
Abstract
Branching enzymes (BEs) confer to α-glucans, the primary energy-storage reservoir in nature, a variety of features, like slow digestion. The full catalytic cycle of BEs can be divided in six steps, namely two covalent catalytic steps involving glycosylation and transglycosylation, and four noncatalytic steps involving substrate binding and transfers (SBTs). Despite the ever-growing wealth of biochemical and structural information on BEs, clear mechanistic insights into SBTs from an industrial-performance perspective are still missing. Here, we report a Rhodothermus profundi BE (RpBE) endowed with twice as much enzymatic activity as the Rhodothermus obamensis BE currently used in industry. Furthermore, we focus on the SBTs for RpBE by means of large-scale computations supported by experiment. Engineering of the crucial positions responsible for the initial substrate-binding step improves enzymatic activity significantly, while offering a possibility to customize product types. In addition, we show that the high-efficiency substrate-transfer steps preceding glycosylation and transglycosylation are the main reason for the remarkable enzymatic activity of RpBE, suggestive of engineering directions for the BE family.
Similar content being viewed by others

Introduction
α-glucans, mainly referring to starch and glycogen, are the predominant carbohydrates accumulated for energy storage in nature1; their utilization, for instance, as main human diet constituents and food-industry raw materials, is increasing globally because of their low cost, wide distribution, and renewability2,3. Branching enzymes (BEs; EC number 2.4.1.18 at https://www.brenda-enzymes.org/), which possess distinctive functions of creating α−1,6-glucosidic linkages on the α−1,4-glucoside-linked glucan chains, can alter the physicochemical properties of α-glucans, and accordingly modulate their biological functions4,5. It would be, therefore, extremely valuable to have a complete understanding of the functional mechanism of BEs to help develop mechanism-based enzyme-engineering strategies aimed at improving enzymatic performance and at synthesizing specific products.
BEs are distributed ubiquitously in nature6,7,8. The first primary structure information on BE was on the enzyme from Escherichia coli (E. coli) defined in 19869 and subsequently discovered to be homologous to amylolytic enzymes10. To date, numerous BEs have been characterized, including the elucidation of their three-dimensional structure, and they have been categorized in the glycoside-hydrolase (GH) family 13 and 5711,12,13 in the carbohydrate-active enzymes database (CAZy; www.cazy.org)14. BEs possess α−1,4-glucoside-linkage hydrolytic activity and α−1,6-glucoside-linkage creating activity, producing branching patterns, i.e., the transglycosylation function15. Although the covalent catalytic steps have been elucidated16, the product properties of BEs from different species and their variants are very diverse; the mechanistic basis for their functional discrepancy in this respect remains somewhat unclear17.
In a directed evolution study of cyclodextrin glucanotransferase into an α-amylase, Kelly et al. have provided an early evidence for the potential correlation of the enzyme-substrate binding mode with product specificities18; the findings of Ban et al. further supported this correlation19. By means of crystallographic experiments, a number of research groups, e.g., Feng et al. (PDB codes 4LPC and 5E6Z)20,21, Chaen et al. (PDB code 3VU2)22, Froese et al. (PDB code 5CLW)23, Gavgani et al. (PDB code 7ML5)24, and Fawaz et al. (PDB code 8SDB)25 have explored possible binding modes of substrates on the outside of the active region of BEs. The only experimental evidence of polysaccharide binding to the active-site cleft came from Hayashi et al17. Based on their findings, one possible substrate binding mode during transglycosylation was proposed. However, although much attention has been given in recent years to the noncovalent interactions of BEs with polysaccharides, crystallographic data only provide limited substrate binding clues, and the detailed SBTs through the entire catalytic process have not yet been decrypted.
In this contribution, we report a Rhodothermus profundi BE (RpBE), which possesses twice as much activity (total activity, including hydrolysis and branching activity) as the commercial BE from Rhodothermus obamensis (RoBE; also known as RoGBE; Branchzyme®, Novozymes)26. Based on the modeled RpBE structure, we unveil its distinctive SBTs mechanism by means of large-scale enzyme-substrate docking, molecular dynamics (MD) simulations, as well as one- and two-dimensional free-energy calculations. We also perform biochemical studies to support the proposed mechanism, and further conduct mechanism-based enzyme engineering to improve the performance of RpBE. In general, the present work, at the confluence of theory and experiment, brings a fundamental understanding of noncovalent interactions of BE with their substrates; the engineering avenues brought to light by the SBTs are expected to promote further development of these industrially important BEs.
Results
Characteristics of RpBE
Thermostability and activity are some of the key parameters for the industrial use of BEs1. We performed a sequence search with Blastp through the NCBI web server (https://www.ncbi.nlm.nih.gov/) to find highly homologous sequences with the industrial RoBE in thermophilic bacteria (see homology analysis in Supplementary Fig. 1). Nine uncharacterized BEs, consisting of enzymes secreted by R. profundi DSM 22212 (RpBE), Desulfurococcaceae archaeon (DaBE), Chloroflexi bacterium (CbBE), Thermosynechococcus elongatus (TeBE), Hydrogenobacter hydrogenophilus (HhBE), Myxococcales bacterium (MbBE), Thermoplasmata archaeon (TaBE), Limisphaera ngatamarikiensis (LnBE), and Thermobifida halotolerans (ThBE) were chosen and produced recombinantly. We then measured the thermostability of the above BEs, and further compared them with three characterized BEs, i.e., RoBE and enzymes from Aquifex aeolicus (AaBE) and Butyrivibrio fibrisolvens (BfBE).
As shown in Fig. 1, RpBE possesses the highest activity at 60 oC and around neutral pH (the detailed information is provided in Supplementary Fig. 2 and Fig. 3a); it has approximately twice the activity of the commercial RoBE. Furthermore, the branching degree of amylose after catalysis with RpBE reached 14.3% (see nuclear magnetic resonance spectroscopy results in Supplementary Fig. 3b), which is much higher than that of the wild-type (WT) RoBE and of its variants19. Additionally, it has been reported that the degree of polymerization (DP) of the α−1,6-glucoside-linked branching chain affects digestibility27,28. As illustrated in Supplementary Fig. 3c, with RpBE catalysis, the DP distributes in 2–8, which contributes to a healthy, slow digestion, suggesting excellent performance of RpBE.

Activity measurement for the twelve BEs at their optimal reaction conditions. The vocabulary activity (ordinate) denotes the total activity, consisting of hydrolysis and branching activity. The bars and the error bars are the average and standard deviation of triplicates (n = 3 independent experiments), respectively. Source data are provided as a Source Data file.
Structural feature
Nowadays, computer-based structure prediction has become a powerful tool to provide three-dimensional protein structure models to help decipher enzymatic biological functions29. AlphaFold30, the state-of-the-art artificial intelligence (AI) algorithm developed by DeepMind, can predict three-dimensional protein structures with atomic accuracy. Powered by AlphaFold v2.0, the openly accessible AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk)31 contains over 200 million predicted structures, the accuracy of which has been demonstrated to be competitive with the ones obtained by traditional crystallographic methods32. Among the determined BE crystals to date, as reported in Supplementary Table 1, RpBE has the highest sequence homology of 93.4% with RoBE (PDB code 6JOY). Such a high homology will contribute to improving the accuracy of model construction. Consequently, the three-dimensional structure of RpBE constructed with AlphaFold v2.0 possesses a remarkable average model confidence of 96.4. The unified estimation data in Supplementary Table 2 analyzed by the ResQ server (https://zhanggroup.org/ResQ/)33 further suggests the soundness of the constructed RpBE model. The 3D RpBE structure, as depicted in Fig. 2a, indicates that the enzyme is composed of three structural domains, in which domain A is the catalytic area, containing the active-site cleft and catalytic site. Furthermore, at the center of domain A, there is a (β/α)8 barrel (see Fig. 2b), according to which, RpBE is classified in the GH 13 family1. The catalytic triad, which is located at the top of the (β/α)8 barrel, and consists of residues D305, E356, and D424, is responsible for the hydrolysis and transglycosylation reactions.

a Three domains (N, A and C) of RpBE, in which the catalytic domain is known as the A domain, containing the active-site cleft and catalytic residues. The vertical lines represent the approximate partition of the three domains. b (β/α)8 barrel structure. D305, E356, and D424, shown in a licorice representation and colored in yellow, constitute the classical catalytic triad in the GH13 family.
As shown in Supplementary Fig. 4, three loops (loop 1: 105–114, loop 2: 257–274, and loop 3: 314–328; RpBE numbering) lie on the side of the active-site cleft and may affect the enzyme-substrate association. RpBE loops 2 and 3 are very similar to the relevant ones of the BE crystal from Cyanothece sp. ATCC 51142 (CceBE1, PDB code 5GQU; see Supplementary Fig. 4,5). In RoBE (PDB code 6JOY), loops 2 and 3, however, adopt different conformations and exhibit the highest flexibility in the three BEs, especially loop 3. We, therefore, reason that when the enzyme interacts with the substrate, RpBE and CceBE1 loops 2 and 3 do not undergo significant conformational changes like the corresponding loops in RoBE. Furthermore, we found that the conformation and flexibility of loop 1 vary between the three enzymes, suggesting significant structural differences across species.
We further conducted crystallographic experiments in the hope of providing experimental evidence for the conformation of the important loop 2. We have obtained the RpBE crystal with a 2.9 Å resolution (PDB code 8ZQA, see Supplementary Table 3). Our findings are in line with the structural prediction of AlphaFold v2.0 (see Supplementary Fig. 6a,b); the conformation of loop 2 in the RpBE crystal is similar to that from the AlphaFold modeling of RpBE and CceBE1, albeit quite different from that in RoBE (see Supplementary Fig. 6c). Therefore, despite the high sequence similarity between RpBE and RoBE, the conformation of loop 2 can prove to be different between the two structures. The pronounced flexibility of loop 3, however, precluded access to its electron density. Given the similarity between modeled and experimental structures, as well as the low resolution of the crystal structure, it is reasonable to perform computational work using the modeled structure.
Donor-substrate initial binding (step i)
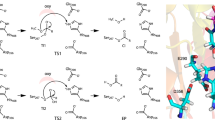
On the basis of the constructed RpBE structure described above, we first defined the process of generating the branching product from a linear polysaccharide with BE catalysis, which consists of (i) donor-substrate initial binding, (ii) donor-substrate transferring to the catalytic position, (iii) glycosylation, (iv) acceptor-substrate initial binding, (v) acceptor-substrate transferring to the catalytic position, and (vi) transglycosylation (see Fig. 3). Steps (iii) and (vi) are covalent catalysis obeying a well-defined mechanism, while the noncovalent SBTs investigated in the present work involve the other four steps, i.e., (i), (ii), (iv), and (v). To explore as many enzyme-substrate initial binding modes as possible (step i), we defined a broad substrate binding surface on the active-site cleft side throughout the three domains (N, A, and C) of RpBE (see Supplementary Fig. 7), which contains 138 residues used in the molecular docking process (see Supplementary Table 4). Regarding the substrate selection for docking, maltoheptaose (G7)—an oligosaccharide, is the most common linear substrate model obtained in BE-substrate crystal complexes17,20,23, and the only polysaccharide that binds to the active-site cleft to date17. Additionally, as shown in Supplementary Fig. 3c, maltodecaose (G10) is the minimum glucan length necessary for the RpBE reaction to be observed. We, therefore, employed both G7 and G10 as substrates to dock with RpBE, and compared their binding poses.

In (a), step i, the substrate is G7. The constructed RpBE was employed as the structural model; For clarity, the enzyme is displayed with an opaque representation in a, but with a semi-transparent one in b,d and e. In (b), step ii, D305, E356, and D424 form the catalytic triad in RpBE. ES I denotes the noncovalent enzyme-substrate complex before glycosylation. In (c), step iii, TS1 and CI denote the transition state in the course of glycosylation and the formed enzyme-substrate covalent intermediate (CI) after glycosylation, respectively. In (d), step iv, the acceptor chain (cyan) binds to the enzyme. In (e), step v, the acceptor chain moves to the catalytic position and prepares for transglycosylation, wherein the donor chain (green) will be linked as a branch to the hydroxyl on C6 of the cyan acceptor chain. ES II denotes the enzyme-substrate complex before transglycosylation. In (f), step vi, TS2 and EP denote the transition state in transglycosylation and the noncovalent enzyme-product complex after catalysis, respectively; the product is the α−1,6-glucosidic branching glucan.
Docking can generate useful and testable experimental hypotheses, albeit many challenges in enzyme-glycan docking remain to be addressed. The HADDOCK web server34,35, GlycoTorch Vina36, and Vina-Carb37 are the main software currently available to perform glycan docking. We chose the HADDOCK web server based on the following reasons: (i) the refinement step using MD simulations greatly contributes to optimizing the binding pose of the substrate on the enzyme, which is not available in other docking software; (ii) the clustering information, which reflects the number of polysaccharides lying at a similar location, is an important indicator to evaluate different poses; and (iii) the user-friendly interface enables protein engineers and biotechnologists to run docking procedure without the need of programming skills and local high-performance computers. The HADDOCK web server, with version 2.4 of the program, was therefore employed to perform the 50,000 enzyme-substrate rigid docking attempts to get a sufficient number of polysaccharide binding poses. The top 1000 generated structures of the complex were selected, and subsequently subjected to semi-flexible refinement as a preamble to a final refinement with short MD simulations in water. Root mean square deviation (RMSD) values for assessing structural stability, and the energies, such as van der Waals and electrostatic force were calculated for these refined structures. Structural clustering based on the distance RMSD over atomic positions38 reflects the number of polysaccharides lying at a similar location. The optimal binding poses in clusters i-iv are named accordingly poses i–iv.
As illustrated in Supplementary Fig. 8a,b, the optimal conformation of G7 binding to RpBE is very similar to that of G10. Supplementary Fig. 8c, d, however, indicate that G10 adopts a twisted conformation, wherein three glucosyl rings do not interact with the enzyme. The electrostatic interaction energy in the optimal RpBE-G10 complex is much lower than that in the optimal RpBE-G7 complex (see Supplementary Table 5 and Table 6). These results suggest that compared to G10, using G7 as a substrate is more conducive to exploring enzyme-substrate association. Furthermore, in a previous work, Ban et al. employed G7 binding to RoBE in order to examine potential mutation sites that affect enzyme activity19. These theoretical investigations of G7 binding to BEs, together with the numerous BE-G7 crystallographic complexes (PDB codes 5GQX, 4LPC, 5CLW, to cite a few) obtained experimentally17,20,23 led us to focus on RpBE-G7 association in the present study.
As depicted in Fig. 4a, we align the RpBE-G7 complex (pose i) with the CceBE1-G7 complex. Additionally, Fig. 4b indicates that in the RpBE-G7 complex, seven residues interact with the polysaccharide chain, wherein residue W270 (RpBE numbering) is the conserved residue, corresponding to residue W399 in the CceBE1-G7 complex. We further detected the binding affinity between RpBE and G7 using isothermal titration calorimetry (ITC) experiment, which is −4.1 ± 0.2 kcal/mol (see Supplementary Fig. 9). Ban et al. also aligned their docking RoBE-G7 complex with the CceBE1-G7 complex, and deduced a possible conformation of a long-chain substrate binding to RoBE through the poses of the two G7 substrates. In light of the result of our alignment, we suggest the possible conformation of a long-chain donor substrate binding to RpBE (see Supplementary Fig. 10; named CceBE1 G7-pose i here).

a Alignment of pose i with the substrate in the CceBE1-G7 complex. The carbon atoms of the substrates in the CceBE1-G7 complex (PDB code 5GQX) and the RpBE-G7 complex (pose i) are colored in blue and green, respectively. In the center of RpBE, the catalytic triad is highlighted in pink. b Enzyme-substrate interaction in pose i. The polysaccharide chain and its adjacent residues are displayed as a licorice representation, the carbon atoms of which are colored in green and cyan, respectively. c Average interaction energy of the polysaccharide chain with the adjacent residues in pose i. d Time series of the interaction energies of residues D269 and W270 with the polysaccharide chain in pose i. e Interaction energies of the catalytic triad with the polysaccharide chain in pose i. Source data for c–e are provided as a Source Data file.
Donor-substrate transfer for glycosylation (step ii)
To identify the key residues that play a noteworthy role in the step ii, we perform µs-scale MD simulations and calculated the interaction energies of the polysaccharide chain with the adjacent residues in binding pose i. Among the seven interacting residues, as shown in Fig. 4c, D269 and W270 are the two ones that interact the strongest with the polysaccharide chain. We further found that the interaction energy of residue W270 with the polysaccharide chain is constant, while dynamic and extremely strong interactions were observed between residue D269 and the polysaccharide chain (see Fig. 4d). We reason that residue D269 is the decisive residue for the substrate transfer. To investigate the importance of residue D269 experimentally, we performed alanine replacement at this position, and found that only 16.0% relative activity remained (see Supplementary Fig. 11). We further conducted circular dichroism (CD) spectroscopy experiments and found that the variant is properly folded (see Supplementary Fig. 12a). The decrease in enzyme activity is, therefore, due to the disruption of the interaction between D269A and the substrate.
In the active site of RpBE, residues D305, E356, and D424 form the catalytic triad of the enzyme. We analyzed the interaction energies of the catalytic triad with the polysaccharide chain in pose i and found that residue D305 possesses the strongest affinity for the substrate (see Fig. 4e). To confirm experimentally the importance of residue D305 in catalysis, we performed alanine replacement at this position, which, as expected, resulted in an almost complete loss of activity (5.6% relative activity remained; see Supplementary Fig. 11). The CD spectroscopy experiment indicates that the variant is properly folded (see Supplementary Fig. 12b). Given the key functions of residues D269 and D305 in substrate transfer, we propose that after the initial step i (initial donor-substrate-enzyme binding), step ii consists first in residue D269 grabbing the substrate and releasing it in the active site region, i.e., the ‘G-R’ actions. Then, D305 competes for substrate interactions with D269 and finally stabilizes it in the catalytic position, i.e., the ‘C-S’ actions. The competing action of D305 is thought to occur simultaneously with the releasing action by D269.
To decipher the detailed mechanism of ‘G-R/C-S’ actions mediated by residues D269 and D305, we performed two-dimensional free-energy calculations by defining the distance reaction coordinates, namely the distance separating the center of mass (COM) of the –1 glucosyl ring from residues D269 and D305 (RCs; see Fig. 5a). The free-energy landscape shown with a minimum free-energy path (see Fig. 5b) indicates an extremely favorable free energy for residue D269 interacting with the polysaccharide chain (site I). This result also implies that the grabbing action by this residue could be the dominant force for the polysaccharide chain approaching the active site. Furthermore, the processes from site I to site II (‘R/C’ actions), and from site II to site III (‘C-S’ actions) overcome approximately 0.3 kcal/mol and 1.8 kcal/mol free-energy barrier, respectively (see Fig. 5b, c).

a Defined RCs. d1 denotes the distance between the COM of the –1 glucosyl ring and residue D305. ξ1 denotes the distance between the COM of the –1 glucosyl ring and residue D269. Two-dimensional free-energy landscape (b) and (c) potential of mean force characterizing the polysaccharide chain approaching the catalytic position. The thick red line in (b) highlights the least free-energy pathway66 for the polysaccharide chain movement to the catalytic position. In (b), S0 (site I) corresponds to the initial position that residue D269 interacts with G7, and S1 (site III), the final stable position that G7 has reached the catalytic position and stabilized by residue D305. d Two-dimensional free-energy landscape characterizing the polysaccharide chain approaching the catalytic position with P268A replacement. e One-dimensional free-energy profile, or potential of mean force, characterizing residue D269 approaching residue D305 in the polysaccharide-free model. d2 denotes the distance of the COM between residues D269 and D305. Source data for c,e are provided as a Source Data file.
To explore the universality of ‘G-R/C-S’ actions in the BE family, we performed a structure alignment of RpBE with CceBE1 (PDB code 5GQU)17. Residues W198 and W270 in RpBE are the frame residues that stabilize the polysaccharide chain, which correspond to residues W327 and W399 in CceBE1 (see Supplementary Fig. 13a). However, the residues are of a different nature at position 268 (P268 in RpBE corresponding to K397 in CceBE1). We infer that residue replacement at position 268 may influence the ‘G-R’ actions of D269 and further modulate enzyme activity. To verify this hypothesis, we performed saturation mutagenesis at this position in RpBE, and found that the variants are properly folded (see CD spectroscopy results in Supplementary Fig. 12c–e for three randomly selected variants), but the activity in each variant has been impaired (see Supplementary Fig. 14).
To explain this phenomenon from a mechanistic perspective, we carried out two-dimensional free-energy calculations for the P268A variant (the best replacement in the nineteen negative ones, which still retained 26.9% relative activity) following the identical parameters of Fig. 5b. As shown in Fig. 5d, there are no free-energy low-lying lands at the sites of I, II, and III, which imply that the ‘G-R/C-S’ actions mediated by residues D269 and D305 have been destroyed. To further clarify the cause behind it, we performed one-dimensional free-energy calculations for the WT RpBE and the P268A variant to investigate the process of D269 approaching D305 in a substrate-free model (there is no polysaccharide in the active-site cleft). As depicted in Fig. 5e, the free energy is favorable for D269 approaching D305, which will help D269 deliver the polysaccharide to D305 to facilitate the subsequent releasing action of D269 and the competing action of D305. In contrast, in the P268A variant, the free energy is very unfavorable for D269 approaching D305, thereby hindering the “R/C” actions. P268 is, therefore, considered to play a role in the process of D269 approaching D305.
We aligned the WT RpBE with the P268A variant, and found that the orientation of D269 is not altered by mutagenesis. Loop 2, including P268 and D269, plays an important role in enzyme-substrate association. Furthermore, as shown in Supplementary Fig. 5, loop 2 possesses a high flexibility. Proline, which has the feature of conferring structural stability, can stabilize loop 2. We, therefore, infer that P268 can modulate the motion of loop 2 in enzyme-substrate interaction to promote the approach of D269 towards D305. In addition, we aligned the RpBE with RoBE (PDB code 6JOY)39, whereas the latter possess a long, flexible loop (see Supplementary Fig. 13b). The sequence alignment results at that region suggested that ten out of the set of twelve BEs possess the P and D assembly, including the RoBE (see Supplementary Fig. 15). Therefore, we reason that P268 and D269 in conjunction with the short loop 2 may be the result of natural evolution, endowing the RpBE with superior activity.
Acceptor-substrate initial binding for transglycosylation (step iv)
After glycosylation, as depicted in Fig. 3d, a new polysaccharide (shown in cyan) binds the enzyme as an acceptor chain. The donor polysaccharide chain (shown in green) remained at the active-site cleft will act as a branch to link to the acceptor chain by the transglycosylation. To elaborate on step iv, as shown in Supplementary Fig. 16 and Table 7, we chose 85 residues as the docking surface and docked a new G7 chain to the enzyme-donor substrate complex. As illustrated in Fig. 6a, pose I which possesses the largest cluster, containing 479 structures, is the optimal form, while the cluster of pose II consists of 110 structures (see Supplementary Table 8). Furthermore, we found that the polysaccharide chain (white) in the optimal complex structure of pose II did not reach the catalytic position. The above results suggest that pose I corresponds to the most likely acceptor-substrate binding mode prior to transglycosylation. Additionally, as depicted in Fig. 6b, twelve residues are found to form hydrogen-bonding interactions with the polysaccharide chain (pose I), namely D314, S316, R317, H385, R392, D424, E425, G429, K430, G431, T435, and K436.

a Docking results of the acceptor polysaccharide chains (cyan and white) for transglycosylation. The carbon atoms of the donor polysaccharide chain in the active-site cleft are colored in green. b Hydrogen-bonding interactions of the acceptor polysaccharide chain (pose I) with the adjacent twelve residues. The polysaccharide chain and the adjacent residues are shown in a licorice representation, and their carbon atoms are colored in cyan and orange, respectively. c Distance (Å) separating the COM of the polysaccharide chain and the catalytic triad with the time course in each assembly. A distance greater than 17 Å means that the polysaccharide chain has separated from the active-site cleft. d and e Interactions of the polysaccharide chain with the protein and the adjacent residues, respectively. Source data for c–e are provided as a Source Data file.
Acceptor-substrate transfer for transglycosylation (step v)
We performed MD simulations to investigate the dynamic movement of the acceptor polysaccharide chains in poses I and II. As depicted in Fig. 6c, although the polysaccharide chain in pose I interacts with the enzyme much longer time than the one in pose II, the distance separating the COM of the polysaccharide chain and the catalytic triad both reach 17 Å within 100 ns, which reflects physical separation of the polysaccharide chains from the active-site cleft. Furthermore, Fig. 6d indicates that the interaction energy of the polysaccharide chain with the enzyme decreases gradually with time. As shown in Fig. 6e, we calculated the interaction energies of the polysaccharide chain with the twelve adjacent residues, and found that residue E425 is undoubtedly the most important residue, and dominates step v.
In the constructed CI complex, as illustrated in Fig. 7a, we found that the position and orientation of E425 are conducive to attract the acceptor polysaccharide chain and further stabilize it in the catalytic position (named ‘A-S’ actions here). To investigate the role of residue E425 more in depth, we explore the process of the polysaccharide chain approaching the catalytic position through one-dimensional free-energy calculations (see the defined RC in Fig. 7b). The free-energy profile in Fig. 7c suggests that the transfer process of the acceptor polysaccharide chain to the catalytic position overcomes 3.4 kcal/mol free-energy barrier. Finally, the polysaccharide chain has been stabilized in the catalytic position by residue E425. To further understand the function of this residue, we performed saturation mutagenesis on it. The CD spectroscopy results in Supplementary Fig. 12f–h indicate that the three randomly selected variants are properly folded, whereas the experimentally measured activity decreased in all cases (see Supplementary Fig. 17). The least damaging replacement out of the nineteen variants is by aspartate, which possesses similar properties as the original glutamate, but only retained 27.2% relative activity. The sequence-alignment results in Supplementary Fig. 15 indicate that all the twelve BEs possess the same E at that site, suggesting that E425 in RpBE is the optimal solution by natural evolution.

a Diagrammatic sketch of the constructed CI, wherein residue E425 interacts with the polysaccharide chain (cyan) by hydrogen bonds. b Defined RC, i.e., d3, denotes the distance of the COM between residue E425 and the glucosyl ring. c Free-energy profile characterizing the polysaccharide chain in pose I transferring to the catalytic position. In (c), the long black arrow denotes the direction of the acceptor polysaccharide chain approaching the catalytic position. The lowering in energy at the position pinpointed by the small black arrow indicates that the polysaccharide chain has been stabilized by residue E425. Source data for c are provided as a Source Data file.
SBTs mechanism-based enzyme engineering
In RpBE, residues P268, D269, D305, and E425, as well as the short loop 2 enable the enzyme to execute efficiently the crucial ‘G-R/C-S’ and ‘A-S’ actions and mediate substrate transfer, which accordingly could point to further engineering for the BE family. In view of the proposed mechanism, we turned our attention to step i, the initial donor-substrate binding for glycosylation. The Rosetta software (https://www.rosettacommons.org)40 was employed to improve enzyme-substrate association (see Supplementary Table 9). The experimental results for all the forty-seven variants are reported in Supplementary Table 10, wherein the top ten are shown in Fig. 8a. As depicted in Fig. 8b, seven mutation sites, i.e., A243, K436, G431, G429, P420, L421, and H385, lie around the CceBE1 G7-pose i, which includes four of the five top variants, i.e., A243T (1.7-fold increase), K436R (1.5-fold increase), G431N (1.5-fold increase), and G429M (1.4-fold increase). Our enzymatic kinetic experiments indicate that in the top two variants (A243T and K436R), the Km values decrease by 44.2% and 46.5%, respectively, suggesting enhanced enzyme-substrate affinity (see Supplementary Table 11). We, therefore, conclude that enhancing the enzyme-substrate affinity around the CceBE1 G7-pose i is a potent strategy to improve enzyme activity.

a Top ten variants amid the forty-seven ones. The WT RpBE is regarded as possessing 100% enzyme activity. b Mutation sites (shown in sphere) around the CceBE1 G7-pose i. The catalytic triad is shown in a licorice representation, and colored in yellow (carbon atoms). c Distribution of the glycan chain length. The data without enzyme catalysis are not shown for clarity. The two-tailed p values were obtained by the unpaired t test analysis using Graphpad Prism 8. No adjustment was made for multiple comparisons. The bars and the error bars are the average and standard deviation of triplicates (n = 3 independent experiments), respectively. Source data for a, c are provided as a Source Data file.
We further measured DP of products for the top two variants and found that the ratios of DP 4–9 have been improved markedly (see Fig. 8c). The largest significant differences are found in DP 5 (27.3% increase with A243T, ***p < 0.001) and DP 8 (21.4% increase with K436R, ***p < 0.001). Intriguingly, the increased ratios in DP 4 and DP 5 with A243T catalysis are higher than those with K436R. The two variants have a similar increased ratio in DP 6. However, in DP 7–9, the increased ratios with K436R catalysis are higher than those by A243T. The present results, therefore, imply that we can engineer the residues around A243 to produce high-level DP 4–5 branching chains, while design the residues around K436 to acquire products with abundant DP 7–9, which could help product customization with specific DP.
Discussion
The feature of controlling the branching point and length in an α-glucan chain is the foundation for industrial applications for BEs, playing an irreplaceable role in the food industry1. Unveiling the key functional mechanism of BEs and further identifying directions for enzyme engineering are desirable for promoting their industrial use, which is admittedly still in infancy. Many initiatives, for instance, those of Hayashi et al17., Ban et al19,41., and Fawaz et al25. have explored possible enzyme-substrate initial association to guide BE engineering, while a systematic understanding of the underlying molecular mechanism is still lacking. Here, we characterized the RpBE and deciphered its SBTs for the entire process of product generation by means of computational methods, contributing a substantial knowledge advancement in noncovalent enzyme-substrate interactions, which are crucial for activity. Mechanism-guided mutagenesis produced variants of augmented enzymatic activity, thereby reinforcing previous findings that identification of the optimal initial enzyme-substrate association and further modulation of the enzyme-substrate affinity are reliable strategies to improve enzyme performance. In addition, we found that the specific DP ranges, such as the high-abundance DP 4–5 or 7–9, can be modulated, which paves the way for producing specific α-glucans. In general, the present work, resting upon a synergistic combination of theory and experiment, is envisioned to help drive the use of BEs as the decisive enzymes to promote α-glucan utilization in the food and health industries.
Methods
Enzyme production
The gene sequences of twelve BEs (Supplementary Data 1) were synthesized by GENEWIZ (Suzhou, China) and cloned into the pET28a expression vector (Thermo Fisher Scientific, Waltham). The recombinant proteins were expressed in E. coli BL21 (DE3). The variants of RpBE were generated by Rosetta Cartersian_ddg following the software protocols and created experimentally by site-specific mutagenesis via the primers listed in Supplementary Table 12. The inducible expression of DE3 and purification of the His-tag labeled enzymes were carried out following the manufacturer’s protocols26. Enzyme concentration of the purified enzymes was measured by Quick Start™ Bradford Protein Assay kit (Bio-Rad Laboratories, Veenendaal, Netherlands) and purity verified by SDS-PAGE.
Activity assessment
The enzymatic activity was detected via the iodine-staining protocol with appropriate modifications26,42,43. Potato amylose type III (Sigma-Aldrich) was dissolved in pure DMSO at a concentration of 100 g/L and used as the mother liquid. 1.25 mg/mL amylose and the enzyme were mixed in sodium phosphate buffer (pH 7.0, 50 mM) and incubated at relevant temperatures for 20 min. The reaction was terminated by boiling for 15 min; the mixture was cooled to room temperature subsequently. Then 10 μL reaction solution was mixed with 150 μL iodine solution in a 96-well plate (aqueous solution of 0.0127% I2 (w/v) and 0.035% KI (w/v)) and kept at room temperature for 10 min to allow the color development. The absorbance at 660 nm was measured for the mixed solution. Furthermore, to maintain the absorbance within a reasonable range of 0.2–0.8, enzymatic dosages in the reaction system were adjusted appropriately. One unit of enzyme activity was defined as the amount of enzyme required to reduce the absorbance value by 1% per minute.
Branching degree
The branching density of BEs was measured by 1H-NMR spectroscopy. After dissolving the samples in 0.6 mL of D2O, the NMR spectra were recorded through a Varian Inova 600 spectrometer at 313 K. Then the acquired data was processed by MestReNova software and underwent Whittaker Smoother baseline correction and zero filling up to 32 k complex points. The branching density was calculated by the division of the peak area of α-1,6-linkage signal at 4.98 ppm on the combined peak areas of the signals at 4.98 and 5.39 ppm.
DP analysis
Lyophilized samples of amylopectin (10 mg) were dispersed in 2.0 mL sodium acetate buffer (50 mM, pH 4.5) and then treated with 1 U/mL pullulanase and 2 U/mL isoamylase at 40 °C for 16 h. The reaction was halted by boiling for 15 min. Then the samples were centrifuged at 20,000 × g for 10 min. The supernatants were further filtered through a 0.22 μm membrane to obtain the debranched amylopectin, i.e., linear dextrin or short-chain amylose. Then the debranched amylopectin was catalyzed by BEs to add α-1,6-glucoside-linked branching chains. The branching products were further debranched. Dionex ICS-6000 system (Thermo Scientific, USA), equipped with a pulsed amperometric detector and a Dionex CarboPac™ PA1 column (250 × 2 mm), was employed to analyze the oligosaccharides through the high-performance anion exchange chromatography. The elution of oligosaccharides was achieved by performing a NaOH gradient in sodium acetate buffer. To determine the elution time of each component qualitatively, malto-oligosaccharides with a DP 1–7 were used as reference standards.
Crystal structure determination
Crystallization experiments were conducted at 25 °C using the sitting-drop vapor-diffusion method. 10 mg/mL protein was mixed with reservoir solution in a 1:1 ratio. The crystals were obtained using the “1–28” condition of the Morpheus II screen kit (Molecular Dimensions; 0.1 M buffer System 4, 31% Precipitant Mix 8, and 4 mM Alkalis). Crystals were tested and X-ray diffraction data collected at beamlines BL10U2, BL17B, BL18U1, and BL19U1 of the National Facility for Protein Science in Shanghai (NFPS). The crystals were mounted in a cryoloop and soaked with 20% glycerol before data collection at 100 K. The diffraction images were processed using HKL200044. Crystals were solved by the molecular replacement (MR) method with the Phaser program45 from the Phenix46 suite using the AlphaFold modeling of RpBE. Further refinement was carried out using programs of Phenix Refine47 and Coot48. Before structural refinements, 5% of randomly selected reflections were set aside for calculating Rfree49 as a monitor. The data collection, processing, and refinement statistics can be found in Supplementary Table 3.
Kinetic Assay
Potato amylose type III (Sigma-Aldrich) was used as a substrate to perform the kinetic Assay. The enzyme activity was measured at the substrate concentrations from 0.1 to 5 mg/mL, following the protocol described above. GraphPad Prism 8 (GraphPad Software Inc., San Diego, CA, USA) was employed to analyze the results and obtain the enzyme kinetic constants.
ITC test
ITC experiments were conducted on a MicroCal PEAQ-ITC system (Malvern Panalytical Ltd) at 25 °C. To determine the binding affinity between G7 and RpBE, 2 μl 8 mM G7 was titrated into 45 μM enzyme solution. As the control group, 2 ul 8 mM G7 solution was titrated to the 10 mM Phosphate Buffer (pH 7.4). The results were analyzed using the ITC evaluation software provided by the manufacturer.
CD spectroscopy experiment
CD spectropolarimetry (Applied Photophysics, Britain) was employed to detect whether the proteins are properly folded. The WT enzyme and the variants are dissolved in 10 mM Phosphate Buffer (pH 7.4), and the final concentration for each sample is 0.2 mg/mL. Three scans from 180 to 260 nm at 1 nm intervals were accumulated and averaged. The background of the Phosphate Buffer was subtracted from the CD data.
Enzyme-substrate docking
All the enzyme-substrate associations were investigated using the HADDOCK Web Server (https://bianca.science.uu.nl/haddock2.4/). The docking procedures were carried out following the HADDOCK protocols.
MD simulations
The computational assays built for the investigation of the substrate movement after initial binding were simulated following protocols published previously50,51,52,53. The molecular assemblies were solvated in an equilibrated box of water and neutralized by adding Na+ ions to the solution. During glycosylation, E356 is protonated. Each enzyme-substrate complex was pretreated by 5000 steps of energy minimization and 100 ps MD simulation, gradually relaxing harmonic restraints: (i) restraint of enzyme and substrate; and (ii) restraint of substrate. Then, a 2.2-μs MD simulation was performed, devoid of geometric restraints, for the five assemblies. The particulars for each molecular assembly are provided in Supplementary Table 13. NAMD 2.1454 was employed to perform the MD simulations with the CHARMM36 force field55,56,57 and the TIP3P water model58. All the simulations were carried out at 300 K and 1 atm. The temperature and the pressure were controlled by Langevin dynamics and the Langevin piston method, respectively59,60. The Shake/Rattle and Settle algorithms were utilized to constrain the length of the covalent bonds involving hydrogen atoms to their equilibrium value61,62,63. The interaction energy is defined as the sum of the electrostatic interaction energy and the van der Waals interaction energy. Visualization and analysis of the trajectories were achieved with the VMD 1.9.4 program64 and PyMOL molecular graphics system (version 2.4 Schrödinger, LLC).
Free-energy calculations
The computational assays built for the investigation of the substrate transferring to the catalytic position during the whole process of enzyme interacting substrate and the process of D269 approaching D305 were simulated following the protocols described above. The free-energy profiles underlying these processes were determined using the well-tempered meta-eABF (WTM-eABF) algorithm65. A 2-μs free-energy calculation was performed for the five assemblies. The details for each molecular assembly are provided in Supplementary Table 14.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw data generated in this study, the interaction-energy scripts, and simulation files have been deposited in the Figshare database under https://doi.org/10.6084/m9.figshare.26500141. Crystallographic data for the WT RpBE has been deposited at the Protein Data Bank with accession code 8ZQA. Source Data are provided with this paper as source data file. Source data are provided with this paper.
References
Ban, X. et al. Bacterial 1,4-alpha-glucan branching enzymes: characteristics, preparation and commercial applications. Crit. Rev. Biotechnol. 40, 380–396 (2020).
Chakraborty, R., Kalita, P. & Sen, S. Natural starch in biomedical and food industry: perception and overview. Curr. Drug. Discov. Technol. 16, 355–367 (2019).
Punia, S. Barley starch modifications: physical, chemical and enzymatic - A review. Int. J. Biol. Macromol. 144, 578–585 (2020).
Li, X. et al. Partial branching enzyme treatment increases the low glycaemic property and α-1,6 branching ratio of maize starch. Food Chem. 164, 502–509 (2014).
Tetlow, I. J. & Emes, M. J. A review of starch-branching enzymes and their role in amylopectin biosynthesis. IUBMB Life 66, 546–558 (2014).
Bilyard, M. K. et al. Palladium-mediated enzyme activation suggests multiphase initiation of glycogenesis. Nature 563, 235–240 (2018).
Huynh, N., Ou, Q., Cox, P., Lill, R. & King-Jones, K. Glycogen branching enzyme controls cellular iron homeostasis via Iron Regulatory Protein 1 and mitoNEET. Nat. Commun. 10, 5463 (2019).
Bürgy, L. et al. Coalescence and directed anisotropic growth of starch granule initials in subdomains of Arabidopsis thaliana chloroplasts. Nat. Commun. 12, 6944 (2021).
Baecker, P. A., Greenberg, E. & Preiss, J. Biosynthesis of bacterial glycogen. primary structure of Escherichia coli 1,4-alpha-D-glucan:1,4-alpha-D-glucan 6-alpha-D-(1, 4-alpha-D-glucano)-transferase as deduced from the nucleotide sequence of the glg B gene. J. Biol. Chem. 261, 8738–8743 (1986).
Svensson, B. Regional distant sequence homology between amylases, alpha-glucosidases and transglucanosylases. FEBS Lett. 230, 72–76 (1988).
Palomo, M. et al. Thermus thermophilus glycoside hydrolase family 57 branching enzyme: crystal structure, mechanism of action, and products formed. J. Biol. Chem. 286, 3520–3530 (2011).
Bax, H. H. M., van der Maarel, M. & Jurak, E. Alpha-1,4-transglycosylation activity of GH57 glycogen branching enzymes is higher in the absence of a flexible loop with a conserved tyrosine residue. Polym. (Basel) 15, 2777 (2023).
Xiang, G., Leemhuis, H. & van der Maarel, M. Structural elements determining the transglycosylating activity of glycoside hydrolase family 57 glycogen branching enzymes. Proteins 90, 155–163 (2022).
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M. & Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42, D490–D495 (2014).
Ye, X. et al. Glycogen branching enzyme with a novel chain transfer mode derived from corallococcus sp. strain EGB and its potential applications. J. Agric. Food Chem. 70, 4735–4748 (2022).
Janeček, Š. & Svensson, B. Amylolytic glycoside hydrolases. Cell. Mol. Life Sci. 73, 2601–2602 (2016).
Hayashi, M. et al. Bound substrate in the structure of cyanobacterial branching enzyme supports a new mechanistic model. J. Biol. Chem. 292, 5465–5475 (2017).
Kelly, R. M., Leemhuis, H. & Dijkhuizen, L. Conversion of a cyclodextrin glucanotransferase into an alpha-amylase: assessment of directed evolution strategies. Biochemistry 46, 11216–11222 (2007).
Ban, X. et al. Alternations in the chain length distribution of polysaccharides by adjusting the active sites of the 1,4-α-glucan branching enzyme. Food Res. Int. 162, 112119 (2022).
Feng, L. et al. Crystal structures of Escherichia coli branching enzyme in complex with linear oligosaccharides. Biochemistry 54, 6207–6218 (2015).
Feng, L. et al. Crystal structures of Escherichia coli branching enzyme in complex with cyclodextrins. Acta Crystallogr. D. Struct. Biol. 72, 641–647 (2016).
Chaen, K., Noguchi, J., Omori, T., Kakuta, Y. & Kimura, M. Crystal structure of the rice branching enzyme I (BEI) in complex with maltopentaose. Biochem. Biophys. Res. Commun. 424, 508–511 (2012).
Froese, D. S. et al. Structural basis of glycogen branching enzyme deficiency and pharmacologic rescue by rational peptide design. Hum. Mol. Genet. 24, 5667–5676 (2015).
Gavgani, H. N. et al. A structural explanation for the mechanism and specificity of plant branching enzymes I and IIb. J. Biol. Chem. 298, 101395 (2022).
Fawaz, R. et al. The structure of maltooctaose-bound escherichia coli branching enzyme suggests a mechanism for donor chain specificity. Molecules 28, 4377 (2023).
Zhang, X., Leemhuis, H. & van der Maarel, M. Synthesis of highly branched alpha-glucans with different structures using GH13 and GH57 glycogen branching enzymes. Carbohydr. Polym. 216, 231–237 (2019).
Li, D. et al. A cold-active 1,4-alpha-glucan branching enzyme from Bifidobacterium longum reduces the retrogradation and enhances the slow digestibility of wheat starch. Food Chem. 324, 126855 (2020).
Sorndech, W. et al. Synergistic amylomaltase and branching enzyme catalysis to suppress cassava starch digestibility. Carbohydr. Polym. 132, 409–418 (2015).
Huang, J. et al. Discovery of deaminase functions by structure-based protein clustering. Cell 186, 3182–3195 (2023).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 50, D439–D444 (2022).
Laurents, D. V. AlphaFold 2 and NMR spectroscopy: partners to understand protein structure, dynamics and function. Front. Mol. Biosci. 9, 906437 (2022).
Yang, J., Wang, Y. & Zhang, Y. ResQ: an approach to unified estimation of B-factor and residue-specific error in protein structure prediction. J. Mol. Biol. 428, 693–701 (2016).
Honorato, R. V. et al. Structural biology in the clouds: the WeNMR-EOSC Ecosystem. Front Mol. Biosci. 8, 729513 (2021).
Honorato, R. V. et al. The HADDOCK2.4 web server for integrative modeling of biomolecular complexes. Nat Protoc (2024).
Boittier, E. D., Burns, J. M., Gandhi, N. S. & Ferro, V. GlycoTorch Vina: docking designed and tested for glycosaminoglycans. J. Chem. Inf. Model 60, 6328–6343 (2020).
Nivedha, A. K., Thieker, D. F., Makeneni, S., Hu, H. & Woods, R. J. Vina-Carb: Improving glycosidic angles during carbohydrate docking. J. Chem. Theory Comput. 12, 892–901 (2016).
Rodrigues, J. P. et al. Clustering biomolecular complexes by residue contacts similarity. Proteins 80, 1810–7 (2012).
Wang, Z. et al. Expression and characterization of an extremely thermophilic 1,4-α-glucan branching enzyme from Rhodothermus obamensis STB05. Protein Expr. Purif. 164, 105478 (2019).
Löffler, P., Schmitz, S., Hupfeld, E., Sterner, R. & Merkl, R. Rosetta:MSF: a modular framework for multi-state computational protein design. PLoS Comput. Biol. 13, e1005600 (2017).
Ban, X. et al. The amino acid on the top of the active groove allosterically modulates product specificity of the 1,4-α-glucan branching enzyme. Food Chem. 384, 132458 (2022).
Jiang, H. et al. Flexible loop in carbohydrate-binding module 48 allosterically modulates substrate binding of the 1,4-alpha-glucan branching enzyme. J. Agric. Food Chem. 69, 5755–5763 (2021).
Takata, H. et al. Properties and active center of the thermostable branching enzyme from Bacillus stearothermophilus. Appl. Environ. Microbiol. 60, 3096–3104 (1994).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D. Struct. Biol. 75, 861–877 (2019).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D. Biol. Crystallogr. 68, 352–367 (2012).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 (2004).
Brünger, A. T. et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D. Biol. Crystallogr. 54, 905–921 (1998).
Zong, Z. et al. Mechanism and biomass association of glucuronoyl esterase: an α/β hydrolase with potential in biomass conversion. Nat. Commun. 13, 1449 (2022).
Knott, B. C., Crowley, M. F., Himmel, M. E., Stahlberg, J. & Beckham, G. T. Carbohydrate-protein interactions that drive processive polysaccharide translocation in enzymes revealed from a computational study of cellobiohydrolase processivity. J. Am. Chem. Soc. 136, 8810–8819 (2014).
Zong, Z. et al. Lysine mutation of the claw-arm-like loop accelerates catalysis by cellobiohydrolases. J. Am. Chem. Soc. 141, 14451–14459 (2019).
Zong, Z., Liu, X., Ye, Z. & Liu, D. A double-switch pHLIP system enables selective enrichment of circulating tumor microenvironment-derived extracellular vesicles. Proc. Natl Acad. Sci. 120, e2214912120 (2023).
Phillips, J. C. et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 (2005).
Vanommeslaeghe, K. et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690 (2010).
MacKerell, A. D. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998).
MacKerell, A. D. Jr., Feig, M. & Brooks, C. L. 3rd Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 126, 698–699 (2004).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Uhlenbeck, G. E. & Ornstein, L. S. On the theory of the Brownian motion. Phys. Rev. 36, 823–841 (1930).
Feller, S. E., Zhang, Y., Pastor, R. W. & Brooks, B. R. Constant pressure molecular dynamics simulation: The langevin piston method. J. Chem. Phys. 103, 4613–4621 (1995).
Miyamoto, S. & Kollman, P. A. Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 (1992).
Ryckaert, J.-P., Ciccotti, G. & Berendsen, H. J. C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Andersen, H. C. Rattle: a “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 52, 24–34 (1983).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–8 (1996).
Fu, H., Shao, X., Cai, W. & Chipot, C. Taming rugged free energy landscapes using an average force. Acc. Chem. Res. 52, 3254–3264 (2019).
Marcos-Alcalde, I., Setoain, J., Mendieta-Moreno, J. I., Mendieta, J. & Gómez-Puertas, P. MEPSA: minimum energy pathway analysis for energy landscapes. Bioinformatics 31, 3853–5 (2015).
Acknowledgements
The authors thank Prof. Zhaofeng Li from Jiangnan University for the guidance on assay methods and the useful discussion. This study was supported by Strategic Priority Research Program of the Chinese Academy of Sciences (XDC0120303; Y.S), National Key R&D Program of China (2023YFC3403600; Z.Z), National Natural Science Foundation of China (22005157; Z.Z), and Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (TSBICIP-CXRC-077; Z.Z). Furthermore, the FP7 WeNMR (project# 261572), H2020 West-Life (project# 675858), the EOSC-hub (project# 777536) and the EGI-ACE (project# 101017567) European e-Infrastructure projects are acknowledged for the use of their web portals, which make use of the EGI infrastructure with the dedicated support of CESNET-MCC, INFN-LNL-2, NCG-INGRID-PT, TW-NCHC, CESGA, IFCA-LCG2, UA-BITP, TR-FC1-ULAKBIM, CSTCLOUD-EGI, IN2P3-CPPM, CIRMMP, SURFsara and NIKHEF, and the additional support of the national GRID Initiatives of Belgium, France, Italy, Germany, the Netherlands, Poland, Portugal, Spain, UK, Taiwan and the US Open Science Grid.
Author information
Authors and Affiliations
Contributions
Y.S., Z.Z., and X.Z. designed the research; Z.Z. carried out the calculations.; X.Z., Z.F., P.C., Y.Z., and Q.W. characterized the enzymes; Z.Z. drafted the manuscript. Y.S., L.L., and C.C. edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zong, Z., Zhang, X., Chen, P. et al. Elucidation of the noncovalent interactions driving enzyme activity guides branching enzyme engineering for α-glucan modification. Nat Commun 15, 8760 (2024). https://doi.org/10.1038/s41467-024-53018-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53018-6
This article is cited by
-
Direct generation of nitrogen-centered radicals via non-covalent interaction between Cu complexes and BiVO4 photoanodes
Nature Communications (2025)
-
Computational design of allulose-responsive biosensor toolbox for auto-inducible protein expression and CRISPRi mediated dynamic metabolic regulation
Nature Communications (2025)
