
Abstract
The aetiology of childhood motor speech disorders of dysarthria and apraxia has been poorly understood. Recent evidence suggests a moderate genetic contribution for these rare and severe speech disorders. To date, however, no studies have examined genetic diagnostic yield for childhood apraxia of speech (CAS) and dysarthria in a clinical setting. Here, we used a clinically accredited genomics pipeline to investigate genetic diagnostic yield and variables predictive of a genetic diagnosis in a tertiary hospital speech clinic. A cohort of 153 children (range 2;7-16;5 years, 42 female) ascertained for motor speech disorder were assessed by a clinical geneticist and speech pathologist and underwent chromosomal microarray, Fragile X and exome sequencing. Odds ratios identified predictors of genetic diagnosis. 44/153 (29%, 15 female) had pathogenic variants (30 de novo), encompassing monogenic conditions (n = 35) and copy number variants (n = 9) across 38 distinct disorders. Delayed walking, fine and gross motor disorder, receptive language impairment and/or cognitive impairment, and dysmorphism were associated with a genetic diagnosis. The presence of CAS and dysarthria was more commonly associated with a genetic diagnosis than CAS alone. Autism spectrum disorder was less commonly associated with a genetic diagnosis. No child had a Fragile X diagnosis. The clinical genetic diagnostic yield for motor speech disorders is comparable to epilepsy and cerebral palsy, conditions where genetic testing is routine in most centres, unlike for motor speech disorders. Children with motor speech disorder with co-occurring motor, language and/or learning deficits, should be prioritised for genomic testing.
Similar content being viewed by others


Introduction
Speech disorders are a common presenting concern for children referred to paediatricians and primary care physicians [1]. Associated poor language, literacy, educational, employment and social outcomes mean speech disorders come with significant individual, societal and health system burden [2,3,4].
Up to 5% of children develop common speech disorders, including articulation and phonological impairments [1]. These conditions are highly tractable and typically resolve by 7 years of age, with or without intervention [4, 5]. By contrast, 1 in 1000 children follow a severely disrupted developmental path to intractable motor speech disorders of childhood apraxia of speech (CAS) and/or dysarthria [6]. CAS affects the ability to plan and sequence speech movements, decreasing the precision, consistency and intelligibility of speech [7]. Children with CAS need explicit teaching and practice of new sounds and words, often requiring speech therapy into adolescence or adulthood [8]. A diagnosis of CAS implicates disruption of inferior frontal lobe and basal ganglia pathways, whereas dysarthria is a disorder of the control and execution of neuromuscular movements for speech implicating the ‘final common’ motor pathway or corticobulbar tract [9,10,11].
A major challenge of managing motor speech disorders has been a lack of aetiological knowledge. Families search for explanatory causes in a long diagnostic journey [12], attending multiple primary care physician, paediatric, neurology, and other specialists appointments, involving serial investigations (e.g. MRI, metabolic testing) with significant costs and psychological stress and without explanatory results [13]. In the past five years, genomic sequencing, performed in a research setting, has shown a substantial genetic contribution to these conditions [14,15,16,17], even in the absence of intellectual disability [15, 16], with a diagnostic yield of 30% [3]. This research yield is comparable to that seen in cerebral palsy, epilepsy and intellectual disability, conditions for which genomic clinical testing is routine in many centres [3]. However, clinical genomic testing is not yet the standard of care for motor speech disorders, despite such testing being highly valued by families with lived experience [13].
In this study, we offered clinical genomic testing, consisting of chromosomal microarray, fragile X PCR and exome sequencing, to a clinically ascertained population of children referred to a speech genomics clinic at a paediatric tertiary hospital. We examined diagnostic yield and clinical variables predictive of a genetic diagnosis.
Materials and methods
Ethical consent
The study was approved by the Human Research Ethics Committee of The Royal Children’s Hospital, Melbourne, Australia (#37353). Written informed consent was obtained from parents or legal guardians.
Study design
This prospective single-centre cohort study recruited probands referred to the Genetics of Speech Disorders clinic at the Royal Children’s Hospital in Melbourne, Australia, between August 2020 and December 2023.
Recruitment
Children were referred for motor speech testing by their treating primary care physician, paediatrician, paediatric sub-specialist (metabolic, genetics, neurology) or speech pathologist. Probands were triaged for eligibility by the clinical nurse co-ordinator with input from the clinical team where indicated (e.g., psychologist to interpret cognitive assessments; clinical geneticist to interpret past genetic reports). Inclusion criteria were: (1) probands aged 2;7 to 17 years with CAS or dysarthria in the absence of known or previously diagnosed intellectual disability, autism spectrum disorder (ASD) level III, or acquired brain injury; and (2) parent, speech pathologist and treating physician reporting the primary presenting feature and clinical concern as motor speech disorder [15, 16].
Phenotyping
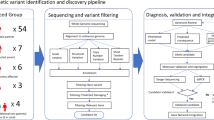
Probands were prospectively assessed by a speech pathologist (AM, ML, CB); paediatrician and clinical geneticist (DA, dual-qualified); and psychologist (EB, CB) using a protocol outlined below; see clinical pipeline Fig. 1a.

a Clinical workflow. PCR Polymerase chain reaction. Referral received by clinical co-ordinator (nurse, LO) at Motor Speech Disorders Clinic. Triage by nurse (LO) and speech pathologist (AM). Proband seen by speech pathologist (AM, ML, CB, AG) to confirm speech motor disorder. Medical work-up by paediatrician and clinical geneticist (DA, dual-qualified). Psychologists (EB, CB) determined IQ status at the further appointment. A genetic counsellor (ML) or a clinical geneticist (DA) performed pre-test genetic counselling and genetic consent. Saliva samples for chromosomal microarray and exome sequencing were collected using paediatric. Oracollect kits from DNAGenotek. Clinically accredited genomic sequencing was performed at the Victorian Clinical Genetics Service, including: initial computational bioinformatics analysis; meeting to prioritise variants for assessment attended by senior medical genomic scientists, clinical geneticists, and genetic counsellors; variant curation as per American College of Medical Genetics and Genomics guidelines. Next, a review of variant and phenotypic data was undertaken by a multidisciplinary speech clinic team, with laboratory scientists, clinical geneticists and genetic counsellors independent to the speech clinic, to reach consensus and ensure genotype concordant with phenotype prior to reporting. Proband attended further clinic appointment (in-person or telehealth) for the return of results with post-test genetic counselling and segregation encouraged where appropriate. b Flowchart of recruitment and genetic results. CNV Copy number variant; *Monogenic findings include single nucleotide variants (SNVs) and intragenic deletions.
A detailed family history and pedigree were recorded and prospective phenotyping documented medical and developmental history, including the presence of dysmorphism [15, 16] (Table 1, Supplemental Tables 1, 2a, b). All secondary neurodevelopmental outcomes were defined using standardised testing and a professional report for validation, e.g., multi-disciplinary assessment standardised testing for a formal diagnosis of ASD; a physiotherapist confirmed fine or gross motor disorder. Where children had not yet received a formal ASD or attention deficit hyperactivity disorder (ADHD) diagnosis, the paediatrician (DA) recorded the presence of ASD/ADHD features (Table 1; Supplemental Tables 1, 2a, b).
For speech, CAS was confirmed if the American Speech-Language-Hearing Association (ASHA) criteria for CAS were met: (1) inconsistent errors on consonants and vowels in repeated productions of syllables or words, (2) lengthened and disrupted coarticulatory transitions between sounds and syllables, and (3) inappropriate prosody; all relative to age and stage of development [7]. ASHA criteria were operationally defined and rated from: phonetic transcriptions of 55 single-word speech tests designed to elicit all sounds of English, a test of speech consistency (children repeat 15/55 stimuli words twice) and a 5 min conversational speech sample as described previously [15, 16, 18]. Dysarthria was diagnosed as an oral tone or coordination disturbance, examined by a paediatrician and speech pathologist and speech dysarthric features were identified during the conversation sample, using the Mayo Clinic Dysarthria rating scale [15, 16, 18, 19]. Receptive and expressive language was assessed where time and resources were available [20,21,22,23] (Table 1, Supplemental Tables 1, 2a, b). Cognition was assessed by a psychologist using standardised testing [24,25,26].
Genetic and genomic analysis
A genetic counsellor (ML) and/or clinical geneticist (DA) provided pre and post-genetic counselling. Chromosomal microarray analysis (CMA, Illumina GSA, San Diego, USA) and Fragile X PCR sizing were performed on saliva or blood DNA for all probands prior to exome sequencing (ES). Saliva samples were collected using paediatric Oracollect kits from DNAGenotek. For ES, genomic DNA was isolated from saliva samples with trio testing (probands together with parents) performed where possible. ES was performed by Victorian Clinical Genetics Services (VCGS), Melbourne, see Supplemental Fig. 1 for an overview of the analysis pipeline. Coding regions were enriched using TWIST exome capture (TWIST Bioscience). Sequencing was performed on Illumina sequencers to an average target depth of 80×. Data were analysed using an in-house validated version of Cpipe [27], followed by variant filtering and analysis using Alissa Interpret analysis software (Agilent). Data were analysed for variants linked to disorders relevant to the proband phenotype following a tiered approach. Initially, variants in genes associated with intellectual disability were evaluated (https://panelapp-aus.org/panels/250/). Analysis was then expanded to a virtual panel of ~6000 genes known to cause Mendelian disorders (https://panelapp-aus.org/panels/137/). Variants were classified according to the current American College of Medical Genetics and Genomics (ACMG) guidelines for variant interpretation [28].
Statistical analysis
We report the proportion and type of pathogenic variants causative for motor speech disorder from CMA, Fragile X and ES testing. For variables predictive of a genetic diagnosis, baseline clinical variables are recorded as frequencies (n, %) and medians (range), as dictated by data type. Odds ratios have been used to identify variables associated with a genetic diagnostic result. The variables of ASD, ADHD, dysmorphism, intelligence quotient (IQ) and receptive and expressive language were all dichotomised for the odds ratio analysis.
Results
Genetic diagnostic yield and genomic findings
172 probands met the inclusion criteria, of whom 153 (88.9%; 111 males, 42 females) completed genetic testing (153 CMA, 143 ES), at an average age of 5 years 8 months (range 2 years 7 months to 16 years 5 months). Two probands withdrew from the study and 4 declined genetic testing. Despite three reminders to do so, the remaining 13 either did not respond to the genetic testing appointment invitation, did not send back their consent form, or did not return their saliva sample (Fig. 1b).
Overall, 44/153 (29%) probands received a genetic diagnosis (15 females; median 5 years 1 month; Tables 1, 2). 30 findings were confirmed de novo and 9 inherited (5 maternal; 4 paternal). Inheritance status could not be confirmed for 5 pathogenic variants due to missing parental DNA (Table 2, Supplemental Fig. 1a). 9 were aneuploidies or copy number variants (CNVs) (all >50 kb) detected on CMA: 15q13.2q13.3 del, 22q11.21 del, 22q11.21q11.22 dup, 1q21.1q21.2 dup, 16p12.2 del, 17q12, 47 dup, 47,XXX, 48,XXYY, 48,XYYY. There were 35 monogenic diagnoses, including single-nucleotide variants (SNVs) and intragenic deletions (< 50 kb). 34/35 were identified from ES: ADGRL1, ANK2, BPTF, CACNA1A (n = 3), CAMK2A, CAMTA1, EBF3, EHMT1 (n = 2), FBXW7, FOXP1, GNAI1, KCND3, KDM5C, NSD1, PPP2R5D, RAF1, SCN8A, SET, SETBP1 (n = 2), SETD1A, SETD2, SETD5, SLC6A1, SLC6A8, SMARCA2 (n = 2), SPTBN1, SRRM2, TAB2, TRIM8. 1/35 was identified from CMA:
CUX1 (Tables 1, 2). Variant types for SNVs (n = 32) included missense (n = 13), frameshift (n = 9), nonsense (n = 7), in-frame deletions (n = 2), and splicing (n = 1) (Table 2). All variants were deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/; accession numbers SCV002557839.2- SCV005399598.1).
13 candidate variants of uncertain significance (VUS) were identified in probands without a diagnosis; comprising SNVs in the following genes: AP1G, BCORL1, BRPF1, CRY, DPF2, FGF13, IQSEC2, USP9X (n = 2); and the following CNVs: 14q12 del (de novo), 7p22.3 del (maternal), 3p14.2 del (de novo), and 2q37.2q37.3 del (maternal) (Supplemental Table 3). Two additional VUSs were identified in solved probands ID8 FOXP2, and ID5 TRRAP [28].
All (likely) pathogenic variants identified were in genes previously associated with syndromic or non-syndromic intellectual disability, epilepsy, ASD and cerebral palsy, Supplemental Table 4. No child received a diagnosis of Fragile X syndrome or Fragile X-associated disorder/pre-mutation carrier status.
Phenotyping: variables predictive of a genetic diagnosis
Phenotypes of 44 probands with a genetic diagnosis, and 109 without a genetic diagnosis, are presented in Fig. 2 and Supplemental Tables 1, 2, 5. Compared to those without a genetic diagnosis, a higher proportion of children with genetic diagnoses had dysarthria (with and without CAS; 18.2% vs 8.3%), gross motor delays (68.2% vs. 37.6%), fine motor delays (65.9% vs. 40.4%), or a delayed age of walking (52.3% vs. 6.4%), dysmorphism (45.5% vs. 5.5%) and/or borderline or mild intellectual disability (55.9% vs. 15.2%); and a lower proportion had confirmed ASD (9.1% vs 18.3%). No participant with a genetic diagnosis had seizures or a diagnosis of epilepsy, although participant 6 had staring spells with a normal EEG. Of those without genetic diagnoses, only two had seizures; participant 147 had absence seizures; participant 71 had a history of febrile seizures.

ADHD ADHD diagnosis or ADHD features, ASD ASD diagnosis or ASD features, Dysmorphism Dysmorphic or dysmorphic features, Intellectual impairment: borderline ID or mild ID; Expressive language impairment: mildly impaired, moderately impaired, severely impaired, minimally verbal or could not be assessed due to severity; Receptive language impairment: mildly impaired, moderately impaired or severely impaired.
No potential environmental factors contributing to CAS were identified. No probands had a history of congenital infection, teratogen exposure, birth injury, severe prematurity or acquired brain injury. Six probands had a history of moderate to late prematurity (32–26 weeks) and none of these had a genetic diagnosis.
Consistent with these descriptive findings, the odds of having a genetic diagnosis were highest for individuals with delayed age of walking (OR:15.96, 95% CI: 6.34, 44.86), receptive language impairment (OR: 7.14, 95% CI: 2.96, 18.62), borderline or mildly impaired intellect (OR: 7.07, 95% CI: 2.89, 18.18), dysmorphism (present/features) (OR: 5.59, 95% CI: 2.66, 12.19), expressive language impairment (OR: 3.62, 95% CI: 1.28, 13.04), gross motor delay/disorder (OR: 3.55, 95% CI: 1.72, 7.65); and fine motor delay/disorder (OR: 2.86, 95% CI: 1.39, 6.06), respectively (Supplemental Table 5). The odds of having a genetic diagnosis were lowest for children with confirmed diagnoses of ADHD or ASD. Wide confidence intervals for the OR reflect the relatively small sample size for children with a genetic diagnosis.
Discussion
We identified a specific genetic diagnosis in 29% of children referred to a motor speech disorders clinic at a paediatric tertiary hospital. This yield replicates research findings in a clinical setting for the first time. Previous prospective research-based cohorts ascertained for a primary motor speech diagnosis have identified genetic diagnoses in 42%, 33% and 26% respectively [14,15,16], but may have been subject to ascertainment bias. Importantly, this diagnostic yield is comparable to that of genomic testing in other neurodevelopmental conditions, such as epilepsy, cerebral palsy and intellectual disability, for which genomic testing is increasingly routine [29,30,31,32].
Mendelian diagnoses were present in 23% (35/153) of the cohort. We implicate 15 Mendelian disorder genes as novel explanations for CAS and dysarthria: ADGRL1, ANK2, BPTF, CAMK2A, CUX1, FBXW7, KCND3, NSD1, RAF1, SETD2, SLC6A8, SPTBN1, SRRM2, TAB2, TRIM8). We also confirm involvement of CACNA1A, CAMTA1, EBF3 EHMT1, GNAI1, KDM5C, PPP2R5D, SCN8A, SET, SETBP1, SETD1A, SETD5, SLC6A1, SMARCA2 and FOXP1, now recurrent in unrelated cases across cohorts ascertained for CAS and dysarthria [14,15,16,17, 33]. Past work led to the development of the first publicly available clinical speech panel in July 2024, which we have now expanded to include newly implicated genes with sufficient evidence for inclusion (https://panelapp-aus.org/panels/4290/). We highlight that two of our implicated genes are associated with multiple speech disorder phenotypes: SPTBN1 in CAS, stuttering [34] and speech delay [35]; and SETD1A in CAS and speech delay [35].
Although we excluded children with a pre-existing diagnosis of intellectual disability, our subsequent assessment indicated that 9.7% had mild and 17.7% had borderline intellectual disability. Probands with a genetic diagnosis were more likely to have cognitive impairment compared to those without a genetic diagnosis. This result was not unexpected, given that all genes associated with CAS to date are also associated with intellectual disability [3]. Of those children with a genetic diagnosis and normal IQ, most had an IQ at the lower end of the normal range; although, there were some exceptions: an IQ above 100 was recorded in female participant 47 with 48,XXX (Trisomy X), male participant 10 with an EBF3 variant, and male participant 38 with 16p12.2 deletion. Children with a genetic diagnosis were also more likely to have impairments in motor domains, with delays in walking and fine and gross motor function commonly reported (52.3%, 65.9%, 68.2%, respectively). Overall, these data support the hypothesis that in monogenic forms of CAS, the speech phenotype is part of a broader neurodevelopmental impairment. We also noted that children with CAS and co-occurring dysarthria were more likely to have a genetic diagnosis than children with CAS alone. This is the first evidence of this observation to our knowledge, however requires independent replication.
It is notable that children with a genetic diagnosis ascertained via CAS and/or dysarthria in our cohort were typically towards the milder end of the reported severity spectrum for their disorder. This is likely explained by ascertainment biases favouring (1) severe cases in the published literature, and (2) milder cases in our CAS cohort, also seen in our past studies [15, 16]. For example, we identified CACNA1A variants in three unrelated probands in the absence of epilepsy or intellectual disability [36]. By extension from these data, milder forms of monogenic neurodevelopmental disorders are likely to be more common than currently recognised, and a case can be made for expanding the application of genomic sequencing to children (and adults) with mild and borderline ID.
In the 71% of our cohort without a specific genetic diagnosis, a different phenotypic profile emerged. A significant majority of this group had average FSIQ and expected age of walking, and nearly half (47.7%) had either features of ASD or a formal ASD diagnosis. The genetic basis of these undiagnosed cases remains unclear, but the clinical overlap with ASD implicates polygenic inheritance [37, 38]. Based on these observations, we hypothesise that the genetic architecture of CAS comprises at least two distinct causal pathways: (1) monogenic CAS, characterised by variable cognitive impairment, motor impairment and syndromic features, and (2) polygenic CAS, in which cognition and motor skills are relatively spared, but ASD features are more common.
The association between CAS abnormalities on CMA has yet to be fully characterised. Only 4% of our cohort had a copy number variant. All six CNVs in our study are recognized recurrent CNVs with incomplete penetrance, and each was detected in only one patient. An additional 2% of patients had sex chromosome aneuploidies, including two patients with four sex chromosomes (XXYY and XYYY). The association between sex chromosome trisomies and quadrisomies has been previously recognized [39]. An overall yield of 6% for CMA in CAS is lower than reported previously in an Italian CAS cohort (13%) [17] and for intellectual disability, but comparable to the yield in some other neurodevelopmental phenotypes, such as autism and epilepsy, and microarray remains an important front-line test for motor speech disorders. By contrast, we found no cases of fragile X (neither premutation nor full mutation), in line with past prospective studies in the field, suggesting that this test may be less useful in children with CAS [14,15,16,17]. Past retrospective chart review studies of large cohorts reporting on genotype-CAS phenotype associations have also failed to identify any association with fragile X [33, 40].
We found no significant evidence for environmental factors contributing to CAS, including pregnancy-related and neonatal complications. Moderate to late prematurity (32-36 weeks) was reported in 5.5% of patients without a genetic diagnosis, comparable to the general population. Prematurity was not seen in any participants with a genetic diagnosis. Our data are in line with past studies showing no evidence of CAS or dysarthria diagnoses in children born pre-term [41].
The range of genetic diagnoses in our cohort underscores the role of chromatin modifiers and transcriptional regulators (BPTF, CAMTA1, CUX1, EBF3, EHMT1, FOXP1, KDM5C, NSD1, SET, SETD1A, SETD2, SETD5, SMARCA2, SETBP1, SRRM2) in speech disorder pathogenicity, consistent with our prior reports [3]. We highlight the commonality of genes encoding SET domain proteins (SETD1A, SETD2, SETD5, NSD1, EHMT1), as these are part of a methyltransferase superfamily integral in post-translational protein regulation with a well-established role in neurodevelopmental disorders [15, 16]. Other protein classes enriched for pathogenic variants were ion channels and solute carriers (ANK2, SCN8A, KCND3, CACNA1A, SLC6A1, SLC6A8) and cell signalling pathway members (FBXW7, PPP2R5D, TAB2, RAF1, SET).
Whilst further replication and verification of results is warranted, given the modest size of our cohort, motor speech disorders are extremely rare (1 in 1000) when compared to other neurodevelopmental conditions (1 in 100 for intellectual disability, autism; 4-10 per 1000 for epilepsy) and greater collaboration will be necessary to build sizeable cohorts for independent replication.
While this research focused on a clinical diagnostic setting, future gene discovery research outside of a clinical pipeline remains important to explore novel variants associated with CAS, with functional characterisation.
Conclusion
Our findings confirm a diagnostic yield for motor speech disorder that is comparable to other neurodevelopmental conditions. We have identified predictors of genetic diagnosis that can be used to guide advances in clinical practice and health services for motor speech disorder. We suggest prioritisation of clinical genomic testing for children with a primary motor speech disorder on a background of delayed walking, persistent fine and gross motor delays/disorder, receptive language impairment, dysmorphism, and/or borderline IQ. The presence of ASD appears to be less commonly associated with a genetic diagnosis. Motor speech conditions are distinctive, socially debilitating disorders, and clinical genomic testing should be warranted to end the diagnostic odyssey and position them for precision therapies.
Data availability
The datasets are included in this study and in supplementary information files or are available from the corresponding author on reasonable. Variant data is available in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
References
Morgan A, Ttofari Eecen K, Pezic A, Brommeyer K, Mei C, Eadie P, et al. Who to refer for speech therapy at 4 years of age versus who to “Watch and Wait”? J Pediatr. 2017;185:200–4.e1. https://doi.org/10.1016/j.jpeds.2017.02.059.
US Preventative Service Task Force, Barry MJ, Nicholson WK, Silverstein M, Chelmow D, Coker TR, et al. Screening for speech and language delay and disorders in children: US Preventive Services Task Force Recommendation Statement. JAMA. 2024;331:329–34. https://doi.org/10.1001/jama.2023.26952.
Morgan AT, Amor DJ, St John MD, Scheffer IE, Hildebrand MS. Genetic architecture of childhood speech disorder: a review. Mol Psychiatry. 2024;29:1281–92. https://doi.org/10.1038/s41380-024-02409-8.
Eadie P, Morgan A, Ukoumunne OC, Ttofari Eecen K, Wake M, Reilly S. Speech sound disorder at 4 years: prevalence, comorbidities, and predictors in a community cohort of children. Dev Med Child Neurol. 2015;57:578–84. https://doi.org/10.1111/dmcn.12635.
Reilly S, McKean C, Morgan A, Wake M. Identifying and managing common childhood language and speech impairments. BMJ. 2015;350:h2318. https://doi.org/10.1136/bmj.h2318.
Shriberg LD, Aram DM, Kwiatkowski J. Developmental apraxia of speech: I. Descriptive and theoretical perspectives. J Speech Lang Hear Res. 1997;40:273–85. https://doi.org/10.1044/jslhr.4002.273.
American Speech-Language-Hearing Association. Childhood Apraxia of Speech. 2007. http://www.asha.org/policy/TR2007-00278/.
Lewis B, Ekelman B. Literacy Problems Associated With Childhood Apraxia of Speech. Perspectives Language Learning and Education. 2007;14:10–17. https://doi.org/10.1044/lle14.3.10.
Liegeois FJ, Turner SJ, Mayes A, Bonthrone AF, Boys A, Smith L, et al. Dorsal language stream anomalies in an inherited speech disorder. Brain. 2019;142:966–77. https://doi.org/10.1093/brain/awz018.
Liegeois FJ, Hildebrand MS, Bonthrone A, Turner SJ, Scheffer IE, Bahlo M, et al. Early neuroimaging markers of FOXP2 intragenic deletion. Sci Rep. 2016;6:35192. https://doi.org/10.1038/srep35192.
Liegeois F, Tournier JD, Pigdon L, Connelly A, Morgan AT. Corticobulbar tract changes as predictors of dysarthria in childhood brain injury. Neurology. 2013;80:926–32. https://doi.org/10.1212/WNL.0b013e3182840c6d.
Atkinson C, Lee YQ, Lauretta ML, Jarmolowicz A, Amor DJ, Morgan AT Parental attitudes and experiences in pursuing genetic testing for their child’s motor speech disorder. Eur J Hum Genet. 2024; https://doi.org/10.1038/s41431-024-01755-z.
Meng Y, Best S, Amor DJ, Braden R, Morgan AT, Goranitis I. The value of genomic testing in severe childhood speech disorders. Eur J Hum Genet. 2024;32:440–7. https://doi.org/10.1038/s41431-024-01534-w.
Eising E, Carrion-Castillo A, Vino A, Strand EA, Jakielski KJ, Scerri TS, et al. A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol Psychiatry. 2019;24:1065–78. https://doi.org/10.1038/s41380-018-0020-x.
Hildebrand MS, Jackson VE, Scerri TS, Van Reyk O, Coleman M, Braden RO, et al. Severe childhood speech disorder: Gene discovery highlights transcriptional dysregulation. Neurology. 2020;94:e2148–e2167. https://doi.org/10.1212/WNL.0000000000009441.
Kaspi A, Hildebrand MS, Jackson VE, Braden R, van Reyk O, Howell T, et al. Genetic aetiologies for childhood speech disorder: novel pathways co-expressed during brain development. Mol Psychiatry. 2023;28:1647–63. https://doi.org/10.1038/s41380-022-01764-8.
Formicola D, Podda I, Dirupo E, Andreucci E, Giglio S, Cipriani P, et al. Expanding the molecular landscape of childhood apraxia of speech: evidence from a single-center experience. Front Neurosci. 2024;18:1396240. https://doi.org/10.3389/fnins.2024.1396240.
Mei C, Fedorenko E, Amor DJ, Boys A, Hoeflin C, Carew P, et al. Deep phenotyping of speech and language skills in individuals with 16p11.2 deletion. Eur J Hum Genet. 2018;26:676–86. https://doi.org/10.1038/s41431-018-0102-x.
Morison LD, Kennis MGP, Rots D, Bouman A, Kummeling J, Palmer E, et al. Expanding the phenotype of Kleefstra syndrome: speech, language and cognition in 103 individuals. J Med Genet. 2024;61:578–85. https://doi.org/10.1136/jmg-2023-109702.
Wiig EH, Secord WA, Semel E Clinical Evaluation of Language Fundamentals Preschool Third Edition (CELF P-3). Bloomington, MN: Pearson, 2020.
Wiig EH, Semel E, Secord WA Clinical Evaluation of Language Fundamentals Fifth Edition (CELF-5). Bloomington, MN: Pearso 2013.
Semel E, Wiig EH, Secord WA Clinical Evaluation of Language Fundamentals Fourth Edition (CELF-4). San Antonio, TX: Pearson 2003.
Zimmerman IL, Steiner VG, Pond RE Preschool Language Scale Fifth Edition (PLS-5). San Antonio, TX: Pearson; 2011.
Kaufman AS, Kaufman ML. Kaufman Assessment Battery for Children Second Edition (KABC-II). Circle Pines, MN: American Guidance Service 2004.
Wechsler D Wechsler Intelligence Scale for Children, Fifth Edition (WISC-V). Bloomington, MN: Pearson 2014.
Wechsler D Wechsler Preschool & Primary Scale of Intelligence Fourth Edition (WPPSI-IV). San Antonio, TX: Pearson; 2012.
Sadedin SP, Dashnow H, James PA, Bahlo M, Bauer DC, Lonie A, et al. Cpipe: a shared variant detection pipeline designed for diagnostic settings. Genome Med. 2015;7:68. https://doi.org/10.1186/s13073-015-0191-x.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. https://doi.org/10.1038/gim.2015.30.
Sheidley BR, Malinowski J, Bergner AL, Bier L, Gloss DS, Mu W, et al. Genetic testing for the epilepsies: A systematic review. Epilepsia. 2022;63:375–87. https://doi.org/10.1111/epi.17141.
Stefanski A, Calle-Lopez Y, Leu C, Perez-Palma E, Pestana-Knight E, Lal D. Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: A systematic review and meta-analysis. Epilepsia. 2021;62:143–51. https://doi.org/10.1111/epi.16755.
Lewis SA, Chopra M, Cohen JS, Bain JM, Aravamuthan B, Carmel JB, et al. Clinical actionability of genetic findings in cerebral palsy. medRxiv. Sep 11 2023; https://doi.org/10.1101/2023.09.08.23295195.
Manickam K, McClain MR, Demmer LA, Biswas S, Kearney HM, Malinowski J, et al. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23:2029–37. https://doi.org/10.1038/s41436-021-01242-6.
Mitchel MW, Oetjens M, Berry ASF, Johns A, Moreno-De-Luca A, Torene RI, et al. Monogenic disorders associated with motor speech phenotypes in children and adolescents undergoing clinical exome sequencing. Genet Med. 2025:101374. https://doi.org/10.1016/j.gim.2025.101374.
Eising E, Vino A, Mabie HL, Campbell TF, Shriberg LD, Fisher SE. Genome Sequencing of Idiopathic Speech Delay. Human Mutation. 2024;2024:1–14.
Eising E, Dzinovic I, Vino A, Stipdonk L, Pavlov M, Winkelmann J, et al. De novo protein-coding gene variants in developmental stuttering. 2024.
Lipman AR, Fan X, Shen Y, Chung WK. Clinical and genetic characterization of CACNA1A-related disease. Clin Genet. 2022;102:288–95. https://doi.org/10.1111/cge.14180.
Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 2019;51:431–44. https://doi.org/10.1038/s41588-019-0344-8.
Trost B, Thiruvahindrapuram B, Chan AJS, Engchuan W, Higginbotham EJ, Howe JL, et al. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell. 2022;185:4409–27.e18. https://doi.org/10.1016/j.cell.2022.10.009.
Samango-Sprouse C, Keen C, Mitchell F, Sadeghin T, Gropman A. Neurodevelopmental variability in three young girls with a rare chromosomal disorder, 48, XXXX. Am J Med Genet A. 2015;167A:2251–9. https://doi.org/10.1002/ajmg.a.37198.
Magielski JH, Ruggiero SM, Xian J, Parthasarathy S, Galer PD, Ganesan S, et al. The clinical and genetic spectrum of paediatric speech and language disorders. Brain. 2025;148:663–74. https://doi.org/10.1093/brain/awae264.
Northam GB, Liegeois F, Chong WK, Baker K, Tournier JD, Wyatt JS, et al. Speech and oromotor outcome in adolescents born preterm: relationship to motor tract integrity. J Pediatr. 2012;160:402–8.e1. https://doi.org/10.1016/j.jpeds.2011.08.055.
Funding
This work was primarily funded by a Royal Children’s Hospital Foundation grant (AM, IS, DA). The work was also supported by National Health and Medical Research Council (NHMRC) Centre of Research Excellence Grants (CRE-SLANG: 1116976, AM, MH, IS, DA; and Translational Centre for Speech Disorders: 2015727, AM, IS, MH, DA) and an NHMRC project grant (1160893, AM, DA, MH). AM was supported by an NHMRC Practitioner Fellowship (1105008) and an Investigator grant (1195955). DA is supported by the Lorenzo and Pamela Galli Research Trust. IS was supported by an NHMRC Investigator grant (1172897). Additional funding was provided by the Independent Research Institute Infrastructure Support Scheme and the Victorian State Government Operational Infrastructure Program. Open Access funding is enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Contributions
Professors Morgan and Amor conceptualized and designed the study, coordinated and supervised the study, collected data, carried out analyses, drafted the initial manuscript, and reviewed and revised the manuscript. Professors Scheffer, Hildebrand and Stark contributed to the genetic design and analysis of the study, interpreted data, and reviewed and revised the manuscript. Dr. Brenchley, Dr. Baker, Ms. Van Niel, Ms. Lauretta, Ms. O’Donnell, Ms. Boulton collected data, carried out analyses, and reviewed and revised the manuscript. Professor Coman, Dr. Goel, Dr. Thompson, Dr. Webster, Dr. Paxton collected and interpreted data, and reviewed and revised the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The study was approved by the Human Research Ethics Committee of The Royal Children’s Hospital, Melbourne, Australia (#37353). Written informed consent was obtained from all parents or legal guardians for their child to take part in our study and to publish their data. We have archived this data in our REDCap database held at the Murdoch Children’s Research Institute at the Royal Children’s Hospital campus.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Van Niel, H., Lauretta, M., Baker, E. et al. Childhood motor speech disorders: who to prioritise for genetic testing. Eur J Hum Genet 34, 639–648 (2026). https://doi.org/10.1038/s41431-025-01993-9
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-025-01993-9
