
Abstract
Purpose
Primary open-angle glaucoma (POAG) is the leading cause of irreversible blindness worldwide and mutations in known genes can only explain 5–6% of POAG. This study was conducted to identify novel POAG-causing genes and explore the pathogenesis of this disease.
Methods
Exome sequencing was performed in a Han Chinese cohort comprising 398 sporadic cases with POAG and 2010 controls, followed by replication studies by Sanger sequencing. A heterozygous Ramp2 knockout mouse model was generated for in vivo functional study.
Results
Using exome sequencing analysis and replication studies, we identified pathogenic variants in receptor activity-modifying protein 2 (RAMP2) within three genetically diverse populations (Han Chinese, German, and Indian). Six heterozygous RAMP2 pathogenic variants (Glu39Asp, Glu54Lys, Phe103Ser, Asn113Lysfs*10, Glu143Lys, and Ser171Arg) were identified among 16 of 4763 POAG patients, whereas no variants were detected in any exon of RAMP2 in 10,953 control individuals. Mutant RAMP2s aggregated in transfected cells and resulted in damage to the AM-RAMP2/CRLR-cAMP signaling pathway. Ablation of one Ramp2 allele led to cAMP reduction and retinal ganglion cell death in mice.
Conclusion
This study demonstrated that disruption of RAMP2/CRLR-cAMP axis could cause POAG and identified a potential therapeutic intervention for POAG.
Similar content being viewed by others


INTRODUCTION
Glaucoma is a degenerative disease of the optic nerve that is characterized by the death of retinal ganglion cells (RGCs), with an increase in the vertical cup-to-disc ratio and losses in the visual field.1 This condition is a leading cause of irreversible blindness worldwide; indeed, estimates suggest that by 2020 there will be 79.6 million individuals affected with glaucoma.2
Primary open-angle glaucoma (POAG) is the most common type of glaucoma, and is diagnosed among a wide range of ethnic groups, including whites, blacks, and Asians.2 Both genetic and environmental factors play critical roles in the development of POAG. From a genetic perspective, POAG can be caused by mutations in a single gene (monogenic form) or by combined action of genetic and environmental factors (polygenic form). A mutation in MYOC (encoding myocilin),3 OPTN (encoding optineurin),4 WDR36 (encoding WD repeat domain 36),5 TBK1 (encoding TANK-binding kinase 1),6 and ASB10 (encoding ankyrin repeat- and SOCS box-containing 10)7 could result in POAG. The genetic risk factors for POAG have been studied in the past decade using genome-wide association studies (GWAS), which have identified numerous associated genes/loci. These genes/loci include CAV1-CAV2,8,9 TMCO11,10 CDKN2B-AS1,10 SIX1-SIX6,11 ABCA1,9,12,13,14 PMM2,12 FNDC3B,13,15 AFAP1,14 GMDS,14 TGFBR3,16 TXNRD2,17 ATXN2,17 FOXC1,17 and the chromosomal regions include 8q22 and 11p11.2.11,13 However, genetic variants associated with these known genes/loci account for only 5–6% of all patients with POAG,18,19 and the pathogenic mechanisms underpinning their role in the development of POAG remain unclear. Although sporadic occurrences account for at least 28% of all reported cases of POAG,20 the genes responsible for such cases are largely unknown due to several limitations: (1) traditional genetic studies for monogenic forms depend on large pedigrees, (2) genetic variants identified by GWAS using chip arrays generally are not in the coding regions of specific genes and represent genetic risk factors. Recent advances in sequencing techniques and algorithms have made it possible to identify pathogenic variants in the disease-related genes from sporadic cases.
The aims of the present study were to identify genes associated with sporadic POAG and to determine how disruption of their normal biological functions might cause this condition. We performed exome sequencing (ES) analysis using samples collected from 398 unrelated patients with POAG and 2010 unrelated control individuals without POAG, all of whom were Han Chinese ancestry. Then, the pathogenesis of POAG resulting from gene pathogenic variants identified in this study was further investigated.
MATERIALS AND METHODS
Study participants
This study included four cohorts of patients with POAG, characterized by high intraocular pressure (IOP), derived from Han Chinese, German, and Indian populations (Supplementary Table 1). Written informed consent was obtained from all subjects. This project was approved by the Institutional Review Board of ophthalmic clinics at the following centers: Eye and Ear Nose Throat (ENT) Hospital, Fudan University; Hong Kong Eye Hospital at the Chinese University of Hong Kong; Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital; Center for Ophthalmology, University of Tuebingen; Department of Ophthalmology at Universitätsklinikum Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg; and India Aravind Hospital. All participants underwent an extensive ophthalmic examination and were recruited according to standardized criteria (see details in Supplementary Materials), and as described in a previous study.12
Exome sequencing
We sequenced 2408 subjects at the first stage of the project, including 398 POAG cases and 2010 controls, using Illumina Truseq Enrichment System Capture (62 M) and following the manufacturer’s instructions. The data filtering strategy was described in our previous study21 and a data production summary of individuals in this study is shown in Supplementary Table 2.
Sanger sequencing to identify other pathogenic variants in the replication stage
We first validated the two RAMP2 pathogenic variants in three POAG patients by Sanger sequencing and then sequenced more samples to identify other mutant RAMP2s. Sequencing primers for the coding sequences of RAMP2 (NM_005854) were designed using Primer 5.0 (Supplementary Table 3).
Multiple sequence alignment of the human RAMP2 protein was performed along with other RAMP2 proteins across different species to examine the conservation of the residues. The possible damaging effects of the pathogenic variant on the structure and function of RAMP2 were predicted using SIFT, PolyPhen2_HDIV, PolyPhen2_HVAR, LRT, Mutation Taster, Mutation Assessor, M-CAP, Fathmm-MKL, and CADD.
Expression constructs and mutagenesis
Human calcitonin receptor-like receptor (CRLR, NM_00579) and RAMP2 complementary DNA (cDNA) ligated into the SgfI/MluI sites of a mammalian expression plasmid (pCMV6-myc/DDK) were obtained from OriGene. The RAMP2 plasmid was used as a template to generate specific mutations of RAMP2 using the QuikChange II Site-Directed Mutagenesis kit (Stratagene) (Supplementary Table 4).
Cell culture and transfection
COS-7 monkey kidney fibroblast-like cells and 293 T cells were maintained in 60-mm culture dishes (Corning) with Eagle’s Minimal Essential Medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and antibiotics (1% penicillin–streptomycin; Invitrogen). Cells (105/cm2) were transfected with the wild-type (WT) or mutant RAMP2 constructs using Lipofectamine® 3000 Reagent (Invitrogen) in Opti-MEM® I (Invitrogen).
Expression analysis in human ocular tissues
A deceased 55-year-old Han Chinese male graciously donated human ocular tissues. Total RNA from the optical nerve, retina, iris, corneal, and trabecular meshwork was extracted with TRIzol (Invitrogen). Semiquantitative polymerase chain reaction (PCR) was performed for RAMP2. A housekeeping gene (GAPDH; NM_001256799.2) was used as an internal control, with its transcripts amplified in parallel reactions. The sequences of the primers used for semiquantitative PCR are listed in Supplementary Table 5.
Immunofluorescence and western blotting
COS-7 cells (American Tissue Culture Collection, Manassas, VA) and retinal sections were fixed, permeabilized, and followed by immunostaining (see details in Supplementary Materials). Samples were imaged with a confocal microscope (Leica). Mouse retinas were lysed for transferring to a nitrocellulose membrane (Millipore) and followed by antibody incubation (see details in Supplementary Materials). Image Quant LAS4000 mini Luminescent image analyzer was used for image acquisition (GE Healthcare). The comparison of RAMP expression in each group was analyzed by t test.
Generation of Ramp2 knockout mice
The Ramp2 knockout mice were generated using the TALEN technique. To disrupt the Ramp2 gene in a mouse, a pair of TALEN plasmids was constructed to target exon 2 of the mouse Ramp2 gene. A total of 35 offspring was derived by microinjection. After DNA sequencing, one mouse was found to carry a heterozygous frameshift pathogenic variant within exon 2 of the Ramp2 gene. In this mouse, an A residue was missing within TCTCTTCCGGAGTC and a C residue was missing at the position immediately preceding TCTCTTCCGGAGTC. The heterozygous Ramp2 knockout (Ramp2+/−) mice were bred for three generations to dilute potential off-target pathogenic variants before they were used in experiments.
Animal husbandry
All animals were treated according to the guidelines of the Association for Research in Vision and Ophthalmology for the use of animals in research. The Animal Care and Use Committee of the Sichuan Provincial People’s Hospital approved all experimental protocols.
Measurement of intracellular cAMP
Wild-type (WT) and mutant RAMP2s were cotransfected with WT CRLR in 293 T cells (American Tissue Culture Collection, Manassas, VA). Transfectants grown in 24-well culture plates were incubated for 15 minutes at 37 °C with different concentrations of adrenomedullin (AM) in Hanks’ buffer containing 20 mM HEPES, 0.2% BSA, 0.5 mM 3-isobutyl-1-methylxanthine (IBMX; Sigma). The reactions were terminated by the addition of lysis buffer (GE Healthcare), after which the cAMP content was determined with a commercial enzyme immunoassay kit according to the manufacturer’s instructions (GE Healthcare) using a nonacetylation protocol. The difference for cAMP concentration in each group was analyzed by t test.
RGC death analysis and cAMP assay in vivo
The WT and Ramp2+/− mice were killed at three months of age and their eyeballs fixed using 4% paraformaldehyde. Cryosection and immunostaining procedures were performed as described above. The retinal sections were stained using a TUNEL assay kit (Roche). The retinal sections were stained with a rabbit anti-cAMP antibody (1:200, Millipore, 116820-1ST) (see details in Supplementary Materials) and viewed using a confocal microscope (Leica).
RESULTS
Variants in the RAMP2 gene were identified among patients with POAG
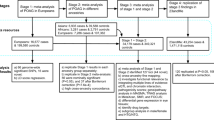
By using the IlluminaTruSeq enrichment system capture and the HiSeq 2000/2500 Sequencer (Supplementary Fig. 1 and 2), an average of 9.97 Gb of raw sequence data was generated with 112× depth of exome target regions for each individual as paired-end 101 base pair reads in the discovery cohort (Table 1 and Supplementary Table 2). In total, samples from 398 Han Chinese patients with POAG and 2010 unrelated Han Chinese control individuals were successfully sequenced and analyzed (Supplementary Fig. 1).
We used both joined call and individual call methods for variant calling. The average summary data output of each sample is shown in Supplementary Table 2. Overall, 677,000 variants were detected in the samples, including 328,000 coding variants, among which 201,000 were not silenced and 143,000 were rare coding variants with a minor allele frequency (MAF) of <0.001. Each participant had an average of 75,000 variants, including 20,000 variants in coding regions, of which there were 9515 nonsynonymous changes and 404 coding indels. No population stratification was found by principal component analysis of their genotypes (Supplementary Fig. 3).
We selected candidate genes that met all the following three standards to be considered as causative of POAG. First, candidate genes represented nonsilent variants only in the 398 POAG cases but not in the 2010 control individuals;22,23 second, candidate genes harbored at least one “disruptive” variant (nonsense, frameshifts, or in splice sites);24 and third, the variant was present in at least two cases of POAG. In all, three genes including receptor activity-modifying protein 2 (RAMP2), serine incorporator 1 (SERINC1), and histone cluster 1 H4 family member D (HIST1H4D) were identified to meet our stringent criteria (Table 1 and Supplementary Table 6). We further validated the three genes in the replication cohorts and found that the variants identified in the ES discovery stage in the SERINC1 and HIST1H4D genes were present in controls in the replication stage, and thus the two genes were excluded. Further replication of the RAMP2 gene showed that the variants identified in the ES discovery stage were present only in the POAG patients and not in the controls in the different cohorts including Han Chinese, German, and Indian populations. Results of the functional study further evidenced RAMP2 as a potential POAG gene. For these variants in the RAMP2 gene, patient K182 had an insertion variant (c.338_339insA, p.Asn113Lysfs*10), and patients K148 and K441 were found to carry a missense variant in this gene (c.117G>C, p.Glu39Asp, Table 1, Fig. 1a, d). The two variants in the three POAG patients were confirmed using Sanger sequencing. All of the RAMP2 variants identified in our ES analysis are shown in Supplementary Table 7.

Chromatograms of the pathogenic variants detected in the RAMP2 gene and multiple sequence alignment of RAMP2 proteins across species. (a) Glu39Asp(c.117G>C), (b) Glu54Lys (c.160G>A), (c) he103Ser (c.308T>C), (d) sn113Lysfs*10 (c.338_339insA), (e) Glu143Lys (c.427G>A), (f) Ser171Arg (c.511A>C). Vertical red arrows denote nucleotide changes. The red boxed amino acid residues indicate that most of them occurred at highly conserved positions among all species analyzed.
Then, we screened variants by direct Sanger sequencing of the whole exomes of RAMP2 in three replication cohorts: a Han Chinese cohort (3194 cases and 7880 controls), a German cohort (721 cases and 653 controls), and an Indian cohort (450 cases and 410 controls) (Supplementary Table 1). Among these participants, no close relatives were included based on calculation of genome-wide identity-by-state and identity-by-descent among all the samples by Plink. In the Han Chinese cohort, the p.Asn113Lysfs*10 and p.Glu39Asp variants, plus a novel heterozygous variant (p.Glu143Lys), were observed in 8 of the 3194 POAG cases (Table 1 and Fig. 1). In the German cohort, the p.Glu143Lys variant, along with two novel heterozygous variants (p.Glu54Lys and p.Ser171Arg), were found in 4 of the 721 POAG cases (Table 1 and Fig. 1). In the Indian cohort, 1 novel variant (p.Phe103Ser) was identified in 1 of the 450 POAG cases (Table 1 and Fig. 1c). No amino acid changes in the RAMP2 protein were detected among the control groups included in either the discovery or the replication cohorts. In total, six variants that affected the RAMP2 protein (p.Glu39Asp, Asn113Lysfs*10, Glu54Lys, Phe103Ser, Ser171Arg, and Glu143Lys) were identified among 16 of the 4763 POAG cases analyzed in the present study (Table 1 and Fig. 1). The main features of the POAG patients with the RAMP2 gene variants are listed in Supplementary Table 8. Of the six RAMP2 variants, two had CADD score of >20 and one was a frameshift mutant, suggesting that the three variants may affect RAMP2 function. The remaining three RAMP2 variants were very rare and were not found in the controls, whilst they did not have a high CADD score. Thus, we speculated that these variants identified in our study may affect RAMP2 function (Supplementary Table 9).
Mutant RAMP2 proteins aggregated in transfected cells and disrupted cAMP signaling pathway
The genetic analysis indicated that variants in RAMP2 were strongly associated with POAG. To examine if the RAMP2 variants found in POAG might potentially affect the protein function, we performed multiple sequence alignments among orthologs and functional predictions for different species of RAMP2 using different prediction tools (Fig. 1 and Supplementary Table 7). This analysis showed that the amino acids corresponding to most of the RAMP2 variants were highly conserved during evolution (Fig. 1).
RAMP2 encodes a single-domain transmembrane protein.25 Five of the variants identified in this study were located within the extracellular region of RAMP2, whereas the sixth variant (Ser171Arg) was located in the cytoplasmic region of this protein (Supplementary Fig. 4). To explore the mechanisms through which the mutant forms of RAMP2 are involved in the pathogenesis of POAG, we first investigated the expression profile of RAMP2 in different human tissues. The semiquantitative PCR results indicated that RAMP2 was ubiquitously expressed in all human tissues, including optic nerve head and retina (Supplementary Fig. 5). We then investigated the intracellular distribution of RAMP2 in the COS-7 cells transfected with the recombinant myc-DDK (Flag)-tagged RAMP2 plasmids. Compared with the diffused distribution of WT RAMP2 in cytoplasm, all six RAMP2 mutants aggregated in the cells (Fig. 2), implying that the variant may disrupt the normal trafficking of RAMP2 and thus have adverse effects on the function of RAMP2.

Mutant RAMP2 proteins formed intracellular aggregates. A lower magnification (myc staining, green) showed the expression of wild-type (WT) RAMP2 (a) and mutant RAMP2s (b–g) in COS-7 cells. (a) WT RAMP2 was diffusely distributed within the cytoplasm; (b–g) mutant RAMP2s formed intracellular aggregates. Each experiment was replicated in triplicate. Nuclei were stained with 4’-6-diamidino-2-phenylindole (DAPI). Scale bar: 10 μm.
Upon binding AM, the RAMP2-CRLR (calcitonin receptor-like receptor) complex activates the cAMP signaling pathway25,26 and leads to accumulation of cAMP in cultured cells.27,28 The presence of cAMP seems to be beneficial toward the survival of retinal ganglion cells (RGCs).29,30 Therefore, we hypothesized that RAMP2 variants could cause POAG, affecting the RAMP2-CRLR complex and thus interrupting the cAMP signaling. To investigate whether the observed RAMP2 variants disrupt the normal biological functions of RAMP2, we cotransfected mutant RAMP2 proteins with CRLR and analyzed intracellular cAMP expression in 293 T cells stimulated with AM. In the cells expressing mutant RAMP2 proteins, the cAMP concentration in the cells expressing mutant RAMP2 Asn113Lysfs*10 and Phe103Ser was significantly lower compared with that in the cells expressing WT protein after AM (10−7 mol/L) treatment (P = 0.001) (Supplementary Fig. 6). Therefore, the mutant RAMP2 proteins indeed disrupt the AM-RAMP2/CRLR-cAMP signaling pathway in vitro.
Ramp2 +/− mice displayed glaucoma features of RGC death
Immunohistochemical analysis also indicated that RAMP2 was strongly expressed within the mouse RGC layer, where it colocalized with the RGC marker Brn3a (Fig. 3a). This is consistent with previous reports,31,32 and suggests that RAMP2 might have an important function in RGC survival. Subsequently, we investigated whether disruption of Ramp2 in mice would mimic the clinical features of glaucoma. We used TALEN technology to generate a Ramp2+/− mouse model (the homozygous Ramp2 knockout [Ramp2−/−] mice displayed embryonic lethality31). As shown by western blotting and immunofluorescence, the expression of RAMP2 protein was decreased in the Ramp2+/− mice compared with WT littermate controls (P = 0.01, Fig. 3b–d). Meanwhile, we found that 74.7% of RGCs were Brn3A-positive in each staining section of the retinas in Ramp2+/− mice, whereas 78.4% of RGCs were Brn3A-positive in the WT mice, indicating loss of RGCs in the Ramp2+/− mice.

RAMP2 expression in mouse retinal layers.(a, b) Immunofluorescence analysis showed that RAMP2 (green) is expressed in the RGC layer and internucleus layer in WT mice and is colocalized with Brn3A (red) in the RGC layer. RAMP2 expression was largely reduced in the retina of Ramp2+/− mice, compared with WT mice. (c, d) Western blotting analysis showed reduction of RAMP2 expression in the retina of Ramp2+/− mice when compared with that in WT (P = 0.01). The asterisk indicates a P < 0.05 by t test. The expression of the housekeeping protein β-actin was shown as a reference; transfected and nontransfected 293 T cells were used for positive and negative controls. Each experiment was replicated in triplicate. Nuclei were stained with 4’-6-diamidino-2-phenylindole (DAPI). RGC retinal ganglion cell, WT wild-type. Scale bar: 20 μm.
Haploinsufficiency has been demonstrated for the Ramp2 gene, with the ablation of one allele leading to embryonic lethality among mice with a C57/BL6 background.31 To avoid this problem, we used the CD-1 genetic background to generate Ramp2+/− mice (see “Materials and Methods”). Because ablation of one Ramp2 allele could affect survival of mice, we hypothesized that the retina of Ramp2+/− mice would display reduced sensitivity in response to stimulation by AM. Our results showed that upon an in vivo treatment of the retina with AM, less cAMP was present in the RGCs of Ramp2+/−, compared with the WT mice (P = 0.01, Fig. 4a, b), suggesting that both alleles of Ramp2 are required for cAMP function in the retina in vivo, which is consistent with the in vitro results.

Lower cAMP levels in the Ramp2+/− retina treated with AM and RGC death were observed in the Ramp2+/−mice. (a,b) the cAMP levels were significantly lower in the RGC layer in the Ramp2+/− retina treated with AM compared with the WT mice (P = 0.01). The asterisk indicates a P < 0.05 by t-test. (c) RGC apoptosis (green) was observed in the Ramp2+/− retina, compared with negative staining in WT retina. The red arrows indicated detection of an apoptosis signal. Each experiment was replicated in triplicate. Nuclei were stained with 4’-6-diamidino-2-phenylindole (DAPI). INL inner nuclear layer, ONL outer nuclear layer, RGC retinal ganglion cell, WT wild-type. Scale bar: 20 μm.
RGC death is a characteristic feature of glaucoma. We thus examined Ramp2+/− mouse retina by TUNEL assay. Six of the ten Ramp2+/− mice tested at three months of age were found to have RGC death, whereas no apoptosis was found in WT controls. AM can regulate normal vascular development, and AM−/− mice exhibit defective vascular development.31,33 Furthermore, the AM-RAMP2 system protects nerve cells from both acute and chronic cerebral ischemia.31,34 AM has been shown to be up-regulated in retina under hypoxic conditions, and may play a protective role during retina ischemia.35 Normal oxygen levels are critical for proper development of mouse retina and RGCs are sensitive to oxygen fluctuation. RAMP2, as an AM receptor, may also protect RGCs from ischemic injury. To address the impact of hypoxic stress on RGCs of Ramp2+/−, the mice were placed under a hypoxic condition with 9% O2 for three months. The death of RGCs (green signal indicated by red arrows) was evident in all the treated Ramp2+/− mice as shown by the TUNEL assay (Fig. 4c). We found that 5.1% of all RGCs were TUNEL-positive in each staining section of the retinas of the Ramp2+/− mice. By contrast, no apoptosis was observed among the WT control mice subjected to the same treatment. Taken together, the data suggest that disruption of one Ramp2 allele in mice could lead to RGC death and display a glaucoma-like phenotype.
DISCUSSION
In this study, RAMP2 was identified as a novel POAG gene by exome sequencing and our results showed that RAMP2 pathogenic variants account for approximately 0.34% of all POAG cases. Among the 16 mutations, it is noted that p.Glu39Asp and p.Glu143Lys each occurred in four and five patients, respectively, while p.Glu54Lys, another missense mutation that also involves glutamic acid, was found in two patients (Table 1). All were due to a glutamic base change in the nucleotide sequence that affected the coding for glutamic acid. The local sequence context could cause mutational bias of genes.36 The respective positions in exons 2 and 4 of the RAMP2 gene are likely susceptible sites of mutational changes and give rise to hotspot mutations. We further investigated alterations in the biological function of the mutated RAMP2 protein that potentially underpin the pathogenesis of sporadic POAG by in vitro and in vivo assays.
The RAMP family comprises a group of single-transmembrane domain proteins, including RAMP1, RAMP2, and RAMP3. RAMP2, ubiquitously expressed in various human tissues, is highly conserved across orthologs and is required to transport CRLR to the plasma membrane.25 In the presence of RAMP2, CRLR functions as an AM receptor that can activate the cAMP signaling pathway. Among the various Ramp knockout mice reported to date, only the Ramp2−/− mouse has an embryonic lethal phenotype with cardiovascular abnormalities that closely resemble AM−/− mice, suggesting that the AM-RAMP2 pathway plays a vital part in the cardiovascular system.31,34
Interaction of CRLR with RAMP2 is critical for AM binding and activation of downstream cAMP signaling pathway. In the present study, mutant RAMP2 proteins aggregated in cytoplasm in the cultured cells, which might lead to affecting its interaction with CRLR and disruption of CRLR-mediated signaling pathway. As shown, a decrease of cAMP was found in the RAMP2 mutant-expressing cells upon stimulation by AM. Apart from its important function in the cardiovascular system, RAMP2 has been demonstrated to function in RGC survival. A recent study indicated an overlapping distribution of CRLR, RAMP2, and AM in the retina, especially in the somata of the inner nuclear and RGC layers among normal mice.32 This finding suggests that AM, CRLR, and RAMP2 colocalize in normal RGCs to exert their usual biological functions. Additionally, a clinical study showed that mean AM levels in aqueous humor in POAG patients were substantially higher compared with control individuals, implying the AM-RAMP2/CRLR-cAMP axis may play an important role in the development of POAG.
Oxygen level is a factor that may affect RGC survival. This could be due to the fact that RGCs are relatively high energy–demanding cells and are more sensitive to metabolism change.35 We observed increased RGC death among Ramp2+/− versus WT mice. This finding suggests that RGCs in individuals carrying RAMP2 pathogenic variants may be more susceptible to ischemia and hypoperfusion. The disruption of one allele of the Ramp2 gene in mice may affect the biological functions of RGCs, thereby leading to increased apoptosis of these cells and the development of POAG. The RGC death might be due to inadequate activation of the RAMP2-cAMP signaling pathway in the RGCs of the mutant mice, as cAMP levels were decreased in the RGC of Ramp2+/− mice after AM treatment. Our results further supported previous studies indicating cAMP might be important for RGC survival.37 We measured the IOP of these Ramp2+/− mice and found that the IOP was not elevated in the Ramp2+/− mice, suggesting that the mutant Ramp2s could cause disease independent of IOP. This could explain in part why some patients carrying these RAMP2 variants displayed normal tension glaucoma (Supplementary Table 8).
Two previous independent studies reported embryonic lethality in mouse when one allele of Ramp2 was ablated in the C57Bl6 background, suggesting that a normal dosage of RAMP2 protein is essential for maintaining normal function of RAMP2. Therefore, deletion of one Ramp2 allele in mouse may affect its function in RGCs and thereby lead to RGC death. Similarly, our results in this study showed that RGC death was found in Ramp2+/− mice and RAMP2 mutants exhibit a dominant genetic pattern in POAG patients. RGC death, as a major clinical feature of POAG, poses an appreciable clinical problem that can lead to irreversible visual impairment. Unfortunately, no effective therapy for POAG currently exists. Our results provide evidence that the pathogenesis of POAG could be associated with cAMP reduction in RGC death, as demonstrated in Ramp2+/– mice. These findings increase the possibility that cAMP could be used therapeutically to prevent the apoptosis of RGCs in POAG. Indeed, forskolin, a potent adenylatecyclase agonist to induce production of cAMP,38 has been shown to protect RGCs from death both in vitro and in vivo.29,30
In conclusion, this study demonstrated that genetic disruption of RAMP2 might disturb the AM-RAMP2/CRLR-cAMP axis and prevent RGCs from responding to AM stimulation, resulting in RGC death. This finding supports that cAMP plays an important role in RGC health, thus it could offer a potential target for the treatment of POAG.
References
Quigley HA. Number of people with glaucoma worldwide. Br J Ophthalmol. 1996;80:389–393.
Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90:262–267.
Stone EM, et al. Identification of a gene that causes primary open angle glaucoma. Science. 1997;275:668–670.
Rezaie T, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295:1077–1079.
Footz TK, et al. Glaucoma-associated WDR36 variants encode functional defects in a yeast model system. Hum Mol Genet. 2009;18:1276–1287.
Fingert JH, et al. Copy number variations on chromosome 12q14 in patients with normal tension glaucoma. Hum Mol Genet. 2011;20:2482–2494.
Pasutto F, et al. Variants in ASB10 are associated with open-angle glaucoma. Hum Mol Genet. 2012;21:1336–1349.
Thorleifsson G, et al. Common variants near CAV1 and CAV2 are associated with primary open-angle glaucoma. Nat Genet. 2010;42:906–909.
Huang L, et al. Genome-wide analysis identified 17 new loci influencing intraocular pressure in Chinese population. Sci China Life Sci. 2019;62:153–164.
Burdon KP, et al. Genome-wide association study identifies susceptibility loci for open angle glaucoma at TMCO1 and CDKN2B-AS1. Nat Genet. 2011;43:574–578.
Wiggs JL, et al. Common variants at 9p21 and 8q22 are associated with increased susceptibility to optic nerve degeneration in glaucoma. PLoS Genet. 2012;8:e1002654.
Chen Y, et al. Common variants near ABCA1 and in PMM2 are associated with primary open-angle glaucoma. Nat Genet. 2014;46:1115–1119.
Hysi PG, et al. Genome-wide analysis of multi-ancestry cohorts identifies new loci influencing intraocular pressure and susceptibility to glaucoma. Nat Genet. 2014;46:1126–1130.
Gharahkhani P, et al. Common variants near ABCA1, AFAP1 and GMDS confer risk of primary open-angle glaucoma. Nat Genet. 2014;46:1120–1125.
Shiga, Y et al. Genome-wide association study identifies seven novel susceptibility loci for primary open-angle glaucoma. Hum Mol Genet 2018;27:1486–1496.
Li Z, et al. A common variant near TGFBR3 is associated with primary open angle glaucoma. Hum Mol Genet. 2015;24:3880–3892.
Bailey JN, et al. Genome-wide association analysis identifies TXNRD2, ATXN2 and FOXC1 as susceptibility loci for primary open-angle glaucoma. Nat Genet. 2016;48:189–194.
Fingert JH. Primary open-angle glaucoma genes. Eye (Lond). 2011;25:587–595.
Allingham RR, Liu Y, Rhee DJ. The genetics of primary open-angle glaucoma: a review. Exp Eye Res. 2009;88:837–844.
Gong G, et al. Inherited, familial and sporadic primary open-angle glaucoma. J Natl Med Assoc. 2007;99:559–563.
Gong B, et al. Exome sequencing identified a recessive RDH12 mutation in a family with severe early-onset retinitis pigmentosa. J Ophthalmol. 2015;2015:942740.
Walter K, et al. The UK10K project identifies rare variants in health and disease. Nature. 2015;526:82–90.
Tang R, et al. Candidate genes and functional noncoding variants identified in a canine model of obsessive-compulsive disorder. Genome Biol. 2015;15:R25.
Novarino G, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014;343:506–511.
McLatchie LM, et al. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature. 1998;393:333–339.
Kusano S, et al. Structural basis for extracellular interactions between calcitonin receptor-like receptor and receptor activity-modifying protein 2 for adrenomedullin-specific binding. Protein Sci. 2012;21:199–210.
Sato A, Canny BJ, Autelitano DJ. Adrenomedullin stimulates cAMP accumulation and inhibits atrial natriuretic peptide gene expression in cardiomyocytes. Biochem Biophys Res Commun. 1997;230:311–314.
Nishikimi T, et al. Effect of adrenomedullin on cAMP and cGMP levels in rat cardiac myocytes and nonmyocytes. Eur J Pharmacol. 1998;353:337–344.
Russo R, et al. Intravitreal injection of forskolin, homotaurine, and L-carnosine affords neuroprotection to retinal ganglion cells following retinal ischemic injury. Mol Vis. 2015;21:718–729.
Santos RC, Araujo EG. Cyclic AMP increases the survival of ganglion cells in mixed retinal cell cultures in the absence of exogenous neurotrophic molecules, an effect that involves cholinergic activity. Braz J Med Biol Res. 2001;34:1585–1593.
Ichikawa-Shindo Y, et al. The GPCR modulator protein RAMP2 is essential for angiogenesis and vascular integrity. J Clin Invest. 2008;118:29–39.
Blom J, Giove TJ, Pong WW, et al. Evidence for a functional adrenomedullin signaling pathway in the mouse retina. Mol Vis. 2012;18:1339–1353.
Shindo T, et al. Vascular abnormalities and elevated blood pressure in mice lacking adrenomedullin gene. Circulation. 2001;104:1964–1971.
Koyama T, et al. Vascular endothelial adrenomedullin-RAMP2 system is essential for vascular integrity and organ homeostasis. Circulation. 2013;127:842–853.
Fujita Y, et al. Involvement of adrenomedullin induced by hypoxia in angiogenesis in human renal cell carcinoma. Int J Urol. 2002;9:285–295.
Schroeder JW, Hirst WG, Szewczyk GA, Simmons LA. The effect of local sequence context on mutational bias of genes encoded on the leading and lagging strands. Curr Biol. 2016;26:692–697.
Shaw PX, et al. Soluble adenylyl cyclase is required for retinal ganglion cell and photoreceptor differentiation. Invest Ophthalmol Vis Sci. 2016;57:5083–5092.
Wagh VD, Patil PN, Surana SJ, Wagh KV. Forskolin: upcoming antiglaucoma molecule. J Postgrad Med. 2012;58:199–202.
Acknowledgements
We thank all the patients with POAG and their families for participating in this study. This research project was supported by National Key R&D Program of China (2016YFC20160905200), the National Natural Science Foundation of China (81430008 [Z.Y.], 81790643 [Z.Y.], 81842021 [Z.Y.], 81670853 [B.G.], 81300802 [L.H.], 81670895 [L.H.], 81170882 [Y.S], 81790641 [X.S.], and 81371030 [H.Z.]); the grant from Department of Science and Technology of Sichuan Province, China (2014SZ0169 [Z.Y.], 2015SZ0052 [Z.Y.], 2019JDJQ0031 [B.G.], 2014FZ0124 [D.Y.L.], 2015JQO057 [L.H.], 2017JQ0024 [L.H.], 2016HH0072 [L.H.], and 2014JZ0004 [Y.S]); foundation for Technology & Science & Technology Bureau of Chengdu (2018-YF05-00348-SN [B.G.]); and research grants from the General Research Fund, Hong Kong (14100917 [C.-P.P.]); We thank Olga Zwenger for special assistance with sequencing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Gong, B., Zhang, H., Huang, L. et al. Mutant RAMP2 causes primary open-angle glaucoma via the CRLR-cAMP axis. Genet Med 21, 2345–2354 (2019). https://doi.org/10.1038/s41436-019-0507-0
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41436-019-0507-0
Keywords
This article is cited by
-
Exploring the role of VAV2 rare variants in primary open-angle glaucoma: genetic insights and pathophysiological mechanisms
Science China Life Sciences (2025)
-
Cholesterol homeostasis regulated by ABCA1 is critical for retinal ganglion cell survival
Science China Life Sciences (2023)
-
Identification of the potential biological target molecules related to primary open-angle glaucoma
BMC Ophthalmology (2022)
-
A Comparison of Genomic Advances in Exfoliation Syndrome and Primary Open-Angle Glaucoma
Current Ophthalmology Reports (2021)
-
Identification of lncRNA–miRNA–mRNA regulatory network associated with primary open angle glaucoma
BMC Ophthalmology (2020)
