
Abstract
Therapy-induced senescence (TIS) is a major challenge in cancer therapy as senescent cancer cells provoke local and systemic inflammation and might be the cause of recurrences. Elucidation of pathways leading to TIS is of utmost importance for establishing strategies to counteract this. Previously we have shown that temozolomide (TMZ), an alkylating drug used forefront in glioma therapy, causes majorly cellular senescence, which is triggered by the primary damage O6-methylguanine, activating the mismatch repair dependent ATR/ATM-CHK1/CHK2-p53 damage response pathway. The downstream pathways leading to TIS remained to be explored. Here, we show that TMZ-induced TIS in glioma cells does not require activation of the DREAM complex, but is bound on a G2-specific response. We show that the CDK inhibitor p21CIP1 does not interact with CDK4, but with CDK1 and CDK2 causing abrogation of the B-Myb and FOXM1-signaling pathway and subsequently arrest of cells in the G2-phase. The induced G2-arrest is incomplete as DNA synthesis can be resumed leading to endoreduplications. This process, which is inhibited by the CDK4-blocking drug palbociclib, is preceded by reactivation of the G1/S-specific E2F1-signaling pathway due to lack of functional DREAM activation. These findings provide an explanation for the polyploidization and giant cell phenotype of anticancer drug-induced senescent cells. Incomplete DREAM activation may also explain the observation that downregulation of DNA repair is a transient phenomenon, which goes along with the entrance of cells into the senescent state.
Similar content being viewed by others


Introduction
Cellular senescence is defined as an irreversible cell cycle arrest and inability of cells to respond to proliferative conditions (for review see [1,2,3,4]). During normal aging, replicative senescence occurs in cells that reach the Hayflick limit [5] and is caused by telomere shortening during cell divisions. Cellular senescence is also induced during tumorigenesis through proliferation-associated signals (oncogenic senescence) and by genomic stress induced by external and internal DNA-damaging agents. A specific type of DNA damage-induced senescence is therapy-induced senescence (TIS), which is triggered by genotoxic anticancer drugs and ionizing radiation during cancer therapy [6].
Hallmarks of cellular senescence are sustained activation of the DNA damage response (DDR) resulting in a permanent cell cycle arrest via inhibition of cyclin-dependent kinases (CDKs) through the CDK inhibitors CDKN1a/p21CIP1, CDKN2a/p14ARF and CDKN2a/p16INK4a. An important downstream target of p21CIP1 is the DREAM complex, which arrests cells in the G0-phase and controls p53-mediated gene repression. In this case, p53 does not bind directly to promoter regions of the repressed genes, but causes through transcriptional induction of p21CIP1 an activation of the dimerization partner, RB-like, E2F, and multi-vulval class B (DREAM) complex [7,8,9,10,11]. The core DREAM complex is formed by the MuvB complex. During cell cycle progression, B-Myb (encoded by MYBL2) and/or FOXM1 bind to the MuvB complex and thus activate genes involved in proliferation. Upon p21CIP1 induction, CDK4 becomes inactivated, leading to dephosphorylation of p105 and p130 that bind together with E2F4 and E2F5 to the MuvB complex, forming the DREAM complex. In contrast to E2F1, E2F4 and E2F5 are transcriptional repressors, which silence the expression of proliferation-associated factors. Besides CDK4, p21CIP1 can also inhibit CDK1 and CDK2 [12] and is thereby involved in cell cycle arrest in the G1- and G2-phase, independent of DREAM.
We have shown that the anticancer drug temozolomide (TMZ), via induction of O6-methylguanine (O6MeG), triggers apoptosis [13], autophagy, and senescence in glioma cells [14, 15], with senescence being the main trait [16, 17]. TMZ is administered in the first-line therapy of high-grade gliomas [18,19,20], with glioblastomas (GBM) being incurable due to acquired resistance and formation of recurrences [21]. To study the mechanism of TMZ-induced senescence in GBM cells, we analyzed the transcriptional response, interaction of p21CIP1 with CDKs, and post-translational modification of cell cycle control mechanisms. Our data revealed that TMZ does not activate the DREAM complex, but rather induces an incomplete G2-arrest, which enables cells to re-enter the S-phase, causing endoreduplications and the formation of polyploid giant cells. Additional inhibition of CDK4 by palbociclib caused a stable G2-arrest and prevented polyploidy. These findings bear therapeutic implications as CDK4/CDK6 inhibitors may foster TMZ-induced senescence and thereby reduce senescence evasion.
Materials and methods
Cell culture, drug treatment, pharmacological inhibition and p21CIP1 knockdown
The glioma cell line LN229 (RRID:CVCL_0393) was obtained from LGC Standards (Wesel am Rhein, Germany). MCF7 (RRID:CVCL_0031), A172 (RRID:CVCL_0131), and U87MG (RRID:CVCL_0022; since misidentified, it refers to as “glioblastoma of unknown origin”) were purchased from Cell Line Service (Eppelheim, Germany). RPE1 cells (RRID:CVCL_4388) were obtained from ATCC. LN308 cells were a generous gift of Prof. Michael Weller (University of Zürich, Switzerland). All cell lines were cultivated in Dulbecco’s minimal essential medium (DMEM) supplemented with 10% fetal bovine serum at 37 °C in a humidified atmosphere containing 5% CO2 and were regularly checked for mycoplasma contamination (Venor GeM Cassic kit, Minerva Biolabs). All cell lines besides MCF7 are deficient for MGMT and besides LN308 are proficient for p53 [22, 23]. TMZ was kindly provided by Prof. Geoff Margison, Centre for Occupational and Environmental Health, University of Manchester, United Kingdom, and the CDK4 inhibitor palbociclib (S4482) was purchased from Selleckchem. Oxaliplatin and irinotecan (CPT-11) were prepared by the pharmacy of the University Medical Center Mainz, Germany. For gene silencing, predesigned siRNAs specific for p21CIP1 (sc-29427, Santa Cruz Biotechnology) were used; control human non-silencing siRNA (Silencer Select Predesigned siRNA Negative Control #1 siRNA; Ambion) was used as negative control. Transfections were performed using Lipofectamine RNAiMAX Reagent (Invitrogen).
Cell cycle progression and senescence
To quantify TMZ-induced cell cycle distribution, cells were stained with propidium iodide (PI). The DNA content was determined by flow cytometry using BD FACSCanto™ II. Senescence was measured microscopically by SA(senescence-associated)-β-Gal staining or using C12FDG stainig and flow cytometry as described before [14]. Experiments were performed as biological triplicates.
Cell proliferation and DNA synthesis
To measure cell proliferation, LN229 cells were seeded at a density of 50.000 cells in 6-cm dishes. Six hours later, the attached cells were harvested and counted to obtain the starting number for cell counts. In parallel, cells were treated with 50 µM TMZ or left untreated. After additional 24–144 h, cells were harvested and counted. To measure DNA synthesis, LN229 cells were seeded into a 96-well microtiter plate. Six hours later, cells were treated with 50 µM TMZ or left untreated. After additional 24–144 h, DNA synthesis was measured by the colorimetric BrdU ELISA assay (Roche). Experiments were performed as biological triplicates.
Preparation of RNA, real-time qPCR, methylation specific PCR and RNA-Seq
Total RNA was isolated using the NucleoSpin® RNA Kit from Macherey-Nagel (Düren, Germany). Utilizing the Verso cDNA Kit (Thermo Scientific, Dreieich, Germany), 1 µg total RNA was transcribed into cDNA and qPCR was performed in technical triplicates using the GoTaq® qPCR Master Mix Protocol (Promega, Madison, USA) and the CFX96 Real-Time PCR Detection System (Biorad, München, Germany). Analysis was performed using the CFX ManagerTM software, and the expression was normalized to gapdh and β-actin. Gene espression of the untreated control was set to one. Experiments were performed as technical triplicates. Primers sequences are listed in Table S1. For methylation specific PCR (MSP), DNA was extracted using phenol-chloroform extraction. Modification of the DNA was performed using the EZ DNA Methylation Kit (Zymo Research) as described [23]. p14ARF (Esteller et al. [24]) and p16INK4A (Herman et al. [25]) specific primers have been published before. As positive control for the reaction, the β-actin promoter was amplified. All primers sequences are listed in Table S1. For RNA-Seq, NGS library prep was performed with Illumina’s TruSeq stranded mRNA LT Sample Prep Kit following TruSeqStrandedmRNAReferenceGuide (Oct. 2017) (Document # 1000000040498v00). Libraries were prepared with a starting amount of 1000 ng and amplified in 10 PCR cycles. Libraries were profiled in a High Sensitivity DNA on a 2100 Bioanalyzer (Agilent technologies) and quantified using the Qubit dsDNA HS Assay Kit, in a Qubit 2.0 Fluorometer (Life technologies). All 12 samples were pooled in equimolar ratio and sequenced on NextSeq 500 Highoutput FC, PE for 2 × 150 cycles plus 7 cycles for the index read.
Gene sets used for evaluation of NGS data
The following gene sets were obtained from the Molecular Signatures Database (MSigDB database v7.5.1) [26]: FISCHER_DREAM_TARGETS, FISCHER_DIRECT_P53_TARGETS_META_ANALYSIS, FISCHER_G1_S_CELL_CYCLE, FISCHER_G2_M_CELL_CYCLE [27]; E2F4DP1_01; E2F5_TARGET_GENES [28]; SHEPARD_BMYB_TARGETS [29], GOBP_MITOTIC_NUCLEAR_DIVISION, GOBP_MICROTUBULE_CYTOSKELETON_ORGANIZATION. FOXM1 and E2F-RB target genes were directly obtained from Fischer et al. [27]. Venn diagrams were drawn using the program from (http://bioinformatics.psb.ugent.be/webtools/Venn).
Preparation of protein extracts, western blot analysis, co-immunoprecipitation and interactomics
Whole-cell extracts were prepared as described [30]. The specific antibodies are listed in Table S2. Co-immunoprecipitation was performed as already described [30, 31].
Xenograft experiments
The experiments were performed in accordance with relevant institutional and national guidelines and regulations, approved by the Landesuntersuchungsamt Rheinland-Pfalz, Germany (23 177-07/041-15V2). The experimental details were published previously [14] and summarized in the Legend to Fig. S11.
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism version 6.01 for Windows, GraphPad Software, La Jolla, California USA (www.graphpad.com). Data were evaluated using Student’s t-test and were expressed as a mean ± SD; *p ≤ 0.05 was considered statistically significant, **p ≤ 0.01 very significant, and ***p ≤ 0.001 highly significant.
Results
Phenotype of TMZ-induced senescence
The alkylating drug TMZ induced cell death (Fig. 1A, C) and, predominantly, cellular senescence (Fig. 1B, D). Senescence was associated with an arrest of cells in the G2-phase and an increase in the DNA content (Fig. 1A). The G2-arrest was observed already 48 h after TMZ treatment (Fig. 1C), whereas the number of cells with increased DNA content (>2n) went up later with time. They were first observed after 72 h and reached a level of 45% 144 h after treatment with 50 µM (Fig. 1C). This went along with the appearance of the senescence phenotype (Fig. 1D). Whereas the initial G2-arrest following TMZ treatment was not affected by knockdown of p21CIP1 and, therefore, is independent of p21CIP1 (Fig. 1E), the senescence phenotype was strongly affected and thus is p21CIP1 dependent (Fig. 1F). In line with this, knockdown of p21CIP1 had no impact on proliferation, but slightly increased the cell death level after late times (2 weeks), indicating killing of senescent cells (Fig. S1A–C). Proliferation assays indicated that cells could still perform two cycles of proliferation before they were blocked 48-72 h after TMZ exposure (Fig. 1G). However, despite the proliferation arrest, cells were able to restart DNA-synthesis 72 h after TMZ (Fig. 1H) and, as a consequence, showed an increased DNA content (Fig. 1I). There are two major pathways that can explain the observed over-replication, namely endoreduplication and re-replication [32]. During re-replication, origins can fire more than once within a single S-phase while during endoreduplication, cells circumvent mitosis entering a G1-like state and re-enter the S-phase, leading to polyploid mononucleated cells. Endoreduplication gives rise to discrete peaks in DNA content corresponding to 4n and 8n, which is different from re-replication. In the case of TMZ-induced senescence, a clear 4n and even an 8n peak (Fig. 1J) as well as polyploid mononucleated cells (Fig. S1D) were observed, clearly pointing to endoreduplication, and not re-replication to occur.

A–D LN229 cells were exposed to different concentrations of TMZ for 144 h (A, B) or to 50 µM TMZ for different time periods (C, D). Experiments were performed in triplicates. A, C Cell death was measured by flow cytometry using PI staining. B, D Frequency of senescent cells was detected microscopically by SA-β-Gal staining. E, F LN229 cells were transfected with p21CIP1 specific siRNA or nonspecific siRNA and 24 h later exposed to 50 µM TMZ for 144 h. Cell death was measured by flow cytometry using PI staining (E) and frequency of senescent cells was detected by flow cytometry using C12FDG staining (F). G–I LN229 cells were exposed to 50 µM TMZ for different time periods. G Proliferation was measured by cell counting. H DNA synthesis was measured by BrdU assay. I DNA content per cell was measured using the NanoDrop 1000 Spectrophotometer. J LN229 cells were exposed to 50 µM TMZ for 144 h, and cell cycle distribution was measured by flow cytometry using PI staining. A representative experiment is shown. B, D, I Differences between treatment and control were statistically analyzed using Student’s t test (p < 0.001). F Differences between treatment and control, as well as between ns-si/TMZ and p21-si/TMZ were statistically analyzed using Student’s t test (**p < 0.01***p < 0.001).
TMZ does not cause activation of DREAM
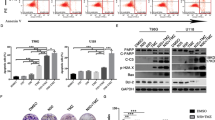
To analyze whether the DREAM complex is activated following TMZ treatment, the expression and phosphorylation of p130 and RB as well as the expression of p21CIP1, E2F1, E2F4, E2F5, B-Myb, FOXM1, LIN9 and p27 were analyzed 96 and 144 h after TMZ exposure, when cells reached the senescent stage (Fig. 2A). The cells in the population showed no significant reduction in the phosphorylated form of RB and p130, indicating the DREAM complex was not activated. This result was unexpected, giving the fact that p21CIP1 was strongly induced after genotoxic exposure. For E2F1, E2F4, E2F5, LIN9, and p27 no changes in the expression were observed and, in case of FOXM1 and B-Myb, decreased expression was observed 144 h after TMZ treatment.

A LN229 cells were exposed to 50 µM TMZ for 96 or 144 h. Expression and phosphorylation of p130 (pp130), Rb (pRb), as well as expression of p21CIP1, E2F1, E2F4, E2F5, B-Myb, FOXM1, LIN9, and p27 were measured by immunoblotting. β-Actin or HSP90 was used as internal loading control. Quantification of the immunoblot indicates x-fold induction in TMZ-exposed cells compared to untreated cells. B, C LN229 cells were exposed to 50 µM TMZ for 48 or 144 h. B Expression of FOXM1, MYBL2, CDK1, CDKN1A, CDKN1B, E2F1, E2F4 and E2F5 was measured by qPCR. C Expression of CCNA1, CCNA2, CCNB1, CCNB2, CCND1, CCND2, CCNE1 and CCNE2 was measured by qPCR. B, C Experiments were performed in triplicates, ACTB and GAPDH were used as internal loading control. Differences between the control and TMZ treatment were statistically analyzed using Student’s t test (non-labeled = not significant; *p < 0.1; **p < 0.01; ***p < 0.001).
Besides protein expression, we analyzed the expression of FOXM1, MYBL2, CDK1, CDKN1A, CDKN1B, E2F1, E2F4 and E2F5 48 and 144 h after TMZ on transcriptional level (Fig. 2B), revealing a strong induction of CDNK1A at all time points and a weak induction of E2F1, E2F4, and E2F5. The expression of MYBL2 and CDKN1B was unaltered, and FOXM1 was repressed 144 h after treatment. To gain further insight into the cell cycle regulation following TMZ, we also analyzed the expression of different cyclins (Fig. 2C). The data showed a strong repression of CCNA2 and CCNB1, which is in line with the arrest of cells in G2. They also showed induction in CCNA1, CCND2, CCNE1, and CCNE2, indicating an increased G1/S activity, which could explain the endoreduplications observed in the cell population.
Transcriptomics reveals a G2/M-specific response
To gain more insight into the transcriptional response upon TMZ, we performed NGS-based transcriptomics 48 and 144 h after TMZ treatment of LN229 cells. The results indicate 198 genes to be induced after 48 h and 688 genes after 144 h, including 137 genes to be upregulated at both time points (Fig. 3A). Pathway analysis identified cellular response to stimuli as sole induced pathway after 48 h. After 144 h a strong induction of the immune system and of extracellular matrix organization was observed (Fig. 3B, upper table). Concerning down-regulated genes, 57 were repressed after 48 h and 277 after 144 h, including 48 genes repressed at both time points (Fig. 3A). Concomitantly, a drastic repression of genes belonging to the cell cycle pathway was observed (Fig. 3B, lower table). Additional pathway analysis using the WEB-based GEne SeT AnaLysis Toolkit (http://www.webgestalt.org) and the Reactome and Kegg database are shown in Fig. S2.

LN229 cells were exposed to 50 µM TMZ for 48 or 144 h. RNA was isolated and subjected to RNA-Seq. A The genes commonly up-regulated (upper panel) or down-regulated (lower panel) after 48 and 144 h exposure are displayed as Venn diagrams. B Significantly regulated pathways were identified by the Reactome pathway browser (https://reactome.org/PathwayBrowser/). C The up-regulated genes and overlap with p53 targets are displayed as Venn diagrams. D The down-regulated genes and overlap with DREAM targets are displayed as Venn diagrams. E LN229 cells were exposed to 50 µM TMZ for 48 or 144 h. Expression of WEE1, PLK1, CENPA, MYT1, BIRC5, AURKA, AURKB, CDC20 and CDH1 was measured by qPCR. Differences between the control and TMZ treatment were statistically analyzed using Student's t test (non labelled = not significant; *p < 0.001 and log2 fold change > 1.5 are included.
Among the upregulated genes, several p53-regulated genes were identified (Fig. 3C), among them CDKN1A, MDM2, and TP53INP1 (Fig. S3A). Concerning DREAM targets, several gene sets exist. Using the DREAM-set derived from Fischer [27] (DREAM-F), 39 genes were repressed after 48 h and 77 after 144 h, including 38 at both time points (Figs. 3D and S3B). Using the DREAM-set derived from Engeland [33] (DREAM-E), 30 genes were repressed after 48 h and 53 after 144 h, including 29 at both time points and using the DREAM-set derived from Uxa [11] (DREAM-U), 31 genes were repressed after 48 h and 56 after 144 h, including 30 at both time points. Among the DREAM targets repressed at both time points, we identified important cell-cycle regulating factors like AURKA, BIRC5, BUB1, CCNA1, CCNB1, CDC20, CENPA, and PLK1. After 144 h, also AURKB, BORA, CCNB2, and FOXM1 were repressed (Fig. S3B).
The observed repression of important regulators of the G2/M-phase was further analyzed by qPCR (Fig. 3E). Similar to the results of the transcriptomic approach, a strong repression of AURKA, CDC20, CENPA, PLK1, and a weaker repression of BIRC5 was observed. Opposite, an increased expression of CDH1 was observed after 48 and 144 h and of MYT1 after 144 h; the expression of AUKB and WEE1 changed only marginal.
Similar to the expression data of the cyclins, the transcriptomic data suggested specific alterations in the G2/M-phase. Thus, among the repressed genes, only 5 were G1/S-specific whereas 59 were G2/M-specific (Fig. 3F). Importantly, most of the repressed G2/M, but not G1/S genes, were DREAM targets (Fig. S4A), which contradicts the normal distribution among G1/S and G2/M-specific genes (Fig. S4B). Upon TMZ exposure, using all DREAM-sets, only one gene was G1/S-specific, whereas 44 (DREAM-E/F) and 45 (DREAM-U) genes were G2/M-specific (Figs. S4C and S5). At the first glance, the high number of repressed DREAM targets hints at a DREAM response, however, this response is not characterized by the typical repression of cardinal G1/S-specific DREAM targets.
These non-repressed G1/S-specific DREAM targets included important cell cycle regulators like CDK1 and E2F1. Moreover, B-Myb was also described as G1/S-specific gene [34], however is not part of the used G1/S-specific gene set. Interestingly, a high frequency of repressed genes was associated with mitosis. Thus 22 genes were associated with microtubule cytoskeleton organization and 34 with mitotic nuclear division, which include AURKA, AURKB, BUB1, BORA, CCNB1, CDC20, and PLK1 (Figs. 3G and S6).
The missing activation of the DREAM complex as well as the missing repression of G1/S-specific genes in TMZ-treated LN229 cells was quite unexpected, giving the strong activation of p21CIP1 and the observed repression of multiple G2/M-specific DREAM targets. Therefore, we analyzed the interaction between p21CIP1 and the main CDKs by co-immunoprecipitation (Fig. 4A). The results showed binding of p21CIP1 to CDK1 and less pronounced to CDK2, but not to CDK4. Of note, this binding was only observed at late time points (144 h), but not early (48 h) after TMZ, indicating that the initial cell cycle arrest does not depend on p21CIP1 mediated CDK inhibition. Concomitantly, we also analyzed the expression of the CDKs and observed a slightly decreased expression of all CDKs at late times (96 h, 144 h) after exposure (Fig. 4B).

A LN229 cells were exposed to 50 µM TMZ for 48 or 144 h. Interaction between p21CIP1 and CDK1, CDK2, and CDK4 was measured by co-immunoprecipitation. B LN229 cells were exposed to 50 µM TMZ for 48, 96, or 144 h. Expression of CDK1, CDK2, and CDK4 was measured by immunoblotting. β-Actin was used as internal loading control. C LN229 cells were exposed to 50 µM TMZ for 24, 48, or 72 h. D LN229 cells were exposed to 50 µM TMZ for 96 or 144 h. C, D Expression of p21CIP1, CDC25c and PLK1, as well as expression and phosphorylation of CDK1 (pCDK1), CHK1 (pCHK1), WEE1 (pWEE1) and B-Myb (pB-Myb) was measured by immunoblotting. β-Actin or HSP90 were used as internal loading control. B, C Quantification of the immunoblot indicates x-fold induction in TMZ exposed cells compared to untreated cells.
TMZ-induced senescence is regulated by silencing of B-Myb and FOXM1 signaling
The missing involvement of the DREAM complex raised the question of how the cell cycle arrest was provoked and maintained after TMZ treatment. Therefore, protein expression and phosphorylation of important cell cycle regulators were analyzed 24–72 h after TMZ exposure (Fig. 4C). In the undisturbed cell cycle, CDK2 together with Cyclin E forms a complex at the end of the G1-phase that allows entry into the S-phase (for review see [35]). On one hand, it supports the function of Cyclin D/CDK4/CDK6 by additional phosphorylation of RB, and on the other hand, activates proteins involved in DNA replication via activation of the transcription factor B-Myb. If CDK2 is defective, CDK1 can take over the function. In this case, progression through the G2-phase and onset of mitosis is regulated via CDK1, which depends on its dephosphorylation mediated by the phosphatase CDC25c, as well as on interaction with Cyclin B1. Activation of CDC25c is mediated via PLK1-mediated phosphorylation. Both, Cyclin B1 and PLK1 are transcriptionally activated by B-Myb. In response to DNA damage, CDC25c undergoes proteasomal degradation upon phosphorylation by CHK1. Activated CHK1 can also phosphorylate/activate WEE1, which acts as CDK1 inhibitor.
In accordance to this background information, we observed phosphorylation/activation of CHK1 and WEE1 and degradation of CDC25c (Fig. 4C). Moreover, reduced phosphorylation of CDK1 and B-Myb was observed. This was in contrast to p21CIP1, which only showed a weak induction at early times (<72 h) after TMZ exposure. The results clearly indicate that the early cell cycle arrest is mediated by posttranslational modifications of CHK1 and WEE1, which is provoked by the DDR pathway independent of p21CIP. Dephosphorylation of CDK1 and B-Myb as well as degradation of CDC25c and repression of PLK1 was still observed at late times after damage induction (96 and 144 h), whereas the activated form of pWEE1 was missing (Fig. 4D). As indicated by co-immunoprecipitation experiments, at this time point p21CIP1 takes over the role from WEE1 in inhibiting CDK1 and CDK2.
Overall, the data indicate that upon TMZ-induced DNA damage, dephosphorylation and thereby inactivation of B-Myb as well as repression of FOXM1 is of outmost importance for arresting cells in the cell cycle and for transcriptional repression. During an undisturbed cell cycle progression, B-Myb is phosphorylated by CDK2 and acts together with FOXM1 as transcription factor for G2/M-specific genes (for review see [35]). At later times, B-Myb is removed from the MuvB complex and FOXM1 acts as the sole transcription factor regulating mitosis. This is also reflected by our transcriptomics data. It is well known that the DREAM complex silences genes normally induced by E2F1, B-Myb, and FOXM1, explaining the overlapping target-gene sets (Fig. S7A). Therefore, the transcriptional response in TMZ-treated cells can be explained by DREAM-independent silencing of the B-Myb and FOXM1 response. Indeed, the majority of targets of the observed transcriptional response are also targets of B-Myb and FOXM1 (Fig. S7B).
To test whether the missing activation of the DREAM response is a common event in glioblastoma, we extended the study to two other glioma cell lines (A172 and U87MG). Similar to LN229 cells, A172 and U87MG cells express functional p53 and show a robust induction of p21CIP1 upon TMZ (Fig. S8A–D) [14, 36]. Furthermore, all cell lines are deficient for CDKN2A (p14ARF and p16INK4a) due to deletion mutations (Fig. S8E). In these cells, TMZ strongly induced cellular senescence while only a low amount of apoptosis was detected (Fig. 5A, B). Similar to LN229, a high level of cells harboring a polyploid DNA content (>2n) was observed. We also analyzed the expression of factors involved in the senescence-associated secretory phenotype (SASP). In all cell lines, a strong upregulation of CCL2, CCL8, CCXL1, IL1α, IL1β, IL6, and IL8 was observed in the senescent population (Fig. S9). The analysis of cell cycle regulating genes revealed an expression pattern similar to LN229 cells. Thus, a repression of AURKA, CCNA2, CCNB1, CENPA, FOXM1, and PLK1 and an induction of E2F1 were observed in A172 and U87MG cells (Fig. 5C).

A, B LN229, U87MG, and A172 cells were exposed to 50 µM TMZ for 144 h. A Cell death and cell cycle distribution were measured by flow cytometry using PI staining. B Frequency of senescent cells was measured by flow cytometry using C12FDG staining. C A172 and U87MG cells were exposed to 50 µM TMZ for 144 h. Expression of FOXM1, E2F1, MYBL2, PLK1, WEE1, AURKA, AURKB, MYT1, CDKN1A, BIRC5 and CENPA was measured by qPCR. A–C Experiments were performed in triplicates. B–C Differences between treatment and control were statistically analyzed using Student’s t test (not-labeled = not significant; *p < 0.1; **p < 0.01; ***p < 0.001).
Similar to LN229, the initial cell cycle arrest in U87MG and A172 cells was mediated by post-translational modifications (Fig. 6A), whereas the final senescence-associated cell cycle arrest was dependent on p21CIP1 as indicated by co-immunoprecipitation with CDK1 and CDK2, but not with CDK4 during the senescence maintenance phase (Fig. 6B). Also, in A172 and U87MG cells, the inhibition of CDK1 and CDK2 was accompanied by repression of CCNA1/2 and CCNB1/2, whereas induction of CCND1, CCND2 and, in U87MG, also of CCNE1 and CCNE2 was found (Fig. S10).

A U87MG and A172 cells were exposed to 50 µM TMZ for 96 or 144 h. Expression of p21CIP1, CDC25c, and PLK1, as well as expression and phosphorylation of CDK1 (pCDK1), CHK1 (pCHK1), WEE1 (pWEE1) and B-Myb (pB-Myb) were measured by immunoblotting. β-Actin or HSP90 was used as an internal loading control. Quantification of the immunoblot indicates x-fold induction in TMZ-exposed cells compared to untreated cells. B U87MG and A172 cells were exposed to 50 µM TMZ for 48 or 144 h. Interaction between p21CIP1 and CDK1, CDK2, and CDK4 was measured by co-immunoprecipitation. C LN229, U87MG, and A172 cells were exposed to 50 µM TMZ for 48 or 144 h. In addition, the cells were exposed to 1 µM Palbociclib for 144 h or the cells were exposed to 50 µM TMZ, and 48 h later 1 µM Palbociclib was added for additional 96 h (TMZ/Palbo). Cell death and cell cycle distribution were measured by flow cytometry using PI staining; experiments were performed in triplicates. Differences between the frequency of cells in the G2-phase or showing a DNA content >2n after TMZ and TMZ/Palbo treatment were statistically analyzed using Student’s t test (***p < 0.001).
To substantiate the data, the activation of the DREAM complex and regulation of DREAM targets were analyzed in mouse U87MG xenograft experiments 96 h after TMZ exposure (Fig. S11). Immunodetection revealed the induction of p21CIP1, but no reduced phosphorylation of p130 and RB1. Furthermore, repression of G2/M-specific DREAM targets (e.g., AURKA, CCNB1, PLK1), but not G1/S-specific DREAM targets (e.g., E2F1, MSH2, MSH6) was observed. Overall, the data indicate that also in vivo, in human glioma xenografts, the DREAM complex is not fully activated.
Lack of DREAM activation leads to endoreduplications
To test whether missing inhibition of CDK4 and abrogated DREAM activation might explain the formation of endoreduplications and polyploid cells, we inhibited CDK4/6 by palbociclib, 48 h after drug treatment. At this time point the cells were arrested in G2, but did not show endoreduplication yet (Fig. 6C). Indeed, pharmacological CDK4/6 inhibition completely abrogated the expression of the cell cycle regulators E2F1, FOXM1 and MYBL2 in all cell lines (Fig. S12) and strongly reduced the number of cells performing endoreduplications (Fig. 6C).
An important question is whether missing activation of DREAM and accompanied endoreduplication is cell type- or agent-specific. Therefore, we compared the response of LN229 and the breast cancer cell line MCF7, as well as non-tumorigenic RPE1 (hTERT-immortalized retinal pigment epithelial) cells to TMZ, irinotecan, and oxaliplatin. With all agents examined, LN229 but not MCF7 or RPE1 cells showed endoreduplications that indicated missing DREAM activation (Fig. S13A). Most importantly, senescence-inducing drug concentrations (Fig. S13B) led to repression of DREAM targets only in MCF7 and RPE1, but not in LN229 cells (Fig. S14). The data suggest that the missing activation of DREAM and the subsequent endoreduplications are specific for glioblastoma cells and independent of the agent.
Transcriptional silencing of DNA repair is not a hallmark of TMZ-induced senescence
An important hallmark of senescent cells is downregulation of DNA repair genes [37]. However, analyzing the transcriptional response to TMZ in senescent glioblastoma cells revealed that DNA repair genes were not transcriptionally repressed (Fig. 7A). The missing repression was quite surprising, since we previously observed a transcriptional repression of the MMR genes EXO1, MSH2, and MSH6 at early time points upon TMZ treatment [14]. Therefore, we re-analyzed the transcriptional expression of MSH2, MSH6, and EXO1 following TMZ. As observed before, the genes were transcriptionally repressed at early times after treatment (48–72 h), however they recovered later when cells reached the senescent state (Fig. 7B). This missing repression at late time points was confirmed in A172 and U87MG cells (Fig. 7C). Of note, nearly all DNA repair genes that are DREAM targets are G1/S-specific (Fig. S15).

A LN229 cells were exposed to 50 µM TMZ for 144 h. RNA was isolated and subjected to RNA-Seq. Number of downregulated DNA repair genes is displayed as Venn diagram. B LN229 cells were exposed to 50 µM TMZ for different time periods, and the expression of MSH2, MSH6, and EXO1 was analyzed by qPCR. C A172 and U87MG cells were exposed to 50 µM TMZ for 144 h, and the expression of MSH2, MSH6, and EXO1 was analyzed by qPCR. B–D Differences between the control and TMZ treatment were statistically analyzed using Student’s t test (not-labeled = not significant; *p < 0.1; **p < 0.01; ***p < 0.001. D Expression of MSH2, MSH6, and MGMT was analyzed in LN229 cells and four LN229-derived clones escaping/evading TMZ-induced senescence by qPCR. B–D Experiments were performed in triplicates, ACTB and GAPDH were used as internal loading control, and the control was set to 1. E Expression of MSH2, MSH6, and MGMT was analyzed in LN229 cells and four LN229-derived clones escaping/evading TMZ-induced senescence by immunodetection. HSP90 was used as an internal loading control.
To prove whether lack of DREAM is responsible for the re-expression of MSH2, MSH6, and EXO1 in the senescence maintenance phase, we inhibited CDK4/6 by palbociclib 48 h after drug treatment. Under these conditions, stable repression of MSH2, MSH6, and EXO1 was achieved (Fig. S12). Obviously, the initial transcriptional repression of MSH2, MSH6, and EXO1 is lost during senescence maintenance. Decreased expression of MMR factors was described in glioblastoma relapses [38, 39]. Therefore, the question arose as to whether re-expression of these genes occurs in cells that were regrown from the senescent population. To address this, we generated a large number of cell clones that grew up as survivors from a TMZ-exposed cell population. The frequency of these clones was 0.02% of the senescent cell population. In none of them transcriptional repression of MSH2 or MSH6 was observed (Figs. 7D and S16), indicating that alterations in MMR activity in recurrences are not directly associated with senescence evasion. Same results were obtained on protein level (Fig. 7E). Since MGMT is a most important determinant of TMZ resistance, we also analyzed a potential change in MGMT expression, however no reactivation of MGMT was observed (Figs. 7D, E and S16).
Discussion
Here we show that the methylating anticancer drug TMZ strongly activates p21CIP1-dependent cellular senescence in glioblastoma cells, which is independent of DREAM activation. Our data revealed that p21CIP1 does not interact with CDK4, and the DREAM complex is not formed. As a consequence, only a subset of potential DREAM targets was repressed, namely G2/M-specific genes. In contrast, G1/S-specific DREAM targets (notably E2F1 and MYBL2) were not repressed. The findings are opposed to the transcriptional response we observed following B[a]P and IR in breast cancer cells, which led to DREAM formation and repression of G1/S and G2/M-specific DREAM-regulated genes [40].
In the TMZ-triggered O6-methylguanine response, futile mismatch repair on O6MeG/T mismatches causes gaps and subsequent replication fork collapse leading to the formation of DSBs [41, 42]. As shown here, this results in inactivation of CDK1 by two mechanisms: (i) Activated CHK1 phosphorylates and thus activates WEE1, which acts as a direct CDK1 inhibitor, and (ii) CHK1 phosphorylates CDC25c resulting in its proteasomal degradation. Inactivation of CDK1 leads to missing phosphorylation/activation of B-Myb and thereby repression of B-Myb targets, including CCNB2, PLK1, and FOXM1. At later times, p21CIP1 inhibits CDK1 and CDK2, enhancing the transcriptional repression of B-Myb and FOXM1 targets and thus arresting the cells in the G2-phase. However, the suppression of B-Myb and FOXM1 activity seems not to be sufficient and, therefore, the G2/M-arrest is leaky, allowing E2F1-mediated restart of DNA synthesis, resulting in endoreduplications and polyploidy. Of note, the CDK4/6 inhibitor palbociclib, which is clinically used in adjuvant cancer therapy, reduced the expression of E2F1, MYBL2, and FOXM1 and of endoreduplications. The DREAM response was also not observed in GBM cells after irinotecan and oxaliplatin treatment. On opposite, MCF7 and RPE1 cells responded with a full DREAM response, indicating that the missing DREAM response is specific for glioma cells treated with genotoxic drugs.
Our data show that p21Cip1 is not essential for the activation of the G2/M-arrest, since the G2/M-arrest occurred in both p53-proficient and p53-deficient cells (not upregulating p21CIP1) following TMZ treatment and was abrogated upon inhibition of CHK1 [14, 43, 44]. Overall, our data suggest that p21Cip1 is involved in turning the G2/M-arrest irreversible, which seems to be a bona fide prerequisite for the onset and maintenance of senescence. This and the question of why p21CIP1 does not interact with CDK4 in GBM cells are important topics to be addressed in future research.
Among the DREAM targets, DNA repair genes play an important role. Thus, downregulation of DNA repair triggered by DREAM is thought to be a hallmark of senescent cells [37, 45]. Actually, in GBM cells, early after TMZ exposure, we observed repression of DNA repair genes which, however, recovered in the senescent population. This is explained by the finding that MSH2, MSH6, and EXO1 as well as many other DNA repair genes are G1/S-specific targets of DREAM, which was not fully activated. It seems that TMZ induces a specific senescence phenotype in GBM cells that is in sharp contrast to mammary cancer cells and non-transformed cells following genotoxic stress [40]. Overall, our data support the notion that downregulation of DNA repair genes is a hallmark of DREAM-mediated senescence, which appears to be tumor-specific.
What are the clinical implications? We would like to propose that the unstable G2/M-arrest and endoreduplications observed following TMZ exposure, causing the formation of giant cells might contribute to therapy resistance. Genotoxin-induced polyploid giant cells have been observed in other studies. Thus, using a set of four human cancer cell lines, a direct relation between invasiveness and polyploidy was observed [46]. Importantly, it has been suggested that polyploidy may confer cells the ability to escape from senescence. Therefore, killing of polyploid cells is anticipated to improve anticancer treatment (for review see [47]). In line with this, our data suggest that CDK4/6 inhibitors, which are already in the clinic [48, 49], might foster TMZ-induced senescence via additional inhibition of the E2F1 pathway and thereby prevent endoreduplications and the formation of therapy-refractory polyploid cells.
Data availability
Raw transcriptomics data are available at GEO (Gene expression omnibus) under GSE276693.
References
d’Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–22.
Muller M. Cellular senescence: molecular mechanisms, in vivo significance, and redox considerations. Antioxid Redox Signal. 2009;11:59–98.
Fridman AL, Tainsky MA. Critical pathways in cellular senescence and immortalization revealed by gene expression profiling. Oncogene. 2008;27:5975–87.
Cichowski K, Hahn WC. Unexpected pieces to the senescence puzzle. Cell. 2008;133:958–61.
Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–36.
Saleh T, Bloukh S, Carpenter VJ, Alwohoush E, Bakeer J, Darwish S, et al. Therapy-induced senescence: an “old” friend becomes the enemy. Cancers. 2020;12:822.
Grant GD, Brooks L 3rd, Zhang X, Mahoney JM, Martyanov V, Wood TA, et al. Identification of cell cycle-regulated genes periodically expressed in U2OS cells and their regulation by FOXM1 and E2F transcription factors. Mol Biol Cell. 2013;24:3634–50.
Quaas M, Muller GA, Engeland K. p53 can repress transcription of cell cycle genes through a p21(WAF1/CIP1)-dependent switch from MMB to DREAM protein complex binding at CHR promoter elements. Cell Cycle. 2012;11:4661–72.
Fischer M, Quaas M, Steiner L, Engeland K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 2016;44:164–74.
Fischer M, Quaas M, Nickel A, Engeland K. Indirect p53-dependent transcriptional repression of Survivin, CDC25C, and PLK1 genes requires the cyclin-dependent kinase inhibitor p21/CDKN1A and CDE/CHR promoter sites binding the DREAM complex. Oncotarget. 2015;6:41402–17.
Uxa S, Bernhart SH, Mages CFS, Fischer M, Kohler R, Hoffmann S, et al. DREAM and RB cooperate to induce gene repression and cell-cycle arrest in response to p53 activation. Nucleic Acids Res. 2019;47:9087–103.
Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–4.
He Y, Roos WP, Wu Q, Hofmann TG, Kaina B. The SIAH1-HIPK2-p53ser46 damage response pathway is involved in temozolomide-induced glioblastoma cell death. Mol Cancer Res. 2019;17:1129–41.
Aasland D, Gotzinger L, Hauck L, Berte N, Meyer J, Effenberger M, et al. Temozolomide induces senescence and repression of DNA repair pathways in glioblastoma cells via activation of ATR-CHK1, p21, and NF-kappaB. Cancer Res. 2019;79:99–113.
Knizhnik AV, Roos WP, Nikolova T, Quiros S, Tomaszowski KH, Christmann M, et al. Survival and death strategies in glioma cells: autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS One. 2013;8:e55665.
Reich TR, Schwarzenbach C, Vilar JB, Unger S, Muhlhausler F, Nikolova T, et al. Localization matters: nuclear-trapped Survivin sensitizes glioblastoma cells to temozolomide by elevating cellular senescence and impairing homologous recombination. Cell Mol Life Sci. 2021;78:5587–604.
Beltzig L, Schwarzenbach C, Leukel P, Frauenknecht KBM, Sommer C, Tancredi A, et al. Senescence is the main trait induced by temozolomide in glioblastoma cells. Cancers. 2022;14:2233.
Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66.
Wick A, Felsberg J, Steinbach JP, Herrlinger U, Platten M, Blaschke B, et al. Efficacy and tolerability of temozolomide in an alternating weekly regimen in patients with recurrent glioma. J Clin Oncol. 2007;25:3357–61.
Wick A, Pascher C, Wick W, Jauch T, Weller M, Bogdahn U, et al. Rechallenge with temozolomide in patients with recurrent gliomas. J Neurol. 2009;256:734–41.
Vilar JB, Christmann M. Tomicic, MT. Alterations in molecular profiles affecting glioblastoma resistance to radiochemotherapy: where does the good go? Cancers. 2022;14:2416.
Wischhusen J, Naumann U, Ohgaki H, Rastinejad F, Weller M. CP-31398, a novel p53-stabilizing agent, induces p53-dependent and p53-independent glioma cell death. Oncogene. 2003;22:8233–45.
Christmann M, Nagel G, Horn S, Krahn U, Wiewrodt D, Sommer C, et al. MGMT activity, promoter methylation and immunohistochemistry of pretreatment and recurrent malignant gliomas: a comparative study on astrocytoma and glioblastoma. Int J Cancer. 2010;127:2106–18.
Esteller M, Tortola S, Toyota M, Capella G, Peinado MA, Baylin SB, et al. Hypermethylation-associated inactivation of p14(ARF) is independent of p16(INK4a) methylation and p53 mutational status. Cancer Res. 2000;60:129–33.
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–26.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50.
Fischer M, Grossmann P, Padi M, DeCaprio JA. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016;44:6070–86.
Kolmykov S, Yevshin I, Kulyashov M, Sharipov R, Kondrakhin Y, Makeev VJ, et al. GTRD: an integrated view of transcription regulation. Nucleic Acids Res. 2021;49:D104–D111.
Shepard JL, Amatruda JF, Stern HM, Subramanian A, Finkelstein D, Ziai J, et al. A zebrafish bmyb mutation causes genome instability and increased cancer susceptibility. Proc Natl Acad Sci USA. 2005;102:13194–9.
Christmann M, Kaina B. Nuclear translocation of mismatch repair proteins MSH2 and MSH6 as a response of cells to alkylating agents. J Biol Chem. 2000;275:36256–62.
Christmann M, Tomicic MT, Kaina B. Phosphorylation of mismatch repair proteins MSH2 and MSH6 affecting MutS{alpha} mismatch-binding activity. Nucl Acids Res. 2002;30:1959–66.
Zhang J, Qiao Q, Xu H, Zhou R, Liu X. Human cell polyploidization: the good and the evil. Semin Cancer Biol. 2022;81:54–63.
Engeland K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018;25:114–32.
Musa J, Aynaud MM, Mirabeau O, Delattre O, Grunewald TG. MYBL2 (B-Myb): a central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis. 2017;8:e2895.
Matthews HK, Bertoli C, de Bruin RAM. Cell cycle control in cancer. Nat Rev Mol Cell Biol. 2022;23:74–88.
Sallbach J, Woods M, Rasenberger B, Christmann M, Tomicic MT. The cell cycle inhibitor p21(CIP1) is essential for irinotecan-induced senescence and plays a decisive role in re-sensitization of temozolomide-resistant glioblastoma cells to irinotecan. Biomed Pharmacother. 2024;181:117634.
Collin G, Huna A, Warnier M, Flaman JM, Bernard D. Transcriptional repression of DNA repair genes is a hallmark and a cause of cellular senescence. Cell Death Dis. 2018;9:259.
Felsberg J, Thon N, Eigenbrod S, Hentschel B, Sabel MC, Westphal M, et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer. 2011;129:659–70.
Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB, et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res. 2007;13:2038–45.
Schmidt A, Allmann S, Schwarzenbach C, Snyder P, Chen JX, Nagel G, et al. The p21CIP1-CDK4-DREAM axis is a master regulator of genotoxic stress-induced cellular senescence. Nucleic Acids Res. 2024;
Ochs K, Kaina B. Apoptosis induced by DNA damage O6-methylguanine is Bcl-2 and caspase-9/3 regulated and Fas/caspase-8 independent. Cancer Res. 2000;60:5815–24.
Quiros S, Roos WP, Kaina B. Processing of O6-methylguanine into DNA double-strand breaks requires two rounds of replication whereas apoptosis is also induced in subsequent cell cycles. Cell Cycle. 2010;9:168–78.
Hirose Y, Berger MS, Pieper RO. Abrogation of the Chk1-mediated G(2) checkpoint pathway potentiates temozolomide-induced toxicity in a p53-independent manner in human glioblastoma cells. Cancer Res. 2001;61:5843–9.
Hirose Y, Berger MS, Pieper RO. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001;61:1957–63.
Frey Y, Haj M, Ziv Y, Elkon R, Shiloh Y. Broad repression of DNA repair genes in senescent cells identified by integration of transcriptomic data. Nucleic Acids Res. 2025;53:
Mercapide J, Anzanello F, Rappa G, Lorico A. Relationship between tumor cell invasiveness and polyploidization. PLoS ONE. 2012;7:e53364.
Saleh T, Carpenter VJ, Bloukh S, Gewirtz DA. Targeting tumor cell senescence and polyploidy as potential therapeutic strategies. Semin Cancer Biol. 2022;81:37–47.
Bronner SM, Merrick KA, Murray J, Salphati L, Moffat JG, Pang J, et al. Design of a brain-penetrant CDK4/6 inhibitor for glioblastoma. Bioorg Med Chem Lett. 2019;29:2294–301.
Zhao W, Zhang L, Zhang Y, Jiang Z, Lu H, Xie Y, et al. The CDK inhibitor AT7519 inhibits human glioblastoma cell growth by inducing apoptosis, pyroptosis and cell cycle arrest. Cell Death Dis. 2023;14:11.
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (German Research Foundation)— Project-ID 393547839 – SFB 1361 - TP05 to MC, as well as by the Wilhelm Sander Stiftung—Project-ID 2019.154.1 and by the Deutsche Forschungsgemeinschaft (German Research Foundation) Project-ID 508828494 to MTT, and Project-ID 470145176 to MC. We acknowledge that this work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project-ID 393547839—SFB 1361, through the support of the IMB Genomics Core Facility and the use of its NextSeq500.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
MC and MTT designed the experiments and analyzed the data. ASchm, ASchoe, BR, CS, JR, JBV, JS, MC, and LT conducted the experiments. MC, BK, and MTT wrote the original and revised version of the manuscript. MC and MTT supervised this research work. All authors reviewed the results and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
(a) All methods included in the manuscript were performed in accordance with the relevant guidelines and regulations. (b) The mouse experiments were performed in accordance with relevant institutional and national guidelines and regulations, approved by the Landesuntersuchungsamt Rheinland-Pfalz, Germany (23 177-07/041-15V2).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Boris Zhivotovsky
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schwarzenbach, C., Rinke, J., Vilar, J.B. et al. Therapy-induced senescence of glioblastoma cells is determined by the p21CIP1-CDK1/2 axis and does not require activation of DREAM. Cell Death Dis 16, 357 (2025). https://doi.org/10.1038/s41419-025-07651-8
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41419-025-07651-8
This article is cited by
-
Kaempferol triggers cellular senescence via CDK1 ubiquitination in HCC cells
Cancer Cell International (2025)
-
Integrative analysis of single-cell and bulk transcriptome data reveals age-related immune cell alterations in primary glioblastoma associated with prognosis
Cancer Immunology, Immunotherapy (2025)
-
Senotherapeutics in Malignant Brain Cancer Therapy
Current Oncology Reports (2025)
