
Abstract
RNAs and their encoded proteins intricately regulate diverse cell types and states within the human body. Dysregulated RNA expressions or mutations can lead to various diseased cell states, including tumorigenesis. Detecting and manipulating these endogenous RNAs offers significant promise for restoring healthy cell states and targeting tumors both in research and clinical contexts. This study presents an RNA-IN and RNA-OUT genetic circuit capable dynamically sensing and manipulating any RNA target in a programmable manner. The RNA-IN module employes a programmable CRISPR-associated protease (CASP) complex for RNA detection, while the RNA-OUT module utilizes an engineered protease-responsive dCas9-VPR activator. Additionally, the CASP module can detect point mutations by harnessing an uncovered dual-nucleotide synergistic switching effect within the CASP complex, resulting in the amplification of point-mutation signals from initially undetectable levels (1.5-fold) to a remarkable 94-fold. We successfully showcase the circuit’s ability to rewire endogenous RNA-IN signals to activate endogenous progesterone biosynthesis pathway, dynamically monitor adipogenic differentiation of mesenchymal stem cells (MSCs) and the epithelial-to-mesenchmal trans-differentiation, as well as selective killing of tumor cells. The programmable RNA-IN and RNA-OUT circuit exhibits tremendous potential for applications in gene therapy, biosensing and design of synthetic regulatory networks.
Similar content being viewed by others

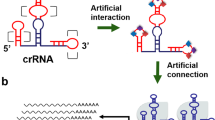

Introduction
Single-cell RNA sequencing technology has revolutionized our understanding of cellular diversity, uncovering a myriad of cell types and states along with their distinct physiological characteristics and developmental trajectories, both in healthy and pathological tissues1,2,3. Initiatives like the Human Cell Atlas and Cancer Genome Atlas projects have generated >4000 transcriptomic datasets (www.ncbi.nlm.nih.gov/geo/), offering comprehensive insights into the dynamic expression profiles of RNA molecules across various spatial and temporal dimensions4,5,6,7,8,9. Leveraging the transcriptomic data as signaling inputs and outputs holds immense potential for advancing both basic research and clinical applications10. To effectively rewire the regulation of endogenous input- and output-RNA expressions, a programmable RNA-IN and RNA-OUT regulatory circuit must be developed.
Three indispensable components comprise an RNA-IN/RNA-OUT circuit: a programmable RNA sensor for detecting any input RNAs, a programmable actuator for activating desired output RNAs, and a signal processor for linking the sensor to actuator modules. Various in vivo RNA sensors have been designed, including micro RNA (miRNA)11, switchable gRNA-based Cas912,13, eukaryotic toehold switch14, and programmable adenosine deaminases acting on RNA (ADAR)-based switches15,16,17,18. Unfortunately, existing sensors exhibit limitations in detecting point-mutated RNA targets, necessitating the development of high-resolution RNA-sensing technologies. For the actuator module, engineered dCas9-VPR and other programmable transcription activators could activate any desired RNA expression, thus suitable as actuators of the RNA-IN/RNA-OUT circuit. For the last but most important module, the signal processor must be carefully designed to properly transduce the activity from the upstream sensor to the downstream actuator (Fig. 1).

Three indispensable components comprise an RNA-IN/RNA-OUT circuit: 1) a programmable RNA sensor for detecting any input RNAs as RNA-IN module; 2) a programmable actuator for activating desired output-RNAs as RNA-OUT module; 3) a signal processor for linking the sensor to actuator modules.
Here, we report a high-resolution RNA-IN/RNA-OUT genetic circuit to rewire endogenous RNA regulatory network in a programmable manner. A highly sensitive RNA sensor as RNA-IN module is designed to detect insensitive point mutations by introducing another designed auxiliary mutation and creating a synergistic switching effect. The optimized synergistic sensor generates about a 94-fold response for point-mutated sequences compared to wild-type ones. A protease-responsive RNA-OUT module is developed to process the RNA-IN signal and activate any endogenous and exogenous RNA by conditionally inhibiting the dCas9-VPR activator with a protease-sensitive AcrIIA4 effector. The advanced RNA-based circuit is successfully applied in various directions, including rewiring an endogenous-mRNA expression to activate three mRNA expressions of a progesterone biosynthesis pathway, dynamically sensing the mRNA expression in the adipogenic differentiation of mesenchymal stem cells (MSCs) and epithelial-to-mesenchymal transition (EMT) of keratinocytes (HaCat), and selectively killing tumor cells harboring specific point-mutation mRNAs. We anticipate that the advanced RNA-based genetic circuit would find widespread applications in cell state sensing, cell fate transformation tracking, cancer cell-specific killing, and artificial control network synthesis and reconstruction.
Results
Design of programmable RNA sensors
In order to develop RNA-IN and RNA-OUT genetic circuits, we first need to build a programmable RNA sensor to detect any input RNAs. We proposed a “binding-and-signaling” strategy to create a programmable RNA sensor named the CASP sensor that enabled dynamic sensing in mammalian cells. The sensor consisted of two parts: a programmable RNA-binding protein and a signaling effector. We chose the type III-E Cas7-11 from Desulfonema ishimotonii (DiCas7-11)19,20,21 as a programmable RNA-binding protein for RNA recognition (Fig. 2a). The signaling effector was composed of a transcription factor (CI434) and an activator domain (VP64). The CASP sensors followed a crRNA-guided mechanism to activate a protease activity, releasing the signaling effectors from the cell membrane (Fig. 2a).

a Schematics of the CASP sensor. b The fluorescence outputs for the CASP sensor with or without target RNAs in HEK293T cell lines, the target and nontargets sequences were listed in Supplementary Data 2. Significance determined by two-tailed Student’s t test (P = 0.0008).****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. c The output distribution of the flow cytometry data for the CASP sensor, collecting at least 10,000 cells. d Performance comparison of the wild type (DiCas7-11(wt)) and RNase-deactivated (DiCas7-11(mut)) of CASP sensor. Fold Change is calculated as the fluorescence outputs in the presence of targeting-crRNA (crRNA (+)) vs non-targeting-crRNA (crRNA (–)). e The dose-response curves between CASP sensor output and the amounts of transfected plasmids of target RNA. Fold Change is calculated as the fluorescence outputs in the presence of target (Target RNA (+)) vs non-target (Target RNA (–)). f Detection of endogenous RNAs of 20 genes in the HEK293T cell lines by CASP sensor. Gene expression (transcripts per million, TPM) is shown in log-scale ranging from 3105 TPM (RSP2) to 1 TPM (CACNA1G) for the 20 genes. For each gene, 2–3 crRNAs are engineered to target different sites on the transcribed mRNAs. Fold Change is calculated as same as the ones in (d). Data are the average of three biological replicates ± s.e.m. Source data are provided as a Source Data file.
We tested the CASP sensor in human embryonic kidney 293T (HEK293T) cells. Firstly, we evaluated different membrane translocation sequences (MTS)22,23 and found that the N-terminal MTS yielded the higher performance (Supplementary Fig. 1). Then, we constructed the sensor using DiCas7-11, Csx29, MTS-Csx30-CI434-VP64 genes driven by the cytomegalovirus (CMV) promoter, a crRNA driven by the U6 promoter, and an mCherry reporter gene activated by the signaling effector (CI434-VP64). With the expression of exogenous non-target and target mRNA, we observed a 12.1-fold change of the fluorescence output (Fig. 2b, c). We observed that CASP sensor exhibited superior switching performance and lower background activity, and could be easily engineered by substituting a small crRNA sequence. These encouraging findings motivated us to delve deeper into researching and enhancing the capabilities of the CASP sensor.
Improving the sensitivity of CASP sensor
Previous research suggested that binding to target RNA was sufficient to activate the Csx29 protease activity, while the protease activity would turn off once the target RNA was cleaved20. This finding led us to speculate that the ribonuclease-deactivated DiCas7-11(mut)-Csx29-crRNA may exhibit more sustained protease activity than the DiCas7-11(wild-type)-Csx29-crRNA. To verify this hypothesis, we created a deactivated DiCas7-11D429A/D654A(DiCas7-11(mut)) mutant and designed crRNAs against both exogenous and endogenous mRNAs. Subsequently, HEK293T cells were transfected with the CASP sensor with or without targeting crRNAs. We calculated the fold change of the fluorescence outputs for each target with vs without targeting crRNAs in the sensor. Our results showed that in most cases, the deactivated DiCas7-11 mutant exhibited larger fold change than the wild-type one, indicating that the DiCas7-11(mut) had better RNA-sensing capacity compared to the DiCas7-11(wt) in the sensors (Fig. 2d and Supplementary Fig. 2).
We then quantitatively investigated the sensitivity of the DiCas7-11(wt) and DiCas7-11(mut) systems. We titrated the amounts of transfected plasmids of the targets (from 0.01 to 500 ng) in HEK293T cells, which were expected to linearly corelate with transcribed target mRNA levels24. For any given amount of the transfected plasmids, the DiCas7-11(mut) system consistently generated higher fold change than the DiCas7-11(wt) system (Fig. 2e). Our quantitative fitting curves revealed that the sensitivity index (EC50 value) was increased 20-fold (14.73 ng for the mutant vs 279.4 ng for the wild type), indicating that the ribonuclease-deactivated DiCas7-11 (mut) sensor is much more sensitive than the wild-type one (Supplementary Fig. 3).
To assess the endogenous-mRNA-sensing capabilities of the CASP sensor, we selected 20 endogenous mRNAs from HEK293T cells based on transcriptomic data (ranging from 1 to 3104 transcripts per million (TPM))25. For each target mRNA, we designed two or three crRNAs. Our results showed that the fold change for each mRNA varied considerably, but their maximal values could be fitted by the same sigmoid curve (Fig. 2f). Notably, the CASP sensor demonstrated sensitivity to a broad range of endogenous mRNAs (Fig. 2f). We also evaluated the performance of the CASP sensor across various human cell lines and observed that it remained robust performance in diverse contexts (Supplementary Fig. 4). These results indicated that the CASP sensor has great potential for various biological applications, such as identification of cell types and states, biosensing, disease diagnosis, drug screening, and cellular RNA imaging.
Synergistic effect for high-resolution CASP sensor
Genetic point mutations and aberrant RNA expressions are responsible for multiple diseases, especially cancers26, neurological diseases27 and viral infections28. However, sensing single-base-mutations is challenging due to the minimal difference between mutants and wild-types. To explore whether the CASP sensor could sense the single-base-mutation, we measured all point mutations from position 1 to 31 of a defined target mRNA. We observed almost all position mutations resulted in less than twofold change (Fig. 3a, Supplementary Fig. 5), indicating the CASP sensor was not sensitive enough. Considering position 21 was in the core seed region of the ternary DiCas7-11-Csx29-crRNA-tgRNA complex29, we attempted to introduce another auxiliary mismatch beside the position 21 point-mutation and observed that position 17 to 18 as an auxiliary site substantially increased up to 4-fold change compared to the auxiliary-site mutation only (Supplementary Fig. 6).

a Systematically scanning the single mutation (position 1 to 31) on a target RNA. Fold Change is calculated as the fluorescence outputs of perfectly matched vs point-mutated crRNAs. Blue arrows indicate two local maximal sites (position 3 and 21). b Schematics of the mechanism of synergistic switch for dual-mutation. c Systematically scanning the auxiliary site (position 1 to 31) of the dual-mutated crRNAs with the fixed target point-mutation on position 3. Fold Change is calculated as the fluorescence outputs of auxiliary mutation vs dual-mutated crRNAs. d Heat map of Fold Change for different dual-mutation combinations. e Synergistic effects for dual-mutation when comparing with matched (0 mismatch), single-mutated (1 mismatch), dual-mutated (2-mismatch) and non-targeting (31 mismatch) RNA targets from reporter genes or tumor driver genes. Fluorescence outputs are normalized by non-targeting points. Data are the average of three biological replicates ± s.e.m. Source data are provided as a Source Data file.
We hypothesized that some combinations of mutation and auxiliary site in the parallel pairing region (position 1 to 13) of crRNA could dramatically improve the fold change for point-mutation detection (Fig. 3b). To test this, we assigned position 3 as the point-mutation site based on its higher sensitivity in Fig. 3a and screened for auxiliary sites from position 1 to 31 (Fig. 3c and Supplementary Fig. 7). Our results showed that some dual-mutation combinations, specifically those involving position 1, 2, 5, 6, 7, 8, and 9 as auxiliary sites, produced a 1–40-fold increase of induction, while other regions did not induce or showed weak induction (Fig. 3c). Interestingly, the fold change distribution for the screened auxiliary sites was surprisingly consistent with the structural information of the ternary DiCas7-11-Csx29-crRNA-tgRNA complex. We observed that position 4, 10, and 14 as auxiliary sites did not significantly induce fold change (Fig. 3c and Supplementary Fig. 7). These sites corresponded to flipped unmatched or helical pairing bases in the DiCas7-11-Csx29-crRNA-tgRNA complex structure, and could be used to divide other auxiliary sites into four blocks: block-1 (position 1–3), block-2 (position 5 to 9), block-3 (position 11 to 13) and block-4 (position 14 to 31).
To further explore the design spaces of a synergistic CASP sensor, we systematically investigated the dual-mutation combinations across block-1 to block-4. After analyzing the fold change between point mutations and dual-mutations (Fig. 3d and Supplementary Fig. 8), we made several noteworthy observations about the synergistic effect. Firstly, the diagonals of the matrix, where only single mutations were introduced, showed that a single mutation was insufficient to switch off the CASP sensor. Secondly, the block-3 region was less sensitive to the synergistic effect. Thirdly, the combinations of block-1 and block-2 resulted in a high-fold change for the point-mutation sensing, supporting the importance of these two regions in synergistically turning off the CASP sensor. Fourthly, the best synergistic combination was the 3–7 dual-mutation, which was consistent with our previous results in Fig. 3c. Fifthly, dual-mutation combinations with position 21 were less effective due to its high background. Based on these results, we speculated the synergistic dual-mutations could dramatically deactivate the protease activity in the CASP complex, providing some valuable guidelines for future practical applications.
Furthermore, to enhance the effectiveness of the synergistic CASP sensor, we aimed to increase the activated output signals or to decrease the leaked output signals by changing the regulatory components (Supplementary Figs. 9 and 10). We first attempted to increase the activated outputs by replacing the weak activation domain (VP64) with stronger ones (VP64-P65, P65-Rta, VTR3, VRP) (Supplementary Fig. 10a). The VPR domain proved to be the most effective, with point-mutation detection capability reaching 40-fold. We further improved the activated outputs by increasing the number of transcription factor binding sites (TFBS) and truncating the Csx30 protease-substrate (Supplementary Figs. 9a, b and 10b, c). Both approaches significantly enhanced sensing performance, with more TFBSs or a shorten Csx30 resulting in increased fold change for the point-mutation detection. To decrease the leaked outputs, we employed two strategies to reduce the cellular concentration of the fused MTS-Csx30-CI434-VP64 signaling effector. Firstly, we added an N-terminal dihydrofolate reductase (DHFR) degron30 to promote the degradation of the intact effector proteins without affecting the cleaved CI434-VP64 signaling effector (Supplementary Figs. 9c, d and 10d). Secondly, we reduced the amount of transfected DNA of the fused effector from 250 ng to 150, 100, 50 or 10 ng. Reducing the amount of transfected DNA proved to be critical as the fold changes for point-mutation exhibited a peak value at 50 ng (Supplementary Fig. 10e). Finally, we combined the two best strategies (the VPR activation domain and 50 ng plasmid DNA of the effector) successfully achieved 94-fold change for point-mutation detection (Supplementary Fig. 10f).
Subsequently, we investigated the potentials of the synergistic CASP sensor to detect tumor-related single point mutations. We selected seven point mutations from four cancer driver genes, including KRAS (c.35G>T (G12V); c.35G>A (G12D); c.182A>G (Q61R))31, TP53 (c.742C>T (R248W); c.818G>A (R273H))32, BRAF (c.1799T>A (V600D))33, and PIK3CA (c.3145G>C (G1049R))34, which are frequently mutated in human pancreatic adenocarcinomas35, small cell lung cancer36, melanoma37 and breast cancers38. For each mutation, HEK293T cells were transiently transfected with plasmids of the fluorescence CASP sensor, the cancer driver gene, and plasmids expressing perfectly matched crRNAs (0 mismatch), point-mutated crRNAs (1 mismatch), dual-mutated crRNAs (2-mismatch) or non-targeting crRNAs (31 mismatch). The fold change of single-base-mutation could be calculated by the ratio of the 0 mismatch crRNA to 1 mismatch RNA (named 0–1 switch), or by the ratio of the 1 mismatch crRNA to 2-mismatch RNA (named 1–2 switch) (Fig. 3e). We observed that the “0–1 switch” generated only 1.5- to 2.2-fold change for the point mutations, while the “1–2 switch” generated 15- to 20-fold change for the same point mutations. The 2-mismatch crRNA totally abrogated CASP sensor function, similar to the effects of non-targeting crRNAs (Fig. 3e). These results supported that the dual-mutations at position 3 and 7 had the synergistic effect in switching the activity of the CASP sensor, which could be applied in the precision therapeutics, pathogen diagnosis (Supplementary Fig. 11) and other fields.
Design of RNA-IN/RNA-OUT genetic circuit to rewire endogenous RNA regulatory network
To construct an RNA-IN/RNA-OUT genetic circuit, the CASP sensor needs to be linked with a programmable and switchable dSpCas9-VPR-gRNA activator to direct the expression of a target output RNA. In a free dSpCas9-VPR-gRNA activator, the gRNA can guide the dSpCas9-VPR protein to bound the upstream region of the promoter of the target RNA, and the VPR domain could recruit the host RNA polymerase II to transcribe the output RNA. The Csx29 protease activity within the CASP complex serves as an ideal interface to connect an input RNA signal to a protease-responsive form of the dSpCas9-VPR-gRNA activator. We thus designed the protease-responsive dSpCas9-VPR-gRNA by fusing Csx30 and dSpCas9 inhibitor (AcrIIA4) tandemly to the N-terminal of the dSpCas9 protein. This design allows AcrIIA4 to interact with the DNA binding pocket of dSpCas9-VPR-gRNA complex with high specificity, preventing it from recognizing its targeted DNA sequence39 (Fig. 4a). Cleavage of Csx30 by Csx29 releases the dSpCas9-VPR-gRNA activator from the AcrIIA4 inhibitor (Fig. 4a), ultimately activating the output-RNAs in a programmable manner.

a Schematics of the RNA-IN/RNA-OUT genetic circuit. Programmable CASP sensor as the RNA-IN module, dSpCas9-VPR-gRNA as the RNA-OUT module, and the AcrIIA4 inhibitor fused with Csx30 and dSpCas9-VPR as signal processor. b The relative expression of endogenous RNA (MIAT) under three different endogenous RNAs (RPS2, HSP90AB1, CANX) as input in HEK293T cells transfected. Blank HEK293T cells without genetic circuits as negative group. Fold change is calculated as the relative MIAT expression under endogenous RNA as input vs the relative MIAT expression without any endogenous RNAs as input. c, d Optimization of RNA-IN/RNA-OUT genetic circuits, adding N-terminal dihydrofolate reductase (DHFR)-degron to reduce the concentration of free AcrIIA4 inhibitor (c). Weakened version of the AcrIIA4 with different key mutations to decrease the binding affinity of AcrIIA4 to dSpCas9 (d). e Schematics of heat shock responsive RNA-IN/RNA-OUT circuit to activate the expression of endogenous RNA (MIAT). f The relative expression of heat shock family gene (HSP70) at 42 °C or 37 °C. g The relative expression of MIAT of cells with HSP70-sensing genetic circuits transfected at 42 °C or 37 °C. Significance determined by two-tailed Student’s t test (P = 0.0009). ***P < 0.001, **P < 0.01, *P < 0.05. Data are the average of three biological replicates ± s.e.m. Source data are provided as a Source Data file.
Experimental data indicate that the transcriptional activity of constantly expressed protease-responsive activator was fully suppressed the fused AcrIIA4 inhibitor (Fig. 4b), while three different endogenously expressed RNAs (RPS2: 3104 TPM; HSP90AB1: 874 TPM; CANX: 616 TPM) successfully activated an endogenous target output RNA (MIAT: 0.12 TPM) (Fig. 4b). In particular, the most highly expressed RPS2 induced nearly 10-fold increase of MIAT. The expression of the RNA-OUT, however, remains notably low compared to that of a free dSpCas9-VPR-gRNA activator (Supplementary Fig. 12), suggesting that the cleaved AcrIIA4 still bound and inhibited the dSpCas9 protein. To mitigate this, we employed two strategies: 1) reducing the concentration of the free AcrIIA4 by adding N-terminal dihydrofolate reductase (DHFR) degron, and 2) decreasing the binding affinity of AcrIIA4 to dSpCas9 by mutating key residues. Incorporating the DHFR degron increased the activation expression of the output RNA (Fig. 4c). Subsequently, we introduced three weakened versions of AcrIIA4 mutants as a previous study40, which led to a further increase in the expression of the output RNA although the activation fold not significantly improved (Fig. 4d). Additionally, we constructed other versions of the protease-responsive dSpCas9-VPR-gRNA activator that demonstrated a similar activation effect (Supplementary Fig. 13).
To showcase the environmentally responsive capability of the RNA-IN/RNA-OUT circuit, we designed a circuit to sense dynamically changed transcripts of heat shock protein Hsp70 and control the expression of MIAT transcript. Upon exposure to higher temperatures (37°C to 42°C), we observed 12.9-fold increase in the Hsp70 gene expression. Only the high levels of Hsp70 mRNA, in combination with its cognate crRNA, could activate the MIAT mRNA expression, increasing 12.6-fold. Conversely, the absence of either Hsp70 mRNA or crRNA led to no increase in RNA-OUT expression (Fig. 4e–g). In summary, RNA-IN/RNA-OUT genetic circuits can be designed to rewire any constantly or dynamically expressed endogenous gene to alter other RNA expressions, demonstrating their programmable rewiring capability in cellular endogenous RNA regulatory networks.
Potential applications of the RNA-based genetic circuits
To harness the endogenous RNA sensing and manipulating capability of the RNA-IN/RNA-OUT circuit, we chose several different types of cells to rewire and reprogram their transcriptional regulatory networks, including: (1) rewiring constantly expressed RNAs to activate an endogenous biosynthesis pathway of progesterone, (2) dynamically monitoring cell state changes of cell differentiation and trans-differentiations, and (3) selectively killing pancreatic cancer and liver tumor cells by recognizing their featured point-mutated RNAs. First, we designed the RNA-IN/RNA-OUT genetic circuits to respectively sense one of three input RNAs (RPS2, CHSP90AB1, and CANX), and activate unrelated output mRNA (STAR, CYP11A1, and HSD3B2) for biosynthesis of progesterone from cholesterol in human HEK293T cells (Fig. 5a). We found that the expression of each output RNA was positively correlated with the expression levels of the sensed input RNAs (Fig. 5b), indicating the circuits could respond the dose change of the input RNAs. More interestingly, the yields of progesterone also depended on the concentration of the input RNAs, consistent with the biosynthesis gene expression levels (Fig. 5c). It is worthy to note that dose-dependent sensing and responding capability would empower the RNA-IN/RNA-OUT circuit to implement more sophisticated biological regulatory functions of cellular networks.

a The workflow to build RNA-sensing progesterone-producing cell factories in HEK293T cells. b The relative expression of STAR, CYP11A1 and HSD3B2 gene under different endogenous gene (RPS2, HSP90AB1, CANX) as input RNAs. c The biosynthesis pathway of progesterone and secreted progesterone levels in different RNA-IN and RNA-OUT genetic circuits. d Schematic of monitoring adipogenic differentiation of mesenchymal stem cells (MSCs). e Lipid droplet presence within the cells in brightfield and Oil Red O staining field, all experiments were repeated 2 times independently with similar results. f The microscopy images for the genetic circuits with or without crRNAs in adipogenic differentiation of MSCs. g The relative expression of adipocyte-related mRNA in undifferentiated and differentiated MSC cells. h The fluorescence of the genetic circuits with or without crRNAs targeted adipocyte-related mRNA in adipogenic differentiation of MSCs. i Schematic for monitoring trans-differentiation of HaCat from an epithelial-to-mesenchymal state. j Characterization of epithelial-to-mesenchymal transition (EMT) of keratinocytes (HaCat). Top: The cell morphology of HaCat with or without TGFβ factor induction. Bottom: The microscopy images for the genetic circuits with or without crRNAs in EMT induction of HaCat. k The relative expression of upregulated mRNAs of mesenchymal cell states in HaCat with or without TGFβ factor induction. l The fluorescence of the genetic circuits with or without crRNAs targeted mesenchymal-specific mRNAs with or without TGFβ factor induction in HaCat. m–p Sensing and killing two types of tumor cells with specific point mutations. Schematic of point-mutation-sensing-based tumor cell death (m). Point-mutation detection of endogenous TP53 R273H mutation (c.818G>A) in PANC1 tumor cells and Point-mutation detection of endogenous TP53 Y220C mutation (c.659A>G) in HuH7 tumor cells (n). Significance determined by two-tailed Student’s t test (Pleft = 0.004; Pright = 0.01). ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. Cell viability of HEK293T, PANC1, and HuH7 cells after transfection of different CASP sensor expressing HSV-tk (o, p). Non-targeting CASP sensor contains a scramble crRNA. Data are the average of three biological replicates ± s.e.m. Source data are provided as a Source Data file.
Dynamic changes in RNA profiles play a pivotal role in delineating closely related cell states/types during cell differentiation and trans-differentiation. Monitoring the process of cell differentiation and trans-differentiation through RNA sensing and responding to genetic circuits can provide profound insights into manipulating cell fate. The models of adipogenic differentiation of MSCs and EMT of HaCat were used to demonstrate the ability of the RNA-based genetic circuits to monitor the process of cell differentiation and trans-differentiation (Fig. 5d, i, Supplementary Fig. 14). MSCs can differentiate into adipocytes under specific induction medium (Fig. 5d), and their differentiation potential holds significant implications in stem cell therapy. We engineered genetic circuits targeting adipocyte-related mRNAs (Adipoq, Fabp4, PPARG) and validated successful differentiation through the significant lipid droplet presence and upregulated mRNA expression levels (Fig. 5e, g). The reporter gene of the genetic circuit was activated in response to elevated target mRNA levels (Fig. 5f, h). Likewise, the trans-differentiation of HaCat from an epithelial to a mesenchymal state plays a critical role in wound healing, skin regeneration, as well as tumor invasion and metastasis. The developed genetic circuits specifically targeted upregulated mRNAs of mesenchymal cell states, Fibronectin and Vimentin (Fig. 5i). Following TGFβ factor induction in vitro, observable changes in cell morphology (Fig. 5j) and increased mesenchymal-related mRNA level (Fig. 5k) were detected, indicating transcriptional changes driving cell state alterations. The reporter gene showed significant activation in response to increased target mRNA level (Fig. 5l, Supplementary Fig. 14). Our study underscored the potential of RNA-based genetic circuits in effectively sensing and responding to transcriptional alterations in cell types and states, offering a valuable tool for monitoring cell differentiation and trans-differentiation processes.
Specific point mutations in driver genes can also be used as a sensing target for the killing of tumor cells, which lack specific surface antigens. We introduced a fluorescence CASP into the Pancreatic Adenocarcinoma cell line PANC1 (TP53 R273H) and the Hepatocellular Carcinoma cell line HuH7 (TP53 Y220C) to assess the detection capability of endogenous point mutations. Our results demonstrated the successful identification of endogenous point mutations in tumor cells using the CASP sensor (Fig. 5m, n). Then, the toxic cargo (HSV-tk gene) regulated by the CASP sensor was delivered into HEK293T (TP53 R273/Y220), PANC1 (TP53 R273H/Y220) and HuH7 (TP53 R273/Y220C) cells to assess the specific tumor cell killing capability. To evaluate cell viability, we employed the CCK8 assay41 and observed that the genetic circuit targeting the R273H point-mutation selectively eliminated PANC1 cells, while the circuits for Y220C point-mutation selectively killed HuH7 cells, exhibiting minimal toxicity towards other cell types. Moreover, the control cells lacking targeted mutation exhibited no significant cell death (Fig. 5m, o, p). To further enhance the efficacy of the RNA-based genetic circuits in these two tumor cell lines, we employed an optimized CASP sensor and observed an enhanced killing effect similar to previous studies15 (Fig. 5m, o, p). These findings established a basis for effectively targeting previously undruggable tumor markers utilizing the RNA-sensing and responding system.
Discussion
In this study, we have developed a programmable RNA-IN/RNA-OUT genetic circuit composed of a CASP sensor, an actuator, and a signal processor. The programmable CASP sensor module could detect a broad spectrum of RNA signals in a living cell, including exogenously expressed RNA signals, endogenous expressed RNA signals, dynamically changing RNA signals, and point-mutated RNA signals. The point-mutation detection capability of the CASP sensor was harnessing the uncovered dual-nucleotide synergistic switching effect within the CASP complex, resulting in the amplification of point-mutation signals from initially undetectable levels (1.5-fold) to a remarkable 94-fold. To design the actuator and cognate signal processor, a protease-responsive dSpCas9-VPR-gRNA was designed to activate any output RNA in a programmable manner, and the protease activity as signaling connector linked the switchable Csx29 protease of CASP and protease-responsive dSpCas9 in the RNA-IN/RNA-OUT genetic circuit. To demonstrate the programmability of the RNA-based circuit, we designed a couple of circuits to sense different endogenous RNA signals and activate unrelated mRNA expression or to activate an endogenous progesterone biosynthesis pathway. Potential applications demonstrated in monitoring the mRNA expression in the adipogenic differentiation of MSCs and the EMT of HaCat, as well as selectively killing tumor cells by the point-mutation sensitive RNA circuits. This application highlights the ability of mammalian cell state monitoring and fate manipulation based on RNA signals, which has a wide range of application prospects in biology, biotechnology, and medicine.
Although the RNA-IN/RNA-OUT genetic circuits demonstrated high sensitivity and effectiveness in detecting high- and medium-expression endogenous gene targets, they face challenges in detecting very low-expression genes and the relevant point mutations. However, the sensitivity can be improved by incorporating additional positive feedback or enzymatic amplifier modules42,43 in the sophisticated design. Furthermore, the low delivery efficiency of plasmids into more cell types presents another important obstacle for the genetic circuits. We anticipate that the efficiency will be improved by utilizing large-cargo viral delivery vectors, such as adenovirus and HSV-1 vectors, in future studies.
Moreover, the RNA-based circuits for remarkable point-mutation detection can be harnessed by in vitro and in vivo identification of virus subtypes. This extends beyond influenza and HPV viruses to encompass emerging viruses like COVID-19. Accurate identification of virus subtypes enables a comprehensive understanding of their prevalence and pathogenicity, leading to more effective strategies for diagnosis, treatment, and prevention. Overall, the programmable high-resolution RNA-IN/RNA-OUT gene circuits developed in living cells can be used as an efficient and universal technology platform, which is expected to be used to study the entire RNA regulatory network of mammalian cells, realize cell state monitoring and cell fate manipulation, and be widely used in gene therapy, biosensing, and the design of synthetic regulatory networks.
Methods
Cloning of CASP sensor constructs
DiCas7-11 protein and Csx29 protein were attached to the BB1 via Golden Gate cloning using the enzyme SapI (NEB), respectively. mCherry as the reporter was expressed under OR434-TRE 3G promoter, and the MTS-Csx30-CI434-VP64 fusion protein was expressed under CMV promoter as a signaling effector. The genes were linked to each other by GS linker (GSSSS). Both the reporter and the signaling effector were attached to BB2. The crRNA was expressed under the U6 promoter, and the target gene was expressed under the CMV promoter. The crRNA and target gene were both attached to BB2. A complete list of plasmids and associated sequences were provided in Supplementary Data 1 and 2.
Plasmids were transformed into Trans10 Chemically competent cells (TransGen Biotech), and these cells were cultured in LB medium with 100 ug/ml ampicillin for selection. All plasmids were extracted from cells using QIAprep Spin Miniprep.
Mammalian cell culture
Human embryonic kidney cells (HEK293T, ATCC), human pancreatic cancer cells (PANC1, ATCC), human hepatoma cells (HuH7, ATCC) were cultured in Dulbecco’s modification Eagle’s medium (DMEM-high glucose, Hyclone), supplemented with an additional 10% fetal bovine serum (FBS, GIBCO) and 1% penicillin-streptomycin (PS, Hyclone). All cell cultures were kept under standard culture conditions (5% CO2 and 37 °C). Other types of cell lines used in this study: Intracholecystic Papillary Neoplasm cell lines (NOZ, ATCC), Esophagogastric Adenocarcinoma cell lines (AGS, ATCC), Hepatocellular Carcinoma cell lines (HuH7, ATCC), Diffuse Glioma cell lines (HS683, ATCC), Endometrial Carcinoma cell lines (RL952, ATCC), Invasive Breast Carcinoma cell lines (T47D, ATCC), Colorectal Adenocarcinoma cell lines (SW48, ATCC), Pancreatic Adenocarcinoma cell lines (MIAPaCa2, ATCC), Colorectal Adenocarcinoma cell lines (NCIH508, ATCC), Melanoma cell lines (WM115, ATCC), Invasive Breast Carcinoma cell lines (MCF7, ATCC), Diffuse Glioma cell lines (SF126, ATCC) were also cultured under the standard methods.
Cell transfections
According to the manufacturer’s instructions, all plasmids were transfected into cells by Lipofectamine 3000 (Invitrogen) and Lipo8000 (Beyotime). Typically, cells (seeded 2 × 105 per well) were seeded on 12-well plates coated with polyD-lysine. When the confluency of the cell was ~70–90%, transfection was performed. Among them, 250 ng per plasmid of the CASP Sensor was co-transfected in 12-well plates. For Lipofectamine 3000 transfection, we diluted 2 μl P3000 reagent into 50 μl of Opti-MEM (GIBCO), as well as 1.5 μl Lipofectamine 3000 and all plasmids into 50 μl of Opti-MEM per well. For Lipo8000 transfection, we diluted 2 μl Lipo8000 reagent and all plasmids into 50 μl of Opti-MEM per well. We analyzed the cells 72 h after transfection.
Microscopy images
For the imaging of cells, plates were removed from the incubator after 72 h transfection, and all wells were measured via EVOS M7000 (Invitrogen) at room temperature. Cells were imaged using a ×10 objective to photograph fluorescence under brightfield, GFP, RFP, BFP filters, respectively. The M7000 software was used for the quantitative and image analysis.
Flow cytometry and data analysis
Seventy-two hours after transfection, cells were harvested for flow cytometry analysis. To prepare samples into 96-well plates. We aspirated the medium, washed the cells with 100 μl PBS, and added 60 μl of 0.25% trypsin-EDTA (GIBCO) to digest the cells, harvested the cells, and resuspended them in flow buffer (HBSS + 2.5 mg/ml bovine serum albumin). Cells were analyzed by LSRFortessaTM flow cytometer (BD Biosciences) running FACSDiva software and the data were processed using the FlowJo (V10).
Scanning of point-mutation
Gluc was chosen to validate the point-mutation scanning. The crRNA contained 31-nt spacer that can pair with the target RNA sequence. The perfectly matched crRNA (Gluc) sequence was TGCAGCCAGCTTTGGGCATTGGCTTCCAT. 31 different types of crRNA plasmids were constructed by mutating each base in the crRNA sequence according to the principles of A→T/ T→A/C→G/G→C mutation. A complete list of sequences was provided in Supplementary Data 5. In each group, the crRNA plasmid and CASP system plasmids were co-transfected into HEK293T cells. After 72 hours, fluorescence analysis was performed.
Scanning of synergistic dual-mutation combinations based on position 21/3 point-mutation
In accordance with the point-mutation scanning strategy, the synergistic dual-mutation scanning was to introduce additional auxiliary mutation based on position 3/21 point-mutation. 30 different dual-mutation combination plasmids were constructed by mutating each base in the crRNA sequence according to the principles of A→T/ T→A/C→G/G→C mutation, respectively. A complete list of sequences was provided in Supplementary Data 5. In each group, the crRNA plasmid and CASP system plasmids were co-transfected into HEK293T cells. After 72 hours, fluorescence analysis was performed.
Systematic scanning of synergistic effect
The spacer was divided into four blocks: block-1 (position 1–3), block-2 (position 5–9), block-3 (position 11–13) and block-4 (position 14–31). We thoroughly scanned the sequence in the block-1, -2, and -3 regions, which were deemed more critical, while the sequences of the block-4 region were selectively tested. A complete list of sequences was provided in Supplementary Data 5. In each group, the crRNA plasmid and CASP system plasmids were co-transfected into HEK293T cells. After 72 hours, fluorescence analysis was performed.
Extraction of total RNA and quantitative PCR
The cells grown in the 12-well plate were lysis by TRIzol Reagent (Invitrogen), mixed with 200 μl chloroform, incubated at room temperature for 15 min, and then centrifuged at 4 degrees and 12,000 × g for 15 min. Add 0.5 mL of isopropanol to the aqueous phase and incubate for 10 minutes and then centrifuge at 4 degrees and 12,000 × g for 10 min. Total RNA precipitate formed a white gel-like pellet at the bottom of the tube. Wash them with 75% ethanol, and then dissolve with 30 μl of water to obtain RNA.
1ug total RNA was used for cDNA synthesis with random primers (GenStar). Primer pair sequences of different genes were list in in Supplementary Data 10. The cDNA was added to a qPCR mixture that contained 2X SYBR Green PCR master mix (GenStar). All qPCR reactions were performed in a 20 μl reaction with three technical replicates in 96-well format and read using the Qtower instrument. The expression level was calculated by subtracting the housekeeping control (GAPDH) period threshold (Ct) value from the target Ct value to normalize the total input, resulting in the ΔCt level. ΔΔCt was obtained by subtracting ΔCt from the control group sample ΔCt, and the relative transcript abundance was calculated as 2−ΔΔCt. All replicates were performed as biological replicates. Primer sequences are available in the Supplementary Data 10.
Heat shock-related mRNA-sensing genetic circuit testing
The HSP70-sensing RNA-IN/RNA-OUT genetic circuits were transfected into cells. At 24 h.p.i., the cells in the experimental group were moved to 42 °C for 24 h, while the cells in the control group remained at 37 °C for 24 h. Cell samples were collected for RNA extraction and RT-PCR analysis.
Differentiation of MSCs into adipocytes
MSCs were seeded into a six-well plate at the density of 2–3 × 104 cells per square centimeter. Each well was supplemented with 2 ml of complete culture medium for cultivation until the cell confluence reached 100%. The adipogenic induction medium ADP1 (supplemented with 10% fetal bovine serum, 1% glutamine, 1% antibiotics, 2‰ insulin, 1‰ IBMX, 1‰ rosiglitazone, 1‰ dexamethasone) for 3 days of induction and ADP2 (supplemented with 10% fetal bovine serum, 1% glutamine, 1% antibiotics, 2‰ insulin) for 1 day of induction were used alternately.
After 6–7 days of induction, observe the cell morphology and lipid droplet formation by Oil Red O staining followed the standard experimental procedure provided by Pronase Corporation for the Oil Red O reagent kit purchased. Remove the culture medium from the six-well plate, wash with PBS 1–2 times, and then add 4% neutral formaldehyde solution to fix the cells for 30 minutes. During this period, prepare the Oil Red O working solution by mixing saturated Oil Red O solution with distilled water at a ratio of 3:2. After 30 minutes, remove the formaldehyde solution, wash with PBS 1–2 times, and add 1 ml of the Oil Red O working solution to each well. Incubate for 30 minutes at room temperature. After staining, remove the Oil Red O working solution, wash away background impurities with PBS, and observe the induction and staining effects under a microscope. RT-PCR was used to analysis the target RNA expression level. The MSCs in the control group were cultured using a complete culture medium.
EMT induction
The human HaCat cell line and human hepatocellular carcinoma cell line (HuH7) were seeded into 12-well plates at a density of 1 × 105 cells per well. Each well was supplemented with 1 ml of complete culture medium (containing 10% fetal bovine serum and 1% Penicillin-Streptomycin). After culturing the cells for 8–12 hours, the medium was changed to a starvation medium (without fetal bovine serum, supplemented with 1% Penicillin-Streptomycin) for starvation treatment. After 24 hours, the medium was changed to an induction medium (without fetal bovine serum, supplemented with 1% Penicillin-–Streptomycin, 10 ng/ml TGFβ, and 100 ng/ml EGF or HGF). After 3 days of induction, the cell morphology was observed, and samples were collected for RT-PCR analysis. The cells in the control group were cultured with a starvation medium for adhesion culture.
Cell viability assay
HSV-tk was selected as reporter genes in replace of mCherry. HEK293T, PANC1, HuH7 cells were transfected with HSV-tk cargo CASP sensor. Forty-eight hours after transfection, we obtained the successfully transfected cells by flow cytometry sorting. 20,000 cells per well were seeded in 96-well plate. 500 μg/ml Ganciclovir (Cayman) was added for induction after six hours. After 48 hours of additional growth, cells were assayed for viability by CCK8 assay (Livning) according to the manufacturer’s protocol. Absorbance was determined at 450 nm, and cells in the control group were transfected with empty carrier plasmids as controls to calculate cell viability.
ELISA of progesterone
The progesterone was measured according to the standard instructions of the progesterone ELISA kit (Beyotime. no. SEKSM-0002). At 48 h post-transfection of control gRNA (NT) or the gRNA array with RNA-IN/RNA-OUT genetic circuits into HEK293T cell line, cell culture supernatant was collected. 90 μl cell culture supernatant was used for ELISA as per the manufacturer’s instructions. Measure the absorbance at 450 nm using an ELISA reader. Draw a standard curve based on the provided standard samples, and calculate the progesterone content based on the absorbance.
Statistical analysis
The reported values served as averages of at least three biological replicates and represent two independent biological experiments with similar results. GraphPad Prism was used for data processing and graphing, and two-tailed Student’s t test was used for p value testing.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available in the paper (and in its supplementary information files). The data generated in this study are provided in the Source Data file. DNA sequences are available in the Supplementary Sequences section of the Supplementary Data. Source data are provided with this paper.
References
Guruprasad, P., Lee, Y. G., Kim, K. H. & Ruella, M. The current landscape of single-cell transcriptomics for cancer immunotherapy. J. Exp. Med. 218, e20201574 (2021).
Karlsson, M. et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 7, eabh2169 (2021).
Qiu, X. et al. Mapping transcriptomic vector fields of single cells. Cell 185, 690–711.e645 (2022).
Souza-Santos, P. T. et al. Mutations, differential gene expression, and chimeric transcripts in esophageal squamous cell carcinoma show high heterogeneity. Transl. Oncol. 11, 1283–1291 (2018).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Group, P. T. C. et al. Genomic basis for RNA alterations in cancer. Nature 578, 129–136 (2020).
Urbanski, L. et al. MYC regulates a pan-cancer network of co-expressed oncogenic splicing factors. Cell Rep. 41, 111704 (2022).
Ma, Y. et al. Identification of mutations, gene expression changes and fusion transcripts by whole transcriptome RNAseq in docetaxel resistant prostate cancer cells. SpringerPlus 5, 1861 (2016).
Martínez-Jiménez, F. et al. Pan-cancer whole-genome comparison of primary and metastatic solid tumours. Nature 618, 333–341 (2023).
Wang, D., Liu, B. & Zhang, Z. Accelerating the understanding of cancer biology through the lens of genomics. Cell 186, 1755–1771 (2023).
Xie, Z., Wroblewska, L., Prochazka, L., Weiss, R. & Benenson, Y. Multi-input RNAi-based logic circuit for identification of specific cancer cells. Science 333, 1307–1311 (2011).
Wang, W. J. et al. Establishing artificial gene connections through RNA displacement-assembly-controlled CRISPR/Cas9 function. Nucleic Acids Res. 51, 7691–7703 (2023).
Lin, J., Wang, W. J., Wang, Y., Liu, Y. & Xu, L. Building endogenous gene connections through RNA self-assembly controlled CRISPR/Cas9 function. J. Am. Chem. Soc. 143, 19834–19843 (2021).
Zhao, E. M. et al. RNA-responsive elements for eukaryotic translational control. Nat. Biotechnol. 40, 539–545 (2022).
Jiang K. et al. Programmable eukaryotic protein synthesis with RNA sensors by harnessing ADAR. Nat. Biotechnol. 41, 698–707 (2022).
Kaseniit K. E. et al. Modular, programmable RNA sensing using ADAR editing in living cells. Nat. Biotechnol. 41, 482–487 (2022).
Qian, Y. et al. Programmable RNA sensing for cell monitoring and manipulation. Nature 610, 713–721 (2022).
Gayet, R. V. et al. Autocatalytic base editing for RNA-responsive translational control. Nat. Commun. 14, 1339 (2023).
Özcan, A. et al. Programmable RNA targeting with the single-protein CRISPR effector Cas7-11. Nature 597, 720–725 (2021).
Kato, K. et al. RNA-triggered protein cleavage and cell growth arrest by the type III-E CRISPR nuclease-protease. Science 378, 882–889 (2022).
Strecker, J. et al. RNA-activated protein cleavage with a CRISPR-associated endopeptidase. Science 378, 874–881 (2022).
Megason, S. G. In toto imaging of embryogenesis with confocal time-lapse microscopy. Methods Mol. Biol. 546, 317–332 (2009).
Heo, W. D. et al. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 314, 1458–1461 (2006).
Muldoon, J. J. et al. Model-guided design of mammalian genetic programs. Sci. Adv. 7, eabe9375 (2021).
Yang, M. et al. Integrated analysis of the altered lncRNAs and mRNAs expression in 293T cells after ionizing radiation exposure. Int. J. Mol. Sci. 20, 2968 (2019).
Dasa, O. & Pearson, T. A. Somatic mutations in “benign” disease. N. Engl. J. Med. 385, e34 (2021).
Proukakis, C. Somatic mutations in neurodegeneration: an update. Neurobiol. Dis. 144, 105021 (2020).
Lyons, D. M. & Lauring, A. S. Mutation and epistasis in influenza virus evolution. Viruses 10, 407 (2018).
Huo, Y., Zhao, H., Dong, Q. & Jiang, T. Cryo-EM structure and protease activity of the type III-E CRISPR-Cas effector. Nat. Microbiol. 8, 522–532 (2023).
Gao, X. J., Chong, L. S., Kim, M. S. & Elowitz, M. B. Programmable protein circuits in living cells. Science 361, 1252–1258 (2018).
Luo, J. KRAS mutation in pancreatic cancer. Semin. Oncol. 48, 10–18 (2021).
Olivier, M., Hollstein, M. & Hainaut, P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2, a001008 (2010).
Ritterhouse, L. L. & Barletta, J. A. BRAF V600E mutation-specific antibody: a review. Semin. Diagn. Pathol. 32, 400–408 (2015).
Kumar, D. T. & Doss, C. G. Investigating the inhibitory effect of wortmannin in the hotspot mutation at codon 1047 of PIK3CA kinase domain: a molecular docking and molecular dynamics approach. Adv. Protein Chem. Struct. Biol. 102, 267–297 (2016).
Huang, L., Guo, Z., Wang, F. & Fu, L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct. Target. Ther. 6, 386 (2021).
Chen, X. et al. Mutant p53 in cancer: from molecular mechanism to therapeutic modulation. Cell Death Dis. 13, 974 (2022).
Dankner, M., Rose, A. A. N., Rajkumar, S., Siegel, P. M. & Watson, I. R. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene 37, 3183–3199 (2018).
Willis, O. et al. PIK3CA gene aberrancy and role in targeted therapy of solid malignancies. Cancer Gene Ther. 27, 634–644 (2020).
Dong, D. et al. Structural basis of CRISPR-SpyCas9 inhibition by an anti-CRISPR protein. Nature 546, 436–439 (2017).
Aschenbrenner, S. et al. Coupling Cas9 to artificial inhibitory domains enhances CRISPR-Cas9 target specificity. Sci. Adv. 6, eaay0187 (2020).
Fan, Y. et al. CircNR3C2 promotes HRD1-mediated tumor-suppressive effect via sponging miR-513a-3p in triple-negative breast cancer. Mol. Cancer 20, 25 (2021).
Blüthgen, N. et al. Effects of sequestration on signal transduction cascades. FEBS J. 273, 895–906 (2006).
Buchler, N. E. & Cross, F. R. Protein sequestration generates a flexible ultrasensitive response in a genetic network. Mol. Syst. Biol. 5, 272 (2009).
Acknowledgements
We thank professor Shiyong Liu from Huazhong University of Science and Technology for helpful discussions. This work was supported by the Ministry of Science and Technology of China [no. 2021YFC2101700 to Q.W., no. 2021YFF1200500 and 2020YFA0907101 to C.B.L.], the Natural Science Foundation of China [no. 31961133019 to Q.W., no. 32071412 to C.B.L.], the Chinese Academy of Sciences [no. XDB0480000 of the Strategic Priority Research Program to C.B.L.] and CAS Youth Interdisciplinary Team. This work was also supported by the Vanke Special Fund for Public Health and Health Discipline Development, Tsinghua University (2022Z82WKJ006).
Author information
Authors and Affiliations
Contributions
Q.W., C.B.L., M.Z., X.Z., and Y.Y.X. designed the study. M.Z., X.Z., Y.Y.X., Y.H.X., and B.Z. performed and analyzed most of the experiments. M.Z., X.Z., Q.W., and C.B.L. wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xiaojing Gao, and Anna Sokolovska for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, M., Zhang, X., Xu, Y. et al. High-resolution and programmable RNA-IN and RNA-OUT genetic circuit in living mammalian cells. Nat Commun 15, 8768 (2024). https://doi.org/10.1038/s41467-024-52962-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-52962-7
