
Abstract
B chromosomes (Bs) are supernumerary genomic elements found in many eukaryotes, yet their full sequence composition, functional potential, and regulatory impact on the host genome remain unclear. Here, we present a chromosome-level genome assembly of the cichlid fish Astatotilapia latifasciata, integrating PacBio long reads, Illumina short reads, and Hi-C chromatin contact maps to resolve both A and B chromosomes. The 0.93 Gb assembly (N50 = 36.2 Mb) includes a 34 Mb B chromosome containing 789 predicted protein-coding genes and a markedly higher density of transposable elements (TEs), especially long terminal repeats (LTR) retrotransposons. Transcriptome profiling revealed that B-linked genes are predominantly transcriptionally repressed relative to their A chromosome paralogs. Hi-C–based chromatin modeling uncovered distinct 3D structural configurations associated with the B chromosome, including fewer topologically associating domains (TADs), reduced loop formation, and altered compartmentalization. These changes are linked to long-range chromatin interactions and genomic rearrangements, suggesting that the B chromosome reshapes the nuclear architecture of the host genome. Our study proposes a potential regulatory role of Bs in genome and provides a genomic resource for investigating chromosome evolution in cichlids.
Similar content being viewed by others


Introduction
B chromosomes (Bs) are supernumerary, non-essential chromosomes present in a wide range of plant and animal taxa. Typically, heterochromatic and enriched in repetitive sequences, Bs are often considered selfish genetic elements that persist through non-Mendelian inheritance mechanisms such as meiotic drive (Camacho et al. 2000; Jones and Houben 2003; Houben 2017). Despite being dispensable for viability and development of the organism, the recent emergence role of Bs have been implicated in sex determination, and potential influence on A chromosomes gene expression, yet their precise evolutionary origin and functional impact remain unresolved (Camacho 2005; Jones and Houben 2003; Valente et al. 2017; Makunin et al. 2016). Current evolutionary models suggest that Bs originate from A chromosomes or from interspecific hybridization events, followed by amplification of repeats, segmental duplications, and structural rearrangements (Jones and Houben 2003; Houben et al. 2014; Valente et al. 2014; Ahmad and Martins 2019). In cichlids, the maintenance of B chromosomes has been linked to drive mechanisms, such as female-biased transmission and escape from elimination during gametogenesis (Yoshida et al. 2011; Clark et al. 2017; Clark and Kocher 2019). Although Bs were traditionally viewed as inert genomic elements, increasing evidence points toward functional roles, including the expression of B-specific transcripts and modulation of host gene networks (Banaei-Moghaddam et al. 2013; Trifonov et al. 2013; Valente et al. 2014; Houben et al. 2019; Oliveira et al. 2024).
The cichlid fish A. latifasciata provides an emerging model to investigate B chromosome biology. This species harbors one or two morphologically identical Bs that coexist with 22 pairs of standard A chromosomes. Previous cytogenetic studies have identified ribosomal DNA clusters (Poletto et al. 2010), satellite DNA, and duplicated gene fragments on the B chromosome (Fantinatti et al. 2011; Carmello et al. 2017). Moreover, transcriptional comparative analysis between individuals with and without Bs (1B vs 0B) suggest potential regulatory impacts on gene expression (Ramos et al. 2017; Nascimento-Oliveira et al. 2021; Oliveira et al. 2022). While B chromosomes have been shown to harbor transcriptionally active sequences, the lack of a fully assembled B chromosome and the absence of Hi-C chromatin conformation data have limited efforts to define their regulatory landscape and spatial interactions with the A chromosome complement. Additionally, the high abundance of repetitive DNA on B chromosomes presents further limitations in characterizing their regulatory features and interactions.
Hi-C sequencing has transformed our understanding of genome architecture, enabling the identification of topologically associating domains (TADs) and chromatin loops, at kilobase to megabase resolution (Lieberman-Aiden et al. 2009; Rao et al. 2015). TADs delineate functional domains that constrain enhancer-promoter interactions, and their disruption can impact transcriptional regulation. To date, no study has explored whether B chromosomes alter 3D genome topology or whether their structural features affect host nuclear organization. In this study, we generated the first chromosome-scale reference genome of A. latifasciata with a nearly assembled B chromosome, using deep coverage long-read sequencing and Hi-C chromatin contact maps. We assembled a 930 Mb genome, including a 34 Mb B chromosome, and performed transcriptomic, repeatome, and chromatin architecture analyses. Our results reveal that the B chromosome is transcriptionally less active, enriched in LTRs, and physically reorganizes chromatin interactions, supported from Hi-C comparative analysis. These findings provide new insights into the functional relevance of B and its evolutionary integration into host genome.
Materials and methods
Long-read PacBio and Hi-C genome sequencing
We obtained 20 adults Astatotilapia latifasciata individuals from a local aquarium store in Botucatu, São Paulo, Brazil. Karyotyping was performed at the Integrative Genomics Laboratory (UNESP–São Paulo State University) to determine the presence of B chromosomes. The presence or absence of B chromosomes was further confirmed by genotyping using primers developed by Fantinatti and Martins (2016). High molecular weight genomic DNA was extracted from the muscle tissue of a single adult male carrying one B chromosome (1B) using the Qiagen MagAtrract HMW DNA kit (cat no. 67563), with gDNA extraction for B chromosomes genotyping was performed using phenol-chloroform method following Green and Sambrook (2012).
Sequencing libraries were prepared and sequenced on the PacBio Sequel II platform using SMRT Cell v2.0. For Hi-C sequencing, muscle tissues from one individual with a B chromosome (1B) and one without (0B) were used. Cross-linked chromatin was prepared and libraries constructed by Phase Genomics (Seattle, WA, USA). DNA was digested using the Sau3AI restriction enzyme, and proximity ligation was performed using biotinylated nucleotides to generate chimeric molecules reflecting spatial proximity. These were processed into paired-end sequencing libraries. Chimeric contacts represent loci that are physically proximal in vivo but not necessarily genomically adjacent. All experimental protocols were approved by the Ethics Committee of the Institute of Biosciences, UNESP (Protocol No. 769-2015).
Chromosome-scale genome assembly
Long-read data from the 1B male sample were independently assembled using two tools: Falcon v0.3.0 (Chin et al. 2016) and wtdbg2 v2.5 (Ruan and Li 2020). Customized parameters were applied for read correction, trimming, overlap detection, and de novo contig/scaffold construction. The assembly generated using wtdbg2 was selected for downstream analysis due to its superior continuity (Table S3).
We polished the assembly using short-read Illumina data previously generated for the same individual (Jehangir et al. 2019) with Pilon v1.22 (Walker et al. 2014). Redundant sequences were removed using Redundans (Pryszcz and Gabaldón 2016). The curated and polished draft genome was then scaffolded to the chromosome scale using Hi-C data with Juicer (Durand et al. 2016). Final assembly quality was assessed using gVolante (Nishimura et al. 2017), BUSCO (Simão et al. 2015), and LTR Assembly Index (LAI) (Ou et al. 2018).
Genome annotation
Repeat annotation was performed using the Extensive de-novo TE Annotator (EDTA; Ou et al. 2019) and RepeatMasker version 4 using metazoa and crossmatch engine (Smit et al. 2013). Protein-coding genes were predicted using BRAKER (Hoff et al. 2016; Brůna et al. 2021), incorporating transcriptomic evidence mapped by GMAP (Wu and Watanabe 2005) from A. latifasciata RNA-seq data of 0B and 1B transcriptomes. Predicted genes were also compared to the Metriaclima zebra genome via BLASTp to identify putative orthologs. BRAKER was run in ab initio mode using GeneMark (Lomsadze et al. 2014) and AUGUSTUS (Stanke et al. 2004). Functional annotation of predicted coding genes on the B chromosome was conducted using BLAST2GO (Conesa et al. 2005), incorporating multiple protein databases for homolog detection.
B chromosome identification
To identify B chromosome–linked sequences, we used male and female short-read Illumina data from 0B, 1B, and 2B individuals. Reads were aligned to the Asta_v3 assembly using Bowtie2 (Langmead and Salzberg 2012) with the “--very-sensitive --end-to-end” alignment mode, and per base coverage was calculated with Bedtools (Quinlan and Hall 2010). Log2-normalized coverage ratios were compared between 1B and 0B samples to detect B-enriched scaffolds, following the previous adapted strategy by Valente et al. (2014). Multicoverage reads coverage were computed with Bedtools to identify scaffolds enriched in 1B samples.
To further improve B chromosome identification, we analyzed Hi-C contact profiles. Hi-C data from 0B and 1B individuals were aligned to the Asta_v3 genome using HiC-Pro (Servant et al. 2015). Unplaced scaffolds showing significantly higher Hi-C interaction frequencies with the B super-scaffold were identified using Wilcoxon rank-sum tests and incorporated into the B chromosome assembly. Validation of B chromosome sequences was performed by aligning previously published B-specific markers validated by FISH mapping experiments (Valente et al. 2014; Ramos et al. 2017) using BLASTn. Significant hits with high sequence identity (>95%) were considered evidence of correct B chromosome assembly.
3D genome organization and TAD analysis
To explore chromatin structure, Hi-C reads from 0B and 1B samples were aligned to the Asta_v3 genome using Juicer (Durand et al. 2016). Differences in 3D conformation were visualized in Juicebox. The in-silico genome was modeled using Chrom3D (Paulsen et al. 2018). The resulting structures were visualized in Chimera (Pettersen et al. 2004). Hi-C matrices were generated and corrected using HiCExplorer v3.0 (Ramírez et al. 2018). Hi-C reads were mapped upstream of HiCExplorer using Bowtie2 in local mode with “--local --reorder” to preserve read order for downstream matrix construction. Contact matrices were generated with hicBuildMatrix using a bin size of 10 kb (“--binSize 10000”), the default minimum mapping-quality threshold of 15, and an input buffer size of 400,000 (“--inputBufferSize 400000”). Matrix quality-control statistics were collected using the “—Qcfolder” option. TADs were identified using “hicFindTADs” at 20 kb and 50 kb resolutions with FDR correction. TAD boundaries were visualized using HiCPlotter (Akdemir and Chin 2015). Chromatin loops were detected using “hicDetectLoops” from HiCExplorer, applying a negative binomial model and Wilcoxon test for loop significance. We compared TAD sizes, loop sizes, and duplication breakpoints using Bedtools and regioneR (Gel et al. 2016). Duplication events were inferred using Syri and intersected with TAD boundaries to examine disruption patterns. We also compared TAD profiles of A. latifasciata with those of other cichlids (O. niloticus and A. citrinellus), analyzing Hi-C data with the same pipeline and parameters. Hi-C sequencing data of representative cichlid species including O. niloticus and A. citrinellus were retrieved from NCBI SRA (Accession: SRR11744829 and SRR12137490) respectively. The Hi-C reads of both representative cichlid genomes data were aligned with their respective assembled chromosome level genome (Assembly NCBI accessions: GCA_013435755.1; GCA_922820385.1). Same set of tools and parameters for Hi-C alignments, matrices correction and prediction of TADs and loops were applied as described for our A. latifasciata TADs analysis. We then counted total number of TADs in each species and compared with TADs of 0B and 1B genomes.
Transcriptome and gene expression analysis
RNA-seq data from brain, muscle, and gonad of 0B and 1B individuals were aligned to the genome using STAR v2.75 (Dobin et al. 2013). BAM files were sorted and indexed with Samtools (Li et al. 2009). Read counts for B chromosome–localized genes were obtained using Bedtools “multicov”. Log-transformed normalized read counts were analyzed, and ANOVA test was performed to assess significant differences of mean gene expression across chromosomes. Differential expression of B-linked genes was assessed using edgeR v3.36.0 (Robinson et al. 2010), applying CPM > 1, logFC = 1.5, and FDR = 0.05 thresholds. Genes with significant expression changes were identified across tissues.
Comparative genomics and gene family analysis
Orthogroup clustering was performed across 11 cichlids and 5 outgroup species using OrthoFinder2 (Emms and Kelly 2019), incorporating DIAMOND (Buchfink et al. 2015) and clustered with OrthoMCL (Li et al. 2003). Phylogenetic trees were reconstructed following multiple sequence alignment with MAFFT (Katoh et al. 2002), alignment trimming using TrimAl (Capella-Gutiérrez et al. 2009), and tree inference with RAxML (Stamatakis 2014) and IQ-TREE (Minh et al., 2022). Divergence times were estimated with MCMCtree (Yang, 2007), and convergence assessed in Tracer (Rambaut et al. 2018). Gene family expansions and contractions were assessed with CAFE (De Bie et al. 2006). Enrichment analysis of expanded/contracted families was performed in BLAST2GO using Fisher’s exact test (FDR < 0.05).
Structural variants (SVs) between the 0B and 1B genomes were identified using whole-genome alignments generated by NUCmer (Kurtz et al. 2004) and analyzed with SyRI (Goel et al. 2019), a tool designed to detect a broad spectrum of structural rearrangements including inversions, translocations, duplications, and unaligned or divergent regions. Since SyRI requires both the reference and query genomes to be in chromosome-scale assembly format, we first anchored our previously published draft 0B genome (Jehangir et al. 2019) to chromosome scale. To achieve this, we employed a hybrid strategy using Hi-C scaffolding and reference-guided alignment. Specifically, the 0B draft assembly was aligned to the 1B reference genome using RagTag (Alonge et al. 2022), which enables misassembly correction and reference-guided scaffolding based on homology with a high-quality reference. The RagTag-anchored scaffolds were further improved using Hi-C data to resolve ambiguities and finalize chromosome-level continuity (Table S3). This chromosome-scale 0B genome was then compared with the 1B assembly using SyRI, enabling the detection of genome-wide structural differences, including 1B genome-specific rearrangements potentially associated with B chromosome presence.
Results
Chromosome-scale assembly of the A. latifasciata genome
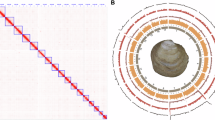
We generated a chromosome-level reference genome for A. latifasciata, integrating PacBio long reads (~118 Gb), Illumina short reads (~51 Gb), and Hi-C chromatin interaction data (~58 Gb) to produce a contiguous and accurate assembly of 930 Mb (Table S1). K-mer-based genome size estimation using Illumina data (1B male) predicted a genome of 902 Mb with low heterozygosity (0.49%) (Table S2). PacBio scaffolds integrated with Hi-C mapping were anchored into 22 A chromosomes and a single B chromosome, covering 94% (872 Mb) of the genome and yielding an improved scaffold N50 of 36.2 Mb (Table S3). Assembled chromosomes were ordered based on size (Chr1 to Chr22) and were assigned to cichlid linkage groups (LGs) based on their sequence homology to Oreochromis niloticus and Amphilophus citrinellus (Figs. 1a; S1a and Table S4). The chromosome scale assembly substantially filled the gaps in the previous draft genome (Fig. S1b). Hi-C contact maps at high resolution (10–50 kb) revealed strong intra-chromosomal interactions in A chromosome, whereas Chr B exhibited comparatively sparse interaction density (Fig. 1a), a pattern that may reflect both its distinct chromatin organization and reduced unique mappability associated with its repeat-rich sequence composition. 3D genome in silico modeling showed Chr B localized toward the nuclear periphery with reduced chromatin compaction (smaller bead count), whereas A chromosomes (e.g., Chr 1) were centralized, consistent with active euchromatic regions (Fig. 1b). To benchmark assembly quality, we employed multiple metrics. K-mer spectra plots (Fig. S2a) revealed accurate haplotype resolution with minimal artifactual duplication. BUSCO analysis (Actinopterygii dataset, n = 3640) recovered 96% complete orthologs (Fig. S2b), indicating high genome completeness. The LTR Assembly Index (LAI) averaged 8.5, with a maximum of 19.17, denoting high quality assembly for repetitive elements (Fig. S2c and Table S5). The genome assembly was performed in a stepwise manner, progressing from initial draft versions (Asta_v1 and Asta_v2) to the final chromosome-scale assembly, Asta_v3. Compared to earlier drafts, Asta_v3 recovered approximately 150 Mb of previously missing or fragmented regions and improved assembly contiguity over 300-fold, increasing the scaffold N50 from 25 Kb to 36.4 Mb (Figs. S2d, e and S1b; Table S3).

a Hi-C contact matrix (500 kb bins) showing chromatin interactions within and between chromosomes of A. latifasciata, including chromosomes 1–22 and the assembled “B_superscaffold” representing the B chromosome. Strong intrachromosomal interactions are evident along the diagonal, particularly between euchromatic arms of A chromosomes. Color intensity reflects the frequency of contact between pairs of 100-kb loci. White rows and columns represent bins with no valid interaction data. A magnified view (250 kb resolution) comparing the chromatin architecture of LG8 (Chr 1), LG3 (Chr 2), and Chr B is shown on the right. b Chrom3D structural model of the diploid genome (including the B chromosome). Each chromosome occupies a distinct territory within the nucleus (represented by different colors), with chromosomes modeled as continuous chains of beads, each bead corresponding to a predicted topologically associating domain (TAD). A separate 3D view highlights LG8, LG3, and Chr B in blue, green, and red, respectively.
Genome annotation and repeatomics analysis revealed a low gene density and expansion of LTRs on B
Repeatome analysis revealed that approximately 39% of the A. latifasciata genome consists of repetitive DNA, an 11% increase over previous estimates based on short-read assemblies (Coan and Martins 2018; Jehangir et al. 2019). This improvement reflects the enhanced resolution of complex repeats achieved through long-read sequencing. Most repeats were TEs, with DNA transposons comprising the largest fraction (23.1%). Among retroelements (13.4%), LINEs were the most abundant (~6%), followed by LTR retrotransposons, with Gypsy elements accounting for 5.3% of the genome (Table S6). Insertion time analysis of LTRs indicated a relatively recent burst, beginning ~300 Kya and peaking at a median age of 269 Kya (Fig. S3 and Table S7). The genome also showed evidence of recent insertions from Gypsy, Copia, and unclassified LTR families (Table S8). Overall, LTR elements were distributed at a mean density of 10.45 per 10 Kb across the genome. Strikingly, Chr B exhibited a much higher LTR density, averaging 47.6 per 10 Kb, highlighting extensive LTR expansion on the B chromosome (Fig. 2 and Table S9).

The circos plot displays multiple layers of genome annotation and structure across LGs (assembled chromosomes), including the B_superscaffold. Outermost ring: Topologically associating domains (TADs) shown as black tiles. Second ring: Assembled chromosomes scaled by size (Mb), with A chromosomes in gray and the B_superscaffold in red. Third and fourth rings: Density of LTR retrotransposons and DNA transposons, respectively, calculated per 100 kb window. Red peaks denote regions with transposon enrichment (>30 TEs per 100 kb), notably concentrated on the B chromosome. Fifth ring: Gene density visualized as a heatmap—blue (30–60 genes/100 kb), red (>60 genes/100 kb) indicating gene-rich regions, and light yellow (<7 genes/100 kb) marking gene-poor regions, prominently observed on the B chromosome. Sixth ring: GC content displayed as a scatterplot (100 kb bins). Gray dots represent GC% between 40 and 45%, green dots <40%, and red dots >45%, with high GC variability particularly notable on the B chromosome. Innermost links: Syntenic duplications inferred from SyRI analysis. Red lines highlight duplicated regions involving the B chromosome.
Protein-coding genes were annotated on a repeat-masked version of Asta_v3 using an integrated approach that combined ab initio prediction with the A. latifasciata transcriptomes and proteomes from reference cichlid species. This resulted in 35,755 predicted protein-coding genes, with 33,146 (92.7%) assigned to the chromosomes, while remaining as unplaced scaffolds. Gene density was skewed toward smaller chromosomes, with LG19 (Chr 21) and LG8/LG9 (Chr 22) being the most gene-rich (Fig. 2 and Table S8). The overall genome-wide mean gene density was 36.8 genes per 100 Kb, peaking on LG3/LG4 (Chr 17) (85.7 genes/100 Kb) and lowest on LG2 (Chr 12) (22.4 genes/100 Kb) (Table S8). Chr B had the lowest gene density at 7.9 genesx per 100 Kb and carried 789 protein-coding genes in total (Fig. 2 and Table S8). Additionally, GC content profiling in 10 Kb windows revealed highly variable patterns across Chr B, in contrast to the more uniform profiles observed on A chromosomes (Fig. 2).
An integrative genomic approach identified and validated B chromosome sequences
The B chromosome was identified by comparing normalized read coverage between 1B (genome with B) and 0B (genome without B) individuals; scaffolds with high 1B/0B ratios were classified as B-linked. These were assembled into a 34.3 Mb long super-scaffold and validated using Hi-C interactions and known B-specific markers. While our chromosome-scale assembly resolved 22 A chromosomes, the identity of B chromosome scaffolds among unplaced sequences remained uncertain. To isolate and validate B-linked sequences, we implemented a three-tiered integrative strategy:
First, we performed a log₂ coverage ratio analysis using Illumina short-read data from 1B and 0B individuals, to recover scaffolds with significantly higher representation of mapped reads in 1B genome (Fig. 3a). Second, we applied a Hi-C–based comparative interaction frequency analysis between 1B and 0B contact maps. This identified a total of 283 unplaced scaffolds linked to B chromosome (Table S9) with significantly higher Hi-C interaction density in the 1B genome (Fig. 3b, c), consistent with their integration into a distinct chromosomal territory (Fig. 1). Furthermore, global coverage map of 0B and 1B genomic reads revealed B chromosome specific coverage peaks (highlighted as gray regions in Fig. 3d). Together, these two strategies allow us to recover a total of 34.4 Mb of B super-scaffold, placing the unplaced regions of the 1B assembly. Additionally, we mapped the remaining unplaced scaffolds against all assembled chromosomes, including the 22 A chromosomes and the B super-scaffold. This analysis revealed that most unplaced scaffolds shared homology with the B super-scaffold (Fig. S4), suggesting they are predominantly composed of repetitive sequences, a hallmark feature of the B chromosome. In the third approach, we validated B super-scaffold, by aligning previously characterized B-linked molecular markers, ihhb and BncDNA, previously experimentally confirmed and known for their localization on the B chromosome in A. latifasciata (Ramos et al. 2017; Jehangir et al. 2019). We observed the highest number of alignment hits for both ihhb (1284 hits) and BncDNA (505 hits) on the assembled B super-scaffold (Fig. 3e). Other B-associated genes were also mapped to B super-scaffold, confirming the chr B assembly (Tables S10, S11). Of particular interest, LG3 (Chr 2) displayed several genomic mappings, and interaction features mirroring Chr B, including low gene density, high TE content, elevated mapping of B markers, and enrichment in unplaced scaffold alignments, suggesting a potential ancestral or structural relationship between LG3 and the B chromosome (Fig. 3f, g).

a, b Boxplots showing the distribution of log₂(2B/0B) Illumina read ratios and log₂(1B/0B) Hi-C read ratios across LG8, LG3, and Chr B, respectively, indicating B chromosome-specific enrichment. c Distribution of Hi-C reads mapped to the genome assemblies of 0B and 1B individuals. d Comparative coverage plot of Illumina reads mapped to the assembled B chromosome, with 0B (blue peaks) and 2B (red peaks) samples. Each peak represents read depth across 10 kb windows. Regions with substantial differences in coverage—lower in 0B and higher in 2B—are highlighted in gray. The X-axis indicates genomic position, and the Y-axis represents read depth. e Line plot comparing Hi-C interaction frequencies along the B chromosome between 0B (blue line) and 1B (red line) genomes, demonstrating increased interaction density in 1B. f, g Bar charts displaying the distribution of BLAST hits for B chromosome marker genes BncDNA and ihhb, confirming their enrichment on the B chromosome.
TAD and loop architecture are altered in the 1B genome
To investigate the impact of B chromosome presence on chromatin organization in host genome, we analyzed Hi-C data from both 0B and 1B individuals, mapping them to the chromosome-scale 1B genome assembly. We assessed chromatin contact profiles, TADs and loops, to compare 3D chromatin organization between the two genomes. Analysis of contact probability decay as a function of genomic distance revealed that both 0B and 1B genomes follow the expected power-law scaling, consistent with broadly conserved chromatin folding principles. Relative to 0B, the 1B genome showed reduced short-range (<1 Mb) and increased long-range (>1 Mb) interactions, suggesting a shift in contact distribution. This pattern was further reflected by differences in the short-to-long contact ratio across 50-kb bins and was also apparent at the level of individual chromosomes (Figs. 4a, b and S5). Because the B chromosome is highly enriched in repetitive DNA, however, we note that reduced unique mappability of Hi-C reads in repeat-rich regions may contribute in part to the observed decrease in short-range contacts.

a Boxplot comparison of short-range versus long-range chromatin contact frequencies in 0B and 1B genomes. b Hi-C contact decay curve showing contact frequency as a function of genomic distance at 50 kb resolution. The 0B genome (blue line) is enriched for short-range contacts (<1 Mb), while the 1B genome (red line) displays more long-range interactions (>1 Mb). c Correlation between the number of TADs (y-axis) and chromosome size (x-axis) in both 0B and 1B genomes. The 1B genome features longer but fewer TADs compared to the 0B genome. d Distribution of TAD sizes in 0B and 1B genomes. e Number of duplicated regions predicted by SyRI between the two genome types, and f correlation between TAD size (x-axis) and the number of duplications (y-axis). Each point represents a TAD, colored by genome (blue = 0B, red = 1B) and scaled by TAD size. g High-resolution in situ Hi-C heatmaps of LG8 (25 Mb region) for 0B (top panel) and 1B (bottom panel) genomes at 50 kb resolution. TADs appear as “mountains,” with fewer, larger TADs in 1B. TAD separation scores (see Methods) are shown as tracks, with vertical lines indicating boundary positions. h Comparative Hi-C contact maps at 40 kb resolution showing loop structures (red dots) in 0B and 1B genomes. i Bar chart summarizing the total number of TADs and chromatin loops identified in each genome.
Using our defined overlap criteria, 170 TADs were shared between the 0B and 1B genomes. The 1B genome contained fewer but larger TADs than the 0B genome (mean size 2.1 Mb versus 0.7 Mb; Fig. 4c, d). We also detected a modest positive correlation between chromosome size and TAD number (R = 0.53, P < 0.001), indicating that larger chromosomes generally harbor more TADs (Fig. 4c). In addition, the 1B genome showed a shift toward broader domains with reduced interaction density and a narrower, less negative insulation score range (Fig. 4d), consistent with weaker or less sharply defined boundaries. Given that TAD calling and inferred boundary strength may be affected by contact-map resolution, overlap stringency, and reduced unique mappability in repeat-rich regions, independent validation will be necessary to confirm whether these patterns reflect reorganization of domain architecture in the 1B genome. We next examined whether apparent TAD reorganization in the 1B genome was associated with structural duplications. Using SyRI (Goel et al. 2019), we identified 9659 duplications in the 1B genome, and these regions overlapped inferred TAD boundaries significantly more often than in the 0B genome (Fig. 4e, f). These findings are consistent with an association between duplicated regions and local changes in domain organization, although the reduced unique mappability of duplicated sequences may also influence boundary detection in these regions. To further investigate the structural TADs organization, we examined TAD separation scores and found clear differences in domain insulation strength between 0B and 1B, with 1B exhibiting a narrower and less negative insulation score range (–1.7 to 1.0) compared to 0B (–1.9 to 1.6), indicative of weaker TAD boundaries (Fig. 4g). We identified 207 chromatin loops in 0B and only 67 in 1B, indicating a reduced number of detectable loop interactions in 1B. (Fig. 4h, i). Additionally, comparison with Hi-C data from other cichlid species revealed a higher number of long-range interchromosomal interactions in 1B than in 0B or other genomes (Fig. S6 and Table S12). These observations suggest that the B chromosome contributes to chromatin reorganization in the host genome. These chromatin changes are also marked by highly rearranged 1B genome, with a total of 1885 inversions (INV), 2278 translocations (INVTR), and other SVs variations (Fig. S7). Comparison of B- and B+ genomes further identified that 1B genome has higher number of duplications as compared to 0B suggesting the B chromosome sequences might have duplicated at large scale during its evolution (Table S13). We also recognized multiple intra-chromosomal INVTRs on LG8 (Chr 1), a large intra-chromosomal INV on LG3, as well as inter-chromosomal INVs between different chromosomes (Fig. S7). Nevertheless, these rearrangement patterns are related to A chromosomes, and may in part reflect intraspecific variation, and more completely resolved telomere-to-telomere assembly comparisons will be important to clarify whether they are linked to chromatin reorganization.
B chromosome genes exhibits transcriptional repression
To evaluate the transcriptional expression of genes located on the B chromosome, we analyzed RNA-seq expression data from brain, muscle, and gonadal tissues of 1B and 0B individuals (both male and female). On average, genes located on Chr B exhibited markedly lower expression (mean logCPM = 1.84) compared to those on A chromosomes (mean logCPM > 4.58) across all tissues (Fig. 5 and Table S14). When comparing expression levels across representative chromosomes (LG8, LG3, and Chr B), we observed consistently lower expression level (p < 0.05) for Chr B genes, with LG3 also showing lower activity than LG8 (Fig. 5a–f).

a–f Boxplots showing log₂-transformed RNA-seq read counts for genes located on LG8, LG3 (representative A chromosomes), and Chr B across various tissues from 1B individuals. In all cases, genes on the B chromosome exhibit significantly lower expression levels (p < 0.05) compared to A chromosomes. Similar patterns were observed for the remaining autosomes (not shown). g Volcano plot of differentially expressed genes (DEGs) located on the B chromosome between 0B and 1B brain samples. The x-axis shows log₂ fold change (FC), and the y-axis shows –log₁₀(adjusted p-value). Genes significantly downregulated in 1B (padj < 0.001, log₂ FC < –2) are highlighted in dark red. Additional DEG results for gonad and muscle tissues are provided in the Supplementary Data. h Lollipop plot illustrating enriched functional categories of genes annotated on the B chromosome. Dot size corresponds to fold enrichment, and color intensity represents –log₁₀(FDR) values, providing a functional overview of the transcriptionally active regions on Chr B.
To further assess the regulatory impact of B-linked genes across different tissues, we performed differential expression analysis between 0B and 1B individuals using the edgeR package (Robinson et al. 2010). Consistent with the global repression of B chromosome genes, only a small number of differentially expressed genes (DEGs) were identified in muscle and gonadal tissues (Fig. S8 and Table S15). This supports the interpretation that the B chromosome is largely transcriptionally inactive. If Chr B were broadly transcribed, we would expect widespread expression differences between 0B and 1B transcriptomes. Notably, a distinct pattern emerged in brain tissue, where several genes were significantly downregulated in 1B individuals (Fig. 5g and Table S16). This suggests that B chromosome gene expression may be cell type–specific, with functions related to hemostasis, spermatogenesis, skin pigmentation, defense response to hatching behavior and others. These findings suggest the selective transcriptional repression of B-linked genes and highlight tissue-specific regulatory effects associated with the presence of the B chromosome (Oliveira et al. 2024).
To explore the global functional repertoire of Chr B genes, we annotated all 789 protein-coding genes using Blast2GO, involving BLASTx homology search, GO term mapping, and annotation. Of these, 649 genes showed homology to known proteins, while 140 (17.7%) had no hits in current databases, indicating potential novel or B-specific genes. GO mapping assigned functional terms to 115 genes (19.2%), primarily sourced from the UniProtKB database. The top-hit species distribution showed Oreochromis niloticus as the closest homolog for most annotated genes. In the molecular function category, glycogen phosphorylase activity had the highest representation (88%), followed by RNA–DNA hybrid ribonuclease activity (Table S17). In the biological process category, GO enrichment highlighted functions related to DNA transposition (16%), DNA integration, and metabolic and recombination-related processes (Fig. 5h and Table S18). Together, these findings indicate that the B chromosome in A. latifasciata harbors largely transcriptionally silenced genes. However, the presence of a subset of active and potentially functional genes, some of which are potential novel, suggests that the B chromosome may behave as regulatory influence within the host genome.
Comparative genomics reveals evolutionary dynamics and B chromosome–linked gene family expansion
To contextualize the evolutionary history of A. latifasciata and assess the potential evolutionary adaptation signatures, we performed comparative phylogenomics and gene family evolution analyses across 16 teleost genomes, including 11 cichlids and 5 non-cichlid outgroups. A species tree constructed using 384 single-copy orthologs confirmed the monophyly of A. latifasciata, Pundamilia nyererei, Metriaclima zebra, and Astatotilapia calliptera. A. latifasciata and P. nyererei formed the most closely related pair, with a divergence time estimated at ~4.83 Mya (Figs. 6a and S9). The Victoria and Malawi lake lineages diverged ~6.14 Mya, while African and American cichlids split ~65.6 Mya (Fig. 6a).

a Time-calibrated phylogenetic tree of 11 representative cichlid species, with teleost outgroups. Gene family expansions (blue) and contractions (red), as identified by CAFE analysis, are indicated along branches and nodes. Numbers marked with asterisks denote predicted gene duplications inferred from gene clustering at each ancestral node. Divergence times are indicated with shaded boxes in units of million years ago (Ma). MRCA: most recent common ancestor. b UpSet plot showing the number of unique and shared gene families across species groups. A. latifasciata is highlighted in red. Bar plots showing Gene Ontology (GO) enrichment of expanded (c) and contracted (d) gene families. The x-axis represents fold enrichment, while the y-axis lists GO terms, color-coded by –log₁₀(FDR) values. e Genome-wide collinearity among three cichlid species assemblies. Syntenic blocks highlight chromosomal rearrangements, with green and red regions on LG8 and LG3 of A. latifasciata representing fusion like events. LG23 is highlighted in purple and carries amh, homologous to the sex-determining LG23 in other cichlid species.
Orthogroup analysis clustered 97.1% of all genes into 30,304 gene families, including 4443 species-specific orthogroups. A. latifasciata exhibited the highest number of unique gene families (n = 565), followed by O. niloticus (n = 359), suggesting substantial lineage-specific diversification (Fig. 6b and Tables S18–S19). A total of 29,679 genes from A. latifasciata were assigned to 16,666 gene families. Among these, 7395 gene families were conserved and shared across all analyzed species, reflecting a core set of orthologous genes.
To model gene family dynamics, we used a stochastic birth–death process (see Materials and Methods) to infer lineage-specific gene expansions and contractions. In A. latifasciata, 3820 gene families were contracted, while 408 expanded indicating a net gene family loss relative to related lineages such as Lake Victoria and Malawi cichlids (Fig. 6a). In contrast, O. niloticus showed substantial gene family expansion (n = 6099) and minimal contraction (n = 734). At the teleost most recent common ancestor (MRCA), we identified 1074 duplicated gene families.
Gene ontology enrichment of significantly expanded families (FDR < 0.05) in A. latifasciata revealed functional enrichment for genes involved in ion homeostasis, oxygen transport, and chromatin assembly (Fig. 6c and Table S20). These categories suggest possible roles in osmoregulatory adaptation and epigenetic remodeling. Conversely, contracted gene families were enriched in functions related to vesicle transport, catabolism, and developmental regulation (Fig. 6d and Table S21).
Remarkably, 16 of the rapidly expanded gene families were localized to the B chromosome (Table S22). Many of these genes lacked homology to reference databases, indicating possible novel B-linked genes. Others, such as slc24a5, smg1, rgs2, atp5f1e, and histone1-like genes, exhibited partial homology and were enriched in functions related to morphogenesis, chromatin organization, and DNA conformation (Fig. S10 and Table S23). These chromatin and DNA related enriched functions further support the link between B chromosome–specific gene expansion and the large-scale 3D chromatin remodeling observed in 1B individuals (Fig. 4).
To explore chromosome evolution and structural rearrangements in cichlids genome, we performed interspecies synteny analyses using genome assemblies of A. citrinellus and O. niloticus. We observed high chromosomal synteny (>83%) between A. latifasciata and these reference genomes. Notably, LG3 of A. latifasciata appeared to result from ancestral fusion events involving multiple linkage groups (LGs) of A. citrinellus, while LG8 likely derived from a fusion of LGs present in O. niloticus (Fig. 6e). Chr 3 of A. latifasciata was syntenic with LG23 in both species, a region harboring the sex-determining gene amh (Table S24). Further comparison identified conserved scaffolds and ancestral rearrangement signatures, including chromosomal inversions and fusions, shared with M. zebra and A. aureus (Fig. S11). These syntenic relationships underscore the dynamic nature of cichlid genome evolution, shaped by lineage-specific fusions and duplications events.
Discussion
This study presents the chromosome-level genome assembly of an important cichlid species being extensively studies as model for chromosome biology and evolution, providing new insights into the structural, regulatory, and evolutionary roles of supernumerary chromosomes. Using a hybrid sequencing approach combining PacBio long reads, Hi-C chromatin conformation capture, and Illumina short reads, we assembled a 930 Mb reference genome for A. latifasciata, which includes 22 A chromosomes and a 34.3 Mb B chromosome. This high-contiguity assembly resolved ~150 Mb of previously fragmented or missing regions, marking a significant improvement over earlier draft genomes (Jehangir et al. 2019) and enabling in-depth exploration of the B chromosome’s sequence content and biological impact. Inter-chromosomal homology analysis further revealed that many B-linked sequences had substantial homology with LG3 (assembly ID Chr 2). This supports a model in which the A. latifasciata B chromosome originated primarily from an LG3-derived fragment, followed by sequence accretion from other A chromosomes, rearrangement, divergence and extensive TE accumulation (Camacho 2005; Valente et al. 2014). This LG3 association is noteworthy in light of other cichlid systems: in Lake Victoria cichlids, B chromosomes have been proposed to derive from a chromosome corresponding to tilapia LG3 and to influence sex determination, whereas in oreochromines the giant LG3 sex chromosome has been suggested to involve fusion with a B chromosome (Conte et al. 2021). Taken together, these findings raise the possibility that LG3-related sequence has been recurrently co-opted during the evolution of supernumerary and sex-linked chromosomes in cichlids, while the contrasting transmission dynamics reported for Lake Malawi B chromosomes and A. latifasciata suggest that B chromosome evolution in cichlids has proceeded through lineage-specific trajectories rather than through a single conserved evolutionary trajectory (Clark et al. 2017; Cardoso et al. 2022). B chromosomes from Lake Malawi mbuna cichlids also show a repeat-rich, gene-bearing and mosaic sequence composition, suggesting that independently evolved cichlid B chromosomes may converge on broadly similar genomic architectures even if their detailed sequence content, transmission dynamics and evolutionary origin differ (Clark et al. 2018).
Our results support the view that B chromosomes are largely heterochromatic and enriched in repetitive sequences, particularly LTR retrotransposons, consistent with previous reports across diverse taxa (Jones and Rees 1982; Camacho 2005; Coan and Martins 2018; Ramos et al. 2017; Ahmad et al. 2020). Although the B chromosome encodes 789 protein-coding genes, it displays markedly lower gene density and transcriptional activity compared to A chromosomes (mean logCPM = 1.84 vs. >4.58), suggesting widespread transcriptional repression. This likely reflects dosage regulation or epigenetic silencing mechanisms that limit the impact of B-linked sequences on host genome function. Notably, differential expression analysis revealed that B-linked genes were significantly expressed only in brain tissue, indicating that this repression is not uniform and may be relaxed in a tissue-specific manner. These findings highlight the potential for context-dependent regulatory activity of B chromosomes.
Hi-C analysis revealed that the presence of the B chromosome is associated with large-scale reorganization of 3D genome structure, particularly at the level of TADs. The 1B genome exhibited fewer but significantly larger TADs compared to the 0B genome, indicative of weakened domain insulation and altered chromatin compartmentalization. This shift in TAD organization was accompanied by an increase in long-range chromatin interactions and a decrease in short-range contacts, as quantified by SVL contact ratios. These results suggest that the B chromosome alters the folding landscape of the host genome, potentially through disruption of preexisting domain boundaries. Notably, TADs in the 1B genome frequently overlapped with duplicated genomic regions, consistent with models in which structural variation drives TAD fusion or boundary erosion (Franke et al. 2016; Bonev and Cavalli 2016; Szabo et al. 2019). Such architectural changes may have functional consequences by reshaping regulatory domains and altering the spatial organization of the genome. Moreover, the identification of species related expanded gene families on Chr B particularly those related to chromatin assembly, and DNA conformation, hints for B chromosome linked role in chromatin architecture modulation
From an evolutionary perspective, our orthogroup and gene family comparative analysis revealed a net loss of gene families in A. latifasciata, but also lineage-specific expansion of 408 families, including 16 localized on the B chromosome. These expanded gene families were enriched in homeostatic and oxygen transport functions, suggesting potential roles in species related environmental adaptation. Importantly, many of the B-linked expanded genes lacked homologs in current databases, indicating a reservoir of novel or rapidly evolving sequences. These results echo findings in other organisms where B chromosomes have been proposed for their potential role in genome innovation and adaptation (Miao et al. 1991; Ahmad et al. 2020; Johnson Pokorná and Reifová 2021; Liu et al. 2025).
While these findings collectively support the hypothesis that B chromosomes are not merely passive passengers but potentially dynamic contributors to genome architecture and evolution, our study has limitations. Despite the high contiguity of our assembly, there is cytological evidence that the B chromosome of A. latifasciata has a similar size to Chr 1 (Poletto et al. 2010; Fantinatti et al. 2011; Cardoso et al. 2022), which indicates that the B chromosome was not fully resolved due to its extreme repeat content, ampliconic regions, and structural complexity. Additional long-read technologies such as Oxford Nanopore ultra-long reads or targeted optical mapping may be required to resolve remaining gaps or collapsed duplications within B-linked regions. Similarly, although Hi-C analysis revealed notable altered chromatin landscape in the 1B genome, these findings are based on single-sample datasets and require further validation. Recent advances in long-read chromatin conformation profiling, particularly methods that combine chromosome conformation capture (3C) with nanopore sequencing such as Pore-C, offer new opportunities to resolve complex genome architecture (Deshpande et al. 2022). Future studies incorporating biological replicates, cell-type–specific Hi-C, or single-cell 3D genome profiling will be essential to validate and refine these observations, providing a more nuanced understanding of the chromatin effects associated with B chromosome presence.
In conclusion, this study provides a detailed genomic characterization of B chromosome of the important model species, integrating structural, transcriptomic, and 3D architectural dimensions. The high-quality reference genome of A. latifasciata offers a valuable resource for future investigations into B chromosome function, chromatin regulation, and genome evolution in cichlids and beyond. Our findings open new avenues for investigating how non-essential, supernumerary chromosomes influence host genome architecture, by mediating structural rearrangements and potentially shaping aspects of genome function. Taken together, our study highlights the complex and dynamic aspects of B chromosomes although non-essential yet influential genomic elements, as potential contributor to shape chromatin reorganization and carrying extra genetic content, raising the possibility that their persistence reflects either functional co-option by the host genome or selfish evolutionary survival.
Data availability
Sequencing data have been deposited to NCBI’s SRA database under the project accession number PRJNA1283115 with SRA accession IDs SRR34268038, SRR34268037 and SRR34268036. The A. latifasciata genome assembly with B chromosome is available under PRJNA1268020https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_051989935.1/. The repeat annotations, gene annotations, and supporting datasets are available on Zenodo (https://doi.org/10.5281/zenodo.15522222).
References
Ahmad SF, Martins C (2019) The modern view of B chromosomes under the impact of high scale omics analyses. Cells 8:156. https://doi.org/10.3390/cells8020156.
Ahmad SF, Jehangir M, Cardoso AL, Wolf IR, Margarido VP, Cabral-de-Mello DC et al. (2020) B chromosomes of multiple species have intense evolutionary dynamics and accumulated genes related to important biological processes. BMC Genom 21: 1. https://doi.org/10.1186/s12864-020-07072-1.
Akdemir KC, Chin L (2015) HiCPlotter integrates genomic data with interaction matrices. Genome Biol 16: 198. https://doi.org/10.1186/s13059-015-0767-1.
Alonge M, Lebeigle L, Kirsche M, Jenike K, Ou S, Aganezov S et al. (2022) Automated assembly scaffolding using RagTag elevates a new tomato system for high throughput genome editing. Genome Biol 23:258. https://doi.org/10.1186/s13059-022-02823-7.
Banaei-Moghaddam AM, Meier K, Karimi-Ashtiyani R, Houben A (2013) Formation and expression of pseudogenes on the B chromosome of rye. Plant Cell 25:2536–2544. https://doi.org/10.1105/tpc.113.111856.
De Bie T, Cristianini N, Demuth JP, Hahn MW (2006) CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22:1269–1271. https://doi.org/10.1093/bioinformatics/btl097.
Bonev B, Cavalli G (2016) Organization and function of the 3D genome. Nat Rev Genet 17:661–678. https://doi.org/10.1038/nrg.2016.112.
Brůna T, Hoff KJ, Lomsadze A, Stanke M, Borodovsky M (2021) BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom Bioinform 3:1–11. https://doi.org/10.1093/nargab/lqaa108.
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60. https://doi.org/10.1038/nmeth.3176.
Camacho JPM (2005) B chromosomes in the eukaryote genome. Cytogenet Genome Res 106:153–160.
Camacho JPM, Sharbel TF, Beukeboom LW (2000) B-chromosome evolution. Philos Trans R Soc B Biol Sci 355:163–178. https://doi.org/10.1098/rstb.2000.0556.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. https://doi.org/10.1093/bioinformatics/btp348.
Cardoso AL, Venturelli NB, da Cruz I, Patroni FMS, Moraes D, Oliveira RA et al. (2022) Meiotic behavior, transmission and active genes of B chromosomes in the cichlid Astatotilapia latifasciata: new clues about nature, evolution and maintenance of accessory elements. Mol Genet Genom 297:1151–1167. https://doi.org/10.1007/s00438-022-01911-4.
Carmello BO, Coan RLB, Cardoso AL, Ramos E, Fantinatti BEA, Marques DF et al. (2017) The hnRNP Q-like gene is retroinserted into the B chromosomes of the cichlid fish Astatotilapia latifasciata. Chromosome Res 25:277–290. https://doi.org/10.1007/s10577-017-9561-0.
Chin CS, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A et al. (2016) Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13:1050–1054. https://doi.org/10.1038/nmeth.4035.
Clark FE, Kocher TD (2019) Changing sex for selfish gain: B chromosomes of Lake Malawi cichlid fish. Sci Rep 9:20213. https://doi.org/10.1038/s41598-019-55774-8.
Clark FE, Conte MA, Kocher TD (2018) Genomic characterization of a B chromosome in Lake Malawi cichlid fishes. Genes 9:610. https://doi.org/10.3390/genes9120610.
Clark FE, Conte MA, Ferreira-Bravo IA, Poletto AB, Martins C, Kocher TD (2017) Dynamic sequence evolution of a sex-associated B chromosome in Lake Malawi cichlid fish. J Hered 108:53–62. https://doi.org/10.1093/jhered/esw059.
Coan RLB, Martins C (2018) Landscape of transposable elements focusing on the B chromosome of the cichlid fish Astatotilapia latifasciata. Genes 9:602. https://doi.org/10.3390/genes9060269.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676. https://doi.org/10.1093/bioinformatics/bti610.
Conte MA, Gammerdinger WJ, Bartie KL, Penman DJ, Kocher TD (2021) Origin of a giant sex chromosome. Mol Biol Evol 38:1554–1569. https://doi.org/10.1093/molbev/msaa319.
Deshpande AS, Ulahannan N, Pendleton M, Dai X, Ly L, Behr JM et al. (2022) Identifying synergistic high-order 3D chromatin conformations from genome-scale nanopore concatemer sequencing. Nat Biotechnol 40:1488–1499. https://doi.org/10.1038/s41587-022-01289-z.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. https://doi.org/10.1093/bioinformatics/bts635.
Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES et al. (2016) Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst 3:99–101. https://doi.org/10.1016/j.cels.2015.07.012.
Emms DM, Kelly S (2019) OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20:238. https://doi.org/10.1186/s13059-019-1832-y.
Fantinatti BEA, Martins C (2016) Development of chromosomal markers based on next-generation sequencing: the B chromosome of the cichlid fish Astatotilapia latifasciata as a model. BMC Genet 17:112
Fantinatti BEA, Mazzuchelli J, Valente GT, Cabral-de-Mello DC, Martins C (2011) Genomic content and new insights on the origin of the B chromosome of the cichlid fish Astatotilapia latifasciata. Genetica 139:1273–1282. https://doi.org/10.1007/s10709-012-9629-x.
Franke M, Ibrahim DM, Andrey G, Schwarzer W, Heinrich V, Schöpflin R et al. (2016) Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538:265–269. https://doi.org/10.1038/nature19800.
Gel B, Díez-Villanueva A, Serra E, Buschbeck M, Peinado MA, Malinverni R (2016) RegioneR: an R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics 32:289–291. https://doi.org/10.1093/bioinformatics/btv562.
Goel M, Sun H, Jiao W-B, Schneeberger K (2019) SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol 20:277. https://doi.org/10.1186/s13059-019-1911-0.
Green MR, Sambrook J (2012) Molecular cloning: a laboratory manual, 4th edn. Cold Spring Harbor Laboratory Press, New York
Hoff KJ, Lange S, Lomsadze A, Borodovsky M, Stanke M (2016) BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32:767–769. https://doi.org/10.1093/bioinformatics/btv661.
Houben A (2017) B chromosomes—a matter of chromosome drive. Front Plant Sci 8:210. https://doi.org/10.3389/fpls.2017.00210.
Houben A, Banaei-Moghaddam AM, Klemme S, Timmis JN (2014) Evolution and biology of supernumerary B chromosomes. Cell Mol Life Sci 71:467–478. https://doi.org/10.1007/s00018-013-1437-7.
Houben A, Jones N, Martins C, Trifonov V (2019) Evolution, Composition and Regulation of Supernumerary B Chromosomes. Genes 10:161.
Jehangir M, Ahmad SF, Cardoso AL, Ramos E, Valente GT, Martins C (2019) De novo genome assembly of the cichlid fish Astatotilapia latifasciata reveals a higher level of genomic polymorphism and genes related to B chromosomes. Chromosoma 128:81–96. https://doi.org/10.1007/s00412-019-00707-7.
Johnson Pokorná M, Reifová R (2021) Evolution of B chromosomes: from dispensable parasitic chromosomes to essential genomic players. Front Genet 12: 727570. https://doi.org/10.3389/fgene.2021.727570.
Jones, RN, Rees, H, 1982. B chromosomes. Academic Press, London.
Jones RN, Houben A (2003) B chromosomes in plants: escapees from the A chromosome genome? Trends Plant Sci 8:417–423. https://doi.org/10.1016/S1360-1385(03)00187-0.
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. https://doi.org/10.1093/nar/gkf436.
Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C et al. (2004) Versatile and open software for comparing large genomes. Genome Biol 5: R12. https://doi.org/10.1186/gb-2004-5-2-r12.
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. https://doi.org/10.1038/nmeth.1923.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. (2009) The sequence alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. https://doi.org/10.1093/bioinformatics/btp352.
Li L, Stoeckert JrCJ, Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189. https://doi.org/10.1101/gr.1224503.
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A et al. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326:289–293. https://doi.org/10.1126/science.1181369.
Liu Q, Liu Y, Yi C, Gao Z, Zhang Z, Zhu C et al. (2025) Genome assembly of the maize B chromosome provides insight into its epigenetic characteristics and effects on the host genome. Genome Biol 26:47. https://doi.org/10.1186/s13059-025-03517-6.
Lomsadze A, Burns PD, Borodovsky M (2014) Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res 42:e119. https://doi.org/10.1093/nar/gku557.
Makunin AI, Kichigin IG, Larkin DM, O’Brien PCM, Ferguson-Smith MA, Yang F et al. (2016) Contrasting origin of B chromosomes in two cervids (Siberian roe deer and grey brocket deer) unravelled by chromosome-specific DNA sequencing. BMC Genoms 17:618. https://doi.org/10.1186/s12864-016-2933-6.
Miao VP, Covert SF, VanEtten HD (1991) A fungal gene for antibiotic resistance on a dispensable (“B”) chromosome. Science 254:1773–1776. https://doi.org/10.1126/science.1763326.
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A et al. (2020) IQ-TREE 2: New models and efficient methods for phylogenomic inference in the genomic era. Mol Biol Evol 37:1530–1534.
Nishimura O, Hara Y, Kuraku S (2017) GVolante for standardizing completeness assessment of genome and transcriptome assemblies. Bioinformatics 33:3635–3637. https://doi.org/10.1093/bioinformatics/btx445.
Oliveira JIN, Cabral-de-Mello DC, Valente GT, Martins C (2024) Transcribing the enigma: the B chromosome as a territory of uncharted RNAs. Genetics 227:1. https://doi.org/10.1093/genetics/iyae026.
Oliveira JIN, Cardoso AL, Wolf IR, Oliveira RA, Martins C (2022) First characterization of PIWI-interacting RNA clusters in a cichlid fish with a B chromosome. BMC Biol 20:204. https://doi.org/10.1186/s12915-022-01403-2.
Oliveira JIN, Fantinatti BEA, Wolf IR, Cardoso AL, Ramos E, Rieder N et al. (2021) Differential expression of miRNAs in the presence of B chromosome in the cichlid fish Astatotilapia latifasciata. BMC Genom 22:1. https://doi.org/10.1186/s12864-021-07651-w.
Ou S, Chen J, Jiang N (2018) Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res 46:e126. https://doi.org/10.1093/nar/gky730.
Ou S, Su W, Liao Y, Chougule K, Agda JRA, Hellinga AJ et al. (2019) Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol 20:275. https://doi.org/10.1186/s13059-019-1905-y.
Paulsen J, Liyakat Ali TM, Nekrasov M, Delbarre E, Baudement MO, Kurscheid S et al. (2018) Chrom3D: three-dimensional genome modeling from Hi-C and nuclear lamin-genome contacts. Genome Biol 19:21. https://doi.org/10.1186/s13059-018-1393-1.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC et al. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. https://doi.org/10.1002/jcc.20084.
Poletto AB, Ferreira IA, Martins C (2010) The B chromosomes of the African cichlid fish Haplochromis obliquidens harbour 18S rRNA gene copies. BMC Genet 11:1. https://doi.org/10.1186/1471-2156-11-1.
Pryszcz LP, Gabaldón T (2016) Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res 44:e113. https://doi.org/10.1093/nar/gkw294.
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. https://doi.org/10.1093/bioinformatics/btq033.
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904. https://doi.org/10.1093/sysbio/syy032.
Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J et al. (2018) High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun 9:189. https://doi.org/10.1038/s41467-017-02525-w.
Ramos É, Cardoso AL, Brown J, Marques DF, Fantinatti BEA, Cabral-de-Mello DC et al. (2017) The repetitive DNA element BncDNA, enriched in the B chromosome of the cichlid fish Astatotilapia latifasciata, transcribes a potentially noncoding RNA. Chromosoma 126:313–323. https://doi.org/10.1007/s00412-016-0601-x.
Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT et al. (2015) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 162:687–688. https://doi.org/10.1016/j.cell.2015.07.024.
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616.
Ruan J, Li H (2020) Fast and accurate long-read assembly with wtdbg2. Nat Methods 17:155–158. https://doi.org/10.1038/s41592-019-0669-3.
Servant N, Varoquaux N, Lajoie BR, Viara E, Chen C-J, Vert J-P et al. (2015) HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16:259. https://doi.org/10.1186/s13059-015-0831-x.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. https://doi.org/10.1093/bioinformatics/btv351.
Smit AFA, Hubley R, Green P (2013) RepeatMasker Open-4.0. Available at: http://www.repeatmasker.org
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313.
Stanke M, Steinkamp R, Waack S, Morgenstern B (2004) AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res 32:W309–W312. https://doi.org/10.1093/nar/gkh379.
Szabo Q, Bantignies F, Cavalli G (2019) Principles of genome folding into topologically associating domains. Sci Adv 5:eaaw1668. https://doi.org/10.1126/sciadv.aaw1668.
Trifonov VA, Dementyeva PV, Larkin DM, O’Brien PCM, Perelman PL, Yang F et al. (2013) Transcription of a protein-coding gene on B chromosomes of the Siberian roe deer (Capreolus pygargus). BMC Biol 11:90. https://doi.org/10.1186/1741-7007-11-90.
Valente GT, Conte MA, Fantinatti BEA, Cabral-de-Mello DC, Carvalho RF, Vicari MR et al. (2014) Origin and evolution of B chromosomes in the cichlid fish Astatotilapia latifasciata based on integrated genomic analyses. Mol Biol Evol 31:2061–2072. https://doi.org/10.1093/molbev/msu148.
Valente GT, Nakajima RT, Fantinatti BEA, Marques DF, Almeida RO, Simões RP et al. (2017) B chromosomes: from cytogenetics to systems biology. Chromosoma 126:73–81. https://doi.org/10.1007/s00412-016-0613-6.
Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S et al. (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e112963. https://doi.org/10.1371/journal.pone.0112963.
Wu TD, Watanabe CK (2005) GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21:1859–1875. https://doi.org/10.1093/bioinformatics/bti310.
Yang Z (2007) PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591. https://doi.org/10.1093/molbev/msm088.
Yoshida K, Terai Y, Mizoiri S, Aibara M, Nishihara H, Takahashi H et al. (2011) B chromosomes have a functional effect on female sex determination in Lake Victoria cichlid fishes. PLoS Genet. 7:e1002203. https://doi.org/10.1371/journal.pgen.1002203.
Acknowledgements
Assemblies and all bioinformatics and genomics data analyses were conducted using high-performance computing (HPC) resources at Kasetsart University and UNESP. We thank the members the Integrative Genomics Laboratory at UNESP, and the AGB Research Unit at Kasetsart University for their valuable feedback, early suggestions, and their kind support in this study.
Funding
The project for was supported by São Paulo Research Foundation, FAPESP as Thematic Project Grant (process number: 2015/16661-1) funded to C.M. through the UNESP. FAPESP funded sequencing and experimental materials costs. MJ received financial support as doctoral fellowship awards from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, CAPES (process number: 88882.433287/2019.01) and Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq Ph.D. sandwich program (process number: 201042/2020-7). FAPESP, CAPES and CNPq are acknowledged for supporting this research. These funding agencies did not contribute to the design of the study or collection, analysis and interpretation of data and writing the manuscript. The Article Processing Charge (APC) for the publication of this research was funded by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) (ROR identifier: 00x0ma614).
Author information
Authors and Affiliations
Contributions
All authors discussed the results and contributed to the final manuscript. M.J. and C.M. conceived the study and, together with S.F.A. and J.I.N.O., designed the experiments and co-wrote the manuscript. M.J. and C.M. secured funding for the project. A.L.C. collected and prepared biological samples. J.I.N.O. extracted DNA and prepared materials for sequencing. M.J. performed genome assembly. S.F.A. and I.R.W. carried out repeat and gene annotation analyses, with support from G.T.V. and M.J. All remaining data analyses and visualizations were conducted by S.F.A. and M.J. K.S. co-supervised the project and provided bioinformatics support for high-performance computing and data processing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All animal procedures were conducted in compliance with the ethical guidelines established by the Brazilian College of Animal Experimentation. Experimental protocols were reviewed and approved by the institutional ethics committees of the Institute of Biosciences at UNESP (Protocol no. 769/2015) and CEEAAP at UNIOESTE (Protocol no. 13/09).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Stacey Hanlon.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jehangir, M., Ahmad, S.F., Oliveira, J.I.N. et al. Genome assembly of Astatotilapia latifasciata uncovers B chromosome–linked chromatin reorganization. Heredity (2026). https://doi.org/10.1038/s41437-026-00847-4
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41437-026-00847-4
