
Abstract
Acute kidney injury (AKI) is a common and critical clinical condition with complex pathogenesis and limited early intervention options. Ferroptosis, an iron-dependent form of cell death driven by lipid peroxidation, plays a pivotal role in AKI development. This study aimed to investigate ferroptosis-related gene expression, spatial distribution, and immune interactions in AKI to identify potential therapeutic targets. We analyzed gene expression changes in a mouse model of ischemia–reperfusion-induced AKI and constructed a machine learning–based diagnostic model. This model identified five ferroptosis-related genes (TFRC, TXNRD1, SLC39A14, GCLM, and HMOX1) closely associated with immune cell infiltration. Integration of single-cell and spatial transcriptomics revealed that these genes were predominantly expressed in proximal tubule cells. Notably, TFRC exhibited distinct spatial proximity to infiltrating macrophages. In vivo, administration of the ferroptosis inhibitor NSC306711 significantly reduced macrophage infiltration and renal injury, as confirmed by immunofluorescence. In vitro, co-culture experiments showed that TfR1 degradation alleviated hypoxia–reoxygenation injury in tubular cells and attenuated immune cell activation. This study highlights the central role of ferroptosis in AKI pathogenesis and its interaction with immune components in the renal microenvironment. Targeting ferroptosis, particularly TFRC, may offer a promising strategy to mitigate kidney injury and immune activation in AKI.
Similar content being viewed by others

Introduction
Acute kidney injury (AKI) is a common and severe clinical condition with high morbidity and mortality rates [1]. It is primarily characterized by a rapid decline in kidney function over a short period, accompanied by the retention of uremic waste products such as creatinine and urea nitrogen, as well as disturbances in water, electrolyte, and acid-base balance [2]. The incidence of AKI is around 10–15% in hospitalized patients, and it affects up to 50% of patients in intensive care units [3,4,5,6]. Globally, approximately 13.3 million new cases of AKI occur each year, leading to 1.7 million deaths [7]. The major causes of AKI include ischemia-reperfusion (IR) injury, nephrotoxic substances, sepsis, and the use of contrast agents [7, 8]. Based on the underlying cause, AKI can be classified into prerenal (51.8%), renal (27.6%), and postrenal (8.8%) types [9]. Renal ischemia-reperfusion injury is primarily caused by prerenal factors, including hypovolemia, peripheral vasodilation, decreased cardiac output, and renal vasoconstriction [10, 11]. Although recent randomized controlled trials on AKI early warning have reported negative results [12, 13], there are still some promising preventive strategies yielding up for AKI, such as ischemic preconditioning [14] and intravenous amino acid injection [15]. Therefore, it is still of significant value to explore earlier and more accurate AKI biomarkers and models via multi-omics approaches to achieve precise prediction of AKI, and to gain a deeper understanding of the pathophysiological characteristics of AKI.
Ferroptosis has emerged as a new type of programmed cell death and is characterized by iron accumulation and lipid peroxidation, leading to the oxidative destruction of cell membranes and ultimately cell death [16, 17]. Ferroptosis has been reported to participate in the pathophysiological processes of various diseases, including cancers, nervous system diseases such as Huntington’s disease, Alzheimer’s disease, and Parkinson’s disease [18], acute liver injury [19], as well as acute kidney injury [20]. The regulation of ferroptosis has become a hotspot in AKI etiological research and treatment [21]. Synergistically with autophagy and necroptosis, ferroptosis regulates cell death in AKI and triggers inflammation through immunogenicity. Damage-associated molecular patterns (DAMPs) and alarmins are released in this process, which further alert the innate immune system and shape the inflammatory response to cell death [21]. However, limited literature about the molecular and signaling pathways through which ferroptosis regulates the immune microenvironment in AKI has been reported.
This study aimed to reveal the expression pattern of the ferroptosis gene signature in AKI human and mouse samples by bulk-RNA sequencing, single-cell RNA sequencing, and spatial transcriptome. We also developed a robust diagnostic model to identify early AKI patients using ferroptosis-related genes with receiver operator characteristic curve and calibration curve analysis. Further, we explored the relationship between ferroptosis-related genes and immune microenvironment at the scRNA-seq and spatial transcriptomic level, and revealed that Tfrc+ proximal tubule cells (PTCs) demonstrated both closer proximity and stronger communication, implying that TFRC acted as a crucial role in ferroptosis and immunomodulation in AKI. Finally, by inhibiting TFRC in the IR-AKI mice model, we validated the intercellular cooperation between PTCs and macrophages. Our study demonstrated the crucial genes of ferroptosis in AKI development and immune activation from multi-omics perspectives.
Methods
Design of the study
This study aims to investigate the role of dysregulated ferroptosis-related gene expression in IR-AKI and its association with immune activation. Leveraging machine learning models, single-cell RNA sequencing (scRNA-seq), and spatial transcriptomics, we conducted a comprehensive analysis based on spatiotemporal transcriptomics, validated through experimental data. The workflow of the study is as Fig. 1a showed.

a Schematic representation of the experimental workflow employed in this study, depicting the key steps involved in data collection, analysis, and interpretation. b Comparison of ferroptosis scores obtained in GSVA between the AKI and normal kidney samples. c The volcano plot depicting the differentially expressed ferroptosis-related genes, with colors indicating upregulation or downregulation, and the size of circles representing fold changes. d The heatmap comparing the expression patterns of ferroptosis-related genes in the AKI and normal kidney samples, with the color blocks representing the levels of gene expression. e The boxplot showing the statistical comparison of ferroptosis-related gene expression between the AKI and normal kidney samples. The Mann–Whitney–Wilcoxon test was applied between the two groups. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. BIR: bilateral IR.
Data acquisition and processing
Single-cell RNA sequencing dataset (GSE139506) was downloaded from the Gene Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo). Cell types were identified and clustered by the Seurat pipeline [22]. The expression profiles and survival data of the GSE43974 dataset containing both human IR-AKI biopsy samples(n = 101) and control kidney biopsies from living donors(n = 37) were downloaded from the GEO database. The ferroptosis gene list (WP_FERROPTOSIS.v2024.1.Hs.gmt) and local acute inflammatory response gene list (BIOCARTA_LAIR_PATHWAY.v2024.1.Hs.gmt) were obtained from the GSEA|MSigDB (https://www.gsea-msigdb.org/gsea/msigdb) database (Supplementary Tables 1 and 2). Finally, 64 ferroptosis genes and 17 local acute inflammatory response genes were included in the following analysis.
Calculation of ferroptosis scores
In bulk-RNA sequencing datasets, ssGSEA was performed to obtain the ferroptosis score using the GSVA algorithm [23]. In scRNA sequencing datasets, the ferroptosis score for each cell type was computed based on a ferroptosis gene set, calculated using the “AddModuleScore” algorithm as previously described [24].
Differential analysis of ferroptosis genes between AKI and control human samples
Differential gene analysis was performed using the “limma” package in R [25]. Genes with an adjusted p-value (adj.P.Val) less than 0.05 and an absolute log2 fold change (log2fc) threshold of 0.5 were considered differentially expressed. The visualization of differentially expressed genes was achieved using the “ggplot2” package, generating volcano plots and heatmaps. For volcano plots, a more stringent threshold of adj.P.Val < 0.01 was applied specifically for ferroptosis-related genes. Heatmaps were utilized to intuitively compare the expression patterns of differentially expressed ferroptosis-related genes across AKI and control samples. The “ggpubr” package was employed for comparing the expression levels of key ferroptosis-related genes between the two groups, followed by statistical analysis [26].
Construction and evaluation of an AKI diagnostic model with differentially expressed ferroptosis genes
The “svmRadial” function in the “caret” package was applied [27]. The screened gene variables were further reduced by the “randomForest” algorithm [28]. Genes with MeanDecreaseGini scores greater than 0 were selected as ferroptosis-related genes. These selected genes were then subjected to multicox logistic regression analysis to explore their predisposition towards AKI, leading to the construction of a glm predictive model. The “forestploter” algorithm was employed to visualize the hazard ratios of the genes obtained from the multicox regression. The accuracy of the model in diagnosing AKI, represented by the Area Under the Curve (AUC) value, was evaluated using the “pROC” package.
The ferroptosis-related AKI diagnostic model was internally validated through repeated bootstrap resampling using the “boot” package to ensure comprehensive accuracy [29]. The “decision_curve” function was employed to compare the standardized net benefit of single versus multiple genes for joint predictions, determining the optimal diagnostic model [30]. Additionally, the “regplot” package was utilized to create a nomogram predicting individualized AKI propensity [31].
Immune infiltration and inflammatory cytokine analysis
CIBERSORT was used for the immune cell analysis of the bulk-RNA expression data [32]. FPKM values were used due to their superiority for deconvolution analysis. CIBERSORT will automatically choose the record with the highest mean expression across the mixtures during analysis. The created gene expression file with the 138 cases was uploaded to CIBERSORT as a mixture file, and CIBERSORT was run with the following options: relative and absolute modes together, LM22 signature gene file, 100 permutations, and quantile normalization disabled. One hundred permutations were used as the recommended minimum, but the use of 1000 permutations showed no change in the returned absolute proportion of each cell type. The inflammatory cytokine dataset (BIOCARTA_INFLAM_PATHWAY.v7.5.1) was downloaded from GSEA|MSigDB (https://www.gsea-msigdb.org/gsea/msigdb) database (Supplementary Table 3). The correlation analysis between immune infiltration or the expression of inflammatory cytokine and model ferroptosis genes was performed by the Pearson test in the “psych” package and visualized in a heatmap by the “ggplot” function in the “ggsci” package.
RNA-seq bulk clustering of AKI patients
The k-Means algorithm in the “ConsensusClusterPlus” package was applied to partition AKI patients into three subgroups based on the expression of ferroptosis genes [33]. The optimized cluster number (with k ranging from 2 to 10) was selected by consensusScore. The procedure was repeated 100 times to ensure the reproducibility of the results. The results of the k-Means clustering of AKI patients were visualized by the “pheatmap” package.
Cell-to-cell crosstalk analysis
We analyzed intercellular communication using the CellChat package, which determines intercellular interactions by assessing the expression of ligand and receptor pairs within a cell population [34,35,36]. In principle, CellChat applies network analysis and pattern recognition methods to predict the signal inputs and outputs of cells and how these signals regulate cellular function, and thus assess intercellular crosstalk [37].
Single-cell pseudotime analysis
To explore the potential influence of ferroptosis-related gene signature on the development of proximal tubule cells in the AKI progression, we employed the “CytoTRACE” and “Monocle2” R packages to conduct pseudotime analysis on the scRNA-seq AKI dataset. CytoTRACE is a powerful computational framework for predicting cell differentiation states using scRNA-seq data. By capturing, smoothing, and calculating the expression levels of genes that are highly correlated with single-cell gene counts in single-cell RNA sequencing (scRNA-Seq) data, each single cell receives a score ranging from 0 to 1 that represents its stemness within a specific dataset. A higher score indicates greater stemness (lower differentiation) and vice versa. The R package CytoTRACE v0.3.3 was used to calculate the CytoTRACE scores for proximal tubule cells [38]. Size factors and dispersions were estimated, and highly variable features were identified within the Monocle object [39]. Cell differentiation states were determined using the DDRtree method and the “reduceDimension” function within the monocle package [40]. The “plot_cell_trajectory” function was applied to visualize the trajectory of cell differentiation. The results from CytoTRACE were clearly depicted on the cell developmental trajectories obtained from Monocle analysis.
Spatial transcriptomic analysis
Seurat pipeline for spatial transcriptomic analysis was employed. Initial data preprocessing involved filtering out mitochondrial and ribosomal genes, as well as genes expressing fewer than 10 spots [41]. The expression profile was subsequently subjected to SCTransform. To visualize the section’s landscape, the SpatialDimPlot and SpatialFeaturePlot functions were utilized. Gene set evaluation was conducted through the AddModuleScore function. Spatial deconvolution was done by the RCTD method; the aforementioned single-cell RNA sequencing data was used as references [42]. The spatial correlations among the deconvolved spots were assessed by SPOTlight [43].
Spatial cell–cell interaction analysis
The intercellular communication networks within spatial transcriptomic sequencing data were investigated by the stLearn CCI (Cell–Cell Interaction) pipeline [44]. Ligand-receptor-mediated interactions between different annotated spots were examined by the “st.tl.cci.run” function. The “st.pl.ccinet_plot” function was employed to visually represent the quantity and potency of the communication among the annotated spots. The assessment of major ligand-receptor pairs across all AKI cell spots was conducted utilizing connectomeDB2020lit.Human database [45].
Weighted gene co-expression network analysis (WGCNA) and enrichment analysis
Gene co-expression networks were constructed using the “WGCNA R” package [46]. Raw expression data were normalized and filtered to retain genes with high variability. A signed weighted adjacency matrix was generated by raising the pairwise Pearson correlation coefficients between genes to a soft thresholding power (β). The optimal β-value was determined using the scale-free topology criterion (R² > 0.85) to ensure a biologically meaningful network structure 48. The topological overlap matrix (TOM) was computed to minimize noise and enhance module detection 8 [47]. Within key modules, intramodular connectivity (kME) and gene significance (GS) were calculated to prioritize hub genes. Genes with kME > 0.8 and high GS (p < 0.01) were considered central drivers of module function [48]. The hub genes identified by WGCNA analysis were subjected to enrichment analysis using https://metascape.org/ [49].
IR injury animal model for validation
Male C57BL/6 mice aging 6–8 weeks were anesthetized and underwent aseptic surgical procedures. Male mice were exclusively used in this study to avoid the influence of hormonal fluctuations associated with the estrous cycle in females, which may affect renal ischemia-reperfusion outcomes [50]. Through a midline abdominal incision, bilateral renal arteries were gently clamped using non-traumatic vascular clamps, inducing ischemia. The duration of ischemia was 27 min, and subsequent reperfusion was initiated by releasing the clamps. Sham-operated mice underwent identical anesthesia and surgical procedures without vascular occlusion, serving as control counterparts. During ischemia, body temperature was maintained at 36.5–37.5 °C using an electric heating pad with continuous monitoring of rectal temperature. The depth of anesthesia was continuously monitored to ensure the animals’ well-being. Post-surgery, mice received appropriate post-operative care. Post-surgery, mice received appropriate post-operative care. Kidney tissues and blood samples were collected 24 h after reperfusion for further analyses. The successful establishment of the IR model was validated through meticulous assessment methods, including renal function parameters, serum biomarker analysis, and histological examination [51]. Mice treated with the TfR1 degradation agent underwent surgery 30 min after an intraperitoneal injection of 10 mg/kg NSC306711 [52].
Western blot
Cell or tissue samples were homogenized in lysis buffer, and protein concentrations were determined using the BCA assay. Equal amounts of proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose or polyvinylidene fluoride (PVDF) membrane. After blocking with non-fat milk or bovine serum albumin, the membrane was probed with specific primary antibodies, including neutrophil gelatinase-associated lipocalin (NGAL) (abcam, ab125075, 1:1000), TfR1 (abcam, EPR20584, 1:1000), and GAPDH (proteintech, 10494-1-AP, 1:5000), overnight at 4 °C. Following incubation with corresponding secondary antibodies, protein bands were visualized using chemiluminescence detection reagents. Quantification of band intensity was performed by GraphPad 9.1.1, and target protein levels were normalized against GAPDH. Each experiment was repeated independently to ensure the reliability and reproducibility of the obtained results. Figi 1.0 was employed to conduct the image and statistical analysis [53].
Hematoxylin and eosin (HE) staining, immunohistochemistry, immunofluorescence, and DHE staining
Kidney tissues were fixed in 4% paraformaldehyde overnight at 4 °C, dehydrated through a graded ethanol series, cleared in xylene, and embedded in paraffin. Sections of 4 μm thickness were cut, deparaffinized, and rehydrated. The sections were stained with hematoxylin solution for 5 min, rinsed in running tap water, differentiated in 1% acid alcohol, and blued in 0.2% ammonia water. After counterstaining with eosin solution for 2 min, the sections were dehydrated, cleared, and mounted with neutral resin. Histological changes were observed and imaged under a light microscope.
Paraffin-embedded tissue sections were baked at 60 °C for 20 min to 1–2 h, followed by deparaffinization in xylene and rehydration through a graded alcohol series. Antigen retrieval was performed by heating the sections in retrieval buffer until boiling, then maintaining at medium–low temperature for 15–20 min, followed by natural cooling and PBS washes. Sections were blocked with serum and incubated overnight at 4 °C with primary antibodies: TfR1 (Abcam, EPR20584, 1:100), F4/80 (CST, 70076, 1:800), and AQP1 (Abcam, ab168387, 1:50). Appropriate HRP-conjugated or fluorescent secondary antibodies were then applied at room temperature. For immunohistochemistry, DAB was used for color development, followed by hematoxylin counterstaining [54]. For immunofluorescence, TSA amplification was applied, nuclei were stained with DAPI, and slides were mounted with antifade medium [55].
Intracellular lipid reactive oxygen species (ROS) levels were detected using frozen tissue sections stained with dihydroethidium (DHE). Sections were allowed to equilibrate to room temperature and air-dried, then washed three times in PBS (pH 7.4) for 5 min each. Sections were circled with a hydrophobic barrier pen to prevent reagent runoff and incubated with DHE solution, fully covering the tissue, at room temperature in the dark for 30 min. After three additional PBS washes (5 min each), DAPI staining was performed at room temperature in the dark for 10 min. Sections were washed and mounted with antifade medium. Fluorescence was observed and imaged using an inverted fluorescence microscope [56].
Intracellular ROS detection
Intracellular ROS levels in HK2 cells were measured using the DCFH-DA Reactive Oxygen Species Assay Kit (Beyotime, S0034) following the manufacturer’s instructions. Briefly, cells were seeded in 6-well plates and subjected to the experimental treatments. After treatment, cells were washed twice with serum-free DMEM and incubated with 10 μM DCFH-DA in serum-free medium at 37 °C for 20–30 min in the dark. Following incubation, cells were washed three times with serum-free medium to remove excess probe.
For fluorescence microscopy, cells were observed under an inverted fluorescence microscope (FITC channel, excitation 485 nm/emission 530 nm), and representative images were captured. For flow cytometry analysis, cells were trypsinized, resuspended in PBS, and analyzed on a flow cytometer to determine the mean fluorescence intensity and the percentage of DCF-positive cells. Data were quantified using FlowJo software.
Cell culture and treatment
Human renal proximal tubular epithelial cells (HK2; Cell Bank of the Chinese Academy of Sciences, Shanghai, China) were cultured in DMEM/F12 medium (Gibco) supplemented with 10% fetal bovine serum (FBS; FDCC, Shanghai, China) and 1% penicillin-streptomycin (Sigma–Aldrich) at 37 °C under 5% CO₂ [53]. For hypoxia/reoxygenation (H/R) induction, cells were placed in a modular hypoxic chamber (MGC, #D-07AnaeroPack) flushed with a gas mixture (1% O₂, 94% N₂, 5% CO₂) for 12 h, followed by 6 h of reoxygenation under normoxic conditions (21% O₂) [57]. To evaluate pharmacological effects, HK2 cells were pretreated with 10 μM NSC306711 (ProbeChem, #PC-61207) for 6 h prior to H/R exposure [58].
THP-1 monocytes (ATCC) were maintained in RPMI-1640 medium (HyClone) containing 10% FBS and 0.05 mM β-mercaptoethanol (Thermo Fisher Scientific). Cells were passaged every 2–3 days and seeded at 5 × 10⁵ cells/mL in 6-well plates (Corning) for co-culture experiments [59].
A co-culture system was established to explore the impact of conditioned medium (CM) on THP-1 cells. CM was collected from HK2 cells after 12-h hypoxia, centrifuged at 1500 × g for 10 min, and filtered through a 0.22 μm membrane to remove debris. THP-1 cells were first differentiated by stimulation with phorbol 12-myristate 13-acetate (PMA; 100 ng/mL, Sigma) for 48 h, and subsequently incubated for 6 h in a medium composed of 50% CM and 50% fresh complete medium, as previously described [60].
Reverse transcription-PCR and real-time quantitative PCR
Total RNA was isolated from tissues or cells using TRIzol reagent (Sigma–Aldrich). For cDNA synthesis, 500 ng of purified RNA was reverse-transcribed with the PrimeScript RT Master Mix (Takara Bio). Real-time quantitative PCR (qPCR) was performed using the SYBR Premix Ex Taq kit (Takara Bio) on a real-time PCR system, following the manufacturer’s protocol.
The relative mRNA expression levels of target genes were calculated using the 2^−ΔΔCt method, normalized to the endogenous reference gene GAPDH [53]. Primer sequences are listed in Tables 1 and 2.
Cell viability assay
HK2 cells were seeded in 96-well plates at a density of 5 × 103 cells per well and allowed to adhere overnight. After the indicated treatments, the culture medium was replaced with fresh medium containing 10 μL of CCK-8 reagent (Beyotime, Cell Counting Kit-8, C0037) per well. Cells were incubated at 37 °C for 0.5–2 h, and the absorbance at 450 nm was measured using a microplate reader. Relative cell viability was calculated by comparing the absorbance of treated wells to that of control wells according to the manufacturer’s instructions.
Statistical analyses
For in vitro experiments, all assays were performed with at least three independent biological replicates (n = 3) to ensure reproducibility, consistent with standard practice in cell-based studies [61, 62]. For in vivo mouse experiments, each experimental group included six animals (n = 6), which is commonly used in preliminary studies to detect biologically meaningful effects while adhering to ethical considerations regarding animal use [62, 63]. Sample sizes were chosen based on previous literature and feasibility, and all experiments were repeated independently to confirm the findings. No blinding was performed during animal allocation or outcome assessment. All statistical analyses in our study were performed using appropriate and justified tests. The data met the assumptions required for these tests, including normal distribution where applicable. Estimates of variation within each group are presented as mean ± SEM (or SD as specified), and the variance between groups being compared was confirmed to be similar. Statistical significance was set at P < 0.05.
Results
Ferroptosis-related differentially expressed genes in IR-AKI and control human samples
Utilizing the Gene Set Variation Analysis (GSVA) algorithm, we observed a significant increase in the enrichment scores of the ferroptosis-related gene set in IR-AKI patients (Fig. 1b). The volcano plot illustrated a distinct expression pattern of ferroptosis-related genes in AKI group (Fig. 1c). Differentially Expressed Genes (DEGs) analysis identified 17 genes(Fig. 1d). Among the upregulated genes, TFRC (transferrin receptor protein 1), TXNRD1 (thioredoxin reductase 1), SLC39A14 (solute carrier family 39 member 14), GCLM (glutamate-cysteine ligase modifier subunit), and HMOX1 (heme oxygenase 1) were notably prominent. Conversely, the downregulated genes included SLC11A2 (solute carrier family 11 member 2), POR (cytochrome P450 oxidoreductase), and CISD1 (CDGSH iron-sulfur domain-containing protein 1). The correlations among these DEGs are shown in Supplementary Fig. 1a, b. These genes exhibited statistically significant differences in expression between human IR-AKI and control samples (Fig. 1e). Together, these results demonstrated a unique ferroptosis-related profile in IR-AKI patients.
Construction and validation of the ferroptosis-related IR-AKI diagnostic model
To further investigate the significance of ferroptosis-related genes in acute kidney injury (AKI), we applied machine learning approaches to prioritize these genes. Specifically, we utilized the Random Forest algorithm and the Support Vector Machine-Recursive Feature Elimination (SVM-RFE) method. The Random Forest model performance curve is shown in Fig. 2a, and based on importance rankings, the top ten ferroptosis-related genes were identified (Fig. 2a, b). Subsequent SVM-RFE analysis further refined this list to five key genes, with their expression validation results shown in Fig. 2c. A Venn diagram illustrating the overlap among genes identified by differential expression analysis, Random Forest, and SVM-RFE highlighted five hub genes: SLC39A14, TFRC, HMOX1, TXNRD1, and GCLM (Fig. 2d).

a Performance curve of the Random Forest model used to evaluate model fitting. b Bar plot showing the top ten ferroptosis-related genes ranked by importance scores in the Random Forest algorithm. c Expression validation of five ferroptosis-related genes identified by Support Vector Machine-Recursive Feature Elimination (SVM-RFE). d Venn diagram illustrating the overlapping feature genes identified by differential expression analysis, Random Forest, and SVM-RFE algorithms. e Forest plot of the multivariate logistic regression analysis demonstrating odds ratios and 95% confidence intervals for the five ferroptosis-related genes; only TFRC showed statistical significance. f Receiver operating characteristic (ROC) curve showing the diagnostic performance of the five-gene model, with area under the curve (AUC) = 0.908. g ROC curve generated from internal bootstrap validation. h Calibration curve comparing the predicted probabilities versus observed outcomes, demonstrating excellent model calibration. i Nomogram constructed based on the five ferroptosis-related genes to estimate individual AKI risk. j Decision curve analysis (DCA) evaluating the net clinical benefit of the nomogram across a range of threshold probabilities. k Schematic representation of the in vivo experimental workflow. l Western blot analysis validating increased protein expression of NGAL and TfR1 in kidney tissues from IR-AKI mice. *p < 0.05; ****p < 0.0001.
Multivariate logistic regression analysis revealed a significant association between ferroptosis-related genes and AKI susceptibility. Among the five genes, TFRC emerged as the only statistically significant risk factor, with an odds ratio (OR) of 7.94 (95% CI: 3.25–23.44) (Fig. 2e). These five genes were subsequently incorporated into the construction of a diagnostic nomogram. Receiver operating characteristic (ROC) analysis demonstrated that the model achieved a high area under the curve (AUC) of 0.908 (Fig. 2f). Internal validation using bootstrap resampling and calibration curve analysis consistently confirmed the model’s robust predictive performance (Fig. 2g, h; Supplementary Fig. 2a, b). Notably, the nomogram substantially outperformed any single gene in diagnostic performance (Fig. 2i). Furthermore, decision curve analysis (DCA) demonstrated that, within a defined threshold range, the nomogram provided a greater net clinical benefit (Fig. 2j).
Importantly, correlation analysis showed that TFRC was not significantly associated with other ferroptosis-related genes such as FTH1 and GPX4 in the control group, whereas in AKI patients, TFRC exhibited a strong positive correlation with these genes (Supplementary Fig. 1c). These findings prompted us to focus further investigations on TFRC. To validate transferrin receptor (TfR1), encoded by TFRC, at the protein level, we established an IR-AKI mouse model (workflow diagram shown in Fig. 2k). Western blot analysis was performed to assess the expression of neutrophil gelatinase-associated lipocalin (NGAL) and TfR1 in renal tissues. NGAL levels were significantly elevated in the IR group, confirming successful AKI model establishment. Quantitative analysis further revealed that TfR1 expression was also significantly upregulated in the IR-AKI group (Fig. 2l; Supplementary Material 1, 2).
The association between ferroptosis and local acute inflammatory response
Additionally, we explored the immunomodulatory role of the five model genes. We applied the CIBERSORT algorithm to analyze the correlation between the five model genes and immune infiltration, as well as the expression of inflammatory factors. Unlike the other four genes, TFRC showed a strong positive association with macrophages, including both M1 and M2 subtypes, as well as with CD8+ T cells, osteoblasts, plasma cells, and smooth muscle cells. In contrast, TFRC demonstrated a significant negative relationship with various immune cell types, such as CD4+ T cells, NKT cells, Treg cells, Th2 cells, memory B cells, and dendritic cells (DCs), including subtypes pDC, cDC, aDC, iDC, and DC in general. Notably, the other four genes mainly showed positive correlations with dendritic cells, highlighting the unique immunological association of TFRC (Fig. 3a). In terms of cytokines and chemokines, TFRC showed a significant positive correlation with the inflammatory factor HLA-DRB3, which is essential for antigen-presenting cells such as macrophages, B lymphocytes, and dendritic cells (Fig. 3b). In contrast, TXNRD1, SLC39A14, GCLM, and HMOX1 demonstrated a positive correlation with M2 macrophages, and exhibited a positive correlation with inflammatory factors like CXCL8 and IL7 (Fig. 3a, b).

a The heatmap showing the Pearson correlation between the expression of five model ferroptosis-related genes and immune infiltration calculated by CIBERSORT. *p < 0.05; **p < 0.01; ***p < 0.001. b The heatmap showing the Pearson correlation between the expression of five model ferroptosis-related genes and the expression of inflammatory cytokines. The shades of the color blocks indicate correlation coefficients. *p < 0.05; **p < 0.01; ***p < 0.001. c The clustering heatmap showing AKI patient subtypes when k-means equaled three by k-means methods. d The comparison of the local acute inflammatory response level calculated by GSVA among the three AKI subtypes. The Mann–Whitney–Wilcoxon test was adopted between the three subtypes. e The comparison of ferroptosis levels calculated by GSVA among the three AKI subtypes. The Mann–Whitney–Wilcoxon test was adopted between the three subtypes. f 13 modules revealed by the WGCNA. g The correlation between the 13 modules and the 3 patient clusters. h Enrichment analysis of the hub genes in the yellow and black modules. i The comparison of TFRC expression levels among the three AKI subtypes. j The correlation between TFRC expression level and ferroptosis level. k The correlation between TFRC expression level and local acute inflammatory response level.
Subsequently, we classified AKI patients into three clusters based on the expression levels of ferroptosis-related genes and analyzed the differences among these clusters (Fig. 3c). Compared to the other two clusters, patients in cluster 1 exhibited the highest levels of local acute inflammatory responses (Fig. 3d) and ferroptosis (Fig. 3e). The heatmap shows the expression of ferroptosis-related genes in the three clusters of patients, as well as the predicted probability of AKI based on the model (Supplementary Fig. 2c). Additionally, the boxplot illustrates the expression levels of 17 differentially expressed genes across the three patient clusters (Supplementary Fig. 2d). WGCNA analysis was further performed, starting with hierarchical clustering and generating a dendrogram to remove outlier samples from the dataset (Supplementary Fig. 3a, b). The scale independence plot evaluates the network’s topological structure independence at different power values. The mean connectivity plot is used to assess the average connectivity of the network at different power values. WGCNA analysis identified 13 gene modules (Fig. 3f), among which cluster 1 patients were significantly negatively correlated with the yellow and black modules. Enrichment analysis revealed that the yellow and black modules were significantly enriched in pathways related to damage repair, including cell population proliferation, response to growth factor, negative regulation of cell population proliferation, blood vessel development, positive regulation of programmed cell death, and response to wounding. Further analysis indicated that TFRC levels in cluster 1 patients were the highest among the three clusters (Fig. 3i), and correlation analysis showed that TFRC was not only significantly positively correlated with ferroptosis (Spearman ρ = 0.53, p = 1.5e-08), but also with local acute inflammatory responses (Spearman ρ = 0.21, p = 0.033) (Fig. 3j, k).
Ferroptosis-related proximal tubule cells exhibited an enhanced interaction with the immune microenvironment
We explored the ferroptosis-related characteristics of AKI at a single-cell level. In the scRNA-seq dataset GSE139506, cells were categorized into distinct subtypes, including proximal tubule cells, collecting duct epithelial cells (CDEC), fibroblast, endothelial cells, podocytes, and macrophages (Fig. 4a, Supplementary Fig. 4a–f). The overall level of ferroptosis in the AKI group was significantly higher than in the sham group (Fig. 4b, Supplementary Fig. 4g). Notably, PTCs exhibited the highest ferroptosis scores among all cell types in AKI samples (Fig. 4c).

a Cell type annotations clustered by the Seurat uniform manifold approximation and projection (UMAP) in GSE139506 in AKI and sham samples. b The violin plot shows the overall ferroptosis levels in the IR-AKI and Sham groups. c Ferroptosis levels in various kidney cells of the IR-AKI group mice. d TFRC expression levels in various kidney cells of the IR-AKI group mice. e The number of interaction pathways among various cell types is displayed. f Interaction signaling pathways among various cells are presented. ECDC: Epithelium collecting duct cell. g Heatmap displaying signaling pathways emitted and received among various cells. h Spatial transcriptomics localization of Tfrc+ PTCs. i Spatial transcriptomics localization of Tfrc- PTCs. j Spatial transcriptomics localization of macrophages. k Heatmap showing intercellular interaction analysis based on spatial transcriptomics. l Heatmap of cell–cell communication strength among different cell populations in the spatial transcriptomics dataset. m Network visualization of intercellular communication in the spatial transcriptomics dataset. n Heterogeneous cell–cell interaction network analysis. o Macrophage enrichment score analysis. CDEC: collecting duct epithelial cell.
Cell localization analysis revealed that TFRC was predominantly expressed in PTCs of the AKI kidney (Fig. 4d). We classified the PTCs from AKI mice kidneys into Tfrc+ PTCs and Tfrc-PTCs based on Tfrc expression. CellChat analysis indicated that Tfrc+ PTCs had more interaction pathways with macrophages compared to Tfrc- PTCs (Fig. 4e). Receptor-ligand analysis showed that, compared to the pair between Tfrc-PTC and macrophage, significant activation of the C4a - C3aR1 and Mdk - Ncl pathway between Tfrc+ PTCs and macrophages was detected, suggesting that Tfrc+ PTCs played a crucial role in regulating immune responses, especially complement system, promoting inflammation, and attracting immune cells to the site of inflammation (Fig. 4f). Signaling pathway analysis indicated that Tfrc+ PTCs exhibited higher intensity in both sending and receiving CXCL and complement pathways compared to Tfrc- PTCs (Fig. 4g).
Furthermore, Tfrc+ PTCs, Tfrc− PTCs, and macrophages were mapped onto the spatial transcriptomics dataset GSE171406 (Fig. 4h–j, Supplementary Fig. 5a–f). Notably, Tfrc+ PTCs exhibited a closer association with macrophages compared with Tfrc− PTCs (Supplementary Fig. 5g, h). The heatmap generated by mistyR analysis revealed stronger interaction strength between Tfrc+ PTCs and macrophages (Fig. 4k). Consistently, further cell–cell interaction analysis demonstrated that, compared with Tfrc− PTCs, Tfrc+ PTCs exhibited more prominent interactions with macrophages (Fig. 4l). In the interaction network, Tfrc+ PTCs established clear connections with macrophages, whereas Tfrc− PTCs showed no direct interactions with them (Fig. 4m). Colocalization network analysis revealed that Tfrc+ PTCs had more connections with macrophages than Tfrc− PTCs, suggesting stronger communication potential. (Fig. 4n). Consistently, macrophages showed higher enrichment scores in the Tfrc+ PTC context (Fig. 4o).
Tfrc expression varies at different stages of proximal tubule cell differentiation after AKI
To explore the role of Tfrc in the process of tubular injury, we conducted a more in-depth analysis of PTCs during AKI in terms of ferroptosis. We isolated 667 PTCs from AKI mice and conducted dimensionality reduction clustering, identifying 8 PTC subtypes (PTC (0–7)) (Fig. 5a). The ferroptosis score indicated that PTC (4) and PTC (0) had the highest levels of ferroptosis, while PTC (5) and PTC (6) had the lowest (Fig. 5b). CytoTRACE analysis ranked the stemness of the 8 PTC subtypes, revealing that PTC (1) and PTC (4) exhibited the highest stemness, while PTC (0) and PTC (6) showed the lowest (Fig. 5c). This indicated that PTC (1) and PTC (4) were in an earlier stage of differentiation compared to PTC (0) and PTC (6) (Fig. 5d).

a Further clustering of PTC subtypes in IR-AKI samples using Seurat UMAP. b The violin plot shows the ferroptosis scores of PTC cell subtypes, with scores decreasing from left to right. c The boxplot displays the cell stemness of each PTC subtype based on CytoTRACE analysis. d The UMAP plot shows the differentiation levels of each PTC cell subtype. e Monocle analysis presents the cell developmental trajectory based on CytoTRACE results. f The boxplot displays the Tfrc expression levels of each PTC subtype. g Monocle analysis shows the cell developmental trajectories of various PTC subtypes.
Monocle analysis assisted in revealing the PTC differentiation trajectories after AKI, which presented two distinct trajectories with special PTC subtypes (Fig. 5e–g). Tfrc expression across PTC subtypes (excluding those that did not express Tfrc) presented the highest Tfrc expression level in PTC (0) (Fig. 5f). Trajectory 1 culminated in PTC (0) and PTC (4), which had higher levels of ferroptosis. In contrast, Trajectory 2 was primarily characterized by PTC (2) and PTC (5), which exhibited lower levels of ferroptosis (Fig. 5b, g). These results indicated that PTCs with high Tfrc expression led to the differentiation pathway with high ferroptosis levels after AKI. Combined with findings in Part 4, it was determined that this subset of PTCs was also associated with a high level of local acute inflammatory response.
Validation of ferroptosis-related Tfrc + PTCs and their interaction with macrophages in IR-AKI
Subsequently, we experimentally validated the role of ferroptosis-related genes by treating IR-AKI mice with the TfR1 degrader NSC306711, followed by Western blot, immunohistochemistry, and immunofluorescence analyses, as illustrated in the workflow (Fig. 6a). HE staining revealed marked tubular injury in the IR group compared to the sham group, characterized by tubular epithelial cell disruption and the formation of tubular casts, whereas treatment with NSC306711 markedly alleviated these pathological changes (Fig. 6b). Quantitative assessment of pathological injury scores further confirmed that NSC306711 significantly ameliorated IR-induced kidney injury (Fig. 6c). In addition, serum creatinine and blood urea nitrogen levels were both significantly reduced in the NSC306711-treated group compared to untreated IR mice, indicating functional renal protection (Fig. 6d, e).

a Workflow of experimental validation in IR-AKI mice treated with the TfR1 degrader NSC306711. b H&E staining showing tubular injury and cast formation in sham, IR, and IR + NSC306711 groups. c Quantification of renal pathological injury scores. d Serum creatinine levels in sham, IR, and IR + NSC306711 groups. e Blood urea nitrogen (BUN) levels in sham, IR, and IR + NSC306711 groups. f qPCR analysis of GPX4 mRNA expression in kidney tissues. g qPCR analysis of SLC7A11 mRNA expression in kidney tissues. h Immunofluorescence (DHE, red) showing ROS levels in kidney tissues. i ELISA analysis of MDA levels indicating lipid peroxidation. j Western blot analysis of TfR1 and NGAL protein levels. k Immunohistochemistry staining of TfR1 in kidney sections. l Immunofluorescence staining of TfR1 (magenta) and F4/80 (cyan) showing proximal tubular cell expression and macrophage infiltration.
We next examined the impact of NSC306711 treatment on ferroptosis. qPCR analysis demonstrated that, relative to the sham group, the mRNA expression of GPX4 and SLC7A11 was significantly downregulated in the IR group, whereas NSC306711 treatment markedly restored their expression levels (Fig. 6f, g). Immunofluorescence staining further showed increased DHE fluorescence (red) in the IR group, consistent with elevated ROS production, which was significantly attenuated after NSC306711 treatment (Fig. 6h). Similarly, ELISA analysis of MDA revealed significantly increased lipid peroxidation in the IR group, which was reversed by NSC306711 treatment (Fig. 6i).
Western blot analysis confirmed that NSC306711 markedly reduced TfR1 protein levels in the IR group, along with decreased expression of the kidney injury marker NGAL (Fig. 6j, Supplementary Material 3–7). These results were further corroborated by immunohistochemistry (Fig. 6k). Moreover, immunofluorescence staining revealed that both TfR1 (magenta) and F4/80 (cyan) were markedly elevated in the IR group compared to the sham group, indicating increased TfR1 expression in proximal tubular cells and enhanced macrophage infiltration following IR injury. NSC306711 treatment significantly reduced both TfR1 and F4/80 levels, suggesting that ferroptosis and macrophage infiltration were alleviated (Fig. 6l).
TFRC + PTCs exacerbate AKI damage by promoting the release of inflammatory cytokines from macrophages
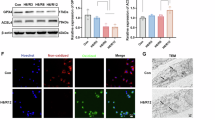
Finally, we validated the effects of NSC306711 in vitro using HK2 cells and further assessed its impact on THP-1–derived macrophages. In HK2 cells subjected to H/R, NSC306711 treatment was applied, followed by qPCR, fluorescence microscopy, and flow cytometry analyses (workflow shown in Fig. 7a). qPCR revealed that TFRC mRNA levels were markedly elevated after H/R, whereas NSC306711 treatment significantly reduced their expression (Fig. 7b). A similar pattern was observed for the injury marker NGAL, which was strongly induced by H/R but attenuated upon NSC306711 supplementation, indicating a protective effect against H/R-induced injury (Fig. 7c). Regarding ferroptosis-related markers, H/R resulted in a pronounced decrease in GPX4 and SLC7A11 mRNA levels, both of which were restored by NSC306711, suggesting that the compound mitigates ferroptotic stress in HK2 cells (Fig. 7d, e). Consistently, ROS levels assessed using the DCFH-DA probe showed a marked increase following H/R (green fluorescence), which was reduced by NSC306711 treatment (Fig. 7f). Flow cytometry further confirmed these findings, demonstrating elevated mean fluorescence intensity and an increased proportion of ROS-positive cells in the H/R group, both of which were significantly decreased by NSC306711 (Fig. 7g). In addition, ELISA results indicated that MDA levels, a marker of lipid peroxidation, were markedly increased in H/R-treated HK2 cells compared with the control group, whereas NSC306711 significantly reduced MDA accumulation (Fig. 7h). HK2 cell viability was markedly reduced following H/R, and NSC306711 administration effectively restored cell viability (Fig. 7i).

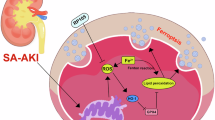
a Experimental workflow for HK2 cell H/R and NSC306711 treatment assays. b qPCR analysis of TFRC mRNA levels in HK2 cells after H/R treatment and NSC306711 intervention. c qPCR analysis of NGAL mRNA levels in HK2 cells after H/R treatment and NSC306711 intervention. d qPCR analysis of GPX4 mRNA levels in HK2 cells after H/R treatment and NSC306711 intervention. e qPCR analysis of SLC7A11 mRNA levels in HK2 cells after H/R treatment and NSC306711 intervention. f Fluorescence microscopy showing ROS levels in HK2 cells after H/R treatment and NSC306711 intervention. g Flow cytometry analysis of DCFH-DA mean fluorescence intensity and positive cell percentage in HK2 cells after H/R treatment and NSC306711 intervention. h ELISA measurement of MDA levels in HK2 cells after H/R treatment and NSC306711 intervention. i Cell viability in HK2 cells. j Schematic workflow of co-culture experiments: THP-1-derived macrophages co-cultured with NSC306711-treated HK2 cells. k IL1B mRNA levels in THP-1 macrophages after co-culture. l IL6 mRNA levels in THP-1 macrophages after co-culture. m TNF mRNA levels in THP-1 macrophages after co-culture. n TFRC links ferroptosis and inflammatory injury in IR-AKI by facilitating iron transfer into proximal renal tubules, which exacerbates local inflammation and IR damage, but its inhibition reduces ferroptosis and mitigates acute inflammatory responses. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns not significant.
To further investigate the influence of NSC306711-treated HK2 cells on macrophage responses, we performed co-culture experiments using conditioned medium from HK2 cells pretreated with NSC306711 and THP-1–derived macrophages (workflow shown in Fig. 7j). H/R-treated HK2 cells markedly enhanced macrophage secretion of inflammatory cytokines, including IL1B, IL6, and TNF. However, this pro-inflammatory effect was significantly attenuated when HK2 cells were pretreated with NSC306711. Importantly, direct exposure of macrophages to NSC306711 did not significantly affect cytokine production (Fig. 7k–m). Together, these findings suggest that Tfrc⁺ PTCs exacerbate AKI by promoting macrophage-mediated inflammatory responses through the release of IL1B, IL6, and TNF, whereas pharmacological inhibition of TFRC with NSC306711 effectively dampens this inflammatory amplification.
Discussion
Ferroptosis is a regulated form of cell death characterized by iron-dependent lipid peroxidation and intracellular accumulation of ROS, which has been implicated in the onset and progression of AKI [64]. In this study, we observed enrichment of ferroptosis-related gene expression in AKI, which enabled the construction of a robust early diagnostic model and patient classification tool. Notably, we identified a strong correlation and spatial interplay between critical ferroptosis genes, particularly TFRC, and aberrant immune activation, suggesting novel pathogenesis mechanisms and potential therapeutic targets for AKI.
The TFRC gene encodes transferrin receptor 1 (TfR1), a key regulator of cellular iron uptake and homeostasis [65]. TFRC has been implicated in regulating ferroptosis through its effects on intracellular iron availability and oxidative stress [66, 67]. Abnormal expression of TFRC is associated with various diseases, mainly including cancer [68] and contrast-induced nephropathy [69], making it a potential therapeutic target [70]. This association might be owed to TFRC-related ferroptosis. Studies have found that the demethylase activity of the m6A demethylase (Fat Mass and Obesity-Associated Protein, FTO) can alleviate hepatic ischemia-reperfusion injury in the elderly by reducing the expression of ACSL4 and TFRC [71]. Another study suggests that inhibiting TFRC can significantly suppress ferroptosis in neuronal cells, thereby delaying the onset and progression of Amyotrophic Lateral Sclerosis (ALS) [72]. In AKI, our ionomic mass spectrometry analysis revealed elevated urinary iron ions, and existing literature indicates that TFRC is primarily expressed on the luminal side of proximal tubular cells (PTCs), suggesting that it may exacerbate local ferroptosis by facilitating iron uptake [73, 74].
Using CIBERSORT analysis, TFRC expression was strongly positively correlated with both M1 and M2 macrophages and CD8+ T cells, while negatively correlated with fibroblasts, indicating its involvement in immune modulation and potential impact on kidney repair or AKI-to-CKD progression [75,76,77,78]. Spatial transcriptomics further revealed that Tfrc+ PTCs exhibited closer interactions with endothelial cells, podocytes, macrophages, and fibroblasts compared to Tfrc− PTCs, suggesting that TFRC mediates intercellular crosstalk and may influence macrophage polarization and iron homeostasis during IR-AKI [36, 79]. Interestingly, discrepancies between bulk and single-cell analyses highlight the complex role of TFRC across different cell types.
Novel biomarkers, such as NGAL, kidney injury molecule-1 (KIM-1), and Tissue Inhibitor of Metalloproteinase-2 (TIMP2), Insulin-like Growth Factor Binding Protein 7 (IGFBP7), are the most accepted early detection targets of AKI. The above biomarkers still reflect the structural injury of PTCs during AKI, and lag behind the subcellular functional injury. Our study has developed a novel and robust diagnostic model of early AKI based on the expression of ferroptosis genes by bioinformatics and machine learning methods, further advancing the diagnostic time window of AKI.
From a translational perspective, TFRC may represent a therapeutic target to mitigate ferroptosis-driven injury in AKI. Modulating TFRC activity could reduce iron-dependent lipid peroxidation and ROS accumulation, thereby protecting PTCs and limiting inflammation [69]. Careful evaluation of dosing, timing, and safety is essential due to TFRC’s systemic role in iron homeostasis. Future preclinical studies should test TFRC-targeted interventions across diverse AKI models and assess whether patient stratification based on TFRC expression could guide personalized therapy [80]. Furthermore, combining TFRC modulation with current clinical management strategies, such as renal replacement therapy or anti-inflammatory treatment, may enhance therapeutic efficacy and accelerate recovery [81].
The involvement of ferroptosis-related genes in immune regulation in AKI has become a subject of keen interest. For instance, ferroptosis-related genes, including HMOX1, MAPK14, and TLR4, showed diagnostic and predictive values in children's sepsis and immune responses[82]. An increasing body of literature suggests the relationship between ferroptosis and inflammation in AKI, and the persistent presence of ferroptosis and inflammation can lead to the progression from AKI-to-CKD by stimulating fibrosis in myofibroblasts [83,84,85]. Unraveling the mutual regulation between ferroptosis and immune infiltration holds promising implications for novel immune-modulating treatments for AKI.
Our research has innovatively identified that PTCs with elevated expression of ferroptosis-related genes exhibited closer interactions with macrophages, accompanied by higher levels of inflammatory factors. Specifically, TFRC was highlighted in our research for its contribution to the closer relationship between PTCs and macrophages in AKI.
Finally, our study has limitations. First, the diagnostic model has not yet been validated in clinical cohorts. Second, the molecular pathways linking TFRC, ferroptosis, and macrophage function remain to be elucidated, which may be critical for determining PTC trajectory and clinical outcomes, including complete or partial recovery, progression to CKD, or dialysis dependency.
Conclusions
In this study, we found a crucial ferroptosis-related genes enabled the construction of a robust model for AKI early diagnosis. Tfrc+ PTCs showed a positive correlation with the immune activation, especially macrophages (Fig. 7n). By TfR1 degradation, ferroptosis, and macrophage infiltration were reduced, and kidney injury was relieved. Altogether, these findings might contribute to a deeper understanding of the complex interplay between PTCs, ferroptosis-related genes, and immune cells, shedding light on potential therapeutic interventions for AKI.
Data availability
All data can be obtained from the GEO (https://www.ncbi.nlm.nih.gov/geo/) database. Other queries for data and code could be directly sent to the corresponding author.
Code availability
The custom scripts used for data processing and analysis in this study are available from the corresponding author upon reasonable request. All scripts were written in R (version 4.1.3) (http://www.R-project.org/). No restrictions apply to the use of the code by academic researchers.
References
Gaudry S, Palevsky PM, Dreyfuss D. Extracorporeal kidney-replacement therapy for acute kidney injury. N Engl J Med. 2022;386:964–75.
Wang S, Chen Y, Han S, Liu Y, Gao J, Huang Y, et al. Selenium nanoparticles alleviate ischemia reperfusion injury-induced acute kidney injury by modulating GPx-1/NLRP3/Caspase-1 pathway. Theranostics. 2022;12:3882–95.
Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet. 2012;380:756–66.
Ronco C, Bellomo R, Kellum JA. Acute kidney injury. Lancet. 2019;394:1949–64.
Rewa O, Bagshaw SM. Acute kidney injury-epidemiology, outcomes and economics. Nat Rev Nephrol. 2014;10:193–207.
Xu S, Jia P, Fang Y, Jin J, Sun Z, Zhou W, et al. Nuclear farnesoid X receptor attenuates acute kidney injury through fatty acid oxidation. Kidney Int. 2022;101:987–1002.
Mehta RL, Cerda J, Burdmann EA, Tonelli M, Garcia-Garcia G, Jha V, et al. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet. 2015;385:2616–43.
Moore PK, Hsu RK, Liu KD. Management of acute kidney injury: core curriculum 2018. Am J Kidney Dis. 2018;72:136–48.
Yang L, Xing G, Wang L, Wu Y, Li S, Xu G, et al. Acute kidney injury in China: a cross-sectional survey. Lancet. 2015;386:1465–71.
Palevsky PM, Liu KD, Brophy PD, Chawla LS, Parikh CR, Thakar CV, et al. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for acute kidney injury. Am J Kidney Dis. 2013;61:649–72.
Kidney Disease: Improving Global Outcomes CKDWG. KDIGO 2024 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. 2024;105:S117–S314.
Aklilu AM, Menez S, Baker ML, Brown D, Dircksen KK, Dunkley KA, et al. Early, individualized recommendations for hospitalized patients with acute kidney injury: a randomized clinical trial. JAMA. 2024;332:2081–2090.
Li T, Wu B, Li L, Bian A, Ni J, Liu K, et al. Automated electronic alert for the care and outcomes of adults with acute kidney injury: a randomized clinical trial. JAMA Netw Open. 2024;7:e2351710.
Jia P, Ji Q, Zou Z, Zeng Q, Ren T, Chen W, et al. Effect of delayed remote ischemic preconditioning on acute kidney injury and outcomes in patients undergoing cardiac surgery: a randomized clinical trial. Circulation. 2024;150:1366–76.
Landoni G, Monaco F, Ti LK, Baiardo Redaelli M, Bradic N, Comis M, et al. A randomized trial of intravenous amino acids for kidney protection. N Engl J Med. 2024;391:687–98.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25:424–42.
Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–25.
Luo L, Mo G, Huang D. Ferroptosis in hepatic ischemia-reperfusion injury: regulatory mechanisms and new methods for therapy (Review). Mol Med Rep. 2021;23:225.
Wang Y, Zhang M, Bi R, Su Y, Quan F, Lin Y, et al. ACSL4 deficiency confers protection against ferroptosis-mediated acute kidney injury. Redox Biol. 2022;51:102262.
Ni L, Yuan C, Wu X. Targeting ferroptosis in acute kidney injury. Cell Death Dis. 2022;13:182.
Wang X, Miao J, Wang S, Shen R, Zhang S, Tian Y, et al. Single-cell RNA-seq reveals the genesis and heterogeneity of tumor microenvironment in pancreatic undifferentiated carcinoma with osteoclast-like giant-cells. Mol Cancer. 2022;21:133.
Liang L, Yu J, Li J, Li N, Liu J, Xiu L, et al. Integration of scRNA-seq and bulk RNA-seq to analyse the heterogeneity of ovarian cancer immune cells and establish a molecular risk model. Front Oncol. 2021;11:711020.
Deng C, Deng G, Chu H, Chen S, Chen X, Li X, et al. Construction of a hypoxia-immune-related prognostic panel based on integrated single-cell and bulk RNA sequencing analyses in gastric cancer. Front Immunol. 2023;14:1140328.
Korshunov A, Okonechnikov K, Stichel D, Schrimpf D, Delaidelli A, Tonn S, et al. Gene expression profiling of Group 3 medulloblastomas defines a clinically tractable stratification based on KIRREL2 expression. Acta Neuropathol. 2022;144:339–52.
Wang Y, Shen Z, Mo S, Dai L, Song B, Gu W, et al. Construction and validation of a novel ten miRNA-pair based signature for the prognosis of clear cell renal cell carcinoma. Transl Oncol. 2022;25:101519.
Jiang L, Wang S, Ai Z, Shen T, Zhang H, Duan S, et al. Development and external validation of a stability machine learning model to identify wake-up stroke onset time from MRI. Eur Radiol. 2022;32:3661–9.
Guan S, Chen X, Chen Y, Xie W, Liang H, Zhu X, et al. FOXM1 variant contributes to gefitinib resistance via activating Wnt/β-catenin signal pathway in patients with non-small cell lung cancer. Clin Cancer Res. 2022;28:3770–84.
Henderson AR. The bootstrap: a technique for data-driven statistics. Using computer-intensive analyses to explore experimental data. Clin Chim Acta Int J Clin Chem. 2005;359:1–26.
Fitzgerald M, Saville BR, Lewis RJ. Decision curve analysis. JAMA. 2015;313:409–10.
Sui Z, Wu X, Du L, Wang H, Yuan L, Zhang JV, et al. Characterization of the immune cell infiltration landscape in esophageal squamous cell carcinoma. Front Oncol. 2022;12:879326.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7.
Zhang C, Ding J, Xu X, Liu Y, Huang W, Da L, et al. Tumor microenvironment characteristics of pancreatic cancer to determine prognosis and immune-related gene signatures. Front Mol Biosci. 2021;8:645024.
Hou R, Denisenko E, Ong HT, Ramilowski JA, Forrest ARR. Predicting cell-to-cell communication networks using NATMI. Nat Commun. 2020;11:5011.
Vento-Tormo R, Efremova M, Botting RA, Turco MY, Vento-Tormo M, Meyer KB, et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature. 2018;563:347–53.
Wang Y, Shen Z, Mo S, Zhang H, Chen J, Zhu C, et al. Crosstalk among proximal tubular cells, macrophages, and fibroblasts in acute kidney injury: single-cell profiling from the perspective of ferroptosis. Hum Cell. 2024;37:1039–55.
Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun. 2021;12:1088.
Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ, et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science. 2020;367:405–11.
Ionkina AA, Balderrama-Gutierrez G, Ibanez KJ, Phan SHD, Cortez AN, Mortazavi A, et al. Transcriptome analysis of heterogeneity in mouse model of metastatic breast cancer. Breast Cancer Res. 2021;23:93.
Shi M, Wang J, Zhang C. Integration of cancer genomics data for tree-based dimensionality reduction and cancer outcome prediction. Mol Inf. 2020;39:e1900028.
Kuppe C, Ramirez Flores RO, Li Z, Hayat S, Levinson RT, Liao X, et al. Spatial multi-omic map of human myocardial infarction. Nature. 2022;608:766–77.
Cable DM, Murray E, Zou LS, Goeva A, Macosko EZ, Chen F, et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat Biotechnol. 2022;40:517–26.
Elosua-Bayes M, Nieto P, Mereu E, Gut I, Heyn H. SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 2021;49:e50.
Peng L, He X, Peng X, Li Z, Zhang L. STGNNks: identifying cell types in spatial transcriptomics data based on graph neural network, denoising auto-encoder, and k-sums clustering. Comput Biol Med. 2023;166:107440.
Lin Z, Lin X, Sun Y, Lei S, Cai G, Li Z. Melanoma molecular subtyping and scoring model construction based on ligand-receptor pairs. Front Genet. 2023;14:1098202.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559.
Feng S, Xu Y, Dai Z, Yin H, Zhang K, Shen Y. Integrative analysis from multicenter studies identifies a WGCNA-derived cancer-associated fibroblast signature for ovarian cancer. Front Immunol. 2022;13:951582.
Rezaei Z, Ranjbaran J, Safarpour H, Nomiri S, Salmani F, Chamani E, et al. Identification of early diagnostic biomarkers via WGCNA in gastric cancer. Biomed Pharmacother. 2022;145:112477.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523.
Tonnus W, Maremonti F, Gavali S, Schlecht MN, Gembardt F, Belavgeni A. et al. Multiple oestradiol functions inhibit ferroptosis and acute kidney injury. Nature. 2025;645:1011–1019.
Lee D, Nakai A, Miwa Y, Tomita Y, Kunimi H, Chen J, et al. Retinal degeneration induced in a mouse model of ischemia-reperfusion injury and its management by pemafibrate treatment. FASEB J. 2022;36:e22497.
Cheng Y, Qu W, Li J, Jia B, Song Y, Wang L, et al. Ferristatin II, an iron uptake inhibitor, exerts neuroprotection against traumatic brain injury via suppressing ferroptosis. ACS Chem Neurosci. 2022;13:664–75.
Gu Y, Chen J, Zhang H, Shen Z, Liu H, Lv S, et al. Hydrogen sulfide attenuates renal fibrosis by inducing TET-dependent DNA demethylation on Klotho promoter. FASEB J. 2020;34:11474–87.
Chen J, Gu Y, Zhang H, Ning Y, Song N, Hu J, et al. Amelioration of uremic toxin indoxyl sulfate-induced osteoblastic calcification by SET domain containing lysine methyltransferase 7/9 protein. Nephron. 2019;141:287–94.
Taube JM, Akturk G, Angelo M, Engle EL, Gnjatic S, Greenbaum S, et al. The Society for Immunotherapy of Cancer statement on best practices for multiplex immunohistochemistry (IHC) and immunofluorescence (IF) staining and validation. J Immunother Cancer. 2020;8:e000155.
Daiber A, Oelze M, Steven S, Kroller-Schon S, Munzel T. Taking up the cudgels for the traditional reactive oxygen and nitrogen species detection assays and their use in the cardiovascular system. Redox Biol. 2017;12:35–49.
Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. 2011;17:1391–401.
Bernkop-Schnurch AD, Hermann M, Leitner D, Talasz H, Descher HA, Hohloch S. et al. Transferrin receptor-mediated cellular uptake of fluorinated chlorido[N,N’-bis(salicylidene)-1,2-phenylenediamine]iron(III) complexes. ACS Omega. 2024;9:35394–407.
Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int J Cancer. 1980;26:171–6.
Lu CS, Shiau AL, Su BH, Hsu TS, Wang CT, Su YC, et al. Oct4 promotes M2 macrophage polarization through upregulation of macrophage colony-stimulating factor in lung cancer. J Hematol Oncol. 2020;13:62.
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034.
Lazic SE, Clarke-Williams CJ, Munafo MR. What exactly is ‘N’ in cell culture and animal experiments?. PLoS Biol. 2018;16:e2005282.
Festing MF, Altman DG. Guidelines for the design and statistical analysis of experiments using laboratory animals. ILAR J. 2002;43:244–58.
Shi Y, Wang Y, Dong H, Niu K, Zhang W, Feng K, et al. Crosstalk of ferroptosis regulators and tumor immunity in pancreatic adenocarcinoma: novel perspective to mRNA vaccines and personalized immunotherapy. Apoptosis. 2023;28:1423–35.
Feng G, Arima Y, Midorikawa K, Kobayashi H, Oikawa S, Zhao W, et al. Knockdown of TFRC suppressed the progression of nasopharyngeal carcinoma by downregulating the PI3K/Akt/mTOR pathway. Cancer Cell Int. 2023;23:185.
Liu X, Chen C, Han D, Zhou W, Cui Y, Tang X, et al. SLC7A11/GPX4 inactivation-mediated ferroptosis contributes to the pathogenesis of triptolide-induced cardiotoxicity. Oxid Med Cell Longev. 2022;2022:3192607.
Luo T, Gao J, Lin N, Wang J. Effects of two kinds of iron nanoparticles as reactive oxygen species inducer and scavenger on the transcriptomic profiles of two human leukemia cells with different stemness. Nanomaterials. 2020;10:1951.
Jabara HH, Boyden SE, Chou J, Ramesh N, Massaad MJ, Benson H, et al. A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nat Genet. 2016;48:74–8.
Zhu Z, Li J, Song Z, Li T, Li Z, Gong X. Tetramethylpyrazine attenuates renal tubular epithelial cell ferroptosis in contrast-induced nephropathy by inhibiting transferrin receptor and intracellular reactive oxygen species. Clin Sci. 2024;138:235–49.
Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572:402–6.
Li R, Yan X, Xiao C, Wang T, Li X, Hu Z, et al. FTO deficiency in older livers exacerbates ferroptosis during ischaemia/reperfusion injury by upregulating ACSL4 and TFRC. Nat Commun. 2024;15:4760.
Wang D, Liang W, Huo D, Wang H, Wang Y, Cong C, et al. SPY1 inhibits neuronal ferroptosis in amyotrophic lateral sclerosis by reducing lipid peroxidation through regulation of GCH1 and TFR1. Cell Death Differ. 2023;30:369–82.
Shen Z, Lin J, Teng J, Zhuang Y, Zhang H, Wang C, et al. Association of urinary ionomic profiles and acute kidney injury and mortality in patients after cardiac surgery. J Thorac Cardiovasc Surg. 2020;159:918–26.e5.
Zhang D, Meyron-Holtz E, Rouault TA. Renal iron metabolism: transferrin iron delivery and the role of iron regulatory proteins. J Am Soc Nephrol. 2007;18:401–6.
Kormann R, Kavvadas P, Placier S, Vandermeersch S, Dorison A, Dussaule JC, et al. Periostin promotes cell proliferation and macrophage polarization to drive repair after AKI. J Am Soc Nephrol. 2020;31:85–100.
Niculae A, Gherghina ME, Peride I, Tiglis M, Nechita AM, Checherita IA. Pathway from acute kidney injury to chronic kidney disease: molecules involved in renal fibrosis. Int J Mol Sci. 2023;24:14019.
Rabb H, Griffin MD, McKay DB, Swaminathan S, Pickkers P, Rosner MH, et al. Inflammation in AKI: current understanding, key questions, and knowledge gaps. J Am Soc Nephrol. 2016;27:371–9.
Zhang YL, Tang TT, Wang B, Wen Y, Feng Y, Yin Q, et al. Identification of a novel ECM remodeling macrophage subset in AKI to CKD transition by integrative spatial and single-cell analysis. Adv Sci. 2024;11:e2309752.
Gaetano C, Massimo L, Alberto M. Control of iron homeostasis as a key component of macrophage polarization. Haematologica. 2010;95:1801–3.
Walker VJ, Agarwal A. Targeting iron homeostasis in acute kidney injury. Semin Nephrol. 2016;36:62–70.
Soofi A, Li V, Beamish JA, Abdrabh S, Hamad M, Das NK, et al. Renal-specific loss of ferroportin disrupts iron homeostasis and attenuates recovery from acute kidney injury. Am J Physiol Ren Physiol. 2024;326:F178–F88.
Li Z, Zhang C, Liu Y, Wang F, Zhao B, Yang J, et al. Diagnostic and predictive values of ferroptosis-related genes in child sepsis. Front Immunol. 2022;13:881914.
Maremonti F, Meyer C, Linkermann A. Mechanisms and models of kidney tubular necrosis and nephron loss. J Am Soc Nephrol. 2022;33:472–86.
Belavgeni A, Meyer C, Stumpf J, Hugo C, Linkermann A. Ferroptosis and necroptosis in the kidney. Cell Chem Biol. 2020;27:448–62.
Guerrero-Mauvecin J, Villar-Gomez N, Rayego-Mateos S, Ramos AM, Ruiz-Ortega M, Ortiz A, et al. Regulated necrosis role in inflammation and repair in acute kidney injury. Front Immunol. 2023;14:1324996.
Funding
This study was supported by research grants from the Natural Science Foundation of Shanghai (25ZR1402072) and the China National Natural Scientific Foundation (82170752). The funders had no roles in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The experimental platform for this study was supported by the national major science and technology projects of the National Health Commission (2025ZD0547600).
Author information
Authors and Affiliations
Contributions
Yulin Wang provided the original idea of the paper, analyzed the data, and wrote the draft of the manuscript. Cheng Zhu polished the manuscript and guided the analysis. Xiaoyan Zhang and Ziyan Shen reviewed and revised the manuscript. Shiqi Lv, Xinhui Huang, Yue Yang, Jiayi Wang, and Shuangxin Yuan polished the article. Ziyan Shen provided the grant Natural Science Foundation of Shanghai (25ZR1402072). Xiaoyan Zhang provided the grant China National Natural Scientific Foundation (82170752). Xiaoqiang Ding provided the grant National Major Science and Technology Projects of the National Health Commission (2025ZD0547600). Xiaoyan Zhang, Ziyan Shen, and Xiaoqiang Ding were co-investigators and supervisors of the study. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was conducted in accordance with the ethical guidelines for animal research as outlined in the Institutional Animal Care and Use Committee (IACUC) guidelines. Ethical approval was granted by the Animal Ethics Committee of Fudan University (2020.12/No.202011024S).
Consent for publication
Every co-author has reviewed the final version of the manuscript and approved it for publication.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Zhu, C., Lv, S. et al. Spatiotemporal transcriptomic insights into ferroptosis and TFRC-linked immune interactions in ischemia-reperfusion acute kidney injury. Genes Immun (2025). https://doi.org/10.1038/s41435-025-00364-0
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41435-025-00364-0
