
Abstract
Spermiogenesis, the complex transformation of haploid spermatids into mature spermatozoa, relies on precise spatiotemporal regulation of gene expression at the post-transcriptional level. The mechanisms underlying this critical process remain incompletely understood. Here, we identify centrosomal protein 112 (CEP112) as an essential regulator of mRNA translation during this critical developmental process. Mutations in CEP112 are discovered in oligoasthenoteratospermic patients, and Cep112-deficient male mice recapitulate key phenotypes of human asthenoteratozoospermia. CEP112 localizes to the neck and atypical centrioles of mature sperm and forms RNA granules during spermiogenesis, enriching target mRNAs such as Fsip2, Cfap61, and Cfap74. Through multi-omics analyses and the TRICK reporter assay, we demonstrate that CEP112 orchestrates the translation of target mRNAs. Co-immunoprecipitation and mass spectrometry identify CEP112’s interactions with translation-related proteins, including hnRNPA2B1, EEF1A1, and EIF4A1. In vitro, CEP112 undergoes liquid-liquid phase separation, forming condensates that recruit essential proteins and mRNAs. Moreover, variants in patient-derived CEP112 disrupt phase separation and impair translation efficiency. Our results suggest that CEP112 mediates the assembly of RNA granules through liquid-liquid phase separation to control the post-transcriptional expression of fertility-related genes. This study not only clarifies CEP112’s role in spermatogenesis but also highlights the role of phase separation in translational regulation, providing insights into male infertility and suggesting potential therapeutic targets.
Similar content being viewed by others

Introduction
Spermiogenesis, the complex differentiation of post-meiotic round spermatids into mature spermatozoa, is a critical process in male reproduction1. This remarkable transformation relies on precise spatiotemporal regulation of gene expression, particularly at the post-transcriptional level. As spermatids undergo chromatin compaction, transcriptional silence necessitates a shift to post-transcriptional regulatory mechanisms to ensure protein synthesis for sperm maturation2,3. Therefore, the expression of spermiogenic genes depends on the post-transcriptional regulation of messenger ribonucleoproteins (mRNPs), which are composed of mRNAs and RNA-binding proteins (RBPs)4,5. mRNPs are stored in a translationally inactive state until they are transported to specific subcellular locations and activated by various signals5. Despite indications that both activation and repression of mRNP translation play indispensable roles during spermiogenesis, the specific regulatory mechanisms governing these dynamic transitions remain poorly defined.
Centrosomal proteins (CEPs) have emerged as important regulators of spermatogenesis and male fertility. Variants in genes encoding for CEPs such as CEP55 and CEP135 have been linked to morphological abnormalities in sperm and male sterility6,7,8. Centrin 1 may participate in fibrous sheath development and sperm head–neck junctions9. Cep131 loss promoted manchette and flagella defects, leading to male infertility in mice10; CEP78 regulates spermatogenesis by modulating USP16-mediated Tektin function, while CEP128 is involved in male reproduction by regulating gene expression and/or phosphorylation of TGF-β/BMP-signaling members11,12. Among these CEPs, the 112 kDa Centrosomal Protein (CEP112) has been implicated in male infertility, yet its specific functions remain largely unexplored. CEP112 localizes to the centrosome13 and has been identified as a putative RNA-binding protein (RBP) that interacts with the noncoding RNA Ginir in cancer cells14. Additionally, CEP112 has been assigned to the basal foot, a conical structure extending from the microtubule wall of the basal body in primary and motile cilia15. Recent studies have linked mutations in CEP112 to the human acephalic spermatozoa phenotype, characterized by the absence of sperm heads16. This finding suggests that CEP112 may play a crucial role in the proper development of sperm morphology. Despite these initial findings, the molecular mechanisms underlying CEP112’s involvement in spermatogenesis and male fertility are not well understood. Its potential functions as an RNA-binding protein suggest that CEP112 may participate in the post-transcriptional regulation of genes critical for sperm development.
Liquid-liquid phase separation (LLPS) is increasingly recognized as a fundamental mechanism for organizing cellular biochemistry17,18. This process, mediated by proteins often rich in intrinsically disordered regions, drives the formation of dynamic, membraneless organelles that facilitate efficient and localized biochemical reactions17. Recent studies indicate LLPS plays an important role in reproduction. MAGE-B2 maintains spermatogonial stem cell stemness by reducing G3BP protein expression to inhibit stress granule formation19. In meiosis, LLPS can drive the formation of synaptonemal complexes20, meiotic sex chromosome inactivation, and subsequent XY-body formation21. Acentrosomal spindle assembly is efficiently driven by LLPS within mammalian eggs22. LLPS is crucial for Piwi-interacting RNA-mediated gene silencing and various RNA processing activities within chromatoid bodies and piP-bodies of round spermatids23. Intriguingly, FXR1, a gene related to Fragile X syndrome, has been shown to facilitate the amalgamation of messenger ribonucleoprotein granules with translational machinery through LLPS, thereby initiating mRNA translation during spermiogenesis24. Despite these insights, the full spectrum of LLPS functions in spermatogenesis remains largely uncharted. Furthermore, whether defective phase separation of germ cells directly contributes to male infertility has not been reported. Therefore, a deeper understanding of LLPS’s role in this context is essential for elucidating the molecular intricacies of spermatogenesis.
In this study, we present compelling evidence for CEP112 as a critical regulator of mRNA translation during spermiogenesis, operating through a mechanism of liquid-liquid phase separation. Our investigation was sparked by the discovery of CEP112 mutations in men with oligoasthenoteratozoospermia, a finding that broadens the spectrum of CEP112-associated male infertility phenotypes beyond the previously reported acephalic spermatozoa16. Through a comprehensive approach combining genetic analysis, multi-omics studies, and advanced imaging techniques, we demonstrate that CEP112 forms phase-separated condensates that sequester and regulate the translation of key spermiogenesis-related mRNAs. Our findings reveal a function for CEP112 in coordinating the post-transcriptional regulation of fertility genes, distinct from its known structural roles at the centrosome and in sperm head-tail attachment. We show that CEP112 interacts with crucial translation factors and RNA-binding proteins, forming a specialized translational regulatory complex during spermiogenesis. The ability of CEP112 to undergo LLPS and form biomolecular condensates provides a mechanistic basis for its role in mRNA regulation, contributing to our understanding of how spatial organization of cellular components can influence gene expression during gametogenesis. Moreover, our study bridges the gap between basic molecular mechanisms and clinical relevance by demonstrating how mutations in CEP112 disrupt its phase separation properties and impair translation efficiency, directly linking molecular dysfunction to infertility phenotypes. This work not only advances our understanding of the molecular underpinnings of spermatogenesis but also opens potential avenues for diagnostic and therapeutic approaches in male infertility.
Results
Bi-allelic CEP112 variants in men with oligoasthenoteratozoospermia
To explore the etiology of male infertility, we performed whole-exome sequencing (WES) on 568 unrelated men diagnosed with oligoasthenoteratozoospermia. Bi-allelic variants in the CEP112 gene (NM_001199165.4, NP_001186094.1) were identified in two cases (0.35% of the cohort) through filtering of the WES data (Fig. 1a). The variants were validated by PCR and Sanger sequencing in the probands and available family members, which showed that the bi-allelic variants were inherited from heterozygous carrier parents (Fig. 1a). This is consistent with an autosomal recessive inheritance pattern. Interestingly, one proband (Y9438) exhibited recurrent upper respiratory tract infections, suggesting a broader phenotypic impact of CEP112 variants. Analysis of population databases including 1000 Genomes, GnomAD, and GnomAD-EAS showed that four CEP112 variants detected in this study were either present at low allele frequencies or absent in the general population. These mutations are predicted to be deleterious, with the p.R76X mutation expected to generate premature stop codons leading to truncated proteins, and the missense mutations (p.S137L; p.G90R; p.H766R) located in evolutionarily conserved regions of the gene (Supplementary Table S1, Fig. 1b, c). None of the identified CEP112 variants were present in a control cohort of 392 fertile men. These findings expand the spectrum of CEP112-associated male infertility phenotypes beyond the previously reported acephalic spermatozoa syndrome16. The identification of CEP112 mutations in patients with oligoasthenoteratozoospermia suggests that CEP112 dysfunction may lead to diverse sperm defects. This phenotypic variability likely reflects the different functional consequences of distinct mutations within CEP112, highlighting the complex genotype-phenotype relationships in male infertility.

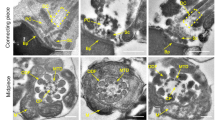
a Pedigrees and Sanger sequencing confirmation of bi-allelic CEP112 variants in two unrelated men with oligoasthenoteratozoospermia (F1-Y3937 and F2-Y9438). Affected individuals: filled symbols; unaffected: open symbols. Probands screened by whole-exome sequencing (WES): black arrows. Sanger sequencing was performed on numbered individuals to validate the variants. C: control. b Schematic representation of the CEP112 protein structure and location of identified variants. Domains are indicated by colored rectangles: DUF4485 (red) and coiled-coil regions (purple). Patient mutations discovered in this study are shown in red (c.226C>T p.R76X, c.268G>A p.G90R, c.410C>T p.S137L, c.2297A>G p.H766R), while previously reported variants associated with acephalic spermatozoa syndrome are shown in black (c.496C>T p.R166X, c.2074C>T p.R692W, c.2104C>T p.R702C) above the protein structure. The genomic structure of CEP112 is shown below the protein diagram, with exons represented by vertical lines. c Conservation of the missense-mutated amino acids is indicated by the alignment of nine species. The detected missense variants were conserved according to the sequence alignment. Morphological assessment of spermatozoa from a fertile control and CEP112-deficient probands using Papanicolaou staining (d) and scanning electron microscopy (SEM) (e). Spermatozoa from the control exhibit normal morphology with elongated, smooth flagella. In contrast, spermatozoa from probands display a range of head and flagellar abnormalities (Scale bar: 5 μm). f Transmission electron microscopy (TEM) reveals ultrastructural defects in sperm flagella of CEP112-deficient individuals. Cross-sections of control spermatozoa show the canonical “9 + 2” axonemal structure. Spermatozoa from probands exhibit loss of central pair (CP) microtubules (“9 + 0” configuration), disorganization of the “9 + 2” structure, and disordered outer dense fibers (ODFs) in the midpiece. Principal and end piece sections display missing or disorganized central and peripheral microtubules, as well as incomplete fibrous sheaths (Scale bar: 100 nm). g Aberrant ultrastructure of the connecting piece in spermatozoa from CEP112-deficient probands revealed by TEM. Proximal centrioles (PCs) and distal centrioles (DCs) are missing or incomplete in the connecting piece of affected individuals (Scale bar: 100 nm). Experiments were repeated three times independently with similar results; representative images are shown in d–g.
Clinical assessments following World Health Organization guidelines25 showed significantly reduced sperm concentrations and complete lack of motility in both probands (Supplementary Table S2). In our analysis of patient Y9438, a sperm chromatin structure assay (SCSA) revealed a DNA fragmentation index (DFI) of 7.83% and high DNA stainability (HDS) of 0.60%, both within normal ranges. Staining with Papanicolaou Stain coupled with scanning electron microscopy (SEM) revealed considerable sperm morphology defects in their sperm samples, with a high percentage of aberrant tails seen in both men (Fig. 1d, e, Supplementary Fig. S1a, b). Transmission electron microscopy (TEM) further highlighted axonemal ultrastructural defects in the sperm flagella of these men. Compared to control sperm, which exhibited the typical “9 + 2” microtubule structure, the sperm flagella from the CEP112 variant carriers showed a range of abnormalities, including swollen mitochondrial sheaths and disorganized or absent outer dense fibers and dynein arms (Fig. 1f, Supplementary Fig. S1c). These defects were pervasive across the midpiece, principal piece, and end piece of the sperm flagella. Additionally, TEM analysis revealed anomalies in the connecting pieces of the sperm, with incomplete or missing proximal and distal centrioles despite the presence of the basal plate (Fig. 1g, Supplementary Fig. S1d).
To evaluate if male infertility resulting from CEP112 variants could be overcome by intracytoplasmic sperm injection (ICSI), we analyzed the outcome of ICSI cycles in patient Y3937. As presented in Supplementary Table S3, only two of the 11 embryos achieved the 8-cell stage, signaling poor embryonic development. However, the family encountered a miscarriage following implantation.
CEP112 expression and localization during spermatogenesis
To investigate the role of CEP112 in spermatogenesis, we analyzed its expression patterns in both human and mouse tissues. Reverse transcription PCR (RT-PCR) showed higher Cep112 mRNA levels in mouse testes compared to other tissues (Supplementary Fig. S2a). In mice, CEP112 expression was first detectable at postnatal day 7 (P7), gradually increased with a peak at P42, and persisted at high levels thereafter (Supplementary Fig. S2b). Immunofluorescence analysis revealed low CEP112 expression in spermatogonia and spermatocytes, but markedly increased expression during spermiogenesis (Supplementary Fig. S2c). Similar expression was seen in human testis sections (Supplementary Fig. S2d), corroborating single-cell RNA sequencing data (Supplementary Fig. S2e). Additionally, Western blot analysis of sorted mouse spermatogenic cells revealed significantly higher CEP112 protein levels in round and elongating spermatids compared to spermatogonia and spermatocytes (Supplementary Fig. S2f)), indicating a critical role for CEP112 during spermiogenesis. To gain higher resolution insights into CEP112 localization, we employed structured illumination microscopy (SIM) on isolated spermatogenic cell populations. SIM imaging revealed a dynamic distribution of CEP112 throughout spermatogenesis. In spermatogonia and early spermatocytes, CEP112 showed a diffuse localization in both the cytoplasm and nucleus. As cells progressed to the round spermatid stage, CEP112 began to concentrate in distinct cytoplasmic foci. In elongating spermatids, these foci became more pronounced and localized primarily to the cytoplasm, often in close proximity to the developing acrosome and manchette (Fig. 2a, b). Intriguingly, in mature spermatozoa, CEP112 exhibited a highly specific localization pattern. Immunofluorescence and SIM analyses indicated that CEP112 is localized to the neck region of mouse sperm (Fig. 2c) and to atypical centrioles and the base of the axoneme in human sperm (Fig. 2d, Supplementary Movie 1). Notably, atypical centrioles play a vital role in facilitating asymmetric tail beating of sperm and zygotic functions26,27. To further elucidate CEP112’s subcellular localization, we conducted immunogold labeling followed by transmission electron microscopy (IG-TEM). This approach primarily detected gold particles in atypical centrioles and axofilaments beneath the atypical centrioles (Fig. 2e). Quantitative analysis of gold particle distribution revealed that nearly half of the particles were associated with atypical centrioles (49.93 ± 35.90%), while a substantial proportion was linked to the adjacent axonemal base (43.45 ± 41.67%). This distinct distribution pattern underscores the potential significance of CEP112 in the organization and function of sperm flagella (Supplementary Fig. S2g).

a Immunofluorescence (IF) staining of CEP112 expression across various germ cell stages in mice. (green, CEP112; blue, DAPI; Scale bar: 5 μm). b IF staining of CEP112 expression across various germ cell stages in humans. (green, CEP112; red, PNA; blue, DAPI; Scale bar: 5 μm). c Single-sperm IF analysis demonstrates CEP112 localization to the neck region of mouse spermatozoa (green, CEP112; magenta, tubulin; blue, DAPI; Scale bar: 5 μm). d Structured illumination microscopy (SIM)-based 3D reconstruction of human spermatozoa reveals CEP112 localization to atypical centrioles and the axonemal base. Lateral and axial line profiles are indicated by white dotted lines. PC, proximal centriole; DC, distal centriole. (green, CEP112; magenta, Ac-tubulin; blue, DAPI; Scale bar: 5 μm). e Immuno-gold transmission electron microscopy (IG-TEM) analysis of CEP112 subcellular localization in human spermatozoa. PC proximal centriole, DC distal centriole, Ax axoneme, Bp basal plate, Sc segmented column (Scale bar: 500 nm). f Co-immunofluorescence staining of CEP112 with centrosomal and axonemal markers (Ac-tubulin, POC1B, POC5, CEP290, or CEP63) validates CEP112 localization to atypical centrioles and axofilaments in human spermatozoa. Magnified areas are represented by dotted rectangles. Right panels, areas of co-localization represented by line-intensity profiles (green, CEP112; magenta, various markers as indicated; blue, DAPI; Scale bar: 5 μm). g Schematic representation summarizing the localization patterns of CEP112 in human spermatozoa. PC proximal centriole, DC distal centriole, Ax axoneme, Bp basal plate. h Phylogenetic analysis of CEP112 orthologs demonstrates evolutionary conservation across species. Numbers at branch points represent bootstrap values (0–100), indicating the statistical support for each branching event in the evolutionary tree of Cep112. i IF analysis of CEP112 localization in porcine and bovine spermatozoa confirms its presence in atypical centrioles. Magnified areas are indicated by dotted rectangles (green, CEP112; magenta, Ac-tubulin; blue, DAPI; Scale bar: 5 μm). Experiments were repeated three times independently with similar results; representative images are shown in a–f and i.
To contextualize CEP112’s localization relative to other known centriolar proteins, we conducted co-localization studies using several established markers. These included Ac-tubulin (localized to proximal centrioles(PCs), distal centrioles(DCs), and the axoneme), POC1B (preferentially in DCs), POC5 (equally in DCs and PCs), CEP63 (in PCs and DCs), and CEP290 (marking the tip of DCs)26. SIM revealed CEP112 was below CEP63 in the axoneme; co-localized with DCs labeled with POC1B, POC5, and CEP290-labeled DCs; and co-stained with Ac-tubulin, indicating its localization at the base of the axoneme (Fig. 2f, g). Multialignment and phylogenetic analyses of CEP112 orthologs across various species demonstrate its evolutionary conservation, particularly in large mammals (Fig. 2h). IF staining also showed CEP112’s localization to atypical centrioles of porcine and bovine spermatozoa (Fig. 2i), suggesting that CEP112 plays a similar essential role in the reproduction of large mammals. These detailed localization studies provide crucial insights into the potential functions of CEP112 during spermiogenesis and in mature sperm. The dynamic redistribution of CEP112 from a diffuse pattern in early germ cells to distinct cytoplasmic foci in spermatids suggests a transition in its functional role during spermatogenesis. Its ultimate localization to atypical centrioles and the axonemal base in mature sperm points to a potential structural or regulatory role in sperm motility and fertilization.
Cep112 deletion impairs mouse fertility and spermatogenesis
To elucidate the functional significance of CEP112 in spermatogenesis, we engineered Cep112 knockout (Cep112−/−) mice using the CRISPR-Cas9 system targeting exons 4 and 5. Successful deletion was validated by qPCR, Western blotting, Sanger sequencing and immunofluorescence assay (Supplementary Fig. S3a–e). Fertility assessments revealed that homozygous males (Cep112−/−) displayed severely impaired fertility with almost no offspring, whereas heterozygous males (Cep112+/−) remained fertile (Fig. 3a). Notably, female reproduction capacity remained unaffected across all genotypes (Fig. 3a), indicating that CEP112 plays a critical and male-specific role in fertility. Testis weights of knockout mice were similar to wildtype (Cep112+/+) from postnatal day 7 to 42 but slightly lower thereafter (Supplementary Fig. S3f), with no epididymal weight differences (Supplementary Fig. S3g). Computer-assisted sperm analysis (CASA) revealed Cep112−/− mice had significantly lower sperm counts and motility compared to Cep112+/+, lacking forward progressive motility (Table 1, Supplementary Movie 2, 3). Quantitative analysis using the STA-PUT velocity sedimentation method for counting spermatogenic cells indicated a substantial decrease in elongated spermatids within Cep112−/− mouse testes (Fig. 3b), suggesting a defect in the later stages of spermatogenesis. Histological analysis of testicular sections revealed the presence of immature or degenerative spermatogenic cells in the lumen and disrupted cell-to-cell contact within the seminiferous tubules of Cep112−/− mice (Fig. 3c). TUNEL assay showed a significant increase in apoptotic cells in Cep112−/− testes compared to wildtype (Supplementary Fig. S3h). While abundant sperm filled wildtype epididymides, Cep112−/− epididymides contained disorganized sperm clumps (Fig. 3d). Detailed morphological analysis using Papanicolaou staining highlighted severe abnormalities in Cep112−/− sperm, including tail defects like absent, short, bent, or coiled flagella (Fig. 3e, Supplementary Fig. S3i, j).

a Wildtype (WT) mice (Cep112+/+), heterozygous mice (Cep112+/−), and Cep112-knockout mice (Cep112−/−) were bred with WT counterparts to determine average litter sizes. Cep112−/− male mice exhibited severely reduced fertility, with almost no offspring produced, while Cep112+/− male mice remained fertile. In contrast, female reproductive function was unaffected in Cep112−/− mice. n = 12 independent mice per sex per genotype, each mated with two WT counterparts. (Two-sided Student’s t test; error bars, mean ± SEM). The p values are indicated in the graphs. Source data are provided as a Source Data file. b Spermatogenic cell quantification using STA-PUT method: Cep112−/− mice had significantly fewer round and elongating/elongated spermatids. n = 3 independent mice per genotype. (Two-sided Student’s t test; error bars, mean ± SEM). The p values are indicated in the graphs. Source data are provided as a Source Data file. c and d Histological analysis of testes and epididymides from Cep112−/− mice. Hematoxylin and eosin (H&E) staining of testicular sections revealed degenerated spermatogenic cells in the lumen and disrupted cell-to-cell contacts within seminiferous tubules at different stages of spermatogenesis (c). Epididymal sections from Cep112−/− mice showed significantly fewer spermatozoa compared to Cep112+/+ (d) (Scale bar: 50 μm). Papanicolaou staining (e) and SEM analysis (f) showed that Cep112−/− mice had abnormal sperm morphologies, including absent, short, bent and coiled flagella, and defective heads were also visible. SEM analysis also showed abnormal annulus and disorganized midpiece-principal piece junctions in Cep112−/− spermatozoa. Magnified areas are indicated by magenta dashed rectangles (Scale bar: 5 μm). g TEM cross-sectional analysis of Cep112−/− mouse sperm flagella revealed various ultrastructural defects, including mitochondrial sheath (MS) abnormalities, disordered and absent outer dense fibers (ODFs), missing microtubules, disorganized microtubule doublets (MTDs), and absence of central pairs (CPs) (Scale bar: 500 nm). h TEM analysis of spermiogenesis in Cep112−/− mice showed impaired development of the connecting piece and defective conjunctions between sperm head and tail at different developmental stages (Scale bar: 500 nm). Experiments were repeated three times independently with similar results; representative images are shown in c–h.
Scanning electron microscopy (SEM) showed abnormal annuli and disorganized midpiece-principal piece junctions in knockout sperm (Fig. 3f). Immunostaining revealed marginal annulus protein SEPT4 expression compared to wildtype (Supplementary Fig. S3k)). Transmission electron microscopy (TEM) investigations of sperm from the testes and epididymides revealed profound ultrastructural defects in Cep112−/− mice. While normal sperm exhibited the characteristic “9 + 2” axonemal arrangement, encased by outer dense fibers28,29, Cep112−/− sperm showed statistically significant increase in structural abnormalities within the midpiece, principal piece, and end piece (Fig. 3g, Supplementary Fig. S3I)). Deformities included irregular or missing outer dense fibers, and disorganized or absent doublet microtubules. Notably, the central pair was frequently missing in the axoneme’s principal and midpiece sections (Fig. 3g). Further TEM analysis indicated aberrant assembly of the connecting piece and defective conjunctions between sperm head and tail in Cep112−/− testicular sperm. In earlier spermatid stages (9–12), the proximal centriole, segmented column, and basal plate appeared typical, but disarray was evident in the proximal centriole, and the segmented column was misaligned during later stages (13–16), leading to separation of the sperm head and neck. Disorganized axonemal filaments and cytoplasmic remnants were also observed in the neck region of sperm from Cep112−/− epididymides (Fig. 3h). To assess the functional capacity of Cep112−/− sperm, we conducted acrosome reaction assays. Upon induction with progesterone, only 27.96 ± 2.04% of Cep112−/− sperm exhibited acrosome reaction compared to 54.41 ± 8.58% of wildtype sperm (Supplementary Fig. S3m). Together, these defects likely cause the poor motility and morphology, consistent with asthenoteratozoospermia phenotype.
In assessing the fertility of Cep112−/− male mice through IVF and ICSI assays, we observed significantly lower development rates of zygotes, two-cell embryos, and blastocysts in IVF using Cep112−/− sperm compared to Cep112+/+ sperm (Supplementary Fig. S4a, b). However, ICSI showed improved outcomes with Cep112−/− sperm. Zygote development to blastocyst stages in both Cep112+/+ and Cep112−/− groups was similar (Supplementary Fig. S4c, d). TUNEL and γH2AX assays detected no significant DNA damage differences in embryos from both groups (Supplementary Fig. S4e, f). Embryo transfer experiments resulted in successful deliveries of ICSI pups in both Cep112+/+ (n = 11) and Cep112−/− (n = 24) groups (Supplementary Fig. S4g), demonstrating that ICSI can overcome IVF failures with Cep112−/− sperm.
CEP112’s role in post-transcriptional control in spermiogenesis
To further explore the role of CEP112 in spermatogenesis following the observed male infertility in both CEP112 mutation carriers and knockout mice, proteomic analysis was conducted on the testes of Cep112 knockout mice. Principal Component Analysis (PCA) and Pearson correlation validated the consistency of biological replicates from Cep112+/+ and Cep112−/− mouse testes (Supplementary Fig. S5a, b). A significant differential protein expression was noted, with 417 proteins upregulated and 314 downregulated in the Cep112−/− group (Supplementary Fig. S5c). Gene Ontology (GO) analysis revealed a marked enrichment in biological processes and cellular components essential for male reproduction, including spermatogenesis and axonemal structure (Fig. 4a). This finding was further corroborated by Gene Set Enrichment Analysis (GSEA), which demonstrated a significant association of these proteins with male reproductive functions, particularly sperm motility and fertilization (Fig. 4b, Supplementary Fig. S5d). Immunoblotting confirmed that key proteins involved in sperm flagellogenesis and fertilization (CFAP74. CFAP61, FSIP2, QRICH2, DPY19L2, ZPBP1, SPACA1, PRSS55) were significantly downregulated in Cep112−/− mice (Supplementary Fig. S5e, f). To investigate whether the downregulation of crucial proteins involved in male reproduction was a consequence of mRNA expression changes, we conducted RNA sequencing (RNA-Seq) analysis was conducted on testes from Cep112+/+ and Cep112−/− mice. Intriguingly, our integrated analysis of proteomic and transcriptomic data revealed a complex regulatory landscape. The majority of differentially expressed proteins did not exhibit corresponding changes in mRNA levels, with only a few exceptions, such as Serpina3n, showing concordant downregulation at both protein and RNA levels (Fig. 4c). This discrepancy suggests CEP112 may regulate its targets post-transcriptionally, potentially affecting translation. Gene Ontology (GO) analysis of RNA data revealed no significant association with spermatogenesis but demonstrated enrichment for terms related to other developmental processes, including kidney development, inner ear development, and heart development (Supplementary Fig. S6a). While previous studies have linked CEP mutations to a spectrum of ciliopathies30,31, our histological examination of Cep112−/− mice did not show any developmental anomalies in the kidneys or heart (Supplementary Fig. S6b, c), nor were there morphological differences in cochlear kinocilia or auditory function deficits (Supplementary Fig. S6d, f).

a Gene Ontology (GO) analysis of differentially expressed proteins in Cep112−/− testis shows significant enrichment for terms related to spermatogenesis and axonemal structures. Analysis was performed using the clusterProfiler R package (two-sided hypergeometric test; adjusted p-value < 0.05). b Gene Set Enrichment Analysis (GSEA) demonstrates significant enrichment of gene sets associated with male reproduction in Cep112+/+ compared to Cep112−/− testis. Statistical significance was assessed using 1000 permutations (FDR < 0.25, two-sided). c Venn diagram shows minimal overlap between downregulated genes in the transcriptome and proteome of Cep112−/− testis. d RIP-Seq GO analysis reveals enrichment in reproductive processes (two-sided hypergeometric test; adjusted p-value < 0.05). e Motif analysis of CEP112-binding mRNAs shows specificity for known and de novo motifs. f Genome browser tracks display RIP-Seq data for Fsip2, Cfap61, and Cfap74. g RIP-qPCR confirms CEP112 binding to Fsip2, Cfap61, and Cfap74 mRNAs. Three independent experiments were performed. (Two-sided Student’s t test; error bars, mean ± SEM). The p values are indicated in the graphs. Source data are provided as a Source Data file. h RT-qPCR shows consistent mRNA levels of Fsip2, Cfap61, and Cfap74 between Cep112+/+ and Cep112−/− testes. n = 3 independent mice per genotype. (Two-sided Student’s t test; error bars, mean ± SEM). Source data are provided as a Source Data file. i Representative polysome gradient profiles from Cep112+/+ and Cep112−/− mouse testes, showing the positions of 40S, 60S subunits, 80S monosomes, and polysomes. Western blot analysis indicates CEP112 distribution across sucrose gradient fractions, predominantly in the RNP and lighter polysome regions. Three independent experiments were performed. Source data are provided as a Source Data file. j–l Polysome distribution of Cfap61, Cfap74, and Fsip2 mRNAs in Cep112-/ testes indicates reduced translational efficiency, confirmed by quantitative analysis. (Two-sided Student’s t test; error bars, mean ± SEM). Source data are provided as a Source Data file. m TRICK reporter assay in COS7 cells demonstrates CEP112’s role in promoting CFAP61 mRNA translation. Puromycin confirms translation-dependent RFP signals. Experiments were repeated three times independently; representative images are shown.
Given CEP112’s known interactions with long noncoding RNAs14, we hypothesized a regulatory role for CEP112 in post-transcriptional regulation during spermatogenesis. To test this hypothesis, we performed RNA immunoprecipitation sequencing (RIP-Seq). Our analysis confirmed CEP112 binding predominantly to coding sequences and 5′-UTRs (Supplementary Fig. S6g)). Genomic analysis of RIP-Seq data revealed 446 mRNAs coimmunoprecipitated with CEP112 in mouse testes, with significant enrichment observed in the Cep112+/+ group compared to Cep112−/− controls (Supplementary Fig. S6h)). GO analysis indicated that these CEP112-interacting mRNAs are implicated in gamete generation and reproductive cellular processes (Fig. 4d). Motif analysis revealed preferential binding of CEP112 to established motif regions, such as MD59_0.6/RRM/RRM_1, and de novo motifs like ACCG, suggesting a specificity in RNA element interactions (Fig. 4e). RIP-Seq analysis highlighted significant enrichment for mRNAs such as Cfap61, Cfap74 and Fsip2, which are essential for sperm-flagellum formation and fertilization (Fig. 4f). Furthermore, molecular simulations of CEP112 binding to the identified RNA sequences demonstrated favorable binding energies, providing additional support for the specificity of these interactions (Supplementary Table S4). Immunoblot analysis of isolated spermatogenic cells revealed highest expression of CFAP61, CFAP74, and FSIP2 in elongating spermatids compared to earlier stages (Supplementary Fig. S6i, j), coinciding with CEP112’s peak expression and suggesting its role in regulating these proteins during late spermiogenesis. RIP-qPCR confirmed these findings, with significant enrichment of these target mRNAs compared to IgG controls (Fig. 4g). Despite consistent mRNA expression levels, proteomic and immunoblotting analyses indicated protein downregulation in Cep112−/− mice (Fig. 4h, Supplementary Fig. S5e, f). Immunofluorescence staining corroborated lower expression levels of these proteins in spermatozoa from both infertile patients with CEP112 variants and Cep112−/− mice (Supplementary Fig. S7a, b). To further elucidate the spatial relationship between CEP112 and its target mRNAs, we performed combined immunofluorescence and fluorescence in situ hybridization (IF-FISH) assays. These experiments demonstrated CEP112’s interaction with Fsip2 and Cfap61 in granular structures within spermatogenic cells (Supplementary Fig. S8a, b).
To assess the translational impact of CEP112, we examined the ribosome profiles of Cfap61, Cfap74, and Fsip2 mRNAs in the testes of both Cep112+/+ and Cep112−/− mice to assess translational impact. CEP112 predominantly localized to lighter polysome fractions, and global polysome-to-monosome (P/M) ratios remained unchanged between Cep112+/+ and Cep112−/− testes (1.28 ± 0.03 vs 1.28 ± 0.04, p > 0.05). This suggests CEP112 regulates specific mRNAs rather than modulating overall translational activity (Fig. 4i, Supplementary Fig. S8c). Nevertheless, further polysome profiling revealed a notable shift of Cfap61, Cfap74, and Fsip2 mRNAs towards lighter polysome fractions in the Cep112−/− mice, suggesting a diminished translation efficiency (Fig. 4j–l). To directly visualize the impact of CEP112 on mRNA translation, we employed the TRICK (Translating RNA Imaging by Coat protein Knock-off) reporter system in COS7 cells. Co-transfection of CFAP61 mRNA (carrying PP7 and MS2 stem loops) with NLS-PCP-GFP and NLS-MCP-RFP resulted in GFP and RFP labeling of the untranslated mRNA within the nucleus. Introduction of BFP-CEP112 led to the formation of cytoplasmic CEP112 granules containing CFAP61 mRNA, demonstrating that CEP112 sequesters target mRNAs. Notably, these sequestered mRNAs were labeled only with RFP, suggesting active translation. This finding was further validated by puromycin treatment as control, which resulted in dual GFP and RFP labeling within the CEP112 granules (Fig. 4m, Supplementary Fig. S8d). Quantitative analysis revealed that CEP112 expression significantly increased the proportion of translated CFAP61 mRNA (RFP-only granules) from 10.66 ± 3.16% to 73.28 ± 8.20% (Supplementary Fig. S8d).
Collectively, these multi-omics analyses and functional assays provide compelling evidence for CEP112’s role as a post-transcriptional regulator during spermiogenesis. Our findings suggest that CEP112 actively recruits specific target mRNAs into granular structures and promotes their translation, thereby orchestrating the expression of key proteins essential for sperm development and function
CEP112 forms translational complex in spermiogenesis
To elucidate CEP112’s role in mRNA translation during spermatogenesis, we utilized an anti-CEP112 antibody for immunoprecipitation (IP) experiments with total protein extracts from mouse testes, aiming to identify proteins interacting with CEP112. These proteins were then analyzed via liquid chromatography-tandem mass spectrometry (LC-MS/MS). Notably, the CEP112 interactome revealed the presence of key translation factors EIF4A1 and EEF1A1, along with the RNA-binding protein hnRNPA2B1(Supplementary Data 1, Fig. 5a). As an RNA-binding protein that enhances ribosome recruitment, hnRNPA2B1 plays a key role in translation initiation by binding target mRNA 5’-untranslated regions32. EIF4A1 unwinds secondary structures within mRNA 5’-untranslated regions to facilitate ribosome scanning for the start codon33. The elongation factor EEF1A1, together with EFTu-GTP, delivers aminoacyl-tRNAs to the elongating ribosome34. These CEP112 interactions were validated in co-immunoprecipitation assays in mouse testis and were not affected by RNA (Fig. 5b). Notably, Quantitative analysis revealed that the interactions between CEP112 and hnRNPA2B1, EEF1A1, and EIF4A1 were significantly enhanced during spermiogenesis compared to in spermatocytes (Fig. 5c, Supplementary Fig. S9a), suggesting a critical role for these interactions during the translational wave of spermiogenesis. Further, co-IP assays in HEK293T cells expressing tagged versions of these proteins confirmed their in vitro interactions with CEP112, independent of RNA (Fig. 5d–f). Immunofluorescence analysis revealed punctate co-localization of CEP112 and these translation-related proteins in the cytoplasm of elongating spermatids, resembling biomolecular condensates (Fig. 5g–i).

a Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of CEP112 immunoprecipitates from mouse testes reveals hnRNPA2B1, EEF1A1, and EIF4A1 as interacting proteins. The table summarizes the CEP112-interacting proteins identified, their sequence coverage, number of unique peptides, and peptide intensity. b Co-immunoprecipitation (co-IP) assays confirm the interactions between endogenous CEP112 and hnRNPA2B1, eEF1A1, and eIF4A1 in mouse testes. These interactions are not affected by RNase A treatment. c Co-immunoprecipitation of CEP112 and its interacting proteins during spermatogenesis. Lysates from spermatocytes (SC), round spermatids (RS), and elongating spermatids (ES) were immunoprecipitated with anti-CEP112 antibody. Western blots show co-precipitation of hnRNPA2B1, EEF1A1, and EIF4A1, normalized to immunoprecipitated CEP112. The interactions between CEP112 and these proteins progressively strengthen during spermiogenesis. Co-IP assays in HEK293T cells co-transfected with plasmids encoding Myc-tagged CEP112 and His-tagged hnRNPA2B1 (d), HA-tagged EEF1A1 (e), or Flag-tagged EIF4A1 (f) validate the interactions between CEP112 and these translation-related proteins in vitro. The interactions are not disrupted by RNase A treatment. Immunofluorescence staining of CEP112 (green) and hnRNPA2B1 (g), EEF1A1 (h), or EIF4A1 (i) (magenta) in mouse spermatogenic cells at different stages reveals their co-localization within cytoplasmic condensates during spermiogenesis. Nuclei are stained with DAPI (blue). Right panels display line-intensity profiles highlighting areas of co-localization (Scale bar: 5 μm). Lower panels show Pearson correlation coefficient analysis of co-localization at each spermatogenic stage. Data are presented as mean Pearson correlation coefficient values ± SEM. Error bars represent SD. n = 30 cells per spermatogenic stage. Source data are provided as a Source Data file. Experiments were repeated three times independently with similar results; representative images are shown in b–f.
To gain structural insights into the interactions between CEP112 and its binding partners, we performed computational molecular docking studies. These analyses revealed that the N-terminal region of CEP112 (amino acids 1–478) is primarily responsible for binding to hnRNPA2B1 and EIF4A1, facilitated by numerous hydrogen bonds and salt bridges at the respective binding interfaces (Supplementary Data 2, Supplementary Fig. S9b, c)). In contrast, the C-terminal region of CEP112 (amino acids 479–955) harbors the principal binding interface for EEF1A1, mediated by several hydrogen bonds and salt bridge contacts (Supplementary Data 2, Supplementary Fig. S9d)).
CEP112-mediated biomolecular condensates driven by LLPS
Because CEP112 aggregated into condensates during spermiogenesis, we speculated that the formation of CEP112-dependent RNA puncta within spermatogenic cells was driven by LLPS. Intrinsically disordered regions are frequently enriched in phase-separated compartments and are important drivers of phase separation behavior through multivalent interactions35,36. We analyzed the domains present in CEP112 using the PONDR software. We found that except for a few regions that could be folded into ordered structures, most of the CEP112 sequences existed in an intrinsically disordered region form (Supplementary Fig. S10a)). Analysis of endogenous CEP112 protein-expression patterns in GC-1, GC-2, and TM4 cells showed endogenous CEP112 formed cytoplasmic condensates (Supplementary Fig. S10b)). After constructing a GFP-CEP112 plasmid and transfecting it into HEK293 cells, we observed that GFP-CEP112 aggregated into condensates within cells (Fig. 6a, b, Supplementary Movie 4), and the condensates rapidly underwent fusion and fission (Fig. 6c). Fluorescence recovery after photobleaching (FRAP) analysis of GFP-CEP112 puncta disclosed a fast recovery rate in HEK293T cells (Fig. 6d). GFP-CEP112 puncta exhibited sensitivity to 1,6-hexanediol, a disruptor of hydrophobic interactions in phase-separated condensates. Treatment with 1,6-hexanediol resulted in a significant reduction in visible CEP112 condensates within minutes (Supplementary Fig. S10c). These results suggest that GFP-CEP112 forms condensates through LLPS in living cells. To test whether CEP112 proteins form condensates through LLPS in vitro, we purified recombinantly expressed GFP-CEP112 fusion proteins (Supplementary Fig. S10d). Consistently, GFP-CEP112 droplets fused within seconds, and 1,6-hexanediol-sensitivity assays showed they dissolved in 10% 1,6-hexanediol (Supplementary Fig. S10e, f), indicating that the GFP-CEP112 droplets had liquid properties. GFP-CEP112 proteins both phase separated in vitro in a protein- and salt-concentration-dependent manner as expected (Fig. 6e). The FRAP experiment also showed that GFP-CEP112 recovered rapidly in droplets after bleaching (Fig. 6f), indicating that GFP-CEP112 underwent a dynamic exchange between the droplets and the surrounding soluble proteins.

a, b Fluorescence microscopy reveals that GFP-CEP112 forms condensates within HEK293T cells (a). 3D visualization confirms the spherical morphology of these intracellular condensates (b) (Scale bar: 5 μm). c Time-lapse fluorescence microscopy demonstrates that GFP-CEP112 condensates undergo rapid fusion and fission events in HEK293T cells. Magnified areas are indicated by purple dotted boxes. d Fluorescence recovery after photobleaching (FRAP) assay of GFP-CEP112 droplets in HEK293T cells. Representative images (left) show bleaching at the indicated time points and recovery at 37 °C (Scale bar: 5 μm). Fluorescence-intensity analysis (right) of the FRAP data over 300 s. Three independent experiments were performed. Data are presented as mean values ± SEM. Source data are provided as a Source Data file. e GFP-CEP112 undergoes LLPS in vitro in a protein and salt concentration-dependent manner. Fluorescence microscopy images show the formation of GFP-CEP112 droplets at increasing protein concentrations (top) and NaCl concentrations (left) (Scale bar: 10 μm). f FRAP assay of in vitro GFP-CEP112 droplets. Representative images (left) depict bleaching at the indicated time points and recovery at 37 °C (Scale bar: 5 μm). Fluorescence intensity analysis (right) of FRAP data over 300 s. Three independent experiments were performed. Data are presented as mean values ± SEM. Source data are provided as a Source Data file. g Confocal microscopy reveals co-localization of GFP-CEP112 (green) and mCherry-tagged hnRNPA2B1, eIF4A1, or eEF1A1 (red) within droplets in vitro (Scale bar: 10 μm). h Purified GFP-CEP112 selectively recruits its target RNAs, FSIP2-CDS and FSIP2-5’UTR, but not the control METTL5-CDS, into phase-separated droplets in vitro (Scale bar: 10 μm). Experiments were repeated three times independently with similar results; representative images are shown in a, b, e, g and h.
Multivalent interactions between proteins and RNAs also play an important regulatory role in phase separation37. We explored the interplay between CEP112, its binding partners, and emergence of biomolecular condensates. Using exogenous GFP-CEP112 and mCherry-tagged hnRNPA2B1, mCherry-EIF4A1, and mCherry-EEF1A1, we observed formation of condensates with globular/spherical morphologies within HEK293T cells (Supplementary Fig. S10g). To analyze impacts of CEP112’s interactions with binding proteins on phase separation, we employed an in vitro approach using mCherry-tagged fusion proteins of hnRNPA2B1, EIF4A1, and EEF1A1. Intriguingly, CEP112 orchestrated recruitment of hnRNPA2B1, EIF4A1, and EEF1A1 into granules in vitro. By co-mixing GFP-CEP112 with mCherry-tagged binding partners at varying molar ratios, we observed co-localization within droplets, with droplet sizes dynamically scaling with increasing binding partner concentrations (Fig. 6g, Supplementary Fig. S10h, j). Furthermore, we delved into the role of RNA in this process. Combining total RNAs from mouse testes with GFP-CEP112-positive droplets, we observe a concentration-dependent increase in droplet formation, underscoring RNA’s active participation in LLPS within these condensates (Supplementary Fig. S10k). Notably, using Cy3-labeled RNA sequences identified from our RIP-seq analysis, we demonstrated that purified GFP-CEP112 exhibits preferential recruitment of specific target transcripts, such as Fsip2-CDS and Fsip2-5’UTR, while excluding the control transcript Mettl5-CDS (Fig. 6h).
Increasing FSIP2-CDS concentration enhanced GFP-CEP112 phase separation (Supplementary Fig. S10I). In summary, our integrated approach reveals that CEP112 orchestrates binding partners and RNA cargos to undergo LLPS during spermiogenesis, resulting in biomolecular condensates involved in localized regulation of essential fertility mRNAs.
CEP112 mutations impair phase separation and translation
We further explored the functional impact of patient-derived CEP112 mutations using overexpression studies in 293T cells. RT-PCR showed that expression of the mutant plasmid (R76X) was greatly reduced compared to wild type CEP112, while expression of the mutant plasmids (S137L, G90R, H766R) did not change significantly (Fig. 7a). Western blot results showed that the R76X mutant plasmid did not express CEP112, which might be due to degradation of the truncated protein in cells. In contrast, the mutant plasmids (S137L, G90R, H766R) showed no difference in expression compared to wild type, suggesting these mutations may affect CEP112 function through other pathways (Fig. 7b). To assess the impact of the missense mutations (S137L, G90R, and H766R) on CEP112 structure and function, we performed computational modeling and molecular dynamics simulations using PyMOL and GROMACS. Superposition of the G90R mutant structures at 0 ns and 50 ns revealed significant conformational differences, with a Cα-RMSD value of 45.012 (10,138 atoms). Similar structural deviations were observed for the H766R (RMSD = 64.202; 10,099 atoms) and S137L (RMSD = 53.721; 9,760 atoms) mutants (Fig. 7c–e). Although the morphology of condensates formed by S137L, G90R, and H766R mutants resembled that of the wild-type protein, a significant reduction in condensate number was observed (Fig. 7f, g). FRAP analysis revealed the H766R mutation did not alter condensate dynamics. However, S137L and G90R exhibited significantly slowed recovery, indicating impaired liquid-like behaviors (Fig. 7h). Furthermore, TRICK reporter assays demonstrated that all three missense mutations compromised CEP112’s ability to promote the translation of target mRNAs (Fig. 7i).

a RT-PCR analysis of wild-type (WT) and mutant CEP112 in HEK293T cells shows significantly reduced expression of the R76X mutant, while S137L, G90R, and H766R mutants have comparable expression levels to WT. Three independent experiments were performed. (Two-sided Student’s t test; error bars, mean ± SEM). The p values are indicated in the graphs. Source data are provided as a Source Data file. b Western blot analysis of CEP112 proteins in HEK293T cells. Representative blot and quantification of CEP112 relative to GAPDH. Three independent experiments were performed. (Two-sided Student’s t test; error bars, mean ± SEM). Source data are provided as a Source Data file. c–e Structural impact of missense mutations (S137L, G90R, H766R) predicted by computational modeling, with mutated residues highlighted in purple. f Fluorescence microscopy shows WT and mutant CEP112 (S137L, G90R, H766R) forming spherical condensates in HEK293T cells (Scale bar: 10 μm). g Quantification of condensates per cell with WT or mutant CEP112 in HEK293T cells. n = 50 cells per group. (Two-sided Student’s t test; error bars, mean ± SEM). The p values are indicated in the graphs. Source data are provided as a Source Data file. h FRAP analysis of condensates in HEK293T cells reveals that the S137L mutant recovers similarly to WT, while G90R and H766R mutants show slower recovery, indicating altered liquid-like properties. Three independent experiments were performed. Data are presented as mean values ± SEM. Source data are provided as a Source Data file. i TRICK reporter assay in COS-7 cells shows WT CEP112 sequesters CFAP61 mRNA into granules, promoting translation (RFP signal only). Mutant CEP112 disrupts this, with GFP and RFP signals indicating impaired translational regulation (left panel, Scale bar: 10 μm). Quantification of the proportion of untranslated mRNA granules is shown. n = 30 cells per group. (Two-sided Student’s t test; error bars, mean ± SEM). The p values are indicated in the graphs. Source data are provided as a Source Data file.
Taken together, these results strongly implicate pathogenic CEP112 mutations in disrupting both phase separation and mRNA translational regulation-two interconnected processes coordinated by biomolecular condensates. Aberrant liquid-liquid demixing provides a putative mechanism linking genetic variation at the CEP112 locus to infertility phenotypes in human patients. Strategies restoring the dynamics of these membraneless organelles could offer routes to rescue the underlying molecular pathology.
Discussion
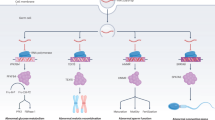
Spermiogenesis culminates in the intricate transformation of haploid spermatids into specialized motile spermatozoa. This complex process relies on precise spatiotemporal regulation mediated by molecular pathways and regulatory networks1. Our study provides evidence for an important role of CEP112 in spermiogenesis, suggesting its involvement in regulating the expression of spermiogenesis-related mRNAs through LLPS (Fig. 8). Our investigation into CEP112 mutations expands the spectrum of CEP112-related male infertility phenotypes, demonstrating an association with oligoasthenoteratozoospermia. Examination of Cep112 knockout mice revealed reduced sperm count, impaired motility, and morphological abnormalities, features that align with the phenotype observed in our human patients. This consistency provides additional support for the critical role of CEP112 in spermatogenesis. While Sha et al. reported CEP112 mutations primarily associated with acephalic spermatozoa syndrome16, our study identifies different mutations (R76X, S137L, G90R, and H766R) leading to a distinct yet related phenotype. This discovery underscores the complexity of genotype-phenotype relationships in male infertility caused by CEP112 dysfunction. Sha et al. identified a homozygous nonsense mutation (R166X) and compound heterozygous mutations (R692W and R702C) in CEP112, resulting predominantly in acephalic spermatozoa16. However, a closer examination of their supplementary data reveals similarities with our findings. Their patients also exhibited significantly reduced sperm concentration, a feature we observed in our oligoasthenoteratozoospermic patients. Interestingly, while the primary phenotype differed between the two studies, both observed a spectrum of sperm abnormalities. In Sha et al.‘s study, a small percentage of sperm (1.2% in P1 and 2.3% in P2) displayed normal head-tail junctions, although potential tail abnormalities in these sperm were not reported. Similarly, our patients exhibited a range of sperm defects, including reduced count, motility, and morphological abnormalities, rather than a uniform phenotype. This phenotypic variability is not uncommon in reproductive genetics. For instance, mutations in FSIP2 have been associated with both multiple morphological abnormalities of the sperm flagella38 and acrosomal defects39. Similarly, CFTR mutations can lead to a range of conditions from congenital absence of the vas deferens to defects in sperm production and motility40. Our findings on CEP112 contribute to this growing body of evidence, demonstrating how different mutations in a single gene can manifest in a spectrum of male infertility phenotypes. Importantly, Our study provides mechanistic insights into CEP112 function beyond genetic association. We demonstrate CEP112 undergoes LLPS, a previously undescribed property, revealing its role in translational regulation of key spermiogenesis-related mRNAs. This finding advances our understanding of CEP112’s molecular function in spermatogenesis and sperm development.

Schematic illustration depicting CEP112’s dual functions in spermiogenesis. Left: CEP112 undergoes liquid-liquid phase separation during spermiogenesis, forming biomolecular condensates that recruit target mRNAs and translation factors to facilitate efficient translation. Right: CEP112 also localizes to sperm atypical centrioles, crucial for zygotic development.
The identification of CEP112 as a component of atypical centrioles in human sperm suggests its potential involvement in embryonic development and reproductive health (Fig. 8). Previous findings have demonstrated that defective centriole-associated proteins in human sperm can lead to poor ICSI outcomes in the clinical management of infertile patients. For instance, fertilized eggs obtained from infertile patients with CEP128 deficiencies failed to develop into viable embryos during the ICSI treatment cycle11. Similarly, ICSI was performed for an infertile patient with a mutation of CEP135 and the embryos were transplanted; however, the wife of the patient did not become pregnant due to poor embryo quality8. In our study, we tracked the ICSI outcome of one patient (Y3937) harboring CEP112 mutations. Despite successful fertilization, only two out of eleven embryos reached the 8-cell stage, indicating severely compromised embryonic development. Furthermore, this patient experienced a miscarriage following implantation. These results suggest that CEP112 mutations may negatively impact early embryo development. However, a significant limitation of this study is the small sample size for ICSI outcomes. While our results indicate that CEP112 deficiency may lead to poor ICSI outcomes, a larger cohort investigation is necessary to definitively assess this relationship. Future studies should aim to correlate CEP112 mutations, centriole abnormalities, and embryonic development outcomes in a more extensive group of patients. To gain a more comprehensive understanding of the role of human sperm centrioles in fertility, we propose a multifaceted approach combining advanced techniques for infertility patients with centriolar defects or centriole-related gene mutations. For example, techniques for sperm centriole evaluation, like the Fluorescence-based Ratiometric Assessment of Centrioles (FRAC) developed by Turner et al. 41, should be employed. This comprehensive analysis would provide a more nuanced understanding of how centriole-associated proteins and centriole defects impact sperm function and reproductive outcomes, even in cases where conventional sperm parameters appear normal. Interestingly, in contrast to our human data, ICSI treatment successfully overcame male sterility caused by CEP112 deficiency in mice, which is understandable because the involvement of sperm-derived centrioles is not required for rodent embryogenesis42.
The phase-separation system can enrich actin and promote its assembly, suggesting that phase separation can facilitate dynein assembly and cytoskeleton formation43,44,45. Similarly, previous studies have shown that BuGZ, SPD-5, and Tau associated with granules that can recruit tubulin, promote its multimerization, and promote spindle apparatus assembly, microtubule aster formation in centrosomes, and microtubule-bundle formation in neurons, respectively46,47,48. abLIM1 is an LLPS-dependent self-organizer of actin networks, which facilitates actin nucleation and network formation49, suggesting that LLPS is important for the formation of cilia and flagella. Recent findings highlighted a key function of FXR1 LLPS in directly activating translation of stored mRNAs during spermiogenesis24. Phase separation appears to be central to the regulatory function of CEP112 in spermiogenesis. The formation of RNA granules through LLPS, a process that we demonstrate is mediated by CEP112, contributes to our understanding of the spatiotemporal control of gene expression. The clinical implications of our work are underscored by the functional disruptions caused by patient-derived CEP112 mutations. These mutations disrupt both LLPS and subsequent mRNA translation, furnishing a molecular basis for the infertility phenotypes. This correlation underscores the potential of targeting CEP112 and its associated pathways for therapeutic interventions in cases of male infertility rooted in these molecular defects. There is growing evidence of compounds being developed to modulate LLPS in various pathological conditions, from neurodegenerative diseases to cancers50,51,52. These agents, aimed at stabilizing or destabilizing specific biomolecular condensates, present a novel strategy to rectify the aberrant phase transitions at the root of certain disorders. Given the critical role of LLPS in male reproduction, similar approaches could be envisioned for infertility treatments. The precise targeting of LLPS, particularly by addressing the post-translational modifications that regulate phase separation in proteins pivotal to spermatogenesis, like CEP112 and FXR1, holds promise for restoring the delicate equilibrium necessary for sperm maturation and function.
While our study established the importance of CEP112-mediated liquid-liquid phase separation in facilitating RNA granule assembly and regulating translation during spermiogenesis, several questions remain unanswered. The precise molecular triggers initiating this phase transition process and the full repertoire of RNA species incorporated into these condensates warrant deeper investigation. Additionally, exploring how post-translational modifications of CEP112 and its protein interactions modulate biomolecular condensation dynamics presents another promising research direction. Our findings significantly advances the understanding of spermiogenesis by uncovering CEP112’s multifaceted functions, ranging from atypical centriole formation to translational control via RNA granules. The demonstration of a phase separation-dependent mechanism governing mRNA translation provides crucial insights into male reproductive biology. These discoveries not only enhance our comprehension of male germ cell development but also highlight the importance of biomolecular condensates in reproduction. Furthermore, this work contributes to the broader understanding of how phase separation phenomena regulate cellular processes in disease. The expanded understanding of the molecular pathology related to aberrant CEP112 function could potentially catalyze the development of targeted therapeutic strategies to address certain forms of male infertility.
Methods
Study subjects
In this study, 568 idiopathic infertile men with oligoasthenoteratozoospermia and asthenozoospermia (with or without teratozoospermia) were recruited between June 2017 and March 2022 in the Reproductive and Genetic Hospital of CITIC-Xiangya (Changsha, Human, China) and the West China Second University Hospital. The individuals were recruited according to the WHO Laboratory Manual for the Examination and Processing of Human Semen53. All individuals were idiopathic with a normal 46, XY karyotype and without a Y chromosome AZF microdeletion. Other causes of infertility (i.e., latrogenic injury, reproductive-tract infection, testicular inflammation, and drug exposure) were excluded. Physical examinations showed normal results, including height, weight, hair distribution, mental state, testis size, and external genital organs. As controls, we recruited 392 fertile men with normal semen parameters who had fathered at least one healthy child. Participants were not compensated for their involvement. Sex and gender were considered in the study design, with data collected and analyzed for male participants only, as the study focused on male infertility. This study complies with all relevant ethical regulations and was conducted in accordance with the Declaration of Helsinki. The study was approved by the Ethics Committee of the Reproductive and Genetic Hospital of CITIC-Xiangya (LL-SC-2017-025 and LL-SC-2019-034) and the Ethical Review Board of West China Second University Hospital (2021006). All human participants provided written informed consent.
WES and variant filtering
Genomic DNA from whole peripheral-blood samples was extracted using a QIAamp DNA Blood Midi Kit (Qiagen, 51106). Genomic DNA was prepared using the Agilent SureSelect Human All Exon V6 Kit and sequenced using the Illumina HiSeq 2000 platform54. Original data were mapped to human genome GRCh37/hg19 using Burrows-Wheeler Aligner software.
Candidate variants were identified as described previously55, and the pathogenicity of the detected variants was evaluated as recommended by the American College of Medical Genetics and Genomics. Briefly, candidate variants had a frequency of <1% in public databases (1000 Genomes and gnomAD databases), were predicted to be deleterious and impact normal protein functions, were bi-allelic or hemizygous, were associated with a relevant phenotype, had high testis expression, were enriched for the GO term “biological process associated with spermatogenesis,” and model organism data were available (male infertility phenotype observed in animal models).
Generating animal models
Cep112-knockout mice were generated using clustered regularly interspaced short palindromic repeats-Cas9 genome-editing technology. Guide RNAs were designed against exons 4–5 of Cep112 (sgRNA-1: 5′-AGGGATTTGTAGCTGATTTAAGG-3′, sgRNA-2: 5′-AAAACAGTGAGCACCACTCAGGG-3′). Cas9 mRNA and gRNA were transcribed in vitro and co-microinjected into the cytoplasm of fertilized C57BL/6 mouse oocytes. A frameshift mutation in Cep112 was identified in the founder mice and their offspring by PCR, qPCR, and immunoblotting analyzes. The primers used for PCR and qPCR are listed in Supplementary information, Table S5. Mice were housed under standard conditions (12 h light/dark cycle, 22 ± 2 °C, 50 ± 10% humidity) with ad libitum access to food and water. All animal experiments were performed according to the recommendation of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Animal experiments were also approved by the Experimental Animal Management and Ethics Committee of the West China Second University Hospital.
Sperm chromatin structure assay (SCSA)
SCSA was performed as previously described56. Briefly, semen samples were diluted in TNE buffer (0.01 M Tris-HCl, 0.15 M NaCl, 1 mM EDTA, pH 7.4). Samples were treated with acid detergent solution (0.1% Triton X-100, 0.15 M NaCl, and 0.08 M HCl, pH 1.2) for 30 s, followed by staining with acridine orange (6 μg/mL in phosphate-citrate buffer, pH 6.0). Flow cytometry analysis was performed using a FACScan flow cytometer (Becton Dickinson). The DNA Fragmentation Index (DFI) and High DNA Stainability (HDS) were calculated using FCS Express 6 Flow Cytometry Software.
H&E staining
Testis, epididymis, kidney, and heart tissue samples from Cep112+/+ and Cep112−/− mice were fixed with 4% paraformaldehyde for 24 h at room temperature, dehydrated in graded ethanol and xylene, embedded in paraffin, and sliced to obtain 5 µm-thick sections. The tissue sections were routinely stained with H&E and photographed under a microscope (Olympus Slideview VS200, Japan).
SEM, TEM, and IG-TEM analyses
Fresh spermatozoa from humans and mice were washed thrice with 1 × PBS and fixed with 2.5% glutaraldehyde overnight at 4 °C. For SEM, the immobilized spermatozoa were fixed on poly-l-lysine-coated coverslips and washed in 1 × PBS. Subsequently, the specimens were dehydrated with an ascending gradient of cold ethanol (30%, 50%, 60%, 70%, 80%, 95%, and 100%) and dried at the critical point using a Leica EM CPD300 Critical Point Dryer (Leica Microsystems, Japan). The specimens were then coated with gold particles using an MC1000 Ion Sputter Coater (Hitachi, Japan). Finally, the specimens were observed under an SEM (Hitachi S3400, Japan) at an accelerating voltage of 15 kV.
For TEM, the samples were fixed in 3% glutaraldehyde and osmium tetroxide and then dehydrated with an ethanol gradient. Subsequently, the samples were embedded in Epon 812, dodecenylsuccinic anhydride, methylnadic anhydride, and dimethylaminomethyl phenol. Ultrathin sections (70–90 nm) were cut and double-stained with lead citrate and uranyl acetate. For IG-TEM analysis, ultrathin sections were collected on nickel grids and simultaneously blocked with 1% BSA for 5 min at 37 °C. The sections were incubated with an anti-CEP112 primary antibody for 2 h at 37 °C in a humid chamber. After washing thrice in 1 × PBS with 0.005% Tween-20, the samples were incubated with a secondary goat anti-rabbit IgG antibody (10 nm; Bioss, bs-0295G-Gold) for 1 h at 37 °C. As a negative control, the anti-CEP112 primary antibody was omitted. The samples were sequentially washed thrice in 1 × PBS with 0.005% Tween-20, washed twice in bi-distilled water, and air dried. Finally, images were obtained by TEM (TECNAI G2 F20, Philips, Japan) at an accelerating voltage of 80 kV. For quantitative analysis of CEP112 localization, gold particles were counted and categorized based on their association with different ultrastructural features (e.g., distal centrioles, proximal centrioles, axoneme, or other regions). At least 51 cross-sections from 3 individuals were analyzed.
Isolation of spermatogenic cells at different stages
Spermatogenic cells were obtained at different stages through cell diameter/density at unit gravity using the STA-PUT velocity sedimentation method56. Briefly, testes from Cep112+/+ and Cep112−/− mice were dissected and decapsulated. Seminiferous tubules were minced and incubated in 1×PBS containing collagenase (Invitrogen, 17104-019) and hyaluronidase (Sigma-Aldrich, H6254) at 37 °C for 15 min with gentle shaking. The undigested tissue was discarded after centrifugation at 4 °C and 200 × g for 3 min. After washing with 1 × PBS, the cells were incubated in 1 × PBS with 0.25% Trypsin-EDTA (Gibco, 15050065) and 1 mg/mL DNase I (Sigma-Aldrich, 11284932001) at 37 °C for 15 min with gentle shaking. After filtration, the spermatogenic cells were resuspended in a buffer containing 0.5% BSA and loaded into an STA-PUT Velocity Sedimentation Cell Separator (ProScience) for gradient separation. Spermatogenic cells were isolated at different stages for further analysis.
RNA extraction and quantitative RT-PCR
Total RNA was extracted from samples using TRIzol (Invitrogen, 15596-026) and converted to cDNA using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, 4368813). RT-PCR was conducted in 96-well plates using a CFX96 Touch Real-Time PCR Detection System (Bio-Rad, USA) in 10 μL reactions containing 1 μL cDNA template, 0.5 μL forward primer, 0.5 μL reverse primer, 5 μL SYBR Green (Bimake, B21202), and 3 μL DEPC-H2O. Gapdh mRNA expression was detected as a quantitative control, and relative amplicon expression was calculated using the 2−ΔΔCt method. All RT-PCR primers used are listed in Supplementary Table S5.
Immunoblotting and co-IP experiments
Cell and tissue samples were lysed for 45 min at 4 °C in RIPA buffer (Beyotime, P0013B) containing protease inhibitor cocktail (Thermo Fisher Scientific, 78425) and Phosphatase Inhibitor Cocktail (Sigma-Aldrich, P5726). Total protein concentrations were determined using the BCA Assay Kit (Thermo Fisher Scientific, 23227). Lysates were mixed with SDS-PAGE Sample Loading Buffer (Beyotime, P0015) and boiled for 5 min, after which total proteins (20–50 μg) were separated by 7.5–12.5% SDS-PAGE and transferred to PVDF membranes (Millipore, ISEQ00010, IPVH00010). The membranes were blocked in 5% fat-free milk for 30 min at room temperature, incubated with primary antibodies overnight at 4 °C, incubated with secondary antibodies for 1 h at room temperature, and visualized with the ECL HRP substrate (Millipore, WBKLS0500).
For the co-IP assays, extracted total proteins were incubated with an appropriate primary antibody overnight at 4 °C and then with Protein A/G magnetic beads (Thermo Fisher Scientific, 88804) for 1 h at room temperature. After washing with washing buffer (50 mM Tris-HCl [pH, 7.4], 0.1% Triton X-100, and 500 mM NaCl) and eluting with 1× SDS loading buffer, the samples were boiled for 10 min at 75 °C and analyzed by immunoblotting assay (described above). The primary and secondary antibodies used for immunoblotting and co-IP are described in Supplementary Table S6.
Immunofluorescence staining
For spermatozoa or spermatogenic cell immunostaining, samples were washed in 1 × PBS, fixed with 4% paraformaldehyde for 15 min at room temperature, coated on slides pre-coated with 0.1% poly-l-lysine, permeabilized with 0.3% Triton X-100 (Thermo Fisher Scientific, 85111) for 15 min, and blocked with 5% BSA (BioFroxx, 4240GR100) for 1 h at room temperature. The slides were sequentially incubated overnight with primary antibodies at 4 °C and for 1 h with secondary antibodies at room temperature. Nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI) (Beyotime, C1002) for 10 min at room temperature and the slides were observed under SIM (Nikon N-SIM S, Japan).
For staining, testicular tissues were first fixed with Bouin’s solution (Sigma-Aldrich, HT101128), paraffin-embedded, and sectioned into 5 μm slices. After deparaffination and rehydration, the sections were processed with sodium citrate for antigen repair, washed thrice in 1 × PBS, blocked with goat serum (Thermo Fisher Scientific, 31872) at room temperature for 30 min, and incubated with primary antibodies at 4 °C overnight. Subsequently, the sections were incubated with secondary antibodies for 1 h at room temperature and sealed with a fluorescence decay-resistant medium containing DAPI. The prepared slides were examined by confocal microscopy (FV3000, Olympus Corporation). The antibodies used are described in Supplementary Table S6. For high-resolution imaging of CEP112 localization, we employed structured illumination microscopy (SIM). Samples were prepared as described above and imaged using a Nikon N-SIM S system. Z-stacks were acquired with a step size of 0.12 μm, and 3D reconstructions were generated using NIS-Elements software.
RNA-Seq analysis
Total RNA was extracted from the testes of 8-week-old mice using the Dynabeads RNA Purification Kit (Thermo Fisher Scientific, 61006). The mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. RNA integrity was assessed using the RNA Nano 6000 Assay Kit and the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Synthetic cDNA fragments and library fragments were purified with the AMPure XP system (Beckman Coulter, Beverly, USA). PCR was performed with DNA polymerase, universal PCR primers, and the Index (X) primer; the products were purified; and the library quality was assessed on the Bioanalyzer 2100 system. Libraries were sequenced (paired-end, 150 bp) using the NovaSeq platform (Illumina).
Raw reads in FASTQ format were subjected to quality-control analysis using FASTQC. Clean reads were obtained by removing low-quality reads and trimming adapters using Trim Galore (0.6.7; https://github.com/FelixKrueger/TrimGalore). All downstream analyses were based on high-quality clean data. Paired-end clean reads were aligned to the reference genome (GRCm38) with HISAT2 (2.0.5)57. FeatureCounts (1.5.0-p3; subread package)58 was used to count the reads mapped to each gene, and FPKM values were calculated for each gene using R script based on gene lengths and read counts. Differential-expression analysis between Cep112-knockout and WT mice was performed using the DESeq2 Bioconductor R package59. The resulting P-values were adjusted using the Benjamini–Hochberg method to control the false-discovery rate (FDR). Genes with a fold-change (FC) of ≥2 and a P-value of <0.05 (determined using DESeq2) were considered differentially expressed.
Proteomic analysis
The testicular tissues of 8-week-old male mice were ground in liquid nitrogen into powder form. Four volumes of lysis buffer (8 M urea, 1% protease inhibitor cocktail) were added to the powder, and each sample was sonicated thrice on ice using a High-Intensity Ultrasonic Processor (Scientz). The remaining debris was removed by centrifugation at 12,000 × g at 4 °C for 10 min, the supernatant was collected, and the protein concentration was determined with a BCA Kit. After trypsin digestion and purification, peptides were reconstituted in 0.5 M Triethylammonium bicarbonate and processed using the TMT Kit. A total of 6 samples (n = 3 biological replicates per condition) were analyzed. The separated peptides were identified by LC-MS/MS and quantified in a Q ExactiveTM HF-X instrument (Thermo Fisher Scientific) with a nano-electrospray ion source (electrospray voltage applied, 2.0 kV; full MS scan resolution, 60000; scan range, 350–1600 m/z). Up to 20 of the most abundant precursors were selected for further MS/MS analyses with 30 s dynamic exclusion. The resulting MS/MS data were processed using the MaxQuant Search Engine (v.1.6.15.0). Tandem MS data were searched against the human SwissProt Database (20,422 entries) concatenated with the reverse-decoy database. The false discovery rate (FDR) was set to 1% for both peptide and protein identification.
RIP-Seq analysis
Testicular tissues from 8-week-old male mice were subjected to RIP-Seq analysis. The tissues were treated with lysis buffer. Then 10% of each sample was stored (“input” sample), 80% was used in IP reactions with an anti-Cep112 antibody (“IP” sample), and 10% was incubated with rabbit IgG (Cell Signaling Technology) as a negative control (“IgG” sample). The RNA from the input and IP samples was extracted using TRIzol (Invitrogen, 15596026). RNA-Seq libraries were constructed using the KC-DigitalTM Stranded mRNA Library Prep Kit for Illumina® (Wuhan Seqhealth Co., Ltd. China, DR08502). The kit eliminates duplication bias generated in the PCR and sequencing steps with a unique molecular identifier (comprising eight random bases) to label the pre-amplified cDNA molecules. Library products (200–500 bp) were enriched, quantified, and sequenced on a DNBSEQ-T7 sequencer (PE150 model, MGI Tech Co., Ltd. China).
The clean reads were mapped to the mouse reference genome (mm10) using STAR software (2.5.3a)60 with the default parameters. Fold-differences in expression (log FC > 1) between the IP and input samples and significance levels (P-value < 0.05) were used to identify upregulated genes in IP samples. Peak calling was performed using exomePeak software (3.8)61. Peaks were annotated using BEDTools (2.25.0)62 and ChIPseeker63. deepTools (2.4.1)64 was used for the peak-distribution analysis. Differential binding peaks were identified with a Python script, using Fisher’s test. HOMER (4.10)65 was used to search for motifs. We determined the relationships between the peaks corresponding to these motifs and the associated genes to identify genes corresponding to the motifs. The igraph package (https://cran.r-project.org/web/packages/igraph/index.html) (1.0.0)66 was used for network analysis. Difference analysis was performed for peaks in the treatment and control groups, and the different peaks were annotated.
Functional-enrichment analysis
ClusterProfiler (4.0.5)67 and Metascape (https://metascape.org)68 were used for the GO and Kyoto Encyclopedia of Genes and Genomes pathway-enrichment analyses. Functional enrichment was conducted for GO terms in the CC, BP, and molecular function categories. GO terms with a P-value of <0.01 and at least three enriched genes were considered statistically enriched. Hypergeometric tests and Benjamini–Hochberg-based FDR-control procedures enabled enrichment determinations.
GSEA69,70 was used to identify classes of over-represented proteins with the proteomics data. Proteins showing subtle expression changes were explored for important biological implications by GSEA. Pathways with FDRs of <0.25 and P-values of <0.05 were considered significant.
Sequence alignments and phylogenetic analysis
Multiple-sequence alignments were performed using ClustalW. Evolutionary histories were inferred using the neighbor-Joining method71 and bootstrap analysis with 1000 replicates72. Evolutionary distances (i.e., number of base substitutions/site) were computed using the maximum-composite likelihood method73. Evolutionary analyses were conducted with MEGA774.
Polysome profiling
Polysome profiling was performed as previously described24. Briefly, mouse testes were homogenized in lysis buffer containing cycloheximide. Lysates were fractionated on 10–60% sucrose gradients by ultracentrifugation. Twelve fractions were collected using a gradient fractionator with continuous absorbance monitoring at 254 nm. RNA was extracted from each fraction, with polysome-associated fractions pooled for analysis. To assess global translation efficiency, polysome-to-monosome (P/M) ratios were calculated. The area under the curve for polysome peaks was divided by the area under the monosome peak. For each genotype, three independent experiments were performed.
Plasmid construction
The full-length human CEP112 cDNA (NM_001199165.4) was synthesized and cloned into the pCMV-MCS-Myc vector to generate the Myc-tagged CEP112 plasmid. The plasmid encoding human hnRNPA2B1 (NM_031243) was synthesized and cloned into the pENTER-His vector to produce the His-tagged hnRNPA2B1 plasmid. The human EEF1A1 (NM_001402) and EIF4A1 (NM_001416) cDNAs were synthesized and cloned into the pCMV-HA and pCMV-Flag vectors, respectively, to generate the HA-tagged EEF1A1 and Flag-tagged EIF4A1 plasmids. Site-directed mutagenesis was performed on the Myc-CEP112 plasmid to introduce the mutations identified in patients (R76X, S137L, G90R, and H766R) using the Q5® Site-Directed Mutagenesis Kit (New England Biolabs, E0554S) according to the manufacturer’s instructions. The mutagenesis primers were designed using the NEBaseChanger tool (New England Biolabs). The mutated plasmids were verified by DNA sequencing.
For the TRICK assay, the coding sequence of human CFAP61 (NM_015585) was modified by inserting PP7 and MS2 stem-loop sequences while preserving the reading frame. The modified CFAP61 sequence was synthesized and cloned into a mammalian expression vector. Plasmids expressing the fusion proteins GFP-NLS-PCP and RFP-NLS-MCP were generated by cloning the corresponding sequences into suitable mammalian expression vectors. All constructed plasmids were verified by DNA sequencing and purified using the EndoFree Mini Plasmid Kit (TIANGEN, DP118) for transfection experiments.
Cell culture and transfections
HEK293T, TM4, GC-1, GC-2 and COS-7cells were obtained from the American Type Culture Collection (CRL-11268™, CRL-1715™, CRL-2053™, CRL-2196™ and CRL-1651™ respectively). HEK293T, GC-1, GC-2 and COS-7 cells were grown in Dulbecco’s modified Eagle medium (Gibco, 11965092) containing 10% FBS (Gibco, 10091148). TM4 cells were grown in Dulbecco’s modified Eagle medium / F12 (Gibco, 11330057) containing 2.5% FBS and 5% horse serum (Gibco, 26050088). Cells were maintained in 5% CO2 and a 95% humidified air atmosphere at 37 °C. Transfections were performed with the Lipofectamine™ 3000 Transfection Reagent (Thermo Fisher Scientific, L3000015).
TRICK reporter assay
We performed the TRICK (Translational Regulation by Internally Controlled Kinetics) reporter assay as previously described to study the role of CEP112 in translation24,75. In brief, the coding region of the target mRNA, CFAP61, was modified by insertion of PP7 binding sites (PBS) while keeping the reading frame intact. The 3’ untranslated region of CFAP61 remained unchanged. COS-7 cells were transfected with the CFAP61 TRICK reporter construct along with plasmids expressing the fusion proteins GFP-NLS-PCP and RFP-NLS-MCP. These fusion proteins bind to the PP7 binding sites in the CFAP61 mRNA. BFP-CEP112 and its mutant form were co-transfected to examine the impact of CEP112 on translation. ImageJ software enabled quantitative analysis of the TRICK reporter fluorescence in cells expressing wild-type or mutant CEP112.
Recombinant proteins
For the expression of recombinant GFP-CEP112, the coding sequence was cloned into the pFastBac1 vector using the Bac-to-Bac baculovirus expression system. The construct was verified by DNA sequencing, and a positive clone containing the recombinant baculovirus plasmid was obtained. Sf9 insect cells were transfected with the recombinant bacmid to generate the baculovirus stock. High Five insect cells were infected with the amplified baculovirus stock for protein expression. The infected cells were harvested, resuspended in lysis buffer, and sonicated. The clarified lysate was purified by Ni-NTA affinity chromatography, and the purity and molecular weight of the recombinant GFP-CEP112 were verified by SDS-PAGE and immunoblotting.
To purify recombinant mCherry-hnRNPA2B1, mCherry-EEF1A1, and mCherry-EIF4A1 proteins, the corresponding coding sequences were cloned into the pET28a vector for expression in the Escherichia coli system. The constructs were verified by DNA sequencing, and positive clones were transformed into BL21(DE3) competent cells. Protein expression was induced with 0.5 mM IPTG at 16 °C for 18 h. The bacterial cells were harvested by centrifugation, resuspended in lysis buffer, and sonicated. The clarified lysates were purified using Ni-NTA affinity chromatography to obtain recombinant mCherry-hnRNPA2B1, mCherry-EEF1A1, and mCherry-EIF4A1. The purity and molecular weight of the purified proteins were verified by SDS-PAGE and immunoblotting.
FRAP experiments
FRAP experiments were performed on the Olympus IXplore SpinSR10 confocal microscope system. For the in vitro experiment, samples were applied to a glass-bottom 8-well chamber slides (Millipore, PEZGS0816) to image at ×60 magnification and ×6 optical zooming, and the droplets were outlined with a circular overlay object. The area was bleached for 1 s with 20% of the maximum laser power of a 488-nm laser (1 Airy unit). The recovery was recorded at a rate of 10 s/frame for 300 s. For the in vivo experiment, transfected HEK293T cells were cultured in glass-bottom culture dishes (φ15 mm) (NEST, 801002) and at 37 °C in a live-cell-imaging chamber. Before the FRAP experiment, the culture medium was exchanged to Opti-MEM (Gibco, 31985062). The puncta in cells were photobleached for 2 s with 20% maximum laser using a 488 nm laser (1 Airy unit). The recovery was recorded at a rate of 10 s/frame for 300 s.
The images were processed with FIJI/Image J and the analysis of the recovery curves was performed with GraphPad Prism 9.3.0. The FRAP experiments were repeated three independent times.
IVF, ICSI, and ET experiments
Mature oocytes were prepared from ICR mice (8–12 weeks of age). Mice were injected with human Chorionic Gonadotropin (Easycheck, M2530) at 44–46 h post-injection with pregnant mare serum gonadotropin (ProSpec, HOR-272), and the oocyte–cumulus complexes were harvested after another 12–14 h. For IVF, cauda epididymal sperm cells from male mice were capacitated by incubating them in HTF medium (Easycheck, M1150) for 1 h in 5% CO2 at 37 °C. Next, a drop of the capacitated sperm suspension was diluted with HTF medium containing OCCs to a final sperm concentration of 1 × 105/mL. Subsequently, the mixtures were incubated at 37 °C under 5% CO2 to calculate the fertilization and embryo-development rates.
For ICSI, spermatozoa from male Cep112+/+ and Cep112−/− mice were decapitated by mild sonication and sperm cells were collected by centrifugation in 70% Percoll (Sigma, P4937). After three washes in M2 medium (Easycheck, M1250), a single-sperm head was injected into MII oocytes using a Piezoelectric actuating pipette and a micromanipulator. After injection, the oocytes were rapidly transferred to KSOM medium (Easycheck, M1430), covered with mineral oil, and cultured at 37.5 °C in 5% CO2 and 95% air. The development was recorded for 96 h after ICSI. Developing embryos and pseudopregnant mice (recipient females) were prepared before ET. Pseudopregnancy occurred in mature females after mating with vasectomized males. After 2 days of pseudopregnancy, 20–25 embryos (two-cell stage) were transferred to the oviducts of ICR females. Full-term ICSI pups were delivered naturally. The animal experiments were approved by the Experimental Animal Management and Ethics Committee of West China Second University Hospital (Sichuan University).
Computational modeling and molecular dynamics simulations
Structural models of wild-type and mutant CEP112 proteins were generated using PyMOL. Molecular dynamics simulations were performed using GROMACS with the CHARMM36m force field. Systems were solvated in TIP3P water and neutralized with Na+ and Cl- ions. Energy minimization was performed using the steepest descent algorithm. The systems were equilibrated in the NVT and NPT ensembles for 100 ps each, followed by 50 ns production runs. Trajectories were analyzed using built-in GROMACS tools and visualized with VMD. For CEP112-mRNA interaction modeling, we utilized the Alphafold3 and PISA (Proteins, Interfaces, Structures and Assemblies) tool in PyMOL to analyze potential RNA-binding interfaces on CEP112. This analysis provided insights into the structural features that may be involved in CEP112-mRNA interactions.
Statistical analysis
Statistical analyses were performed using GraphPad Prism (9.3.0) and SPSS (19.0) software. All data are presented as means ± SEM. P < 0.05 was considered statistically significant. Statistical significance between two groups was calculated using an unpaired, parametric, two-tailed Student’s t test.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The proteomic data generated in this study have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository under accession code PXD037982. The RNA-seq data generated in this study have been deposited in the NCBI Sequence Read Archive (SRA) database under accession code PRJNA898827. The RIP-seq data generated in this study have been deposited in the NCBI Gene Expression Omnibus (GEO) database under accession code GSE216936. All other data supporting the findings of this study are available within the article and its Supplementary Information files. Source data are provided with this paper. Any additional information required to reanalyze the data reported in this paper is available from the corresponding author upon request. Source data are provided with this paper.
References
Tanaka, H. & Baba, T. Gene expression in spermiogenesis. Cell Mol. Life Sci. 62, 344–354 (2005).
Santiago, J., Silva, J. V., Howl, J., Santos, M. A. S. & Fardilha, M. All you need to know about sperm RNAs. Hum. Reprod. Update 28, 67–91 (2021).
Bettegowda, A. & Wilkinson, M. F. Transcription and post-transcriptional regulation of spermatogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365, 1637–1651 (2010).
Heidaran, M. A., Showman, R. M. & Kistler, W. S. A cytochemical study of the transcriptional and translational regulation of nuclear transition protein 1 (TP1), a major chromosomal protein of mammalian spermatids. J. Cell Biol. 106, 1427–1433 (1988).
Kleene, K. C. Patterns of translational regulation in the mammalian testis. Mol. Reprod. Dev. 43, 268–281 (1996).
Sinha, D. et al. Cep55 overexpression causes male-specific sterility in mice by suppressing Foxo1 nuclear retention through sustained activation of PI3K/Akt signaling. FASEB J. 32, 4984–4999 (2018).
Shen, Y. et al. Identification and Potential Value of Candidate Genes in Patients With Non-obstructive Azoospermia. Urology 164, 133–139 (2022).
Sha, Y. W. et al. A homozygous CEP135 mutation is associated with multiple morphological abnormalities of the sperm flagella (MMAF). Gene 633, 48–53 (2017).
Avasthi, P. et al. Germline deletion of Cetn1 causes infertility in male mice. J. Cell Sci. 126, 3204–3213 (2013).
Hall, E. A. et al. Acute versus chronic loss of mammalian Azi1/Cep131 results in distinct ciliary phenotypes. PLoS Genet 9, e1003928 (2013).
Zhang, X. et al. CEP128 is involved in spermatogenesis in humans and mice. Nat. Commun. 13, 1395 (2022).
Zhang, X. et al. Loss-of-function mutations in CEP78 cause male infertility in humans and mice. Sci. Adv. 8, eabn0968 (2022).
Jakobsen, L. et al. Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J. 30, 1520–1535 (2011).
Panda, S. et al. Noncoding RNA Ginir functions as an oncogene by associating with centrosomal proteins. PLoS Biol. 16, e2004204 (2018).
Nguyen, Q. P. H. et al. Comparative Super-Resolution Mapping of Basal Feet Reveals a Modular but Distinct Architecture in Primary and Motile Cilia. Dev. Cell 55, 209–223 e207 (2020).
Sha, Y. et al. Loss-of-function mutations in centrosomal protein 112 is associated with human acephalic spermatozoa phenotype. Clin. Genet 97, 321–328 (2020).
Zhang, H. et al. Liquid-liquid phase separation in biology: mechanisms, physiological functions and human diseases. Sci. China Life Sci. 63, 953–985 (2020).
Hyman, A. A., Weber, C. A. & Julicher, F. Liquid-liquid phase separation in biology. Annu Rev. Cell Dev. Biol. 30, 39–58 (2014).
Lee, A. K. et al. Translational Repression of G3BP in Cancer and Germ Cells Suppresses Stress Granules and Enhances Stress Tolerance. Mol. Cell 79, 645–659.e649 (2020).
Rog, O., Kohler, S. & Dernburg, A. F. The synaptonemal complex has liquid crystalline properties and spatially regulates meiotic recombination factors. Elife 6, https://doi.org/10.7554/eLife.21455 (2017)
Alavattam, K. G., Maezawa, S., Andreassen, P. R. & Namekawa, S. H. Meiotic sex chromosome inactivation and the XY body: a phase separation hypothesis. Cell Mol. Life Sci. 79, 18 (2021).
So, C. et al. A liquid-like spindle domain promotes acentrosomal spindle assembly in mammalian oocytes. Science 364. https://doi.org/10.1126/science.aat9557 (2019)
Dodson, A. E. & Kennedy, S. Phase Separation in Germ Cells and Development. Dev. Cell 55, 4–17 (2020).
Kang, J. Y. et al. LLPS of FXR1 drives spermiogenesis by activating translation of stored mRNAs. Science 377, eabj6647 (2022).
Bjorndahl, L. The sixth edition of the WHO Laboratory Manual for the Examination and Processing of Human Semen: ensuring quality and standardization in basic examination of human ejaculates. Fertil. Steril. 117, 246–251 (2022).
Fishman, E. L. et al. A novel atypical sperm centriole is functional during human fertilization. Nat. Commun. 9, 2210 (2018).
Khanal, S. et al. A dynamic basal complex modulates mammalian sperm movement. Nat. Commun. 12, 3808 (2021).
Lehti, M. S. & Sironen, A. Formation and function of sperm tail structures in association with sperm motility defects. Biol. Reprod. 97, 522–536 (2017).
Kumar, N. & Singh, A. K. The anatomy, movement, and functions of human sperm tail: an evolving mystery. Biol. Reprod. 104, 508–520 (2021).
Braun, D. A. & Hildebrandt, F. Ciliopathies. Cold Spring Harb. Perspect. Biol. 9. https://doi.org/10.1101/cshperspect.a028191 (2017)
Breslow, D. K. & Holland, A. J. Mechanism and Regulation of Centriole and Cilium Biogenesis. Annu. Rev. Biochem. 88, 691–724 (2019).
Liu, Y. & Shi, S. L. The roles of hnRNP A2/B1 in RNA biology and disease. Wiley Interdiscip. Rev. RNA 12, e1612 (2021).
Schmidt, T. et al. eIF4A1-dependent mRNAs employ purine-rich 5’UTR sequences to activate localised eIF4A1-unwinding through eIF4A1-multimerisation to facilitate translation. Nucleic Acids Res. 51, 1859–1879 (2023).
Hamey, J. J. & Wilkins, M. R. Methylation of Elongation Factor 1A: Where, Who, and Why? Trends Biochem. Sci. 43, 211–223 (2018).
Martin, E. W. & Holehouse, A. S. Intrinsically disordered protein regions and phase separation: sequence determinants of assembly or lack thereof. Emerg. Top. Life Sci. 4, 307–329 (2020).
Darling, A. L., Liu, Y., Oldfield, C. J. & Uversky, V. N. Intrinsically Disordered Proteome of Human Membrane-Less Organelles. Proteomics 18, e1700193 (2018).
Garcia-Jove Navarro, M. et al. RNA is a critical element for the sizing and the composition of phase-separated RNA-protein condensates. Nat. Commun. 10, 3230 (2019).
Martinez, G. et al. Whole-exome sequencing identifies mutations in FSIP2 as a recurrent cause of multiple morphological abnormalities of the sperm flagella. Hum. Reprod. 33, 1973–1984 (2018).
Zheng, R. et al. FSIP2 plays a role in the acrosome development during spermiogenesis. J. Med. Genet. 60, 254–264 (2023).
Bieniek, J. M., Lapin, C. D. & Jarvi, K. A. Genetics of CFTR and male infertility. Transl. Androl. Urol. 10, 1391–1400 (2021).
Turner, K. A. et al. Fluorescence-Based Ratiometric Analysis of Sperm Centrioles (FRAC) Finds Patient Age and Sperm Morphology Are Associated With Centriole Quality. Front. Cell Dev. Biol. 9, 658891 (2021).
Avidor-Reiss, T., Carr, A. & Fishman, E. L. The sperm centrioles. Mol. Cell Endocrinol. 518, 110987 (2020).
Feng, X. et al. Myosin 1D and the branched actin network control the condensation of p62 bodies. Cell Res. 32, 659–669 (2022).
Huizar, R. L. et al. A liquid-like organelle at the root of motile ciliopathy. Elife 7, https://doi.org/10.7554/eLife.38497 (2018)
King, S. M. Cytoplasmic factories for axonemal dynein assembly. J. Cell Sci. 134, https://doi.org/10.1242/jcs.258626 (2021)
Jiang, H. et al. Phase transition of spindle-associated protein regulate spindle apparatus assembly. Cell 163, 108–122 (2015).
Woodruff, J. B. et al. The Centrosome Is a Selective Condensate that Nucleates Microtubules by Concentrating Tubulin. Cell 169, 1066–1077.e1010 (2017).
Hernandez-Vega, A. et al. Local Nucleation of Microtubule Bundles through Tubulin Concentration into a Condensed Tau Phase. Cell Rep. 20, 2304–2312 (2017).
Yang, S. et al. Self-construction of actin networks through phase separation–induced abLIM1 condensates. Proc. Natl Acad. Sci. 119, e2122420119 (2022).
Liu, Z. et al. Liquid‒liquid phase separation: roles and implications in future cancer treatment. Int. J. Biol. Sci. 19, 4139–4156 (2023).
Li, T., Zeng, Z., Fan, C. & Xiong, W. Role of stress granules in tumorigenesis and cancer therapy. Biochim. Biophys. Acta Rev. Cancer 1878, 189006 (2023).
Ferreira, P. A. Nucleocytoplasmic transport at the crossroads of proteostasis, neurodegeneration and neuroprotection. FEBS Lett. 597, 2567–2589 (2023).
Sanchez-Alvarez, J., Cano-Corres, R. & Fuentes-Arderiu, X. A Complement for the WHO Laboratory Manual for the Examination and Processing of Human Semen (First Edition, 2010). EJIFCC 23, 103–106 (2012).
Tu, C. et al. Bi-allelic mutations of DNAH10 cause primary male infertility with asthenoteratozoospermia in humans and mice. Am. J. Hum. Genet. 108, 1466–1477 (2021).
Tan, Y. Q. et al. Loss-of-function mutations in TDRD7 lead to a rare novel syndrome combining congenital cataract and nonobstructive azoospermia in humans. Genet. Med. 21, 1209–1217 (2019).
Bryant, J. M., Meyer-Ficca, M. L., Dang, V. M., Berger, S. L. & Meyer, R. G. Separation of spermatogenic cell types using STA-PUT velocity sedimentation. J. Vis. Exp. https://doi.org/10.3791/50648 (2013)
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Meng, J., Cui, X., Rao, M. K., Chen, Y. & Huang, Y. Exome-based analysis for RNA epigenome sequencing data. Bioinformatics 29, 1565–1567 (2013).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Yu, G., Wang, L. G. & He, Q. Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015).
Ramírez, F., Dündar, F., Diehl, S., Grüning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Csardi, G. & Nepusz, T. The igraph software. Complex syst 1695, 1–9 (2006).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Mootha, V. K. et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 (2003).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Saitou, N. & Nei, M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evolution 4, 406–425 (1987).
Felsenstein, J. Confidence Limits On Phylogenies: An Approach Using The Bootstrap. Evolution 39, 783–791 (1985).
Tamura, K., Nei, M. & Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl Acad. Sci. USA 101, 11030–11035 (2004).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evolution 33, 1870–1874 (2016).
Halstead, J. M. et al. Translation. An RNA biosensor for imaging the first round of translation from single cells to living animals. Science 347, 1367–1671 (2015).
Acknowledgements
We thank patients, their family members, and normal control subjects for their support and contribution to this study. The authors thank the Analytical and Testing Center of Sichuan University for the morphology characterization. The authors thank Guiping Yuan and Yi He for assistance in generating the TEM and SEM images. This work was supported partly by the National Key R&D Program of China (2022YFC2702603 to W.X.), National Natural Science Foundation of China (82371624 to W.X.).
Author information
Authors and Affiliations
Contributions
W.X. designed and supervised the experiments. Y.T. and L.M. collected the data and conducted the clinical evaluations. X.Z., G.H., T.L., and T.J. performed most experiments and analyzed the data. G.Z., B.Z., X.C., Nana L., N.W., S.W., J.G., T.J., and R.Z. performed experiments including immunofluorescence, Western blotting, and cell culture experiments. X.Z. wrote the manuscript. D.D., Z.J., Z.X., Z.W., and N.L. assisted with the experiments. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mofang Liu and Tomer Avidor-Reiss for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, X., Huang, G., Jiang, T. et al. CEP112 coordinates translational regulation of essential fertility genes during spermiogenesis through phase separation in humans and mice. Nat Commun 15, 8465 (2024). https://doi.org/10.1038/s41467-024-52705-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-52705-8
This article is cited by
-
The spermatozoon neck role in infertility and intracytoplasmic sperm injection outcomes
Journal of Assisted Reproduction and Genetics (2026)
-
Function of manchette and intra-manchette transport in spermatogenesis and male fertility
Cell Communication and Signaling (2025)
-
Reproductive physiological impacts of high ambient temperature on animals: the impaired testicular function and compromised sperm quality in C57BL/6 mice
Biological Research (2025)
-
Mutations in CFAP57 disrupt the localization of MYH10 and IFT88, leading to flagellogenesis failure in humans and mice
Human Genomics (2025)
-
Single-cell transcriptomic atlas of the human testis across the reproductive lifespan
Nature Aging (2025)
