
Abstract
SARS-CoV-2 JN.1 with an additional L455S mutation on spike when compared with its parental variant BA.2.86 has outcompeted all earlier variants to become the dominant circulating variant. Recent studies investigated the immune resistance of SARS-CoV-2 JN.1 but additional factors are speculated to contribute to its global dominance, which remain elusive until today. Here, we find that SARS-CoV-2 JN.1 has a higher infectivity than BA.2.86 in differentiated primary human nasal epithelial cells (hNECs). Mechanistically, we demonstrate that the gained infectivity of SARS-CoV-2 JN.1 over BA.2.86 associates with increased entry efficiency conferred by L455S and better spike cleavage in hNECs. Structurally, S455 altered the mode of binding of JN.1 spike protein to ACE2 when compared to BA.2.86 spike at ACE2H34, and modified the internal structure of JN.1 spike protein by increasing the number of hydrogen bonds with neighboring residues. These findings indicate that a single mutation (L455S) enhances virus entry in hNECs and increases immune evasiveness, which contribute to the robust transmissibility of SARS-CoV-2 JN.1. We further evaluate the in vitro and in vivo virological characteristics between SARS-CoV-2 BA.2.86/JN.1 and EG.5.1/HK.3, and identify key lineage-specific features of the two Omicron sublineages that contribute to our understanding on Omicron antigenicity, transmissibility, and pathogenicity.
Similar content being viewed by others


Introduction
SARS-CoV-2 Omicron emerged in late 2021 and is characterized by robust immune evasion, attenuated pathogenicity, and markedly increased transmissibility1,2,3,4,5,6,7,8,9,10,11. The virus continues to evolve as it becomes endemic among the human population. In mid-2023, the dominant circulating SARS-CoV-2 variants were descendants of the recombinant XBB lineage, including XBB.1.5, XBB.1.16, and EG.5.1. Thereafter, BA.2.86, a descendant of BA.2, was detected in July 2023 and received international attention since BA.2.86 spike contains more than 30 changes when compared with that of BA.2. Recent studies demonstrated that BA.2.86 is strongly immune evasive12,13,14,15,16, efficient at infecting the bronchial adenocarcinoma Calu3 cells12,17,18, but remain attenuated in pathogenicity when compared to BA.219.
Shortly after, a descendent lineage of BA.2.86, JN.1, was first detected on August 25th, 2023. Although JN.1 has only one additional change (L455S) in spike protein when compared with BA.2.86, it disseminates quickly and has replaced BA.2.86 and other XBB descendants to become the dominant variant worldwide. Due to its rapidly increasing spread, WHO has classified JN.1 as a separate variant of interest (VOI) from its parent lineage BA.2.86. Recent studies have characterized the immune evasion potential of JN.120,21,22,23. However, other features of JN.1, such as its intrinsic pathogenicity, protease usage, cell entry, spike cleavage, and infectivity in human nasal epithelial cells, have not been thoroughly investigated.
In this work, we comprehensively investigated the in vivo and in vitro virological features of JN.1 and compared the results with that of BA.2, BA.2.86, XBB.1, EG.5.1, and HK.3. Our study identified a gained cell entry advantage in addition to the increased immune evasiveness conferred by the L455S mutation on JN.1 spike. In addition, our findings demonstrated distinct lineage-specific features between the BA.2 descendent variants BA.2.86/JN.1 and the XBB descendent variants EG.5.1/HK.3.
Results
Evolution of SARS-CoV-2 JN.1
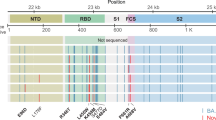
Among the circulating variants in mid-late 2023, XBB.1, EG.5.1, and HK.3 are descendants of the XBB lineage that emerge from the recombination of the BJ.1 and BM.1.1.1 (Fig. 1a–c). BA.2.86 and JN.1 are descendants of BA.2 that are phylogenetically distinct from the XBB lineage (Fig. 1a–c). The spike protein of EG.5.1 differs from that of XBB.1 at N-terminal domain (NTD) Q52H and receptor-binding domain (RBD) F456L, while HK.3 spike RBD contains an additional L455F mutation when compared to EG.5.1. BA.2.86 spike differs from BA.2 spike at over 30 amino acid positions. When compared to BA.2.86 spike, JN.1 contains one additional mutation at L455S in spike RBD (Fig. 1b, c), as well as an R252K mutation in nsp6 and an F19L mutation in ORF7b (Supplementary Fig. 1). In late 2023, the circulating SARS-CoV-2 variants in Africa were dominated by BA.2.86 and XBB.1, while Asia and Oceania were dominated by HK.3 and EG.5.1. At the same time, Europe, North America, and South America were dominated by EG.5.1 and XBB.1 (Fig. 1d). Starting from December 2023, JN.1 has grown rapidly and has become the most prevalent SARS-CoV-2 variant in all six regions until today (Fig. 1d).

a A representative phylogenetic tree of SARS-CoV-2 from September 1, 2023, to March 1, 2024. Pre-Omicron variants are labeled as other variants. b Step-wise accumulation of key spike mutations in Omicron XBB.1, EG.5.1, HK.3, BA.2.86, and JN.1 compared to BA.2. Key mutations are color barcoded, and represented as colored bars when present or white bars if absent. Different amino acids are shown in colored bars. The number of key mutations of each variant is summarized at the top. Mutation events are labeled on arrows. R493Q is a reversion mutation. c Summary of the spike mutations of SARS-CoV-2 lineages BA.2*, BA.2.86*, JN.1*, XBB.1*, EG.5.1*, and HK.3* compared to the ancestral SARS-CoV-2 spike protein. Color represents the proportion of each mutation in each variant. d Variant dynamics of Omicron lineages BA.2*, BA.2.86*, JN.1*, XBB.1*, EG.5.1*, and HK.3* worldwide and in different regions in terms of variant frequency. The genome data from September 1, 2023, to March 1, 2024 were analyzed. Note that BA.2* excludes BA.2.86*, XBB.1* does not include EG.5.1*, EG.5.1* does not include HK.3*, and BA.2.86* does not include JN.1* for sequence and mutation analysis.
Immune evasion of SARS-CoV-2 JN.1
To assess the immune evasion characteristics of BA.2.86/JN.1 and EG.5.1/HK.3 together with their ancestral variants BA.2 and XBB.1, we performed pseudovirus neutralization assays on these variants against a panel of monoclonal antibodies (mAbs). Omicron subvariants are known to exhibit marked escape of mAbs1,2,8,10. Consistent with recent studies, clinically authorized mAb LY-CoV140424 was found to be inactive against the pseudovirus of all tested Omicron variants, with the exception of the parental variant BA.2 (Fig. 2a). Furthermore, the mAb S3H325, targeting the SD1 domain, demonstrated efficacy against BA.2, XBB.1, EG5.1, and HK.3 pseudoviruses, while its neutralization activity against BA.2.86 and JN.1 pseudoviruses was limited (Fig. 2a). Notably, while we found that mAb S30926 remained effective against all evaluated variants, it is more effective against XBB descendent variants compared to BA.2.86 and JN.1, which was evidenced by their IC50 values of 0.104 μg/ml, 0.073 μg/ml, 0.126 μg/ml, 2.440 μg/ml and 1.993 μg/ml against XBB.1, EG.5.1, HK.3, BA.2.86 and JN.1 pseudoviruses, respectively (Fig. 2a). Interestingly, SA5527 remained effective against all tested Omicron variants including BA.2.86 and JN.1 pseudoviruses (Fig. 2a).

a Neutralization curves (left) and heatmap showing the IC50 values (right) of mAbs with Omicron-spike pseudoviruses (n = 3). The n number represents biological repeats. b Neutralization of the indicated Omicron-spike pseudoviruses by sera collected from individuals between days 14 and 28 after XBB reinfection following prior infection with BA.5 virus after receiving two to three doses of inactivated vaccine (n = 16). The horizontal dashed line indicates the detection limit (40-fold). c Antigenic map based on all the breakthrough infection serum neutralization data from Fig. 2b. Virus positions are represented by closed circles, whereas serum positions are shown as open squares. d Neutralization of the indicated Omicron-spike pseudoviruses by sera collected from individuals between days 14 and 28 after JN.1 reinfection following prior infection with BA.5 virus after receiving three doses of inactivated vaccine (n = 16). The horizontal dashed line indicates the detection limit (20-fold). e Antigenic map based on all the breakthrough infection serum neutralization data from Fig. 2d. Virus positions are represented by closed circles, whereas serum positions are shown as open squares. f Sera from infected hamsters were used for neutralization of the indicated Omicron-spike pseudoviruses (n = 6 for BA.2 and JN.1, n = 5 for BA.2.86, XBB.1, EG.5.1, and HK.3). The horizontal dashed line indicates the detection limit (10-fold). g Antigenic map based on the hamster serum neutralization data from Fig. 2f. Virus positions are represented by closed circles, whereas serum positions are shown as open squares. h Principal components analysis (PCA) map based on the hamster serum neutralization data from Fig. 2f. i Sera was obtained from mice immunized with the spike trimer of BA.2, BA.2.86, JN.1, and XBB.1 (n = 8). The horizontal dashed line indicates the detection limit (20-fold). Statistical significance in (b) was determined with the two-tailed Wilcoxon matched-pairs signed rank test. Statistical significance in (f) and (i) was determined with the Kruskal-Wallis test with post hoc Dunn’s multiple comparisons tests. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. NS is not statistically significant. Source data are provided as a Source Data file.
We next evaluated the humoral immune evasion of these Omicron variants with plasma from individuals who received inactivated vaccines before experiencing an XBB or JN.1 breakthrough infection. We recruited two cohorts one group (n = 16) with post-vaccination BA.5 breakthrough infection followed by JN.1 reinfection and another group (n = 16) that was reinfected with XBB after BA.5 breakthrough infections (Supplementary Table 1). In keeping with recent studies21,23,28, sera from XBB reinfection displayed a shifted neutralization pattern, with the highest geometric mean titer (GMT) of 1454 against BA.2 pseudoviruses (Fig. 2b and Supplementary Fig. 2a). These sera were remarkably decreased in titers against XBB descendent (XBB.1, EG.5.1, HK.3) pseudoviruses, as well as BA.2.86 and JN.1 pseudoviruses (Fig. 2b). JN.1 pseudovirus displayed significantly enhanced immune escape compared to BA.2.86 pseudoviruses, as evidenced by a 1.6-fold decrease in GMT among individuals who were reinfected with XBB post-BA.5 infection (Fig. 2b). Similarly, HK.3 pseudoviruses demonstrated significantly enhanced immune evasion when compared with EG.5.1 pseudoviruses, as evidenced by a 1.4-fold decrease in GMT within this cohort (Fig. 2b). The serum neutralization data from this cohort was further utilized to construct an antigenic map. The emerging subvariants JN.1 and BA.2.86 clustered together, whereas HK.3 and EG.5.1 constituted another group (Fig. 2c). These subvariants notably diverged from their ancestral strains, demonstrating not only their antigenic similarity but also their greater antibody resistance compared with their predecessors. In the second cohort, we found that individuals who experienced breakthrough infections with JN.1 exhibited high levels of neutralizing titers against BA.2, BA.2.86, and JN.1 pseudoviruses, with GMTs exceeding 2500. However, neutralizing titers against XBB.1, EG.5.1, and HK.3 pseudoviruses were significantly less robustly triggered, suggesting that the XBB descendent variants are antigenically distanced from JN.1 (Fig. 2d and Supplementary Fig. 2b). In keeping with this observation, antigenic mapping revealed clustering of BA.2, JN.1, and BA.2.86, while HK.3 and EG.5.1 formed a separate cluster. Notably, XBB.1 was distant from both clusters, indicating significant evolutionary divergence within the XBB subvariants (Fig. 2e).
Antigenicity of SARS-CoV-2 JN.1
To further evaluate the antigenicity of these Omicron subvariants, we infected hamsters with SARS-CoV-2 BA.2, BA.2.86, JN.1, XBB.1, EG.5.1, and HK.3, and harvested sera from infected hamsters at 16 days post-infection (dpi). Our results indicated that both JN.1 and BA.2.86 pseudoviruses demonstrated pronounced resistance to sera obtained from hamsters infected with SARS-CoV-2 BA.2, XBB.1, EG.5.1, and HK.3 (Fig. 2f). Interestingly, while SARS-CoV-2 JN.1 and BA.2.86 infection primarily elicited autologous antibody responses, their ancestral strain BA.2 as well as the XBB sub-lineages induced a broader spectrum of protection (Fig. 2f and Supplementary Fig. 3). The antigenic distance map (Fig. 2g) and principal component analysis (PCA) map (Fig. 2h) based on hamster sera data, again but more clearly, grouped JN.1 and BA.2.86 together, with XBB.1, HK.3, and EG.5.1 formed another group, demonstrating that cross-reactivity among SARS-CoV-2 subvariants aligns with their evolutionary relationships.
We further assessed the humoral immune evasion of these SARS-CoV-2 variants using sera obtained from mice immunized with the indicated spike trimer. The neutralization assay using mice sera showed that JN.1 and BA.2.86 pseudoviruses were more significantly resistant to BA.2 and XBB.1 spike-immunized sera when compared to XBB.1, HK.3, and EG.5.1 pseudoviruses (Fig. 2i). In keeping with results obtained in infected hamsters, JN.1 and BA.2.86 spike immunization predominantly triggered an autologous antibody response (Fig. 2i and Supplementary Fig. 4). Together, these results indicate that JN.1 and BA.2.86 are antigenically distinct from the XBB sublineages.
Virological features of SARS-CoV-2 BA.2.86/JN.1 and EG.5.1/HK.3 in vitro
To investigate the virological features of SARS-CoV-2 BA.2.86/JN.1 in comparison to EG.5.1/HK.3, we first performed a split GFP-based cell-cell fusion assay to assess the fusogenicity of their spike proteins in 293T cells. Our results showed that BA.2.86 and JN.1 spike appeared to mediate cell-cell fusion less efficiently than the spike of XBB descendent variants XBB.1, EG.5.1, and HK.3 (Fig. 3a). Interestingly, by comparing BA.2.86 and JN.1 spike-mediated fusion, we found that the L455S mutation in JN.1 spike significantly increased fusogenicity by 3.4-folds (P = 0.0325). Similarly, by comparing EG.5.1 and HK.3 spike-mediated fusion, the L455F mutation in HK.3 spike significantly increased fusogenicity by 2.2-folds (P < 0.0001) (Fig. 3a). We evaluated the surface expression of the spike proteins on 293T cells with flow cytometry and found that the increased fusion mediated by L455S and L455F was not due to increased surface expression of the expressed spike proteins (Supplementary Fig. 5). In addition, we investigated cell-cell fusion between 293T and Calu3 cells and found that L455S and L455F similarly promoted fusion by 1.5-fold and 1.2-fold, respectively, albeit the differences did not reach statistical significance (Supplementary Fig. 6). In parallel, we evaluated pseudovirus entry using VSV-based pseudoviruses carrying the spike proteins of B.1, BA.2, BA.2.86, JN.1, XBB.1, EG.5.1, and HK.3. Our results demonstrated that the L455S mutation significantly promoted pseudovirus entry in VeroE6-TMPRSS2, VeroE6, Caco2, and 293T cells (Fig. 3b), which was evidenced by the increase in JN.1 pseudovirus entry over BA.2.86 pseudovirus entry in these cell types. Similarly, the L455F mutation in HK.3 spike significantly promoted pseudovirus entry of HK.3 over EG.5.1 in VeroE6 and Calu3 cells (Fig. 3b). We next infected VeroE6 cells with SARS-CoV-2 BA.2, BA.2.86, JN.1, XBB.1, EG.5.1, and HK.3, and quantified plaque formation at 96 and 120 hpi. We found that the BA.2 descendent variants developed smaller plaque sizes than the XBB descendent variants. At both 96 and 120 hpi, the plaque sizes of BA.2.86 and JN.1 were significantly smaller than those of EG.5.1 and HK.3 (Fig. 3c). In addition, we evaluated protease usage of pseudoviruses carrying the spike proteins of B.1, BA.2, BA.2.86, JN.1, XBB.1, EG.5.1, and HK.3. The evaluated cells were treated with the pan-serine protease inhibitor, Camostat, or the pan-cathepsin inhibitor, E64D, for 2 h before pseudovirus transduction. We found that in VeroE6-TMPRSS2 and Calu3 cells, where SARS-CoV-2 entry predominantly occurs at the plasma membrane29,30,31, the entry of BA.2.86 and JN.1 pseudoviruses were significantly more sensitive to Camostat treatment than that of EG.5.1, and HK.3 pseudoviruses (Fig. 3d and Supplementary Fig. 7). In parallel, in 293T cells, where SARS-CoV-2 primarily enter through the endosomal entry route30,32, the entry of EG.5.1, and HK.3 pseudoviruses were more sensitive to E64D treatment than that of BA.2.86 and JN.1 pseudoviruses (Fig. 3d and Supplementary Fig. 7). In keeping with these findings, in Caco2 cells that supports both entry pathways30,33, Camostat treatment was more effective against BA.2.86 and JN.1 pseudovirus entry, while E64D treatment was more effective against the entry of EG.5.1 and HK.3 pseudoviruses (Fig. 3d and Supplementary Fig. 7). Overall, these results demonstrate a lineage-specific preference of the route of virus entry, which indicate that the BA.2 descendent variants BA.2.86 and JN.1 are more dependent on TMPRSS2-mediated cell surface entry pathway than the XBB descendent variants EG.5.1 and HK.3. In addition, the L455S and L455F mutations both promote cell-cell fusion and pseudovirus entry of JN.1 and HK.3 when compared to their parental variants.

a SARS-CoV-2 spikes mediated cell-cell fusion in vitro. 293T cells (effector cells), which were co-transfected with the indicated spike and GFP1-10, were co-cultured with 293T cells co-transfected with hACE2 and GFP11 (target cells) (n = 10). The cells were fixed after 24 hours of incubation. GFP signal intensity was measured by ImageJ. The n number represents biological repeats. b Entry efficiency of pseudoviruses in cell lines. Pseudoviruses entry were quantified by measuring the luciferase signal at 24 h post-transduction (n = 6 for B.1 and n = 7 for other variants). Fold change in the luciferase signal was normalized to the mean luciferase readout of BA.2-spike pseudoviruses. The n number represents biological repeats. c Plaque size of Omicron BA.2, BA.2.86, JN.1, XBB.1, EG.5.1, and HK.3 in VeroE6 cells. VeroE6 cells were challenged with 60 PFU of Omicron BA.2, BA.2.86, JN.1, XBB.1, EG.5.1, and HK.3. The infected cells were fixed with 4% paraformaldehyde at 96 and 120 hpi and stained with crystal violet. Plaque size was measured by Adobe Photoshop (n = 10). The n number represents biological repeats. d Protease usage by pseudoviruses in cell lines. VeroE6-TMPRSS2, Calu3, Caco2, and 293T cells were pre-treated with 10 μM Camostat or E64D for 2 h, followed by transduction with B.1-, BA.2-, BA.2.86-, JN.1-, XBB.1-, EG.5.1-, and HK.3-spike pseudoviruses. At 24 h post-transduction, the level of pseudovirus entry was quantified by measuring the luciferase signal. The fold change was normalized to the mean luciferase readout of cells treated with DMSO for each variant (n = 6). The n number represents biological repeats. Data represent mean ± SEM. Statistical significance in (a–d) was determined with one-way ANOVA with post hoc Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. NS is not statistically significant. Source data are provided as a Source Data file.
Virological features of SARS-CoV-2 BA.2.86/JN.1 and EG.5.1/HK.3 in human nasal epithelial cells
Results from us and others revealed that the L455S mutation in the JN.1 spike and the L455F mutation in the HK.3 spike contributed to enhanced immune escape when compared to their parental variants BA.2.86 and EG.5.121,34. However, it remains to be determined whether the high transmissibility of SARS-CoV-2 JN.1 in humans is also associated with enhanced fitness in primary human nasal epithelial cells (hNECs). To this end, we first evaluated the replication of SARS-CoV-2 JN.1 in Calu3 and hNECs, with BA.2, BA.2.86, XBB.1, EG.5.1, and HK.3 included as comparison groups. In keeping with our findings on pseudovirus entry and protease dependency in Calu3 cells (Fig. 3b and Fig. 3d), the entry and replication of SARS-CoV-2 BA.2.86 and JN.1 in Calu3 cells were more efficient than those of EG.5.1 and HK.3 in early time points (2 and 24 hpi). However, SARS-CoV-2 EG.5.1 and HK.3 replicated efficiently in Calu3 cells. By 72 hpi, the viral replication intermediate, subgenomic envelope (sgE) gene copy of SARS-CoV-2 EG.5.1 and HK.3 has surpassed that of BA.2.86 and JN.1 (Fig. 4a). Interestingly, at the same time point post-infection, SARS-CoV-2 JN.1 replicated significantly more efficiently in hNECs than other evaluated Omicron variants, including BA.2 (4.7-folds, P < 0.0001), BA.2.86 (2.7-folds, P < 0.0001), XBB.1 (8.6-folds, P < 0.0001), EG.5.1 (6.0-folds, P < 0.0001), and HK.3 (3.0-folds, P < 0.0001) (Fig. 4b). To understand whether the difference at 72 hpi might be associated with discrepant levels of virus entry, we evaluated pseudovirus entry of JN.1 in hNECs and compared the result with that of BA.2.86, EG.5.1, and HK.3. In keeping with the replication results, we found that BA.2.86-pseudoviruses entered hNECs at a higher efficiency than that of EG.5.1- and HK.3-pseudoviruses, while JN.1-pseudoviruses entered hNECs more robustly than that of BA.2.86- (1.6-fold, P = 0.0082), EG.5.1- (3.9-folds, P < 0.0001), and HK.3-pseudoviruses (3.2-folds, P < 0.0001) (Fig. 4c). To pinpoint the role of L455S on pseudovirus entry in hNECs, we introduced L455S in the background of XBB.1, EG.5.1, and HK.3 spike protein. Our data demonstrated that the presence of L455S increased the entry of XBB.1, EG.5.1, and HK.3 pseudoviruses in hNECs by 7.7-, 1.3-, and 1.9-folds, respectively (Supplementary Fig. 8). In addition, to gain further insight on the increased hNEC entry by JN.1, we assessed protease usage and spike cleavage of JN.1 in hNECs. We found that while both SARS-CoV-2 BA.2.86 and JN.1 infection in hNEC are dependent on TMPRSS2 but not cathepsins, which is consistent with other Omicron variants35, there is a limited difference in the efficiency of TMPRSS2 usage between BA.2.86 and JN.1 (Fig. 4d). Next, we harvested hNEC cell lysates at 48 hpi and investigated spike cleavage of SARS-CoV-2 JN.1. Interestingly, we found that the cleavage of SARS-CoV-2 JN.1 spike at S1/S2 is more efficient than that of BA.2.86, while spike cleavage of BA.2.86 is more efficient than those of EG.5.1, and HK.3 (Fig. 4e, f), which is in keeping with the pseudovirus entry result in hNECs. Furthermore, we performed immunostaining on SARS-CoV-2 BA.2.86-, JN.1-, EG.5.1-, and HK.3-infected hNECs harvested at 24 hpi and quantified nucleocapsid (N) signal. Our results detected significantly higher N signals in SARS-CoV-2 JN.1-infected hNECs when compared with BA.2.86-, EG.5.1-, or HK.3-infected cells (Fig. 4g, h). Together, these results demonstrate a better fitness of BA.2.86 and JN.1 over the XBB descendent variants EG.5.1 and HK.3 in hNECs and indicate that the further elevated infectivity of JN.1 in hNECs conferred by the L455S mutation may contribute to its dominance over other variants in humans.

a Replication of BA.2, BA.2.86, JN.1, EG.5.1 and HK.3 in Calu3 cells (n = 6). Cell lysates were quantified for viral subgenomic E gene (sgE) copies. The n number represents biological repeats. b Replication of BA.2, BA.2.86, JN.1, EG.5.1 and HK.3 in hNECs (n = 3). Cell lysates were quantified for viral sgE copies. The n number represents biological repeats. c Pseudovirus entry in hNECs. hNECs were transduced with BA.2.86- (n = 7), JN.1- (n = 7), EG.5.1- (n = 4), and HK.3-spike (n = 4) pseudoviruses. Pseudovirus entries were quantified by measuring the luciferase signal. The n number represents biological repeats. d Preference of protease usage by BA.2.86 and JN.1 in hNECs (n = 3). The supernatant was quantified for viral RdRp gene level with qRT-PCR. The n number represents biological repeats. e Spike cleavage of BA.2.86, JN.1, EG.5.1, and HK.3 in hNECs. A representative image of the spike was shown with β-actin added as a sample processing control. Spike and β-actin were run on different gels and detected on different membranes. The cleavage ratio of spike proteins was quantified by ImageJ. f Quantification of spike cleavage as performed in Fig. 4e (n = 4). The n number represents biological repeats. g Infectivity of BA.2.86, JN.1, EG.5.1, and HK.3 in hNECs. Infected hNECs were fixed at 48 hpi for the visualization of ciliated cell marker beta-tubulin (red) and SARS-CoV-2 nucleocapsid protein (green). The DAPI channel was included in the merged images. Scale bar, 20 μm. h Immunofluorescence signal intensity of SARS-CoV-2 N BA.2.86 (n = 5), JN.1 (n = 9), EG.5.1 (n = 7), and HK.3 (n = 5) in Fig. 4g was quantified with ImageJ. The n number represents biological repeats. Scale bar = 20 µm. Data represent mean ± SEM. Statistical significance in (a–c and f, h) was determined with one-way ANOVA with post hoc Tukey’s multiple comparisons test. Statistical significance in (d) was determined with two-way ANOVA with Sidak’s multiple comparison tests. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. NS is not statistically significant. Source data are provided as a Source Data file.
Cryo-EM structures of SARS-CoV-2 BA.2.86/JN.1 spike in complex with ACE2
To evaluate whether JN.1 spike binds ACE2 differently when compared with BA.2.86 spike, we determined the structures of ACE2 in complex with stabilized prefusion ectodomain of BA.2.86 S (6 P) and JN.1 S (6 P) trimers individually by cryo-electron microscopy (cryo-EM) (Fig. 5). The purified spike trimers were mixed with ACE2 at the molar ratio of 1:1.2, and vitrified for cryo-EM study after 10 min. Cryo-EM analysis revealed that both BA.2.86 spike-ACE2 (S-ACE2) and JN.1 S-ACE2 complexes had only one conformational state: three up-RBDs bound with three ACE2 (Fig. 5a, c) which were determined to the resolution of 2.70 Å and 3.06 Å, respectively (Supplementary Figs. 9, 10). To improve the density of the spike-ACE2 interface, the NTD_RBD_ACE2 regions of BA.2.86 and JN.1 were locally refined to 3.29 Å and 3.07 Å, respectively (Fig. 5b, d and Supplementary Figs. 9, 10), allowing the unambiguous model building of the RBD-ACE2 region.

a Cryo-EM structure of the BA.2.86 spike in complex with ACE2. Two perpendicular views of BA.2.86 spike-ACE2 are depicted as surface, with ACE2 in cornflower blue and the trimeric spike in pink, medium purple, and purple. b Structure of BA.2.86 RBD-ACE2. c Cryo-EM structure of the JN.1 spike in complex with ACE2. Two perpendicular views of JN.1 spike-ACE2 are depicted as surface, with ACE2 in cornflower blue and the trimeric spike in orange, gold, and pale violet. d Structure of JN.1 RBD-ACE2. e Detailed interactions between the BA.2.86 spikeL455-ACE2 and the local refinement cryo-EM maps. f Detailed interactions between the JN.1 spikeS455-ACE2 and the local refinement cryo-EM maps. g Comparation of the RBD-ACE2 interface region in other recent variants: WT RBD-ACE2(PDB:6LZG), XBB.1 RBD-ACE2(PDB:8IOV), EG.5.1 RBD-ACE(PDB:8XLN).
The interfaces of BA.2.86 S-ACE2 and JN.1 S-ACE2 are nearly identical with the exception that the L455S mutation caused a local change. In BA.2.86 spike-ACE2 structure, the side chain of ACE2H34 has two alternative orientations (Fig. 5e). In the favored orientation A, ACE2H34 establishes van der Waals interactions with spikeL455 and forms a hydrogen bond with spikeY453 (Fig. 5e). In orientation B, the side chain of ACE2H34 swings to the opposite direction, forming a hydrogen bond with S494 (Fig. 5e). Importantly, in JN.1 spike-ACE2 structure, the side chain of ACE2H34 only exists in orientation B as the L455S mutation breaks the interaction between ACE2H34 and spikeL455 (Fig. 5f). In both orientations, spikeQ493 forms van der Waals interactions with ACE2H34 (Fig. 5e, f). In summary, ACE2H34 binds to BA.2.86 spike with its side chain adopting two alternative orientations, while in the JN.1 spike-ACE2 structure, interaction occurs in orientation B when the side chain of ACE2H34 forms a hydrogen bond with S494. Further structural analysis of the RBD-ACE2 interface regions of BA.2.86, JN.1, WT, XBB.1, and EG.5.1 variants revealed that most variants have only one conformation at ACE2H34 and no binding mode similar to BA.2.86, which may explain the strong binding force between BA.2.86. S and ACE2 (Fig. 5g)36. Notably, there were significant differences in the region adjacent to the specific mutation site of JN.1 RBDL455S compared to other variants. In this region, RBDS455 can form 5 pairs of hydrogen bonds with neighboring residues F456, Q493, P491, and Y453, but RBDL455 in other variants can only form 2 pairs of hydrogen bonds with P491 and Y453 (Supplementary Fig. 11). Based on the structural analysis, it is inferred that the hydrogen bond network changes as a result of the L455S mutation, which may explain why JN.1 has an enhanced ineffectiveness, although it has only one additional L455S mutation when compared with BA.2.86 at the spike protein.
Intrinsic pathogenicity of SARS-CoV-2 BA.2.86/JN.1 and EG.5.1/HK.3 in vivo
Next, we investigated the intrinsic pathogenicity of SARS-CoV-2 BA.2.86 and JN.1 together with the XBB descendant variants EG.5.1 and HK.3 (Fig. 6). We first infected hamsters with 1×104 PFU SARS-CoV-2 B.1, BA.2, BA.2.86, JN.1, XBB.1, EG.5.1, and HK.3, and harvested hamster lungs at 2 days post-infection (dpi) for virological assessments. We found that the ancestral B.1 variant replicated more efficiently than all emerging Omicron subvariants, as evidenced by its significantly higher sgE gene copy and infectious titer in hamster lungs (Fig. 6a, b). Interestingly, when compared with their parental subvariant BA.2, BA.2.86 and JN.1 appeared to be further attenuated in sgE gene copy and infectious titer in hamster lungs. In contrast, when compared with XBB.1, we observed a trend of increase in sgE gene copy and infectious titer for EG.5.1 and HK.3 in hamster lungs (Fig. 6a, b). In parallel, we performed immunostaining against N protein and quantified viral antigen expression in the infected hamster lungs. The pattern of N expression was largely in agreement with that of sgE gene copy and infectious titer. While we detected a clear decrease in N expression in SARS-CoV-2 BA.2.86- and JN.1-infected hamster lungs when compared to BA.2, N expression in EG.5.1- and HK.3-infected hamster lungs was significantly higher than that of XBB.1 (Fig. 6c, g). In addition, N expression in SARS-CoV-2 BA.2.86- and JN.1-infected hamster lungs was significantly lower than that of EG.5.1 and HK.3, demonstrating a lineage-specific difference in antigen expression in the lungs (Fig. 6c, g). We next examined the histopathological changes in hamster lungs upon virus infection with haematoxylin and eosin (H&E) staining and quantified lung pathology scores based on our semi-quantitative protocol35,37. We found that while the pathological changes of lungs from SARS-CoV-2 B.1-infected hamsters were featured by perivascular edema, alveolar wall collapse, alveolar wall congestion, and epithelium damage, the pathological manifestations were milder in EG.5.1- and HK.3-infected hamsters, which were further attenuated in BA.2.86- and JN.1-infected hamsters (Fig. 6d, h). In addition to the hamster model, we similarly challenged wildtype C57 mice and K18-hACE2 mice with SARS-CoV-2 BA.2.86, JN.1, EG.5.1, and HK.3, and assessed virus replication at 2 dpi. In keeping with results from the hamster model, BA.2.86 and JN.1 have a trend of less efficient replication than EG.5.1 and HK.3 in mice lungs (Fig. 6e, f). Overall, these results indicated a lineage specific difference in replication and intrinsic pathogenicity between BA.2.86/JN.1 and the XBB descendant variants EG.5.1 and HK.3 in vivo.

a, b 6-to-8-week-old male and female Syrian golden hamster were challenged with 1 × 104 PFU B.1, BA.2, BA.2.86, JN.1, XBB.1, EG.5.1, and HK.3 (n = 5). Animals were euthanized at 2 dpi for collection of lung tissues for detection of viral sgE copies (a) and infectious titers (b) by probe-specific RT-qPCR and TCID50 assay, respectively. c, g Infected hamster lungs were immunolabelled with anti-SARS-CoV-2-N antibody and visualized with a microscope. Cell nuclei were revealed with a DAPI stain. The immunofluorescence signal intensity of N was quantified with ImageJ software (n = 8) (c). Representative images are shown in (g). Scale bar, 200 μm. d, h Hamster lungs were stained with hematoxylin and eosin staining, and histology score was estimated (n = 5) (d). Representative images are shown in (h). Bold thick arrows, alveolar wall collapse; arrows, epithelium damage; dotted arrows, perivascular edema; arrowheads, alveolar wall congestion. Scale bar, 200 μm. e 6-to-8-week-old male and female C57BL/6 mice were challenged with 1 × 105 PFU BA.2 (n = 6), BA.2.86 (n = 7), JN.1 (n = 7), XBB.1 (n = 6), EG.5.1 (n = 7) and HK.3 (n = 7). Animals were euthanized at 2 dpi for collection of lung tissues for detection of viral burden. Viral sgE gene copies were detected with probe-specific RT-qPCR. f 6-to-8-week-old male and female K18-hACE2 transgenic mice were challenged with 1×104 PFU BA.2.86, JN.1, EG.5.1, and HK.3 (n = 7). Animals were euthanized at 2 dpi for collection of lung tissues for detection of viral burden. Viral sgE gene copies were detected with probe-specific RT-qPCR. Data represent mean ± SEM. Statistical significance in (a–f) was determined with one-way ANOVA with post hoc Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. NS is not statistically significant.
Discussion
In mid-2023, the circulating SARS-CoV-2 variants were dominated by XBB descendants, including XBB.1, XBB.1.5, XBB.1.6, and EG.5.1. A descendant of BA.2, BA.2.86, which emerged in July 2023, did not outcompete the XBB sublineages despite demonstrated an increased efficiency in using the plasma membrane route of entry12,17. Intriguingly, JN.1, a descendent variant of BA.2.86 that contained an additional L455S mutation on a spike when compared to BA.2.86, quickly replaced all other circulating variants to become the dominant variant across the world. Recent studies, including the current one, have demonstrated enhanced immune evasion of JN.1, which is believed to contribute to its rapid growth in numbers20,21. However, it remains to be answered whether the high transmissibility of JN.1 in humans is also associated with additional factors. In the current study, we found that JN.1 has a higher infectivity than BA.2.86 in hNECs. Mechanistically, we demonstrated that the gained infectivity of JN.1 over BA.2.86 is associated with increased entry efficiency conferred by L455S and better spike cleavage in hNECs. These results indicate that the robust transmissibility of JN.1 is associated with increased infectivity in hNECs in addition to its gained immune escape. Structurally, we demonstrated that ACE2H34 binds to BA.2.86 spike with its side chain adopting two alternative orientations, while in the JN.1 spike-ACE2 structure, interaction occurs in one orientation when the side chain of ACE2H34 forms a hydrogen bond with S494 as the L455S mutation breaks the interaction between ACE2H34 and spikeL455. In addition, we revealed significant differences in the region adjacent to the specific mutation site of JN.1 RBDL455S when compared to other variants. Specifically, we found that RBDS455 can form 5 pairs of hydrogen bonds with neighboring residues F456, Q493, P491, and Y453, while RBDL455 in other variants can only form 2 pairs of hydrogen bonds with P491 and Y453. The increased number of hydrogen bonds and a modified internal structure of JN.1 RBDL455S region may explain why JN.1 has an enhanced infectiveness over BA.2.86. However, the exact mechanism of how this change altered the virus-host interaction that increased the infectivity of JN.1 should be evaluated in future studies.
Since BA.2.86 and JN.1 are derived from the BA.2 lineage, while EG.5.1 and HK.3 are descendants of the XBB lineage, we compared the virological characteristics of these two independently evolved Omicron sublineages in a series of in vitro and in vivo experiments. We found that while EG.5.1 and HK.3 remain sensitive to mAb S3H3 and S309, BA.2.86 and JN.1 have escaped the neutralization effect of S3H3 and are only partially sensitive to S309, which is in keeping with recent findings23. In a cohort of BA.5 breakthrough infection followed by XBB reinfection, we found that the XBB reinfection induced limited neutralization against both BA.2-descendent variants (BA.2.86 and JN.1) and XBB-descendent variants (XBB.1, EG.5.1, HK.3). Among the evaluated subvariants, the lowest GMT was detected against HK.3, which may potentially due to its dual FLip mutations at L455F and F456L34. Since JN.1 has remained the predominant circulating variant, we evaluated serum neutralization upon JN.1 breakthrough infection. We found that while JN.1 breakthrough infection induced high GMT against BA.2, BA.2.86, and JN.1, it triggered substantially lower neutralization titer against XBB.1, EG.5.1, and HK.3. Similarly, hamsters infected with JN.1 or mice vaccinated with JN.1 spike primarily elicited autologous antibody responses against the BA.2.86/JN.1 lineage and is severely reduced in bidirectional cross-reactivity with other XBB sublineages. Together, the limited cross-protection between Omicron sublineages that are antigenically distanced should be taken into consideration in the development of future COVID-19 vaccines38,39.
In the hNECs, our results demonstrated a clear infection advantage by the BA.2-descendent variants (BA.2.86 and JN.1) over the XBB-descendent variants (EG.5.1 and HK.3), which was evidenced by the higher virus replication, more robust pseudovirus entry, better spike cleavage, and increased number of infected cells. By further introducing the L455S mutation in the background of XBB.1, EG.5.1, and HK.3 spike, we demonstrated that the L455S mutation was capable of enhancing the pseudovirus entry of all three variants in hNECs. In addition, the most recent variants under monitoring (VUM), including JN.1.7, KP.2. KP.3, JN.1.18, and LB.1, all carry L455S, further indicating the impact of L455S on enhanced fitness. A number of recent studies have investigated the infectiveness of JN.1 on various cell types18,21,22,40. It is important to note that differences in the protocol of pseudovirus preparation, virus culture, and cell culture may have an impact on the obtained results. Therefore, findings across laboratories should be compared and contrasted to arrive at a convincing conclusion. Interestingly, in the lung of infected animals, infection by BA.2-descendent variants (BA.2.86 and JN.1) resulted in lower virus replication and attenuated histological changes when compared with the XBB-descendent variants (EG.5.1 and HK.3), which is in keeping with earlier findings comparing the replication and pathogenicity of BA.2.86 and EG.5.1 in hamsters19 and may be associated with the non-spike mutations carried by these variants.
Taken together, our study indicates that the L455S mutation on the JN.1 spike facilitates the infection of JN.1 in hNECs, which may contribute to the high transmissibility of JN.1 among humans. In addition, our findings demonstrate lineage-specific virological characteristics between the BA.2 descendent variants BA.2.86/JN.1 and the XBB descendent variants EG.5.1/HK.3 which contribute to our understanding of their transmissibility and pathogenicity (Supplementary Fig. 12).
Methods
Human serum collection
Peripheral blood samples were collected from individuals who had previously experienced BA.5 infection and XBB or JN.1 reinfection following two to three doses of inactivated vaccines and were collected at the Nanjing Hospital of Chinese Medicine. Sera were isolated from centrifuged blood samples and then stored at − 80 °C. All collections were conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of the Ethics Committee of Nanjing Hospital of Chinese Medicine Affiliated to Nanjing University of Chinese Medicine (KY2023073). All participants provided written informed consent. Background information of the convalescent donors is summarized in Supplementary Table 1.
Virus challenge in hamster and mice
The use of all animals in the study was approved by The Committee on the Use of Live Animals in Teaching and Research (CULATR) of The University of Hong Kong (HKU) under CULATR 22-397. The golden Syrian hamsters and C57BL/6 J mice were obtained from the Center for Comparative Medicine Research (CCMR) at HKU. Heterozygous K18-hACE2 C57BL/6 J mice (2B6.Cg-Tg(K18-ACE2)2Prlmn/J) were obtained from The Jackson Laboratory. The hamsters and mice were housed in cages equipped with individual ventilation systems, maintained at a humidity level of 65%, and kept at an ambient temperature of 21–23 °C with a 12-h day-night cycle for proper care and management. Group sizes were chosen based on statistical power analysis and our prior experience in examining viral titers in SARS-CoV-2-infected animals37,41. Gender- and age-matched animals were randomized into different experimental groups. For hamster infections, 4-6 weeks old male hamsters were anesthetized with ketamine and xylazine, inoculated with 1 × 104 PFU SARS-CoV-2 B.1, Omicron BA.2, BA.2.86, JN.1, XBB.1, EG.5.1 or HK.3 pre-diluted in 50 μL PBS intranasally. For infection of wild-type C57BL/6 J mice, 8–10 weeks old mice were anesthetized with ketamine and xylazine, followed by intranasal inoculation with 20 µl per mouse of Omicron BA.2, BA.2.86, JN.1, XBB.1, EG.5.1 or HK.3 at 1 × 105 PFU. For virus infection of K18-hACE2 transgenic mice, 6–8 weeks old K18-hACE2 mice were anesthetized with ketamine and xylazine, followed by intranasal inoculation with 20 µl per mouse of Omicron BA.2.86, JN.1, EG.5.1 or HK.3 at 1 × 104 PFU. Virus-infected hamsters and mice were euthanized at 2 dpi to collect lung tissues for virological assessment, histological examination, and pathology scoring.
Mouse serum collection
7-weeks female BALB/c mice were randomly assigned to different groups and subjected to a total of two immunizations at 14-day intervals. Each immunization involved the use of 25 μg of the indicated Omicron BA.2, BA.2.86, JN.1, and XBB.1 variant spike trimer through the intraperitoneal route. Two weeks after the second immunization, blood was collected, and sera were collected by centrifugation.
Hamster serum collection
4-weeks male Syrian hamsters were inoculated with 3 × 104 PFU of Omicron BA.2, BA.2.86, JN.1, XBB.1, EG.5.1 and HK.3 via the intranasal route under anesthesia. After 16 days, the serum was harvested, centrifuged, and incubated at 56 °C for 30 min for inactivation.
Cell culture and virus infection
293T, Caco2, Huh7, and VeroE6 were obtained from ATCC and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (11965-092, Gibco, Texas, USA) supplemented with 10% fetal bovine serum (FBS), 100 units penicillin, and 100 µg/ml streptomycin, according to supplier’s instructions. VeroE6-TMPRSS2 was obtained from the Japanese Collection of Research Bioresources (JCRB) Cell Bank and cultured in DMEM supplemented with 10% fetal FBS, 100 units penicillin,100 µg/ml streptomycin, and 2% G418 (ant-gn-5, InvivoGen, China). Calu3 cells were cultured in DMEM/F12 (10565042, Gibco) supplemented with 10% FBS, 100 units of penicillin, and 100 µg/ml streptomycin. All cells were cultured at 37 °C in an incubator with 5% CO2. All cell lines used are routinely tested for mycoplasma and are maintained mycoplasma-free. For comparison of virus replication in vitro, Calu3 cells were seeded in 96-well plates and then infected with SARS-CoV-2 B.1 and Omicron subvariants at 2 multiplicity of infection (MOI).
Human nasal epithelial cell air-liquid interface culture and virus infection
The human nasal epithelial cells (hNEC) in an air-liquid interface (ALI) culture were purchased from Epithelix (EP02MP, Epithelix, Switzerland) and maintained with MucilAir culture medium (EP04MM, Epithelix) until virus challenge. During the virus challenge experiment, cells were washed with a MucilAir culture medium to remove the apical mucin. Subsequently, the virus was inoculated at the apical side with a 2 MOI. Cells were incubated for 2 h at 37 oC to facilitate efficient virus entry. The residual inoculum was removed and rinsed away after incubation. Supernatants and cell lysates were harvested from the apical side for viral genome copy quantification with RNA extraction followed by one-step RT-qPCR at the designated time points. For immunofluorescence staining, cells were fixed with neutral-buffered formalin followed by permeabilization with 0.1% Triton-X-100 (11332481001, Sigma-Aldrich). The SARS-CoV-2 nucleocapsid protein and ciliated cells were detected with in-house rabbit anti-SARS-CoV-2 nucleocapsid serum and mouse anti-beta-tubulin (T7941, Sigma-Aldrich), respectively. Primary antibodies were visualized with Alexa Fluor 594-conjugated goat anti-mouse secondary antibody (A-11005, Thermo Fisher Scientific, USA) and Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody (A-11034, Thermo Fisher Scientific), followed by mounting with ProLong™ Diamond Antifade Mountant with DAPI (P36962, Thermo Fisher Scientific). Images were acquired with an LSM980 confocal microscopy system (Zeiss, USA). To acquire the monolayer image of hNECs, a Z-stack was applied to capture each layer in the z-axis of hNECs. The orthogonal projection was used to stack the Z-stack into one image. Images were processed and analyzed by ZEISS Zen (blue edition) software using the maximum intensity projection setting.
Viruses and biosafety
SARS-CoV-2 D614G (B.1) (GISAID: EPL_ISL_497840), Omicron BA.2 (GISAID: EPI_ISL_9845731), BA.2.86 (GISAID: EPI_ISL_18986956), JN.1 (GISAID: EPL_ISL_18841631), XBB.1 (GISAID: EPI_ISL_15602393), EG.5.1 (GISAID: EPI_ISL_18461518), and HK.3 (GISAID: EPI_ISL_18986955) were isolated from laboratory-confirmed COVID-19 patients in Hong Kong. All variants of SARS-CoV-2 were cultured and titrated by plaque assays using VeroE6-TMPRSS2 cells. In vivo and in vitro experiments with infectious SARS-CoV-2 were performed according to the approved standard operating procedures of the Biosafety Level 3 facility at the Department of Microbiology, the University of Hong Kong (HKU)42,43.
Phylogenetic, mutation, and epidemic analysis
A total of 3836 SARS-CoV-2 genome sequences and pair-wised phylogenetic trees were downloaded from nextstrain (https://nextstrain.org/) with the GISAID data (https://www.gisaid.org/) from December 2019 to February 2024. We accessed recent 6-month global data on March 10, 2024, which highlights the Omicron variants. We re-colored the phylogenetic tree by highlighting BA.1, BA.2, BA.4/5, XBB, XBB.1*, XBB.2*, EG.5.1, HK.3, BA.2.86, JN.1, WT, and other variants (all non-WT pre-Omicron variants). We analyzed the mutation and epidemic by using data from covSPECTRUM (https://cov-spectrum.org/) based on the GISAID data (https://www.gisaid.org/) on March 11, 2024. The mutation proportion of each variant was based on all genome sequences from January 6, 2020, to March 6, 2024, obtained on March 11, 2024. Here, only mutations with > 10% frequency were included. The variants frequency of each variant was calculated based on genome sequences from September 1, 2023, to March 1, 2024 obtained on March 11, 2024. In the analysis of the step-wise accumulation of key Spike mutations, we used the color barcode to represent the convergent mutations. In this study, we included R346T, K356T, V445P/H, G446S, N450D, L452W, F456L, N460K, F486P/S, F490S/V, and R493Q based on key convergent mutations44, as well as the “FLip” mutations L455F/S.
Protein expression and purification
The constructs used for expression of stabilized soluble BA.2, XBB.1, BA.2.86, and JN.1 S6P spike trimer proteins were prepared as we previously described45. In brief, each spike ectodomain was separately cloned into mammalian expression vector pCMV3 with an 8x His tag and 2x Strep-tag II tags at the C terminus. To obtain the protein, Expi293 cells were used for transient transfection with the suitable S6P stabilized S-expression plasmids by using 1 mg/mL polyethylenimine (PEI, Polysciences). The supernatant was harvested, and the S trimer was purified using Ni-Smart resin from Smart Lifesciences (Ni Smart Beads 6FF: SA036100, Changzhou, China) in accordance with the manufacturer’s protocol five days after transfection. Prior to use, all proteins were further evaluated for size and purity through SDS-PAGE.
Antibody expression and purification
Monoclonal antibodies, LY-1404, S3H3, S309 and SA55, were obtained from our previous studies46. Briefly, variable genes were codon optimized for human cell expression and synthesized by HuaGeneTM (Shanghai, China) into plasmids (gWiz) that encoded the constant region of human IgG1 heavy or light chain. Antibodies were expressed in Expi293F (A14527, ThermoFisher) by co-transfection of heavy and light chain expressing plasmids using polyethylenimine (Polyscience), and cells were cultured at 37 °C with shaking at 125 rpm and 8% CO2. Supernatants were also collected on day 5 for antibody purification using MabSelectTM PrismA (17549801, Cytiva) affinity chromatography.
Production of SARS-CoV-2-spike pseudoviruses
Pseudoviruses were prepared as we previously described47,48,49. Plasmids encoding the spike of BA.2, BA.2.86, JN.1, XBB.1, EG.5.1, and HK.3 variants were synthesized. Expi293F cells were grown to 3 × 106/mL before transfection with the indicated spike gene using polyethylenimine. Cells were cultured overnight at 37 °C with 8% CO2 and VSV-G pseudo-typed ΔG-luciferase (G*ΔG-luciferase, Kerafast) was used to infect the cells in DMEM at a multiplicity of infection of 5 for 4 h before washing the cells with PBS/2% FBS three times. The next day, the transfection supernatant was collected and clarified by centrifugation at 300 × g for 10 min. Each viral stock was then incubated with 20% I1 hybridoma (anti-VSV-G; CRL-2700, ATCC) supernatant for 1 h at 37 °C to neutralize the contaminating VSV-G pseudotyped ΔG-luciferase virus before measuring titers and making aliquots to be stored at − 80 °C.
Pseudovirus neutralization
Neutralization assays were performed by incubating pseudoviruses with serial dilutions of monoclonal antibodies or sera and scored by the reduction in luciferase gene expression as we described previously50. In brief, VeroE6 cells were seeded in a 96-well plate at a concentration of 2 × 104 cells per well. On the following day, pseudoviruses were incubated with serial dilutions of the test samples in triplicate for 30 min at 37 °C. The mixture was then added to cultured cells and incubated for an additional 24 hours. The luminescence was measured by the Luciferase Assay System (Beyotime). IC50 was defined as the dilution at which the relative light units were reduced by 50% compared with the virus control wells (virus + cells) after subtraction of the background in the control groups with cells only. The IC50 values were calculated using nonlinear regression in GraphPad Prism.
Omicron-spike pseudovirus entry efficiency in cell lines and hNECs
293T, Caco2, Calu3, Huh7, VeroE6, A549, and VeroE6-TMPRSS2 cells seeded in 96 well plates were transduced with B.1-, BA.2-, BA.2.86-, JN.1-, XBB.1, EG.5.1-, and HK.3- spike pseudoviruses. Differentiated hNECs were transduced with BA.2.86-, JN.1-, EG.5.1-, and HK.3- spike pseudoviruses. At 24 h post-transduction, transduced cells were washed three times with PBS and lysed by the lysis buffer (RG129M), luminescence signal was measured with the Luciferase Assay System (Beyotime). For the pseudoviruses entry inhibition experiment, VeroE6-TMPRSS2, Calu3, Caco2, and 293T cells were seeded in 96 well plates. After 24 h, the target cells were pretreated with 10 µM Camostat (MedChem Express HY-13512, Monmouth Junction, NJ, USA) or E64D (MedChem Express HY-100229, Monmouth Junction, NJ, USA) for 2 h, followed by inoculation with B.1-, BA.2-, BA.2.86-, JN.1-, XBB.1-, EG.5.1-, and HK.3-spike pseudoviruses. At 24 h post-transduction, the level of pseudovirus entry was quantified by measuring the luciferase signal as mentioned above.
Antigenic maps
The constructed antigenic maps were based on serum neutralization data utilizing the antigenic cartography methods, which are implemented in the Racmacs package (https://acorg.github.io/Racmacs/). The antigenic maps were generated in R with 10000 optimization steps and other default parameters in a 2-dimensional space. The distances between positions of sub-lineages and serum on the antigenic map were optimized so that distances approach the fold decreases in neutralizing ID50 titer, relative to the maximum titer for each serum. Each unit of distance in arbitrary directions in the antigenic map represents a 2-fold change in the ID50 titer. The antigenic differences among the sub-lineages are calculated based on the antigenicity distances between different sub-lineages and serums, determined as follows:
Where Dij represents the antigenicity distance between sub-lineage i and serum j, bj denotes the maximum titer against one specific strain (normally, it is strain j itself) for serum generated by strain j as the base of antigenic distance. Hij is the ID50 measurement of neutralizing titer of serum generated by j against strain i51.
Histology and immunofluorescence staining of hamster lungs
Infected hamster lungs were fixed by 4% paraformaldehyde for 24 h. Then, samples were washed with 70% ethanol and a TP1020 Leica semi-enclosed benchtop tissue processor (Leica Biosystems, Buffalo Grove, IL, USA) was applied to embed lung samples in paraffin. Embedded samples were sectioned with a microtome (Thermo Fisher Scientific) and placed on microscope slides for drying at 37 °C overnight. Sample slides were dewaxed by washing in serially diluted xylene, ethanol, and double-distilled water. For antigens unmasking, diluted antigen unmasking solution (H-3300, Vector Laboratories) was heated until 85 °C, then slides were put in solution and boiled for 90 s. Unmasked slides were stained with Sudan black B and then blocked with 5% FBS for 30 min. The in-house rabbit anti-SARS-CoV-2-N immune serum was applied as the primary antibodies and incubated at 4 °C overnight against SARS-CoV-2 nucleocapsid. Goat anti-rabbit immunoglobulin G (H + L) fluorescein isothiocyanate (65-6111), which functioned as a secondary antibody, was purchased from Thermo Fisher Scientific. Slides were mounted, and meanwhile, the cell nucleus was stained with the antifade mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; H-1200, Vector Laboratories, Burlingame, CA, USA). Images were taken with the Olympus BX53 fluorescence microscope (Olympus Life Science, Tokyo, Japan). Quantification of the fluorescence signal was conducted with ImageJ. For H&E staining, tissue sections were stained with Gill’s haematoxylin and eosin. Images were acquired using the Olympus BX53 light microscope (Olympus Life Science, Japan). Five hamsters were sampled in each group (as specified in the figure legends), and three sections from each animal were used for histology analysis. Histopathological scores were quantified using a semi-quantitative method we previously described35,52.
RNA extraction and quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Viral RNA was extracted from homogenized animal tissues using RNeasy Mini Kit (74106, Qiagen). Viral RNA of supernatants collected from infected Calu3, or hNECs, was extracted using Viral RNA Mini Kit (52906, Qiagen) or QIAsymphony RNA Kit (931636, Qiagen). After RNA extraction, qRT-PCR was performed using QuantiNova Probe RT-PCR Kit (208354, Qiagen) or QuantiNova SYBR Green RT-PCR Kit (208154, Qiagen) with the LightCycler 480 Real-Time PCR System (Roche, Basel, Switzerland). The primer and probe sequences are provided in Supplementary Table 4.
Quantification of infectious titer from hamster lungs
To quantify the infectious titer of virus-infected hamster lung tissues, a monolayer of VeroE6-TMPRSS2 cells in a 96-well plate was challenged with 10-fold serial diluted supernatants of homogenized tissues. Cytopathic effect (CPE) was observed at four days post-infection to quantify the median tissue culture infectious dose as we previously described53.
Cell-cell fusion assay
For the 293T-based cell-cell fusion assay, 293T cells were co-transfected with different SARS-CoV-2 spike plasmids with GFP1-10 plasmid (cat#68715, Addgene) as effector cells. Another population of 293T cells was co-transfected with human ACE2 (hACE2), and GFP11 (cat#68716, Addgene) as target cells as previously described53. After 24 h post-transfection, the effector and target cells were digested by EDTA-Trypsin (25200072, Gibco) and mixed at a 1:1 ratio. The mixed cells were co-cultured in an 8-well chamber slides at a 37 °C incubator for another 24 h. The mixed cells were fixed in 10% formalin and then permeabilized with 0.1% Triton-X100 (11332481001, Sigma, USA) at room temperature. The antifade mounting medium with 4′,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI, H-1200, Vector Laboratories) was used for mounting and DAPI staining. Images were taken with the Olympus BX73 fluorescence microscope (Olympus Life Science, Tokyo, Japan). The fusion area of images was quantified by ImageJ. For the Calu3 cell-cell fusion assay, 293T cells co-transfected with different SARS-CoV-2 spike plasmids and the GFP plasmid (cat#13031, Addgene) were co-cultured with Calu3 cells. The co-cultured cells were fixed after 24 h of incubation and GFP signal intensity was quantified by ImageJ.
Flow cytometry
293T cells were transfected with B.1, BA.2, BA.2.86, JN.1, XBB.1, EG.5.1, and HK.3 spike expression plasmids. Then cells were performed as we previously described48. Cells were detached with 0.5 mM EDTA in PBS on day 2 post-transfection and fixed in 4% paraformaldehyde. The fixed cells were then stained with 1:200 diluted polyclonal rabbit anti-S1 antibody (Sino Biological, 40591-T62) for 1.5 h and washed three times with 2% FBS/PBS. The Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody (A-11034, Thermo Fisher Scientific) was used as the secondary antibody. Flow cytometry was performed using a BD FACSCanto II flow cytometer (BD Biosciences), and data was analyzed using FlowJo X 10.0.7 (BD). The gating strategy was demonstrated in Supplementary Fig. 13.
Western blot analysis of spike cleavage
Differentiated hNECs were challenged with Omicron BA.2.86, JN.1, EG.5.1, and HK.3 at 2 MOI and lysed in RIPA buffer (89901, Thermo Fisher Scientific) at 48 hpi for Western blot analysis. The membranes were blocked with 5% milk for 2 h at room temperature and incubated with a rabbit anti-SARS-CoV-2 spike S2 antibody (40590-T62, Sino Biological, China) at 4 °C for overnight incubation, followed by detection with horseradish peroxidase (HRP) conjugated secondary antibodies (31460, Thermo Fisher Scientific) for 1 h at room temperature. The signal was developed using SuperSignal West Pico PLUS Chemiluminescent Substrate (34580, Thermo Scientific) and detected using Alliance Imager apparatus (Uvitec, Cambridge, UK). β-actin was detected with a β-actin antibody (clone AC-74, A5316, Sigma-Aldrich) (1:5000). The uncropped Western blot images were demonstrated in Supplementary Fig. 14.
Cryo-EM data collection and processing
The JN.1 spike trimer (or BA.2.86 spike) at 5 mg/mL was mixed with 5 mg/mL ACE2 at the molar ratio of 1:1.2, and incubated at 4 °C for 10 min. Dilute the mixture to 0.7 mg/mL in 20 mM Tris, pH 8.0, and 200 mM NaCl. The 3 μL composite was added to a newly light-emitting porous amorphous Ni-Ti alloy film supported by 400 mesh gold mesh. The sample was immersed in liquid ethane using Vitrobot IV (FEI, Thermo Fisher Scientific) and blotted for 2 s, -3blot force, 10 s waiting time.
Cryo-EM data was collected on a TITAN Krios G4 transmission electron microscope (Thermo Fisher Scientific) running at 300 kV. The microscope is equipped with Falcon 4i and Selectris X-ray imaging filters (Thermo Fisher Scientific) with a slit width of 20 eV. The EPU software is used to automatically collect data in 300 kV AFIS mode. EER movie stacks were collected in super-resolution mode, JN.1 S-ACE2 and BA.2.86 S-ACE2 magnification of 105,000x, corresponding physical pixel size of 1.19 Å, dose fractioned to 1737 frames. The defocusing range is − 1.0 to − 3.0 μm, and the total dose is about 50e−/Å2 54.
For the dataset of BA.2.86 spike protein and ACE2, all dose-fractioned images were motion-corrected and dose-weighted by RELION software and their contrast transfer functions were estimated by cryoSPARC patch CTF estimation55. The raw particles were auto-picked using the blob picker job and template picker job, and the particles were extracted with a box size of 320 pixels and Fourier crop to a box size of 160 pixels, and the following 2D, 3D classifications, and refinements were all performed in cryoSPARC55 (Supplementary Fig. 9a). 597,656 particles were selected after 2D classification, and these particles were used to do ab initio reconstruction heterogeneous refinement. This process yielded 446,892 converged particles, corresponding to a “3-up” conformation with three ACE2 bound to each RBD. Next, these particle sets were re-extracted with a box size of 320 pixels and used to perform non-uniform refinement, yielding a resolution of 2.70 Å. To elucidate the structural details of the receptor interaction region, a mask encompassing NTD, RBD, and ACE2 was employed for no-alignment 3D classification and local refinement following global particle convergence and C3 symmetry expansion. The provided particles depict a local reconstruction of the NTD-RBD-ACE2 region, resulting in a 3.29 Å map generated using 67,266 particles (Supplementary Fig. 9b).
For the dataset of JN.1 S protein and ACE2, all dose-fractioned images were motion-corrected and dose-weighted by RELION software and their contrast transfer functions were estimated by cryoSPARC patch CTF estimation. The raw particles were auto-picked using the blob picker job and template picker job, and the particles were extracted with a box size of 320 pixels and Fourier crop to a box size of 160 pixels, and the following 2D, 3D classifications, and refinements were all performed in cryoSPARC55 (Supplementary Fig. 10a). Following 2D classification, 973,795 particles were selected for ab-initio reconstruction and subsequent heterogeneous refinement. This process yielded 642,104 converged particles, corresponding to a “3-up” conformation with three ACE2 bound to each RBD. Next, this particle set was re-extracted with a box size of 320 pixels and used to perform non-uniform refinement, yielding a resolution of 3.06 Å. To elucidate the structural details of the receptor interaction region, a mask encompassing NTD, RBD, and ACE2 was employed for no-alignment 3D classification and local refinement following global particle convergence and C3 symmetry expansion. The provided particles depict a local reconstruction of the NTD-RBD-ACE2 region, resulting in a 3.07 Å map generated using 258,064 particles (Supplementary Fig. 10b).
Model building and refinement
The sharpened maps were generated by EMReady56. For model building, the initial model was generated by swiss-model57 and fitted to the map using UCSF Chimera58. The model was then manually adjusted in COOT and furthered refined using PHENIX59. Figures were produced by UCSF Chimera and UCSF ChimeraX60. Data collection and model refinement statistics are shown in Supplementary Table 3.
Statistical analysis
Data on figures represented means and standard error of the mean. Comparison between the two groups was performed with two-sided Student’s t tests. Comparison between three or more experimental groups was performed with one-way or two-way ANOVA. Differences were considered statistically significant when p < 0.05. Data analysis was performed with GraphPad Prism 8.0.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Coordinates and maps associated with data reported in this manuscript was deposited to the Electron Microscopy Data Bank (EMDB) and Protein Data Bank (PDB) with accession numbers EMD-39689 and PDB 8YZC (BA.2.86 spike-ACE2), EMD-39688 and PDB 8YZB (BA.2.86 NTD-RBD-ACE2 local refinement), EMD-39691 and PDB 8YZE (JN.1 spike-ACE2), EMD-39690 and PDB 8YZD (JN.1 NTD-RBD-ACE2 local refinement). Source data are provided in this paper.
References
Liu, L. et al. Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature 602, 676–681 (2022).
Iketani, S. et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 604, 553–556 (2022).
Halfmann, P. J. et al. SARS-CoV-2 Omicron virus causes attenuated disease in mice and hamsters. Nature 603, 687–692 (2022).
McMahan, K. et al. Reduced pathogenicity of the SARS-CoV-2 omicron variant in hamsters. Med 3, 262–268.e4 (2022).
Shuai, H. et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 603, 693–699 (2022).
Yuan, S. et al. Pathogenicity, transmissibility, and fitness of SARS-CoV-2 Omicron in Syrian hamsters. Science 377, 428–433 (2022).
Cao, Y. et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 602, 657–663 (2022).
Dejnirattisai, W. et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell 185, 467–84.e15 (2022).
Hoffmann, M. et al. The Omicron variant is highly resistant against antibody-mediated neutralization: Implications for control of the COVID-19 pandemic. Cell 185, 447–56.e11 (2022).
Meng, B. et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 603, 706–714 (2022).
Planas, D. et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 602, 671–675 (2022).
Qu, P. et al. Immune evasion, infectivity, and fusogenicity of SARS-CoV-2 BA.2.86 and FLip variants. Cell 187, 585–95 (2024).
Lasrado, N. et al. Neutralization escape by SARS-CoV-2 Omicron subvariant BA.2.86. Vaccine 41, 6904–6909 (2023).
Uriu, K. et al. Transmissibility, infectivity, and immune evasion of the SARS-CoV-2 BA.2.86 variant. Lancet Infect Dis 23, e460–e461 (2023).
Wang, Q. et al. Antigenicity and receptor affinity of SARS-CoV-2 BA.2.86 spike. Nature 624, 639–644 (2023).
Yang, S. et al. Antigenicity and infectivity characterisation of SARS-CoV-2 BA.2.86. Lancet Infect. Dis. 23, e457–e459 (2023).
Zhang, L. et al. SARS-CoV-2 BA.2.86 enters lung cells and evades neutralizing antibodies with high efficiency. Cell 187, 596–608 (2024).
Li, P. et al. Distinct patterns of SARS-CoV-2 BA.2.87.1 and JN.1 variants in immune evasion, antigenicity, and cell-cell fusion. mBio 15, e0075124 (2024).
Tamura, T. et al. Virological characteristics of the SARS-CoV-2 BA.2.86 variant. Cell Host Microbe 32, 170–80 (2024).
Jeworowski, L. M. et al. Humoral immune escape by current SARS-CoV-2 variants BA.2.86 and JN.1, December 2023. Euro Surveill 29, 2300740 (2024).
Kaku, Y. et al. Virological characteristics of the SARS-CoV-2 JN.1 variant. Lancet Infect. Dis. 24, e82 (2024).
Planas, D. et al. Distinct evolution of SARS-CoV-2 Omicron XBB and BA.2.86/JN.1 lineages combining increased fitness and antibody evasion. Nat. Commun. 15, 2254 (2024).
Yang, S. et al. Fast evolution of SARS-CoV-2 BA.2.86 to JN.1 under heavy immune pressure. Lancet Infect. Dis. 24, e70–e72 (2024).
Westendorf, K. et al. LY-CoV1404 (bebtelovimab) potently neutralizes SARS-CoV-2 variants. Cell Rep. 39, 110812 (2022).
Xu, S. et al. Mapping cross-variant neutralizing sites on the SARS-CoV-2 spike protein. Emerg. Microbes Infect. 11, 351–367 (2022).
Pinto, D. et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 583, 290–295 (2020).
Cao, Y. et al. Rational identification of potent and broad sarbecovirus-neutralizing antibody cocktails from SARS convalescents. Cell Rep. 41, 111845 (2022).
Wang, X., Jiang, S., Ma, W., Zhang, Y. & Wang, P. Robust neutralization of SARS-CoV-2 variants including JN.1 and BA.2.87.1 by trivalent XBB vaccine-induced antibodies. Signal Transduct. Target. Ther. 9, 123 (2024).
Hoffmann, M., Kleine-Weber, H. & Pöhlmann, S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 78, 779–84.e5 (2020).
Hoffmann, M. et al. SARS-CoV-2 Cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–80 (2020).
Laporte, M. et al. The SARS-CoV-2 and other human coronavirus spike proteins are fine-tuned towards temperature and proteases of the human airways. PLoS Pathog. 17, e1009500 (2021).
Mykytyn, A. Z. et al. SARS-CoV-2 entry into human airway organoids is serine protease-mediated and facilitated by the multibasic cleavage site. Elife 10, e64508 (2021).
Volcic, M. et al. Attenuated replication and damaging effects of SARS-CoV-2 Omicron variants in an intestinal epithelial barrier model. J. Med. Virol. 96, e29783 (2024).
Kosugi, Y. et al. Characteristics of the SARS-CoV-2 omicron HK.3 variant harbouring the FLip substitution. Lancet Microbe 5, e313 (2024).
Shuai, H. et al. The viral fitness and intrinsic pathogenicity of dominant SARS-CoV-2 Omicron sublineages BA.1, BA.2, and BA.5. EBioMedicine 95, 104753 (2023).
Liu, C. et al. A structure-function analysis shows SARS-CoV-2 BA.2.86 balances antibody escape and ACE2 affinity. Cell Rep. Med. 5, 101553 (2024).
Chu, H. et al. Coronaviruses exploit a host cysteine-aspartic protease for replication. Nature 609, 785–792 (2022).
Du, P. et al. The omicron BA.2.86 subvariant as a new serotype of SARS-CoV-2. Lancet Microbe https://doi.org/10.1016/s2666-5247(23)00411-1 (2024).
Hu, S. et al. Classification of five SARS-CoV-2 serotypes based on RBD antigenicities. Sci Bull. 68, 3003–3012 (2023).
Hu, J. et al. Enhanced Reverse Zoonotic Potential and Immune Evasion by Omicron JN.1 Variant. Available at SSRN: https://ssrn.com/abstract=4874431.
Chan, J. F. et al. Altered host protease determinants for SARS-CoV-2 Omicron. Sci. Adv. 9, eadd3867 (2023).
Chu, H. et al. Targeting highly pathogenic coronavirus-induced apoptosis reduces viral pathogenesis and disease severity. Sci. Adv. 7, eabf8577 (2021).
Shuai, H. et al. Emerging SARS-CoV-2 variants expand species tropism to murines. EBioMedicine 73, 103643 (2021).
Cao, Y. et al. Imprinted SARS-CoV-2 humoral immunity induces convergent Omicron RBD evolution. Nature 614, 521–529 (2023).
Zhao, X. et al. Potent and broadly neutralizing antibodies against sarbecoviruses induced by sequential COVID-19 vaccination. Cell Discov. 10, 14 (2024).
Ai, J. et al. Antibody evasion of SARS-CoV-2 Omicron BA.1, BA.1.1, BA.2, and BA.3 sub-lineages. Cell Host Microbe 30, 1077–1083 (2022).
Chan, J. F. et al. Virological features and pathogenicity of SARS-CoV-2 Omicron BA.2. Cell Rep. Med. 3, 100743 (2022).
Chu, H. et al. Host and viral determinants for efficient SARS-CoV-2 infection of the human lung. Nat. Commun. 12, 134 (2021).
Wang, X. et al. Homologous or heterologous booster of inactivated vaccine reduces SARS-CoV-2 Omicron variant escape from neutralizing antibodies. Emerg Microbes Infect. 11, 477–481 (2022).
Wang, X. et al. Neutralization of distinct Omicron sublineages by longitudinal vaccination sera. J. Med. Virol. 94, 5090–5092 (2022).
Smith, D. J. et al. Mapping the antigenic and genetic evolution of influenza virus. Science 305, 371–376 (2004).
Hu, B. et al. Divergent trajectory of replication and intrinsic pathogenicity of SARS-CoV-2 Omicron post-BA.2/5 subvariants in the upper and lower respiratory tract. EBioMedicine 99, 104916 (2024).
Hu, B. et al. Spike mutations contributing to the altered entry preference of SARS-CoV-2 omicron BA.1 and BA.2. Emerg Microbes Infect. 11, 2275–2287 (2022).
Hsieh, C. L. et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 369, 1501–1505 (2020).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
He, J., Li, T. & Huang, S. Y. Improvement of cryo-EM maps by simultaneous local and non-local deep learning. Nat. Commun. 14, 3217 (2023).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 74, 531–544 (2018).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Acknowledgements
We gratefully acknowledge all data contributors, including the authors and their originating laboratories responsible for obtaining the specimens, and their submitting laboratories for generating the genetic sequence and metadata and sharing via the GISAID Initiative, on which this research is based. We also thank the Center of Cryo-Electron Microscopy, Core Facility of Shanghai Medical College, Fudan University, for the support on cryo-EM data collection. Hin Chu acknowledge supports by funding from National Natural Science Foundation of China Excellent Young Scientists Fund (Hong Kong and Macau) (32122001); the Health and Medical Research Fund (COVID1903010-14, 23220522, CID-HKU1-5), the Food and Health Bureau, The Government of the Hong Kong Special Administrative Region; the General Research Fund (17118621, 17119122), Collaborative Research Fund (C7103-22G, C7060-21G), and Theme-Based Research Scheme (T11-709/21-N), the Research Grants Council of the Hong Kong Special Administrative Region; Health@InnoHK, Innovation and Technology Commission, the Government of the Hong Kong Special Administrative Region; Partnership Program of Enhancing Laboratory Surveillance and Investigation of Emerging Infectious Diseases and Antimicrobial Resistance for the Department of Health of the Hong Kong Special Administrative Region Government; Sanming Project of Medicine in Shenzhen, China (SZSM201911014); the High Level-Hospital Program, Health Commission of Guangdong Province, China; National Key Research and Development Program of China (projects 2021YFC0866100 and 2023YFC3041600); Emergency Collaborative Project of Guangzhou Laboratory (EKPG22-01); General Program, Guangdong Provincial National Science Foundation, China (2023A1515012325, 2023A1515011891); the University of Hong Kong Li Ka Shing Faculty of Medicine Enhanced New Staff Start-up Fund; the University of Hong Kong Outstanding Young Researcher Award; the University of Hong Kong Research Output Prize (Li Ka Shing Faculty of Medicine); the Major Science and Technology Program of Hainan Province (ZDKJ202003); the research project of Hainan Academician Innovation Platform (YSPTZX202004); the Hainan Talent Development Project (SRC200003); and the donations from Richard Yu and Carol Yu, May Tam Mak Mei Yin, Richard Yu and Carol Yu, the Shaw Foundation Hong Kong, Michael Seak-Kan Tong, Lee Wan Keung Charity Foundation Limited, Providence Foundation Limited (in memory of the late Lui Hac-Minh), Hong Kong Sanatorium and Hospital, Hui Ming, Hui Hoy and Chow Sin Lan Charity Fund Limited, The Chen Wai Wai Vivien Foundation Limited, Chan Yin Chuen Memorial Charitable Foundation, Marina Man-Wai Lee, the Hong Kong Hainan Commercial Association South China Microbiology Research Fund, Perfect Shape Medical Limited, Kai Chong Tong, Tse Kam Ming Laurence, Foo Oi Foundation Limited, Betty Hing-Chu Lee, Ping Cham So, and Lo Ying Shek Chi Wai Foundation. This study was also supported by funding from the National Key Research and Development Program of China (2023YFC2605400 and 2023YFC3404000 to X.Z.), National Natural Science Foundation of China (32270142 to P.W.; 32300121 to X.Z.), Shanghai Rising-Star Program (22QA1408800 to P.W.), Shanghai Pujiang Program (23PJD007 to X.Z.), the Program of Science and Technology Cooperation with Hong Kong, Macao and Taiwan (23410760500 to P.W.). Pengfei Wang acknowledges support from AI for Science project of Fudan University (XM06231724), Open Research Fund of State Key Laboratory of Genetic Engineering, Fudan University (No. SKLGE-2304) and Xiaomi Young Talents Program. Lei Sun acknowledges support from the Ministry of Science and Technology of China (2021YFC2302500) and the R&D Program of Guangzhou Laboratory (SRPG22-003). Bao-Zhong Zhang, Hin Chu, and Ye-Fan Hu also thank support from the Major Program of the National Natural Science Foundation of China (Grant No. 92369201). All donors and funding sources had no role in the study design, data collection, analysis, interpretation, or writing of the manuscript.
Author information
Authors and Affiliations
Contributions
Y.L., J.S., H.L., B.H., H.S., T.T.-T.Y., Y.C., F.L., L.W., M.H., T.Z., Y.H., X.H., C.Y., Y.W., Y.H., M.Z., L.D., X.Z., W.-M.C., L.-L.C., J.-P.C., S.Y., J.Z., K.-Y.Y., K.K.-W.T., J.F.-W.C., and H.C. performed experiments, analyzed the data, or provided key resources for the in vitro and in vivo pseudovirus and authentic virus assays. X.Z., J.L., X.W., S.J., Y.Z., X.L., and P.W. performed experiments, analyzed the data, or provided key resources for the neutralization assays, generated spike trimers, and constructed the pseudoviruses. Y.W., X.Z., and L.S. performed experiments, analyzed the data, or provided key resources for the Cryo-EM determination of spike-hACE2 interaction. Y.-F.H., H.-R.G., J.-D.H., and B.-Z.Z. performed experiments analyzed the data, or provided key resources for bioinformatic analysis and SPR assays. Y.L., X.Z., Y.W., Y.-F.H., B.-Z.Z, L.S., P.W., and H.C. wrote the manuscript. H.C. conceptualized and supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Y., Zhao, X., Shi, J. et al. Lineage-specific pathogenicity, immune evasion, and virological features of SARS-CoV-2 BA.2.86/JN.1 and EG.5.1/HK.3. Nat Commun 15, 8728 (2024). https://doi.org/10.1038/s41467-024-53033-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53033-7
This article is cited by
-
SARS-CoV-2 variants: biology, pathogenicity, immunity and control
Nature Reviews Microbiology (2026)
-
YL1004 is a SARS-CoV-2 papain-like protease inhibitor with immunomodulatory and antiviral activity in mice
Nature Communications (2026)
-
Inducible CD147 up-regulation boosts extended SARS-CoV-2 infection triggering severe COVID-19 independent of ACE2
Signal Transduction and Targeted Therapy (2026)
-
Immune evasion, infectivity, and membrane fusion of the SARS-CoV-2 JN.1 variant
Virology Journal (2025)
-
SARS-CoV-2 spike protein: structure, viral entry and variants
Nature Reviews Microbiology (2025)
