
Abstract
The prevalence of urological malignancies remains a significant global health concern, particularly given the challenging prognosis for patients in advanced disease stages. Consequently, there is a pressing need to explore the molecular mechanisms that regulate the development of urological malignancies to discover novel breakthroughs in diagnosis and treatment. Ferroptosis, characterized by iron-ion-dependent lipid peroxidation, is a form of programmed cell death (PCD) distinct from apoptosis, autophagy, and necrosis. Notably, lipid, iron, and glutathione metabolism intricately regulate intracellular ferroptosis, playing essential roles in the progression of various neoplasms and drug resistance. In recent years, ferroptosis has been found to be closely related to urological malignancies. This paper provides an overview of the involvement of ferroptosis in the pathogenesis and progression of urological malignancies, elucidates the molecular mechanisms governing its regulation, and synthesizes recent breakthroughs in diagnosing and treating these malignancies. We aim to provide a new direction for the clinical treatment of urological malignancies.
Similar content being viewed by others

Facts
-
Ferroptosis plays a crucial role in the progression and drug resistance of various tumors by modulating lipid, iron, and glutathione metabolism.
-
Ferroptosis is closely associated with the pathogenesis and progression of urologic malignancies.
-
Ferroptosis may represent a novel therapeutic direction for the treatment of urologic malignancies.
Open questions
-
What mechanisms underlie the promotion of urologic malignancies and drug resistance by ferroptosis?
-
Have reliable markers of ferroptosis in urologic malignancies been identified?
-
What strategies can be employed to target ferroptosis to enhance therapeutic efficacy against urologic malignancies?
Introduction
According to GLOBOCAN 2020, patients with urinary malignancies constitute approximately 13% of the total number of cancer patients. The top three cancers in this category are prostate cancer (PCa), bladder cancer (BCa), and renal cell carcinoma (RCC) [1]. Despite advancements in diagnostic and therapeutic methods for urologic tumors that have led to notable enhancements in patient prognosis [2, 3], the mortality rate for individuals in the advanced stages of genitourinary cancer remains high [4]. This imposes a considerable burden on public health services and necessitates ongoing efforts to address the challenges associated with these conditions [4].
In multicellular organisms, regulatory cell death (RCD) is an essential homeostatic mechanism that maintains tissue morphology and function. Ferroptosis is a distinctive form of iron-dependent RCD that differs from apoptosis, necrosis, and autophagy in terms of cell morphology and biochemical characteristics [5]. Ferroptosis is intricately mediated by the interplay of iron metabolism, lipid metabolism, and glutathione (GSH) metabolism and involves a complex network of interactions within diverse metabolic pathways [6]. Ferroptosis, which is characterized by iron-mediated oxidative damage, is triggered by the high expression of unsaturated fatty acids on the cell membrane. This iron-dependent process results in lipid peroxidation, ultimately leading to cell death [7].
Increasing evidence suggests that a deeper understanding of the potential molecular mechanisms of urinary malignancies may be a promising way to enhance treatment strategies and improve prognostic outcomes [8, 9]. Recently, an increasing number of studies have shown that ferroptosis may also participates in the pathophysiological processes of malignancies of the urinary system and has attracted increasing attention from urologists. Therefore, this review aims to summarize current studies regarding the impact of ferroptosis on urinary system malignancies.
Mechanisms of ferroptosis
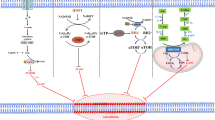
Ferroptosis is a multifaceted biological process primarily instigated by the dysregulated metabolism of iron, GSH, and polyunsaturated fatty acids (PUFAs) (Fig. 1).

The conversion of PUFAs to PL-PUFAs can be mediated by ACSL4 and LPCAT3. PL-PUFAs can be easily converted to PL-PUFA-OOH through various pathways, which can cause lipid peroxidation and ferroptosis. Extracellular Fe3+ enters the cell by binding to TF and undergoes reduction or ferritinophagy, ultimately converting into Fe2+ and forming the LIP. When LIP becomes overloaded with iron, it becomes prone to a Fenton reaction, facilitating lipid peroxidation and thereby triggering ferroptosis. The cysteine transported into cells by SLC7A11 can be converted into cystine, which is an essential component of GSH. GSH, mediated by GPX4, can be transformed into GSSG while reducing PL-PUFA-OOH to PL-PUFA-OH, thereby inhibiting ferroptosis. Abbreviations: PUFAs, polyunsaturated fatty acids; CoA, coenzyme A; PL-PUFAs, phospholipid-containing PUFAs; ACSL4, acyl-CoA synthetase long-chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; TF, transferrin; TFR1, transferrin receptor 1; STEAP3, iron oxide reductase steam 3; DMT1, divalent metal transporter 1; NCOA4, nuclear receptor coactivator 4; LIP, labile iron pool; SLC7A11, solute carrier family 7 member 11; SLC3A2, solute carrier family 3 member 2; GSH, glutathione; GSSG, Oxidized glutathione; GPX4, glutathione peroxidase 4.
Lipid metabolism
Lipid peroxidation and iron metabolism are pivotal biochemical processes that culminate in ferroptosis [10]. Moreover, lipid peroxidation is considered a marker of ferroptosis [11]. PUFAs in the body are metabolized by acyl-coenzyme A (CoA) synthetase long-chain family member 4 (ACSL4) into CoA-PUFAs, which subsequently undergo conversion to phospholipid-containing PUFAs (PL-PUFAs) mediated by lysophosphatidylcholine acyltransferase 3 (LPCAT3) [12]. Due to the dienyl groups in PUFAs, PL-PUFAs are susceptible to the generation of PL-PUFA-OOH through peroxidation catalyzed by iron ions via the Fenton reaction [13, 14]. Additionally, oxidation by lipid oxidases (ALOXs) or cytochrome P450 oxidoreductases (PORs) can also result in the production of substantial quantities of peroxidized phospholipids (PLOOHs) and breakdown products of lipid peroxidation [15, 16]. These products contribute to the disruption of cell membrane integrity, ultimately leading to cell death.
Iron metabolism
Iron plays a pivotal role in various metabolic pathways in the human body, contributing significantly to the regulation of numerous cellular biological functions [17]. Typically, iron ions in the circulation are present in the form of Fe3+. When extracellular Fe3+ binds to transferrin (TF), it is recognized by transferrin receptor 1 (TFR1) on the cell membrane, facilitating its transportation into the cell. The reduction of Fe3+ to Fe2+ in the endosome is mediated by iron oxide reductase steam 3 (STEAP3). Subsequently, divalent metal transporter 1 (DMT1) facilitates the transport of divalent iron into the cytoplasm, where it forms a labile iron pool (LIP). Excess iron will be present in the form of ferritin. Therefore, under normal physiological conditions, iron in the body typically exists in the form of Fe2+, Fe3+ and ferritin and maintains metabolic homeostasis [18]. When iron metabolism is disrupted by the activation of nuclear receptor coactivator 4 (NCOA4)-mediated ferritinophagy for various reasons, intracellular Fe2+ reaches an overloaded state [19]. Fe2+, which is unstable, readily engages in a Fenton reaction with hydrogen peroxide. This interaction leads to the production of hydroxyl radicals, which possess potent oxidizing properties, and subsequently generates substantial amounts of lipid peroxides [20]. Moreover, excess iron augments reactive oxygen species (ROS) production by activating ROS-generating enzymes, including nicotinamide adenine dinucleotide phosphate oxidase and lipoxygenase [21, 22]. The excessive accumulation of ROS and lipid peroxidation ultimately results in ferroptosis.
GPX4/GSH homeostatic system
The system Xc-/GSH/GPX4 regulatory axis is the primary intracellular defense mechanism against the initiation of ferroptosis [23]. System Xc-, a member of the heterodimeric amino acid transporter protein family, comprises solute carrier family 3 member 2 (SLC3A2) and solute carrier family 7 member 11 (SLC7A11), which are linked by disulfide bonds within the cellular membrane. Notably, SLC7A11 serves as the major functional subunit in system Xc-. This system facilitates the transport of cysteine into the cell while simultaneously exporting glutamate out of the cell at a 1:1 ratio [24, 25]. Cystine is reduced to cysteine upon entry into the cell, which is involved in the composition of GSH. GPX4, the exclusive reductive enzyme in the GPX family, plays a pivotal role in reducing phospholipid hydroperoxides (LOOHs) to nontoxic lipohydrols (LOHs), effectively inhibiting iron death. The concerted action of GPX4 with the cofactor GSH is instrumental in mitigating oxidative stress by depleting lipid peroxides and neutralizing ROS [26]. Inhibition of system Xc impedes intracellular GSH biosynthesis, thereby compromising the lipid membrane repair capacity of GPX4. This disruption increases the accumulation of toxic lipid free radicals and ROS, fostering lipid peroxidation and ultimately culminating in ferroptosis [27, 28].
The molecular mechanism of ferroptosis in urological malignancies
Recent studies have demonstrated that ferroptosis may be involved in the initiation of urological malignancies and can contribute to the progression of urological malignancies via distinct mechanisms (Table 1).
Molecular mechanism of ferroptosis in RCC
The regulation of ferroptosis can impact RCC through multiple mechanisms, including the modulation of GSH levels, lipid metabolism, iron metabolism, the NRF2 signaling pathway, and the HIF-2α signaling pathway (Fig. 2).

The modulation of ferroptosis can exert significant effects on RCC through various mechanisms, encompassing the regulation of GSH levels, lipid metabolism, iron metabolism, the NRF2 signaling pathway, the HIF-2α signaling pathway, and the ROS signaling pathway. Abbreviations: PUFAs, polyunsaturated fatty acids; CoA, coenzyme A; PL-PUFAs, phospholipid-containing PUFAs; ACSL4, acyl-CoA synthetase long-chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; TF, transferrin; TFR1, transferrin receptor 1; STEAP3, iron oxide reductase steam 3; DMT1, divalent metal transporter 1; NCOA4, nuclear receptor coactivator 4; LIP, labile iron pool; SLC7A11, solute carrier family 7 member 11; SLC3A2, solute carrier family 3 member 2; GSH, glutathione; GSSG, Oxidized glutathione; GPX4, glutathione peroxidase 4; KLF, Kruppel-like factor; ICS II, Icariside II; DPP4, dipeptidyl-peptidase-4; NRF2, nuclear factor erythroid 2-related factor 2; OGT, O-GlcNAc transferase; HIF-2α, hypoxia-inducible factor-2α; SDH, succinate dehydrogenase; ROS, Reactive Oxygen Species.
GPX4/GSH steady-state system
GSH-related metabolism significantly affects the activity of the system Xc-/GPX4 antioxidant axis and consequently the sensitivity of cells to ferroptosis. Lu et al. reported that one of the transcription factor families, Kruppel-like Factor 2 (KLF2), is sufficient to induce ferroptosis by suppressing the transcriptional repression of GPX4. The decreased expression of KLF2 detected through immunohistochemistry in primary metastatic ccRCC was strongly associated with poor clinical outcomes [29]. SLC7A11 can promote GSH synthesis and GPX4 activity and inhibit ferroptosis in RCC cells [21]. Kang et al. identified a positive correlation between PDIA4 and the PERK/ATF4/SLC7A11 signaling pathway through bioinformatics analyses of clinical RCC samples and databases [30]. Li et al. reported that the inhibition of the long noncoding RNA (lncRNA) SLC16A1-AS1 could result in ferroptosis through the miR-143-3p/SLC7A11 signaling pathway in RCC [31]. Yu et al. reported that Icariside II (ICS II) induces ferroptosis in RCC cells by modulating the miR-324-3p/GPX4 axis, which is characterized by the accumulation of Fe2+, MDA and ROS and a reduction in GSH levels [32]. Zhou et al. reported that KLF11 induced ferroptosis by downregulating the protein expression of GPX4, ferritin, and system Xc-cystine/glutamate antiporter (xCT) [33].
Iron metabolism
Increased intracellular free iron leads to ferroptosis, which can be regulated by regulating intracellular free iron levels through iron uptake, utilization, storage, and translocation. The VHL gene is the most commonly mutated gene in RCC. Inactivation of the VHL gene leads to downstream accumulation of hypoxia-inducible factor (HIF)-1α and HIF-2α [34]. Green et al. discovered that the production of HIF-1α and HIF-2α can be inhibited by targeting iron sulfur cluster assembly 2 (ISCA2). Mechanistically, the inhibition of ISCA2 initiates the iron starvation response, leading to overload. of iron and other metals, ultimately resulting in ferroptosis [35]. SUV39H1, encoding a histone H3 lysine 9 methyltransferase, is frequently upregulated in ccRCC tumors. Wang et al. reported that decreasing the expression of SUV39H1 could induce iron accumulation and lipid peroxidation, leading to ferroptosis that inhibits ccRCC cell growth. They also reported that SUV39H1 deficiency resulted in the upregulation of dipeptidyl-peptidase-4 (DPP4), which leads to ferroptosis [36]. For hereditary leiomyomatosis and renal cell cancer (HLRCC), Kerimoglu et al. showed that the combination of cyst(e)inase and rapamycin could induce ferroptosis. Mechanistically, cyst(e)inase induces ferroptosis through depletion of the exogenous cysteine/cystine supply, while rapamycin diminishes cellular ferritin levels by facilitating the degradation of ferritins through ferritinophagy [37].
Lipid metabolism
Lipid peroxidation is considered a marker of ferroptosis. The absent in melanoma 2 (AIM2) protein complex plays a crucial role in the pathogenesis of RCC through its dysregulated expression and activation [38]. Wang et al. reported that AIM2 promoted FOXO3a phosphorylation and proteasome degradation, thereby reducing its transcriptional effect on ACSL4 and inhibiting ferroptosis [39]. ACSL4 and LPCAT3 are involved in the conversion of PUFAs into PUFA-PLs, which in turn are involved in the process of lipid peroxidation [40]. Using a multiomics approach, Tan et al. revealed that chemerin restrained fatty acid oxidation, thus inhibiting ferroptosis [41]. Klasson et al. reported that acyl-CoA synthetase 3 (ACSL3) enhances ferroptosis sensitivity through the composition of exogenous fatty acids to drive lipid droplet deposition [42]. Zhou et al. reported that, mediated by malonyl-CoA decarboxylase (MLYCD)-facilitated fatty acid oxidation, inhibition of lipid droplet accumulation disrupted endoplasmic reticulum and mitochondrial homeostasis and increased ROS levels, ultimately culminating in the induction of ferroptosis [43].
NRF2 signaling pathway
The transcription factor nuclear factor erythroid 2-related Factor 2 (NRF2) targets genes that can suppress lipid peroxidation and the accumulation of free iron, so NRF2 significantly transcriptionally regulates the expression of antiferroptotic genes [44]. Activation of the NRF2 pathway can lead to a rapid increase in ROS, which can promote ferroptosis. Chang et al. determined that dipeptidyl peptidase 9 (DPP9) could bind to KEAP1 via a conserved ESGE motif and then regulate the KEAP1/NRF2/SLC7A11 axis, resulting in the suppression of ferroptosis [45]. Ni et al. demonstrated that disulfiram/copper treatment prolonged the half-life of NRF2, reducing its degradation and reducing the expression of nuclear protein localization homolog 4 (NPL4), which is a ubiquitin protein-proteasome system involved in NRF2 degradation, ultimately leading to oxidative stress and ferroptosis. However, it is noteworthy that surmounting the compensatory increase in NRF2 triggered by NPL4 inhibition amplifies disulfiram/copper-induced oxidative stress and ferroptosis in RCC [46].
HIF-2α signaling pathway
Unlike HIF-1α, HIF-2α functions as an oncogenic factor to promote the progression of RCC [47]. Yang et al. reported that O-GlcNAc transferase (OGT) increased the expression of HIF-2α by repressing degradation mediated through the ubiquitin‒proteasome system. Furthermore, the OGT/HIF-2α axis increases the sensitivity of ccRCC to ferroptosis [48].
Mitochondrial metabolism
Mitochondria are sites of ROS production, and excess ROS can trigger ferroptosis via the Fenton reaction. Yang et al. demonstrated that succinate dehydrogenase (SDH) inhibition dampened oxidative phosphorylation, which was characterized by decreased mitochondrial ROS levels, decreased cellular ROS and decreased peroxide accumulation, thus reducing ferroptotic events and restoring ferroptotic cell death [49].
Molecular mechanism of ferroptosis in PCa
Ferroptosis can be regulated by GSH, lipids, iron, and mitochondrial metabolism in PCa (Fig. 3).

In PCa, ferroptosis can be regulated through mechanisms involving GSH, lipid metabolism, iron metabolism, the ROS signaling pathway, and the mTOR signaling pathway. Abbreviations: PUFAs, polyunsaturated fatty acids; CoA, coenzyme A; PL-PUFAs, phospholipid-containing PUFAs; ACSL4, acyl-CoA synthetase long-chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; TF, transferrin; TFR1, transferrin receptor 1; STEAP3, iron oxide reductase steam 3; DMT1, divalent metal transporter 1; NCOA4, nuclear receptor coactivator 4; LIP, labile iron pool; SLC7A11, solute carrier family 7 member 11; SLC3A2, solute carrier family 3 member 2; GSH, glutathione; GSSG, Oxidized glutathione; GPX4, glutathione peroxidase 4; NRF2, nuclear factor erythroid 2-related factor 2; ROS, Reactive Oxygen Species; SGK2, serum/glucocorticoid regulated kinase 2; CEMIP, cell migration-inducing protein; HnRNP L, heterogeneous nuclear ribonucleoprotein L; PHGDH, phosphoglycerate dehydrogenase; PPI, Polyphyllin I; TFEB, transcription factor EB; AOC1, amine oxidase copper-containing 1.
GPX4/GSH steady-state system
PCa can also be regulated by ferroptosis through the GPX4/GSH steady-state system. Shi et al. showed that exposure to low doses of antimony promotes cell proliferation in PCa, which is attributed to the inhibition of ferroptosis via the activation of the NRF2/SLC7A11/GPX4 pathway [50]. Zhou et al. revealed that flubendazole induced the expression of P53, which partly accounted for the inhibition of SLC7A11 transcription, and subsequently downregulated GPX4, which is a major antiferroptosis gene [51]. Zhang et al. reported that the lncRNA OIP5-AS1 promoted the progression of PCa and conferred resistance to ferroptosis through the miR-128-3p/SLC7A11 signaling pathway [52]. Cell migration-inducing protein (CEMIP) is an intranuclear protein that is abnormally highly expressed in PCa and promotes tumor invasion and migration [53]. Liu et al. reported that CEMIP inhibited ferroptosis through the ITPR3/CaMKII/NRF2/SLC7A11 pathway [54]. Zhou et al. demonstrated that knockdown of heterogeneous nuclear ribonucleoprotein L (HnRNP L) resulted in increased production of interferon gamma (IFN-γ), which induced ferroptosis in castration-resistant prostate cancer (CRPC) cells through the STAT1/SLC7A11/GPX4 signaling axis [55]. Wang et al. reported that the inhibition of phosphoglycerate dehydrogenase (PHGDH) induced ferroptosis by reducing GSH/GSSG levels, elevating LipROS production and suppressing SLC7A11 expression through the activation of the p53 signaling pathway [56]. Ferroptosis can also be regulated by direct action downstream of GPX4. Cheng et al. reported that the overexpression of serum/glucocorticoid regulated kinase 2 (SGK2) facilitated the translocation of FOXO1 from the nucleus to the cytoplasm, alleviating the inhibitory effect of FOXO1 on GPX4 and consequently inhibiting ferroptosis [57]. Yu et al. reported that evodiamine, a natural alkaloid compound derived from the fruit of Evodiae fructus, can induce ferroptosis by decreasing the expression of GPX4 [58]. Zhang et al. discovered that TQB3720, a second-generation androgen receptor (AR) antagonist, promoted ferroptosis in PCa cells by diminishing the binding of the AR and specificity protein 1 (SP1) transcriptional complex to the GPX4 promoter [59]. In addition, Wang et al. engineered an inorganic metal-free nanoplatform, namely, PSMA-targeted arsenic nanosheets (PMANs), which can enhance GSH consumption, suppress the expression of SLC7A11 and GPX4, and promote the generation of ROS and lipid peroxides (LPOs). These concerted actions synergistically contribute to the promotion of ferroptosis in PCa cells [60].
Lipid metabolism
PCa can also participate in the regulation of ferroptosis through lipid metabolism. Wang et al. demonstrated that loss of the tumor suppressor gene RB1 coupled with transcriptional family E2F activation sensitizes PCa cells to ferroptosis. This sensitivity is attributed to the upregulation of ACSL4 expression and the enrichment of ACSL4-dependent arachidonic acid–containing phospholipids, which are crucial components of ferroptosis execution [61]. Sun et al. demonstrated that acute treatment with the antiandrogen enzalutamide led to a reduction in GSH production and an increase in lipid peroxidation, lending to the induction of ferroptosis in PCa cells. Specifically, the cystine transporter gene SLC7A11 has been identified as a pivotal AR target. Full-length AR (AR-FL) was observed to transactivate SLC7A11 transcription by directly occupying the SLC7A11 promoter and putative enhancer regions. AR variants (AR-Vs) preferentially bind to the SLC7A11 enhancer and upregulate SLC7A11 expression, thereby conferring resistance to ferroptosis induced by ENZ treatment. Therefore, SLC7A11 acts as a direct target gene for both AR-FL and AR-Vs [62]. Nassar et al. discovered that the knockdown of DECR1, a negatively regulated AR target gene, resulted in the cellular accumulation of PUFAs, heightened mitochondrial oxidative stress, and increased lipid peroxidation, ultimately leading to the induction of ferroptosis [63]. Zou et al. reported that Polyphyllin I (PPI), a steroidal saponin in Paris polyphylla, could serve as a ferroptosis inducer to promote ferroptosis in CRPC cells through the ERK/DNMT1/ACSL4 axis [64].
Iron metabolism
PCa may also be involved in the regulation of ferroptosis through lipid metabolism. Kumar et al. reported that supraphysiological levels of testosterone (SupraT) initiate ferroptosis by inducing two parallel autophagy-mediated processes, specifically ferritinophagy and nucleophagy [65]. Similarly, Fu et al. reported that luteolin induces ferroptosis in PCa cells by increasing autophagy and ferritinophagy through the promotion of transcription factor EB (TFEB) nuclear translocation [66].
Mitochondrial metabolism
PCa may also play a role in the regulation of ferroptosis through mitochondrial metabolism. Ding et al. showed that the anticancer effect of amine oxidase copper-containing 1 (AOC1) is mediated via its impact on spermidine, leading to the activation of ROS and subsequent induction of ferroptosis. The expression of AOC1 in PCa was positively regulated by the transcription factor SOX15. Therefore, SOX15 can induce ferroptosis in PCa cells through the SOX15/AOC1/ROS axis [67].
Molecular mechanism of ferroptosis in BCa
Ferroptosis in BCa can be regulated through GSH, lipid metabolism, mitochondrial metabolism, and the NRF2 signaling pathway (Fig. 4).

In BCa, ferroptosis can be regulated through various mechanisms, including GSH levels, lipid metabolism, mitochondrial metabolism, and the MAPK signaling pathway. Abbreviations: PUFAs, polyunsaturated fatty acids; CoA, coenzyme A; PL-PUFAs, phospholipid-containing PUFAs; ACSL4, acyl-CoA synthetase long-chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; TF, transferrin; TFR1, transferrin receptor 1; STEAP3, iron oxide reductase steam 3; DMT1, divalent metal transporter 1; NCOA4, nuclear receptor coactivator 4; LIP, labile iron pool; SLC7A11, solute carrier family 7 member 11; SLC3A2, solute carrier family 3 member 2; GSH, glutathione; GSSG, Oxidized glutathione; GPX4, glutathione peroxidase 4; NRF2, nuclear factor erythroid 2-related factor 2; ROS, Reactive Oxygen Species; PHGDH, phosphoglycerol dehydrogenase; EMP1, epithelial membrane protein 1; FLRT2, fibronectin leucine rich transmembrane protein 2; PCBP1, poly C binding protein 1.
GPX4/GSH steady-state system
The GPX4/GSH steady-state system can also be involved in the regulation of ferroptosis in BCa. PHGDH, a key enzyme in the serine synthesis pathway, is highly expressed in numerous cancers [68]. PHGDH knockdown decreased SLC7A11 expression and subsequently induced ferroptosis in BCa cells. Mechanistically, PHGDH was observed to bind to PCBP2, an RNA-binding protein, and inhibit its ubiquitination-mediated degradation. Consequently, PCBP2 in turn enhances SLC7A11 mRNA stability and increases its expression [69]. Liu et al. demonstrated that epithelial membrane protein 1 (EMP1) deficiency enhanced tumor metastasis by increasing the expression of PPARG, a transcription factor, and promoting its activation, resulting in the upregulation of pFAK (Y397) and SLC7A11, which are involved in cell migration and antiferroptosis, respectively [70]. Sun et al. determined that Fin56, a type 3 ferroptosis inducer, could promote ferroptosis and autophagy in BCa cells. Notably, the induction of ferroptosis by Fin56 was found to mechanistically depend on the autophagy-mediated degradation of GPX4 [71]. The classical tumor suppressor gene p53 can also regulate the expression of SLC7A11 to promote ferroptosis. Li et al. reported that p53 activation stimulated the lipoxygenase activity of ALOX15B by inhibiting SLC7A11 to induce ferroptosis in BCa cells, which provided insight into the molecular mechanism underlying the occurrence and development of BCa [72]. Wang et al. reported that heat shock protein family A (HSP70) member 5 (HSPA5) can inhibit ferroptosis through the p53/SLC7A11/GPX4 pathway [73].
Lipid metabolism
Lipid metabolism can also play a role in the regulation of ferroptosis in BCa. Ding et al. engineered a mitochondrial-targeted liposome loaded with brequinar (BQR) (BQR@MLipo), which was designed to selectively accumulate in mitochondria and inactivate dihydroorotate dehydrogenase (DHODH), resulting in extensive mitochondrial lipid peroxidation and the generation of ROS, ultimately leading to ferroptosis of BCa cells [74]. Fibronectin leucine-rich transmembrane protein 2 (FLRT2) is overexpressed in advanced colorectal cancer and is negatively correlated with patient survival [75]. Jiang et al. discovered that FLRT2 upregulated the expression of ACSL4, leading to increased lipid peroxidation and subsequently promoting ferroptosis in human BCa cells [76].
Mitochondrial metabolism
The metabolism of mitochondria may also play a role in the regulation of ferroptosis in BCa. Luo et al. reported that poly C binding protein 1 (PCBP1) protected BCa cells against mitochondrial injury and ferroptosis through the LACTB/PISD axis. Mechanistic insights revealed that LACTB mRNA is a novel transcript that can bind to PCBP1. The upregulation of LACTB facilitated erastin-induced ferroptosis and induced mitochondrial dysfunction. Moreover, the overexpression of LACTB reversed the ferroptosis protection mediated by PCBP-1, including a decrease in ROS and an enhancement of mitochondrial function [77]. Xu et al. reported that abietic acid (AA)-induced ferroptosis in BCa cells was mediated by a combination of multiple factors. This included the elevation of ROS, intracellular iron, and MDA. Furthermore, AA treatment suppressed GPX4 and enhanced HO-1 [78].
NRF2 signaling pathway
The NRF2 signaling pathway can also play a role in the regulation of ferroptosis in BCa. Xiang et al. showed that erianin facilitated the accumulation of lethal lipid-based ROS and the depletion of GSH, inducing ferroptosis. Mechanistically, NRF2 served as a key determinant of erianin-triggered ferroptosis. The activation of NRF2 through TBHQ treatment protected against erianin-induced ferroptosis and elevated the expression of GPX4, ferritin, xCT and glutaminase. On the other hand, NRF2 knockdown increased the activity of ROS and MDA, decreased GSH levels and decreased the expression of negative regulatory proteins associated with ferroptosis [79]. Wang et al. revealed that the m6A methyltransferase WTAP could methylate the 3’-UTR of the RNA of the endogenous antioxidant factor NRF2, consequently increasing the mRNA stability of NRF2 and thus inhibiting erastin-induced ferroptosis [80].
The role of ferroptosis in the diagnosis of urological malignancies
Patients diagnosed with advanced urological malignancies often have a bleak prognosis and a heightened risk of mortality [81, 82]. Therefore, identifying ferroptosis-related potential prognostic markers is important for the diagnosis and treatment of urological malignancies (Table 2).
RCC
Sixty percent of individuals with RCC are asymptomatic, resulting in late detection of many cancers; more than 25% exhibit evidence of metastasis at the time of diagnosis [83]. Therefore, the development of diagnostic and prognostic markers for RCC is imperative. Xing et al. proposed that five ferroptosis-induced differentially expressed genes (FI-DEGs), namely, CCR4, CMTM3, IFITM1, MX2, and NR3C2, may serve as valuable prognostic and diagnostic biomarkers for patients diagnosed with RCC [84]. Luo et al. combined cuproptosis and ferroptosis and screened six cuproptosis-related ferroptosis genes (CRFGs) that showed promise in predicting the prognosis and therapeutic outcome of patients with RCC [85]. Sun et al. constructed a 7-gene ferroptosis-related prognostic signature by analyzing a panel of ferroptosis-related genes to predict overall survival (OS) in RCC patients [86].
Immunotherapy is a therapeutic strategy that combats malignant tumors by activating or enhancing the body’s immune system to recognize and destroy cancer cells [87]. Although immunotherapy has been applied to a variety of tumors, including urological malignancies, its clinical efficacy remains suboptimal in some patients. The immune system plays a critical role in tumor immunosurveillance through the infiltration of adaptive and innate immune cells into the tumor microenvironment (TME), where they regulate tumor progression. Thus, a comprehensive understanding of immune infiltration within the TME is essential for improving response rates and developing novel immunotherapeutic strategies [88]. The interplay between ferroptosis and tumor immune infiltration is a critical area that warrants further investigation. Tumor immune infiltration, involving various immune cells such as T cells, macrophages, and dendritic cells, plays a pivotal role in shaping the tumor microenvironment and influencing tumor growth and metastasis. Emerging evidence suggests that ferroptosis not only induces cancer cell death but also modulates the immune landscape within tumors. Ferroptotic cancer cells can release damage-associated molecular patterns (DAMPs) and other immunogenic signals that activate and attract immune cells to the tumor site. This process can enhance antitumor immunity by promoting the recruitment and activation of cytotoxic T cells and macrophages, which are essential for attacking and eliminating tumor cells. However, ferroptosis can also lead to the release of lipid peroxidation products, which may have immunosuppressive effects, creating a complex interplay between ferroptosis and immune responses. The study by Xu et al. found that the overexpression of SLC7A11 was associated with poor prognosis in patients with ccRCC and that SLC7A11 expression was positively correlated with the infiltration of immune cells and their corresponding markers, including CD8+ T cells and myeloid dendritic cells. Thus, SLC7A11 could serve as a potential prognostic biomarker for ccRCC and as an indicator of immune cell infiltration within tumors [89]. The study by Zong et al. identified eight ferroptosis-associated lncRNAs in ccRCC that could be used to predict overall patient survival. These lncRNAs were also correlated with the immune microenvironment, immunotherapeutic response, and drug sensitivity in ccRCC, offering a potential method for assessing immunotherapeutic efficacy in ccRCC patients [90].
PCa
Although prostate-specific antigen (PSA) has emerged as the principal screening tool for PCa in clinical settings, it exhibits low specificity and sensitivity [91, 92]. Ji et al. suggested that decreased ARPC1A expression inhibits PCa cell viability and invasion through ferroptosis. The level of ARPC1A may serve as an independent indicator of prognosis in patients with PCa [93]. The ferroptosis-related gene prognostic index (FRGPI), constructed using the genes E2F1, CDC20, TYMS, and NUP85, has been shown to predict disease-free survival (DFS) in patients with PCa [94]. Liu et al. developed a model based on five ferroptosis-related lncRNAs to predict biochemical recurrence in PCa patients [95].
Liu et al. propose a novel strategy for assessing biochemical recurrence in PCa patients based on five ferroptosis-associated lncRNAs. Moreover, they found that the disease severity in the high-risk group identified by this predictive model was positively correlated with the number of infiltrating immune cells. Additionally, these five lncRNAs were linked to key immune checkpoints [95].
BCa
Although the five-year survival rate is high for patients diagnosed with BCa at an early disease stage, survival substantially decreases in patients with muscle-invasive or metastatic disease [96]. Therefore, early detection of their prognosis is of paramount importance. Liu et al. identified five ferroptosis-related genes (FRGs) that demonstrate significant potential in stratifying patients with BCa based on their prognosis [97]. Hou et al. discovered a frlncRNA signature that predicts the prognosis of patients with bladder cancer [98].
Li et al. identified 12 lncRNA pairs linked to immune responses and ferroptosis to develop a risk prediction model for BCa patients. Notably, the high-risk group exhibited significantly worse OS and demonstrated a positive correlation with the majority of tumor-infiltrating immune cells [99].
The roles of ferroptosis in the occurrence and treatment of urological malignancies
Ferroptosis induction has been shown to effectively inhibit tumor growth and metastasis, so ferroptosis has considerable potential for application in the treatment of urological tumors. Recent relevant studies are listed in Table 3.
RCC
As mentioned, ferroptosis is widely believed to play a regulatory role in the progression of RCC [100]. Accordingly, directing therapeutic directions toward regulators associated with ferroptosis has emerged as a highly promising strategy for addressing this disease [101].
SET domain-containing 2 (SETD2) plays a significant role as an epigenetic regulator and has a high mutation rate in RCC [103]. Xue et al. reported that suppression of the epigenetic molecule SETD2 markedly increased the sensitivity of cells to ferroptosis inducers, which facilitates tumor cell death, indicating that SETD2 may be a promising therapeutic target for treating ccRCC [104]. Ye et al. reported that RCC cell lines in which STEAP3 expression was knocked down exhibited increased sensitivity to ferroptosis, and this effect was attributed to the p53/xCT pathway. Therefore, STEAP3 has the potential to become a new biomarker and target for the treatment of RCC [105].
Chemoresistance is a major obstacle in the treatment of RCC and leads to poor prognosis. Recent investigations have illuminated the potential of ferroptosis inducers in overcoming sunitinib resistance. Wang et al. discovered that AIM2 serves as a novel biomarker for RCC and plays a role in promoting both RCC progression and resistance to sunitinib through an inflammasome-independent mechanism [39]. Therefore, this could provide a new therapeutic target for RCC diagnosis and inversion of sunitinib resistance. The ferroptosis inducers RSL3 and erastin may synergize with everolimus by inhibiting the mTOR/4EBP1 axis. This combination strategy holds potential for overcoming resistance to everolimus [106].
However, existing ferroptosis-inducing therapies are limited by a lack of precise targeting. Therefore, it is conceivable to enhance their efficacy by integrating them with targeted nanomedicine delivery systems [107]. Ni et al. designed iron-based metal-organic framework nanoparticles that can deliver RSL3 (MIL-101(Fe)@RSL3) in a targeted manner to ccRCC cells. As a pH-responsive nanodrug, MIL-101(Fe)@RSL3 induces cellular iron overload and promotes hydroxyl radical (•OH) generation via the Fenton reaction. This mechanism targets polyunsaturated fatty acids (PUFAs), resulting in the anomalous accumulation of lipid peroxides (L-OOH). Furthermore, RSL3 directly inhibits GPX4 to detoxify L-OOH. Concurrently, ferrous ions further catalyze the irreversible conversion of highly reactive lipid alkoxyl radicals (L-O•) from L-OOH, initiating a waterfall-like cascade leading to ferroptosis [108].
Phosphorylated NCOA4 has been found to promote ferroptosis by maintaining ferritinophagy [109]. And in ccRCC, NCOA4 deficiency led to impaired immune cell infiltration through the disruption of IFN-γ receptor signaling, which was associated with disease progression and poor prognosis [110]. Therefore, targeting NCOA4 may represent a promising therapeutic strategy that combines ferroptosis induction with immunotherapy. Gong et al. reported that overexpression of the chemokine CX3CL1 inhibited tumor cell proliferation and metastasis by enhancing tumor sensitivity to ferroptosis in ccRCC. Moreover, as the expression level of CX3CL1 is closely correlated with the infiltration of CD8+ T cells in the TME, CX3CL1 can also act as a promising predictor of immunotherapy outcomes in ccRCC patients in the clinic [102].
PCa
Recent studies have demonstrated that treatment-resistant PCa cells exhibit sensitivity to two ferroptosis inducers, erastin and RSL3. Specifically, erastin and RSL3 were effective at inhibiting PCa cell growth, as well as migration in vitro and tumor growth in vivo [111, 112]. These results indicate that the induction of ferroptosis may have the potential to become a new method for treating PCa. As mentioned earlier, SOX15 has the potential to promote ferroptosis in PCa cells through the SOX15/AOC1/ROS axis. Moreover, elevated expression of AOC1 is strongly associated with diminished proliferation and migration in PCa. Therefore, targeting AOC1 and SOX15 is a promising approach for the treatment of PCa [67].
In addition to the above targets, numerous compounds that can inhibit PCa progression by regulating ferroptosis in PCa cells have been identified in recent studies. For example, Xu et al. reported that icariin (ICA), a flavonoid compound isolated from the traditional Chinese medicine Epimedium, and curcumol, a sesquiterpene compound, synergistically regulated the miR-7/mTOR/SREBP1 pathway, inducing ferroptosis and subsequently triggering autophagy in PCa cells [113]. Kim et al. reported that the cytotoxic function of natural killer (NK) cells could be enhanced by ferroptosis, a clinical-grade iron oxide nanoparticle, in cancer cells mediated by ferumoxytol combined with NK cells, which is beneficial for the death of PCa cells [114].
The anti-androgenic drug enzalutamide, which is currently in clinical use, is also associated with ferroptosis. Wang et al. reported a link between high expression of PHGDH and enzalutamide resistance in CRPC cells. Additionally, increased PHGDH levels were found to account for ferroptosis resistance by maintaining redox homeostasis in enzalutamide-resistant CRPC cells, ultimately promoting cell growth and enzalutamide resistance in CRPC cells. Furthermore, the pharmacological inhibition of PHGDH using NCT-503 effectively inhibited cell growth, induced ferroptosis, and overcame enzalutamide resistance in ENZ-resistant CRPC cells. Therefore, the combination of ferroptosis inducers and targeted inhibition of PHGDH has emerged as a promising therapeutic strategy for overcoming enzalutamide resistance in CRPC [56].
The immunotherapeutic drug BEBT-908, a dual inhibitor of PI3K/HDAC, inhibited tumor cell growth by inducing ferroptosis in cancer cells. Mechanistically, this process led to the upregulation of MHC class I and the activation of endogenous IFNγ signaling within the cells [115]. Additionally, inhibiting autophagy could trigger anti-tumor immune memory by increasing MHC-I expression in PCa cells, particularly when combined with PD-L1 blockade, thereby exerting synergistic anti-PCa effects [116]. These findings underscore the potential of leveraging ferroptosis for synergistic immunotherapy in PCa. NK cell-based immunotherapy is a promising therapeutic approach. In combination with ferroptosis, mediated by ferumoxytol-an iron oxide nanoparticle—NK cell therapy has shown enhanced efficacy. This combination treatment leads to the upregulation of ULBPs, ligands for the NK cell-activating receptor NKG2D, as well as increased expression of HMGB1 and PD-L1 in cancer cells. In in vivo experiments, the combination of ferumoxytol-mediated ferroptosis and NK cell therapy resulted in a significant reduction in tumor volume. Thus, ferumoxytol-mediated ferroptosis combined with NK cell therapy demonstrates potential synergistic anticancer effects [114].
BCa
Luo et al. reported that the long noncoding RNA (lncRNA) RP11-89 can enhance cell proliferation, migration and tumorigenesis while inhibiting cell cycle arrest through the miR-129-5p/PROM2 axis. RP11-89 may serve as a prospective biomarker for targeted therapy in BCa [117]. Jiang et al. reported decreased FLRT2 expression in human BCa patients and that increased FLRT2 expression correlated with a decreased survival rate. Functional investigations indicated that downregulation of FLRT2 facilitated tumor cell growth, migration, and invasion, suggesting that FLRT2 could be a tumor suppressor gene [76]. The lncRNA lung cancer-associated transcriptome 1 (LUCAT1) is abnormally expressed in various tumor tissues and is closely related to the proliferation and invasion of tumor cells [118]. Cao et al. revealed that LUCAT1 promoted cell proliferation, migration, and invasion while simultaneously inhibiting ferroptosis in BCa. These findings suggested that LUCAT1 could serve as a promising therapeutic target for BCa [119].
Evodiamine (EVO), a quinazoline alkaloid, has been shown to exert anticancer effects by inhibiting cell proliferation and tumor growth. EVO also functions as a novel inducer that can activate ferroptosis in BCa cells, showing its potential as a therapeutic agent for BCa [120]. Kong N et al. showed that baicalin, a medicinal plant, exerts its anticancer effects by inducing ferroptosis in BCa cells [121].
Checkpoint blockade immunotherapy (CBI) has demonstrated remarkable benefits in cancer therapy [122]. However, the low responsiveness of CBI has hindered its application in the treatment of BCa [122, 123]. Ferroptosis, which has the potential to induce immunogenic cell death, can enhance the responsiveness of CBI [124, 125]. Ding Q et al. developed a mitochondrial-targeted liposome loaded with brequinar (BQR) (BQR@MLipo) to enhance mitochondria-related ferroptosis in situ in BCa. BQR@MLipo not only induces ferroptosis through multiple factors, as mentioned earlier but also significantly accumulates in bladder tumors and successfully initiates the infiltration of CD8+ T cells into the TME, enabling efficient CBI to inhibit bladder tumor growth [74].
Clinical challenges in targeting ferroptosis with therapeutic agents
Targeting ferroptosis presents a promising anti-cancer strategy. However, the clinical translation of ferroptosis-inducing agents faces several significant challenges that require further research to overcome for practical application. Firstly, the activation and regulatory mechanisms of ferroptosis within the body are highly complex and interconnected. Tumorigenesis is a multifaceted process involving metabolic disturbances, where ferroptosis can lead to the death or suppression of tumor cells but, in certain contexts, may paradoxically promote tumor formation [126, 127]. Thus, further research is essential to elucidate the precise mechanisms underlying ferroptosis. Secondly, ferroptosis may exhibit potential synergistic effects with other forms of cell death, such as apoptosis [128] and autophagy [129]. Consequently, more in-depth studies are warranted to elucidate the relationships between ferroptosis and these common cell death pathways. Understanding these correlations is crucial for mitigating potential interferences from other cell death mechanisms and for enhancing the therapeutic efficacy of ferroptosis-based treatments. Thirdly, there is a significant lack of effective and specific drugs capable of safely inducing ferroptosis in cancer cells [126]. Current ferroptosis-inducing agents are hindered by poor targeting and low cellular uptake, leading to potential toxic side effects [7]. As previously noted, ongoing studies aim to enhance drug targeting and minimize side effects by designing targeted nanodelivery systems to encapsulate ferroptosis agonists or inhibitors [74, 108]. Compared to conventional drugs, nanodelivery systems offer superior biocompatibility and targeting capabilities, which reduce toxic side effects while enhancing the stability and bioavailability of the encapsulated agents [130]. Additionally, the development of combination therapeutic strategies based on ferroptosis presents a viable approach to mitigating toxicity. For instance, combining cisplatin with the ferroptosis agonist erastin has demonstrated significant synergistic effects in enhancing anti-tumor activity in lung and colon cancers [131, 132].
To address these challenges, future research should focus on several key issues. First, elucidating the molecular mechanisms underlying ferroptosis pathways, particularly those associated with tumor specificity, to improve the targeting precision of therapeutic agents. Second, developing combination therapy strategies, where ferroptosis inducers are used alongside other anticancer drugs or treatment modalities (e.g., radiotherapy, immunotherapy), may enhance therapeutic efficacy while minimizing adverse effects. Additionally, the design of clinical trials should prioritize personalized treatment approaches, tailoring the therapeutic regimen based on the patient’s genotype and tumor characteristics. Although the clinical application of ferroptosis-related drugs in urological malignancies presents significant challenges, ongoing research into the underlying mechanisms and the development of diverse therapeutic strategies hold promise for establishing ferroptosis as a viable and effective cancer treatment option.
Discussion and perspectives
Ferroptosis, a novel form of PCD that differs from apoptosis and necrosis, plays a pivotal role in the growth inhibition of various tumors and influences the tumor immune microenvironment [21, 133]. Ferroptosis has emerged as a significant factor in tumor suppression and represents a therapeutic target in various cancers, including non-small cell lung cancer, liver cancer, pancreatic cancer, and breast cancer. A notable array of iron death inducers has been developed as prospective cancer treatment modalities. For instance, in triple-negative breast cancer, Compound C18 has been shown to impede tumor cell activity by promoting iron death in tumor cells [134]. The fusion of ferroptosis inducers with nanomaterials facilitates targeted delivery, controlled release, biocompatibility, and minimal toxicity, thereby expanding their potential applications [135]. T cells play a role in promoting ferroptosis in tumor cells, suggesting a novel antitumor mechanism. Consequently, the integration of ferroptosis with tumor immunotherapy holds promise as an innovative approach for cancer treatment [127].
Due to its significant role in regulating the biological functions of tumor cells, ferroptosis has also emerged as a novel avenue for exploring treatments for urologic malignancies and advancing drug development in recent years [101, 136, 137]. In terms of urological tumors, investigators have screened numerous targets, including target genes [138, 139], miRNAs [140], and circRNAs [141], which exhibit the capacity to regulate ferroptosis in tumor cells by modulating pivotal pathways, including GSH metabolism, iron metabolism, lipid metabolism, and other intricate molecular pathways [142]. In addition to the above targets, recent studies have attempted to apply ferroptosis inducers [111], natural compounds [66] and analogous agents in the investigation of urologic malignancies. Moreover, Ding Q et al. [74] and Ni W et al. [108] addressed the challenges related to inadequate targeting and limited specificity of ferroptosis inducers by integrating them with nanomaterials, thereby broadening the potential for ferroptosis in clinical applications. Furthermore, the induction of ferroptosis in tumor cells may reverse drug resistance in tumor cells, offering innovative ideas for the combination of ferroptosis inducers and chemotherapeutic agents [43, 56].
However, the current study has certain limitations, including heterogeneity, inadequate sample sizes, and the absence of in vivo experiments. Future investigations should prioritize addressing these deficiencies to yield more comprehensive and robust results. Additionally, given that current research on the regulatory mechanisms and biological functions of ferroptosis in the urinary system is in its early stages, despite being a trending research topic in the diagnosis and treatment of urinary tumors, there remains a paucity of corresponding clinical experiments to thoroughly validate its efficacy. Moreover, as a prognostic biomarker, its reliability and reproducibility necessitate further validation through extensive studies involving larger sample sizes.
Conclusion
In conclusion, we present a comprehensive overview of the mechanisms governing ferroptosis in urologic malignancies and discuss its potential clinical applications. Despite these limitations, delving more deeply into the molecular intricacies of ferroptosis in urologic malignancies holds promise for generating innovative strategies for prevention, identifying therapeutic targets, and establishing robust prognostic biomarkers for these malignancies.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Tan SK, Hougen HY, Merchan JR, Gonzalgo ML, Welford SM. Fatty acid metabolism reprogramming in ccRCC: mechanisms and potential targets. Nat Rev Urol. 2023;20:48–60.
Li F, Zheng Z, Chen W, Li D, Zhang H, Zhu Y, et al. Regulation of cisplatin resistance in bladder cancer by epigenetic mechanisms. Drug Resist Updat. 2023;68:100938.
Foreman KJ, Marquez N, Dolgert A, Fukutaki K, Fullman N, McGaughey M, et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet. 2018;392:2052–90.
Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2020;66:89–100.
Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–25.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–82.
Zhang ZH, Wang Y, Zhang Y, Zheng SF, Feng T, Tian X, et al. The function and mechanisms of action of circular RNAs in Urologic Cancer. Mol Cancer. 2023;22:61.
Rosellini M, Marchetti A, Mollica V, Rizzo A, Santoni M, Massari F. Prognostic and predictive biomarkers for immunotherapy in advanced renal cell carcinoma. Nat Rev Urol. 2023;20:133–57.
Rochette L, Dogon G, Rigal E, Zeller M, Cottin Y, Vergely C. Lipid peroxidation and iron metabolism: two corner stones in the homeostasis control of ferroptosis. Int J Mol Sci. 2022;24:449.
Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med. 2020;152:175–85.
Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. 2022;82:2215–27.
Cheung JCT, Deng G, Wong N, Dong Y, Ng SSM. More than a duologue: In-depth insights into epitranscriptomics and ferroptosis. Front Cell Dev Biol. 2022;10:982606.
Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. 2019;15:1137–47.
Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17:2054–81.
Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302–9.
Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci. 2016;41:274–86.
Camaschella C. Iron-deficiency anemia. N Engl J Med. 2015;372:1832–43.
Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–32.
Kajarabille N, Latunde-Dada GO. Programmed cell-death by ferroptosis: antioxidants as mitigators. Int J Mol Sci. 2019;20:4968.
Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22:381–96.
Nakamura T, Naguro I, Ichijo H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim Biophys Acta Gen Subj. 2019;1863:1398–409.
Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics. 2019;19:e1800311.
Liu J, Kang R, Tang D. Signaling pathways and defense mechanisms of ferroptosis. FEBS J. 2022;289:7038–50.
Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc. Cell Death Differ. 2020;27:662–75.
Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144–52.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Liu Y, Wan Y, Jiang Y, Zhang L, Cheng W. GPX4: The hub of lipid oxidation, ferroptosis, disease and treatment. Biochim Biophys Acta Rev Cancer. 2023;1878:188890.
Lu Y, Qin H, Jiang B, Lu W, Hao J, Cao W, et al. KLF2 inhibits cancer cell migration and invasion by regulating ferroptosis through GPX4 in clear cell renal cell carcinoma. Cancer Lett. 2021;522:1–13.
Kang L, Wang D, Shen T, Liu X, Dai B, Zhou D, et al. PDIA4 confers resistance to ferroptosis via induction of ATF4/SLC7A11 in renal cell carcinoma. Cell Death Dis. 2023;14:193.
Li YZ, Zhu HC, Du Y, Zhao HC, Wang L. Silencing lncRNA SLC16A1-AS1 induced ferroptosis in renal cell carcinoma through miR-143-3p/SLC7A11 signaling. Technol Cancer Res Treat. 2022;21:15330338221077803.
Yu R, Zhou Y, Shi S, Wang X, Huang S, Ren Y. Icariside II induces ferroptosis in renal cell carcinoma cells by regulating the miR-324-3p/GPX4 axis. Phytomedicine. 2022;102:154182.
Zhou ZQ, Lv X, Liu SB, Qu HC, Xie QP, Sun LF, et al. The induction of ferroptosis by KLF11/NCOA4 axis: the inhibitory role in clear cell renal cell carcinoma. Hum Cell. 2023;36:2162–78.
Kim H, Shim BY, Lee SJ, Lee JY, Lee HJ, Kim IH. Loss of Von Hippel-Lindau (VHL) tumor suppressor gene function: VHL-HIF pathway and advances in treatments for metastatic renal cell carcinoma (RCC). Int J Mol Sci. 2021;22:9795.
Green YS, Ferreira Dos Santos MC, Fuja DG, Reichert EC, Campos AR, Cowman SJ, et al. ISCA2 inhibition decreases HIF and induces ferroptosis in clear cell renal carcinoma. Oncogene. 2022;41:4709–23.
Wang J, Yin X, He W, Xue W, Zhang J, Huang Y. SUV39H1 deficiency suppresses clear cell renal cell carcinoma growth by inducing ferroptosis. Acta Pharm Sin B. 2021;11:406–19.
Kerimoglu B, Lamb C, McPherson RD, Ergen E, Stone EM, Ooi A. Cyst(e)inase-rapamycin combination induces ferroptosis in both in vitro and in vivo models of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Ther. 2022;21:419–26.
Chai D, Shan H, Wang G, Li H, Fang L, Song J, et al. AIM2 is a potential therapeutic target in human renal carcinoma and suppresses its invasion and metastasis via enhancing autophagy induction. Exp Cell Res. 2018;370:561–70.
Wang Q, Gao S, Shou Y, Jia Y, Wei Z, Liu Y, et al. AIM2 promotes renal cell carcinoma progression and sunitinib resistance through FOXO3a-ACSL4 axis-regulated ferroptosis. Int J Biol Sci. 2023;19:1266–83.
Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. 2020;5:108.
Tan SK, Mahmud I, Fontanesi F, Puchowicz M, Neumann CKA, Griswold AJ, et al. Obesity-dependent adipokine chemerin suppresses fatty acid oxidation to confer ferroptosis resistance. Cancer Discov. 2021;11:2072–93.
Klasson TD, LaGory EL, Zhao H, Huynh SK, Papandreou I, Moon EJ, et al. ACSL3 regulates lipid droplet biogenesis and ferroptosis sensitivity in clear cell renal cell carcinoma. Cancer Metabolism. 2022;10:14.
Zhou L, Luo Y, Liu Y, Zeng Y, Tong J, Li M, et al. Fatty acid oxidation mediated by malonyl-CoA decarboxylase represses renal cell carcinoma progression. Cancer Res. 2023;83:3920–39.
Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107.
Chang K, Chen Y, Zhang X, Zhang W, Xu N, Zeng B, et al. DPP9 stabilizes NRF2 to suppress ferroptosis and induce sorafenib resistance in clear cell renal cell carcinoma. Cancer Res. 2023;83:3940–55.
Ni X, Ye C, Yu X, Zhang Y, Hou Y, Zheng Q, et al. Overcoming the compensatory increase in NRF2 induced by NPL4 inhibition enhances disulfiram/copper-induced oxidative stress and ferroptosis in renal cell carcinoma. Eur J Pharm. 2023;960:176110.
Schödel J, Grampp S, Maher ER, Moch H, Ratcliffe PJ, Russo P, et al. Hypoxia, hypoxia-inducible transcription factors, and renal cancer. Eur Urol. 2016;69:646–57.
Yang Z, Wei X, Ji C, Ren X, Su W, Wang Y, et al. OGT/HIF-2alpha axis promotes the progression of clear cell renal cell carcinoma and regulates its sensitivity to ferroptosis. iScience. 2023;26:108148.
Yang J, Zhou Y, Li Y, Hu W, Yuan C, Chen S, et al. Functional deficiency of succinate dehydrogenase promotes tumorigenesis and development of clear cell renal cell carcinoma through weakening of ferroptosis. Bioengineered. 2022;13:11187–207.
Shi J, Ma C, Zheng Z, Zhang T, Li Z, Sun X, et al. Low-dose antimony exposure promotes prostate cancer proliferation by inhibiting ferroptosis via activation of the Nrf2-SLC7A11-GPX4 pathway. Chemosphere. 2023;339:139716.
Zhou X, Zou L, Chen W, Yang T, Luo J, Wu K, et al. Flubendazole, FDA-approved anthelmintic, elicits valid antitumor effects by targeting P53 and promoting ferroptosis in castration-resistant prostate cancer. Pharm Res. 2021;164:105305.
Zhang Y, Guo S, Wang S, Li X, Hou D, Li H, et al. LncRNA OIP5-AS1 inhibits ferroptosis in prostate cancer with long-term cadmium exposure through miR-128-3p/SLC7A11 signaling. Ecotoxicol Environ Saf. 2021;220:112376.
Yu Y, Song Y, Cheng L, Chen L, Liu B, Lu D, et al. CircCEMIP promotes anoikis-resistance by enhancing protective autophagy in prostate cancer cells. J Exp Clin Cancer Res. 2022;41:188.
Liu B, Li X, Wang D, Yu Y, Lu D, Chen L, et al. CEMIP promotes extracellular matrix-detached prostate cancer cell survival by inhibiting ferroptosis. Cancer Sci. 2022;113:2056–70.
Zhou X, Zou L, Liao H, Luo J, Yang T, Wu J, et al. Abrogation of HnRNP L enhances anti-PD-1 therapy efficacy via diminishing PD-L1 and promoting CD8(+) T cell-mediated ferroptosis in castration-resistant prostate cancer. Acta Pharm Sin B. 2022;12:692–707.
Wang J, Zeng L, Wu N, Liang Y, Jin J, Fan M, et al. Inhibition of phosphoglycerate dehydrogenase induces ferroptosis and overcomes enzalutamide resistance in castration-resistant prostate cancer cells. Drug Resist Updat. 2023;70:100985.
Cheng L, He Q, Liu B, Chen L, Lv F, Li X, et al. SGK2 promotes prostate cancer metastasis by inhibiting ferroptosis via upregulating GPX4. Cell Death Dis. 2023;14:74.
Yu Y, Huang X, Liang C, Zhang P. Evodiamine impairs HIF1A histone lactylation to inhibit Sema3A-mediated angiogenesis and PD-L1 by inducing ferroptosis in prostate cancer. Eur J Pharm. 2023;957:176007.
Zhang Z, Xie T, Zhang S, Yin H, Zhang X, Zhang S, et al. Second generation androgen receptor antagonist, TQB3720 abrogates prostate cancer growth via AR/GPX4 axis activated ferroptosis. Front Pharm. 2023;14:1110146.
Wang H, Zhang L, Miao Z, Zhang M, Liu H, He Q, et al. PSMA-targeted arsenic nanosheets: a platform for prostate cancer therapy via ferroptosis and ATM deficiency-triggered chemosensitization. Mater Horiz. 2021;8:2216–29.
Wang ME, Chen J, Lu Y, Bawcom AR, Wu J, Ou J, et al. RB1-deficient prostate tumor growth and metastasis are vulnerable to ferroptosis induction via the E2F/ACSL4 axis. J Clin Invest. 2023;133:e166647.
Sun R, Yan B, Li H, Ding D, Wang L, Pang J, et al. Androgen receptor variants confer castration resistance in prostate cancer by counteracting antiandrogen-induced ferroptosis. Cancer Res. 2023;83:3192–204.
Nassar ZD, Mah CY, Dehairs J, Burvenich IJ, Irani S, Centenera MM, et al. Human DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis. Elife. 2020;9:e54166.
Zou P, Chen Z, He Q, Zhuo Y. Polyphyllin I induces ferroptosis in castration-resistant prostate cancer cells through the ERK/DNMT1/ACSL4 axis. Prostate. 2024;84:64–73.
Kumar R, Mendonca J, Owoyemi O, Boyapati K, Thomas N, Kanacharoen S, et al. Supraphysiologic testosterone induces ferroptosis and activates immune pathways through nucleophagy in prostate cancer. Cancer Res. 2021;81:5948–62.
Fu W, Xu L, Chen Y, Zhang Z, Chen S, Li Q, et al. Luteolin induces ferroptosis in prostate cancer cells by promoting TFEB nuclear translocation and increasing ferritinophagy. Prostate. 2024;84:223–36.
Ding Y, Feng Y, Huang Z, Zhang Y, Li X, Liu R, et al. SOX15 transcriptionally increases the function of AOC1 to modulate ferroptosis and progression in prostate cancer. Cell Death Dis. 2022;13:673.
Rossi M, Altea-Manzano P, Demicco M, Doglioni G, Bornes L, Fukano M, et al. PHGDH heterogeneity potentiates cancer cell dissemination and metastasis. Nature. 2022;605:747–53.
Shen L, Zhang J, Zheng Z, Yang F, Liu S, Wu Y, et al. PHGDH inhibits ferroptosis and promotes malignant progression by upregulating SLC7A11 in bladder cancer. Int J Biol Sci. 2022;18:5459–74.
Liu S, Shi J, Wang L, Huang Y, Zhao B, Ding H, et al. Loss of EMP1 promotes the metastasis of human bladder cancer cells by promoting migration and conferring resistance to ferroptosis through activation of PPAR gamma signaling. Free Radic Biol Med. 2022;189:42–57.
Sun Y, Berleth N, Wu W, Schlutermann D, Deitersen J, Stuhldreier F, et al. Fin56-induced ferroptosis is supported by autophagy-mediated GPX4 degradation and functions synergistically with mTOR inhibition to kill bladder cancer cells. Cell Death Dis. 2021;12:1028.
Li X, Xiong W, Wang Y, Li Y, Cheng X, Liu W. p53 activates the lipoxygenase activity of ALOX15B via inhibiting SLC7A11 to induce ferroptosis in bladder cancer cells. Lab Invest. 2023;103:100058.
Wang Q, Ke S, Liu Z, Shao H, He M, Guo J. HSPA5 promotes the proliferation, metastasis and regulates ferroptosis of bladder cancer. Int J Mol Sci. 2023;24:5144.
Ding Q, Tang W, Li X, Ding Y, Chen X, Cao W, et al. Mitochondrial-targeted brequinar liposome boosted mitochondrial-related ferroptosis for promoting checkpoint blockade immunotherapy in bladder cancer. J Control Release. 2023;363:221–34.
Ando T, Tai-Nagara I, Sugiura Y, Kusumoto D, Okabayashi K, Kido Y, et al. Tumor-specific interendothelial adhesion mediated by FLRT2 facilitates cancer aggressiveness. J Clin Invest. 2022;132:e153626.
Jiang P, Ning J, Yu W, Rao T, Ruan Y, Cheng F. FLRT2 suppresses bladder cancer progression through inducing ferroptosis. J Cell Mol Med. 2023;28:e17855.
Luo Y, Zhang Y, Pang S, Min J, Wang T, Wu D, et al. PCBP1 protects bladder cancer cells from mitochondria injury and ferroptosis by inducing LACTB mRNA degradation. Mol Carcinog. 2023;62:907–19.
Xu Y, Tong Y, Lei Z, Zhu J, Wan L. Abietic acid induces ferroptosis via the activation of the HO-1 pathway in bladder cancer cells. Biomed Pharmacother. 2023;158:114154.
Xiang Y, Chen X, Wang W, Zhai L, Sun X, Feng J, et al. Natural product erianin inhibits bladder cancer cell growth by inducing ferroptosis via NRF2 inactivation. Front Pharm. 2021;12:775506.
Wang K, Wang G, Li G, Zhang W, Wang Y, Lin X, et al. m6A writer WTAP targets NRF2 to accelerate bladder cancer malignancy via m6A-dependent ferroptosis regulation. Apoptosis. 2023;28:627–38.
Wang G, Zhao D, Spring DJ, DePinho RA. Genetics and biology of prostate cancer. Genes Dev. 2018;32:1105–40.
Compérat E, Amin MB, Cathomas R, Choudhury A, De Santis M, Kamat A, et al. Current best practice for bladder cancer: a narrative review of diagnostics and treatments. Lancet. 2022;400:1712–21.
Usher-Smith J, Simmons RK, Rossi SH, Stewart GD. Current evidence on screening for renal cancer. Nat Rev Urol. 2020;17:637–42.
Xing XL, Liu Y, Liu J, Zhou H, Zhang H, Zuo Q, et al. Comprehensive analysis of ferroptosis- and immune-related signatures to improve the prognosis and diagnosis of kidney renal clear cell carcinoma. Front Immunol. 2022;13:851312.
Luo G, Wang L, Zheng Z, Gao B, Lei C. Cuproptosis-related ferroptosis genes for predicting prognosis in kidney renal clear cell carcinoma. Eur J Med Res. 2023;28:176.
Sun Z, Li T, Xiao C, Zou S, Zhang M, Zhang Q, et al. Prediction of overall survival based upon a new ferroptosis-related gene signature in patients with clear cell renal cell carcinoma. World J Surg Oncol. 2022;20:120.
Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov. 2019;18:175–96.
Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol. 2016;27:1482–92.
Xu F, Ji S, Yang L, Li Y, Shen P. Potential upstream lncRNA-miRNA-mRNA regulatory network of the ferroptosis-related gene SLC7A11 in renal cell carcinoma. Transl Androl Urol. 2023;12:33–57.
Zong H, Li A, Huang Y, Che X, Zhang Y, Ma G, et al. Analysis of lncRNAs profiles associated with ferroptosis can predict prognosis and immune landscape and drug sensitivity in patients with clear cell renal cell carcinoma. J Biochem Mol Toxicol. 2023;37:e23464.
Merriel SWD, Pocock L, Gilbert E, Creavin S, Walter FM, Spencer A, et al. Systematic review and meta-analysis of the diagnostic accuracy of prostate-specific antigen (PSA) for the detection of prostate cancer in symptomatic patients. BMC Med. 2022;20:54.
Duffy MJ. Biomarkers for prostate cancer: prostate-specific antigen and beyond. Clin Chem Lab Med. 2020;58:326–39.
Ji J, Li H, Wang W, Yuan B, Shen T. ARPC1A is regulated by STAT3 to inhibit ferroptosis and promote prostate cancer progression. Hum Cell. 2022;35:1591–601.
Wang Y, Fan J, Chen T, Xu L, Liu P, Xiao L, et al. A novel ferroptosis-related gene prognostic index for prognosis and response to immunotherapy in patients with prostate cancer. Front Endocrinol (Lausanne). 2022;13:975623.
Liu C, Gao Y, Ni J, Chen S, Hu Q, Wang C, et al. The ferroptosis-related long non-coding RNAs signature predicts biochemical recurrence and immune cell infiltration in prostate cancer. BMC Cancer. 2022;22:788.
Lenis AT, Lec PM, Chamie K, Mshs MD. Bladder cancer: a review. Jama. 2020;324:1980–91.
Liu X, Qiu Z, Zhang X, Su Z, Yi R, Zou D, et al. Generalized machine learning based on multi-omics data to profile the effect of ferroptosis pathway on prognosis and immunotherapy response in patients with bladder cancer. Environ Toxicol. 2024;39:680–94.
Hou J, Lu Z, Cheng X, Dong R, Jiang Y, Wu G, et al. Ferroptosis-related long non-coding RNA signature predicts the prognosis of bladder cancer. BMC Cancer. 2022;22:719.
Li X, Zhou L, Lu T, Zhang L, Li Y, Xu J, et al. Constructing an immune- and ferroptosis-related lncRNA signature to predict the immune landscape of human bladder cancer. J Clin Lab Anal. 2022;36:e24389.
Li J, Zheng S, Fan Y, Tan K. Emerging significance and therapeutic targets of ferroptosis: a potential avenue for human kidney diseases. Cell Death Dis. 2023;14:628.
Yang L, Fan Y, Zhang Q. Targeting ferroptosis in renal cell carcinoma: potential mechanisms and novel therapeutics. Heliyon. 2023;9:e18504.
Gong Q, Guo Z, Sun W, Du X, Jiang Y, Liu F. CX3CL1 promotes cell sensitivity to ferroptosis and is associated with the tumor microenvironment in clear cell renal cell carcinoma. BMC Cancer. 2022;22:1184.
Liu XD, Zhang YT, McGrail DJ, Zhang X, Lam T, Hoang A, et al. SETD2 loss and ATR inhibition synergize to promote cGAS signaling and immunotherapy response in renal cell carcinoma. Clin Cancer Res. 2023;29:4002–15.
Xue W, Jian W, Meng Y, Wang T, Cai L, Yu Y, et al. Knockdown of SETD2 promotes erastin-induced ferroptosis in ccRCC. Cell Death Dis. 2023;14:539.
Ye CL, Du Y, Yu X, Chen ZY, Wang L, Zheng YF, et al. STEAP3 affects ferroptosis and progression of renal cell carcinoma through the p53/xCT pathway. Technol Cancer Res Treat. 2022;21:15330338221078728.
Yangyun W, Guowei S, Shufen S, Jie Y, Rui Y, Yu R. Everolimus accelerates Erastin and RSL3-induced ferroptosis in renal cell carcinoma. Gene. 2022;809:145992.
Wang H, Qiao C, Guan Q, Wei M, Li Z. Nanoparticle-mediated synergistic anticancer effect of ferroptosis and photodynamic therapy: Novel insights and perspectives. Asian J Pharm Sci. 2023;18:100829.
Ni W, Li Y, Liang L, Yang S, Zhan M, Lu C, et al. Tumor microenvironment-responsive nanodrug for clear-cell renal cell carcinoma therapy via triggering waterfall-like cascade ferroptosis. J Biomed Nanotechnol. 2022;18:327–42.
Wu H, Liu Q, Shan X, Gao W, Chen Q. ATM orchestrates ferritinophagy and ferroptosis by phosphorylating NCOA4. Autophagy. 2023;19:2062–77.
Mou Y, Wu J, Zhang Y, Abdihamid O, Duan C, Li B. Low expression of ferritinophagy-related NCOA4 gene in relation to unfavorable outcome and defective immune cells infiltration in clear cell renal carcinoma. BMC Cancer. 2021;21:18.
Ghoochani A, Hsu EC, Aslan M, Rice MA, Nguyen HM, Brooks JD, et al. Ferroptosis inducers are a novel therapeutic approach for advanced prostate cancer. Cancer Res. 2021;81:1583–94.
Yang Y, Liu T, Hu C, Xia H, Liu W, Chen J, et al. Ferroptosis inducer erastin downregulates androgen receptor and its splice variants in castration‑resistant prostate cancer. Oncol Rep. 2021;45:25.
Xu W, Ding J, Li B, Sun T, You X, He Q, et al. Effects of icariin and curcumol on autophagy, ferroptosis, and lipid metabolism based on miR-7/m-TOR/SREBP1 pathway on prostate cancer. Biofactors. 2023;49:438–56.
Kim KS, Choi B, Choi H, Ko MJ, Kim DH, Kim DH. Enhanced natural killer cell anti-tumor activity with nanoparticles mediated ferroptosis and potential therapeutic application in prostate cancer. J Nanobiotechnol. 2022;20:428.
Fan F, Liu P, Bao R, Chen J, Zhou M, Mo Z, et al. A dual PI3K/HDAC inhibitor induces immunogenic ferroptosis to potentiate cancer immune checkpoint therapy. Cancer Res. 2021;81:6233–45.
Wang Y, Lei H, Yan B, Zhang S, Xu B, Lin M, et al. Tumor acidity-activatable macromolecule autophagy inhibitor and immune checkpoint blockade for robust treatment of prostate cancer. Acta Biomater. 2023;168:593–605.
Luo W, Wang J, Xu W, Ma C, Wan F, Huang Y, et al. LncRNA RP11-89 facilitates tumorigenesis and ferroptosis resistance through PROM2-activated iron export by sponging miR-129-5p in bladder cancer. Cell Death Dis. 2021;12:1043.
Xing C, Sun SG, Yue ZQ, Bai F. Role of lncRNA LUCAT1 in cancer. Biomed Pharmacother. 2021;134:111158.
Cao Y, Zou Z, Wu X, Li W, Lu Z, Hu J, et al. LUCAT1 inhibits ferroptosis in bladder cancer by regulating the mRNA stability of STAT3. Gene. 2024;894:147974.
Hu CY, Wu HT, Shan YS, Wang CT, Shieh GS, Wu CL, et al. Evodiamine exhibits anti-bladder cancer activity by suppression of glutathione peroxidase 4 and induction of ferroptosis. Int J Mol Sci. 2023;24:6021.
Kong N, Chen X, Feng J, Duan T, Liu S, Sun X, et al. Baicalin induces ferroptosis in bladder cancer cells by downregulating FTH1. Acta Pharm Sin B. 2021;11:4045–54.
Topalian SL, Taube JM, Pardoll DM. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science. 2020;367:eaax0182.
He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020;30:660–9.
Conche C, Finkelmeier F, Pešić M, Nicolas AM, Böttger TW, Kennel KB, et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut. 2023;72:1774–82.
Luo H, Wang X, Song S, Wang Y, Dan Q, Ge H. Targeting stearoyl-coa desaturase enhances radiation induced ferroptosis and immunogenic cell death in esophageal squamous cell carcinoma. Oncoimmunology. 2022;11:2101769.
Zhou Q, Meng Y, Li D, Yao L, Le J, Liu Y, et al. Ferroptosis in cancer: from molecular mechanisms to therapeutic strategies. Signal Transduct Target Ther. 2024;9:55.
Zhao L, Zhou X, Xie F, Zhang L, Yan H, Huang J, et al. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun (Lond). 2022;42:88–116.
Ye Z, Zhuo Q, Hu Q, Xu X, Mengqi L, Zhang Z, et al. FBW7-NRA41-SCD1 axis synchronously regulates apoptosis and ferroptosis in pancreatic cancer cells. Redox Biol. 2021;38:101807.
Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10:822.
Zhang M, Guo M, Gao Y, Wu C, Pan X, Huang Z. Mechanisms and therapeutic targets of ferroptosis: Implications for nanomedicine design. J Pharm Anal. 2024;14:100960.
Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res Treat. 2018;50:445–60.
Zeng J, Zhang X, Lin Z, Zhang Y, Yang J, Dou P, et al. Harnessing ferroptosis for enhanced sarcoma treatment: mechanisms, progress and prospects. Exp Hematol Oncol. 2024;13:31.
Zhang C, Liu X, Jin S, Chen Y, Guo R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer. 2022;21:47.
Chen T, Leng J, Tan J, Zhao Y, Xie S, Zhao S, et al. Discovery of novel potent covalent glutathione peroxidase 4 inhibitors as highly selective ferroptosis inducers for the treatment of triple-negative breast cancer. J Med Chem. 2023;66:10036–59.
Luo L, Wang H, Tian W, Li X, Zhu Z, Huang R, et al. Targeting ferroptosis-based cancer therapy using nanomaterials: strategies and applications. Theranostics. 2021;11:9937–52.
Liang J, Liao Y, Wang P, Yang K, Wang Y, Wang K, et al. Ferroptosis landscape in prostate cancer from molecular and metabolic perspective. Cell Death Discov. 2023;9:128.
Zeng F, Lan Y, Wang N, Huang X, Zhou Q, Wang Y. Ferroptosis: a new therapeutic target for bladder cancer. Front Pharm. 2022;13:1043283.
Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D, et al. The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 2019;28:2501–2508 e4.
Zheng Q, Li P, Zhou X, Qiang Y, Fan J, Lin Y, et al. Deficiency of the X-inactivation escaping gene KDM5C in clear cell renal cell carcinoma promotes tumorigenicity by reprogramming glycogen metabolism and inhibiting ferroptosis. Theranostics. 2021;11:8674–91.
Zhu C, Song Z, Chen Z, Lin T, Lin H, Xu Z, et al. MicroRNA-4735-3p facilitates ferroptosis in clear cell renal cell carcinoma by targeting SLC40A1. Anal Cell Pathol (Amst). 2022;2022:4213401.
Wang L, Wu S, He H, Ai K, Xu R, Zhang L, et al. CircRNA-ST6GALNAC6 increases the sensitivity of bladder cancer cells to erastin-induced ferroptosis by regulating the HSPB1/P38 axis. Lab Invest. 2022;102:1323–34.
Wang S, Wei W, Ma N, Qu Y, Liu Q. Molecular mechanisms of ferroptosis and its role in prostate cancer therapy. Crit Rev Oncol Hematol. 2022;176:103732.
Acknowledgements
The authors thank the institute for providing the funding and the individuals for offering help during the manuscript process.
Funding
This work was supported by grants from the National Natural Science Foundation of China (82203842).
Author information
Authors and Affiliations
Contributions
WM and XJ: conceptualization, literature search, writing original draft. YL and RJ: conceptualization, supervision, funding, review and editing. All the authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, W., Jiang, X., Jia, R. et al. Mechanisms of ferroptosis and targeted therapeutic approaches in urological malignancies. Cell Death Discov. 10, 432 (2024). https://doi.org/10.1038/s41420-024-02195-w
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41420-024-02195-w
This article is cited by
-
The promising arsenal of ferroptosis inducers in bladder cancer
Journal of Molecular Medicine (2026)
