
Abstract
Ferroptosis, an iron-dependent form of regulated cell death driven by redox dysregulation, is defined by iron overload, reactive oxygen species overproduction, and subsequent peroxidation of polyunsaturated fatty acid-containing phospholipids, notably glycerophospholipids. This review comprehensively delineates the enzymatic such as lipoxygenases and non-enzymatic including Fenton reaction pathways governing glycerophospholipid peroxidation. Furthermore, we systematically dissect fine regulation of iron ions, including absorption, transport, and redox state transition. Given pathophysiological relevance of ferroptosis to numerous diseases, especially neurodegenerative disorders and various cancers, we evaluate emerging therapeutic strategies targeting key ferroptosis nodes, with a primary focus on the key enzymes involved in lipid peroxidation, transferrin receptor-mediated endocytosis mechanism and traditional Chinese medicine. Our work provides a direction for advancing ferroptosis research and developing combinatorial therapies that synergize ferroptosis induction with conventional treatments.
Similar content being viewed by others
Facts
-
Ferroptosis, a distinct form of regulated cell death, is driven by iron overload and subsequent lipid peroxidation mediated by reactive oxygen species.
-
Lipid peroxidation occurs through enzymatic and non-enzymatic pathways, generating free radicals and protein adducts etc. that directly drive ferroptosis.
-
The metabolism regulation of glycerophospholipid containing polyunsaturated fatty acid chains critical for in ferroptosis.
-
Iron ion homeostasis regulation is critically significant in ferroptosis.
-
Ferroptosis is implicated in numerous diseases, such as neurodegenerative diseases, cardiovascular disorders, and cancers. Therapies targeting ferroptosis, particularly the transferrin receptor-mediated process, hold therapeutic promise. Traditional Chinese herbal medicine, with its multi-target bioactive compounds, has also emerged as a key modulator of ferroptosis.
Outstanding Questions
-
Ferroptosis, a multifactorial cell death mechanism linked to diverse pathologies, exhibits redox duality-either exacerbating or mitigating disease progression. Thus, how to control the balance between oxidation and reduction?
-
Why do certain Traditional Chinese Medicine (TCM) active ingredients exert contradictory bidirectional effects on ferroptosis regulation? Reconciling their multi-target actions (e.g., simultaneous modulation of GPX4, ACSL4, Nrf2) to achieve precise ferroptosis control represents a critical challenge in TCM therapy.
Introduction
Ferroptosis research originated in oncology, driven by 30% mutations in the RAS family of small GTPases in cancer cells [1]. Mechanistically, ferroptosis is initiated by redox activity of iron ions, which catalyze reactive oxygen species (ROXs) generation, leading to lipid peroxidation, this process involved in a variety of cellular processes, including: iron ions transport and transition of redox state, lipids redox homeostasis, metabolism activity and diet structure. In view of the complexity of ferroptosis and crosstalk with diverse signaling networds, ferroptosis dysregulation contributes to multiple pathologies: inflammation [2, 3], ischemic damage [4], cardiovascular diseases [5], kidney injury [6], degenerative pathologies [7, 8] and various cancer [9] (Table 1). Therefore, a systems understanding of its molecular drivers, from iron-lipid interplay to metabolic chaos, is essential for developing precision therapies that exploit ferroptosis modulation.
lipid peroxidation in ferroptosis
Molecular basis of lipid peroxidation
Since its conceptualization in 2012, there has been an explosion of research surrounding ferroptosis [1]. As a distinct type of cell death, ferroptosis differs fundamentally from other types of cell death. Membrane perforins are characteristic features of other cell death modalities, such as BAX/BAK in apoptosis [10], MLKL in necroptosis [11], GSDM and NINJ1 in pyroptosis [12,13,14,15], however this is not the case for ferroptosis. As the name implies, ferroptosis is iron-dependent, and phospholipids that contain polyunsaturated fatty acids (PUFAs) are strongly driven by oxidation, leading to directly damage of cell membrane and ultimately cell death [16]. Superoxide anion is a product obtained by single-electron reduction of oxygen [17], and many proteins and enzymes are involved the process, such as, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [18], purine oxidase [19] and the respiratory chain complex I [20], etc. then superoxide anions can react with reducing substances or biological macromolecules, for example superoxide dismutase (SOD) to produce H2O2 [21]. With the ability to accept and donate electrons, iron was allowed to participate in the Fenton reaction, which generates the hydroxyl radical (HO•) that can indiscriminately attack biomolecules, including lipids (Fig. 1).

Superoxide anion radicals are enzymatically produced by sources such as NADPH oxidase, purine oxidase, mitochondrial complex I etc. These radicals are then converted to hydrogen peroxide (H₂O₂) by superoxide dismutase (SOD). H₂O₂ reacts with Fe²⁺, oxidizing it to Fe³⁺ and producing the highly reactive hydroxyl radical (HO•) via the Fenton reaction. Hydroxyl radicals abstract hydrogen atoms from oxidizable molecules like polyunsaturated lipids (LH), yielding carbon-centered lipid radicals (L•). These rapidly react with oxygen (O₂) to form lipid peroxyl radicals (LOO•). LOO• propagate lipid oxidation by abstracting a hydrogen atom from adjacent polyunsaturated lipids (LH). This forms lipid hydroperoxides (LOOH) and generates a new lipid radical (L•), which rapidly reacts with oxygen to yield another peroxyl radical (LOO•), sustaining the chain reaction. Inhibiter of ferroptosis: GPX4, FSP1, RTAs. GPX4, glutathione peroxidase 4; FSP1, ferroptosis suppressor protein-1; RTAs, radical-trapping antioxidants.
Glycerophospholipids peroxidation in ferroptosis
Phospholipids, key structural components of biological membranes, are primarily classified as glycerophospholipids (GPLs) or sphingomyelin [22]. Regarding GPLs, usually, there is a saturated fatty acyl (SFA), such as palmitic acid (C16:0), stearic acid (C18:0) at the sn-1 position of glycerol backbone, whereas there has a greater diversity at the sn-2 position, could be a SFA; a monounsaturated fatty acyl (MUFA), which has been shown to inhibit ferroptosis [16, 23]; or a polyunsaturated fatty acyl (PUFA), for example arachidonic acid (AA, C20:4), adrenic acid (AdA, C22:4) [24], eicosapentaenoic acids (EPA, C20:5), docosahexaenoic acid (DHA, C22:6), which contains multiple double bonds, are susceptible to peroxidation and particularly able to facilitate ferroptosis. GPLs are categorized by the hydrophilic head groups at the sn-3 position. GPL head groups-including phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidyl glycerol (PG), phosphatidylinositol (PI), and phosphatidic acid (PA) (Fig. 2), PC was mainly present in the extracellular leaflet, whereas PS and PE were exclusively in the cytoplasmic leaflet [25], given that PS acts as a “eat me” signal [26], once flipped into the outside of the phospholipid bilayer, it may indicate that the cell is in poor condition, such as apoptosis [27]. In addition, polyunsaturated glycerophospholipids (PUFA-GPLs) and specifically PUFA-PEs which have been identified as the selective targets of pro-ferroptotic lipid peroxidation [28].

Glycerol serves as the backbone of phospholipids, with two FA chains esterified at the sn-1 and sn-2 positions, and a polar head group attached at the sn-3 position. Common polar head groups in phospholipids include ethanolamine, serine, glycerol, choline, and inositol. PUFAs are typically esterified at the sn-2 position of phospholipids. Their structure, featuring two or more cis-configured double bonds, creates bis-allylic hydrogen atoms with low bond dissociation energies, making these sites highly susceptible to hydrogen abstraction. LP PUFAs undergo oxidation via both enzymatic (e.g., LOXs) and non-enzymatic ways, forming primary oxidized products. These primary products can be further redox to form the second products with intact FA chains (e.g., epoxy-, keto-, hydroxy-containing PLs). Or the electrophilic, chain-shortened products (e.g., carboxy, aldehyde relative PLs) due to the low dissociation energy of the O–O bond. Key byproducts of this process, notably 4-HNE and MDA (middle), readily form covalent adducts with nucleophilic residues on proteins (e.g., cysteine, histidine, and lysine), altering protein function and contributing to oxidative stress pathology (right). PUFA, polyunsaturated fatty acyl; GPL, glycerophospholipid; HpETE, 6 hloride 6 d 6 l-eicosatetraenoic acid; 4-HNE, 4-hydroxynonenal; MDA, malondialdehyde.
Oxidation of PUFAs at the sn-2 position of GPLs proceeds via two distinct mechanisms: enzyme-dependent pathways (lipoxygenases, LOXs) and non-enzymatic dependent modes [29, 30]. For LOXs, are kinds of dioxygenases of iron-dependent, PUFAs are used as their substrates, and are named after the oxidized carbon arrangement number on the PUFA chain, such as 5LOX, 12LOX, 15LOX. Furthermore, functional arachidonate LOX genes have been identified within the human genome, underscoring their critical role in lipid peroxidation and ferroptosis [31]. The peroxidation susceptibility of PUFAs in GPLs is critically determined by their bis-allylic positions-carbon centers flanked by two double bonds, where the low bond dissociation energy facilitates hydrogen abstraction [32]. After oxidation, the main molecular product GPL-OOH is formed (Fig. 2). In mammalian cells AA (C20, 4) is one of the most abundant polyenoic fatty acids (FAs) that serve as substrates for the different mammalian LOXs [33], after oxidized by 15LOX, 12LOX and 5LOX, the HOO-group at the 15th, 12th or 5th carbon of the arachidonoyl residue generates 15/12/5-HpETE-phosphatidylethanolamine, respectively (Fig. 2), these products were detected to accumulate in cells undergoing ferroptosis [24, 34]. This suggests that enzyme-catalyzed lipid peroxidation plays an important role in ferroptosis, especially 15LOX [35, 36].
However, polyunsaturated PLs are inherently unstable due to the weak -O-O bond energy of GPL-OOH and readily undergo secondary free radical reactions, giving way to numerous truncated reactive products, including electrophilic oxidatively truncated lipids (epoxy/keto/hydroxy-lipids), or short leaving fragments without the signatures of parental phospholipid identity, such as 4-hydroxynonenal (4-HNE), 4-oxononenal, acrolein and malondialdehyde (MDA) etc [28]. And then, adduct formation occurs between electrophilic oxidatively truncated lipids and nucleophilic amino acid residues (e.g., histidine, lysine, and notably cysteine) in proteins [28]. These adducts confer increased hydrophobicity and high membrane affinity upon the modified proteins, thereby promoting ferroptosis [6].
Glycerophospholipid (GPL) metabolism and regulation
Phospholipases function in ferroptosis
As peroxide substrates, it is of great significance to investigate the metabolism and regulation of GPLs in ferroptosis [37]. The core structure of GPL comprises three parts: backbone (glycerol), hydrophilic head (ethanolamine/serine/glycerol/choline/inositol/ hydrogen oxide) and two hydrophobic tail (Fig. 2), and they are catalyzed by phospholipase A, phospholipase C, and phospholipase D, respectively [22] (Fig. 3). Among the phospholipases, phospholipase A2 (PLA2) [38] critically regulates ferroptosis by hydrolyzing the sn-2 ester bond of glycerol. For example, Mihee et al proved that darapladib, an inhibitor of lipoprotein-associated phospholipase A2 (LpPLA2), synergistically induces ferroptosis in the presence of GPX4 inhibitors [39]; Ca²⁺-independent phospholipase A2β (IPLA2β) suppresses ferroptosis by hydrolyzing 15-HpETE-PE, while its genetic or pharmacological inactivation sensitizes cells to ferroptotsis [40]; By resisting ferroptosis, PLA2 family members regulate pathogenesis across multiple diseases: cancer [41, 42], brain injury [43], acute liver injury [44], neurodegenerative disease [45]. In addition, it was found that PLD, which is responsible for hydrolyzing the bond between hydrophilic head and phosphoric acid (Fig. 3), regulates ferroptosis signal transduction in mouse spleen hypoxia response [46].

ACSL4 catalyzes PUFA and acyl-CoA to form PUFA-CoA, which then forms GPL with LPL in the presence of LPAAT enzyme. GPLs is hydrolyzed by phospholipases at specific sites: PLA1 cleaves the sn-1 fatty acyl ester, PLA2 the sn-2 fatty acyl ester, PLC the phosphodiester bond between glycerol and the phosphate group, and PLD the phosphodiester bond between phosphate and the head group. PUFA polyunsaturated fatty acid, ACSL4 acyl-CoA synthesis long-chain family member, LPL lysophospholipid, LPLATs lysophospholipid acyltransferases, GPL glycerophospholipid, SFA saturated fatty acid.
ACSL function in ferroptosis
The oxidation of GPL in the process of ferroptosis mainly focus on PUFA, so establishing GPL-PUFA metabolism is particularly critical. Generally, fatty acid (FA) synthesis is initiated by the condensation of malonyl-CoA with acetyl-CoA, followed by multiple elongation steps with two-carbon units to yield long chain FA, then desaturation and elongation of these FA chains result in different FA isomers (for example, ω-7 or ω-9 C18:1) [22, 47]. However, desaturases which generate ω-3 and ω-6 FAs are absent in mammals, therefore, the related precursors must be obtained from dietary.
Acyl-coenzyme A (acyl-CoA) synthases (ACS), which catalyze the conversion of FAs to fatty acyl-CoAs [48], are classified into very long-chain acyl-CoA synthase, acyl-CoA synthase long-chain family member (ACSL; for FAs with 10 to 22 carbons), medium-chain acyl-CoA synthase, and short-chain acyl-CoA synthase based on the carbon chain length of their substrates [49]. ACSL comprises five isoenzymes: ACSL1 and ACSL3-6 [50]. Among ACSL family members, ACSL4 specifically esterifies CoA to PUFAs, including arachidonic acid (AA; forming AA-CoA), adrenic acid (AdA), eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), epoxyeicosatrienoic acids (EETs), and hydroxyeicosatetraenoic acids (HETEs), to generate PUFA-CoAs [51]. These PUFA-CoAs are subsequently esterified into phospholipids (forming PUFA-PLs) by lysophospholipid acyltransferases [52], such as lysophosphatidylcholine acyltransferases (LPCAT) [53, 54] (Fig. 3), PUFA-PLs are readily to be oxidized and trigger ferroptosis [55, 56]. Cheng et al. demonstrated that ACSL4 overexpression elevated levels of ferroptotic markers, including 5‑ hydroxyeicosatetraenoic (HETE), 12‑HETE, and 15‑HETE, and thereby suppressed glioma cell proliferation [57]. Furthermore, accumulating evidence links ACSL4 to the pathogenesis of multiple diseases via ferroptosis [58,59,60,61]. Beyond ACSL4, other family members also modulate ferroptosis. For example: ACSL1 promotes α-eleostearic acid (Αesa) incorporation into neutral lipids (e.g., triacylglycerols); disrupting triacylglycerol biosynthesis suppresses Αesa-induced ferroptosis [62]; Inhibiting ACSL3 reduces ferroptosis susceptibility in clear cell renal cell carcinoma (ccRCC) cells [63].
LPLAT function in ferroptosis
The chemical diversity of phospholipids stems primarily from variations in headgroups at the sn-3 position and FAs at the sn-1 and sn-2 positions of glycerol-based structures. Glycerol-3-phosphate acyltransferases (GPATs) and lysophospholipid acyltransferases (LPLATs) (Fig. 3), working in concert with phospholipase enzymes, contribute to this diversity via selective esterification and deesterification [64]. To date, 14 LPLATs have been identified, all fall into one of two enzyme families: the 1-acylglycerol-3-phosphate O-acyltransferases (AGPATs) or the membrane-bound O-acyltransferases (MBOATs) [65]. Given their role in phospholipid remodeling, LPLATs are closely linked to ferroptosis. Specifically, LPCAT3 (MBOAT5) is responsible for generation of C20:4 phospholipids. Inhibiting LPCAT3 induces rapid remodeling of polyunsaturated phospholipids in human cells, characterized by suppressed C20:4-PLs and a compensatory increase in C22:4-PLs. This lipid rewiring provides a mechanistic basis for the observed partial, yet incomplete, protection from ferroptosis [66]. LPCAT3 is also involved in subarachnoid hemorrhage induced early brain injury and associated with ferroptosis through suppress and upregulate LPCAT3 expression level [67]. Moreover, LPCAT3 is significantly upregulated in various cancers and emerges as a potential prognostic biomarker in acute myeloid leukemia, it may be correlated with immune infiltration and ferroptosis [68]. For LPCAT2, its overexpression enhances ferroptosis susceptibility in HEK293T cells [53]. However, LPLATs incorporate not only PUFAs but also SFAs into GPLs. For example, Hyemin et al. reported that LPCAT1 specifically utilizes SFA-CoA to acylate lysophospholipids, generating SFA-phospholipids that confer ferroptosis resistance by displacing ferroptosis-susceptible PUFAs [69]; similarly, Ziwen Li et al. demonstrated that LPCAT1 confers ferroptosis resistance by enhancing membrane phospholipid saturation via the Lands cycle, thereby reducing PUFA incorporation [70].
Iron transporters and related regulation
Iron absorption, circulation and storage
Iron, a biometal with pleiotropic functions, is central to ferroptosis execution while simultaneously mediating oxygen transport (via hemoglobin), supporting erythropoiesis, and participating in mitochondrial electron transport chains [71], synthesis of iron-sulfur proteins, mitochondrial function, DNA synthesis, etc [72]. In humans, >90% of irons demand is met by recycling existing iron, cause of the total iron flux required to maintain erythropoiesis is about 2–3 × 1015 atoms of iron per second in an adult human [73]. Given the inevitable physiological iron loss, such as perspiration, epithelial desquamation, menstruation, stringent regulation of iron metabolism, including absorption (via DMT1), transport (transferrin-mediated), redox cycling, storage (ferritin-bound), and systemic homeostasis is essential to prevent deficiency [74].
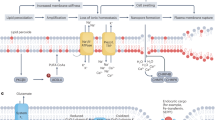
Hepatic and splenic macrophages phagocytosing senescent erythrocytes, extracting iron from heme, and exporting it to the plasma; the remaining systemic iron pool is predominantly replenished from diet [73] (Fig. 4a). Dietary iron is categorized into heme and non-heme forms. Intestinal enterocytes absorb heme iron directly via haem carrier protein 1 (HCP1), while non-heme iron is transported by divalent metal transporter 1 (DMT1) after were reduced by reductase such as duodenal cytochrome b (Dcytb) to ferrous ions (Fe2+) (Fig. 4a); Dcytb, primarily located in enterocyte apical membranes, functions as a ferric reductase that converts ferric ions (Fe³⁺) to absorbable Fe²⁺ [75]. Ferroportin, the sole iron exporter in mammals, transports Fe²⁺ from cells and is critical for systemic iron homeostasis [76]. Multicopper oxidases, including intestinal-enriched hephaestin (HEPH), liver-derived plasma ceruloplasmin (CP), and placental-enriched Zyklopen, oxidize Fe²⁺ to Fe³⁺ through ferroxidase activity [77, 78]; hephaestin presumably works in conjunction with ferroportin [79].

a Systemic iron flux. Macrophages in the liver and spleen take up and digest senescent red blood cells, releasing the iron into the plasma; the remainder comes from dietary sources. Dietary iron comprises heme iron (from lean) and non-heme iron. Intestinal enterocytes absorb heme iron via HCP1, while non-heme iron is absorbed by DMT1 after being reduced to Fe²⁺ by Dcytb, which is primarily localized in the enterocyte’s apical membrane. FPN, the only known iron ions exporter, which mediates Fe2+ export in mammalian cells. Multicopper oxidases oxidize Fe²⁺ to Fe³⁺. HEPH (highly expressed in intestines) and CP (hepatocyte-derived, circulating in plasma) both exhibit ferroxidase activity. HEPH presumably works in conjunction with FPN. b Cellular iron regulation. Tf binds Fe³⁺ and delivers it to cells via TfR-mediated endocytosis, forming endosomes. Within endosomes, Fe³⁺ dissociates from Tf, is reduced to Fe²⁺ by STEAP3, and is transported into the cytosol by DMT1 or ZIP transporters. Human TfR1 was found belonging to cell surface receptor for H ferritin. TfR2 contains mitochondrial targeting motif, facilitates irons delivery to the mitochondria; Lysosomes containing TfR2-α translocate to mitochondria and dock with them, facilitating iron transfer. Cytosolic labile iron is stored within ferritin cages with the assistance of PCBPs. During iron deficiency, ferritin interacts with the receptor NCOA4 and is degraded via ferritinophagy. Furthermore, PCBPs assist in other cellular iron processes: transferring iron from the importer DMT1, exporting iron via FPN, degrading heme through HO1; and regulating iron-dependent enzymes involved in lipid peroxidation (LOXs); other enzymes including DOHH, MO or enzymes in mitochondria. Tf transferrin, TfR transferrin receptor, FPN ferroportin, DMT1 divalent metal transporter 1, Dcytb duodenal cytochrome b, ZIP ZRT/IRT-like protein, STEAP six-transmembrane epithelial antigen of the prostate. HCP haem carrier protein 1, CP Ceruloplasmin, HEPH Hephaestin, LIP Labile ion pool, PCBPs (poly(Rc)-binding proteins 1-4), HO1 heme degradation via heme oxygenase 1, LOXs lipoxygenases, DOHH deoxyhypusine hydroxylase, MO monooxygenases.
Transferrin (Tf) facilitates cellular iron uptake by binding Fe³⁺ and undergoing Tf receptor (TfR)-mediated endocytosis, forming iron-loaded endosomes for furture intracellular transport [80,81,82] (Fig. 4b). Fe3+ dissociates from Tf and is reduced to Fe2+ by metalloreductases six-transmembrane epithelial antigen of the prostate 3 (STEAP3). The Fe2+ are subsequently transported into the cytosol via DMT1 or Zrt- and Irt-like protein (ZIP) transporters [72, 83, 84]. In addition, TfR2 facilitates irons delivery to the mitochondria, which a variety of enzymes are located there, such as, NADH-coenzyme Q oxidoreductase (complex I) and succinate-Q oxidoreductase (complex II), require iron as an essential cofactor; additionally, mitochondria consume iron for heme biosynthesis and iron-sulfur cluster assembly; mitochondrial targeting motif exists in the intracellular domain of TfR2-α, so the lysosomes containing TfR2-α move toward and physically interact with mitochondria to facilitate iron transfer [85,86,87]. The cytosolic labile iron pool (LIP) is regulated through dual mechanisms: under iron-replete conditions, excess labile iron is sequestered within ferritin nanocages via poly(Rc)-binding proteins 1-4 (PCBPs) mediated packaging [88,89,90,91]; conversely, during iron deficiency, NCOA4 acts as a autophagy receptor that directs ferritin to degradation by ferritinophagy [92] (Fig. 4b). PCBPs function as multifunctional chaperones coordinating distinct iron regulatory pathways: first, iron homeostasis: mediate ferritin-dependent storage, DMT1-mediated cellular iron import, and ferroportin-driven export; second, metabolic regulation: facilitate heme catabolism via heme oxygenase 1 (HO1) and modulate lipid peroxidation through LOXs; third, enzymatic coordination: support iron-dependent enzymes including deoxyhypusine hydroxylase (DOHH), monooxygenases (MO), and other enzymes in mitochondria [6, 90] (Fig. 4b).
Transferrin (Tf) and Transferrin Receptors (TfRs)
Tf, a glycoprotein critical for systemic iron transport, comprises three subtypes: serum Tf, lactotransferrin, and melanotransferrin [93], this review focuses on serum Tf. Iron-loaded Tf binds to TfRs which locate on the cell surface, facilitating receptor-mediated endocytosis. This process enables the delivery of Fe³⁺ from the bloodstream into tissues such as the liver, spleen, and bone marrow [74]. Structurally, Tf consists of two lobes [94], and each binds a single Fe³⁺ ion, resulting in three possible iron occupancy states: binding one Fe3+, binding two Fe3+ (holo-Tf), and binding none Fe3+ (apo-Tf) [85]. In addition, the interaction between Fe³⁺ and Tf is pH-dependent: Fe3+ efficiently binds to Tf at Ph 7.4 and dissociates at acidic Ph in endosomes [95, 96]. Mammals express two Tfs, TfR1 and TfR2 [85]: TfR1 is ubiquitously expressed and especially highly in erythroblasts [71], given that over 80% of plasma Tf-bound iron is absorbed by erythroid precursors in the bone marrow, where it is utilized for heme synthesis [97]; however, TfR2 remained unidentified until in 1999 [98], the human TfR2 gene is expressed selectively in hepatocytes and erythroid [99].
Given the critical physiological role of Tf and TfRs in iron transport, their dysregulation has been implicated in the pathogenesis of various diseases, including anemia [100], renal fibrosis [101], systemic lupus erythematosus [102], hemochromatosis [103], among others. Such associations underscore the critical importance of tightly regulating Tf and TfRs, not only to preserve iron balance but also to mitigate disease progression, highlighting their potential as therapeutic targets. TfR expression is regulated through the IRP (iron regulatory protein)/iron-response element (IRE) axis in response to cellular iron status [104]; Under low iron conditions, IRP stabilizes TfR1 mRNA by binding to IREs in the 3’ untranslated region (3’ UTR), enhancing TfR1 protein synthesis and boosting cellular iron uptake [105]. Research demonstrates HIF signaling pathway upregulates TfR expression [106,107,108]. Furthermore, miRNAs are also involved in the TfRs regulation. For instance, miR-148a levels negatively correlate with TfR1 mRNA in hepatocellular carcinoma [109]; Bioinformatic analysis reveals that miR-497-5p inversely correlates with TfR in cervical cancer [110].
Ferroportin function and related regulation
As mentioned above, dietary iron absorbed by intestinal enterocytes and recycled iron from macrophages phagocytosing aged erythrocytes are transported to systemic circulation via ferroportin-mediated export, as detailed below [111].
Ferroportin (FPN/SLC40A1 [79]) the primary cellular iron exporter, is abundantly expressed in key iron-handling tissues: duodenal enterocytes (dietary absorption), hepatocytes (storage), splenic macrophages (erythrocyte recycling), and placental syncytiotrophoblasts (maternal-fetal transfer) [73]. FPN critically regulates systemic iron homeostasis, as evidenced by transgenic models: FPN knockout mice exhibit enterocyte/macrophage/hepatocyte iron overload and embryonic lethality [79]; Hepatic FPN specific knockout mice under iron deficiency develop impaired iron mobilization and severe anemia, with red blood cell and hemoglobin levels much lower, demonstrating FPN’s dual role in hepatocyte iron mobilization and macrophage iron recycling during dietary iron stress [112]. Beyond anemia and hemochromatosis [113], FPN dysregulation contributes to multisystem pathologies: its deficiency induces memory impairment by promoting ferroptosis in Alzheimer disease [114]; while hepatic FPN downregulation drives proliferation and M2-like polarization of macrophages and leading to hepatic fibrosis through increased the levels of the M2 markers CD206, TGF-β, VEGF, MMP-9, Laminin, Collagen, IL-4 and IL-10 [115], etc. So as the sole iron exporter, FPN is strictly regulated.
Hepcidin, a 25 amino acid hepatocyte-derived peptide hormone, critically regulates FPN according to iron concentration [116]. Elevated plasma iron concentration triggers hepcidin upregulation, which binds FPN to induce its internalization and degradation, reducing iron export from enterocytes and macrophages into circulation. Conversely, decreased serum iron suppresses hepcidin production, enhancing dietary iron absorption (enterocytes) and hepatic iron mobilization (hepatocytes) via FPN-mediated efflux into circulation [72, 117]. Structurally, FPN functions as an electroneutral H⁺/Fe²⁺ antiporter, coupling each Fe²⁺ export to counter-transport of two protons; perturbation of either ion binding site uncouples this co-transport [118]. Hepcidin binds FPN in an outward-open conformation and completely occludes the iron efflux pathway to inhibit transport; hepcidin binding to FPN is coupled to iron binding, with an 80-fold increase in hepcidin affinity; the carboxy terminus of hepcidin directly contacts the divalent metal in FPN’s C domain, establishing a degradation targeting mechanism exclusive to iron-loaded FPN [119]. Research confirmed that hepcidin binding triggers rapid FPN ubiquitination in both FPN-overexpressing cell lines and murine bone marrow derived macrophages [120].
In addition, research has found that human FPN binds to and mediates Ca2+ transport; Ca2+ binding site distinct from Fe2+ binding sites; Ca2+ transport is significantly inhibited in the presence of Fe2+ but not vice versa; this indicate function of FPN as a Ca2+ uniporter may allow regulation of iron homeostasis by Ca2+ [121, 122]. Vamifeport (VIT-2763), the first clinical-stage oral FPN inhibitor, binds FPN’s central site, structurally overlapping hepcidin’s interaction site, competitively blocking hepcidin binding, this therapeutic is under clinical evaluation for β-thalassemia and sickle cell disease [123]. Namgaladze et al. demonstrated that Nrf2 (redox-sensitive transcription factor) accumulation coupled with BACH1 (transcription factor) downregulation induces FPN expression, conferring ferroptosis resistance in human macrophages [124] etc. Collectively, these findings validate FPN as a high-potential therapeutic target.
Ferritin function and related regulation
Ferritin, as an iron ion chelating protein, sequesters iron ions to prevent lipid peroxidation caused by Fenton reaction, serving as a critical antioxidant safeguard in ferroptosis [125]. Ferritin, a ubiquitous protein found in all cells, forms multiple isoferritins composed of 24 subunits of two principal types: the H subunit (originally isolated from human heart and the predominant form there, also characterized by its heavier electrophoretic migration) and the L subunit (originally isolated from human liver and the predominant form there, with lighter electrophoretic migration) [126]. While the H subunit gene resides on chromosome 11q and the L subunit gene on chromosome 19q, the H:L ratio incorporated into the assembled ferritin complex varies dynamically depending on tissue type and developmental stage [88, 127].
Within the body’s iron transport system, FPN acts like export vehicles (trucks/ships), transporting irons out of absorption sites (enterocytes) and storage cells (like hepatocytes and macrophages) into the bloodstream. Conversely, the Tf/TfR system functions as import vehicles (delivery vans/trains), transporting irons from the blood into various cells throughout the body for utilization. Meanwhile, ferritin serves as the essential warehouse, storing and stabilizing irons within cells. Its remarkable capacity-each molecule can sequester up to 4500 iron atoms-makes it the central hub for safe, bioavailable iron storage [126]. Addition, ferritin also has ferroxidase activity, converting Fe2+ to Fe3+ with ferritin H chain but not L chain [88]. Ferritin is predominantly localized in the cytoplasm, but small quantities are in serum [128, 129], mitochondrial [130, 131] and nuclear [132, 133]. As macrophages phagocytose senescent red blood cells, releasing large quantities of irons into cytoplasm, and macrophages synthesize ferritin to sequester these irons. Additionally, macrophages secrete a portion of ferritin into the circulation [85, 134]. Beyond above, ferritin exhibits functional parallels with the Tf/ TfR system. Similar to Tf, serum ferritin can deliver iron to cells by binding to dedicated cell surface receptors. Known ferritin receptors include: first, TIM-2 (T cell immunoglobulin-domain and mucin-domain 2) specifically bound ferritin H [135]; second, Scara5 (a scavenger receptor that can bind various ligands) preferentially binds ferritin L [136], then enters cells through internalization; third, surprisingly, human TfR1 was identified as a cell surface receptor for H ferritin [137].
Associated therapy
Internal inhibition mechanism of ferroptosis
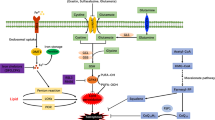
As an iron-dependent and non-apoptotic cell death, ferroptosis represents a promising therapeutic strategy. Modulation, either inhibiting or promoting its induction, holds potential efficacy, according to different diseases and therapeutic goals. The core pathway for ferroptosis inhibition centers on the System Xc⁻/GSH/GPX4 axis; System Xc⁻, an antiporter, imports extracellular cystine in exchange for intracellular glutamate. Cytosolic cystine is rapidly reduced to cysteine, the rate-limiting precursor for glutathione (GSH) synthesis. GSH acts as an essential cofactor for glutathione peroxidase 4 (GPX4), enabling GPX4 to catalyze the reduction of lipid hydroperoxides (LOOH) to corresponding alcohols (LOH), this process is critical for neutralizing membrane lipid peroxidation, the hallmark of ferroptosis [138, 139] (Fig. 1). Furthermore, researches have definitively established that selenium is required for GPX4’s catalytic activity [140, 141]. Critically, seminal research identified two key small-molecule compounds erastin and RSL3 (RAS-selective lethal compound 3) targeting System Xc⁻ SLC7A11 subunit and GPX4 respectively to induce ferroptosis [1, 139, 142]. The ferroptosis suppressor protein 1 (FSP1), operating within the FSP1-CoQ10-NAD(P)H axis, functions as an NAD(P)H-dependent oxidoreductase that catalyzes the reduction of membrane-bound ubiquinone (CoQ10) to ubiquinol (CoQ10H2), the ubiquinol directly reducing lipid peroxyl radicals to terminate lipid peroxidation; concurrently, FSP1 also participates in a noncanonical redox cycle of vitamin K and regenerating the oxidized α-tocopheryl radical (vitamin E) to its non-radical form to against ferroptosis [56, 143]. In addition, squalene and di/tetrahydrobiopterin (BH2/BH4) mediated inhibition of lipid peroxidation, may acting as endogenous radical-trapping antioxidants [56] (Fig. 1). The DHODH-CoQH2 pathway, localized to the outer surface of mitochondrial inner membrane, catalyzes the conversion of dihydroorotate (DHO) to orotate (OA) while simultaneously refreshing CoQH2 to clear lipid radicals [144].
Targeting lipid peroxidation
According to above, therapies targeting lipid peroxidation primarily fall into two categories: radical-trapping antioxidants (RTAs) and inhibitors targeting enzymes such as LOXs and ACSL4.
Radical-trapping antioxidants (RTAs)
Unlike traditional antioxidants, which stabilize free radicals primarily by donating electrons, RTAs physically capture and neutralize radicals by forming stable complexes with them [145]. Upon the conceptualization of ferroptosis, ferrostatin-1 (Fer-1), a RTA was discovered through a small molecules library screening [1] and its function is to scavenge the alkoxyl radicals [146]. Another RTA, liproxstatin-1, H-atom abstracted from liproxstatin-1 by radical occurs preferentially at the aromatic amine site (1’-NH) and liproxstatin-1 radical is easily regenerated to the active reduced form by endogenous ubiquinol, preventing secondary damage by free radicals [147]. Recent studies reveal that 7-dehydrocholesterol (7-DHC, a cholesterol precursor), as a RTA, effectively shields (hloride) lipids from autoxidation and subsequent fragmentation by using the conjugated diene, ultimately inhibiting ferroptosis, thereby presenting a novel metabolic target for therapeutic intervention [145, 148, 149].
Targeting LOXs therapy
Recent advances have revealed a growing repertoire of molecules that critically regulate ferroptosis, including drug candidates from high-throughput screens, rationally designed enzyme-targeting compounds, and bioactive constituents extract from botanical, especially natural Chinese herbs.
LOXs, a class of non-heme iron-containing dioxygenases, mediate PL oxidation in ferroptosis. Humans express six LOX isoforms: ALOX5, ALOX12, ALOX12B, ALOX15, ALOX15B, and ALOXE3 [31]. Inhibitors of LOXs primarily function through three mechanistic classes: (1) directly integrated with LOXs; (2) modulation of post-translational modifications (e.g., acetylation, phosphorylation); (3) disruption of protein-protein interactions in LOX-containing complexes to destabilize functional assemblies (Table 2). ALOX5 promotes pancreatic cancer invasion and metastasis by driving tumor-associated macrophage M2 polarization via the JAK/STAT pathway, however, the ALOX5 inhibitor zileuton, currently the only approved agent in this class, can suppress these effects [150]; Bin Wang et al. reported that Nordy (dl-nordihydroguaiaretic acid compound), another ALOX5 inhibitor, induces differentiation and inhibits self-renewal in glioma stem-like cells [151]; Clau (Clausenamide) was found to inhibit lipid peroxidation and ferroptosis by blocking ALOX5 phosphorylation and nuclear translocation [152]. ALOX12 represents another therapeutic target, IMA-1 treats NASH by disrupting the interaction between ALOX12 and acetyl-CoA carboxylase 1 (ACC1) [153]; ML355, a targeted ALOX12 inhibitor, alleviates lung ischemia-reperfusion injury by reducing endothelial ferroptosis-mediated neutrophil extracellular trap (NET) formation [154]; separately, it impairs leukemia stem/progenitor cell function by disrupting nicotinamide adenine dinucleotide phosphate (NADPH) homeostasis, inducing oxidative stress and damage in CML HSPCs and committed cells by targeting ALOX12–12-HETE [155]; Baicalein protects against cisplatin-induced acute kidney injury (AKI) by inhibiting ALOX12-mediated ferroptosis [156]; hydrogen sulfide was proved to protect myoblasts from ferroptosis by inhibiting ALOX12 acetylation [157]; compound 99089, a potent and selective 12/15-LOX inhibitor, represents a promising candidate for developing anti-stroke therapeutics [158]. For ALOX15, there are many new development: Baicalein, a bioactive flavonoid from Banxia Xiexin Decoction, ameliorates CPT-11-induced gastrointestinal dysfunction by suppressing ALOX15-mediated ferroptosis [159]; Chicoric acid (CA), a natural ALOX15 inhibitor, suppresses ferroptosis in asthmatic by inhibiting ALOX15 expression [160]; Tianma Gouteng Granules (TG), a clinically prescription of traditional Chinese medicine, ameliorate behavioral deficits and dopaminergic neurodegeneration in Parkinson’s disease models by directly inhibiting ALOX15 activity and attenuating lipid peroxidation-driven ferroptosis [161]; Inhibition of ALOX15 by PD146176 suppresses epithelial-mesenchymal transition (EMT) in eosinophilic chronic rhinosinusitis with nasal polyps [162] and protects male germ cells against 4-hydroxynonenal (4-HNE)-induced protein damage [163]; Isochlorogenic acid C, a prominent component in Danzhi Xiaoyao Powder, mitigates macrophage phospholipid peroxidation in the stress-induced tumor microenvironment by inhibiting the ALOX15/PEBP1 complex [164]; FerroLOXINs are specifically designed inhibitors that selectively target the pro-ferroptotic 15LOX-PEBP1 complex, but not 15LOX alone [165]; Scutellarein alleviates chronic obstructive pulmonary disease (COPD) by inhibiting ferroptosis through iron chelation and ALOX15 interaction [166]; α-Tocopherol not only functions as a RTA but also regulates ALOX15: it inhibits ALOX15 activity by binding to 87th Leu and suppresses ALOX15 expression [167].
Targeting ACSL4 therapy
ACSL4, an enzyme that synthesizes PUFA-containing phospholipids, also regulates their production. Baicalein acts not only as a pan-LOX inhibitor but also modulates other enzymatic targets, including ACSL4 [168]; multiple studies confirm that rosiglitazone inhibits ferroptosis by suppressing ACSL4 [58, 169, 170]; like rosiglitazone, pioglitazone and troglitazone-both thiazolidinediones, act as pharmacological inhibitors of ACSL4 [171]; among FDA-approved compounds screened, sertaconazole emerged as a potent ACSL4 inhibitor [172]; PRGL493, an ACSL4 inhibitor developed through homology modeling and virtual screening, demonstrated potent inhibitory activity in validation studies [173]; using nanoparticle-based delivery, compound AS-252424 directly binds glutamine 464 of ACSL4 to inhibit its enzymatic activity, thereby suppressing lipid peroxidation and ferroptosis [60]; Berberine (BBR), an isoquinoline alkaloid historically used to treat diarrhea, has gained attention for its diverse pharmacological effects. Recently, it was found to inhibit ferroptosis in vascular endothelial cells by targeting ACSL4 [174]; post-translational modifications of ACSL4 critically regulate its activity, exemplified by PKCβII-mediated phosphorylation [175]; the natural flavonol fisetin ameliorates fibrotic kidney disease in mice by suppressing transcription activity of ACSL4 mediated-tubular ferroptosis [176].
TfR mediated endocytosis therapy
Given TfR’s role in mediating Tf and ferritin endocytosis, making us to explore the probility of this mechanism as a potential therapeutic strategy. The primary therapeutic strategy involves enhancing drug uptake in target cells to achieve desired therapeutic outcomes. Sorafenib (SOR), a frontline hepatocellular carcinoma (HCC) therapeutic, induces ferroptosis through inhibition of solute carrier family 7 member 11 (SLC7A11). Nanovesicles engineered for surface Tf-Fe³⁺ display and SOR encapsulation (SOR@TF-Fe³⁺ NVs) achieved TfR-directed HCC targeting, enhancing tumoral iron/SOR accumulation and ferroptosis induction while maintaining favorable liver distribution [177]. Increasing intracellular iron levels to promote ferroptosis is another effective treatment. T10@Clav nanodrugs-engineered by conjugating Tf-targeting peptide T10 to cross-linked lipoic acid vesicles, hijack circulatory transferrin Tf for tumor-specific delivery. This system elevates labile Fe²⁺ via: (i) TfR-mediated iron import, and (ii) dihydrolipoic acid mediated Fe²⁺ regeneration from vesicle degradation, synergistically amplifying ferroptosis [178]. Another key therapeutic strategy involves enhancing intracellular accumulation of anticancer agents within malignant cells. Fluorinated 21 hloride[salophene]iron(III) complexes (salophene = N,N′-bis(salicylidene)-1,2-phenylenediamine) exhibit promising anticancer activity. Cellular uptake of the complexes appears to be mediated by TfR1. Critically, all complexes significantly decreased metabolic activity across tested cell lines derived from ovarian cancer, breast cancer, and leukemia [179]. RSL3 induces ferroptosis via GPX4 inhibition [139], while TfR1-mediates ferritin endocytosis promotes ferroptosis by increasing intracellular iron [137]. Consequently, the combination of RSL3 and ferritin therapy holds promise for enhanced therapeutic efficacy by simultaneously inhibiting the GPX4 antioxidant defense and elevating pro-ferroptotic iron levels. A bio-inspired protein nanocomplex was engineered by conjugating naturally occurring bovine serum albumin (BSA) with ferritin via acidity-responsive glutaraldehyde linkers. BSA provides multiple anchoring points for efficient RSL3 loading, while ferritin promoted TfR1-mediated cellular uptake. The nanocomplex significantly enhances antitumor efficacy without inducing observable adverse effects, highlighting its potential for clinical development of synergistic ferroptosis inducers [180].
Ferritin related therapy
Combining different cell death modalities represents an effective strategy for treating diverse cancers. A novel carrier-free nanodrug, nanoparticle ferritin-bound erastin and rapamycin (NFER), was prepared via emulsion. In vitro studies demonstrated NFER’s robust ferroptosis-inducing capability through GPX4 downregulation and concomitant lipid peroxidation accumulation, and rapamycin-mediated autophagy within NFER acted synergistically to amplify ferroptosis [181]. Emerging evidence indicates that combining photothermal therapy (PTT) with ferroptosis induction represents a promising therapeutic strategy against drug-resistant tumors. In a recent study, triple-functional nanoparticle (I@P-ss-FRT) integrating thermal therapy, ferroptosis induction, and magnetic resonance imaging (MRI) capability was developed. Drug-resistant cancer cells exhibited significantly enhanced uptake of I@P-ss-FRT and synergistic sensitivity to PTT/ferroptosis co-activation. Notably, I@P-ss-FRT plus near-infrared (NIR) achieved optimal tumor suppression, accompanied by decline of GSH/GPX4 and accumulation of lipid peroxides at tumor sites in vivo [182]. Analogous to Tf, Ferritin-Hijacking Nanoparticles (Ce6-PEG-HKN15) was engineered by utilizing homing peptide, the nanoparticles spatiotemporally direct endogenous ferroptosis for synergistic anticancer therapy [183]. In addition, the traditional Chinese medicine dihydroartemisinin was revealed to trigger ferroptosis in cancer cells via autophagy-dependent ferritin degradation (ferritinophagy), liberating iron to amplify lipid peroxidation through Fenton reactions [184]. Sono-photodynamic therapy (SPDT) generates cytotoxic reactive oxygen species (ROS) under dual light/ultrasound activation, inducing multimodal cell death. This prompts us to consider the attempt of coupling SPDT and dihydroartemisinin therapy. Yilin Zheng et al. report a facile construction of ferritin-based nanosensitizer FCD through co-encapsulating chlorin e6 (Ce6) and dihydroartemisinin within horse spleen ferritin [185]. GPX4 and FSP1 constitute two dominant defense systems against ferroptosis, and dual inhibition of these two targets significantly enhances therapeutic efficacy against malignancies. A genetically engineered murine heavy-chain ferritin (mHFn) carrier mHFn@RSL3/Ifsp1 was designed to co-deliver RSL3 (GPX4 inhibitor) and iFSP1 (FSP1 suppressor) to tumors via TfR1(highly expressed on malignant cells)-mediated endocytosis. Simultaneous inhibition of both GPX4 and FSP1 antioxidant pathways overwhelms cellular redox homeostasis, triggering catastrophic lipid peroxidation cascades that culminate in synergistic ferroptosis [186].
Traditional Chinese medicine (TCM) related therapy
Complementing conventional treatments, research on natural TCM ingredients targeting ferroptosis has expanded significantly, yielding notable therapeutic effects (Fig. 5 and Table 3). Artemisinin and its derivatives, known as first-line antimalarials, also effectively target ferroptosis-related diseases due to their unique chemical structure [187]. Herbal ingredients exhibit divergent roles in ferroptosis regulation: inhibitors include artemisinin [187], calycosin [188], umbelliferone [189], berberine [190], leonurine [191] etc. while inducers comprise curcumin [192], erianin [193], etc. We further observed that the same herbal ingredient can exert divergent effects on ferroptosis via distinct signaling pathways. Honokiol (HNK), for instance, induces ferroptosis by suppressing GPX4 [194] or upregulating HMOX1 [195], while inhibiting ferroptosis through AMPK/SIRT1/PGC-1α pathway activation [196]; Tanshinone IIA, induces ferroptosis via p53-mediated SLC7A11 down-regulation [197] but inhibits ferroptosis through Nrf2 signaling pathway activation [198]; Schisandrin A, attenuates ferroptosis by AdipoR1/AMPK-ROS/mitochondrial damage [199] while activates ferroptosis via AMPK/mTOR pathway [200]. We further observed that single herbs contain multiple bioactive compounds, for example: astragalus membranaceus, yields both calycosin and quercetin; glycyrrhiza produces compound glycyrrhizin and glabridin; schisandra chinensis contains schisandrin A and schisandrin B (Table 3), consequently, this compositional complexity necessitates cautious in selecting herbal treatments. In addition, multiple studies confirm that nuclear factor erythroid 2-related factor 2 (Nrf2) signaling which regulates core ferroptosis components, including GPX4, system Xc⁻, FSP1 [201], mediates herbal ingredient effects on ferroptosis (Table 3), a mechanism further validated in traditional Chinese herbal therapy.

This figure summarizes 19 traditional Chinese herbs and their associated bioactive compounds, with documented effects on ferroptosis regulation based on current research. Note: As research advances, additional bioactive compounds and regulatory functions in ferroptosis are anticipated to be identified. This diagram should be updated accordingly to reflect new evidence.
In short, ongoing phytochemical and mechanistic studies of TCM will progressively uncover: (1) undiscovered therapeutic compounds, (2) sophisticated action pathways, and (3) expanded clinical applications. Last, expanded ferroptosis research will uncover potent therapeutic strategies, particularly for cancer; however, their clinical application necessitates overcoming persistent translational barriers.
Conclusion
Collectively, all these above significantly advance our understanding of ferroptosis. The peroxidation of glycerophospholipids, generating reactive peroxyl radicals and cytotoxic protein adducts, plays a decisive role in ferroptosis. Accumulating evidence demonstrates that targeting ferroptosis holds significant therapeutic promise, particularly for treating cancers and other ferroptosis-related diseases.
Ferroptosis is a complex event involving multiple cellular processes, such as glycerophospholipids metabolism, redox homeostasis, autophagy, signal transduction, etc. while redox homeostasis regulation is the key to mastering ferroptosis. Therapeutic induction of ferroptosis via promotion of lipid peroxidation is primarily employed against various cancers. In contrast, inhibition of lipid peroxidation to suppress ferroptosis provides a therapeutic avenue for conditions including Parkinson’s disease, fibrosis, fatty liver, and ischemia-reperfusion injury. Key molecular targets for promoting lipid peroxidation include GPX4, FSP1, SLC7A11, Tf/TfR, and LPCAT1 etc.; conversely, targets for inhibiting lipid peroxidation encompass pro-ferroptotic enzymes ALOXs, ACSL4, or administering RTAs, and glutathione precursors (e.g., NAC/D-NAC) etc.
Notably, certain pharmacological agents exhibit context-dependent bidirectional effects on ferroptosis. For instance, TCM compounds including Honokiol, Tanshinone IIA, and Schisandrin A demonstrate both pro-ferroptotic and anti-ferroptotic activities through distinct molecular mechanisms. This functional duality underscores the inherent complexity of TCM-based ferroptosis modulation, necessitating integrated evaluation of disease pathology, drug mechanism of action, and clinical outcomes for therapeutic optimization. Furthermore, nanoparticle-based drug delivery systems for ferroptosis modulation are emerging as a rapidly advancing therapeutic strategy. Concurrently, novel ferroptosis-related molecular targets continue to be identified [202].
In conclusion, targeted modulation of ferroptosis represents a promising therapeutic paradigm for precision disease treatment.
References
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Li X, He J, Gao X, Zheng G, Chen C, Chen Y, et al. GPX4 restricts ferroptosis of NKp46+ILC3s to control intestinal inflammation. Cell Death Dis. 2024;15:687.
Chen Y, Fang Z-M, Yi X, Wei X, Jiang D-S. The interaction between ferroptosis and inflammatory signaling pathways. Cell Death Dis. 2023;14:205.
Yuan Y, Mei Z, Qu Z, Li G, Yu S, Liu Y, et al. Exosomes secreted from cardiomyocytes suppress the sensitivity of tumor ferroptosis in ischemic heart failure. Signal Transduct. Targeted Ther. 2023;8:121.
Fang X, Ardehali H, Min J, Wang F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Rev Cardiol. 2022;20:7–23.
Bayır H, Dixon SJ, Tyurina YY, Kellum JA, Kagan VE. Ferroptotic mechanisms and therapeutic targeting of iron metabolism and lipid peroxidation in the kidney. Nat Rev Nephrol. 2023;19:315–36.
Ryan SK, Zelic M, Han Y, Teeple E, Chen L, Sadeghi M, et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat Neurosci. 2022;26:12–26.
Luoqian J, Yang W, Ding X, Tuo Q-z, Xiang Z, Zheng Z, et al. Ferroptosis promotes T-cell activation-induced neurodegeneration in multiple sclerosis. Cell Mol Immunol. 2022;19:913–24.
Dang Q, Sun Z, Wang Y, Wang L, Liu Z, Han X. Ferroptosis: a double-edged sword mediating immune tolerance of cancer. Cell Death Dis. 2022;13:925.
Yang J, Yao S. JNK-Bcl-2/Bcl-xL-Bax/Bak pathway mediates the crosstalk between matrine-induced autophagy and apoptosis via interplay with Beclin 1. Int J Mol Sci. 2015;16:25744–58.
Davies KA, Czabotar PE, Murphy JM. Death at a funeral: activation of the dead enzyme, MLKL, to kill cells by necroptosis. Curr Opin Struct Biol. 2024;88:102891.
Lawlor KE, Murphy JM, Vince JE. Gasdermin and MLKL necrotic cell death effectors: signaling and diseases. Immunity. 2024;57:429–45.
Dias C, Hornung V, Nylandsted J. A novel NINJ1-mediated regulatory step is essential for active membrane rupture and common to different cell death pathways. Fac Rev. 2022;11:41.
Degen M, Santos JC, Pluhackova K, Cebrero G, Ramos S, Jankevicius G, et al. Structural basis of NINJ1-mediated plasma membrane rupture in cell death. Nature. 2023;618:1065–71.
David L, Borges JP, Hollingsworth LR, Volchuk A, Jansen I, Garlick E, et al. NINJ1 mediates plasma membrane rupture by cutting and releasing membrane disks. Cell. 2024;187:2224–2235.e2216.
Qiu B, Zandkarimi F, Bezjian CT, Reznik E, Soni RK, Gu W, et al. Phospholipids with two polyunsaturated fatty acyl tails promote ferroptosis. Cell. 2024;187:1177–1190.e1118.
Jie Z, Liu J, Shu M, Ying Y, Yang H. Detection strategies for superoxide anion: a review. Talanta 2022;236.
Begum R, Thota S, Abdulkadir A, Kaur G, Bagam P, Batra S. NADPH oxidase family proteins: signaling dynamics to disease management. Cell Mol Immunol. 2022;19:660–86.
Pratomo IP, Noor DR, Kusmardi K, Rukmana A, Paramita RI, Erlina L, et al. Xanthine oxidase-induced inflammatory responses in respiratory epithelial cells: a review in immunopathology of COVID-19. Int J Inflamm. 2021;2021:1–10.
Fujii J, Homma T, Osaki T. Superoxide radicals in the execution of cell death. Antioxidants. 2022;11:501.
Liu M, Sun X, Chen B, Dai R, Xi Z, Xu H. Insights into manganese superoxide dismutase and human diseases. Int J Mol Sci. 2022;23:15893.
Han X. Lipidomics for studying metabolism. Nat Rev Endocrinol. 2016;12:668–79.
Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. 2019;26:420–432.e429.
Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2016;13:81–90.
Sakuragi T, Nagata S. Regulation of phospholipid distribution in the lipid bilayer by flippases and scramblases. Nat Rev Mol Cell Biol. 2023;24:576–96.
Brown GC. Cell death by phagocytosis. Nat Rev Immunol. 2023;24:91–102.
Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013;341:403–6.
Amoscato AA, Anthonymuthu T, Kapralov O, Sparvero LJ, Shrivastava IH, Mikulska-Ruminska K, et al. Formation of protein adducts with Hydroperoxy-PE electrophilic cleavage products during ferroptosis. Redox Biol. 2023;63:102758.
Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. 2019;15:1137–47.
Bayır H, Anthonymuthu TS, Tyurina YY, Patel SJ, Amoscato AA, Lamade AM, et al. Achieving life through death: redox biology of lipid peroxidation in ferroptosis. Cell Chem Biol. 2020;27:387–408.
Funk CD, Chen X-S, Johnson EN, Zhao L. Lipoxygenase genes and their targeted disruption. Prostaglandins Other Lipid Mediators. 2002;68-69:303–12.
Wagner BA, Buettner GR, Burns CP. Free radical-mediated lipid peroxidation in cells: oxidizability is a function of cell lipid bis-allylic hydrogen content. Biochemistry. 2002;33:4449–53.
Kuhn H, Banthiya S, van Leyen K. Mammalian lipoxygenases and their biological relevance. Biochim et Biophys Acta (BBA) - Mol Cell Biol Lipids. 2015;1851:308–30.
Wiernicki B, Dubois H, Tyurina YY, Hassannia B, Bayir H, Kagan VE, et al. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. 2020;11:922.
Sun Q-Y, Zhou H-H, Mao X-Y. Emerging roles of 5-Lipoxygenase phosphorylation in inflammation and cell death. Oxid Med Cell Longev. 2019;2019:1–9.
Wang H, Xu L, Tang X, Jiang Z, Feng X. Lipid peroxidation-induced ferroptosis as a therapeutic target for mitigating neuronal injury and inflammation in sepsis-associated encephalopathy: insights into the hippocampal PEBP-1/15-LOX/GPX4 pathway. Lipids Health Dis. 2024;23:128.
Pope LE, Dixon SJ. Regulation of ferroptosis by lipid metabolism. Trends Cell Biol. 2023;33:1077–87.
Murakami M. The phospholipase A2 superfamily as a central hub of bioactive lipids and beyond. Pharmacol Therapeutics. 2023;244:108382.
Oh M, Jang SY, Lee J-Y, Kim JW, Jung Y, Kim J, et al. The lipoprotein-associated phospholipase A2 inhibitor Darapladib sensitises cancer cells to ferroptosis by remodelling lipid metabolism. Nat Commun. 2023;14:5728.
Sun W-Y, Tyurin VA, Mikulska-Ruminska K, Shrivastava IH, Anthonymuthu TS, Zhai Y-J, et al. Phospholipase iPLA2β averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol. 2021;17:465–76.
Mao C, Lei G, Zhuang L, Gan B. Phospholipase iPLA2β acts as a guardian against ferroptosis. Cancer Commun. 2021;41:1082–5.
Vermonden P, Martin M, Glowacka K, Neefs I, Ecker J, Höring M, et al. Phospholipase PLA2G7 is complementary to GPX4 in mitigating punicic-acid-induced ferroptosis in prostate cancer cells. iScience. 2024;27:109774.
Wang H, Zhou Y, Zhao M, Yu L, Lin Y, Kang D. Ferrostatin-1 attenuates brain injury in animal model of subarachnoid hemorrhage via phospholipase A2 activity of PRDX6. NeuroReport. 2023;34:606–16.
Tang Y, Liu C, Wei R, Li R, Li Z, Zhang K, et al. TRPV1/cPLA2/AA pathway contributes to ferroptosis-mediated acute liver injury in heatstroke. Int Immunopharmacol 2024;138:112539.
Kauther KM, Hoft C, Rissling I, Oertel WH, Moller JC. The PLA2G6 gene in early-onset Parkinson’s disease. Mov Disord. 2011;26:2415–7.
Wang J, Hu Y, Xu Y, Long Q, Gu C, Tang C, et al. Phospholipase D regulates ferroptosis signal transduction in mouse spleen hypoxia response. Brazilian J Med Biol Res 2024;57:e13218.
Nakamura MT, Nara TY. Structure, function, and dietary regulation of Δ6, Δ5, and Δ9 Desaturases. Annu Rev Nutr. 2004;24:345–76.
Steensels S, Ersoy BA. Fatty acid activation in thermogenic adipose tissue. Biochim et Biophys Acta (BBA) - Mol Cell Biol Lipids. 2019;1864:79–90.
Steinberg SJ, Morgenthaler J, Heinzer AK, Smith KD, Watkins PA. Very Long-chain Acyl-CoA Synthetases. J Biol Chem. 2000;275:35162–9.
Yan S. Long-chain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: an update. World J Gastroenterol. 2015;21:3492–8.
Ding K, Liu C, Li L, Yang M, Jiang N, Luo S, et al. Acyl-CoA synthase ACSL4: an essential target in ferroptosis and fatty acid metabolism. Chinese Med J. 2023;136:2521–37.
Hishikawa D, Shindou H, Kobayashi S, Nakanishi H, Taguchi R, Shimizu T. Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc Natl Acad Sci USA. 2008;105:2830–5.
Merkel M, Goebel B, Boll M, Adhikari A, Maurer V, Steinhilber D, et al. Mitochondrial reactive oxygen species formation determines ACSL4/LPCAT2-mediated ferroptosis. Antioxidants. 2023;12:1590.
Zhang Q, Yao D, Rao B, Jian L, Chen Y, Hu K, et al. The structural basis for the phospholipid remodeling by lysophosphatidylcholine acyltransferase 3. Nat Commun. 2021;12:6869.
Gan B. ACSL4, PUFA, and ferroptosis: new arsenal in anti-tumor immunity. Signal Transduct Target Ther. 2022;7:128.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–82.
Cheng J, Fan YQ, Liu BH, Zhou H, Wang JM, Chen QX. ACSL4 suppresses glioma cells proliferation via activating ferroptosis. Oncol Rep. 2019;43:147–58.
Dai Y, Chen Y, Mo D, Jin R, Huang Y, Zhang L, et al. Inhibition of ACSL4 ameliorates tubular ferroptotic cell death and protects against fibrotic kidney disease. Commun Biol. 2023;6:907.
Wang Y, Xia S. Relationship between ACSL4-Mediated ferroptosis and chronic obstructive pulmonary disease. Int J Chronic Obstr Pulm Dis. 2023;ume 18:99–111.
Huang Q, Ru Y, Luo Y, Luo X, Liu D, Ma Y, et al. Identification of a targeted ACSL4 inhibitor to treat ferroptosis-related diseases. Sci Adv. 2024;10:eadk1200.
Li C, Wu Y, Chen K, Chen R, Xu S, Yang B, et al. Gp78 deficiency in hepatocytes alleviates hepatic ischemia-reperfusion injury via suppressing ACSL4-mediated ferroptosis. Cell Death Dis. 2023;14:810.
Beatty A, Singh T, Tyurina YY, Tyurin VA, Samovich S, Nicolas E, et al. Ferroptotic cell death triggered by conjugated linolenic acids is mediated by ACSL1. Nat Commun. 2021;12:2244.
Klasson TD, LaGory EL, Zhao H, Huynh SK, Papandreou I, Moon EJ, et al. ACSL3 regulates lipid droplet biogenesis and ferroptosis sensitivity in clear cell renal cell carcinoma. Cancer Metab. 2022;10:14.
Valentine WJ, Yanagida K, Kawana H, Kono N, Noda NN, Aoki J, et al. Update and nomenclature proposal for mammalian lysophospholipid acyltransferases, which create membrane phospholipid diversity. J Biol Chem 2022;298:35557–63.
Shindou H, Harayama T, Hishikawa D. Lysophospholipid Acyltransferases. Bioactive Lipid Mediators, 2015:3–21.
Reed A, Ichu T-A, Milosevich N, Melillo B, Schafroth MA, Otsuka Y, et al. LPCAT3 inhibitors remodel the polyunsaturated phospholipid content of human cells and protect from ferroptosis. ACS Chem Biol. 2022;17:1607–18.
Hao J, Wang T, Cao C, Li X, Li H, Gao H, et al. LPCAT3 exacerbates early brain injury and ferroptosis after subarachnoid hemorrhage in rats. Brain Res. 2024;1832:148864.
Ke P, Bao X, Liu C, Zhou B, Huo M, Chen Y, et al. LPCAT3 is a potential prognostic biomarker and may be correlated with immune infiltration and ferroptosis in acute myeloid leukemia: a pan-cancer analysis. Transl Cancer Res. 2022;11:3491–505.
Lee H, Zhuang L, Gan B. Rewiring cancer cell death: LPCAT1 shapes lipid composition and ferroptosis resistance. Cell Death Differ. 2024;31:1101–3.
Li Z, Hu Y, Zheng H, Li M, Liu Y, Feng R, et al. LPCAT1-mediated membrane phospholipid remodelling promotes ferroptosis evasion and tumour growth. Nat Cell Biol. 2024;26:811–24.
Richard C, Verdier F. Transferrin receptors in erythropoiesis. Int J Mol Sci. 2020;21:9713.
Zhang DD. Ironing out the details of ferroptosis. Nat Cell Biol. 2024;26:1386–93.
Drakesmith H, Nemeth E, Ganz T. Ironing out Ferroportin. Cell Metab. 2015;22:777–87.
Ogun AS, Adeyinka A. Biochemistry, transferrin. StatPearls: Treasure Island (FL), (2024).
Latunde-Dada GO, Simpson RJ, McKie AT. Duodenal cytochrome B expression stimulates iron uptake by human intestinal epithelial cells. J Nutr. 2008;138:991–5.
Ganz T. Cellular iron: ferroportin is the only way out. Cell Metab. 2005;1:155–7.
Quintanar L, Stoj C, Taylor AB, Hart PJ, Kosman DJ, Solomon EI. Shall We Dance? How a multicopper oxidase chooses its electron transfer partner. Acc Chem Res. 2007;40:445–52.
Prohaska JR. Impact of copper limitation on expression and function of multicopper oxidases (Ferroxidases). Adv Nutr. 2011;2:89–95.
Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1:191–200.
van Renswoude J, Bridges KR, Harford JB, Klausner RD. Receptor-mediated endocytosis of transferrin and the uptake of fe in K562 cells: identification of a nonlysosomal acidic compartment. Proc Natl Acad Sci USA. 1982;79:6186–90.
Collawn JF, Stangel M, Kuhn LA, Esekogwu V, Jing S, Trowbridge IS, et al. Transferrin receptor internalization sequence YXRF implicates a tight turn as the structural recognition motif for endocytosis. Cell. 1990;63:1061–72.
Willingham MC, Hanover JA, Dickson RB, Pastan I. Morphologic characterization of the pathway of transferrin endocytosis and recycling in human KB cells. Proc Natl Acad Sci USA. 1984;81:175–9.
Pinilla-Tenas JJ, Sparkman BK, Shawki A, Illing AC, Mitchell CJ, Zhao N, et al. Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am J Physiol Cell Physiol. 2011;301:C862–C871.
Wang C-Y, Jenkitkasemwong S, Duarte S, Sparkman BK, Shawki A, Mackenzie B, et al. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J Biol Chem. 2012;287:34032–43.
Kawabata H. Transferrin and transferrin receptors update. Free Radic Biol Med. 2019;133:46–54.
Mastroberardino PG, Hoffman EK, Horowitz MP, Betarbet R, Taylor G, Cheng D, et al. A novel transferrin/TfR2-mediated mitochondrial iron transport system is disrupted in Parkinson’s disease. Neurobiol Dis. 2009;34:417–31.
Omura T. Mitochondria-targeting sequence, a multi-role sorting sequence recognized at all steps of protein import into mitochondria. J Biochem. 1998;123:1010–6.
Worwood M. Ferritin. Blood Rev. 1990;4:259–69.
Arosio P, Levi S. Ferritin, iron homeostasis, and oxidative damage1,2 1Guest Editor: Mario Comporti 2This article is part of a series of reviews on “Iron and Cellular Redox Status.” The full list of papers may be found on the homepage of the journal. Free Radic Biol Med. 2002;33:457–63.
Yanatori I, Richardson DR, Toyokuni S, Kishi F. The new role of poly (rC)-binding proteins as iron transport chaperones: Proteins that could couple with inter-organelle interactions to safely traffic iron. Biochim et Biophys Acta (BBA) - General Subjects 2020;1864:129685.
Leidgens S, Bullough KZ, Shi H, Li F, Shakoury-Elizeh M, Yabe T, et al. Each member of the Poly-r(C)-binding Protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J Biol Chem. 2013;288:17791–802.
Ohshima T, Yamamoto H, Sakamaki Y, Saito C, Mizushima N. NCOA4 drives ferritin phase separation to facilitate macroferritinophagy and microferritinophagy. J Cell Biol. 2022;221:e202203102.
Wally J, Buchanan SK. A structural comparison of human serum transferrin and human lactoferrin. Biometals. 2007;20:249–62.
Wally J, Halbrooks PJ, Vonrhein C, Rould MA, Everse SJ, Mason AB, et al. The crystal structure of iron-free human serum transferrin provides insight into inter-lobe communication and receptor binding. J Biol Chem. 2006;281:24934–44.
Steere AN, Byrne SL, Chasteen ND, Mason AB. Kinetics of iron release from transferrin bound to the transferrin receptor at endosomal pH. Biochim et Biophys Acta (BBA) - Gen Subj. 2012;1820:326–33.
Dautry-Varsat A, Ciechanover A, Lodish HF. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc Natl Acad Sci USA. 1983;80:2258–62.
Knutson MD. Non-transferrin-bound iron transporters. Free Radic Biol Med. 2019;133:101–11.
Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem. 1999;274:20826–32.
Deaglio S, Capobianco A, Cali A, Bellora F, Alberti F, Righi L, et al. Structural, functional, and tissue distribution analysis of human transferrin receptor-2 by murine monoclonal antibodies and a polyclonal antiserum. Blood. 2002;100:3782–9.
Krawiec P, Pac-Kożuchowska E. Soluble transferrin receptor and soluble transferrin receptor/log ferritin index in diagnosis of iron deficiency anemia in pediatric inflammatory bowel disease. Digestive Liver Dis. 2019;51:352–7.
Yasumura S, Naito Y, Okuno K, Sawada H, Asakura M, Masuyama T, et al. Effects of Heterozygous TfR1 (Transferrin Receptor 1) deletion in pathogenesis of renal fibrosis in mice. Hypertension. 2020;75:413–21.
Voss K, Sewell AE, Krystofiak ES, Gibson-Corley KN, Young AC, Basham JH, et al. Elevated transferrin receptor impairs T cell metabolism and function in systemic lupus erythematosus. Sci Immunol. 2023;8:eabq0178.
Parrow NL, Fleming RE. Transferrin receptor 1: keeper of HFE. Blood. 2023;141:332–3.
Celma Nos F Iron Regulatory Protein/Iron Responsive Element (IRP/IRE) system: associated diseases and new target mRNAs (PPP1R1B). 2022. https://www.tdx.cat/handle/10803/675437?show=full.
Connell GJ, Abasiri IM, Chaney EH. A temporal difference in the stabilization of two mRNAs with a 3′ iron-responsive element during iron deficiency. RNA. 2023;29:1117–25.
Yu X, Guo Q, Zhang H, Wang X, Han Y, Yang Z. Hypoxia-inducible factor-1α can reverse the Adriamycin resistance of breast cancer adjuvant chemotherapy by upregulating transferrin receptor and activating ferroptosis. FASEB J. 2024;38:e23876.
Petralla S, Saveleva L, Kanninen KM, Oster JS, Panayotova M, Fricker G, et al. Increased expression of transferrin receptor 1 in the brain cortex of 5xFAD mouse model of Alzheimer’s disease is associated with activation of HIF-1 signaling pathway. Mol Neurobiol. 2024;61:6383–94.
Hirota K. An intimate crosstalk between iron homeostasis and oxygen metabolism regulated by the hypoxia-inducible factors (HIFs). Free Radic Biol Med. 2019;133:118–29.
Babu KR, Muckenthaler MU. miR-148a regulates expression of the transferrin receptor 1 in hepatocellular carcinoma. Sci Rep. 2019;9:1518.
Fang X, Hu P, Gao Y, Chen C, Xu J. Transferrin receptor modulated by microRNA-497-5p suppresses cervical cancer cell malignant phenotypes. Adv Clin Exp Med. 2023;33:273–82.
Pietrangelo A. Ferroportin disease: pathogenesis, diagnosis and treatment. Haematologica. 2017;102:1972–84.
Zhang Z, Zhang F, Guo X, An P, Tao Y, Wang F. Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology. 2012;56:961–71.
Liu X-B, Yang F, Haile DJ. Functional consequences of ferroportin 1 mutations. Blood Cells, Molecules, Dis. 2005;35:33–46.
Bao W-D, Pang P, Zhou X-T, Hu F, Xiong W, Chen K, et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021;28:1548–62.
Cai C, Zeng D, Gao Q, Ma L, Zeng B, Zhou Y, et al. Decreased ferroportin in hepatocytes promotes macrophages polarize towards an M2-like phenotype and liver fibrosis. Sci Rep. 2021;11:13386.
Nemeth E, Ganz T. Hepcidin and iron in health and disease. Annu Rev Med. 2023;74:261–77.
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–3.
Pan Y, Ren Z, Gao S, Shen J, Wang L, Xu Z, et al. Structural basis of ion transport and inhibition in ferroportin. Nat Commun. 2020;11:5686.
Billesbølle CB, Azumaya CM, Kretsch RC, Powers AS, Gonen S, Schneider S, et al. Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature. 2020;586:807–11.
Qiao B, Sugianto P, Fung E, del-Castillo-Rueda A, Moran-Jimenez M-J, Ganz T, et al. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012;15:918–24.
Shen J, Wilbon AS, Zhou M, Pan Y. Mechanism of Ca2+ transport by ferroportin. eLife 2023;12:e82947.
Deshpande CN, Ruwe TA, Shawki A, Xin V, Vieth KR, Valore EV, et al. Calcium is an essential cofactor for metal efflux by the ferroportin transporter family. Nat Commun. 2018;9:3075.
Liziczai M, Lehmann EF, Drożdżyk K, Altermatt P, Langini C, Manolova V, et al. Structures of ferroportin in complex with its specific inhibitor vamifeport. eLife. 2023;12:e83053.
Namgaladze D, Fuhrmann DC, Brüne B. Interplay of Nrf2 and BACH1 in inducing ferroportin expression and enhancing resistance of human macrophages towards ferroptosis. Cell Death Discov. 2022;8:327.
Dai E, Chen X, Linkermann A, Jiang X, Kang R, Kagan VE, et al. A guideline on the molecular ecosystem regulating ferroptosis. Nat Cell Biol. 2024;26:1447–57.
Wang W, Knovich MA, Coffman LG, Torti FM, Torti SV. Serum ferritin: Past, present and future. Biochim et Biophys Acta (BBA) - Gen Subj. 2010;1800:760–9.
Worwood M, Brook JD, Cragg SJ, Hellkuhl B, Jones BM, Perera P, et al. Assignment of human ferritin genes to chromosomes 11 and 19q13.3→19qter. Hum Genet. 1985;69:371–4.
Cullis JO, Fitzsimons EJ, Griffiths WJH, Tsochatzis E, Thomas DW. Investigation and management of a raised serum ferritin. Br J Haematol. 2018;181:331–40.
Cavill I. Iron status as measured by serum ferritin: The marker and its limitations. Am J Kidney Dis. 1999;34:s12–s17.
Levi S, Ripamonti M, Dardi M, Cozzi A, Santambrogio P. Mitochondrial ferritin: its role in physiological and pathological conditions. Cells 2021;10:1969.
Levi S, Corsi B, Bosisio M, Invernizzi R, Volz A, Sanford D, et al. A human mitochondrial ferritin encoded by an intronless gene. J Biol Chem. 2001;276:24437–40.
Alkhateeb AA, Connor JR. Nuclear ferritin: a new role for ferritin in cell biology. Biochim et Biophys Acta (BBA) - Gen Subj. 2010;1800:793–7.
Thompson KJ, Fried MG, Ye Z, Boyer P, Connor JR. Regulation, mechanisms and proposed function of ferritin translocation to cell nuclei. J Cell Sci. 2002;115:2165–77.
Cohen LA, Gutierrez L, Weiss A, Leichtmann-Bardoogo Y, Zhang D-l, Crooks DR, et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood. 2010;116:1574–84.
Chen TT, Li L, Chung D-H, Allen CDC, Torti SV, Torti FM, et al. TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J Exp Med. 2005;202:955–65.
Li JY, Paragas N, Ned RM, Qiu A, Viltard M, Leete T, et al. Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev Cell. 2009;16:35–46.
Li L, Fang CJ, Ryan JC, Niemi EC, Lebrón JA, Björkman PJ, et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc Natl Acad Sci USA. 2010;107:3505–10.
Zhang W, Liu Y, Liao Y, Zhu C, Zou Z. GPX4, ferroptosis, and diseases. Biomed. Pharmacother. 2024;174;116512.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172:409–422.e421.
dos Santos AF, Fazeli G, Xavier da Silva TN, Friedmann Angeli JP. Ferroptosis: mechanisms and implications for cancer development and therapy response. Trends Cell Biol. 2023;33:1062–76.
Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–96.
Li W, Liang L, Liu S, Yi H, Zhou Y. FSP1: a key regulator of ferroptosis. Trends Mol Med. 2023;29:753–64.
Liu YE, Lu S, Wu L-L, Yang L, Yang L, Wang J. The diversified role of mitochondria in ferroptosis in cancer. Cell Death Dis. 2023;14:519.
Zhang R, Kroemer G, Tang D. Lipid-derived radical-trapping antioxidants suppress ferroptosis. Life Metab. 2024;3:loae008.
Miotto G, Rossetto M, Di Paolo ML, Orian L, Venerando R, Roveri A, et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020;28:101328.
Sheng X, Shan C, Liu J, Yang J, Sun B, Chen D. Theoretical insights into the mechanism of ferroptosis suppression via inactivation of a lipid peroxide radical by liproxstatin-1. Phys Chem Chem Phys. 2017;19:13153–9.
Freitas FP, Alborzinia H, dos Santos AF, Nepachalovich P, Pedrera L, Zilka O, et al. 7-Dehydrocholesterol is an endogenous suppressor of ferroptosis. Nature. 2024;626:401–10.
Li Y, Ran Q, Duan Q, Jin J, Wang Y, Yu L, et al. 7-Dehydrocholesterol dictates ferroptosis sensitivity. Nature. 2024;626:411–8.
Hu W-M, Liu S-Q, Zhu K-F, Li W, Yang Z-J, Yang Q, et al. The ALOX5 inhibitor Zileuton regulates tumor-associated macrophage M2 polarization by JAK/STAT and inhibits pancreatic cancer invasion and metastasis. Int Immunopharmacol 2023;121:110505.
Wang B, Yu S-c, Jiang J-y, Porter GW, Zhao L-t, Wang Z, et al. An Inhibitor of Arachidonate 5-Lipoxygenase, nordy, induces differentiation and inhibits self-renewal of glioma stem-like cells. Stem Cell Rev Rep. 2010;7:458–70.
Li K, Wang M, Huang Z-H, Wang M, Sun W-Y, Kurihara H, et al. ALOX5 inhibition protects against dopaminergic neurons undergoing ferroptosis. Pharmacol Res. 2023;193:106779.
Zhang X-J, Ji Y-X, Cheng X, Cheng Y, Yang H, Wang J, et al. A small molecule targeting ALOX12-ACC1 ameliorates nonalcoholic steatohepatitis in mice and macaques. Sci Transl Med. 2021;13:eabg8116.
Li C, Gao P, Zhuang F, Wang T, Wang Z, Wu G, et al. Inhibition of ALOX12–12-HETE alleviates lung ischemia–reperfusion injury by reducing endothelial ferroptosis-mediated neutrophil extracellular trap formation. Research 2024;7:0473.
Gao S, Hu J, Li Y. Targeting of the Alox12-12-HETE in blast crisis chronic myeloid leukemia inhibits leukemia stem/progenitor cell function. Cancer Manag Res. 2020;12:12509–17.
Guo S, Zhou L, Liu X, Gao L, Li Y, Wu Y. Baicalein alleviates cisplatin-induced acute kidney injury by inhibiting ALOX12-dependent ferroptosis. Phytomedicine. 2024;130:155757.
Wang Y, Yu R, Wu L, Yang G. Hydrogen sulfide guards myoblasts from ferroptosis by inhibiting ALOX12 acetylation. Cell Signall. 2021;78:109870.
Armstrong MM, Freedman CJ, Jung JE, Zheng Y, Kalyanaraman C, Jacobson MP, et al. A potent and selective inhibitor targeting human and murine 12/15-LOX. Bioorg Med Chem. 2016;24:1183–90.
Pei J, Zou Y, Zhou W, Wang Y. Baicalein, a component of banxia xiexin decoction, alleviates CPT-11-induced gastrointestinal dysfunction by inhibiting ALOX15-mediated ferroptosis. Chem Biol Drug Des. 2023;102:1568–77.
Luo L, Liu K, Deng L, Wang W, Lai T, Li X. Chicoric acid acts as an ALOX15 inhibitor to prevent ferroptosis in asthma. Int Immunopharmacol. 2024;142:113187.
Jiang Y-N, Guo Y-Z, Lu D-H, Pan M-H, Liu H-Z, Jiao G-L, et al. Tianma Gouteng granules decreases the susceptibility of Parkinson’s disease by inhibiting ALOX15-mediated lipid peroxidation. J Ethnopharmacol. 2020;256:112824.
Yan B, Wang Y, Li Y, Wang C, Zhang L. Inhibition of arachidonate 15-lipoxygenase reduces the epithelial–mesenchymal transition in eosinophilic chronic rhinosinusitis with nasal polyps. Int Forum Allergy Rhinol. 2018;9:270–80.
Bromfield EG, Mihalas BP, Dun MD, Aitken RJ, McLaughlin EA, Walters JLH, et al. Inhibition of arachidonate 15-lipoxygenase prevents 4-hydroxynonenal-induced protein damage in male germ cells†. Biol Reprod. 2017;96:598–609.
Luo X, Li D-D, Li Z-C, Li Z-X, Zou D-H, Huang F, et al. Mitigating phospholipid peroxidation of macrophages in stress-induced tumor microenvironment by natural ALOX15/PEBP1 complex inhibitors. Phytomedicine. 2024;128:155475.
Dar HH, Mikulska-Ruminska K, Tyurina YY, Luci DK, Yasgar A, Samovich SN, et al. Discovering selective antiferroptotic inhibitors of the 15LOX/PEBP1 complex noninterfering with biosynthesis of lipid mediators. Proc Natl Acad Sci USA. 2023;120:e2218896120.
Liu L, Zhang Y, Wang L, Liu Y, Chen H, Hu Q, et al. Scutellarein alleviates chronic obstructive pulmonary disease through inhibition of ferroptosis by chelating iron and interacting with arachidonate 15-lipoxygenase. Phytother Res. 2023;37:4587–606.
Zhu R, Kang Y, Li Q, Peng K, Shi X, Yin Z, et al. Alpha-tocopherol inhibits ferroptosis and promotes neural function recovery in rats with spinal cord injury via downregulating Alox15. Biomed Pharmacother. 2024;175:116734.
Li M, Meng Z, Yu S, Li J, Wang Y, Yang W, et al. Baicalein ameliorates cerebral ischemia-reperfusion injury by inhibiting ferroptosis via regulating GPX4/ACSL4/ACSL3 axis. Chemico-Biol Interact. 2022;366:110137.
Shen J, Qian M, Wu M, Tang J, Gong Y, Li J, et al. Rosiglitazone inhibits acyl-CoA synthetase long-chain family number 4 and improves secondary brain injury in a rat model of surgical brain injury. Clin Exp Pharmacol Physiol. 2023;50:927–35.
Wang Y, Zhang M, Bi R, Su Y, Quan F, Lin Y, et al. ACSL4 deficiency confers protection against ferroptosis-mediated acute kidney injury. Redox Biol. 2022;51:102262.
Mauvecín JG, Sanz AB, Ortiz A, Sánchez DM, Gómez NV, Fresnedo O. Ferroptotic proteins ACSL4 and ALOX15 as therapeutic targets in AKI. Nephrol Dialysis Transpl. 2024;39:gfae069–1089.
Marteau R, Ravez S, Mazhari Dorooee D, Bouchaoui H, Porte K, Devedjian J-C, et al. Repositioning of FDA-Approved antifungal agents to interrogate Acyl-CoA synthetase long chain family member 4 (ACSL4) in ferroptosis. Biochem Pharmacol. 2022;204:115239.
Castillo AF, Orlando UD, Maloberti PM, Prada JG, Dattilo MA, Solano AR, et al. New inhibitor targeting Acyl-CoA synthetase 4 reduces breast and prostate tumor growth, therapeutic resistance and steroidogenesis. Cell Mol Life Sci. 2020;78:2893–910.
Hong Y, Feng J, Dou Z, Sun X, Hu Y, Chen Z, et al. Berberine as a novel ACSL4 inhibitor to suppress endothelial ferroptosis and atherosclerosis. Biomedicine & Pharmacotherapy 2024;177:117081.
Zhang H-L, Hu B-X, Li Z-L, Du T, Shan J-L, Ye Z-P, et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol. 2022;24:88–98.
Wang B, Yang L-n, Yang L-t, Liang Y, Guo F, Fu P, et al. Fisetin ameliorates fibrotic kidney disease in mice via inhibiting ACSL4-mediated tubular ferroptosis. Acta Pharmacol Sin. 2023;45:150–65.
Xiao Y, Xu Z, Cheng Y, Huang R, Xie Y, Tsai H-i, et al. Fe3+-binding transferrin nanovesicles encapsulating sorafenib induce ferroptosis in hepatocellular carcinoma. Biomater Res. 2023;27:63.
Zhang S, Wu X, Liao X, Zhang S. Nanodrug hijacking blood transferrin for ferroptosis-mediated cancer treatment. J Am Chem Soc. 2024;146:8567–75.
Bernkop-Schnürch AD, Hermann M, Leitner D, Talasz H, Descher HA, Hohloch S. et al. Transferrin receptor-mediated cellularuptake of Fluorinated Chlorido[N,N′-bis(salicylidene)-1,2-phenylenediamine]iron(III) Complexes. ACS Omega. 2024;9:35394–407.
Xue C, Zhang H, Wang X, Du H, Lu L, Fei Y, et al. Bio-inspired engineered ferritin-albumin nanocomplexes for targeted ferroptosis therapy. J Controlled Release. 2022;351:581–96.
Li Y, Wang X, Yan J, Liu Y, Yang R, Pan D, et al. Nanoparticle ferritin-bound erastin and rapamycin: a nanodrug combining autophagy and ferroptosis for anticancer therapy. Biomater Sci. 2019;7:3779–87.
Chen Y, Li X, Luo K, Wang T, Liu T, Lu E, et al. Hyperthermia/glutathione-triggered ferritin nanoparticles amplify the ferroptosis for synergistic tumor therapy. Mater Today Bio. 2024;26:101085.
Zhu L, You Y, Zhu M, Song Y, Zhang J, Hu J, et al. Ferritin-Hijacking nanoparticles spatiotemporally directing endogenous ferroptosis for synergistic anticancer therapy. Adv Mater. 2022;34:2207174.
Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. 2019;131:356–69.
Zheng Y, Zheng J, Du M, Yang Y, Li X, Chen H, et al. An iron-containing ferritin-based nanosensitizer for synergistic ferroptosis/sono-photodynamic cancer therapy. J Mater Chem B. 2023;11:4958–71.
Cheng J, Yu Q, Li J, Xu Z, Li J, Guan L, et al. Intrinsic tumor-targeted murine ferritin nanocage co-delivers GPX4 and FSP1 inhibitors for synergistic ferroptosis-immunotherapy. Nano Today 2024;58:102411.
Wang Y, Yuan X, Ren M, Wang Z. Ferroptosis: a new research direction of artemisinin and its derivatives in anti-cancer treatment. Am J Chin Med. 2024;52:161–81.
Liu H, Zhao Z, Yan M, Zhang Q, Jiang T, Xue J. Calycosin decreases cerebral ischemia/reperfusion injury by suppressing ACSL4-dependent ferroptosis. Arch Biochem Biophys. 2023;734:109488.
Jin T, Chen C. Umbelliferone delays the progression of diabetic nephropathy by inhibiting ferroptosis through activation of the Nrf-2/HO-1 pathway. Food Chem Toxicol. 2022;163:112892.
Jin Y, Bao L, Han J, Wang W, Qian L, Wu W. Berberine regulates GPX4 to inhibit ferroptosis of islet β cells. Planta Med. 2022;89:254–61.
Hu J, Gu W, Ma N, Fan X, Ci X. Leonurine alleviates ferroptosis in cisplatin-induced acute kidney injury by activating the Nrf2 signalling pathway. Br J Pharmacol. 2022;179:3991–4009.
Tang X, Ding H, Liang M, Chen X, Yan Y, Wan N, et al. Curcumin induces ferroptosis in non-small-cell lung cancer via activating autophagy. Thorac Cancer. 2021;12:1219–30.
Chen P, Wu Q, Feng J, Yan L, Sun Y, Liu S, et al. Erianin, a novel dibenzyl compound in Dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal Transduct Targeted Therapy 2020;5:51.
Guo C, Liu P, Deng G, Han Y, Chen Y, Cai C, et al. Honokiol induces ferroptosis in colon cancer cells by regulating GPX4 activity. Am J Cancer Res. 2021;11:3039–54.
Lai X, Sun Y, Zhang X, Wang D, Wang J, Wang H, et al. Honokiol induces ferroptosis by upregulating HMOX1 in acute myeloid leukemia cells. Front Pharmacol. 2022;13:897791.
Hu M, Jiang W, Ye C, Hu T, Yu Q, Meng M, et al. Honokiol attenuates high glucose-induced peripheral neuropathy via inhibiting ferroptosis and activating AMPK/SIRT1/PGC-1α pathway in Schwann cells. Phytother Res. 2023;37:5787–802.
Guan Z, Chen J, Li X, Dong N. Tanshinone IIA induces ferroptosis in gastric cancer cells through p53-mediated SLC7A11 down-regulation. Biosci Rep. 2020;40:BSR20201807.
Li X, Tang S, Wang H, Li X. Tanshinone IIA inhibits hydrogen peroxide-induced ferroptosis in melanocytes through activating Nrf2 signaling pathway. Pharmacology. 2025;110:141–50.
Wang X, Li Q, Sui B, Xu M, Pu Z, Qiu T, et al. Schisandrin A from Schisandra chinensis Attenuates Ferroptosis and NLRP3 Inflammasome-mediated pyroptosis in diabetic nephropathy through mitochondrial damage by AdipoR1 Ubiquitination. Oxid Med Cell Longev. 2022;2022:1–23.
He LW, Lin CJ, Zhuang LJ, Sun YH, Li YC, Ye ZY. Targeting hepatocellular carcinoma: Schisandrin A triggers mitochondrial disruption and ferroptosis. Chem Biol Drug Design. 2024;104:e70010.
Anandhan A, Dodson M, Schmidlin CJ, Liu P, Zhang DD. Breakdown of an ironclad defense system: the critical role of NRF2 in mediating ferroptosis. Cell Chem Biol. 2020;27:436–47.
Sun S, Shen J, Jiang J, Wang F, Min J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct Target Ther. 2023;8:372.
Nakano H, Minnella A, McCusker KP, Amagata A, Trias B, Weetall M, et al. Targeting ferroptosis with the lipoxygenase inhibitor PTC-041 as a therapeutic strategy for the treatment of Parkinson’s disease. Plos One. 2024;19:e0309893.
Zhao D, Yang K, Guo H, Zeng J, Wang S, Xu H, et al. Mechanisms of ferroptosis in Alzheimer’s disease and therapeutic effects of natural plant products: a review. Biomed Pharmacother. 2023;164:114312.
Li X, Wu L, Sun L, Liu H, Qiao X, Mi N, et al. Ferroptosis-related gene signatures in epilepsy: diagnostic and immune insights. Mol Neurobiol. 2025;62:1998–2011.
Xu Y, Jia B, Li J, Li Q, Luo C. The interplay between ferroptosis and neuroinflammation in central neurological disorders. Antioxidants. 2024;13:395.
Bouchaoui H, Mahoney-Sanchez L, Garçon G, Berdeaux O, Alleman LY, Devos D, et al. ACSL4 and the lipoxygenases 15/15B are pivotal for ferroptosis induced by iron and PUFA dyshomeostasis in dopaminergic neurons. Free Radic Biol Med. 2023;195:145–57.
Li W, Zhao X, Zhang R, Liu X, Qi Z, Zhang Y, et al. Ferroptosis inhibition protects vascular endothelial cells and maintains integrity of the blood-spinal cord barrier after spinal cord injury. Neural Regener Res. 2023;18:2474–81.
Wan K, Jia M, Zhang H, Lan Y, Wang S, Zhang K, et al. Electroacupuncture alleviates neuropathic pain by suppressing ferroptosis in dorsal root ganglion via SAT1/ALOX15 signaling. Mol Neurobiol. 2023;60:6121–32.
Gao S, Zhou L, Lu J, Fang Y, Wu H, Xu W, et al. Cepharanthine attenuates early brain injury after subarachnoid hemorrhage in mice via inhibiting 15-Lipoxygenase-1-Mediated microglia and endothelial cell ferroptosis. Oxid Med Cell Longev. 2022;2022:1–16.
Karuppagounder SS, Alin L, Chen Y, Brand D, Bourassa MW, Dietrich K, et al. N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann Neurol. 2018;84:854–72.
Chen J, Shi Z, Zhang C, Xiong K, Zhao W, Wang Y. Oroxin A alleviates early brain injury after subarachnoid hemorrhage by regulating ferroptosis and neuroinflammation. J Neuroinflamm. 2024;21:116.
Wang J, Cao M, Han L, Shangguan P, Liu Y, Zhong Y, et al. Blood–brain barrier-penetrative fluorescent anticancer agents triggering paraptosis and ferroptosis for glioblastoma therapy. J Am Chem Soc. 2024;146:28783–94.
Wang Y, Wang W, Zhang Y, Fleishman JS, Wang H. Targeting ferroptosis offers therapy choice in sepsis-associated acute lung injury. Eur J Med Chem. 2025;283:117152.
Zhang Z, He Y, Liu H, Liu Y, Wu T, Li R, et al. NLRP3 regulates ferroptosis via the JAK2/STAT3 pathway in asthma inflammation: insights from in vivo and in vitro studies. Int Immunopharmacol. 2024;143:113416.
Yang B, Zhai F, Li Z, Wang X, Deng X, Cao Z, et al. Identification of ferroptosis-related gene signature for tuberculosis diagnosis and therapy efficacy. iScience. 2024;27:110182.
Xu L, Zhang L, Xiang Y, Zhang X. Knockdown of lncRNA NEAT1 suppresses streptococcus pneumoniae-induced ferroptosis in alveolar epithelial cells by regulating the Nrf2-GPX4 pathway. Toxicon. 2024;243:107705.
Yan M, Xu S, Wang H, Dong S, Mo C. Ferroptosis in chronic obstructive pulmonary disease: from cellular mechanisms to therapeutic applications. Chin Med J. 2024;137:1237–9.
Kazmirczak F, Vogel NT, Prisco SZ, Patterson MT, Annis J, Moon RT, et al. Ferroptosis-mediated inflammation promotes pulmonary hypertension. Circ Res. 2024;135:1067–83.
Zheng H, Chen H, Cai Y, Shen M, Li X, Han Y, et al. Hydrogen sulfide-mediated persulfidation regulates homocysteine metabolism and enhances ferroptosis in non-small cell lung cancer. Mol Cell. 2024;84:4016–4030.e4016.
Zhen S, Jia Y, Zhao Y, Wang J, Zheng B, Liu T, et al. NEAT1_1 confers gefitinib resistance in lung adenocarcinoma through promoting AKR1C1-mediated ferroptosis defence. Cell Death Discov. 2024;10:131.
Guo D, Feng Y, Liu P, Yang S, Zhao W, Li H. Identification and prognostic analysis of ferroptosis‑related gene HSPA5 to predict the progression of lung squamous cell carcinoma. Oncol Lett. 2024;27:186.
Yang X, Kawasaki NK, Min J, Matsui T, Wang F. Ferroptosis in heart failure. J Mol Cell Cardiol. 2022;173:141–53.
Conrad M, Proneth B. Broken hearts: iron overload, ferroptosis and cardiomyopathy. Cell Res. 2019;29:263–4.
Lillo-Moya J, Rojas-Solé C, Muñoz-Salamanca D, Panieri E, Saso L, Rodrigo R. Targeting ferroptosis against ischemia/reperfusion cardiac injury. Antioxidants 2021;10:667.
Fan X, Li A, Yan Z, Geng X, Lian L, Lv H, et al. From iron metabolism to ferroptosis: pathologic changes in coronary heart disease. Oxid Med Cell Longev. 2022;2022:1–14.
Wang C, Yuan W, Hu A, Lin J, Xia Z, Yang C, et al. Dexmedetomidine alleviated sepsis‑induced myocardial ferroptosis and septic heart injury. Mol Med Rep. 2020;22:175–84.
Zhang X, Zheng C, Gao Z, Chen H, Li K, Wang L, et al. SLC7A11/xCT prevents cardiac hypertrophy by inhibiting ferroptosis. Cardiovasc Drugs Ther. 2021;36:437–47.
Hu Z, Zhang H, Yang S-k, Wu X, He D, Cao K, et al. Emerging role of ferroptosis in acute kidney injury. Oxid Med Cell Longev. 2019;2019:1–8.
Zhuo W-Q, Wen Y, Luo H-J, Luo Z-L, Wang L. Mechanisms of ferroptosis in chronic kidney disease. Front Mol Biosci 2022;9:975582.
Wu Y, Chen Y. Research progress on ferroptosis in diabetic kidney disease. Front Endocrinol. 2022;13:945976.
Zhou L, Xue X, Hou Q, Dai C. Targeting ferroptosis attenuates interstitial inflammation and kidney fibrosis. Kidney Dis. 2022;8:57–71.
Alli AA, Desai D, Elshika A, Conrad M, Proneth B, Clapp W, et al. Kidney tubular epithelial cell ferroptosis links glomerular injury to tubulointerstitial pathology in lupus nephritis. Clin Immunol 2023;248:109213.
Chen Y, Wang K, Yang J, Zhang A, Dong X, Zhou Z, et al. Mechanism of ferroptosis in hypertensive nephropathy. Transl Androl Urol. 2022;11:617–26.
Feng B, Su W, Guo X, Ding T, Duan Y, Hu L, et al. MDH2 regulates the sensitivity of clear cell renal cell carcinoma to ferroptosis through its interaction with FSP1. Cell Death Discov. 2024;10:363.
Gong Y, Zhang C, Li H, Yu X, Li Y, Liu Z, et al. Ferroptosis-related lncRNA to predict the clinical outcomes and molecular characteristics of kidney renal papillary cell carcinoma. Curr Issues Mol Biol. 2024;46:1886–903.
Zhang T, Wang M-Y, Wang G-D, Lv Q-Y, Huang Y-Q, Zhang P, et al. Metformin improves nonalcoholic fatty liver disease in db/db mice by inhibiting ferroptosis. Eur J Pharmacol. 2024;966:176341.
Li X, Li Y, Zhang W, Jiang F, Lin L, Wang Y, et al. The IGF2BP3/Notch/Jag1 pathway: a key regulator of hepatic stellate cell ferroptosis in liver fibrosis. Clin Transl Med. 2024;14:e1793.
Liang Y, Qiu S, Zou Y, Luo L. Targeting ferroptosis with natural products in liver injury: new insights from molecular mechanisms to targeted therapies. Phytomedicine. 2024;122:155134.
Shao T, Chung RT. Ironing out MAFLD: therapeutic targeting of liver ferroptosis. Cell Metab. 2024;36:2167–9.
Sun Y-W, Zhao B-W, Li H-F, Zhang G-X. Overview of ferroptosis and pyroptosis in acute liver failure. World J Gastroenterol. 2024;30:3856–61.
Du K, Wang L, Jun JH, Dutta RK, Maeso-Díaz R, Oh SH, et al. Aging promotes metabolic dysfunction-associated steatotic liver disease by inducing ferroptotic stress. Nat Aging. 2024;4:949–68.
Hou C-y, Suo Y-h, Lv P, Yuan H-f, Zhao L-n, Wang Y-f, et al. Aristolochic acids-hijacked p53 promotes liver cancer cell growth by inhibiting ferroptosis. Acta Pharmacol Sin. 2025;46:208–21.
Zhang N, Chen P, Liang X, Sun J, Liu Q, Guan S, et al. Luteolin targets the AGE-RAGE signaling to mitigate inflammation and ferroptosis in chronic atrophic gastritis. Aging. 2024;16:10918–30.
Meng X, Liu J, Kang J, Wang M, Guan Z, Tian D, et al. Lamivudine protects mice from gastric ulcer by activating PGK1 to suppress ferroptosis. Biochem Pharmacol. 2024;227:116440.
Yan S, Bao S, Chen T, Chen J, Zhang J, Hu X, et al. Cinnamaldehyde alleviates aspirin-induced gastric mucosal injury by regulating pi3k/akt pathway-mediated apoptosis, autophagy and ferroptosis. Phytomedicine. 2024;132:155791.
Wang L-M, Zhang W-W, Qiu Y-Y, Wang F. Ferroptosis regulating lipid peroxidation metabolism in the occurrence and development of gastric cancer. World J Gastrointest Oncol. 2024;16:2781–92.
Chen H, Qian Y, Jiang C, Tang L, Yu J, Zhang L, et al. Butyrate ameliorated ferroptosis in ulcerative colitis through modulating Nrf2/GPX4 signal pathway and improving intestinal barrier. Biochim et Biophys Acta (BBA) - Mol Basis Dis. 2024;1870:166984.
Lin X, Li Y, Qi B, Zhang S, Li X. Casein-phosphatidylcholine emulsifier remodels LPS-induced intestinal barrier disfunction via regulating ferroptosis and lipid metabolism. Int J Biol Macromol. 2024;254:127595.
Zhang L-l, Ding K, Liao S-s, Zhang Y-g, Liao H-y, Chen R, et al. Sestrin2 reduces ferroptosis via the Keap1/Nrf2 signaling pathway after intestinal ischemia-reperfusion. Free Radic Biol Med. 2024;214:115–28.
Wang C, Chu Q, Dong W, Wang X, Zhao W, Dai X, et al. Microbial metabolite deoxycholic acid-mediated ferroptosis exacerbates high-fat diet-induced colonic inflammation. Mol Metab. 2024;84:101944.
Bi H, Guo S, Wang Y, Liu Z, Wu G, Huo X, et al. Pinobanksin ameliorated DSS-induced acute colitis mainly through modulation of SLC7A11/glutathione-mediated intestinal epithelial ferroptosis. Food Funct. 2024;15:4970–82.
Cui W, Guo M, Liu D, Xiao P, Yang C, Huang H, et al. Gut microbial metabolite facilitates colorectal cancer development via ferroptosis inhibition. Nat Cell Biol. 2024;26:124–37.
Li H, Wu D, Zhang H, Liu S, Zhen J, Yan Y, et al. Autophagy-mediated ferroptosis is involved in development of severe acute pancreatitis. BMC Gastroenterol. 2024;24:245.
Xiang X, Xu M, Liu L, Meng N, Lei Y, Feng Y, et al. Liproxstatin-1 attenuates acute hypertriglyceridemic pancreatitis through inhibiting ferroptosis in rats. Sci Rep. 2024;14:9548.
Suda A, Umaru BA, Yamamoto Y, Shima H, Saiki Y, Pan Y, et al. Polyunsaturated fatty acids-induced ferroptosis suppresses pancreatic cancer growth. Sci Rep. 2024;14:4409.
Ruze R, Chen Y, Song J, Xu R, Yin X, Xu Q, et al. Enhanced cytokine signaling and ferroptosis defense interplay initiates obesity-associated pancreatic ductal adenocarcinoma. Cancer Lett. 2024;601:217162.
Zhang Y, Huang R, Liu X, Cai M, Su M, Cheng Y, et al. Taohong siwu decoction ameliorates abnormal uterine bleeding via inhibiting ACSL4-mediated ferroptosis. J Ethnopharmacol. 2025;339:119130.
Yuan C, Liu L, Zhao Y, Wang K. Puerarin inhibits Staphylococcus aureus-induced endometritis through attenuating inflammation and ferroptosis via regulating the P2X7/NLRP3 signalling pathway. J Cell Mol Med. 2024;28:e18550.
Liu Y, Zhou Y, Hao B, Wu Z, Gao M, Liu L, et al. Inhibition of ferroptosis attenuate lipopolysaccharide-induced early pregnancy loss by protecting against decidual damage of stromal cells. Biochem Biophys Res Commun. 2024;736:150904.
Xing X, Zhang G, Yi F, Xu X. Overexpression of USP22 ameliorates LPS-induced endometrial stromal cells inflammation and modulates cells decidualization by inhibiting ferroptosis. Reprod Biol. 2024;24:100913.
Kobayashi H, Imanaka S, Yoshimoto C, Matsubara S, Shigetomi H. Role of autophagy and ferroptosis in the development of endometriotic cysts (Review). Int J Mol Med. 2024;54:78.
Dai F, Zhang Y, Deng Z, Zhang J, Wang R, Chen J, et al. IGF2BP3 participates in the pathogenesis of recurrent spontaneous abortion by regulating ferroptosis. J Reprod Immunol. 2024;165:104271.
Liu J, Zhu W, Xia L, Zhu Q, Mao Y, Shen Y, et al. Identification of CAPG as a potential prognostic biomarker associated with immune cell infiltration and ferroptosis in uterine corpus endometrial carcinoma. Front Endocrinol. 2024;15:1452219.
Han Q, Shi J, Yu Y, Yuan H, Guo Y, Liu X, et al. Calycosin alleviates ferroptosis and attenuates doxorubicin-induced myocardial injury via the Nrf2/SLC7A11/GPX4 signaling pathway. Front Pharmacol. 2024;15:1497733.
Wang T-T, Yu L-L, Zheng J-M, Han X-Y, Jin B-Y, Hua C-J, et al. Berberine inhibits ferroptosis and stabilizes atherosclerotic Plaque through NRF2/SLC7A11/GPX4 Pathway. Chin J Integr Med. 2024;30:906–16.
Xiang Y, Chen X, Wang W, Zhai L, Sun X, Feng J, et al. Natural product erianin inhibits bladder cancer cell growth by inducing ferroptosis via NRF2 inactivation. Front Pharmacol. 2021;12:775506.
Chen L, Sun R, Fang K. Erianin inhibits tumor growth by promoting ferroptosis and inhibiting invasion in hepatocellular carcinoma through the JAK2/STAT3/SLC7A11 pathway. Pathol Int. 2024;74:119–28.
Yang C, Wu A, Tan L, Tang D, Chen W, Lai X, et al. Epigallocatechin-3-Gallate alleviates liver oxidative damage caused by iron overload in mice through inhibiting ferroptosis. Nutrients 2023;15:1993.
Zhu K, Zhu X, Liu S, Yu J, Wu S, Hei M, et al. Glycyrrhizin attenuates hypoxic-ischemic brain damage by inhibiting ferroptosis and neuroinflammation in neonatal rats via the HMGB1/GPX4 pathway. Oxid Med Cell Longev. 2022;2022:1–18.
Zhu K, Fan R, Cao Y, Yang W, Zhang Z, Zhou Q, et al. Glycyrrhizin attenuates myocardial ischemia reperfusion injury by suppressing Inflammation, oxidative stress, and ferroptosis via the HMGB1-TLR4-GPX4 pathway. Exp Cell Res. 2024;435:113912.
Cui Q, Wang W, Shi J, Lai F, Luo S, Du Y, et al. Glycyrrhizin ameliorates cardiac injury in rats with severe acute pancreatitis by inhibiting ferroptosis via the Keap1/Nrf2/HO-1 Pathway. Digestive Dis Sci. 2024;69:2477–87.
Tan H, Chen J, Li Y, Li Y, Zhong Y, Li G, et al. Glabridin, a bioactive component of licorice, ameliorates diabetic nephropathy by regulating ferroptosis and the VEGF/Akt/ERK pathways. Mol Med. 2022;28:58.
Chen J, Ou Z, Gao T, Yang Y, Shu A, Xu H, et al. Ginkgolide B alleviates oxidative stress and ferroptosis by inhibiting GPX4 ubiquitination to improve diabetic nephropathy. Biomed Pharmacother. 2022;156:113953.
Yang Y, Wu Q, Shan X, Zhou H, Wang J, Hu Y, et al. Ginkgolide B attenuates cerebral ischemia-reperfusion injury via inhibition of ferroptosis through disrupting NCOA4-FTH1 interaction. J Ethnopharmacol. 2024;318:116982.
Zhu X, Chen X, Yin K, Zhang Y, Li Y, Yu X. Leonurine ameliorates diabetic nephropathy through GPX4-mediated ferroptosis of endothelial cells. Front Biosci -Landmark. 2024;29:270.
Wang Y, Quan F, Cao Q, Lin Y, Yue C, Bi R, et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J Adv Res. 2021;28:231–43.
Xie R, Zhao W, Lowe S, Bentley R, Hu G, Mei H, et al. Quercetin alleviates kainic acid-induced seizure by inhibiting the Nrf2-mediated ferroptosis pathway. Free Radic Biol Med. 2022;191:212–26.
Lan D, Qi S, Yao C, Li X, Liu H, Wang D, et al. Quercetin protects rat BMSCs from oxidative stress via ferroptosis. J Mol Endocrinol. 2022;69:401–13.
Tang J-J, Huang L-F, Deng J-L, Wang Y-M, Guo C, Peng X-N, et al. Cognitive enhancement and neuroprotective effects of OABL, a sesquiterpene lactone in 5xFAD Alzheimer’s disease mice model. Redox Biol. 2022;50:102229.
Yu J, Li X, Zhou M, Lu M, Ruan Z, Zou W, et al. Schisandrin B inhibits LPS-induced endometritis through attenuating ferroptosis via AMPK/PGC1α/Nrf2 signalling pathway. J Cell Mol Med. 2024;28:e70281.
Zhou D, Sun L, Li J, Yang Y. Schisandrin B inhibits inflammation and ferroptosis in S.aureus-induced mastitis through regulating SIRT1/p53/SLC7A11 signaling pathway. Int Immunopharmacol. 2024;137:112430.
Shi H, Yan Y, Yang H, Pu P, Tang H, Stanek A. Schisandrin B diet inhibits oxidative stress to reduce ferroptosis and lipid peroxidation to prevent pirarubicin-induced hepatotoxicity. BioMed Res Int. 2022;2022:5623555.
Li X, Wang X, Huang B, Huang R. Sennoside A restrains TRAF6 level to modulate ferroptosis, inflammation and cognitive impairment in aging mice with Alzheimer’s Disease. Int Immunopharmacol. 2023;120:110290.
Wang F, Li W-L, Shen L-J, Jiang T-T, Xia J-J, You D-L, et al. Crocin alleviates intracerebral hemorrhage–induced neuronal ferroptosis by facilitating Nrf2 nuclear translocation. Neurotox Res. 2022;40:596–604.
Hong R, Chen B, Wu H, Ding J. Crocin facilitates osteogenesis and angiogenesis by moderating oxidative stress and ferroptosis via Nrf2/GPX4 pathway. Tissue Cell. 2025;93:102675.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (82122072, 82474033), the Natural Science Foundation of Shanghai (24ZR1493300), the Traditional Chinese Medicine Research Project of Shanghai (2024PT010), and Special Scientific Research Project of Shanghai Fourth People’s Hospital (sykyqd07101).
Author information
Authors and Affiliations
Contributions
S.C. contributed to drafting and editing the manuscript. M.Z. and M.L. helped edit the pictures. S.C., C.L., H.T., and Y.L. participated in the revision of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, S., Zhang, M., Liu, C. et al. Redox mechanism of glycerophospholipids and relevant targeted therapy in ferroptosis. Cell Death Discov. 11, 358 (2025). https://doi.org/10.1038/s41420-025-02654-y
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41420-025-02654-y
