
Abstract
Synthesis of valuable chemicals from CO2 electroreduction in acidic media is highly desirable to overcome carbonation. However, suppressing the hydrogen evolution reaction in such proton-rich environments remains a considerable challenge. The current study demonstrates the use of a hollow fiber silver penetration electrode with hierarchical micro/nanostructures to enable CO2 reduction to CO in strong acids via balanced coordination of CO2 and K+/H+ supplies. Correspondingly, a CO faradaic efficiency of 95% is achieved at a partial current density as high as 4.3 A/cm2 in a pH = 1 solution of H2SO4 and KCl, sustaining 200 h of continuous electrolysis at a current density of 2 A/cm2 with over 85% single-pass conversion of CO2. The experimental results and density functional theory calculations suggest that the controllable CO2 feeding induced by the hollow fiber penetration configuration primarily coordinate the CO2/H+ balance on Ag active sites in strong acids, favoring CO2 activation and key intermediate *COOH formation, resulting in enhanced CO formation.
Similar content being viewed by others
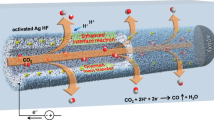

Introduction
The electrochemical conversion of CO2 driven by renewable electricity can produce value-added chemicals and feedstocks while mitigating CO2 emissions1,2,3. Numerous efforts have been made to develop catalysts with high current density (j > 1 A/cm2) and high faradaic efficiency (FE > 90%) toward CO, formate, etc4,5,6,7,8,9. Typically, alkaline or neutral electrolytes are used to suppress the competing hydrogen evolution reaction (HER) while promoting the electrocatalytic CO2 reduction reaction (CO2RR)10,11,12. However, during CO2RR and HER, the rapid consumption of H+ creates a locally alkaline environment close to the catalyst surface. Consequently, rather than being reduced, a major fraction of the input CO2 is consumed in the electrolyte via reaction with hydroxide ion (OH−) to produce (bi)carbonate13,14,15,16,17. In addition, transporting (bi)carbonate to the cathode flow field or anode results in a significant reduction of locally available CO2 and a low CO2 single-pass carbon efficiency (SPCE), impeding the practical applications of CO2 electrolysis18,19,20,21.
One strategy for addressing these issues is conducting CO2RR in an acidic medium22,23,24,25,26,27,28. That is, in a catholyte with a low pH, when the hydronium (H3O+) serves as the proton source for CO2RR and HER, no hydroxide ion (OH–) will be generated, and CO2 conversion can proceed without (bi)carbonate formation; even when H2O is the proton source, any OH– or (bi)carbonate generated locally will be neutralized or converted back to CO2 by protons in the bulk electrolyte, preventing CO2 from transferring to the anode22,29. However, efficient CO2RR in an acidic medium is difficult due to the kinetically superior HER outcompeting the reduction of CO223,30,31. For instance, in a strong acid with a pH ≤ 1, the FE of the CO2RR product is nearly close to zero23. One of the main reasons is that the adsorbed hydrogen (*H) acts as an intermediate for HER, out-competing the adsorption of CO2 (*CO2) overactive sites during CO2RR in an acidic medium23,30,31. Recently, it was discovered that K+ in the electrolyte could shield the electrode electric field and inhibit the transport of hydrogen ions (H+) and that the rapid consumption of surface H+ at high j could increase the pH near active sites, allowing for efficient CO2RR22,23,24,25. Several electrocatalysts, including Au24, Ag32, Cu24, and Ni5@NCN33, have demonstrated the ability to CO2RR in acid, but their high CO2RR selectivity (FECO2RR > 80%) could only be achieved in a limited range of j (≤0.5 A/cm2) with a low rate of product formation, which hinders their scalable applications.
Recently, a hollow fiber penetration electrode (HPE) with a compact structure has shown promising potential for high-rate and efficient CO2 reduction due to enhanced mass transport34,35,36,37,38,39. The unique three-dimensional electrode structure compels gaseous CO2 to permeate its abundant pores; the adequate oriented mass transfer at extensive triphasic reaction interfaces significantly improves the electrocatalytic kinetics. Herein, an Ag2CO3-derived hierarchical micro/nanostructured silver HPE (CD-Ag HPE) was used to investigate the effects of catalyst microenvironments (such as local concentrations of K+/H+ and CO2) on CO2 electrolysis performance in an acidic medium (pH = 1) (Fig. 1). By optimizing catholyte composition (H+, K+ concentration) and input CO2 flow rate, a high CO current density (jCO) of 4.3 A/cm2 with CO FE of 95% and stable electrolysis of 200 h at 2 A/cm2 were achieved in a strongly acidic electrolyte. Furthermore, a CO2 SPCE of over 85% was achieved at 2 A/cm2 by modulating the availability of CO2. In addition, the density functional theory (DFT) calculations indicated that the coexistence of H+ and K+ localized around the Ag sites played a crucial role in the formation of the key intermediates *COOH and *H, which not only suppressed the competitive HER but also promoted the CO2RR.

Schematic of Ag hollow fiber penetration electrode for boosting CO2 electroreduction to CO in a strongly acidic electrolyte (pH = 1) and the left schematic illustration of Ag hollow fiber was reproduced with permission from the reference37. Copyright [2022] [Springer Nature].
Results and discussion
Electrode preparation and characterization
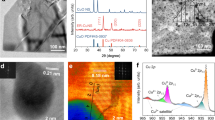
The CD-Ag HPE was fabricated via a two-step approach37 that was based on commercial Ag powder; additionally, it included an industrially viable phase-inversion/sintering process to obtain Ag HPE and an electrochemical redox process to obtain CD-Ag HPE (Supplementary Fig. 1). Compared to Ag HPE, CD-Ag HPE was about 20 μm thick, with partially ordered nanorods evenly coating the outer surface (Fig. 2a–c and Supplementary Fig. 2). This unique hierarchical micro/nanostructured architecture provided an increased electrochemical active surface area (ECSA, Supplementary Fig. 3) that maximized the three-phase reaction interfaces and enabled the efficient transport of reactants and products to/from the active sites for high-efficiency electrocatalytic reaction34,35,36,37,38,39. The X-ray diffraction (XRD) patterns were indexed to metallic Ag (111), (200), (220), (311), and (222) planes (JCPDS no. 04-0783), and there was no obvious crystal face orientation, which was almost the same as those of commercial Ag powder (Fig. 2d and Supplementary Figs. 4a, b). The X-ray photoelectron spectroscopy (XPS) confirmed that the surface compositions of CD-Ag HPE were identical to that of metallic silver (Fig. 2e and Supplementary Fig. 4c). In addition, the selected-area diffraction (SAED) pattern (inset of Fig. 2f) of CD-Ag HPE agreed well with the XRD results. Supplementary Fig. 5 depicted the high-resolution transmission electron microscopy (HRTEM) image and the corresponding fast Fourier transform (FFT) pattern of the marked region, in which only metallic Ag was observed.

SEM images of (a) cross section and (b), (c) outer surface of (a), (c) CD-Ag HPE and b Ag HPE. d XRD patterns, and e XPS spectra of Ag HPE and CD-Ag HPE. f TEM image and corresponding SAED pattern (insert of f) of CD-Ag HF. Optical images of the (g) working electrode of Ag HPE and CD-Ag HPE, (h) electrolyte flow two-compartment electrolysis cell from a side view. i Schematic illustration of the electrolysis system for CO2 electroreduction. WE: working electrode, CE: counter electrode, GC: gas chromatograph.
Electrocatalytic CO2RR in neutral electrolyte
The electrochemical experiment of the CO2RR was conducted in two chamber electrolysis cell with a three-electrode system at room temperature, where the CD-Ag HPE was used as the working electrode and gas diffuser (Fig. 2g–i). During CO2 electroreduction, CO2 penetrated through the porous wall of the CD-Ag HPE into the electrolytes via the copper tube, forming a large amount of bubbles. This unique oriented mass transfer of CO2 could induce the in-situ formation of extensive dynamic CO2(gas)–liquid–catalyst triphasic reaction interfaces, which significantly improve the mass transfer of CO2, electrons, protons, products as well as CO2RR kinetics34,35,36,37,38,39. Subsequently, a mixture of CO produced by CO2RR, H2 produced by HER, and unreacted CO2 flows out through an outlet connected to the top right of the electrolysis cell. The actual outlet flow rate was measured by an independent mass flowmeter and then sent to online gas chromatography (GC) for quantification. And the exhaust from the GC was vented to the outdoor hood (Fig. 2h, i). Unless otherwise specified, a flow rate of 30 standard cubic centimeters per minute (sccm) was used to compare our performance to that of other studies34,35,36. It was shown that only CO and H2 were detected at the j range of 0.1–4.0 A/cm2 in a neutral catholyte (pH ≈ 6.6) or in a strong acidic catholyte (Supplementary Figs. 6–9, pH = 1). The H2 FE remained below 5% as j increased up to 2.0 A/cm2, whereas the CO FE remained as high as 90.0% at j of 2.5 A/cm2, resulting in a high jCO of 2.3 A/cm2. Such CO2RR to CO performance in neutral catholyte distinguishes these electrocatalysts from other recent prominent electrocatalysts (Supplementary Fig. 6 and Supplementary Table 1). Subsequently, the CO FE decreased rapidly with increasing j, falling to 72.2% at 4.0 A/cm2, indicating an increase in HER at further elevated j values. Notably, During both CO2RR and HER, the consumption of H+ will likely produce a locally alkaline environment close to the catalyst surface. As a result, we hypothesized that at high j in a neutral electrolyte, rather than being reduced, a portion of the input CO2 would instead be consumed in the electrolyte through a reaction with OH− to produce (bi)carbonate. Consequently, local CO2 levels are inadequate and HER growth is kinetically more favorable.
Effects of H+ concentration and CO2 carbonation on CO2RR
The calculations based on the reaction and diffusion of species model (Supplementary Fig. 10) within a typical diffusion layer of 50 μm showed that, in the presence of K+ in a strong acidic electrolyte (pH = 1), the surface pH (distance to cathode of 0 μm) was similar to the bulk at j < 100 mA/cm2, while became neutral or basic when current density increase further due to the consumption rate of local protons that exceeds mass transport of protons from the bulk (Fig. 3a, b, j > 100 mA/cm2), effective CO2RR would dominate while HER was inhibited (Figs. 3c, d). Thus, the proton source of CO2RR comes from water on the electrode surface, despite the bulk pH still being in an acidic range (Fig. 3a). As shown in Fig. 3a, although the pH at the cathode surface rapidly increased with the j increase, the pH still remained in an acidic range with the increase in the distance from the cathode. When at a high j of 1 A/cm², although the surface is alkaline, the pH decreased to 7 at 30 μm away from the cathode. In comparison, similar conditions (pH 7 and pH 10.5 at a distance to the cathode of 40 μm for bulk pH 4 and 7, respectively) were reached at much lower j (about 200 mA/cm2) in electrolytes of pH 4 and 7 (Supplementary Fig. 11b, c). That means at high j, the carbonate formation would be more serious in an electrolyte with insufficient H+. In addition, the corresponding variation of CO2 concentration distribution showed that, even in a strong acidic electrolyte with pH 1 (Fig. 3b), the surface available CO2 concentration decreased gradually with the j increased above 200 mA/cm2, which is mainly due to the dual effects of the conversion of CO2 and the carbonation of CO2 caused by the increasing of pH22,23. However, the available CO2 concentration increased to a high level within about 30 μm of the cathode, even at a high j of 1 A/cm2, mainly due to the rapid transport of sufficient H+ and CO2 in the bulk electrolyte. In contrast, in the electrolytes with pH of 4 and 7 (Supplementary Figs. 12b, c), the available CO2 concentrations began to decrease rapidly at a much lower j and recover to a higher level at further distances from the cathode than that at pH 1, implying a greater CO2 carbonation in an electrolyte with insufficient H+. Therefore, in order to pursue efficient CO2RR and high CO2 SPCE at high j, we sought to conduct CO2RR in strongly acidic electrolytes (pH 1), and attempted to explore, including the effect of CO2 flow rate, H+ and K+ concentrations on it.

Modeling of (a) pH, (b) concentration profile of CO2 at different distances to cathode and j in 0.05 M H2SO4 and 3 M KCl. c, e CO and d, f H2 FE over CD-Ag HPE as a function of applied j measured in 3 M KCl and H2SO4 catholytes with different pH values. The input CO2 flow rate was 30 sccm. The error bars in (c)–(f) represent one standard deviation based on five independent tests.
To overcome the problem of (bi)carbonate formation and minimize the effect of CO2 carbonation under high j, we attempted to modulate the H+ concentration in the electrolyte (bulk pH) to neutralize the locally generated OH−, preventing the formation of (bi)carbonate. As shown in Fig. 3c, when j ≤ 0.5 A/cm2, the CO FE decreased with the pH decrease at the same j, whereas the H2 FE exhibited the opposite trend (Fig. 3d and Supplementary Fig. 13). Additionally, the onset j of CO2RR to CO increased gradually with the decrease of pH from 3 to 0.5 (Fig. 3c). This can be attributed to the fact that with the decrease in pH, the local H+ concentration of the electrode increases rapidly, and the HER is more likely to dominate40,41. Thus, a higher value of j is required to consume a substantial amount of H+ and modify the surface pH to make it more favorable for the kinetics of CO2RR. That is the shift of onset j for CO2RR strongly depends on bulk electrolyte pH, which is consistent with the simulation results and other reports (Fig. 3a)22,23,24,25,26. When the applied j was further increased from 0.5–2 A/cm2, all the CO FEs exceeded 95% in the 4–1 pH range, and CO FE exceeded 80% at pH 0.5 (Fig. 3e and Supplementary Fig. 14). Interestingly, when the j > 2 A/cm2, the CO FE increased as the pH decreases from 4 to 1 at the same j. This was irrespective of the decrease of all CO FE in the 4–0.5 pH range, which occurred as j further increased. Notably, at pH 0, almost no CO2RR was observed at any given j (Figs. 3e, f and Supplementary Figs. 13f, 14f). This is because HER always dominated when the surface pH could not be modulated due to the local H+ consumption rate being significantly lower than the mass transport rate of the bulk H+ in the extremely acidic electrolytes22,23.
Concurrently, the CO2 carbonation of different pH electrolytes at different j was further investigated (Fig. 4a and Supplementary Fig. 15). We discovered that in a near-neutral electrolyte (pH 4), the CO2 carbonation percentage increased rapidly from 6.2% to 48.9% as j increased from 0.5 to 4 A/cm2. However, CO2 carbonation decreased significantly as the pH of the electrolyte decreased. Accordingly, when pH ≤ 1, the CO2 carbonation was always < 5%, even when j reached 4 A/cm2 (Fig. 4a). To visualize the change in the trend of CO2 carbonation, the contour mapping distribution of CO2 carbonation on a pH−j plane (Fig. 4b) was plotted based on the date of Fig. 4a. It was clearly demonstrated that the CO2 carbonation increased rapidly in tandem with both the increase in j and the bulk pH (Fig. 4b). Correspondingly, the region with the greatest CO2 carbonation selectivity was located in the upper right of this map, indicating that the higher j as well as higher pH, the more serious CO2 carbonation. Based on the aforementioned findings, we reasoned that for an electrolyte with insufficient H+ concentration, the local OH– was generated rapidly as j increased. Some fraction of the input CO2 would readily react with a large amount of locally generated OH– to form a (bi)carbonate coating on the surface of the electrode resulting in a significant reduction of local available CO2 and CO2RR active sites28,42. As a result, when CO2RR at high j, local CO2 and CO2RR active sites of the electrode were insufficient, and HER was kinetically more favorable. However, an acidic electrolyte with sufficient H+ concentration can provide sufficient H+ to maintain a moderate local pH, effectively prevent CO2 carbonation, and ensure sufficient local CO2 for high-efficiency CO2RR at high j (Fig. 1).

a CO2 carbonation percentage over CD-Ag HPE as a function of applied j measured in 3 M KCl + H2SO4 catholytes with different pH values. b pH−j-dependent mapping distribution of CO2 carbonation percentage over CD-Ag HPE. c CO FE and (e) CO2 SPCE over CD-Ag HPE as a function of input CO2 flow rate in 3 M KCl + H2SO4 catholytes with different pH values at a constant j of 2 A/cm2. The pH−CO2 flow rate-dependent mapping distribution of (d) CO FE and (f) CO2 SPCE over CD-Ag HPE at a constant j of 2 A/cm2. The error bars in (a), (c), (e) represent one standard deviation based on five independent tests.
Effects of H+ concentration and CO2 flow rate on CO2RR
To further confirm this hypothesis, we reduced the flow rate of the input CO2 at a high j of 2 A/cm2 to make the effect of CO2 carbonation on CO2RR more apparent. As depicted in Fig. 4c, when the input CO2 flow rate was reduced from 30 to 25 sccm at pH 4, the H2 FE remained > 3%, and the CO FE remained virtually unchanged (> 97%). This is because a high flow rate of input CO2 could ensure relatively adequate local CO2 for high-efficiency CO2RR, even though ≈ 25% of CO2 was lost (Fig. 4a). However, as the input CO2 flow rate was further reduced from 25 to 5 sccm, the CO FE decreased rapidly (from 97% to 22%), while the H2 FE increased dramatically (Fig. 4c and Supplementary Fig. 16a). Thus, it can be inferred that under the dual influence of CO2 carbonation (Fig. 4a) and decreasing input CO2 flow rate, the local CO2 could not satisfy high-efficiency CO2RR at such a high j. In addition, the locally generated (bi)carbonate on the surface of the electrode may reduce the availability of CO2RR active sites28,42. Although the CO2 SPCE gradually increased by decreasing the CO2 flow rate to limit the availability of CO2 (Fig. 4e and Supplementary Fig. 16i), it was still < 50% of the theoretical limit value for neutral electrolytes22,28.
Then we reduced the input CO2 flow rate of CD-Ag HPE in different pH electrolytes and observed the effect on HER (Supplementary Fig. 16h) and CO2RR (Fig. 4c). In the pH range 1–4 when the CO2 flow rate was ≥ 20 sccm, the HER was effectively suppressed (Supplementary Fig. 16a–d), and the CO FE was always high (Fig. 4c; FECO > 90%). However, when the pH was < 1, an excessively high local H+ concentration increased H2 FE significantly (Supplementary Fig. 16e, f)22,23. Moreover, as the CO2 flow rate fell below 20 sccm, the CO FE decreased, and the H2 FE increased dramatically. However, at identical low input CO2 flow rates, CO FE had a volcano-like distribution in the 0–4 pH range. Consequently, at a high j of 2 A/cm2, the contour mapping distribution of CO FE on the pH−CO2 flow rate plane clearly showed that, at a low CO2 flow rate, the higher CO FE region was located at pH 1–2, in spite of CO FE is basically the same in pH 1–4 at high CO2 flow rate (Fig. 4d). For the distribution of CO2 SPCE (Fig. 4e), which was determined jointly by the jCO and input CO2, the CO2 SPCE increased rapidly at pH 1 and 2 with a decrease in the input CO2 flow rate (from 20–5 sccm). Thus, when combined with the CO FE and input CO2 flow rate, the contour mapping distribution of CO2 SPCE on the pH−CO2 flow rate plane directly showed that the most CO2 SPCE selective area was located in the middle right of the map, where require both low input CO2 flow rate and low pH of 1–2 (Fig. 4f). In other words, at a CO2 flow rate of 5 sccm, the CO2 SPCE at a pH of 1–2 exceeded 80%, which is roughly double that at pH 4 (Fig. 4e). In addition, the j−CO2 flow rate-dependent mapping distribution of theoretical limit of CO FE based on CO2 carbonation in different pH electrolyte also showed a higher theoretical limit of CO FE could be achieved only in a strong acidic electrolyte (Supplementary Fig. 17).
These observations indicated that a high flow rate of CO2 can ensure high local CO2 for high-efficiency CO2RR but limit CO2 SPCE. However, in electrolytes with insufficient H+ concentration, the CO2 SPCE cannot be promoted effectively by merely reducing the input CO2. It is worth noting that the locally generated (bi)carbonate on the surface of the electrode not only causes a reduction of locally available CO2 but also covers the active sites for CO2RR, resulting in the collapse of CO FE at low input CO2 flow rate. When CO2RR was conducted in an acidic electrolyte, the proper H+ concentration in the electrolyte could effectively solve the problem of CO2 carbonation and ensure that the local CO2 concentration was sufficient despite a low input CO2 flow rate. Therefore, while CO2 availability is constrained, high-efficiency CO2RR can be maintained by reducing the input CO2, effectively promoting the CO2 SPCE.
K+ effect on acidic CO2RR
Given the essential role of alkali cation in the activation of CO2 and inhibition of HER22,24,25, we first performed linear voltammetry curve (LSV) analysis (Fig. 5a). In 0.05 M H2SO4 (pH 1) without any K+ electrolyte, the voltammetric properties of the CD-Ag HPE hardly changed regardless of the surrounding atmosphere (Ar or CO2), indicating that only HER occurs. However, HER activity was significantly suppressed by K+ presentation, concurrently, CO2RR occurred. This may be attributed to the fact that the presence of K+ in the acidic electrolyte shielded the electric field in the diffusion layer of the cathode and reduced the H+ concentration around the active site, which not only suppressed the HER, but also stimulated CO2 activation and conversion. (Supplementary Fig. 18a)22,24,41. Subsequently, we conducted Tafel analysis in 0.05 M H2SO4 + KCl catholytes at pH 1 but with different K+ concentrations (Fig. 5b). On the one hand, the CO Tafel slope was found to decrease with increasing K+ concentrations, reaching a minimum (104 mV dec−1) at 3 M K+. This result suggests that the rate-determining step (RDS) for CO formation comprised the adsorption of CO2, which could be altered with the K+ concentrations22,43,44. In other words, the activation energy barrier of the electron transfer over the electrode may be reduced in an electrolyte with a high K+ concentration, which is consistent with the faster initial one-electron transfer step required to form an adsorbed *COO− intermediate. On the other hand, it can be seen that with the increase of K+ concentration, the changing trend of H2 Tafel slope value was opposite to that of CO. That is, the H2 Tafel slope value increased with increasing of K+ concentrations (Supplementary Figs. 19b, c). This is consistent with the results of LSV (Fig. 5a), indicating that the presence of K+ would suppress the HER. In addition, the total Tafel slope values of CO and H2 gradually decreased with increasing of K+ concentrations (Supplementary Fig. 19c), which was consistent with the results in Fig. 5c, where a faster electron transfer was verified by the lowest interfacial charge transfer resistance (Rct) of CD-Ag HPE in 3 M K+ (0.9 Ω cm2, Fig. 5c and Supplementary Table 8). In addition, the higher effective electric double layer capacitance (Cdl) of CD-Ag HPE in acidic electrolyte with high K+ concentration, which was correlated with the electric field strength an enhanced electric field trend (Fig. 5c and Supplementary Table 8). These results are consistent with the hypothesis, that is the hydrated K+ physisorbed on the cathode in the acidic electrolyte modify the distribution of electric field in the double layer, which not only impedes HER by suppression of migration of H+, but also promotes CO2 reduction by stabilization of key intermediates (Supplementary Fig. 18)22,24,41.

a LSV curves of CD-Ag HPE in Ar-saturated or CO2-saturated pure H2SO4 or H2SO4 with 0.1 M K+ catholytes at pH 1. b Tafel slopes and c EIS Nyquist plots obtained in catholytes with different K+ concentrations at pH 1. d CO and (e) H2 FE over CD-Ag HPE as a function of applied j measured in H2SO4 catholytes with different K+ concentrations at pH 1 (input CO2 flow rate: 30 sccm). f j − K+ concentration-dependent mapping distribution of CO FE over CD-Ag HPE (input CO2 flow rate: 30 sccm). The error bars in (d), (e) represent one standard deviation based on five independent tests.
We also examined the CO2RR product distribution in 0.05 M H2SO4 (pH 1) with varying K+ concentrations at different j (Fig. 5d, e and Supplementary Fig. 20). Even at a high j of 4 A/cm2, almost no CO could be detected in 0.05 M H2SO4 without K+ (Fig. 5d and Supplementary Fig. 20a), which was consistent with the LSV results (Fig. 5a). In addition, the HER selectivity (Fig. 5e) decreased as the K+ concentration increased, while the CO2RR selectivity (Fig. 5d) increased for all given values of j. Particularly, at a constant high j of 2 A/cm2, the CO FE increased steadily from 61.2% with 0.1 M K+ to 96.8% with 3 M K+. The contour mapping distribution of CO FE on the j − K+ concentration plane further directly showed that, CO2RR-dominated regions require the presence of high concentrations of K+, while obtaining high CO FE at high j is more dependent on high K+ concentrations (Fig. 5f). In addition, the porous micro/nanostructured Ag (Supplementary Fig. 21) was found to be conducive to increasing the local K+ concentration22,45,46, which can be attributed to the amplified electric field near the pore sites, thereby inhibiting HER and promoting the activity of CO2RR (Supplementary Fig. 22). Note that both controlled experiments and DFT calculations showed that, in an acidic electrolyte, the effect of anions on CO2RR reactivity was not significant; substitution of Br–, SO42– or PO43– for Cl– showed product distribution similar to that of the Cl– case (Supplementary Fig. 23), and the Gibbs free energy of *COOH and *H, the key intermediates from CO2RR to CO and HER, respectively, basically did not change whether there was Cl- adsorption or not (Supplementary Figs. 24, 25).
Effects of CO2 flow rate and j on acidic CO2RR
To pursue a high CO2 SPCE under high CO2RR to CO activity, we analyzed the input CO2 flow rate effect on acidic CO2RR product distribution at various j (Fig. 6a and Supplementary Fig. 26). In strong acidic electrolytes, there is almost no loss of CO2 and CO2RR active sites because, in the absence of (bi)carbonate precipitation, the activation of high K+ concentration and the high input flow rate of CO2 can guarantee sufficient CO2 and CO2RR active sites locally on the catalyst surface for high-efficiency CO2RR. Consequently, the CO FE could maintain a high value (FECO > 95%) even when the j reached 4 A/cm2 (Fig. 6a and Supplementary Fig. 26). As the input flow rate decreased, the local CO2 supply became relatively insufficient, and as j increased, the CO FE began to decline rapidly. Thus, combined with the CO FE and j, the most jCO selective region of the j–CO2 flow rate-dependent mapping distribution of CO FE is located in the top left corner, where the high flow rate of input CO2 could maintain the sufficient CO2 supply to achieve high CO FE at high j due to the unique structure of the HPE (Fig. 6b). These results indicate that a high input CO2 flow rate is necessary for CD-Ag HPE to achieve high-efficiency CO2RR to CO under high j in acidic electrolyte. The detailed performances of the CD-Ag HPE at high CO2 flow rate (30 sccm) revealed that the H2 FE remained < 5% as the j increased, up to 4.5 A/cm2, while the CO FE remained as high as 95.06%, yielding 4.28 A/cm2 jCO at −1.41 V vs. RHE (Fig. 6c and Supplementary Fig. 27). The corresponding CO yield and CO energy efficiency were 80.83 mmol/(h cm2) and 48.25%, respectively (Supplementary Fig. 27 and Supplementary Table 2). Such performance stands out among recently reported prominent electrocatalysts for CO formation from CO2RR (Supplementary Fig. 28 and Supplementary Table 1).

a CO FE and (d) CO2 SPCE over CD-Ag HPE as a function of input CO2 flow rate measured in 3 M KCl + 0.05 M H2SO4 catholytes (pH 1) at different applied j. The j–CO2 flow rate-dependent mapping distribution of (b) CO FE and (e) CO2 SPCE over CD-Ag HPE in 3 M KCl + 0.05 M H2SO4 catholytes (pH 1). c CO, H2 FE and potential over CD-Ag HPE as a function of applied j measured in 3 M KCl + 0.05 M H2SO4 catholytes (pH 1, input CO2 flow rate: 30 sccm). f CO, H2 FE and CO2 SPCE over CD-Ag HPE as a function of input CO2 flow rate measured in 3 M KCl + 0.05 M H2SO4 catholytes (pH 1) at a constant j of 2 A/cm2. g Long-term performance at a constant j of 2 A/cm2 in 3 M KCl + 0.05 M H2SO4 catholytes (pH 1, input CO2 flow rate: 10 sccm). The error bars in (a), (c), (d), (f) represent one standard deviation based on five independent tests.
Regarding CO2 SPCE, it is determined by both the jCO and input CO2 flow rate. At a high input CO2 flow rate of > 20 sccm, the CO FE of CD-Ag HPE could remain above 80% even as j increased to 4 A/cm2 (Fig. 6a), whereas the CO2 SPCE increased only very slowly as j was increased (Fig. 6d, e). However, a moderate CO2 flow rate and a substantial CO FE at high j were more conducive to achieving a high CO2 SPCE. This trend was further visualized in the the j-CO2 flow rate-dependent mapping distribution of CO2 SPCE (Fig. 6e). The most CO2 SPCE selective region was located in the middle right of the map, where high CO2 SPCE of > 80% was only achieved when the j is around 2 A/cm2 and the flow rate is less than 10 sccm. Particularly, when the CO2 flow rate was decreased from 30–5 sccm at a constant high j of 2 A/cm2, the CO2 SPCE increased from 22% to 87% for CO2RR to CO, thereby exceeding the theoretical limit of 50% in neutral/alkaline systems. This was comparable to the record level of 90% at low j of < 0.2 A/cm2 in acidic systems (Fig. 6f and Supplementary Table 6)28.
Although highly challenging, the long-term operation of electrocatalysts under high current density is crucial for their practical applications3,14. Correspondingly, the j was fixed at 2 A/cm2, and the flow rate was set to 10 sccm in the acidic electrolyte stability test. During the 200-h continuous test, the CO FE remained consistently above 90%, while the corresponding CO2 SPCE fluctuated around 70% (Fig. 6g). In addition, the surface structures remained highly stable after the stability test (Supplementary Fig. 29), and the postreaction XRD and XPS further revealed the stable compositions of CD-Ag HPE after electrolysis (Supplementary Fig. 30 and Supplementary Table 10), which were responsible for the steady CO2 electroreduction performance. Thus, the overall CO2RR performance is well-placed among recently reported outstanding electrocatalysts for CO formation from CO2 reduction, including j, CO FE, CO yield (95% CO FE and 80.83 mmol/(h cm2) CO yield at high j of 4.5 A/cm2), CO2 SPCE and stability (87% CO2 SPCE and 200 h long-term test at j of 2 A/cm2) (Supplementary Figs. 26–31 and Supplementary Tables 1, 2, 6), demonstrating great potential for scalable application. And at the current density as high as 4.5 A/cm2, the overpotential of CD-Ag HPE was only 1.3 V, which was comparable to the high-current density electrocatalyst (Supplementary Table 1). In order to demonstrate the scalability for practical applications using hollow fiber penetration electrodes, single-, 2-, 5- and 10-tube arrays of CD-Ag HPE were further adopted and tested in a 2-electrode acidic system (pH = 1). All CD-Ag HPE array electrodes with different tube numbers showed highly similar FE distributions of CO and H2 at given high j range (Supplementary Figs. 32, 33). Thus, the jCO over these CD-Ag HPE array electrodes also exhibited almost same rapidly growth trend with increasing j. Although CO FE and jCO over the CD-Ag HPE array electrodes slightly decreased with increasing tube number and j, 10-tube CD-Ag HPE array still possessed over 90% of CO FE at high j of 4 A cm−2. These results implied the potential scalability for practical applications using hollow fiber penetration electrodes.
Theoretical calculations
The promoting mechanism of K+ and H+ in the aqueous microenvironment surrounding the Ag active site of the CD-Ag HPE was further simulated using DFT calculations. The formation of adsorbed COOH (*COOH) and H (*H) intermediates at the active site were thought to be the RDS for CO2RR and HER (Supplementary Figs. 34, 35)46,47,48, respectively. Thus, we first simulated an Ag (111) surface with a K+ concentration ranging from 0 to 3 (1/18 per H2O molecule), and compared the Gibbs free energy (G) change difference for the formation of *COOH and *H (Fig. 7a). Although both the G(*COOH) and G(*H) decreased with increasing K+ concentration, the G(*COOH–*H) also decreased rapidly (from 1.21 eV to 0.60 eV) with the increase of K+. This was consistent with the experimental findings, and the results indicated that an increase in K+ concentration could promote both CO2RR and HER, with CO2RR being the more dominant effect. On this basis, we further calculated the G for the formation of *COOH and *H at different *H coverages ranging from 0 to 3 (1/9 per site) (Fig. 7b). Correspondingly, we found volcano-shaped relationships between G(*COOH) and *H coverage, which is in stark contrast to the result that G(*H) increases as *H coverage increases. Consequently, the G(*COOH–*H) reached a minimum of 0.04 eV in the optimized structures with a high K+ concentration and moderate *H coverage (3/18 K+−1/9 *H and 3/18 K+−2/9 *H). In addition, we explored the effect of different hydrogen sources on a model of 3 K+ (1/18 / H2O molecule) (Supplementary Figs. 35–38). It was found that the values of G (*COOH), G (*H) and G (*COOH–*H) calculated by the H3O+ model were basically the same as those calculated by the *H model, and they were all much lower than the model values of H2O as hydrogen source (Supplementary Fig. 39). Taken together, these DFT results suggest that the coexistence of H+ and K+ could promote the CO2RR synergistically. A high K+ concentration and a moderate *H coverage are likely to facilitate *COOH formation for CO2RR over *H formation for HER, which is consistent with the experimentally observed high CO2RR to CO conversion in strongly acidic media with a high K+ concentration.

The free energy for the formation of *COOH (G(*COOH)), *H (G(*H)), and their energy difference (G(*COOH)-G(*H)) on Ag (111) plane at various (a) K+ concentrations (1/18 per H2O molecule) and (b) *H coverages (1/9 per site). The light blue, purple, red, gray and white balls represent Ag, K, O, C and H, respectively.
Combined with controlled experiments and theoretical studies, we demonstrated that the presence of K+ in the acidic electrolyte controlled the onset of the CO2RR, and that a moderate concentration of H+ effectively prevented the carbonation of CO2 and CO2RR active sites due to the precipitation of (bi)carbonate, ensuring sufficient CO2 and CO2RR active sites at the catalyst surface for high-efficiency CO2RR at ampere-level current density. Thus, by optimizing the K+ and H+ concentration and CO2 flow rate in a strong acidic electrolyte, a high CO FE > 95% at 4.5 A/cm2 and >200 h of stability testing at 2 A/cm2 are achieved. In addition, by limiting the availability of input CO2, the CO2 SPCE for CO2RR reached 87% at a high j of 2 A/cm2, demonstrating remarkable CO2 conversion capability. This study, therefore, provides a means for high-efficiency CO2 conversion in strong acid by modulating catalyst microenvironments, with great potential for practical application.
Methods
Chemicals and materials
Ag powder (99.9%, 50 nm) was purchased from Ningbo Jinlei Nano Materials Co., Ltd. Polyetherimide (PEI) was purchased from Saudi Basic Industries Corporation (SABIC). N-Methyl-2-pyrrolidone (NMP), sulfuric acid (H2SO4), potassium sulfate (K2SO4), potassium bicarbonate (KHCO3), potassium chloride (KCl) and potassium hydroxide (KOH) were purchased from Sinopharm Chemical Reagent Co., Ltd. Nafion 117 proton exchange membranes (PEMs) were purchased from DuPont. All chemicals were used as received without further purification. Electrolyte solutions were prepared using 18.2 MΩ H2O (ultrapure water, from Master-S30UVF water purification system).
Catalyst preparation
Ag hollow fiber (Ag HF) was fabricated by a combined phase-inversion/sintering process (Supplementary Fig. 1)37. Briefly, commercially available polyetherimide (PEI, 24 g) was added to N-Methyl-2-pyrrolidone (NMP, 96 g), followed by ultrasonic treatment for 1 h to obtain a homogeneous and transparent solution. Then Ag powder (80 g) was added to the above solution. The as-obtained mixture was further treated by the planetary ball-milling (using 250 mL zirconia jar and ϕ5 mm zirconia balls) at 300 rpm for 24 h to form a uniform slurry. After cooling to room temperature, the slurry was vacuumed (1 mbar) for 5 h to remove bubbles and then to obtain a casting solution. Next, the casting solution was extruded through a spinning machine and shaped in a water bath via the phase-inversion process. After spinning, the as-formed tubes were kept in a water bath for 24 h to eliminate the solvent completely, followed by stretching and drying in ambient conditions with a humidity of ~28% for 48 h to obtain a green body. The green body was cut into appropriate lengths and then calcinated in an airflow (100 mL/min) at 600 °C (heating rate: 1 °C/min) for 6 h to remove PEI. After being naturally cooled to room temperature, the calcined green body was then reduced in a 5% H2 (argon balance) flow (100 mL/min) at 300 °C (heating rate: 1 °C/min) for 3 h to obtain Ag HF.
The Ag HF with an exposed length of 4 cm was stuck into a copper tube using conductive silver adhesive for electrical contact (see Fig. 2g for details), while the end of the Ag HF tube as well as the joint between the Ag HF and copper tube were sealed and covered with gas-tight and nonconductive epoxy. After drying at room temperature for 12 h, a working Ag HF penetration electrode (Ag HPE) was obtained with an exposed geometric area of 0.5 cm2 (S = πDoutL = 3.14 × 400 × 10−4 × 4 ≈ 0.5 cm2, where S is the electrode area, Dout is the outer diameter of hollow fiber, and L is the length of hollow fiber).
Ag2CO3-Ag HPE was synthesized from Ag HPE by electrochemical redox activation treatments. Typically, the Ag HPE was subjected to oxidation and reduction treatments on a Biologic VMP3 potentiostat using a three-electrode system in a gas-tight two-compartment electrolysis cell containing a Nafion 117 membrane as the separator, a KCl-saturated Ag/AgCl reference electrode and a platinum mesh (3 cm × 3 cm) counter electrode. The electrolyte solution was CO2-saturated 0.5 M KHCO3, and the CO2 flow rate was kept at 2 mL/min. Prior to the experiments, the electrolysis cell was vacuumized and then purged with CO2 for 30 min. The Ag HPE was electrochemically oxidized at a fixed potential of 2.0 V (vs. Ag/AgCl) for 4 min to obtain Ag2CO3-Ag HPE. Subsequently, the Ag2CO3-Ag HPE was reduced at a fixed potential of −0.50 V (vs. Ag/AgCl) for 10 min to obtain CD-Ag HPE. The CD-Ag HPE possessed the same exposed geometric area of 0.5 cm2 (S=πDoutL = 3.14 × 400 × 10−4 × 4 = 0.5 cm2). For the 10-tube CD-Ag HPE array electrode, the exposure geometric area was 5 cm2 (S = nπDoutL = 10 × 3.14 × 400 × 10−4 × 4 = 5 cm2, where n is the number of hollow fiber tubes). The electrochemical oxidation reaction and reduction reaction obeyed Eqs. (1) and (2), respectively.
In addition, the OD-Ag HPE was also treated in the Ar-saturated 0.5 M KOH with the same electrochemical redox activation treatments as that of CD-Ag HPE. That is, the Ag HPE that underwent 60 s of oxidation and 600 s of reduction in Ar-saturated 0.5 M KOH electrolyte solution, respectively.
Material characterization
The cross-section and surface morphologies of samples were observed via scanning electron microscopy (SEM) with a SUPRRATM 55 microscope using an accelerating voltage of 5.0 kV. Transmission electron microscopy (TEM) investigations were conducted with a JEM-ARM300F microscope operated at 300 kV. X-ray diffraction (XRD) measurements were performed on a Rigaku Ultima 4 X-ray diffractometer using a Cu Kα radiation source (λ = 1.54056 Å) at 40 kV and 40 mA. X-ray photoelectron spectroscopy (XPS) tests were conducted using a Quantum 2000 Scanning ESCA Microprobe instrument with a monochromatic Al Kα source (1486.6 eV). The binding energies in all XPS spectra were calibrated according to the C 1 s peak (284.8 eV).
Electrochemical measurements
All electrochemical measurements were performed using a Biologic VMP3 potentiostat in a two-compartment electrolysis cell at an ambient temperature and pressure. The electrolysis cell comprises two symmetrical compartments made of quartz glass with an inner height of 5.0 cm, an inner length of 5.0 cm and an inner width of 1.5 cm (Fig. 2g, h). The cathodic and anodic compartments were separated by a Nafion 117 membrane, and the electrolysis cell was equipped with a KCl-saturated Ag/AgCl reference electrode in the cathodic compartment and a platinum mesh (3 cm × 3 cm). CO2-saturated H2SO4 containing various concentrations of KCl or different pH was used as catholyte, and 0.5 M K2SO4 + 0.05 M H2SO4 aqueous solution was used as anolyte. Prior to the experiments, the electrolysis cell was vacuumed, and then CO2 was continuously delivered into the cathodic compartment at a constant rate of 30 mL/min for 30 min. During all of the electrochemical measurements, the CO2 flow rate was fixed at 30 mL/min. Note that the input CO2 flow rate was fixed at 30 mL/min during all electrochemical measurements, but the exhaust flow rate was not 30 mL/min at all due to the hydrogen evolution and CO2 consumption. Thus, the outlet flow rate of the electrolysis cell was measured by an independent Alicat® mass flowmeter (Fig. 2i and Supplementary Table 4). The cathodic electrolyte was 3.0 KCl + 0.05 M H2SO4, the anodic electrolyte was 0.5 M K2SO4 + 0.05 M H2SO4, unless otherwise stated.
For electrochemical characterizations, the electrochemical impedance spectroscopy (EIS) measurements were performed at −0.2 A/cm2 with a voltage amplitude of 50 mV, and the frequency limits were typically set in the range from 50 Hz to 500 kHz. The effective double-layer capacitance (Cdl) was obtained from the constant phase element (CPE) parameters and the two resistances using the Brug formula37,49:
Where Rs is the solution resistance, Rct is the charge transfer resistance, T is CPE constant and P is CPE exponent. For the CO2 electroreduction tests, a Biologic VMP3 potentiostat was used for small current situations (≤ 400 mA), while the Biologic VMP3 potentiostat was connected to a VMP3 booster chassis with a 10 A current option to be used under large current situations (≥ 400 mA). The CO2RR performance of the CD-Ag HPE array electrodes with different tube numbers in the 2-electrode acidic system (pH = 1) was performed using an ANS6050D (50 V, 50 A) direct-current source from ANS Power Co., Ltd. with constant current control applied. In addition, both the anolyte and catholyte were cycled in anodic and cathodic compartments with a fixed flow rate of 50 mL/min by using two identical peristaltic pumps (JIHPUMP BT-50EA 153YX). For the long-term performance test of CO2 electroreduction, the current density was fixed at −2 A/cm2 in CO2-saturated 3 M KCl + 0.05 M H2SO4 catholyte, the exhaust from the cathodic compartment was measured by online GC during the entire 200-hour test.
All the current in the main text and supplementary materials were geometrically normalized to the electrode area. All the applied potentials were recorded against the KCl-saturated Ag/AgCl reference electrode and then converted to those versus the reversible hydrogen electrode (RHE) with iR corrections using the following equation:
Where E (vs. Ag/AgCl) is the applied potential, pH indicates the H+ concentrations of the electrolyte solutions (Supplementary Table 7), i is the current density at each applied potential, and Rs is the solution resistance obtained via EIS measurements (Supplementary Table 8). In order to avoid the overcorrected potentials, 85% iR correction was applied as the previous reports17,48. All applied potentials in the main text and Supplementary Information are referred to as RHE, unless otherwise stated. Note that the XY data at different pH values of Figs. 4a, c, e were first converted into XYZ data by the origin® software, to obtain the corresponding contour maps of Figs. 4b, d, f, respectively. Moreover, the XY data at different K+ concentrations of Fig. 5d in manuscript were first converted into XYZ data in origin® software, to obtain the corresponding contour map of Fig. 5f. In addition, the XY data at different current densities of Figs. 6a, d were first converted into XYZ data by the origin® software, to obtain the corresponding contour maps of Fig. 6b, e, respectively. Taking Fig. 4b for example, the X represents current density of Fig. 4a, Y represents pH of Fig. 4a, and Z represents percentage loss of CO2 of Fig. 4a, which were then standard smoothened and transformed back into a virtual matrix. The as-obtained virtual matrix was further presented in the form of a contour map, i.e., Fig. 4b, in consistency with the previous reports28,50.
Product quantifications
Gas-phase products from the cathodic compartment were directly vented into a gas chromatograph (GC-2014, Shimadzu) equipped with a Shincarbon ST80/100 column and a Porapak-Q80/100 column using a flame ionization detector (FID) and a thermal conductivity detector (TCD) during the electroreduction tests and analyzed online. FID was used for CO quantification (as well as CH4, C2H4 and C2H6), while TCD was used for H2 and CO quantification. In addition, the concentration of unreacted CO2 in the outlet gas was analyzed with an independent Agilent 7890B gas chromatograph (Supplementary Fig. 9). A thermal conductivity detector (TCD) with a carbon molecular sieves column (TDX-1) was used for CO2 quantification. All faradaic efficiencies reported were based on at least three different GC runs, and the error bars of all figures in this work were based on the standard deviations of at least five independent electrochemical tests, unless otherwise stated. High-purity argon (99.999%) was used as the GC carrier gas. In all the CO2 electrolysis tests, only H2 and CO were the gas-phase products, and their faradaic efficiencies were calculated as follows:
where Cproduct is the concentration of the gas-phase products (ppm), νoutlet is the outlet flow rate of the electrolysis cell. Note that the exhaust flow rate of the electrolysis cell was not equal to the input CO2 flow rate at all due to the hydrogen evolution and CO2 consumption. Thus, the outlet flow rate of electrolysis cell was measured by an independent Alicat® mass flowmeter (Fig. 2i). The actual measured outlet flow rate was higher than the inlet flow rate due to HER occurrence (Supplementary Table 3 and Fig. 5f). Thus the actual exhaust outlet flow rates (corrected voutlet) were measured by the Alicat® mass flowmeter (Fig. 2i), which possessed a full scale of 50 sccm with the accuracy ± (0.8% of Reading + 0.2% of Full Scale), as shown in the note d of Supplementary Table 3. That is the experiment errors caused by the Alicat® mass flowmeter could be negligible under the CO2 electroreduction conditions. In addition, the Alicat® mass flowmeter results were calibrated using a mixture (including CO, H2, CO2 and water vapor compositions at room temperature) that approximates the actual outlet gas composition (Supplementary Table 4). t is the reaction time, n is the number of transferred electrons for producing CO or H2, F is the Faraday constant, Vm is the gas mole volume, and Q is the total quantity of the electric charge. The CO formation rate was calculated using the following equation:
Where S is the geometric area of the electrode (cm2).
The jCO,limit(gas) is the theoretical limit of CO partial current density with all gas-phase CO2 molecules input into the electrolysis cell were reduced to CO products. The theoretical limits of CO product partial current density, i.e., jCO,lim(gas) were calculated using the following equation37,38:
So, the theoretical limits of CO FE, i.e., FECO,lim(gas) were calculated by the following equation:
The CO2 SPCE, the CO2 carbonation and unreacted CO2 were calculated as follows:
Where the input CO2 (Vinput) of electrolysis cell was controlled by a Alicat® mass flow controller, and the outlet flow rate (Voutlet) of electrolysis cell was measured by an independent Alicat® mass flowmeter. The actual outlet gas of electrolysis cell contains unreacted CO2, produced CO (from CO2RR) and produced H2 (from the HER). The produced CO and H2 at given current density could be quantified by online GC. Furthermore, to verify our calculation of unreacted CO2, the concentration of unreacted CO2 in the outlet gas was measured by an independent online GC (Fig. 2i).
By assuming that the overpotential of oxygen evolution reaction on the anode side is zero, the cathodic energy efficiency for CO was calculated as follows12:
Where ECO is −0.11 V (vs. RHE); 1.23 V is the thermodynamic potential for water oxidation in the anode side.
Possible liquid-phase products from the cathodic compartment after CO2 electrolysis for 1 h were analyzed using another off-line GC-2014 (Shimadzu) equipped with a headspace injector and an OVI-G43 capillary column (Supelco, USA). No liquid-phase product was detected by the off-line GC. The post-reaction catholyte solution was also analyzed by a 600 MHz NMR spectrometer (Bruker) for possible liquid-phase products (especially formate and acetate). After an hour of electrolysis, an aliquot of catholyte solution (0.5 mL) was mixed with 0.1 mL (CH3)3Si(CH2)3SO3Na (DSS) (6 mM) and 0.1 mL D2O for use as internal standards. No liquid-phase product was detected by 1H NMR (Supplementary Figs. 6–9).
COMSOL multiphysics simulations
A reaction-diffusion model (Supplementary Fig. 10) was used to simulate the local pH and CO2 concentration using COMSOL Multiphysics software in a typical 50 μm diffusion layer22,23,25. All the interactions between species in the electrolyte (CO2, HCO3-, CO32-, SO42-, K+, Cl-, OH-, H+ and H2O) were considered (Supplementary Table 11). Specifically, one end of the one-dimensional simulation area is set as the working electrode surface, and the other side is set as the bulk concentration to describe the bulk electrolyte. We used Henry’s law to calculate the CO2 concentration, assuming that the CO2 fugacity is 1 bar.
\({{{{\rm{K}}}}}_{{{{\rm{H}}}}}^{0}\) is the Henry’s constant, which can be calculated by using the equation below, where T is the temperature.
Due to the high concentration of the ions, the saturated concentration of CO2 in an electrolyte is corrected using the following equations.
Cs is the molar concentration and Ks is the Sechenov’s constant.
We considered the following homogeneous and heterogenous reactions in our model, which are based on the previously published works22,23,25. The heterogenous reactions take place in the electrolyte as follow:
In this work, two sets of Butler-Volmer boundary conditions (CO2RR and HER) were set to represent the two main sets of reactions on electrode surface, the kinetic parameters sets for these two reactions were the Tafel slopes of CO (103 mV dec−1) and H2 (56 mV dec−1), respectively, and the FEs of CO and H2 in the model were calculated and simulated based on these parameters.
Over the whole domain, the following homogenous reactions occur:
The bulk concentrations and pH values were measured experimentally and implemented in the model. The thickness of the diffusion layer was assumed to be 50 μm. The electrode surface undergoes a reduction reaction, its current characteristics follow the Bulter-Volmer equation and the Nernst equation, its potential characteristics follow the Nernst equation, the concentration of the opposite body phase is set to the corresponding initial concentration, and the ion migration in the simulation area follows the Nernst Planck equation. The solution process is based on the MUMPS (multiple massively parallel spark direct solver) steady-state solver, and the relative tolerance and residual factor are set to 1E−8 and 1, respectively, eight layers of boundary layer subdivision are set on the simulated electrode surface to ensure the accuracy of the simulation results. The pH and CO2 concentration distribution near the electrode surface was calculated by solving the operating current from 0–1000 mA/cm2 at bulk pH of 1, 4, and 7, respectively.
The electrode surface reaction follows the BV equation:
Where iloc is the local current density at the electrode/electrolyte interface, i0 is the exchange current density, \({{{{\rm{\alpha }}}}}_{{{{\rm{c}}}}}\) and \({{{{\rm{\alpha }}}}}_{{{{\rm{a}}}}}\) is the cathodic and anodic charge transfer coefficients, η is the activation overpotential.
The balance potential follows the Nernst equation:
Where Eeq is the electrode potential, ΔG is the Gibbs free energy, Eeq,ref is the standard electrode potential, \({{{{\rm{a}}}}}_{{{{\rm{i}}}}}\) is the (electrode reactive ion concentration),\(\,{{{{\rm{a}}}}}_{{{{\rm{i}}}},{{{\rm{ref}}}}}\) is the (standard electrode reactive ion concentration), vi is the reaction stoichiometric number.
The transfer of dilute substances follows Fick's law:
Where Ji is the ion flux, Di is the diffusion coefficient (DLi = 1 × 10−7 m2s−1), ci is the concentration of ion, ∇ci is concentration gradient.
The transportation of electricity follows the Nernst Planck relationship:
Where zi is the transfer number (zLi = 1), um,i is the electric mobility coefficient, ϕ is the electrolyte potential.
Computational theoretical calculations
The present first principle DFT calculations are performed by Vienna Ab initio Simulation Package with the projector augmented wave method51. The exchange function is treated using the generalized gradient approximation of Perdew-Burke-Emzerh functional52. For Kohn–Sham wave functions, the cutoff energy of the corresponding plane-wave basis set was set to 450 eV. The K points meshing was obtained from the Monkhorst-Pack scheme53. Grimme’s DFT-D3 methodology was used to describe the dispersion interactions. A vacuum width of of 15 Å along the Z axis was created to ensure negligible interaction. The force convergence criterion was set to 0.02 eV/Å and energy convergence criterion was 10−4 eV. To fully consider the solvation effect, 18 explicit water molecules were optimized, and a local minimum via the hydrogen bond network was formed. The Gibbs free energy change (ΔG) of each step is calculated using the following formula:
where ΔE is the electronic energy difference directly obtained from DFT calculations, ΔZPE is the zero point energy difference, T is the room temperature (298.15 K) and ΔS is the entropy change.
Data availability
The data supporting the findings of the study are available within the paper and its Supplementary Information. Source data are provided in this paper.
References
Davis, S. J., Caldeira, K. & Matthews, H. D. Future CO2 emissions and climate change from existing energy infrastructure. Science 329, 1330–1333 (2010).
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294–303 (2012).
De Luna, P. et al. What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science 364, eaav3506 (2019).
Fang, M., Xu, L., Zhang, H., Zhu, Y. & Wong, W.-Y. Metalloporphyrin-linked mercurated graphynes for ultrastable CO2 Electroreduction to CO with nearly 100% selectivity at a current density of 1.2 A cm–2. J. Am. Chem. Soc. 144, 15143–15154 (2022).
Huang, J.-R. et al. Single-product faradaic efficiency for electrocatalytic of CO2 to CO at current density larger than 1.2 A cm−2 in neutral aqueous solution by a single-atom nanozyme. Angew. Chem. Int. Ed. 134, e202210985 (2022).
Li, Y. et al. Atomically dispersed single Ni site catalysts for high-efficiency CO2 electroreduction at industrial-level current densities. Energy Environ. Sci. 15, 2108–2119 (2022).
Yan, S. et al. Electron localization and lattice strain induced by surface lithium doping enable ampere-level electrosynthesis of formate from CO2. Angew. Chem. Int. Ed. 60, 25741–25745 (2021).
Wen, G. et al. Continuous CO2 electrolysis using a CO2 exsolution-induced flow cell. Nat. Energy 7, 978–988 (2022).
Ma, W. C. et al. Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C-C coupling over fluorine-modified copper. Nat. Catal. 3, 478–487 (2020).
de Arquer, F. P. G. et al. CO2 electrolysis to multicarbon products at activities greater than 1 A cm-2. Science 367, 661–666 (2020).
Möller, T. et al. Efficient CO2 to CO electrolysis on solid Ni–N–C catalysts at industrial current densities. Energy Environ. Sci. 12, 640–647 (2019).
Dinh, C. T. et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 360, 783–787 (2018).
Zhang, X. et al. Selective and high current CO2 electro-reduction to multicarbon products in near-neutral KCl electrolytes. J. Am. Chem. Soc. 143, 3245–3255 (2021).
Ma, M. et al. Insights into the carbon balance for CO2 electroreduction on Cu using gas diffusion electrode reactor designs. Energy Environ. Sci. 13, 977–985 (2020).
Xu, Y. et al. A microchanneled solid electrolyte for carbon-efficient CO2 electrolysis. Joule 6, 1333–1343 (2022).
Rabinowitz, J. A. & Kanan, M. W. The future of low-temperature carbon dioxide electrolysis depends on solving one basic problem. Nat. Commun. 11, 5231 (2020).
Zhao, Y. et al. Industrial-current-density CO2-to-C2+ electroreduction by anti-swelling anion-exchange ionomer-modified oxide-derived Cu nanosheets. J. Am. Chem. Soc. 144, 10446–10454 (2022).
Xie, K. et al. Eliminating the need for anodic gas separation in CO2 electroreduction systems via liquid-to-liquid anodic upgrading. Nat. Commun. 13, 3070 (2022).
Ozden, A. et al. Carbon-efficient carbon dioxide electrolysers. Nat. Sustain. 5, 563–573 (2022).
Shin, H., Hansen, K. U. & Jiao, F. Techno-economic assessment of low-temperature carbon dioxide electrolysis. Nat. Sustain. 4, 911–919 (2021).
Burdyny, T. & Smith, W. A. CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions. Energy Environ. Sci. 12, 1442–1453 (2019).
Huang, J. E. et al. CO2 electrolysis to multicarbon products in strong acid. Science 372, 1074–1078 (2021).
Xie, Y. et al. High carbon utilization in CO2 reduction to multi-carbon products in acidic media. Nat. Catal. 5, 564–570 (2022).
Gu, J. et al. Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium. Nat. Catal. 5, 268–276 (2022).
Qiao, Y. et al. Engineering the local microenvironment over Bi nanosheets for highly selective electrocatalytic conversion of CO2 to HCOOH in Strong Acid. ACS Catal. 12, 2357–2364 (2022).
Löffelholz, M., Osiewacz, J., Weseler, L. & Turek, T. Enhancing carbon efficiency in electrochemical CO2 reduction at silver gas diffusion electrodes-the effect of acidic electrolytes explained via TFFA modeling. J. Electrochem. Soc. 170, 123502 (2023).
Li, H. et al. Tailoring acidic microenvironments for carbon-efficient CO2 electrolysis over Ni-N-C catalyst in a membrane electrode assembly electrolyzer. Energy Environ. Sci. 16, 1502–1510 (2023).
Pan, B. et al. Close to 90% single-pass conversion efficiency for CO2 electroreduction in an acid-fed membrane electrode assembly. ACS Energy Lett. 7, 4224–4231 (2022).
Bondue, C. J., Graf, M., Goyal, A. & Koper, M. T. M. Suppression of hydrogen evolution in acidic electrolytes by electrochemical CO2 reduction. J. Am. Chem. Soc. 143, 279–285 (2021).
Zhang, Y.-J., Sethuraman, V., Michalsky, R. & Peterson, A. A. Competition between CO2 reduction and H2 evolution on transition-metal electrocatalysts. ACS Catal. 4, 3742–3748 (2014).
Cave, E. R. et al. Trends in the catalytic activity of hydrogen evolution during CO2 electroreduction on transition metals. ACS Catal. 8, 3035–3040 (2018).
Yan, Z. F., Hitt, J. L., Zeng, Z. C., Hickner, M. A. & Mallouk, T. E. Improving the efficiency of CO2 electrolysis by using a bipolar membrane with a weak-acid cation exchange layer. Nat. Chem. 13, 33–40 (2021).
Liu, Z. K. et al. Acidic electrocatalytic CO2 reduction using space-confined nanoreactors. ACS Appl. Mater. Interfaces 14, 7900–7908 (2022).
Kas, R. et al. Three-dimensional porous hollow fibre copper electrodes for efficient and high-rate electrochemical carbon dioxide reduction. Nat. Commun. 7, 10748 (2016).
Rabiee, H. et al. Shape-tuned electrodeposition of bismuth-based nanosheets on flow-through hollow fiber gas diffusion electrode for high-efficiency CO2 reduction to formate. Appl. Catal. B-Environ. 286, 119945 (2021).
Li, S. J. et al. Chloride ion adsorption enables ampere-level CO2 electroreduction over silver hollow fiber. Angew. Chem. Int. Ed. 61, e202210432 (2022).
Li, S. J. et al. Hierarchical micro/nanostructured silver hollow fiber boosts electroreduction of carbon dioxide. Nat. Commun. 13, 3080 (2022).
Zhu, C. et al. Ampere-level CO2 reduction to multicarbon products over a copper gas penetration electrode. Energy Environ. Sci. 15, 5391–5404 (2022).
Chen, A. et al. Gas penetrating hollow fiber Bi with contractive bond enables industry-level CO2 electroreduction. Appl. Catal. B-Environ. 333, 122768 (2023).
Sheng, X., Ge, W., Jiang, H. & Li, C. Engineering the Ni-N-C catalyst microenvironment enabling CO2 electroreduction with nearly 100% CO selectivity in acid. Adv. Mater. 34, 2201295 (2022).
Jiang, Z. et al. Molecular catalyst with near 100% selectivity for CO2 reduction in acidic electrolytes. Adv. Energy Mater. 13, 2203603 (2022).
O’Brien, C. P. et al. Single pass CO2 conversion exceeding 85% in the electrosynthesis of multicarbon products via local CO2 regeneration. ACS Energy Lett. 6, 2952–2959 (2021).
Rosen, J. et al. Mechanistic insights into the electrochemical reduction of CO2 to CO on nanostructured Ag surfaces. ACS Catal. 5, 4293–4299 (2015).
Dunwell, M., Luc, W., Yan, Y. S., Jiao, F. & Xu, B. J. Understanding surface-mediated electrochemical reactions: CO2 reduction and beyond. ACS Catal. 8, 8121–8129 (2018).
Ma, Z. et al. CO2 electroreduction to multicarbon products in strongly acidic electrolyte via synergistically modulating the local microenvironment. Nat. Commun. 13, 7596 (2022).
Liu, M. et al. Enhanced electrocatalytic CO2 reduction via field-induced reagent concentration. Nature 537, 382–386 (2016).
Hori, Y., Wakebe, H., Tsukamoto, T. & Koga, O. Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media. Electrochimica Acta 39, 1833–1839 (1994).
Gu, J., Hsu, C. S., Bai, L. C., Chen, H. M. & Hu, X. L. Atomically dispersed Fe3+ sites catalyze efficient CO2 electroreduction to CO. Science 364, 1091–1094 (2019).
Fan, M. et al. Cationic-group-functionalized electrocatalysts enable stable acidic CO2 electrolysis. Nat. Catal. 6, 763–772 (2023).
Wei, D. X. et al. Decrypting the controlled product selectivity over Ag-Cu bimetallic surface alloys for electrochemical CO2 reduction. Angew. Chem. Int. Ed. 62, e202217369 (2023).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Monkhorst, H. J. & Pack, J. D. Special points for brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Acknowledgements
This work was financially supported by the Ministry of Science and Technology of China (National Key R&D Program of China, 2022YFA1504604), the Strategic Priority Research Program (A) of the Chinese Academy of Sciences (XDA0390400), the National Natural Science Foundation of China (nos. 91745114, 21802160, 22302223), the Hundred Talents Program of Chinese Academy of Sciences (no. 2060299), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (no. E224301401), Shanghai Excellent Principal Investigator (no. 23XD1404400), Shanghai Sailing Program (nos. 23YF1453300, 18YF1425700), Science and Technology Innovation Planof Shanghai Science and Technology Commission (Nos. 23DZ1202600.23DZ1201804), the Outstanding Young Talent Project of Shanghai Advanced Research Institute, the Chinese Academy of Sciences (no. E254991ZZ1), the Foundation of Key Laboratory of Low-Carbon Conversion Science & Engineering, Shanghai Advanced Research Institute, Chinese Academy of Sciences (no. KLLCCSE-202207Z, SARI, CAS), Shanghai Functional Platform for Innovation Low Carbon Technology, and the Major Project of the Science and Technology department of Inner Mongolia (no. 2021ZD0020).
Author information
Authors and Affiliations
Contributions
S.L., X.D., G.W., and W.C. conceived the research and designed the experiment. S.L., X.D., G.W., Y.S., J.M., A.C., C.Z., G.L., Y.W., X.L., and J.W. conducted the experiment. S.L., X.D., G.W., and W.C. analyzed the data. S.L., X.D., G.W., Y.S., G.L., and W.C. discussed and interpreted the results. W.C. and W.W. supervised the project. S.L., X.D., and G.W. wrote the manuscript with input from all authors. All authors revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Tiras Lin, Richard Masel, Thomas Turek and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, S., Dong, X., Wu, G. et al. Ampere-level CO2 electroreduction with single-pass conversion exceeding 85% in acid over silver penetration electrodes. Nat Commun 15, 6101 (2024). https://doi.org/10.1038/s41467-024-50521-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-50521-8
This article is cited by
-
Sulfur-doping tunes p-d orbital coupling over asymmetric Zn-Sn dual-atom for boosting CO2 electroreduction to formate
Nature Communications (2025)
-
Two-in-One Integrated CO2/N2 Conversion and Related Systems: Potential, Status, and Future
Electrochemical Energy Reviews (2025)
-
Electrocatalytic CO2 reduction: surface dynamic effects on conversion efficiency
Ionics (2025)
