
Abstract
Homeobox transcription factors CDX1 and CDX2 (hereafter, CDX1/2) play key roles in determining the identity of intestinal epithelial cells and regulating their stem cell functions. However, the role of CDX1/2 in regulating colon cancer stemness and the underlying mechanisms are unclear. Here, we show that complete loss of Cdx1 or concurrent loss of Cdx1/2 increased the stemness and malignancy of intestinal tumors. Consistently, CDX1/2 reduced the expression of cancer stemness-related genes, including LGR5. CDX1/2 bound to the downstream region of the LGR5 transcription start site (TSS), a region where β-catenin also binds. Despite increased H3 acetylation and an open chromatin structure, CDX1/2 reduced the occupancy of DRB sensitivity-inducing factor (DSIF), RNA polymerase II-associated factor 1 (PAF1), and RNA polymerase II (Pol II) complexes around the LGR5 TSS. Through their homeodomains, CDX1/2 inhibited the β-catenin-facilitated formation of active Pol II complexes containing DSIF and PAF1 complexes by preventing the interaction between β-catenin and these complexes, in an additive manner. Our findings suggest that CDX1/2 cooperatively suppressed colonic tumorigenesis and cancer stemness by antagonizing β-catenin via the DSIF and PAF1 complexes. Additionally, DSIF and PAF1 complexes acted as transcriptional platforms that integrated and funneled both tumor-suppressive and oncogenic signals into the expression of genes that control colon cancer stemness.
Similar content being viewed by others

Introduction
The intestine-specific homeoproteins CDX1/2 play a central role in determining the identity of intestinal epithelial cells, promoting their development and differentiation, and maintaining their homeostasis [1,2,3,4,5,6,7,8]. Previous data showed that CDX2 levels were reduced in human colon cancer tissues [9]. Consistently, Cdx2 mutation or reduced Cdx2 expression enhances Apc mutation- or carcinogen-induced colonic tumorigenesis through cell-autonomous and non-cell autonomous mechanisms [10,11,12,13,14,15,16]. However, the cooperative role of CDX1/2 in malignant progression remains unclear.
Mutations in APC, found in ~80% of colon cancer [17,18,19], enhance β-catenin stabilization and induce colonic tumorigenesis [20,21,22]. Hence, Apc mutant mice have been used to genetically model colonic tumorigenesis [23]. In mice, Cdx1 is located on chromosome 18 (Chr 18), approximately 13 centimorgan from Apc. When Apc+/− mice were crossed with Cdx1+/− mice, the resulting Apc+/−Cdx1+/− mice carried mutant Apc and Cdx1 alleles on different Chr 18 homologs (trans-Apc+/−Cdx1+/−). In Apc+/− mice, intestinal tumorigenesis was initiated by the loss of Chr 18 heterozygosity, resulting in the production of adenoma cells homozygous for the mutant Apc allele [24, 25]. In trans-Apc+/−Cdx1+/− mice, adenoma cells possessed only the mutant Apc allele and the wild-type (wt) Cdx1 allele, which rendered genetic analysis of the role of Cdx1 in Apc+/− mice challenging [15, 26]. Thus, a heterozygous Cdx1 mutation did not produce a phenotype in trans-Apc+/−Cdx1+/− mice, and only weak invasiveness was observed in Apc+/−-Cdx2f/f-Villin-CreERT mouse [15].
CDX1/2 can inhibit colon cancer cell proliferation [10, 27, 28], and play roles in the stem cell function of normal intestinal epithelial cells and in colon cancer cell differentiation [29, 30]. CDX1/2 also decrease the reporter activity in TOPflash assays [28, 31]. These results suggest that CDX1/2 suppress β-catenin–T-cell factor (TCF) transcriptional activity and colon cancer stemness. However, there is no direct evidence of the regulation of colon cancer stemness and of expression of β-catenin-target genes by CDX1/2 (along with the underlying mechanisms). CDX2 binds to genomic regions bound by TCF4 as well and is necessary for the expression of TCF-target genes [32, 33]. However, CDX1/2 and β-catenin–TCF4 complex exhibit antagonistic functions, potentially inhibiting each other within the shared genome of colon cancer cells. In this study, we analyzed the roles of CDX1/2 in the malignant progression of colon cancer, regulation of cancer stemness, and β-catenin-mediated transcription in colon cancer cells.
Results
Malignant progression in intestinal tumors induced by Cdx1 and Cdx2 deletion mutations
Cdx1 and Cdx2 mRNA were detectable in the colonic tumor organoids derived from Apc+/− mice [25] but were expressed at significantly lower levels than those observed in normal colonic epithelium (Fig. 1A). To delete Cdx1 in adenomas in Apc+/− mice, we subsequently generated cis-Apc+/−Cdx1+/− mice that carried both Apc and Cdx1 deletion mutations [34] on the same Chr 18 homolog via meiotic recombination. The loss of wt-Cdx1 and wt-Apc in colonic tumor organoids derived from cis-Apc+/−Cdx1+/− mice was confirmed by PCR (Fig. S1A). We also crossed cis-Apc+/−Cdx1+/− mice with Cdx2+/− mice. The loss of Cdx1 did not affect the number or size of intestinal tumors in Apc+/− mice (Fig. 1B, C and Fig. S1B–D). In contrast, the Cdx2 mutation resulted in increased number of colonic tumors and formation of large tumors that were not observed in Apc+/− and cis-Apc+/−Cdx1+/− mice (Fig. 1B, C and Fig. S1D), as reported previously [10]. Tumor epithelial cells in the small intestine of cis-Apc+/−Cdx1+/− mice deeply invaded the submucosa (Fig. 1D, E and Fig. S2A–C, and Table S1). Although no invasion was observed in case of Apc+/−Cdx1+/− colonic tumors (Fig. 1F), an additional Cdx2 mutation induced submucosal invasion (Fig. 1G and Fig. S2D). These results provide genetic evidence that Cdx1, cooperatively with Cdx2, suppressed the malignant progression of intestinal tumors.

A qPCR data showing the relative expression (mean ± SD) of Cdx1, Cdx2, and Lgr5 in colonic tumor organoids derived from Apc+/− mice (compared with those in normal epithelium). P-values were calculated using a Student′s t-test (A–C). Number (B) and size (C) of intestinal tumors in Apc+/−, cis-Apc+/−Cdx1+/−, and Cdx2+/−-cis-Apc+/−Cdx1+/− mice at 10–12 weeks of age (mean ± SD; n = 4–5, excluding dysplastic crypts). Hematoxylin and eosin (H&E)-stained small intestinal tumors in Apc+/− (D) and cis-Apc+/−Cdx1+/− (E) mice. Green arrows indicate invasive tumor cells (E, G). Abbreviations in D–G: mm muscularis mucosa, mp muscularis propria, se serosa. Scale bars, 200 µm (D–G). H&E-stained colonic tumors from cis-Apc+/−Cdx1+/− (F) and Cdx2+/−-cis-Apc+/−Cdx1+/− (G) mice.
Suppression of colon cancer stemness by CDX1 and CDX2
We analyzed the gene expression profiles after expressing mouse wt-Cdx1 or wt-Cdx2 in human colon cancer-derived DLD1-TetOff cells (Fig. 2A, B and Fig. S3A). Microarray analysis showed that 12 and 24 h after expressing wt-Cdx1 and wt-Cdx2, two members of the inhibitor of DNA binding (ID) family (ID1 and ID3), as well as c-MYC were downregulated by 60-90% in DLD1-TetOff cells (Fig. 2C and Table S2). Similar results were observed via qPCR (Fig. 2D, DLD1-TetOff cells; Fig. 2E, LS174T-TetOff cells). Reduced c-MYC expression at the protein level was also confirmed following expression of wt-Cdx1 and wt-Cdx2 (Fig. 2F). Notably, previous studies have shown that ID1, ID3, and c-MYC help maintain the self-renewal capacity of colon cancer stem cells [22, 35, 36]. Concordantly, the expression of cancer stemness-related genes, such as LGR5 and CD44, was significantly reduced at the mRNA and protein levels in TetOff cells upon expressing wt-Cdx1/2 (Fig. 2D–F and Fig. S3B, C). This expression was also reduced upon expression of a transcription-deficient homeodomain (HD) mutant of Cdx2, i.e., Cdx2-R189A (Cdx2-R:A) but not by the other HD mutants, i.e., Cdx2-R189E-N235E-R237E (Cdx2-RNR:3E) and Cdx2-R189A-N235A (Cdx2-RN:2A) (Fig. 2B, D, F and Fig. S3B). Cell-surface expression of CD44 also decreased in TetOff cells upon expressing wt-Cdx1/2 (Fig. 2G and Fig. S4A, B, D), but not by Cdx2-RNR:3E (Fig. 2G and Fig. S4C). Amino acid residues 189, 235, and 237 in Cdx2 HD and their corresponding residues 158, 204, and 206 in Cdx1 HD were found to be critical for DNA binding [27, 37]. These results suggest that CDX1/2 suppressed the expression of genes essential for colon cancer stemness through their HDs.

A Immunoblots showing doxycycline-controlled inducible expression of FLAG-wt-Cdx2 or its homeodomain (HD) mutants in DLD1-TetOff cells. β-Actin was a loading control (A, F). B CDH17 luc reporter activities (mean ± SD) relative to those of the pGL4.10-luc2 control upon expressing wt-Cdx2 or its HD mutants. P-values were calculated using a Student′s t-test (B, D, E). C Microarray data showing the gene expression profiles of DLD1-TetOff cells after expression of wt-Cdx1 (Cy3) for 12 h (compared with those of cells not expressing wt-Cdx1; Cy5). Red arrows denote the relationship between cells with Cdx1 expression and without Cdx1 expression in terms of ID1, ID3, and c-MYC expression. qPCR data showing the relative expression (mean ± SD) of colon cancer stemness-related genes upon expressing wt-Cdx1, wt-Cdx2, or Cdx2 HD mutants for 2 days in DLD1-TetOff cells (D) and for 4 and 7 days in LS174T-TetOff cells (E), when compared with cells without their expression. F Immunoblots showing the expression of LGR5, CD44, and c-MYC upon expressing wt-Cdx1, wt-Cdx2, or Cdx2-RNR:3E in DLD1-TetOff and LS174T-TetOff cells. Note that the basal expression levels of LGR5 were different among the TetOff clones. G Immunocytochemistry showing the expression of CD44 (green) and Cdx1/2 (red) upon expression of wt-Cdx1, wt-Cdx2, or Cdx2-RNR:3E in DLD1-TetOff and LS174T-TetOff cells. The nuclei were stained with DAPI (blue). Scale bars, 20 µm.
Increased colon cancer stemness by Cdx1 and Cdx2 deletion mutations
In colonic tumor organoids derived from cis-Apc+/−Cdx1+/− and Cdx2+/−-cis-Apc+/−Cdx1+/− mice, the expression of Cdx1 and Cdx2 was lower than that in Apc+/− tumor organoids, whereas that of Lgr5 was higher (Fig. 3A). Similarly, higher proliferation rates were observed in the small-intestinal tumor organoids derived from cis-Apc+/−Cdx1+/− mice (Fig. 3B) and in colonic tumor organoids derived from cis-Apc+/−Cdx1+/− and Cdx2+/−-cis-Apc+/−Cdx1+/− mice than in those derived from Apc+/− mice (Fig. 3C, D). These results indicate that reduced expression of Cdx1/2 promoted colon cancer stemness.

A qPCR data showing the relative expression (mean ± SD) of Cdx1, Cdx2, and Lgr5 in colonic tumor organoids derived from cis-Apc+/−Cdx1+/− and Cdx2+/−-cis-Apc+/−Cdx1+/− mice (compared with those from Apc+/− mice). P-values were calculated using a Student′s t-test (A–C). Growth rate (mean ± SD) of organoid cells in normal and tumor epithelial tissues derived from the small intestine (B) and colon (C) of Apc+/−, cis-Apc+/−Cdx1+/−, and Cdx2+/−-cis-Apc+/−Cdx1+/− mice. D Tumor organoids derived from Apc+/−, cis-Apc+/−Cdx1+/−, and Cdx2+/−-cis-Apc+/−Cdx1+/− colonic tumors. Scale bars, 100 µm. E, F Immunohistochemistry of Cd44 expression (brown) in a small intestinal tumor of a cis-Apc+/−Cdx1+/− mouse. F shows the magnified image of the boxed region in (E). The arrows in F indicate cells with elevated Cd44 expression, while arrowheads indicate cells with lower Cd44 expression. The tissue was also counterstained with hematoxylin (blue). Scale bars, 100 µm (E) and 20 µm (F).
The β-catenin-target gene, Cd44, is a marker of cancer stemness [38] and has been implicated in tumor invasion [39]. Cd44 was upregulated in the leading cells at the invasive front of the intestinal tumors in cis-Apc+/−Cdx1+/− mice (Fig. 3E, F and Fig. S5A–K). In contrast, Cd44 expression was lower in cells on the opposite side (Fig. 3E, F and Fig. S5A–K) and in cells in the deeply invaded region (Fig. S5L). These results suggest that a cancer-stem cell feature mediated invasion during colonic tumorigenesis. Proliferating cells were also observed in the non-invasive and invasive regions of cis-Apc+/−Cdx1+/− intestinal tumors (Fig. S5M–O). The role of increased proliferation through CDX1/2 suppression during invasion remains to be further investigated.
Reduction of RNA polymerase II (Pol II), DRB sensitivity-inducing factor (DSIF), and RNA polymerase II-associated factor 1 (PAF1) levels around the TSS of LGR5 induced by CDX1 and CDX2
To elucidate the mechanism whereby CDX1/2 suppressed LGR5 expression, we conducted a chromatin immunoprecipitation-sequencing (ChIP-seq) analysis of FLAG-tagged wt-Cdx1/2 variants expressed in DLD1-TetOff cells. A significant peak was observed approximately 1000 base pairs (bp) downstream of the LGR5 transcription start site (TSS; Fig. 4A), which was confirmed by ChIP-qPCR for both endogenous CDX2 (Fig. 4B) and exogenously expressed wt-Cdx1/2 (Fig. S6A, B). In contrast, the binding of wt-Cdx2 to the LGR5 gene was weakened by the R189A-N235A mutation (Fig. S6C, Cdx2-RN:2A). We recently constructed LGR5 luciferase (luc) reporter plasmids [40], wherein luc was flanked by Cdx1/2 binding regions upstream and downstream of the LGR5 TSS (Fig. 4A, C). As reported [40], a stable β-catenin mutant with an S33Y substitution increased the LGR5 luc reporter activity (Fig. 4C). In contrast, LGR5 luc reporter activity was suppressed by wt-Cdx1/2 in a dose-dependent manner; the activity decreased from 537 to 219 (41%) and from 537 to 86.4 (16%) after transfecting H293T cells with 0.01 μg of a Cdx1-expression vector (plasmid-Cdx1) or a plasmid-Cdx2, respectively (Fig. 4C). LGR5 luc reporter activity decreased from 537 to 68.5 (13%) after co-transfecting 0.01 μg each of plasmid-Cdx1 and plasmid-Cdx2 (Fig. 4C). These results suggest that CDX1 and CDX2 cooperated to suppress LGR5 expression in an additive manner (rather than synergistically). LGR5 luc reporter activity was also suppressed by Cdx2-R:A but not by Cdx2-RN:2A, Cdx2-RNR:3E, or other Cdx2 HD mutants (Fig. 4D). Similar results were obtained for the Cdx1 HD mutants (Fig. S6D). As no significant differences were found between the functions of CDX1 and CDX2, we mainly analyzed CDX2 in subsequent experiments.

A Integrative genome-viewer window showing the occupancy of FLAG-tagged wt-Cdx1 and wt-Cdx2 on the LGR5 gene in DLD1-TetOff cells. Genomic regions analyzed by ChIP-qPCR in B are indicated above, whereas those cloned in the LGR5 luc reporter C, D are indicated below. B ChIP-qPCR data showing the relative occupancy (mean ± SD) of endogenous CDX2 at the indicated positions of LGR5 and CDX2-target gene CDH17 (used as a positive control) in T84 cells. P-values were calculated using a Student′s t-test (B–G). C LGR5 luc reporter activities (mean ± SD) relative to those of the pGL4.10-luc2 control upon expressing wt-Cdx1, wt-Cdx2, or both wt-Cdx1 and wt-Cdx2. The amounts of transfected plasmid DNA used to overexpress wt-Cdx1 and wt-Cdx2 are indicated below the bar graph. D LGR5 luc reporter activities (mean ± SD) relative to those of the pGL4.10-luc2 control upon expressing wt-Cdx2, or its homeodomain (HD) mutants, or β-catenin-S33Y. E ChIP-qPCR data showing the relative occupancy (mean ± SD) of H3K27ac and H3K4me3 at the indicated positions of LGR5 and CDH17 after expressing wt-Cdx2 in DLD1-TetOff cells for 1 day. F qPCR data quantifying MNase protection assays reflecting the chromatin architecture at the indicated positions in LGR5 after expressing wt-Cdx2 in DLD1-TetOff cells for 1 day. G ChIP-qPCR data showing the relative occupancy (mean ± SD) of Pol II, SPT5, and PAF1 at the indicated positions in LGR5 after expressing wt-Cdx2 in DLD1-TetOff cells for 1 day.
Expressing wt-Cdx2 increased H3K27ac and H3K4me levels around the promoter region and first intron of both LGR5 and CDX2-target gene CDH17 (Fig. 4E) [41]. Even after wt-Cdx2 expression, closed chromatin structures were not observed around the LGR5 TSS, as determined in the micrococcal nuclease-protection assays (Fig. 4F). We then analyzed the dynamics of Pol II and its-associated factors, including SPT5, a component of DSIF complex [42], and PAF1, a component of RNA polymerase-associated factor 1 complex (PAF1C) [43]. These molecules are essential for promoter-proximal pausing and regulate mRNA transcription [43,44,45,46]. Wt-Cdx1/2 decreased the occupancy levels of Pol II, SPT5, and PAF1 downstream of the LGR5 TSS (Fig. 4G and Fig. S6E); however, a similar result was not obtained with Cdx2-RN:2A (Fig. S6F). These results suggest that CDX1/2 suppressed Pol II-mediated elongation from the promoter-proximal region of LGR5 through HD.
Suppression of β-catenin-facilitated active Pol II complex formation by CDX1/2 through DSIF and PAF1C
We recently showed that β-catenin regulated LGR5 expression through DSIF and PAF1 complexes (Fig. 5A) [40]. As reported in this study [40], β-catenin-S33Y increased LGR5 luc reporter activity cooperatively with PAF1C components (Fig. 5B). In contrast, wt-Cdx1/2 suppressed LGR5 luc reporter activity (Fig. 5B). Interestingly, wt-Cdx1/2 interacted with PAC1C components (Fig. S7A, B). We then analyzed the role of CDX1/2 in regulating the formation of active Pol II complex via PAF1C by expressing essential components, including SPT5 from DSIF; PAF1 and CDC73 from PAF1C; and RPB2, 3, and 5 from PoI II; and TFIIS (Fig. 5A), based on previously reported findings [40]. In this experiment, we hypothesized that the amount of TFIIS bound to SPT5 or PAF1C correlated with the formation of active Pol II complex (Fig. 5A) [40].

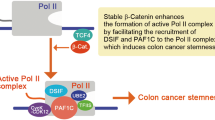
A Mechanism of β-catenin-facilitated formation of the active Pol II complex through PAF1C. Step 1: TCF4 recruits β-catenin to its target genes; Step 2: β-catenin recruits the DSIF complex to Pol II; Step 3: β-catenin facilitates the formation of the Pol II–DSIF–NELF complex; Step 4: β-catenin facilitates the formation of the Pol II–DSIF–PAF1C complex; Step 5: The Pol II–DSIF–PAF1C complex forms a complex with TFIIS, UBE2, and cyclin K (CycK)-CDK12 and initiates mRNA transcription. B LGR5 luc reporter activities (mean ± SD) relative to those of the control pGL4.10-luc2 upon expressing PAF1C components, β-catenin-S33Y, and wt-Cdx2. P-values were calculated using a Student′s t-test. Immunoprecipitation (IP) assays showing the effect of wt-Cdx2 on β-catenin-S33Y (β-Cat.)-facilitated complex formation of C SPT5 (containing PAF1, TFIIS, and RPB5) and D PAF1 (containing SPT5, TFIIS, and RPB5). In the IP assays, the indicated proteins were co-expressed with either FLAG-SPT5 (C) or FLAG-PAF1 (D). The co-immunoprecipitated Myc-tagged proteins were analyzed using immunoblotting. The amounts of plasmid DNA transfected are indicated on the right. E IP assays showing the effects of wt-Cdx1, wt-Cdx2, or both wt-Cdx1 and Cdx2 on β-catenin-S33Y (β-Cat.)-facilitated formation of a complex involving TFIIS, SPT5, PAF1, and RPBs. The indicated proteins were expressed in the IP assays along with FLAG-TFIIS. The co-immunoprecipitated Myc-tagged proteins were analyzed via immunoblotting. The amounts of transfected plasmid DNA used for expressing wt-Cdx1 and wt-Cdx2 are indicated above the gel images.
We expressed FLAG-SPT5 along with Myc-tagged variants of PAF1, TFIIS, and RPBs. β-Catenin-S33Y increased the binding of FLAG-SPT5 to Myc-tagged RPB2, PAF1, and TFIIS with the cooperation with CDC73 (Fig. 5C, lane 4), whereas wt-Cdx2 impaired those interactions (Fig. 5C, lanes 5–7). Similarly, β-catenin-S33Y increased the binding of FLAG-PAF1 to Myc-tagged RPB2, SPT5, and TFIIS with the cooperation with CDC73 or CDK9 (Fig. 5D, lanes 4, 8), whereas wt-Cdx2 suppressed such binding (Fig. 5D, lanes 5-7, 9). Additionally, wt-Cdx1/2 suppressed the β-catenin-S33Y-induced increase in the binding of FLAG-TFIIS to Myc-tagged SPT5, PAF1, and RPBs in a dose-dependent manner (Fig. 5E, lanes 4–7, 8–10). Transfection of 0.1 μg plasmid-Cdx2 suppressed the formation of the Pol II–DSIF–PAF1C complex to a similar extent as transfection of 0.2 μg plasmid-Cdx1 (Fig. 5E, lanes 4, 8). It is noted that both plasmid-Cdx2 and plasmid-Cdx1 increased the CDH17 luc reporter activity to a similar extent (Fig. 2B and Fig. S3A). These results suggest that Cdx2 had an approximately 2-fold more potent inhibitory effect on complex formation than Cdx1, consistent with the results above (Fig. 4C). Co-transfection of 0.1 μg plasmid-Cdx2 and 0.2 μg plasmid-Cdx1 resulted in a similar inhibitory effect on complex formation as transfection of 0.2 μg plasmid-Cdx2 alone (Fig. 5E, lanes 5, 11). Collectively, these results indicate that CDX1/2 additively suppressed β-catenin-facilitated formation of the active Pol II complex by inhibiting the binding of DSIF and PAF1C to Pol II and that the combined levels of CDX1/2 determined the extent of suppression.
Suppression of β-catenin-facilitated Pol II–DSIF–PAF1C complex formation by CDX2 was abrogated by a mutation in its HD
Consistent with the observation that the Cdx2-RN:2A or Cdx2-RNR:3E mutant did not reduce LGR5 expression at the mRNA level (Fig. 2D, F), LGR5 luc reporter activity was not suppressed by Cdx2-RN:2A, Cdx1-RN:2A or Cdx1-RNR:3E (Fig. 6A and Fig. S6D). However, LGR5 luc reporter activity was suppressed by Cdx2-R:A and Cdx1-R:A, albeit less effectively than that by wt-Cdx1/2 (Fig. 6A and Fig. S6D). Likewise, Cdx2-RN:2A or Cdx1-RNR:3A did not reduce the binding of FLAG-SPT5 to Myc-tagged RPB2 or PAF1, or the binding of FLAG-TFIIS to Myc-tagged SPT5, PAF1, and RPBs in the presence of β-catenin-S33Y and CDC73 (Fig. 6B, C and Fig. S8A, lanes 7, 8). However, the Cdx2-R:A and Cdx1-R:A mutants did suppress such binding, although less effectively than that by wt-Cdx2 and wt-Cdx1 (Fig. 6B, C and Fig. S8A, lanes 5, 6, 9, 10). These results suggest that the HD of CDX1/2 helped suppressed β-catenin-facilitated formation of the active Pol II complex through a mechanism independent of transcriptional activation.

A LGR5 luc reporter activities (mean ± SD) relative to those of the pGL4.10-luc2 control upon expressing β-catenin-S33Y, PAF1C components, wt-Cdx2, and its homeodomain (HD) mutants. P-values were calculated using a Student′s t-test. IP assays showing the effects of wt-Cdx2 and its HD mutants on β-catenin-S33Y (β-Cat.)-facilitated complex formation: B SPT5-containing complexes with PAF1, TFIIS, and RPB5 or C TFIIS-containing complexes with SPT5, PAF1, and RPBs. In the IP assays, the indicated proteins were expressed along with FLAG-SPT5 (B) or FLAG-TFIIS (C).
Suppression of β-catenin-facilitated Pol II–DSIF–PAF1C complex formation by CDX2 mediated by its N-terminus and HD
To delineate the regions of CDX2 responsible for suppressing β-catenin-facilitated Pol II activation, we further employed Cdx2-deletion mutants harboring its HD (Fig. 7A). Mutant C2HD contained HD, and mutant C2HD-C2 contained HD and its C-terminally adjacent lysyl-lysine (KK) domain that aids nuclear localization [27]. C2HD-N mutants contained HD with C-terminal KK, and different N-terminal extensions harboring the transactivation domain [27, 47]. C2HD-C mutants contained HD and different C-terminal extensions. Deletion of the N-terminal region impaired the Cdx2-induced suppression of LGR5 reporter activity (Fig. 7A, C2HD-N1). Consistently, the N-terminal deletion mutants of Cdx2 did not reduce the β-catenin-S33Y-induced FLAG-TFIIS-bound Myc-tagged SPT5, PAF1, and RPBs levels (Fig. 7B, lane 7). Interestingly, HD mutations enhanced the interactions of Cdx1/2 with PAF1 and SPT5 complexes (Fig. 7C, lanes 6–9 and Fig. S9A, lanes 9–14). Likewise, the HD interacted with PAF1 and SPT5 complexes (Fig. 7C, lanes 14, 15). Therefore, the N-terminal domain and HD of CDX1/2 mainly contributed to the suppression of β-catenin-facilitated formation of the active Pol II complex.

A LGR5 luc reporter activities (mean ± SD) relative to those of the pGL4.10-luc2 control upon expressing β-catenin-S33Y, CDC73, and wt-Cdx2 and its deletion mutants. P-values were calculated using a Student′s t-test. B IP assays showing the effects of wt-Cdx2 and its deletion mutants on the β-catenin-S33Y (β-Cat.)-facilitated formation of a complex involving TFIIS, SPT5, PAF1, and RPBs. C IP assays showing the interaction between SPT5 and Cdx2 (or its mutants) and between PAF1 and Cdx2 (or its mutants).
Suppression of the interactions of β-catenin with DSIF and PAF1C by CDX1 and CDX2
We further examined the effects of CDX1/2 on the interactions between β-catenin and Pol II, DSIF, and PAF1 complexes. To this end, FLAG-tagged β-catenin-S33Y was co-expressed with Myc-tagged SPT5, PAF1, CDC73, and RPBs (Fig. 8A). Wt-Cdx1/2 suppressed the interaction between β-catenin and the SPT5, PAF1, and Pol II complexes (Fig. 8A, lanes 5, 6, 9, 10), whereas Cdx2-RN:2A and Cdx1-RNR:3A mutations weakened this suppression by wt-Cdx1/2 (Fig. 8A, lanes 7, 8, 11, 12 and Fig. S10A–C).

A IP assays showing the effects of wt-Cdx1/2 and their homeodomain (HD) mutants on the interaction of β-catenin-S33Y (β-Cat.) with SPT5, PAF1 components, and RPBs. The indicated proteins were expressed in the IP assays along with FLAG-β-catenin-S33Y. B IP assays showing the effects of wt-Cdx1/2 and their HD mutants on the interaction of FLAG-TCF4 with PA-β-catenin-S33Y (β-Cat.), Myc-tagged SPT5, PAF1 components, and RPBs. In the IP assays, the indicated proteins were expressed along with FLAG-TCF4. C ChIP-qPCR data showing the relative occupancy (mean ± SD) of β-catenin at the indicated positions in LGR5 after expressing wt-Cdx2 or Cdx2-RN:2A for 1 day in DLD1-TetOff cells, when compared with that in cells with Cdx unexpressed. P-values were calculated using a Student′s t-test. D Transcriptional mechanism underlying the inhibition of stable β-catenin by CDX1/2 via DSIF and PAF1C complexes, resulting in the suppression of colon cancer stemness. First, TCF4 recruits β-catenin to its target gene. β-Catenin then recruits DSIF and PAF1 complexes to the Pol II complex to facilitate the formation of the active Pol II complex, which promotes colon cancer stemness. CDX1/2 suppress these processes. DSIF and PAF1 complexes act as platforms that integrate and funnel oncogenic and tumor-suppressive signals into gene expression, thereby controlling cancer stemness.
We further expressed FLAG-tagged TCF4 along with PA-tagged β-catenin-S33Y, Myc-tagged SPT5, and PAF1C components. β-Catenin-S33Y and CDC73 increased the SPT5 and PAF1C levels in the TCF4 complex (Fig. 8B, lanes 5, 6 and Fig. S10D) but not in a TCF4ΔN mutant lacking the domain required for β-catenin binding (Fig. 8B, lane 13). Due to reduced levels of PA-tagged β-catenin-S33Y in the presence of Cdx1/2 (Fig. S10D), we increased the amount of the transfected plasmid DNA expressing PA-tagged β-catenin-S33Y in cells expressing wt-Cdx1/2 (Fig. 8B, lanes 7, 8, 10, 11). Wt-Cdx1/2 significantly reduced the β-catenin-S33Y- and CDC73-induced SPT5 and PAF1C levels in the TCF4 complex (Fig. 8B, lanes 7, 8, 10, 11), whereas Cdx2-RN:2A and Cdx1-RNR:3A mutations weakened the effects of wt-Cdx1/2 (Fig. 8B and Fig. S10D, lanes 9, 12). Wt-Cdx1/2 also reduced β-catenin levels in the TCF4 complex (Fig. 8B and Fig. S10D), consistent with an earlier study [28]. Likewise, wt-Cdx1/2 reduced the amount of β-catenin bound to LGR5, whereas Cdx2-RN:2A did not (Fig. 8C and Fig. S10E). These results suggest that CDX1/2 suppressed β-catenin-facilitated formation of the active Pol II complex by disrupting the interactions between β-catenin and the DSIF and PAF1 complexes and those between β-catenin and TCF4 (Fig. 8D).
Discussion
The complete loss of Cdx1 resulted in extensive invasion of the small-intestinal tumor cells, whereas the absence of Cdx1 in conjugation with a heterozygous Cdx2 mutation led to the invasion of the colonic tumor cells (Fig. 1). Organoids derived from colonic tumors carrying Cdx1 and Cdx2 mutations exhibited increased proliferation and elevated Lgr5 expression levels (Fig. 3). Additionally, we analyzed data hosted on the Gene Expression Omnibus (GEO) Database and compared the gene expression levels between LGR5high and LGR5low cells from five colorectal cancer (CRC) human organoid clones [48]. In four out of five human CRC organoid clones, the levels of either both CDX1 and CDX2, or one of the genes, were lower in LGR5high cells than in those in LGR5low cells, with no overall increases observed in CDX1/2 expression (Table S3). In the remaining clone, the level of CDX2 expression decreased to 0.79, while that of CDX1 increased to 1.18 in LGR5high cells (Table S3). Moreover, recent data show that in the stem cells of human colon epithelial cells, the levels of CDX1 and CDX2 are reduced to 0.07 and 0.22, respectively (compared with normal epithelial colon cells), while those of LGR5 increased to 3.48 [49]. Collectively, these results suggest that CDX1/2 helped suppress stemness in both normal and cancerous colon epithelial cells of human and mouse origin. Cdx1/2 cooperatively and additively suppressed β-catenin-induced LGR5 luc reporter activity and active Pol II complex formation (Figs. 4 and 5). These findings further suggest that the overall levels of CDX1 and CDX2 determine the suppressive effect on the stemness of colon cancer cells.
Cdx2 bound to the LGR5 gene and increased the levels of H3K27ac and H3K4me3, similar to its target gene, CDH17, resulting in an open chromatin structure around the LGR5 TSS (Fig. 4). These results are consistent with previous data that showed that Cdx2 contributed to the elevated levels of H3K27ac and the expression of intestinal epithelium-specific genes [50]. Therefore, CDX2 may play a role in enhancing the H3K27ac levels and promoting an open chromatin structure around intestinal epithelium-specific genes. However, Cdx1/2 suppressed the binding of Pol II, SPT5, and PAF1 to the LGR5 TSS and reduced LGR5 expression (Fig. 8D). Collectively, these findings suggest that CDX1/2 suppressed LGR5 expression by regulating core transcriptional machinery, distinct from epigenetic regulatory mechanisms. Cdx2 is also reported to inhibit DNA repair by interacting with Ku proteins [51], suggesting that CDX1/2 also participate directly in DNA metabolism.
Recently, we reported that expression of stable β-catenin induced LGR5 transactivation by facilitating the formation of active Pol II complex [40]. CDX1/2 counteracted this β-catenin-facilitated formation of the active Pol II complex by inhibiting the interaction of β-catenin with DSIF and PAF1C (Fig. 8D). The functions of Cdx1/2 were suppressed by Cdx1/2 harboring HD mutations, such as Cdx1-RN:2A, Cdx1-RNR:3A, and Cdx2-RN:2A, but not by Cdx1-R:A or Cdx2-R:A (Figs. 2 and 6). These results suggest that the HDs of CDX1/2 have a different function, separate from transactivation. The alternatively spliced form of CDX2, miniCDX2, has also been reported to exhibit a non-transcriptional function that increases p27Kip1 expression but does not inhibit the β-catenin–TCF transcriptional activity [52]. Collectively, these results suggest that CDX1/2 exhibits some non-transcriptional functions.
Recently, we reported that PAF1C plays a crucial role in maintaining the stemness of colon cancer cells by regulating the expression of genes associated with cancer stemness [40]. The results of this study also indicate that CDX1/2 inhibit the DSIF and PAF1C functions, while β-catenin acts as an activator. Therefore, DSIF and PAF1C function as a transcriptional platform that controls colon cancer stemness by integrating and funneling both tumor-suppressive and oncogenic signals into gene expression (Fig. 8D).
Materials and methods
cis-Apc +/− Cdx1 +/− and Cdx2 +/−-cis-Apc +/− Cdx1 +/− mutant mice
Apc+/−, Cdx1+/−, Cdx2+/−, and Apc+/−Cdx2+/− mice were generated as described previously [6, 10, 25]. To generate cis-Apc+/−Cdx1+/− mice, a Cdx1 null mutation was introduced into Apc+/− mutant mice. Considering that both Cdx1 and Apc are located on Chr 18, cis-Apc+/−Cdx1+/− mice were generated via meiotic recombination with both mutations on the same chromosomal homolog. To generate Cdx2+/−-cis-Apc+/−Cdx1+/− mutant mice, cis-Apc+/−Cdx1+/− mice were crossed with Cdx2+/− mice. All animal experiments were approved by the Animal Care and Use Committee of FUKUI University (approval number R04023).
Number of intestinal tumors
The numbers of intestinal tumors in four to five mice of each mutant strain were determined when the mice were 10–12 weeks of age, as described previously [10].
TetOff cell clones
Cells were cultured in Dulbecco’s Modified Eagle’s medium supplemented with 5% fetal bovine serum. To generate TetOff cell clones, plasmids were transfected into TetOff cells using Lipofectamine LTXTM Reagent with PlusTM Reagent (15338100; Thermo Fisher Scientific). TetOff cell clones were generated by transfecting the human colon cancer cell lines, DLD1 (CCL-221; ATCC) and LS174T (CL188; ATCC) with pTet-Off- (631017; TAKARA)-based plasmid vectors in accordance with the methodology in a previous study [27, 40]. Gene expression was regulated by adding doxycycline (24390-14-5; TGI), a tetracycline analog, to the culture medium at a final concentration of 0.5 µg/mL. pTRE-Tight (631059; TAKARA)-based plasmid vectors capable of inducible expression of Cdx1, Cdx2, and their HD mutants were transfected into DLD1-TetOff and LS174T-TetOff cells in accordance with a previously published methodology [27, 40]. Both Cdx1 and Cdx2 were of mouse origin [27]. These TetOff cells were then exposed to 200 µg/mL hygromycin B gold (ant-hg; InvivoGen) for over 2 weeks after expansion of single-cell clones.
Cdx1 and Cdx2 mutagenesis
Cdx1 and Cdx2 were mutated using the PrimeSTAR® Mutagenesis Basal Kit (R046A; TAKARA) and the oligonucleotide primers used for mutagenesis are listed in the Supplementary Materials and Methods. C2HD mutants have been described previously [27].
DNA microarray analysis
For DNA microarray analysis, RNA was isolated from DLD1-TetOff cells expressing wt-Cdx1 or wt-Cdx2 for 12 and 24 h, using the TRI Reagent® (T9424; Sigma-Aldrich). The cells were treated with DNase, and total RNA was purified using the RNeasy Midi kit® (75144; QIAGEN). RNA quality was analyzed using a Bioanalyzer (Agilent), and gene expression was analyzed by the 3D-Gene platform (TORAY, Japan). As Cdx1 expression in DLD1-TetOff-Cdx1 cells was induced faster than Cdx2 in DLD1-TetOff-Cdx2 cells, RNA was purified at 12 and 24 h post-induction for Cdx1 or 24 h post-induction for Cdx2. The associated GEO accession number is GSE287318.
ChIP-sequencing analysis
To conduct ChIP-sequencing analysis of DNA bound by Cdx1 and Cdx2, samples were prepared using a SimpleChIP® Plus Enzymatic Chromatin IP Kit (9004; Cell Signaling Technology).
For our analysis, 4–5 ×106 DLD1-TetOff cells were seeded onto ten 150-mm dishes, and the expression of FLAG-tagged Cdx1 or Cdx2 was induced by withdrawing DOX from the culture medium. At 20 h post-induction, the cells were fixed by adding 540 µL of formaldehyde (a final concentration, 1%; 064-03843; FUJIFILM) to 20 mL culture medium and incubated for 5 min at 25 °C. Glycine (10×, 2 mL; 077-00735; FUJIFILM) was added to the 10 dishes, which were gently rotated for 5 min at 25 °C. The cells were washed twice with 10 mL ice-cold phosphate-based saline (PBS) and scraped with 1 mL of ice-cold PBS. Cells from the 10 dishes were pooled into a 15 mL centrifuge tube and centrifuged at 3,500 rpm (2,380 ×g; AX-320, TOMY) for 3 min at 4 °C, after which the supernatant was then discarded.
According to the manufacturer’s instructions of the SimpleChIP® Plus Enzymatic Chromatin IP Kit (Agarose Beads; 9004; Cell Signaling Tech.), nuclei were isolated and treated with 0.5 µL micrococcal nuclease solution (MNase; Worthington Bio. Corp.) per sample at 37 °C for 50 min. The MNase digestion reaction was terminated by adding 10 µl of 0.5 M ethylenediaminetetraacetic acid (EDTA). The size of the digested DNA (approximately 150 bp) was confirmed via agarose gel electrophoresis. The samples were centrifuged at 13,000 rpm (15,300 ×g; MX-307, TOMY) for 1 min at 4 °C, after which the supernatant was discarded. Each cell pellet was re-suspended in 100 µL ChIP dilution buffer (150 mM NaCl, 20 mM Tris-HCl, 1% Triton X-100, and 2 mM EDTA), incubated for 10 min on ice, transferred to TPX tubes, and sonicated to disrupt the nuclear membrane at 4 °C using a Biorupter II (Sonicbio). The samples were then centrifuged at 10,000 rpm (9,100 ×g; MX-307, TOMY) for 10 min at 4 °C, and then each supernatant was transferred to a new tube. A 19-fold volume of the ChIP dilution buffer was added to the digested chromatin, and each mixture was incubated with anti-FLAG® M2-antibody-conjugated agarose beads (A2220; Sigma Aldrich) for 12 h at 4 °C, with rotation. The beads were washed six times with ChIP dilution buffer and treated with RNase (final concentration, 20 µg/mL; Worthington Bio. Corp.) for 1.5 h at 4 °C. To elute the FLAG-Cdx1 or FLAG-Cdx2 complexes, the beads were incubated with 200 µL ChIP dilution buffer containing the 3×FLAG peptide (F4799; Sigma Aldrich) at 37 °C for 1 h. The samples were centrifuged at 15,000 rpm (20,400 ×g; MX-307, TOMY) for 1 min at 4 °C, after which 180 µL of each supernatant was transferred to a new tube. A 2× elution buffer consisting of 2% sodium dodecyl sulfate (SDS) and 0.2 M NaHCO3 was added to the samples, and the mixtures were incubated at 65 °C overnight. DNA was purified using the standard phenol–chloroform extraction method and analyzed on the Illumina MiSeq platform (Illumina) by Hokkaido System Science. The GEO-associated accession number is GSE287500.
DATA analysis
Statistical analyses were performed using a Student′s t-test in Microsoft Excel to analyze tumor numbers (Fig. 1A–C and Fig. S1B) and data generated in luciferase reporter assays (Figs. 2B, 4C, D, 5B, 6A, 7A, and Figs. S3A, S6D), qPCR assays (Figs. 2D, E, 3A, and Fig. S3B, C), ChIP-qPCR assay (Figs. 4B, E, G, 8C, and Figs. S6A–C, E, F, S10E), proliferation assays (Fig. 3B, C), and MNase assays (Fig. 4F). The tumor numbers were obtained from four to five mice of each genotype. Luciferase reporter data are presented based on from quadruplicate assays. Data from other experiments, including qPCR, ChIP-qPCR, proliferation, and MNase assays, are generated in triplicate.
Analysis of CDX1/2 expression in human CRC organoids
The expression of CDX1/2 in human CRC organoid clones was analyzed using data from datasets linked to GEO accession numbers GSM2205593–GSM2205602 [48];. The expression levels of CDX1/2 and LGR5 in the stem cells present within normal colonic epithelium in humans have been reported previously (Supplementary Table 2 in [49]).
Data availability
The DNA microarray and ChIP-sequencing data have been deposited in the GEO database under accession numbers GSE287318 and GSE287500, respectively. All relevant data in this study are available from corresponding author upon reasonable request.
References
Mlodzik M, Gehring WJ. Expression of the caudal gene in the germ line of Drosophila: formation of an RNA and protein gradient during early embryogenesis. Cell. 1987;48:465–78.
Silberg DG, Swain GP, Suh ER, Traber PG. Cdx1 and cdx2 expression during intestinal development. Gastroenterology. 2000;119:961–71.
Kakizaki F, Aoki K, Miyoshi H, Carrasco N, Aoki M, Taketo MM. CDX transcription factors positively regulate expression of solute carrier family 5, member 8 in the colonic epithelium. Gastroenterology. 2010;138:627–35.
Chawengsaksophak K, James R, Hammond VE, Köntgen F, Beck F. Homeosis and intestinal tumours in Cdx2 mutant mice. Nature. 1997;386:84–87.
Stringer EJ, Duluc I, Saandi T, Davidson I, Bialecka M, Sato T, et al. Cdx2 determines the fate of postnatal intestinal endoderm. Development. 2012;139:465–74.
Tamai Y, Nakajima R, Ishikawa T, Takaku K, Seldin MF, Taketo MM. Colonic hamartoma development by anomalous duplication in Cdx2 knockout mice. Cancer Res. 1999;59:2965–70.
Gao N, Kaestner KH. Cdx2 regulates endo-lysosomal function and epithelial cell polarity. Genes Dev. 2010;24:1295–305.
Gao N, White P, Kaestner KH. Establishment of intestinal identity and epithelial-mesenchymal signaling by Cdx2. Dev Cell. 2009;16:588–99.
Ee HC, Erler T, Bhathal PS, Young GP, James RJ. Cdx-2 homeodomain protein expression in human and rat colorectal adenoma and carcinoma. Am J Pathol. 1995;147:586–92.
Aoki K, Tamai Y, Horiike S, Oshima M, Taketo MM. Colonic polyposis caused by mTOR-mediated chromosomal instability in Apc+/Δ716Cdx2+/− compound mutant mice. Nat Genet. 2003;35:323–30.
Sakamoto N, Feng Y, Stolfi C, Kurosu Y, Green M, Lin J, et al. BRAFV600E cooperates with CDX2 inactivation to promote serrated colorectal tumorigenesis. eLife. 2017;6:e20331.
Yang L, Tu L, Bisht S, Mao Y, Petkovich D, Thursby SJ, et al. Tissue-location-specific transcription programs drive tumor dependencies in colon cancer. Nat Commun. 2024;15:1384.
Luk IY, Jenkins LJ, Schoffer KL, Ng I, Tse JWT, Mouradov D, et al. Epithelial de-differentiation triggered by co-ordinate epigenetic inactivation of the EHF and CDX1 transcription factors drives colorectal cancer progression. Cell Death Differ. 2022;29:2288–302.
Bonhomme C, Duluc I, Martin E, Chawengsaksophak K, Chenard MP, Kedinger M, et al. The Cdx2 homeobox gene has a tumour suppressor function in the distal colon in addition to a homeotic role during gut development. Gut. 2003;52:1465–71.
Hryniuk A, Grainger S, Savory JGA, Lohnes D. Cdx1 and Cdx2 function as tumor suppressors. J Biol Chem. 2014;289:33343–54.
Balbinot C, Armant O, Elarouci N, Marisa L, Martin E, De Clara E, et al. The Cdx2 homeobox gene suppresses intestinal tumorigenesis through non-cell-autonomous mechanisms. J Exp Med. 2018;215:911–26.
Cornish AJ, Gruber AJ, Kinnersley B, Chubb D, Frangou A, Caravagna G, et al. The genomic landscape of 2,023 colorectal cancers. Nature. 2024;633:127–36.
Nunes L, Li F, Wu M, Luo T, Hammarström K, Torell E, et al. Prognostic genome and transcriptome signatures in colorectal cancers. Nature. 2024;633:137–46.
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7.
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science. 1997;275:1787–90.
Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, et al. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–7.
van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, et al. The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–50.
Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007;120:3327–35.
Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 1998;92:645–56.
Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci USA. 1995;92:4482–6.
Bonhomme C, Calon A, Martin E, Robine S, Neuville A, Kedinger M, et al. Cdx1, a dispensable homeobox gene for gut development with limited effect in intestinal cancer. Oncogene. 2008;27:4497–502.
Aoki K, Kakizaki F, Sakashita H, Manabe T, Aoki M, Taketo MM. Suppression of colonic polyposis by homeoprotein CDX2 through its nontranscriptional function that stabilizes p27Kip1. Cancer Res. 2011;71:593–602.
Guo RJ, Huang E, Ezaki T, Patel N, Sinclair K, Wu J, et al. Cdx1 inhibits human colon cancer cell proliferation by reducing β-catenin/T-cell factor transcriptional activity. J Biol Chem. 2004;279:36865–75.
Yeung TM, Gandhi SC, Wilding JL, Muschel R, Bodmer WF. Cancer stem cells from colorectal cancer-derived cell lines. Proc Natl Acad Sci USA. 2010;107:3722–7.
Ashley N, Yeung TM, Bodmer WF. Stem cell differentiation and lumen formation in colorectal cancer cell lines and primary tumors. Cancer Res. 2013;73:5798–809.
Guo RJ, Funakoshi S, Lee HH, Kong J, Lynch JP. The intestine-specific transcription factor Cdx2 inhibits β-catenin/TCF transcriptional activity by disrupting the beta-catenin–TCF protein complex. Carcinogenesis. 2010;31:159–66.
Ramakrishnan AB, Chen L, Burby PE, Cadigan KM. Wnt target enhancer regulation by a CDX/TCF transcription factor collective and a novel DNA motif. Nucleic Acids Res. 2021;49:8625–41.
Verzi MP, Hatzis P, Sulahian R, Philips J, Schuijers J, Shin H, et al. TCF4 and CDX2, major transcription factors for intestinal function, converge on the same cis-regulatory regions. Proc Natl Acad Sci USA. 2010;107:15157–62.
Subramanian V, Meyer BI, Gruss P. Disruption of the murine homeobox gene Cdx1 affects axial skeletal identities by altering the mesodermal expression domains of Hox genes. Cell. 1995;83:641–53.
O’Brien CA, Kreso A, Ryan P, Hermans KG, Gibson L, Wang Y, et al. ID1 and ID3 regulate the self-renewal capacity of human colon cancer-initiating cells through p21. Cancer Cell. 2012;21:777–92.
Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446:676–9.
Chi YI. Homeodomain revisited: a lesson from disease-causing mutations. Hum Genet. 2005;116:433–44.
Wielenga VJ, Smits R, Korinek V, Smit L, Kielman M, Fodde R, et al. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am J Pathol. 1999;154:515–23.
Hamada J, Sawamura Y, Van Meir EG. CD44 expression and growth factors. Front Biosci. 1998;3:d657–664.
Aoki K, Nitta A, Igarashi A. NELF and PAF1C complexes are core transcriptional machineries controlling colon cancer stemness. Oncogene. 2024;43:566–77.
Hinoi T, Lucas PC, Kuick R, Hanash S, Cho KR, Fearon ER, et al. CDX2 regulates liver intestine-cadherin expression in normal and malignant colon epithelium and intestinal metaplasia. Gastroenterology. 2002;123:1565–77.
Wada T, Takagi T, Yamaguchi Y, Ferdous A, Imai T, Hirose S, et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998;12:343–56.
Jaehning JA. The Paf1 complex: platform or player in RNA polymerase II transcription?. Biochim Biophys Acta. 2010;1799:379–88.
Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–8.
Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13:720–31.
Yu M, Yang W, Ni T, Tang Z, Nakadai T, Zhu J, et al. RNA polymerase II-associated factor 1 regulates the release and phosphorylation of paused RNA polymerase II. Science. 2015;350:1383–6.
Rings EH, Boudreau F, Taylor JK, Moffett J, Suh ER, Traber PG. Phosphorylation of the serine 60 residue within the Cdx2 activation domain mediates its transactivation capacity. Gastroenterology. 2001;121:1437–50.
Shimokawa M, Ohta Y, Nishikori S, Matano M, Takano A, Fujii M, et al. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature. 2017;545:187–92.
Moorman AR, Benitez EK, Cambulli F, Jiang Q, Mahmoud A, Lumish M, et al. Progressive plasticity during colorectal cancer metastasis. Nature. 2025;637:947–54.
Saxena M, Roman AKS, O’Neill NK, Sulahian R, Jadhav U, Shivdasani RA. Transcription factor-dependent ‘anti-repressive’ mammalian enhancers exclude H3K27me3 from extended genomic domains. Genes Dev. 2017;31:2391–404.
Renouf B, Soret C, Saandi T, Delalande F, Martin E, Vanier M, et al. Cdx2 homeoprotein inhibits non-homologous end joining in colon cancer but not in leukemia cells. Nucleic Acids Res. 2012;40:3456–69.
Balbinot C, Vanier M, Armant O, Nair A, Penichon J, Soret C, et al. Fine-tuning and autoregulation of the intestinal determinant and tumor suppressor homeobox gene CDX2 by alternative splicing. Cell Death Differ. 2017;24:2173–286.
Acknowledgements
We thank K. Hori and Y. Deyama for the excellent technical assistance; Drs. M. M. Taketo, M. Oshima, and Y. Tamai for providing Apc and Cdx2 mutant mice; and Dr. P. Gruss for providing Cdx1 mutant mice.
Funding
This work was supported by grants from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (grant numbers 21K06946, 24650619, 24680091 and JPMJPR1181 to KA), the Takeda Science Foundation (to KA), the Princess Takamatsu Cancer Research Fund (to KA), and the Naito Foundation (to KA).
Author information
Authors and Affiliations
Contributions
K. Aoki contributed to the conceptualization and design of the study, performed the experiments, analyzed the data, and wrote the manuscript. A. Nitta and A. Igarashi performed experiments and analyzed the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All methods in this study were carried out in accordance with relevant guidelines and regulations. All animal experiments in this study were approved by the Animal Care and Use Committee of FUKUI University (approval number R04023). This study did not involve human participants or patient-derived materials; therefore, ethical approvals and informed consents were not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Dr Ilio Vitale
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aoki, K., Nitta, A. & Igarashi, A. CDX1 and CDX2 suppress colon cancer stemness by inhibiting β-catenin-facilitated formation of Pol II–DSIF–PAF1C complex. Cell Death Dis 16, 408 (2025). https://doi.org/10.1038/s41419-025-07737-3
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41419-025-07737-3
