpiLC medium: 1 vol of DMEM/F-12 combined with 1 vol of Neurobasal medium, supplemented with 0.5% N2 supplement, 1% B27 supplement, 1% KO serum replacement, 2 mM glutamine (all from Thermo Fisher Scientific), 20 ng/ mL activin A (R&D Systems), and 12 ng/ mL basic fibroblast growth factor (Thermo Fisher Scientific). Within 2 days in these conditions, the cells express epiblast markers. We used the term “EpiLCs” to indicate cells at 3 days of differentiation from ESCs.
Differentiation toward the neuroectodermal lineage was induced by forming SFEBs [6, 21, 22]. SFEBs are generated by culturing ESCs in suspension in a chemically defined, serum-free medium as follows. ESCs were placed at a concentration of 1 × 105 cells/mL in suspension in Petri dishes in the following differentiation medium: KnockOut DMEM supplemented with 2 mM glutamine, 1× nonessential amino acids, 0.1 mM β-mercaptoethanol, and 10% KO serum replacement (all from Thermo Fisher). Unless noted otherwise, the term SFEBs indicates aggregates at 4 days of ESC differentiation. To obtain differentiation into neurons, 4-day-differentiated SFEBs were dissociated with accutase (Thermo Fisher) and plated on poly-D-lysine (Sigma-Aldrich)-coated plates in differentiation medium supplemented with 0.1 µM retinoic acid (Sigma-Aldrich). After 3–4 days, neurite networks were evident by morphological observation.
For the treatment with PRC2 inhibitor, ESCs were induced to differentiate into SFEBs in the presence or in the absence of 10 μM GSK126 (S7061, Selleckchem), and the medium was changed after 2 days, maintaining the same concentration of GSK126 for an additional 2 days.
For endoderm differentiation, 3 × 104 cells/cm2 were plated on gelatin-coated plates in ESC medium and after 24 h, the medium was replaced with RPMI (Gibco) supplemented with 1 μM Retinoic Acid, 3 μM CHIR-99021, 20 ng/ mL Activin, 10 ng/mL LIF, 2 mM glutamine, and 2% of KSR. Cells were harvested at 4 days and 8 days of differentiation.
For cardiomyocyte differentiation, we adopted the hanging drop method previously described [22]. Briefly, a suspension of 40,000 cells/ mL in ESC medium without LIF (using 15% of batch-selected serum (HyClone) was used to generate 20 μl hanging drops to induce the formation of aggregates (EB). After 2 days, the EBs were collected in the same medium and grown in suspension for 3 days. Then, single EBs were placed in a gelatin-coated 48-well plate, EBs showing beating heart areas (95–100%) were counted at 12 days of differentiation.
For transfection, ESCs were plated at density of 80,000 cells/cm2 on gelatin-coated plates. Sixteen hours later, ESCs were transfected with the pCAG-Lin28a-Flag plasmid expressing the FLAG-tagged form of LIN28A (Parisi et al. [6]) or the empty vector, or with siRNAs (Thermo Fisher) using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s protocol.
Generation of Lin28 KO ESCs
To induce specific knock-out of Lin28a in ESCs, we used the SpCas9(BB)2A-Puro vector (PX459) described by Ran et colleagues [71]. The two sgRNA guides were designed using the CRISPOR tool. The targeting regions of the two sgRNAs are reported in Supplementary Fig. 1, the sequences are as follows:
sgLin28#15’-CACCGCTGCGGCTCGTCTGCCGCT-3’
sgLin28#25’-CACCGTCTATGACCGCCCGCGCTG-3’
The sgRNA guides were cloned into the pSpCas9(BB)-2A-puro vector (Addgene) using BbsI (Thermofisher) and the plasmid was then transfected into the ESCs using Lipofectamine 2000. Correct targeting in the selected clones was assessed by PCR and sequencing.
Cell fractionation and Western blot
We adapted a previous published protocol for cell fractionation experiments [72]. Briefly, cells were resuspended in the following ice-cold buffer A: 10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.1% Triton X-100, 1 mM DTT, and 1x protease inhibitors (Sigma-Aldrich). Then, lysates were centrifuged, and the supernatant was stored as the cytoplasmic fraction. The pellet was resuspended in 1X volume of buffer A and 10% of the sample was stored as the total nuclear fraction (including chromatin and non-chromatin bound fraction). Then, the sample was diluted in buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, protease inhibitors, and 0.5% NP-40), vortexed, and incubated for 60 min on ice. The sample was centrifuged and the supernatant was stored as nuclear soluble fraction. The pellet from the previous step was resuspended in 10X volume of B-SDS 1X lysis buffer (composed of 50 mM Tris-Cl pH 7.5, 2 mM EDTA, 2% SDS) to yield the chromatin fraction. The fractions were sonicated with Bioruptor Plus (Diagenode) for 5 min at high power (30 s ON, 30 s OFF) until clarification and diluted in Laemmli 2X. For western blot analysis, proteins were separated by SDS-PAGE under reducing conditions and transferred onto PVDF membranes (Merk Millipore). The membranes were then incubated with the primary antibodies reported in Supplementary Table 2.
Whole cell extracts were obtained using 1X RIPA buffer (Thermo Scientific) supplemented with protease (Sigma-Aldrich) and phosphatase (Roche) inhibitors. After lysis, the samples were sonicated with Bioruptor Plus (Diagenode) and centrifuged at 14,000 × g for 15 min to isolate the total nuclear proteins. Total proteins were quantified using the Bradford Protein Assay (Bradford Reagent, Bio-Rad). For western blot analysis, the proteins were separated by SDS-PAGE under reducing conditions, transferred onto PVDF membranes (Merk Millipore) and incubated with the primary antibodies reported in Supplementary Table 2.
Raw western blot images are reported in the Supplementary File “Original western blots”.
Immunofluorescence, proximity ligation assay (PLA) and microscopy
Undifferentiated ESCs were grown on 8-well chambered coverslip (Ibidi) coated with gelatin, fixed in 4% paraformaldehyde 15’ at room temperature (RT), washed three times in 1X PBS, permeabilized with 0.5% Triton X-100 15’ at RT and incubated with the primary antibodies (listed in Supplementary Table 2) 1 h at RT or overnight at 4 °C in the following incubation solution: 10% FBS and 1% BSA in 1X PBS. Then, the cells were washed three times in 1X PBS for 10 min each, incubated with suitable secondary antibodies (Alexa Fluor, Thermo Fisher) for 1 h at RT in the incubation solution, washed 3 times again with 1X PBS 10 min each and counterstained with 0.2 μg/mL DAPI (Thermo Fisher) for 5 min in 1X PBS.
SFEBs were collected on the indicated differentiation day, fixed in 4% paraformaldehyde dehydrated with increasing percentages of ethanol, embedded in paraffin and sectioned into 7-μm slices. After rehydration and permeabilization (0.2% TX-100 for 5 min) and washing the slices were boiled in citrate buffer. The primary antibodies (listed in Supplementary Table 2) were incubated in 10% FBS/1% BSA/0.1% Tween 20/1× PBS overnight at 4 °C followed by washing and secondary antibody hybridization. Nuclei were counterstained with Dapi (Thermo Fisher).
For whole mount IF on SFEBs (Fig. 4E and Supplementary Fig. 4B), the aggregates were washed twice with 1X PBS, fixed in 4% paraformaldehyde for 2 hon a rotator at 4 °C and permeabilized with 0.5% Triton X-100 30’ on a rotator at 4 °C. Unspecific signals were blocked by incubating the SFEBs for 2 h at 4 °C on rotation in the following solution: 10% FBS/0.2% TX-100 in 1X PBS. All subsequent steps were performed in this solution. The SFEBs were incubated with the primary antibodies (listed in Supplementary Table 2) overnight at 4 °C on rotation. After 3 washes, 15 min each, at 4 °C on a rotator, the SFEBs were incubated with suitable secondary antibodies (Alexa Fluor, Thermo Fisher) for 2 h at 4 °C on a rotator. Finally, after 5X washes, 30 min each, SFEBs were counterstained with 0.2 μg/mL DAPI (Thermo Fisher) for 20 min at 4 °C on rotation. SFEBs were then placed in a PBS glycerol solution at a ratio of 1:1 in an 8-well chambered coverslips (Ibidi) for imaging (see below).
For PLA experiments, ESCs were plated at density of 5 × 105 cells/well on gelatin-coated 8 wells chambered coverslips (Ibidi) and after 24 h, washed with 1X PBS and fixed with 4% paraformaldehyde for 10 min at room temperature. PLA was conducted according to the manufacturer’s protocol (NaveniFlex kit - Navinci). For antigen detection, the cells were incubated with the antibodies reported in Supplementary Table 2. For negative controls, only one of the two primary antibodies was used. The cells were subsequently hybridized with secondary antibodies conjugated with the PLA probes (PLUS and MINUS) and then subjected to ligation and rolling circle amplification using fluorescently labeled oligonucleotides. The cells were then washed and incubated for 5 min in PBS containing 0.2 μg/mL DAPI (D1306, Thermo Fisher Scientific), to allow nuclei detection. Finally, the cells were washed again and placed in a PBS-glycerol solution (1:1). Images were acquired by taking Z-slices from the top to the bottom of the cell using the same settings (LED source power, exposure time) and small-volume computational clearing (SVCC) mode for different cell lines and all experimental conditions.
Images were captured using a Leica Thunder Imaging System (Leica Microsystems) equipped with a LEICA DFC9000 GTC camera and a Lumencor fluorescence LED light source. A 63× or 100× oil immersion objective was used to acquire Z-slice images. Small-volume computational clearing was used to remove background signals derived from out-of-focus blur. When specified, the maximum projection function was used to provide a single, easy-to-interpret 2D view of the entire 3D structure, displaying the brightest pixel value from each depth layer (z-slice) at each x-y position.
For PLA quantitation, images were analyzed using ImageJ software (Fiji). After being imported via the Bio-Formats plugin, the images were segmented, and 2D structures larger than 0.01 μm² were automatically identified using the Analyze Particles command. Figures and statistical analyses related to the quantification of PLA images were conducted using R. The following packages were used: stringr, ggplot2, ggpubr, RColorBrewer, and ggpattern (Supplementary Table 2). For statistical comparisons between experimental conditions, a non-parametric Wilcoxon test was employed, as specified, to assess differences in means. Graphs were generated using the geom_boxplot function to visualize boxplots in the style of Tukey showing the median as a horizontal line and the mean as a red triangle (where shown). Two hinges represent data from the first and third quartiles and two whiskers extend from the smallest to the largest values within 1.5 × IQR. All outlier data are represented as individual spots.
RNA extraction, reverse transcription, and real-time PCR
Total RNA was extracted from undifferentiated and differentiated ESCs, EpiLCs, EBs, and SFEBs using Tri-Sure (Bioline), according to the manufacturer’s protocol. Approximately 1 µg of total RNA was used for reverse transcription with RevertAid Reverse Transcriptase (Thermo Scientific). Quantitative PCR was run on QuantStudio 7 Flex Real Time PCR System using Fast SYBR Green PCR Master Mix (Thermo Fisher Scientific). The expression of Gapdh mRNA was used as an internal control and RNA expression levels were analyzed by performing a comparative analysis using 2−ΔCt method. The gene-specific primers used are reported in Supplementary Table 2.
Alkaline phosphatase staining
For AP staining, ESCs were plated at clonal density (200 cells/cm²) on gelatin-coated dishes and cultured for 5 days. The cells were then fixed with cold 10% neutral buffered formalin (10% formalin, 110 mM Na2HPO4, and 30 mM NaH2PO4 H2O) for 15 min at room temperature. After fixation, the cells were rinsed with distilled water for 15 min. AP staining was performed by incubating the cells for 45 min at room temperature in the following staining solution: 0.1 M Tris-HCl, 0.01% naphthol AS MX-PO4, 0.4% N,N-dimethylformamide, 0.06% red violet LB salt (all from Sigma Aldrich). Staining was blocked by extensive washing with distilled water. Positive AP-stained colonies were identified and counted using phase-contrast microscopy.
Cell viability assay
Cell viability was evaluated using the AlamarBlue® Cell Viability Reagent (Invitrogen). For WT and KO undifferentiated ESCs 12 × 10³ cells were plated onto gelatin-coated 96-well plates. After 18 h, the cells were incubated with resazurin using 1/10th volume of alamarBlue® reagent in the ESC culture medium, following the manufacturer’s instructions. To measure cell viability in SFEBs, WT and Lin28a KO ESCs were induced to aggregate and differentiate in a low-attachment 96-well microplate with round bottom (Greiner) containing 100 µL of differentiation medium as described above (1 × 105 cells/ mL). After 4 days, when the SFEBs were formed, 1/10th volume of alamarBlue® reagent was added directly to the cells. Fluorescence intensity (excitation/emission: 560/590 nm) was measured at different time points using a microplate reader (Synergy H1, BioTek). Three biological replicates were performed for statistical analysis using t-test.
RNA immunoprecipitation
All the immunoprecipitation experiments were performed using the following RIP protocol. Briefly, WT or Lin28a KO (where indicated) ESCs were transfected with the pCAG-Lin28a-Flag plasmid or empty vector (mock) as control. After 48 h of transfection, the undifferentiated cells were harvested and resuspended in polysome lysis buffer supplemented with protease (Sigma-Aldrich) and phosphatase inhibitors (Roche). After centrifugation to remove cell debris, one milligram of total proteins was diluted 1:1 with the following NT2 buffer: 50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.05% NP-40 and immunoprecipitated with anti-FLAG affinity gel (Sigma-Aldrich). The extracts were pre-cleared with 30 µL of Protein G agarose beads (EMD Millipore) for 1 h rotating at 4 °C. To block nonspecific binding before immunoprecipitation, the anti-FLAG affinity gel beads were incubated in 5% BSA in NT2 buffer on a rotator at 4 °C for 1 h. Then, BSA was removed by washing 3 times with NT2 buffer. Then, 50 µL of anti-FLAG affinity gel was added to the cell extracts of both Mock (as negative control) and LIN28A-FLAG expressing cells and incubated for 2 h at 4 °C on rotation. To recover the protein complexes for proteomic analysis and western blot, anti-FLAG affinity gel was washed 5 times with NT2+ buffer containing 0.25-0.4% NP-40 and then resuspended in 2 × Laemmli buffer and boiled for 5 min. To recover the RNA, after washing the beads were treated with proteinase K (10 mg/ mL; Sigma-Aldrich) for 1 h at 55 °C to release bound RNA. The RNA was purified using Tri-Sure (Bioline), and first-strand cDNA synthesis and qPCR were carried out as indicated above. The qPCR results were analyzed by relating the Ct of each sample to the Ct of the input sample (DCt = Ct (input) – Ct (IP)) and then applying 2DeltaCt comparative analysis.
For immunoprecipitation of endogenous EZH2, 5 × 106 of WT and Lin28a KO ESCs were plated in 100-mm gelatin-coated dishes, harvested after 48 h and resuspended in polysome lysis buffer (200 µL/dish) supplemented with protease inhibitors (Sigma-Aldrich) and phosphatase inhibitors (Roche). After centrifugation to remove cell debris, one milligram of total proteins was diluted 1:1 with the following NT2 buffer: 50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.05% NP-40. The extracts were pre-cleared with 30 µL of Protein A agarose beads (EMD Millipore) for 1 h at 4 °C with rotation. Then, the EZH2 antibody (5 µg, Cell Signaling Technologies) was incubated with cell extract overnight at 4 °C with rotation. Where specified in the legend equal amount of rabbit IgGs (Abcam) was used as a negative control. The protein lysates were incubated with protein A Agarose beads (EMD Millipore) for 2 h at 4 °C with on rotation. Then, the beads were washed 5 times with NT2+ buffer and proteins were eluted from the beads by boiling for 5 min in 2X SDS Laemmli loading buffer, followed by SDS-PAGE and Western blot analysis.
For immunoprecipitation experiments with SFEBs, ESCs were transfected with pCAG-Lin28a-Flag plasmid and after 12 h were induced to differentiate into SFEBs. The cells were collected at 4 days of differentiation and immunoprecipitation was performed as described above.
For RNase treatment, the lysates were treated with 50 µg/ mL RNase A for 30 min at RT. RNase was also added to the IP during incubation to a final concentration of 50 µg/ mL. Then, immunoprecipitation was carried out as described above.
Proteomic analysis, protein identification, and bioinformatics analysis
For the proteomic analysis, the immunoprecipitated samples from ESCs and SFEBs expressing LIN28A-FLAG or not (transfection of empty vector indicated as MOCK) were prepared as described in the “RNA immunoprecipitation” section. Affinity purifications were performed on three independent biological replicates per condition. According to sample identity, biological replicates were then pooled and analyzed by 12% T SDS-PAGE. After staining with colloidal Coomassie blue, whole gel lanes were cut into 15 slices, minced and washed with water. The corresponding proteins were separately in-gel reduced, S-alkylated with iodoacetamide and digested with trypsin, as previously reported [73]. The individual protein digests were then analyzed using a nanoLC-ESI-Q-Orbitrap-MS/MS platform consisting of an UltiMate 3000 HPLC RSLC nano system (Thermo Fisher Scientific, USA) coupled to a Q-ExactivePlus mass spectrometer through a Nanoflex ion source (Thermo Fisher Scientific). The peptides were loaded onto an Acclaim PepMapTM RSLC C18 column (150 mm × 75 μm ID, 2 μm particles, 100 Å pore size) (Thermo Fisher Scientific), and eluted using a gradient of solvent B (19.92/80/0.08 v/v/v water/acetonitrile/formic acid) in solvent A (99.9/0.1 v/v water/formic acid), at a flow rate of 300 nL/min. The gradient of solvent B started at 3%, increased to 40% over 40 min, raised to 80% over 5 min, remained at 80% for 4 min, and finally returned to 3% over 1 min, with a column equilibrating step of 30 min before the subsequent chromatographic run. The mass spectrometer operated in data-dependent mode with a full scan m/z range 375–1500, nominal resolution of 70,000, an automatic gain control target of 3000,000, and a maximum target of 50 ms, followed by MS/MS scans of the 10 most abundant ions. MS/MS spectra were acquired in a scan m/z range 200–2000, using a normalized collision energy of 32%, an automatic gain control target of 100,000, a maximum ion target of 100 ms, and a resolution of 17,500. A dynamic exclusion value of 30 s was also used. Triplicate analysis of each sample was performed to increase the number of identified peptides/protein coverage.
The MS and MS/MS raw data files per lane were merged for protein identification into Proteome Discoverer v. 2.4 software (Thermo Scientific), enabling the database search by Mascot algorithm v. 2.4.2 (Matrix Science, UK) with the following parameters: UniProtKB mouse protein database including the most common protein contaminants; carbamidomethylation at Cys as fixed modification; oxidation at Met, deamidation at Asn and Gln, and pyroglutamate formation at Gln as variable modifications. The Peptide mass tolerance and fragment mass tolerance were set to ±10 ppm and ±0.05 Da, respectively. The proteolytic enzyme and the maximum number of missed cleavages were set to trypsin and 2, respectively. Protein candidates that were identified based on at least two sequenced peptides and Mascot score ≥30 were considered confidently identified. Definitive peptide assignment was always associated with manual spectra visualization and verification. The results were filtered to 1% false discovery rate. The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE [74] partner repository with a dataset identifier PXD049332. With the aim to obtain a high-confidence list of LIN28A interactors in ESCs, we subtracted all proteins identified in the mock control immunoprecipitation, i.e., undifferentiated cells transfected with the empty vector from those detected in the corresponding FLAG-LIN28A sample. These subtraction processes removed proteins that were bound non-specifically to the beads or FLAG antibody, thereby ensuring that the remaining components represent specific LIN28A interactors in ESCs or SFEBs. The list of putative LIN28A interactors is reported in Dataset 1. The cumulative list of LIN28A interactors was compared to those analyzed in different types of WT stem cells (mouse EpiLCs), or their derivatives (neural precursors obtained from human iPSCs) cells (Parisi et al., [7]; Yu et al., [13]), and BioGRID (Chatr-Aryamontri et al., [26]), IntAct (Orchard et al., [27]) and STRING databases (Supplementary Table 1). Functional enrichment analysis, including Gene Ontology, was performed using the Metascape and FunRich 3.1.3 tools, setting Mus musculus and UniProtKB Rodents database, respectively. Venn diagrams were obtained using the Bioinformatics & Evolutionary Genomics online tool. The STRING online tool was used to visualize and integrate complex networks of proteomics data.
Chromatin Immunoprecipitation sequencing (ChIP-seq)
Pellets of 3 × 106 cross-linked ESCs or 2 days SFEBs were stored at −80 °C until sonication. Both EZH2 and H3K27me3 ChIP-seq were performed on three independent biological replicates.
For the EZH2 ChIP, each pellet was resuspended in 600 μL of cold Lysis Buffer containing: 50 mM Hepes-KOH, pH 7.5, 10 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40 and 0.25% Triton X-100. After 10 min on a rotator at 4 °C, the nuclei were collected by centrifugation for 5 min at 1350 × g at 4 °C and resuspended in 130 µL of the following sonication buffer: 10 mM Tris-HCl pH 8.0, 2 mM EDTA, 0.1% SDS, 1X Protease Inhibitor Cocktail (Roche), 1 mM PMSF (Sigma-Aldrich). The cells were incubated on ice for 1 h and total extracted chromatin was sonicated in a Covaris M220 focused-ultrasonicator using snap cap microTUBEs (Covaris) with cycles/burst 250, duration 18’. The fragmentation of chromatin to an average size of 200–500 bp was checked on Agilent 2100 Bioanalyzer using High Sensitivity DNA Kit (Agilent). Samples were then diluted by adding 1 volume of equilibration buffer: 0.1% SDS, 10 mM Tris-HCl pH8, 233 mM NaCl, 1.66% Triton X-100, 0.166% DOC, 1 mM EDTA, 1X Protease Inhibitor Cocktail (Roche), 1 mM PMSF (Sigma-Aldrich). To remove insoluble material the samples were centrifuged at 14,000 × g for 10 min at 4 °C and the resulting supernatants were quantified using Nanodrop 1000 spectrophotometer. For each experimental condition 3% of the total chromatin was stored at 4 °C as the input sample. For immunoprecipitation, 150 to 250 µg of chromatin was incubated overnight on a rotator at 4 °C with an EZH2 antibody (Cell Signaling Technology). The next day, protein G beads (Life Technology) were added to each sample and incubated on a rotator for 3 h at 4 °C. Beads were then washed for 10 min on rotator at 4 °C twice with IP buffer, twice with high-salt IP buffer: 10 mM Tris-HCl pH8, 500 mM NaCl, 1.66% Triton X-100, 0.166% DOC, 0.1% SDS, 1 mM EDTA, 1X Protease Inhibitor Cocktail (Roche); 1 mM PMSF (Sigma-Aldrich); once with RIPA-LiCl buffer: (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 250 mM LiCl, 0.5% DOC, 0.5% NP-40, 1X Protease Inhibitor Cocktail (Roche); 1 mM PMSF (Sigma-Aldrich); twice with 10 mM Tris-HCl pH 8.0. Crosslinking was reversed by incubating the beads and input at 65 °C overnight with 100 µL of Elution buffer: 10 mM Tris-HCl pH 8, 0.5 mM EDTA, 300 mM, 0.4% SDS. The next day, all samples were diluted with 100 µL of 1X TE, treated with 2.5 U of RNase cocktail (Ambion) at 37 °C for 120 min, followed by addition of 100 µg of Proteinase K (Invitrogen) at 55 °C for 120 min. DNA was then isolated using phenol/chloroform (Sigma-Aldrich) extraction and precipitation in cold ethanol. The pellets were suspended in nuclease-free water and quantified using Qubit 2.0 fluorometer with Qubit dsDNA HS Assay Kits (Invitrogen).
Libraries were prepared using the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB) and NEBNext Multiplex Oligos for Illumina (NEB). Library quantification and quality assays were done with TapeStation System. The libraries with distinct adapter indexes were normalized to a concentration of 2 nM, equimolarly pooled, and then loaded onto the Illumina NextSeq 2000 instrument. The sequencing was performed with a minimal target of 10 million reads for 100 bases in single-end mode on the Illumina NextSeq 2000 instrument at Ospedale Policlinico in Milan.
For H3K27me3 ChIP, each pellet was resuspended in 300 μL of cold Isotonic Buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 250 mM Sucrose, 5 mM MgCl2, 10 mM KCl, 1% NP-40) supplemented with protease (Sigma-Aldrich) and phosphatase inhibitors (Roche) and incubated on ice for 5’. Then, nuclei were recovered by centrifugation, resuspended in 500 μL of ChIP buffer (0.1% SDS, 1% TRITON X-100, 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA) supplemented with protease and phosphatase inhibitors and incubated on ice for 20’. Then, the lysates were sonicated with Bioruptor Plus (Covance) for 20 cycles (5 cycles at a time) 30” ON/ 30” OFF, high intensity. Samples were then further incubated on ice for 5’ and then centrifuged at 14,000 × g for 10 min at 4 °C to remove insoluble material. Size of chromatin fragments was verified on a 1% agarose gel. The supernatants were diluted by adding 2 volumes of dilution buffer (1% Triton, 2 mM EDTA, 150 mM NaCl, 20 mM Tris HCl pH8) supplemented with protease and phosphatase inhibitors. For each experimental condition 10% of the total chromatin was stored as an input sample. For immunoprecipitation, 350 µg of chromatin were incubated overnight on a rotator at 4 °C with 5 µg of H3K27me3 antibody (EMD Millipore). The next day, protein A beads (EMD Millipore) were added to each sample and incubated on rotator for 2 h at 4 °C. The beads were then washed 5 min on rotator at 4 °C once with low salt buffer (0.1% SDS, 1% Triton, 2 mM EDTA, 20 mM Tris HCl pH8, 150 mM NaCl) once with high salt buffer (0.1% SDS, 1% Triton, 2 mM EDTA, 20 mM Tris HCl pH8, 500 mM NaCl), once with LiCl buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 250 mM LiCl, 0.5% DOC, 0.5% NP-40), twice with TE buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8). Crosslinking was reversed by incubating the beads and input at 65 °C overnight with 500 µL of Elution buffer (1% SDS, 0,1 M NaHCO3). The next day, all samples were treated with 2.5 U of RNA A (Sigma-Aldrich) at 37 °C for 30 min, followed by addition of 100 µg of Proteinase K (Invitrogen) at 55 °C for 60 min. DNA was then isolated using phenol/chloroform (Sigma-Aldrich) extraction and precipitation in cold ethanol. The pellets were resuspended in nuclease-free water and quantified using Qubit 2.0 fluorometer with Qubit dsDNA HS Assay Kits (Invitrogen). The immunoprecipitates were sequenced by GENEWIZ Germany GmbH (Bahnhofstrasse 86 04158 Leipzig, Germany) by requiring a minimal target of 10 million reads for 150 bases in paired-end mode on the Illumina Novaseq instrument.
NGS data processing
The high-throughput EZH2 and H3K27me3 ChIP-seq data generated for this study are available in the NCBI GEO database under the following accession numbers GSE282208 and GSE306869, respectively. The sequences were demultiplexed using bcl2fastq. The ChIP-seq sequencing lanes were merged using GNU parallel software. The raw sequence reads were preliminarily inspected with FASTQC software. The Fastq files were aligned to the mm10 genome using the BWA-0.7.18 software (PMC2705234). PCR duplicates were marked and removed using the Picard (v2.30.0) tool. MACS2 2.2.9.1 software (PMCID: PMC2592715) was used to identify regions of EZH2 and H3K27me3 peaks enrichment by selecting, in each experimental sample, only the peaks identified with a q-value cutoff < 0.05. MACS2 was run with the default parameters, except for H3K27me3 for which the broad parameter was specified. The peaks detected in each biological replicate were intersected by using the intersectBed utility of bedtools 2.31 software (PMCID: PMC2832824). For each condition, only peaks identified in at least two of the three experimental replicates were considered real ChIP-seq peaks. The Metagene profiles reported in Supplementary Fig. 6C were obtained using ngsplot 2.63 (PMCID: PMC4028082) with the following parameters “-G mm10 -R genebody -L 2000”. The peaks obtained in each condition were annotated using the annotatePeak function included in ChIPseeker 1.41.3 package (PMID: 25765347) and specifying as source of annotation the org.Mm.eg.db included in BioConductor 3.20.0. The coverage tracks at specific genes of interest reported in Fig. 5H, Supplementary Fig. 6G and Supplementary Fig. 7D were obtained using the ggcoverage 1.3.0 R package (PMCID: PMC10413535).
The variation in peak enrichment at the promoters of 1866 genes across different conditions in EZH2 ChIP-seq, was estimated by reanalyzing the bigWig tracks with the computeMatrix and plotHeatmap function included in deepTools 3.5.6 (PMCID: PMC4987876).
Bigwig were determined from a single representative biological replicate by using bamCoverage from deepTools 3.5.6 with the following parameters: --outFileFormat bigwig; --blackListFileName ENCFF547MET.bed; --exactScaling; --extendReads 250; --effectiveGenomeSize 2652783500; --normalizeUsing RPKM. The blacklist file was downloaded from ENCODE website (https://www.encodeproject.org/files/ENCFF547MET/).
The computeMatrix tool was used with the “scale-regions” option on window of ±1000 bp, centered on the promoter peaks of the genes being investigated (parameters -b 1000 and -a 1000).
Literature data from Kundu et al. (PMCID: PMC5421375) [37] were downloaded from SRA database (ESC: SRR5027791; SRR5027790; SRR5027798; SRR5027799. NPC: SRR5027797; SRR5027796; SRR5027800; SRR5027801). The raw data were reanalyzed using the same pipeline used for the WT and Lin28a KO samples. Mapped peaks were within ±1000 bp of the TSS. EZH2 and H3K27me3 ChIP-seq were listed in Dataset 2 and Dataset 3, respectively.
Statistical analysis
The number of biological replicates for each experiment is indicated in the figure legends.
The means of at least three independent experiments were used to calculate standard deviation (SD) and to perform statistical analyses. Unless stated otherwise, all P values were calculated using a two-tailed Student’s t test with paired samples.
Data availability
Raw data from proteomic analysis have been deposited in the ProteomeXchange Consortium with a dataset identifier PXD049332. ChIP-seq data have been deposited in the NCBI GEO database with the accession numbers GSE282208 and GSE306869.
References
Schlesinger S, Meshorer E. Open chromatin, epigenetic plasticity, and nuclear organization in pluripotency. Dev Cell. 2019;48:135–50.
Chen Q, Hu G. Post-transcriptional regulation of the pluripotent state. Curr Opin Genet Dev. 2017;46:15–23.
Wright JE, Ciosk R. (2013) RNA-based regulation of pluripotency. Trends Genet. 2013;298:99–107.
Zhang H, Wu Z, Lu JY, Huang B, Zhou H, Xie W, et al. DEAD-Box helicase 18 counteracts PRC2 to safeguard ribosomal DNA in pluripotency regulation. Cell Rep. 2020;30:81–97.e7.
Cho J, Chang H, Kwon SC, Kim B, Kim Y, Choe J, et al. LIN28A is a suppressor of ER-associated translation in embryonic stem cells. Cell. 2012;151:765–77.
Parisi S, Passaro F, Russo L, Musto A, Navarra A, Romano S, et al. Lin28 is induced in primed embryonic stem cells and regulates let-7-independent events. FASEB J. 2017;31:1046–58.
Parisi S, Castaldo D, Piscitelli S, D’Ambrosio C, Divisato G, Passaro F, et al. Identification of RNA-binding proteins that partner with Lin28a to regulate Dnmt3a expression. Sci Rep. 2021;11:2345.
Tan SM, Altschuler G, Zhao TY, Ang HS, Yang H, Lim B, et al. Divergent LIN28-mRNA associations result in translational suppression upon the initiation of differentiation. Nucleic Acids Res. 2014;42:7997–8007.
Wilbert ML, Huelga SC, Kapeli K, Stark TJ, Liang TY, Chen SX, et al. LIN28 binds messenger RNAs at GGAGA motifs and regulates splicing factor abundance. Mol Cell. 2012;48:195.
Viswanathan SR, Daley GQ. Lin28: a microRNA regulator with a macro role. Cell. 2010;140:445–9.
Zhang J, Ratanasirintrawoot S, Chandrasekaran S, Wu Z, Ficarro SB, Yu C, et al. LIN28 regulates stem cell metabolism and conversion to primed pluripotency. Cell Stem Cell. 2016;19:66–80.
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20.
Yu NK, McClatchy DB, Diedrich JK, Romero S, Choi JH, Martínez-Bartolomé S, et al. Interactome analysis illustrates diverse gene regulatory processes associated with LIN28A in human iPS cell-derived neural progenitor cells. iScience. 2021;24:103321.
Chammas P, Mocavini I, Di Croce L. Engaging chromatin: PRC2 structure meets function. Br J Cancer. 2020;122:315–28.
Beltran M, Tavares M, Justin N, Khandelwal G, Ambrose J, Foster BM, et al. G-tract RNA removes Polycomb repressive complex 2 from genes. Nat Struct Mol Biol. 2019;26:899–909.
Beltran M, Yates CM, Skalska L, Dawson M, Reis FP, Viiri K, et al. The interaction of PRC2 with RNA or chromatin is mutually antagonistic. Genome Res. 2016;26:896–907.
Huang X, Bashkenova N, Hong Y, Lyu C, Guallar D, Hu Z, et al. A TET1-PSPC1-Neat1 molecular axis modulates PRC2 functions in controlling stem cell bivalency. Cell Rep. 2022;39:110928.
Kaneko S, Son J, Shen SS, Reinberg D, Bonasio R. PRC2 binds active promoters and contacts nascent RNAs in embryonic stem cells. Nat Struct Mol Biol. 2013;20:1258–64.
Kumar RM, Cahan P, Shalek AK, Satija R, DaleyKeyser A, Li H, et al. Deconstructing transcriptional heterogeneity in pluripotent stem cells. Nature. 2014;516:56–61.
Acampora D, Di Giovannantonio LG, Simeone A. Otx2 is an intrinsic determinant of the embryonic stem cell state and is required for transition to a stable epiblast stem cell condition. Development. 2013;140:43–55.
Musto A, Navarra A, Vocca A, Gargiulo A, Minopoli G, Romano S, et al. miR-23a, miR-24 and miR-27a protect differentiating ESCs from BMP4-induced apoptosis. Cell Death Differ. 2015;22:1047–57.
Navarra A, Musto A, Gargiulo A, Petrosino G, Pierantoni GM, Fusco A, et al. Hmga2 is necessary for Otx2-dependent exit of embryonic stem cells from the pluripotent ground state. BMC Biol. 2016;14:24.
Fuchs C, Scheinast M, Pasteiner W, Lagger S, Hofner M, Hoellrigl A, et al. Self-organization phenomena in embryonic stem cell-derived embryoid bodies: axis formation and breaking of symmetry during cardiomyogenesis. Cells Tissues Organs. 2012;195:377–91.
Sun Z, Yu H, Zhao J, Tan T, Pan H, Zhu Y, et al. LIN28 coordinately promotes nucleolar/ribosomal functions and represses the 2C-like transcriptional program in pluripotent stem cells. Protein Cell. 2022;13:490–512.
Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods. 2013;10:730–6.
Chatr-Aryamontri A, Oughtred R, Boucher L, Rust J, Chang C, Kolas NK, et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017;45:D369–D379.
Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42:D358–D363.
Jin J, Jing W, Lei XX, Feng C, Peng S, Boris-Lawrie K, et al. Evidence that Lin28 stimulates translation by recruiting RNA helicase A to polysomes. Nucleic Acids Res. 2011;39:3724–34.
Peng S, Chen LL, Lei XX, Yang L, Lin H, Carmichael GG, et al. Genome-wide studies reveal that Lin28 enhances the translation of genes important for growth and survival of human embryonic stem cells. Stem Cells. 2011;29:496–504.
Pachano T, Crispatzu G, Rada-Iglesias A. Polycomb proteins as organizers of 3D genome architecture in embryonic stem cells. Brief Funct Genom. 2019;18:358–66.
Tamburri S, Rustichelli S, Amato S, Pasini D. Navigating the complexity of Polycomb repression: enzymatic cores and regulatory modules. Mol Cell. 2024;84:3381–405.
Glancy E, Wang C, Tuck E, Healy E, Amato S, Neikes HK, et al. PRC2.1- and PRC2.2-specific accessory proteins drive recruitment of different forms of canonical PRC1. Mol Cell. 2023;83:1393–1411.e7.
Blanco E, González-Ramírez M, Alcaine-Colet A, Aranda S, Di Croce L. The bivalent genome: characterization, structure, and regulation. Trends Genet. 2020;36:118–31.
Laugesen A, Højfeldt JW, Helin K. Molecular mechanisms directing PRC2 recruitment and H3K27 methylation. Mol Cell. 2019;74:8–18.
Youmans DT, Schmidt JC, Cech TR. Live-cell imaging reveals the dynamics of PRC2 and recruitment to chromatin by SUZ12-associated subunits. Genes Dev. 2018;32:794–805.
Pasini D, Bracken AP, Hansen JB, Capillo M, Helin K. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol Cell Biol. 2007;27:3769–79.
Kundu S, Ji F, Sunwoo H, Jain G, Lee JT, Sadreyev RI, et al. Polycomb repressive complex 1 generates discrete compacted domains that change during differentiation. Mol Cell. 2017;65:432–46.
Noordermeer D, Leleu M, Schorderet P, Joye E, Chabaud F, Duboule D. Temporal dynamics and developmental memory of 3D chromatin architecture at Hox gene loci. Elife. 2014;29:e02557.
Tanay A, O’Donnell AH, Damelin M, Bestor TH. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc Natl Acad Sci USA. 2007;104:5521–6.
Højfeldt JW, Laugesen A, Willumsen BM, Damhofer H, Hedehus L, Tvardovskiy A, et al. Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat Struct Mol Biol. 2018;25:225–32.
Sunwoo H, Dinger ME, Wilusz JE, Amaral PP, Mattick JS, Spector DL. MEN ε/β nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 2009;19:347–59.
Souquere S, Beauclair G, Harper F, Fox A, Pierron G. Highly ordered spatial organization of the structural long noncoding NEAT1 RNAs within paraspeckle nuclear bodies. Mol Biol Cell. 2010;21:4020–7.
Modic M, Grosch M, Rot G, Schirge S, Lepko T, Yamazaki T, et al. Cross-regulation between TDP-43 and paraspeckles promotes pluripotency-differentiation transition. Mol Cell. 2019;74:951–965.e13.
Tan T, Gao B, Yu H, Pan H, Sun Z, Lei A, et al. Dynamic nucleolar phase separation influenced by non-canonical function of LIN28A instructs pluripotent stem cell fate decisions. Nat Commun. 2024;15:1256.
Zeng Y, Yao B, Shin J, Lin L, Kim N, Song Q, et al. Lin28A binds active promoters and recruits Tet1 to regulate gene expression. Mol Cell. 2016;61:153–60.
Piunti A, Shilatifard A. The roles of Polycomb repressive complexes in mammalian development and cancer. Nat Rev Cell Biol. 2021;22:326–45.
Jiang L, Huang L, Jiang W. H3K27me3-mediated epigenetic regulation in pluripotency maintenance and lineage differentiation. Cell Insight. 2024;3:100180.
Zhang J, Zhang Y, You Q, Huang C, Zhang T, Wang M, et al. Highly enriched BEND3 prevents the premature activation of bivalent genes during differentiation. Science. 2022;375:1053–8.
Cifuentes-Rojas C, Hernandez AJ, Sarma K, Lee JT. Regulatory interactions between RNA and polycomb repressive complex 2. Mol Cell. 2014;55:171–85.
Kaneko S, Son J, Bonasio R, Shen SS, Reinberg D. Nascent RNA interaction keeps PRC2 activity poised and in check. Genes Dev. 2014;28:1983–8.
Wang X, Goodrich KJ, Gooding AR, Naeem H, Archer S, Paucek RD, et al. Targeting of polycomb repressive complex 2 to RNA by short repeats of consecutive guanines. Mol Cell. 2017;65:1056–1067.e5.
Wang X, Paucek RD, Gooding AR, Brown ZZ, Ge EJ, Muir TW, et al. Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat Struct Mol Biol. 2017;24:1028–38.
Gail EH, Healy E, Flanigan SF, Jones N, Ng XH, Uckelmann M, et al. Inseparable RNA binding and chromatin modification activities of a nucleosome-interacting surface in EZH2. Nat Genet. 2024;56:1193–202.
Guo JK, Blanco MR, Guttman M. PRC2-RNA interactions: viewpoint from Jimmy K. Guo, Mario R. Blanco, and Mitchell Guttman. Mol Cell. 2024;84:3578–85.
Guo JK, Blanco MR, Walkup WG 4th, Bonesteele G, Urbinati CR, et al. Denaturing purifications demonstrate that PRC2andother widely reported chromatin proteins do not appear to bind directly to RNA in vivo. Mol Cell. 2024;84:1271–1289.e12.
Hall Hickman A, Jenner RG. Apparent RNA bridging between PRC2 and chromatin is an artifact of non-specific chromatin precipitation upon RNA degradation. Cell Rep. 2024;43:113856.
Lee Y, Lee JT. PRC2-RNA interactions: viewpoint from YongWoo Lee and Jeannie T. Lee. Mol Cell. 2024;84:3586–92.
Colognori D, Sunwoo H, Wang D, Wang CY, Lee JT. Xist repeats A and B account for two distinct phases of X inactivation establishment. Dev Cell. 2020;54:21–32.e5.
Wei C, Xiao R, Chen L, Cui H, Zhou Y, Xue Y, et al. RBFox2 binds nascent RNA to globally regulate polycomb complex 2 targeting in mammalian genomes. Mol Cell. 2016;62:875–89.
Healy E, Mucha M, Glancy E, Fitzpatrick DJ, Conway E, Neikes HK, et al. PRC2.1 and PRC2.2 synergize to coordinate H3K27 trimethylation. Mol Cell. 2019;76:437–452.e6.
Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–53.
Jiao L, Liu X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science. 2015;350:aac4383.
Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun. 2016;7:11316.
Denholtz M, Bonora G, Chronis C, Splinter E, de Laat W, Ernst J, et al. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell. 2013;13:602–16.
Wijchers PJ, Krijger PHL, Geeven G, Zhu Y, Denker A, Verstegen MJAM, et al. Cause and consequence of tethering a SubTAD to different nuclear compartments. Mol Cell. 2016;61:461–73.
Eskeland R, Leeb M, Grimes GR, Kress C, Boyle S, Sproul D, et al. Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol Cell. 2010;38:452–64.
Cruz-Molina S, Respuela P, Tebartz C, Kolovos P, Nikolic M, Fueyo R, et al. PRC2 facilitates the regulatory topology required for poised enhancer function during pluripotent stem cell differentiation. Cell Stem Cell. 2017;20:689–705.
Bianchi A, Mozzetta C, Pegoli G, Lucini F, Valsoni S, Rosti V, et al. Dysfunctional polycomb transcriptional repression contributes to lamin A/C-dependent muscular dystrophy. J Clin Investig. 2020;130:2408–21.
Sebestyén E, Marullo F, Lucini F, Petrini C, Bianchi A, Valsoni S, et al. SAMMY-seq reveals early alteration of heterochromatin and deregulation of bivalent genes in Hutchinson-Gilford Progeria Syndrome. Nat Commun. 2020;11:6274.
Davidovich C, Cech TR. The recruitment of chromatin modifiers by long noncoding RNAs: lessons from PRC2. RNA. 2015;21:2007–22.
Ran F, Hsu P, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR Cas9 system. Nat Protoc. 2013;8:2281–308.
Beringer M, Pisano P, Di Carlo V, Blanco E, Chammas P, Vizán P, et al. EPOP functionally links elongin and polycomb in pluripotent stem cells. Mol Cell. 2016;64:645–58.
Ayyildiz D, Antoniali G, D’Ambrosio C, Mangiapane G, Dalla E, Scaloni A, et al. Architecture of the human Ape1 interactome defines novel cancers signatures. Sci Rep. 2020;10:28.
Perez-Riverol Y, Bai J, Bandla C, García-Seisdedos D, Hewapathirana S, Kamatchinathan S, et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552.
Acknowledgements
The authors would like to thank: Tiziana Parisi and Tommaso Russo for the critical reading of the manuscript, the “BIOmedical iMAGIng fAcility” of DMMBM (Naples, Italy); Ludovica D’Auria and Caterina Missero of the Advanced Light Microscopy Facility of CEINGE (Naples, Italy) for help with imaging; Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico (Vittoria Moretti) and Istituto Nazionale Genetica Molecolare (Marco Ghilotti) for the sequencing.
Funding
This research was supported by Ministero dell’Università e della Ricerca (MUR) PRIN 2022 #2022P7R5CJ and PRIN/PNRR2022 #P2022KBAT7 to SPa; Italian Association for Cancer Research (AIRC), Investigator Grant #24976 and by the Ministero dell’Università e della Ricerca (MUR), PRIN/PNRR2022 #P2022F3YRF to PM; AFM (grant #24306); FRRB (grant # 3444218) and Ministero dell’Università e della Ricerca (MUR), PRIN #2022-4RFLLA to CL. Open access funding provided by Università degli Studi di Napoli Federico II within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
Silvia Piscitelli: Experimental design, data collection, analysis and interpretation, manuscript writing and editing. Emanuela Cascone, Giuseppina Divisato, Laura De Lisio, Valentina Rosti: Data collection, validation and analysis. Emilia Giannino: Data collection and analysis for manuscript revisions. Maria Chiara Zizolfi: Data analysis, manuscript writing. Chiara D’Ambrosio: Data collection and analysis, manuscript writing and editing. Guido Leoni: Data analysis and interpretation, manuscript writing and editing. Daniel D’Andrea: ChIP-seq data analysis for revisions, manuscript revision and editing. Danilo Swann Matassa: Experimental design, data interpretation, manuscript writing and editing. Chiara Lanzuolo: Data collection and analysis, manuscript writing and editing. Monica Matuozzo, Emanuele di Patrizio Soldateschi: data analysis. Paolo Maiuri, Andrea Scaloni, Fabiana Passaro: Data analysis and interpretation, manuscript editing. Silvia Parisi: Conceptualization and experimental design, data collection, analysis and interpretation, resources, project administration, manuscript writing and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Piscitelli, S., Cascone, E., D’Ambrosio, C. et al. The RNA binding protein LIN28A mediates chromatin dynamics during neuronal differentiation. Cell Death Differ (2026). https://doi.org/10.1038/s41418-026-01753-2
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41418-026-01753-2
