
Abstract
No study has systematically compared the suitability of DNA methylation (DNAme) profiles in non-invasive samples for the detection of breast cancer (BC). We assess non-tumour DNAme in 1,100 cervical, buccal, and blood samples from BC cases and controls and find that cervical samples exhibit the largest nuber of differentially methylated sites, followed by buccal samples. No sites were significant in blood after FDR adjustment. Deriving DNAme-based classifiers for BC detection in each sample type (WID-buccal-, cervical-, or blood-BC), we achieve validation AUCs of 0.75, 0.66, and 0.51, respectively. Buccal and cervical BC-associated DNAme alterations distinguish between BC cases and controls in both surrogate and breast tissue (AUC > 0.88), yet individual sites and the directionality of methylation changes are not identical between these two sample types, and buccal sample DNAme aligns with breast methylation changes more closely. Pending additional validation, these insights may have the potential to improve non-invasive personalized BC prevention.
Similar content being viewed by others

Introduction
Breast cancer (BC) is the most common and second most fatal cancer affecting women, emphasizing the demand for effective detection methods1. Current screening or detection approaches predominantly revolve around imaging techniques, primarily mammography. While mammography screening has been proven to reduce breast cancer mortality2, it can lead to overdiagnosis and not insignificant false positive rates. Therefore, the combination of existing screening and early detection approaches with molecular biomarkers has been named a key priority in a recent consensus statement for breast cancer prevention3.
In addition to polygenic risk scores4 that aim to stratify the population based on heritable risk, various other tools are being proposed for screening and early detection. The majority of these utilize blood-based measurements of circulating cell-free tumor nucleic acids (DNA or RNA) or their modification (e.g., DNA methylation5,6), proteins, and carcinoma antigens (CAs)7. The emphasis on blood for cancer biomarkers is likely based on multiple factors, including but not limited to practicality (blood samples are routinely collected and are also often stored in biobanks, facilitating biomarker development) and the fact that tumors shed material into the bloodstream that can be detected using “liquid biopsies”. However, the use of other biospecimens may also offer specific advantages and provide further insights into systemic effects causing, or caused by, cancer. For instance, in previous work, we identified that DNA methylation (DNAme) in cervical samples that are routinely collected for cervical cancer screening could be leveraged to assess the risk of being diagnosed with BC. The cervical methylation classifier, called Women’s cancer risk identifier—Breast Cancer (WID-BC) achieved an area under the curve (AUC) of 0.81 in a validation set derived from cervical samples of women with breast cancer or healthy age-matched controls8. DNAme is a relatively stable epigenetic modification that plays a crucial role in regulating gene and protein expression without altering the DNA sequence, and can be modified by external exposures. Thus, the epigenome is hypothesized to capture environmental changes, serving as an important link between genes and the environment. In the absence of cancer tissue present in anatomically distant cervical samples, our prior work indicated that the observed DNAme changes may be indicative of systemic lifetime exposure that may drive cancer in one tissue (breast), but can be read out in a non-invasively collected ‘surrogate’ sample (cervix), rendering it suitable for detection and screening.
No study has systematically compared different surrogate tissues in their potential to detect epigenetic field defects associated with BC, although this investigation could yield further insights into whether, in addition to blood and cervical samples, buccal samples could, for instance, be used to detect BC. Buccal samples may represent an underappreciated but promising surrogate sample for DNAme-based cancer detection due to their relative ease of collection and high suitability for self-sampling that has already been employed for other purposes, such as diagnosing celiac disease in children9 and detecting COVID-19 viral proteins10. Aligned with the fact that both buccal and breast epithelial cells originate from the ectoderm, previous work indicated that buccal samples shared higher variability at breast-variable sites than peripheral blood cells, suggesting that buccal sample DNAme might be a better proxy indicative of breast field cancerization11. The level of physical activity in women has previously been associated with BC risk12, and previous research has provided preliminary evidence of a relationship between exercise behavior, cardiovascular fitness, and methylation on CpG sites linked to breast cancer in buccal cells. These effects seemed specific to genes associated with breast cancer, rather than affecting the general level of methylation of CpG sites across the methylome13. This early evidence indicates that the epigenome of buccal cells might at least in part mirror the risk of being diagnosed with breast cancer.
To further investigate potential epigenetic alterations associated with breast cancer in three easy-to-access surrogate tissues, here we conduct a comparative analysis of the suitability of cervical, buccal, and blood samples (Fig. 1a). We initially perform an epigenome-wide association study (EWAS) for breast cancer in cervical, buccal, and blood samples from women recently diagnosed with BC and cancer-free age-matched controls. We next investigate whether differentially methylated positions (or regions) we identified in cervical, buccal, and blood samples are shared or whether the different surrogate samples have distinct features associated with breast cancer. We, moreover, derive and validate an epigenetic classifier for BC detection in each tissue, comparing their diagnostic accuracy in validation sets and breast tissue itself to assess their potential to reflect epigenetic changes in a different tissue, and evaluate their association with genetic risk.

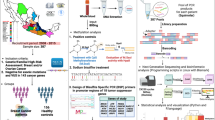
a Overview of the study and datasets. b eFORGE results of significant enrichments of tissue or cell type-specific signals in the subset differentially methylated positions (DMPs) with hypomethylation in buccal samples and hypermethylation in cervical samples. c Mean methylation of a subset of DMPs with hypomethylation in buccal samples and hypermethylation in cervical samples in breast tissue (GSE22584521) by disease status (blue = normal [n = 231], orange = normal-adjacent [n = 140], red = cancer tissue [n = 224]). d Mean methylation of a subset of DMPs with hypermethylation in buccal samples and hypermethylation in cervical samples in breast tissue (GSE22584521). e Mean delta-beta values between BC cases and controls for overlapping DMPs within the buccal and cervical discovery sets, prior to implementing multiple testing correction. n numbers to derive differentially methylated sites in buccal and cervical samples are as follows: buccal sample (cases): n = 200; buccal sample (controls): n = 202; cervical sample (cases): n = 199; cervical sample (controls): n = 199. Annotated is the corresponding odds ratio [95% confidence interval]. f eFORGE results of significant enrichments of tissue or cell type-specific signals in the subset DMPs with hypomethylation in buccal samples and hypomethylation in cervical samples. g Mean methylation of a subset of DMPs with hypomethylation in buccal samples and hypomethylation in cervical samples in breast tissue (GSE22584521). h eFORGE results of significant enrichments of tissue or cell type-specific signals in the subset DMPs with hypermethylation in buccal samples and hypomethylation in cervical samples. i Mean methylation of a subset of DMPs with hypermethylation in buccal samples and hypomethylation in cervical samples in breast tissue (GSE22584521). p values in c, d, g, i are derived from two-sided Wilcoxon tests. No multiple testing correction was applied. Box plots correspond to standard Tukey representation, with boxes indicating mean and interquartile range, and lines indicating smallest and largest values within 1.5 times of the 25th and 75th percentile, respectively. Individual data points are overlaid. BC breast cancer, WID women’s cancer risk identification, ∆ β difference in methylation β value (e.g., between cases and controls). Source data are provided as a Source Data file.
Results
DNAme changes associated with breast cancer in buccal, cervical, and blood samples
We initially performed an epigenome-wide analysis of DNAme changes associated with breast cancer in the three surrogate tissues (buccal, cervical, and blood; Fig. 1a), looking at both individually differentially methylated positions (DMPs) and differentially methylated regions (DMRs), accounting for age and cellular heterogeneity (immune or neutrophil proportion in buccal/cervical and blood samples, respectively). A full overview of sample composition in each dataset is shown in Supplementary Fig. 1, and principal component plots of the top 20% variable CpGs and their association with immune cell proportion is shown in Supplementary Fig. 2d, j, g, revealing a strong dependence of the most variable methylation sites on cellular composition. Overall, sample type and composition were the strongest drivers of variability in the first principal components, as expected (Supplementary Fig. 2a–c). The epigenome-wide study within each surrogate sample type revealed 79,030, 143,986, and 52,147 DMPs significantly associated with breast cancer in buccal, cervical, and blood samples (p < 0.05) of which 585, 21,614, and 0 remained significant after Benjamini–Hochberg correction, respectively (Supplementary Fig. 3; full EWAS results are provided in Supplementary Data 1–3). An overview of the genetic location and region of these sites is shown in Supplementary Fig. 4 and revealed an approximately similar distribution of genomic contexts for differentially methylated sites in buccal and cervical samples, with the majority of CpGs located in Open Sea regions.
For buccal and cervical samples, for whom matched samples were available (Table 1), we assessed overlaps of differentially methylated positions that were shared by both tissues at p < 0.05 and their directionality (Fig. 1e). We found that the directionality was not always shared. Specifically, we observed an underenrichment of shared hypermethylated sites (OR = 0.48 [95% CI: 0.43–0.54]). We also observed that a larger-than-expected proportion of shared significant sites was hypomethylated in cervical cells but hypermethylated in buccal cells (OR = 2.55 [2.50–2.59]). To improve our understanding of the potential functionalities of the shared sites in each quadrant of Fig. 1e (common or opposing directionalities), we applied the eFORGE tool, which aids in the interpretation of groups of CpGs via regulatory elements in specific tissues, specifically DNase I hypersensitive sites (DHS)14 (Fig. 1b, f, h). We also queried the mean methylation of the CpGs in each quadrant in normal breast tissue, normal breast tissue adjacent to a cancer, or breast tumor tissue (Fig. 1c, d, g, i). Specifically, we found that CpGs hypomethylated in buccal cells of breast cancer cases but hypermethylated in cervical sites were enriched for DHS in mammary epithelial cells (Fig. 1b) and exhibited hypomethylation in tumor tissue compared to both normal or normal-adjacent tissue (Fig. 1c). No DHS enrichment was found for sites that shared hypermethylation in both cervical and buccal samples, although these sites exhibited significant hypermethylation in breast tumor tissue compared to normal breast (Fig. 1d). Sites that were hypomethylated in both buccal and cervical samples were enriched for stem cell DHS (Fig. 1f) whereas sites with buccal hypermethylation and cervical hypomethylation were enriched for fetal tissue DHS (Fig. 1h). Interestingly, breast tissue methylation generally followed methylation changes in buccal cells, both when comparing normal tissue versus tumor but also normal tissue versus healthy tissue adjacent to a cancer (“normal-adjacent”; Fig. 1c, d, g, i). As methylation differences could be driven by cellular heterogeneity, we also investigated inferred delta-beta in epithelial or immune cell fractions of buccal and cervical cells, respectively (Supplementary Fig. 5a, b), which revealed that epithelial cell changes (Supplementary Fig. 5a) followed the overall trend of Fig. 1e - revealing a lack of correlation - whereas immune cell changes correlated moderately across the two tissues (Supplementary Fig. 5b; R = 0.4, p < 2.2e−16 for delta-beta immune versus R = −0.047, p = 7 × 1e−10 for delta-beta epithelial).
We identified differentially methylated regions (DMRs), which consist of several CpGs in genomic proximity, using the DMRcate method15. This method fits DNAme measurements spatially across the genome, ranking the most differentially methylated regions based on tunable kernel smoothing of the differential methylation (DM) signal. Again, cervical samples exhibited more pronounced changes than buccal samples, with 1992 and 145 DMRs significantly associated with breast cancer in cervical and buccal samples, respectively. The majority of alterations in cervical samples were associated with a loss of methylation (hypomethylation). We investigated the directionality of differential methylation across regions, and again assessed potential overlaps between buccal and cervical samples (Supplementary Fig. 5c). Twenty-four DMRs were shared by cervical and buccal samples (Supplementary Data 4), e.g., spanning genes LTBP4, RP11-551L14.1, NCK1, and CCDC88C. These included both sites with the same directionality (e.g., RP11-551L14.1, LTBP4) and opposing directionality of differential methylation (e.g., NCK1, CCDC88C) between BC cases and controls, with some examples shown in Supplementary Fig. 5d.
Subsequently, to aid in the interpretation of significantly enriched sites (CpGs) in each surrogate sample, we conducted gene ontology enrichment analysis on the loci that remained significant after Benjamini–Hochberg correction (hence only in buccal and cervical samples). The top 10 significant gene ontology enrichments of biological processes, molecular functions, and cellular functions in buccal and cervical samples are illustrated in Supplementary Figs. 6, 7, respectively. Notably, several enriched biological and molecular processes in buccal and cervical samples were associated with GTPase activity. GTPases are known to regulate cytoskeletal dynamics, which play a crucial role in oncogenic processes such as cellular migration and cell cycle progression16. Aberrant GTPase activity has been established across all subtypes of breast cancer17. In addition, buccal samples exhibited enrichment in Wnt signaling (Supplementary Fig. 6). Dysregulated Wnt signaling has been linked to BC, and expression of Wnt genes has been linked to BC cancer aggressiveness18. Additionally, cervical samples also showed an enrichment for CpGs located in genes associated with developmental pathways, such as gland development (Supplementary Fig. 7).
Development and validation of DNA methylation-based classifiers for breast cancer based on buccal, cervical, and blood samples
We next aimed to compare the performance of buccal, cervical, and blood samples for detecting breast cancer based on DNAme. We trained classifiers using ridge or lasso regression, applying several approaches for feature (pre)selection, including ranking the top 30,000 sites by p value in the original EWAS, by top epithelial or immune delta-beta values as previously described in ref. 8, or inputting the entire beta methylation value matrix (the latter using lasso only). In the internal validation set, we evaluated model fit parameters, including internal validation and out-of-bag estimates of the area under the curve and the calibration intercept and slope. The final models for each sample type were selected based on the number of input CpGs with the optimum number of slope (closest to 1), intercept (closest to 0), and highest out-of-bag AUC estimates; for all surrogate samples, the optimum fit was found to be for ridge regression, inputting the top 30,000 CpGs ranked by p value (lowest to highest) (Supplementary Fig. 8). The final model for each surrogate sample was subsequently trained on the entire discovery set (training and internal validation), and index coefficients and values in the discovery set were saved for computation of the index and scaling in validation datasets.
Following hyperparameter optimization, the final WID indices for each sample type were validated on validation sets (Fig. 2; population characteristics shown in Table 2). Notably, as for the discovery set, within the validation set, matched buccal and cervical samples were available from the same individuals. The WID-buccal-BC index exhibited a slight to moderate dependence on immune cell proportion overall (Fig. 2a), and performance was slightly higher in samples with higher than median immune cell proportion (Fig. 2d and Supplementary Fig. 9a). There was no significant correlation with age (Fig. 2b) or menopausal status, although the performance appeared slightly higher in pre- compared to post-menopausal women (Supplementary Figs. 9b, 10). The WID-buccal-BC index performed significantly better in women with an early age at menarche (defined by median age at menarche (13 years) in the validation set; AUC = 0.81 for early age versus AUC = 0.59 for high age at menarche, respectively; Supplementary Fig. 9c; DeLong’s test for two ROC curves: p = 0.01446). Overall, the WID-buccal-BC index was significantly higher in cases than controls (Fig. 2c, p = 2.179e-09) and after accounting for age and immune cell differences achieved an area under the curve of 0.75 (95% CI: 0.68–0.82) (Fig. 2d). Interestingly the WID-cervical-BC index also exhibited a slightly better performance in samples with higher than median immune cell proportion and post-menopausal women compared to pre-menopausal women (Fig. 2e, d and Supplementary Fig. 9d, e), but in contrast to the WID-buccal-BC, it performed better in women with later age at menarche (>13) than women with earlier age at menarche (Supplementary Fig. 9f; AUCs of 0.72 and 0.63, respectively). The WID-cervical-BC was significantly elevated in cases compared to controls (p = 1.92e-04) and achieved an area under the curve of 0.66 (95% CI: 0.58–0.74) (Fig. 2g, h), lower than the buccal index in the same group of women (Fig. 2d; DeLong’s test for two ROC curves: p = 0.06). The blood validation set comprised DNAme data derived from peripheral blood mononuclear cells (PBMCs). This dataset was obtained from a previous report of BC cases and controls with limited phenotypic information (e.g., no information on age or menopausal status)19. The WID-blood-BC exhibited a strong overall dependence on the proportion of neutrophils, although the discriminative potential was not influenced by neutrophil proportion (Supplementary Fig. 9g), and overall exhibited limited discriminative performance AUC: 0.51, 95% CI: 0.38–0.64) (Fig. 2i–k) relative to WID-buccal-BC and WID-cervical-BC.

a Scatter plot of the WID-buccal-BC index and immune cell proportion or b subject age by disease status (blue = control [n = 93], red = BC case [n = 94]) in the validation set. c Box plot of WID-buccal-BC index residuals after accounting for age and immune cell proportion in the validation set. d ROC curves of the WID-buccal-BC index residuals (after adjusting for age and immune cell proportion) overall and stratifying samples by median immune cell proportion in the validation set. e Scatter plot of the WID-cervical index and immune cell proportion or f subject age by disease status (blue = control [n = 93], red = BC case [n = 91]) in the validation set. g Box plot of the WID-cervical-BC index residuals after accounting for age and immune cell proportion in the validation set. h ROC curves of the WID-cervical-BC index residuals (after adjusting for age and immune cell proportion) overall and stratifying samples by median immune cell proportion in the validation set. i Scatter plot of the WID-blood-BC index with and estimated neutrophil proportion by disease status (blue = control [n = 30], red = BC case [n = 50]), in GSE23703619. j Box plot of the WID-blood-BC index residuals, adjusted for neutrophil proportion in GSE23703619; p = 0.8854. k ROC curves of the WID-blood-BC index residuals, adjusted for neutrophil proportion, overall, and stratifying samples by median neutrophil proportion in GSE23703619. p values in c, g, j are derived from a two-sided Wilcoxon test. Shaded error regions in a, b, e, f, i correspond to 95% confidence intervals for predictions from linear models. Box plots correspond to standard Tukey representation, with boxes indicating mean and interquartile range, and lines indicating smallest and largest values within 1.5 times of the 25th and 75th percentile, respectively. Individual data points are overlaid. No corrections for multiple testing were carried out. BC breast cancer, WID women’s cancer risk identification, AUC area under the (receiver operating characteristic) curve. Source data are provided as a Source Data file.
Association of the classifier with epidemiological and genetic characteristics
To evaluate factors associated with the WID indices in each tissue, we assessed its relationship with various epidemiological, genetic, and sample characteristics (Fig. 3 and Supplementary Fig. 10). As we previously identified a dependence of the index on immune cell proportion and to balance for differences in age, we evaluated residuals after fitting models for age and immune cell proportion in controls (“adjusted indices”, see methods). The WID-buccal-BC index was not significantly different in T1 versus T2 or T3 stages and distinguished across cancer cases from controls regardless of grade, ER, PR, or HER2 status (Fig. 3a–e). HER2 positive (HER2+) cases appeared to exhibit slightly higher values than HER2 negative (HER2-) cancer cases, in line with HER2+ typically corresponding to more aggressive cancer subtypes, but this did not reach significance. Surprisingly, the WID-buccal-BC index did not show any association with a polygenic risk score (PRS313) for breast cancer4 obtained from the same women (Fig. 3f). Likewise, women with increased risk for breast cancer due to BRCA1/2 mutations did not exhibit elevated WID-buccal-BC values (Fig. 3g, h). Similar findings were obtained for cervical samples (Fig. 3i–p), where the index was not strongly dependent on differences in cancer characteristics or genetic risk factors. The WID-buccal-BC, but not WID-cervical-BC, was associated with nodal status, but this association was not linear with increasing nodal stage (Supplementary Fig. 10f, l), requiring further investigation. The WID-blood-BC index was slightly elevated in BRCA1 but not BRCA2 mutation carriers compared to controls, but this did not reach significance (Fig. 3q, r).

All indices shown in this figure represent residuals after adjusting for age and immune cell proportion (WID-buccal- and -cervical-BC) or age and neutrophil proportion (WID-blood-BC). a Box plot of the WID-buccal-BC index grouped by cancer stage, b grade, c estrogen receptor (ER) status, d progesterone receptor (PR) status, and e HER2 status, split by case status (blue = control [n = 93], red = BC case [n = 94]). f Scatter plot of the WID-buccal-BC index and the PRS313 polygenic risk score. g Box plot of the WID-buccal-BC index in buccal samples from cancer-free BRCA1 or BRCA2 mutation carriers and controls. h ROC curves of the WID-buccal-BC index comparing cancer-free BRCA1 or BRCA2 mutation carriers to controls. i Box plot of the WID-cervical-BC grouped cancer stage, j cancer grade, k estrogen receptor (ER) status, l progesterone receptor (PR) status, and m HER2 status age by disease status (blue = control [n = 93], red = BC case [n = 91]). n Scatter plot of the WID-cervical-BC index and the PRS313 polygenic risk score. o Box plot of the WID-cervical-BC index in cervical samples from cancer-free BRCA1 or BRCA2 mutation carriers and controls. p ROC curves of the WID-cervical-BC index comparing cancer-free BRCA1 or BRCA2 mutation carriers to controls. q Box plot of the WID-blood-BC index in blood samples from cancer-free BRCA1 or BRCA2 mutation carriers (green, n = 58 for BRCA1 and n = 53 for BRCA2), and controls (blue, n = 114). r ROC curves of the WID-blood-BC index comparing cancer-free BRCA1 or BRCA2 mutation carriers to controls. p values in a–e, g, i–m, o, q are derived from two-sided Wilcoxon tests. Shaded error regions in f and n correspond to 95% confidence intervals for predictions from linear models. Box plots correspond to standard Tukey representation, with boxes indicating mean and interquartile range, and lines indicating smallest and largest values within 1.5 times of the 25th and 75th percentile, respectively. Individual data points are overlaid. No corrections for multiple tests were carried out. BC breast cancer, WID women’s cancer risk identification, AUC area under the (receiver operating characteristic) curve. T1, T2, T3, cancer stages; I, II, III, cancer grades; ER estrogen receptor, PR progesterone receptor. Source data are provided as a Source Data file.
Application of buccal and cervical indices in blood samples
We were intrigued by the finding that buccal and cervical indices consistently tended to exhibit higher AUCs in samples with higher than median immune cell proportion, which indicated that information about BC status was contained in immune cells (Fig. 2d, h, respectively), yet the blood-specific index performed poorly (Fig. 2k). We applied the buccal and cervical indices to blood samples and found that AUCs were low but significantly higher than 0.5 in the FORECEE study, but not in the GSE237036 validation set (Supplementary Fig. 11).
Validation in breast cancer tissue
Lastly, we were curious as to whether indices trained in the three surrogate tissues (buccal, cervical, blood) could distinguish between normal breast tissue from cancer-free women and normal breast tissue from women with breast cancer (i.e., normal-adjacent breast tissue) and actual breast cancer tissue, and might perhaps be indicative of factors driving cancer progression in the “tissue at risk”. We identified three relevant datasets using the EPIC or 450k array and evaluated values of the WID-buccal-, cervical-, and blood-BC indices. Across all three datasets, the WID-buccal-BC was consistently higher in breast cancer tissue compared to normal control tissue (Fig. 4a–f) and exhibited AUCs of 0.94, 0.9, and 0.88 respectively. Conversely, the WID-cervical-BC index exhibited significantly lower values in cancer tissue compared to control tissue (i.e., an inverted behavior compared to performance in cervical samples, where cancer cases would exhibit higher values), resulting in AUCs of 0.13, 0.04, and 0.12. The behavior of the WID-blood-BC was not consistent across datasets, although breast cancer tissue tended to exhibit significantly lower values than control tissue (Fig. 4a–d), also exhibiting an inverted directionality compared to the surrogate tissue. Normal tissue adjacent to a breast cancer occasionally exhibited significantly higher (or lower) values compared to controls, but this was not consistent across all datasets (Fig. 4a, e). Lastly, normal breast tissue from BRCA1/2 mutation carriers exhibited a significantly lower WID-buccal-BC than control tissue, and significantly higher WID-cervical-BC, indicating it exhibited the opposing directionality compared to breast cancer (Fig. 4a), although sample numbers were small.

a Box plot of WID indices in the tissue at risk set. b ROC curves of the WID indices in the tissue at risk set, normal-adjacent breast tissue (n = 14) versus triple-negative breast cancer tissue (n = 14). c Box plot of the WID indices in the TCGA set. d ROC curves of the WID indices in the TCGA set (n = 97 controls, n = 792 cancer cases). e Assessment of the WID indices in GSE225845. f ROC curves of the WID indices in GSE22584521, normal-adjacent breast tissue versus tumor tissue. p values in a, c, e derived from two-sided Wilcoxon tests; paired Wilcoxon tests were used in (c). Box plots correspond to standard Tukey representation, with boxes indicating mean and interquartile range, and lines indicating smallest and largest values within 1.5 times of the 25th and 75th percentile, respectively. Individual data points are overlaid. No corrections for multiple testing were carried out. BC breast cancer, WID women’s cancer risk identification, AUC area under the (receiver operating characteristic) curve, TCGA the cancer genome atlas. Source data are provided as a Source Data file.
Discussion
Studies on the identification and optimization of biomarkers for earlier cancer detection and stratification of risk of being diagnosed with cancer are a key area of research, and currently often focus on blood samples due to their routine implementation in clinical workflows and availability in many biobanks. Approaches utilizing cell-free DNA methylation for BC detection have so far shown limited success: for instance, in the recent PATHFINDER study, no primary BC was detected20. Our previous work indicated that samples other than blood could provide valuable information for the detection of women’s cancers, importantly cervical samples that contain (hormone-sensitive) epithelial cells with the potential to mirror epigenetic changes in breast tissue8. Additionally, our previous work comparing blood, cervical, and buccal samples as three different types of ‘surrogate’ samples suggested that these tissues share varying levels of information with breast tissue11. Here, we systematically investigated DNAme changes in three surrogate sample types derived from breast cancer cases and controls, and developed and compared DNAm-based classifiers for breast cancer detection. Importantly, in contrast to other approaches, the current study did not investigate cell-free DNA derived from tumor material but intended to identify DNAme changes in surrogate samples (containing limited or no tumor material) that mirror changes in the “at-risk” tissue purportedly due to cumulative clinical/lifetime effects.
To allow for comparability of findings across different sample types, we utilized similar sample numbers across discovery/training sets and, where available, leveraged datasets that consisted of matched surrogate samples from the same individual (i.e., buccal and cervical samples from the same individual). Regrettably, the blood discovery dataset was slightly smaller, and no matched DNAme data for blood samples from the same individuals for buccal and cervical samples were available. However, we were able to leverage a publicly available blood methylation dataset (GSE237036)19 to perform validation of our index. Although fewer CpGs reached formal significance in an epigenome-wide discovery following Benjamini–Hochberg correction in buccal than cervical samples, our results indicated that given similar training sizes, buccal samples exhibited a higher area under the curve in validation (0.75 [95% CI: 0.68–0.82]) than cervical samples (0.66 [95% CI: 0.58–0.74]) (Fig. 2d, h). No CpGs were significantly associated with breast cancer in DNAme from blood samples, and although during index training, out-of-bag estimates of the AUC of up to 0.9 were achieved, the WID-blood-BC was not able to distinguish breast cancer cases from controls in the validation dataset (AUC: 0.46 [95% CI: 0.33–0.6]) (Fig. 2k). It is worth noting that the discovery set was slightly smaller for blood samples compared with buccal and cervical samples (n = 310 for blood, n = 402 for buccal/cervical samples), and the validation set was derived from peripheral mononuclear blood cells (PBMCs) rather than whole blood, which may also have influenced discovery and performance, and it is possible that larger training sizes could lead to slightly improved performance. For instance, the discriminative power of the cervical-BC classifier reported in the current work, trained on 402 samples, exhibits a much lower AUC than a previously reported index trained on 1198 samples8 (AUC: 0.66 versus 0.81, respectively), not surprisingly indicating that performance is linked to training size. It is worth noting that cervical samples typically show high variability in the proportion of immune cells (Supplementary Fig. 1). The pronounced heterogeneity in cell type composition and other factors influencing methylation in cervical samples, in contrast to the less variable immune cell proportion and possibly more homogeneous buccal samples, could make training a classifier in cervical samples more challenging and may therefore necessitate an even larger training set (as previously described by ref. 8, n = 1198 samples for training) compared to that required for buccal samples. However, the relatively lower performance of blood compared to buccal and cervical samples is in line with our previous report that cervical and buccal samples share more variability with breast cancer tissue11 and, for instance, hormone-sensitive epithelial cells in cervical samples may be able to reflect cumulative exposure such as to progesterone8.
Interestingly, while we previously found that buccal and cervical samples with higher inferred epithelial cell proportions share more information (variability) with breast tissue compared to samples with lower epithelial cell proportions, our current and previous8 analysis indicate that detection of BC may be slightly better in samples with higher than median immune cell content (i.e., lower epithelial contents; Fig. 2d, h). This stands in contrast with the finding that blood samples, consisting entirely of immune cells, have generally lower potential for detection (noting certain limitations of the current study design above; Fig. 2k). This could be for several reasons, including a discrepancy of peripheral (blood) and resident (buccal/cervical) immune cells in their relative DNAme information content with relation to breast cancer, or the association of immune content with other unobserved features that can help to detect BC. Future work should more thoroughly assess the potential of purified epithelial, immune, or mixed sample types to detect cancers to better understand the biology of surrogate sample-based cancer detection and potential risk prediction.
Intriguingly, the buccal classifier was consistently elevated in several breast tissue datasets, generated using both the EPIC and 450 K methylation array platforms, whereas the WID-cervical-BC exhibited a significant reduction, i.e., an inverted directionality. This indicated that although both surrogate tissues reflect changes associated with cancer, they do so in different ways and by focusing on different features. The potential for opposing directionality in changes between the surrogate tissues was already hinted at during the initial assessment of differentially methylated regions: Supplementary Fig. 5c highlighted prominent hypomethylation in cervical, but more balanced methylation changes in buccal samples. Notably, among shared DMRs between buccal and cervical samples, these unexpectedly did not always share the same directionality in buccal and cervical samples (Supplementary Fig. 5c). This was particularly intriguing as buccal and cervical samples for discovery originated from the same individuals (matched samples), and provided further evidence for tissue-dependent epigenomic alterations in cancer.
The surrogate-specific classifiers developed in the current manuscript were not elevated in samples from cancer-free women at increased genetic risk, such as due to a BRCA1/2 mutation, or associated with a previously described polygenic risk score for breast cancer (Fig. 3). This suggested that the current classifiers are not strongly associated with genetic features but instead potentially reflect either cumulative risk due to lifetime (including potential in-utero) risk factors, mirrored in the epigenome, or systemic changes elicited by the presence of cancer. In the absence of large-scale DNAme validation datasets derived from buccal or cervical samples collected at or preceding diagnosis, it is challenging to investigate the cause or temporal relationship of the DNAme alterations with BC diagnosis. Our current findings indicate that the WID-BC-buccal index is not elevated in individuals with genetic risk (BRCA1/2 mutation carriers), but at what time in relation to a BC diagnosis, the indices would become elevated remains unclear and will need to be investigated in the future.
While the purpose of the current study was to compare surrogate tissues and not develop optimized classifiers in any given tissue, our findings moreover suggest that there is considerable potential for buccal DNAme-based detection of BC that should be investigated in future studies with larger and more diverse populations. Buccal sample collection is non-invasive, making it a promising tool for simpler detection or risk stratification for breast cancer with the potential for self-collection of samples. Although the sample size in the validation set was limited and our dataset focused on BC cases with at least one poor prognostic feature, the lack of dependence on stage or grade observed in our analysis (Fig. 3a, b) indicates that such an approach may be suitable for early detection and/or screening using buccal samples. Future optimization could also include the addition of epidemiological variables, or other molecular or imaging features into algorithms. A positive test result could have the consequence of earlier, intensified, and/or more sensitive (i.e., magnetic resonance imaging) breast cancer screening measures, although the clinical utility and impact on stage shifts and mortality will need to be further confirmed in clinical studies.
Our study has several limitations, including those inherent to case-control design, and a modest sample size for epigenome-wide discovery, given the practical constraints, outlined above. Nonetheless, we independently validate our buccal and cervical-BC classifiers with AUCs of 0.75 and 0.66, respectively, and demonstrate that classifiers also validate in breast cancer tissue. Our breast tissue evaluation included datasets generated using both the EPIC array (featuring all sites) and the 450 K methylation array (The Cancer Genome Atlas, TCGA; featuring only a subset of all sites). The lower coverage of the 450 K array represents a limitation, however we observe consistent behavior of the WID-buccal-BC and WID-cervical-BC across three independent breast tissue datasets, providing confidence in the observed findings. Our study also has several notable strengths, including a comparative analysis of three non-invasive samples for breast cancer detection and cross-tissue validation. We describe for that cancer-associated differential methylation derived from the same individuals is tissue-specific, identifying potentially opposing differential methylation in buccal and cervical samples of breast cancer cases compared to controls. Future studies of large-scale collections of buccal or cervical samples from diverse and representative populations will be required to further investigate and optimize non-invasive cancer detectors/risk predictors to serve as reliable biomarkers for breast cancer early detection and/or risk stratification. Pending further validation, WID-buccal- or -cervical-BC indices could be applied for regular screening in at-risk populations, and risk monitoring may be combined with (pharmacological) preventive measures in those deemed high risk to either prevent cancers or capture cancers earlier at the point when they become detectable. Thresholds for ‘high risk’ populations will need to be set in large population-based studies with several years follow-up to identify the proportion at highest risk of cancer with appropriate sensitivity and specificity.
Methods
Ethics statement
The multicentre FORECEE study, during which buccal, blood, and cervical samples were collected, received ethical approval from UK Health Research Authority (REC 14/LO/1633) and all contributing centers, including the NRES Committee London (UK), Ethics Committee of the General University Hospital, Prague (Czech Republic), Comitato Etico degli IRCSS Instituto Europeo di Oncologia e Centro Cardiologico Monzino (Itality), Regionale Komiteer for Medisinsk og Helsefaglig Forskningsetikk (Norway), and Ethikkommission bei der LMU München (Germany). All participants were aged >18 years and provided written informed consent. Each prospective study volunteer was given a Participant Information Sheet, as well as a Consent Form, and the rationale for the study was explained.
Study design and data acquisition
A study outline is shown in Fig. 1a, and sample numbers and population characteristics of our discovery and validation sets are reported in Tables 1, 2. Our study population consisted of a case-control study including women with primary BC with at least one poor prognostic feature (defined as >2 cm cancers and/or lymph-node positive and/or hormone-receptor negative and/or grade 3), and age-matched healthy controls (Table 1). We utilized cervical, buccal, and blood sample DNAme data from BC cases and controls collected as part of the multicentre “FORECEE” study in a case-control setting, involving 15 recruitment sites across Europe as previously described8, and publicly available datasets (GSE237036)19. In the FORECEE study, women with symptoms indicative of BC, subsequently confirmed through diagnosis (referred to as “cases”), were approached during outpatient hospital clinics. Healthy volunteers from the general population (referred to as “controls”) were engaged through outreach campaigns, public engagement initiatives, and participation in cervical screening programs. During the FORECEE sample collection, two sets of samples (discovery and validation set, respectively) were collected. Samples within the validation set were exclusively reserved for the validation of the index, i.e., there was no overlap between individuals in the discovery/training and those in the validation phase. However, within the discovery/training and validation sets, matched buccal and cervical samples were available from the same individuals. This allowed us to assess whether, when the two sample types are directly compared from the same individuals, classifiers derived from buccal or cervical samples exhibit better performance. Blood samples were not from matched individuals.
All cervical, buccal, and blood samples were collected at the point of diagnosis and prior to the initiation of any treatment.
A separate sample collection as part of the FORECEE study involved recruitment of cancer-free BRCA1 and BRCA2 mutation carriers, as well as from healthy age-matched controls (BRCA Mutation Carrier Set: Supplementary Table 1), and matched samples for cervical, buccal, and blood samples were available and have been previously described in ref. 11. For blood validation, we utilized a publicly available dataset of peripheral blood-derived mononuclear cell (PBMC) methylation from BC cases and controls (GSE237036), described previously19. A cross-tissue validation was conducted using several independent breast methylation datasets described below.
Buccal, cervical, and blood sample collection
Buccal cells were collected using two Copan 4N6FLOQ Buccal Swabs (Copan Medical Diagnostics, cat #4504 C) by firmly brushing the swab head 5–6 times against the buccal mucosa of each cheek. The swabs were re-capped and left to dry out at room temperature within the sampling tube, which contains a drying desiccant. Cervical and blood sample collections have been described previously in refs. 8,11. Briefly, 2.5 ml of venous whole blood was collected in PAX gene blood DNA tubes (BD Biosciences #761165) and stored locally at 4 ˚C. Cervical sample collection was conducted by trained staff using the ThinPrep system (Hologic Inc, cat #70098-002). A description of blood collected as part of GSE237036 (Blood validation dataset) has been published previously19.
Breast DNA methylation datasets for cross-tissue validation
The current study uses three breast tissue DNA methylation datasets for cross-tissue validation: the “tissue at risk set” (EGAS00001005055, n = 56, previously described in ref. 8, consisting of cancer tissue, matched normal-adjacent tissue, normal breast tissue from cancer-free women without a BRCA1/2 mutation and normal breast tissue from women with a BRCA1/2 mutation; data from The Cancer Genome Atlas (TCGA) (n = 889, IlluminaMethylation450k), consisting of matched cancer tissue and normal-adjacent tissue; and dataset GSE22584521 (n = 595, IlluminaMethylationEPIC), consisting of cancer tissue and matched normal-adjacent samples and data from health controls (reduction mammoplasty) from the NCI-Maryland Breast Cancer Cohort. Details of breast methylation datasets are described in Supplementary Table 2.
DNA methylation array profiling
Cervical and blood sample preprocessing have been described in previous publications8,11. Buccal sample DNAme was specifically generated for the purpose of this study and followed previous procedures. Briefly, buccal sample DNA was normalized to 25 ng/µL, and 500 ng total DNA was bisulfite modified using the EZ-96 DNA Methylation-Lightning kit (Zymo Research Corp, cat #D5047) on the Hamilton Star Liquid handling platform. Eight microliters of modified DNA was subjected to methylation analysis on the Illumina InfiniumMethylation EPIC BeadChip (Illumina, CA, USA) at UCL Genomics according to the manufacturer’s standard protocol. Buccal discovery and validation sets were processed on different occasions (discovery set: January 2019; validation set: March 2021). For additional data, all data from GEO (GSE237036 and GSE225845) were obtained via the GEOquery package, whereas TCGA data were accessed using TCGAbiolinks.
DNA methylation data preprocessing and analysis
With the exception of data from TCGA, all methylation microarray data was processed through the same standardized pipeline that has previously been described in refs. 8,11, packaged as eutopsQC (https://github.com/chiaraherzog/eutopsQC). In brief, raw.idat files are loaded using the R package minfi (current version 1.43.1), samples with median methylated or unmethylated intensity below 9.5 or more than 10% failed probes (detection p value >0.01) are removed, and single sample background intensity- and dye bias correction is carried out using the ssNOOB function (minfi). Beta Mixture Quantile Normalization (BMIQ) is applied to correct for probe type bias (ChAMP, version 2.30.0), and beta values from failed probes are imputed using the impute.knn function function (impute, version 1.72.3). Furthermore, non-CpG probes (n = 2932), SNP-related probes as identified by ref. 22 (n = 82,108), and any probes that map to the Y-chromosome are removed from the dataset. As a result, ~10% of the probes on the EPIC array are removed as part of the quality control (QC) process by default. Beta distributions for each sample are visualized and inspected to identify any samples that exhibit obvious abnormalities. Additionally, principal component analysis (PCA) plots are examined to investigate potential associations with technical or biological factors which emphasized that the major drivers of variability were sample type and composition, and indicated no obvious batch effects (Supplementary Fig. 2). Cell type proportions from processed beta matrices were inferred using the epithelial, fibroblast and immune cell reference dataset in EpiDISH version 2.14.1 (ref.m = centEpiFibIC.m, method = “RPC”). Immune cell subtypes were inferred using the hierarchical EpiDISH function (ref2.m = centBloodSub.m, maxit = 500, h.CT.idx = 3, method = “RPC”).
Epigenome-wide association study (EWAS)
Previous studies have highlighted the importance of accounting for cellular heterogeneity in epigenome-wide studies, and the merit of inferring methylation differences in ‘pure’ immune or epithelial cells of a given sample8,23. We conducted our epigenome-wide association study with breast cancer adjusting for age and the major variable cell type in each tissue (immune cell proportion (ic) in cervical and buccal samples; neutrophil proportion in blood samples) by including them as covariates in linear models. Additionally, we inferred differential methylation, i.e., delta-beta, in pure epithelial cell samples (ic = 0) or pure immune cell samples (ic = 1) by fitting linear regression models separately for cases and controls, and computing the differences of intercepts at ic = 0 and ic = 1, as previously described8,23. To ensure compatibility with future DNAme studies, we restricted our further analysis to probes shared between IlluminaMethylationEPIC version 1 (v1) and the more recent version 2 (v2). Differentially methylated positions (DMPs) were defined as significant sites after false discovery rate (FDR) correction using the Benjamini–Hochberg correction method, with adjusted p value <0.05.
Differentially methylated regions (DMR) were identified using the DMRcate method15 (R package, version 2.14.1), accounting for age and cell heterogeneity [model.matrix(~type + age + ic), where ic is the immune cell proportion in buccal or cervical samples and neutrophil proportion in blood samples]. Briefly, we set the parameter types to use the EPIC array annotation, perform “differential” methylation analysis, and kept default parameters for lambda. No significant DMRs were found for blood samples, even when increasing the false FDR threshold up to 0.2.
Comparison of directionality in buccal and cervical samples and overrepresentation analysis
Differentially methylated CpGs associated with breast cancer at p < 0.05 in buccal and cervical samples were visualized according to delta-beta in the respective sample, either overall or inferred epithelial- or immune-specific differential methylation. Hypo- or hypermethylated CpGs were defined as a delta-beta of below or above 0, respectively. Subsequently, four quadrants were defined: hypomethylated in both, hypermethylated in both, hypomethylated in buccal and hypermethylated in cervical, or hypermethylated in buccal and hypomethylated in cervical. Overrepresentation analysis of shared or opposing hyper- and hypomethylated CpGs was conducted via cross-tabulation of expected and observed overlaps, with the assumption that at random distribution, each quadrant would contain 25% of the total shared significant sites. The odds ratio was computed as follows: [p/(1-p)]/[p0/(1-p0)], where p is the number of CpGs in the quadrant divided by the total number of shared significantly in CpGs, and p0 is 0.25.
eFORGE and gene ontology analysis
For the interpretation of groups of CpGs via regulatory elements (DNase I hypersensitive sites), we used the online implementation of eFORGE 2.014 (https://eforge.altiusinstitute.org/). Where more than 1000 sites were present in one quadrant, the top 1000 sites were selected and analyzed using eFORGE. Gene ontology analysis was conducted only on sites significant after multiple testing correction using the Benjamini–Hochberg method (except for blood where no sites remained after multiple testing correction, and all sites with p < 0.05 were used). Genes within the DMPs were extracted and analyzed using the clusterProfiler R package24, with the background list representing all genes represented on the Illumina Methylation EPIC array. Gene sets with a q value of <0.05 were considered significant.
Classifier training
To train classifiers for breast cancer, we used the R package glmnet (version 4.1.8) with parameter values alpha = 0 (ridge penalty), alpha = 0.5 (elastic net), and alpha = 1 (lasso penalty) with binomial response type, as previously described in ref. 8. Two-thirds of the discovery set (Table 1) were used as training data (buccal: n = 267; ncases = 134, ncontrols = 133. Cervical: n = 263; ncases = 133, ncontrols = 130. Blood: n = 208; ncases = 72, ncontrols = 136). For each tissue type, four ranked lists of CpGs were generated and selected as inputs for the classifier. The ranking of CpGs was determined by delta-beta estimates in epithelial cells (1) and immune cells (2), as well as their combination, each time taking the CpG with the largest epithelial delta-beta, followed by the CpG with the largest immune delta-beta, followed by the next largest epithelial delta-beta and so forth (any duplicates were removed) (3). Finally, a ranked list based on p values, regardless of cell type, was used (4). Tenfold cross-validation was used inside the training set by the cv.glmnet function in order to determine the optimal value of the regularization parameter lambda. Classifier performance was evaluated on an internal validation dataset using the AUC metric versus n, the number of ranked candidate CpGs used as inputs during training. The optimal classifier was selected based on the highest AUCs obtained in the internal validation set (buccal: n = 135; ncases = 66, ncontrols = 69; cervical: n = 135; ncases = 66, ncontrols = 69; blood: n = 102; ncases = 30, ncontrols = 72); part of the discovery set, Table 1), out-of-bag estimates of the AUC as well as calibration slope and intercept obtained from the rms R package (https://hbiostat.org/r/rms/). Training AUC values are shown in Supplementary Fig. 8. Subsequently, the training and internal validation datasets were combined and the classifier was retrained using the entire discovery dataset with alpha and lambda fixed to their optimal values, using the optimal number of ranked input CpGs. In the final index, the top n CpGs are represented as β1,…,βn and the regression coefficients from the trained classifier as w1,…,wn. The WID-BC-index in each tissue is calculated as \({\sum}_{i=1}^n ({w_i}{\beta_i} - \mu)/{\sigma}\), where μ and σ refer to the mean and standard deviation of \({\sum}_{i=1}^n \ {w_i}{\beta_i} \) in the respective training dataset, ensuring that the index is normalized to have a zero mean and unit standard deviation within the training dataset. To ensure comparability across different age ranges and cellular compositions, indices are adjusted after fitting linear models to age (and immune cell proportion in buccal and cervical samples, and the neutrophil proportion in blood samples) in control samples (i.e., adjusted indices are residuals).
Statistical analyses
Statistical analysis was carried out in R version 4.2.3. Areas under the curve of the receiver operating characteristic (AUROCs) and corresponding 95% confidence intervals were calculated using the pROC R package 1.18.4. Code to reproduce the analysis and figures is provided under https://www.github.com/eutops/systems.BC25.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The TCGA used in this study (TCGA-BRCA project [https://portal.gdc.cancer.gov/projects/TCGA-BRCA] are publicly available from the TCGA data portal. The validation set for blood used in this study is available from NCBI Gene Expression Omnibus (GEO) under the accession code GSE23703619. A publicly available breast DNA methylation dataset used in this study is publicly available from NCBI Gene Expression Omnibus (GEO) under the accession number GSE22584521. Raw DNAme and SNP data generated in this study have been deposited in the European Genome-phenome Archive (EGA) database under the study accession codes EGAS00001005055 (breast cancer cervical and buccal discovery methylation data), EGAS00001005070 (breast tissue methylation), EGAS00001005626 (matched methylation data from cervical, buccal, and blood samples from controls and BRCA1/2 mutation carriers), and EGAS00001007658 (buccal validation set methylation data). These data are protected and are not openly available due to data privacy laws. Access can be requested via the EGA portal and raw data may be shared with researchers within EU GDPR-compliant countries while summary statistics may be provided to researchers in other countries. We aim to process requests within 6–8 weeks, and once approved, data will be released within 2–4 weeks and available for the duration of the project, with an annual review. The remaining data were available within the Article, Supplementary Information, or Source Data file. Source data are provided with this paper.
Code availability
Code to reproduce the analysis and figures is provided under https://www.github.com/eutops/systems.BC, and on Zenodo under https://doi.org/10.5281/zenodo.1388525025.
References
Sun, Y.-S. et al. Risk factors and preventions of breast cancer. Int. J. Biol. Sci. 13, 1387–1397 (2017).
Clift, A. K. et al. The current status of risk-stratified breast screening. Br. J. Cancer 126, 533–550 (2022).
Pashayan, N. et al. Personalized early detection and prevention of breast cancer: ENVISION consensus statement. Nat. Rev. Clin. Oncol. 17, 687–705 (2020).
Mavaddat, N. et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet. 104, 21–34 (2019).
Moss, J. et al. Circulating breast-derived DNA allows universal detection and monitoring of localized breast cancer. Ann. Oncol. 31, 395–403 (2020).
Cheng, N. et al. Pre-diagnosis plasma cell-free DNA methylome profiling up to seven years prior to clinical detection reveals early signatures of breast cancer. Preprint at https://doi.org/10.1101/2023.01.30.23285027 (2023).
Li, J. et al. Non-invasive biomarkers for early detection of breast cancer. Cancers 12, 2767 (2020).
Barrett, J. E. et al. The WID-BC-index identifies women with primary poor prognostic breast cancer based on DNA methylation in cervical samples. Nat. Commun. 13, 449 (2022).
Adriaanse, M. P. M. et al. Human leukocyte antigen typing using buccal swabs as accurate and non-invasive substitute for venipuncture in children at risk for celiac disease. J. Gastroenterol. Hepatol. 31, 1711–1716 (2016).
Kalil, M. N. A. et al. Performance validation of COVID-19 self-conduct buccal and nasal swabs RTK-antigen diagnostic kit. Diagnostics 11, 2245 (2021).
Herzog, C. et al. DNA methylation at quantitative trait loci (mQTLs) varies with cell type and nonheritable factors and may improve breast cancer risk assessment. Npj Precis. Oncol. 7, 1–10 (2023).
Guo, W., Fensom, G. K., Reeves, G. K. & Key, T. J. Physical activity and breast cancer risk: results from the UK Biobank prospective cohort. Br. J. Cancer 122, 726–732 (2020).
Bryan, A. D., Magnan, R. E., Hooper, A. E. C., Harlaar, N. & Hutchison, K. E. Physical activity and differential methylation of breast cancer genes assayed from saliva: a preliminary investigation. Ann. Behav. Med. Publ. Soc. Behav. Med. 45, 89–98 (2013).
Breeze, C. E. et al. eFORGE v2.0: updated analysis of cell type-specific signal in epigenomic data. Bioinformatics 35, 4767–4769 (2019).
Peters, T. J. et al. Calling differentially methylated regions from whole genome bisulphite sequencing with DMRcate. Nucleic Acids Res. 49, e109 (2021).
Haga, R. B. & Ridley, A. J. Rho GTPases: regulation and roles in cancer cell biology. Small GTPases 7, 207–221 (2016).
Kazmi, N. et al. Rho GTPase gene expression and breast cancer risk: a Mendelian randomization analysis. Sci. Rep. 12, 1463 (2022).
Castagnoli, L., Tagliabue, E. & Pupa, S. M. Inhibition of the Wnt signalling pathway: an avenue to control breast cancer aggressiveness. Int. J. Mol. Sci. 21, 9069 (2020).
Wang, T. et al. A multiplex blood-based assay targeting DNA methylation in PBMCs enables early detection of breast cancer. Nat. Commun. 14, 4724 (2023).
Schrag, D. et al. Blood-based tests for multicancer early detection (PATHFINDER): a prospective cohort study. Lancet 402, 1251–1260 (2023).
Tang, W. et al. Population-specific mutation patterns in breast tumors from African American, European American, and Kenyan patients. Cancer Res. Commun. 3, 2244–2255 (2023).
Zhou, W., Laird, P. W. & Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 45, e22 (2017).
Barrett, J. E. et al. The DNA methylome of cervical cells can predict the presence of ovarian cancer. Nat. Commun. 13, 448 (2022).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Herzog, C. M., Theeuwes, B. & Widschwendter, M. Systems epigenetic approach towards non-invasive breast cancer detection: systems. https://doi.org/10.5281/zenodo.13885250 (2024).
Acknowledgements
This study was funded by the European Union’s Horizon 2020 Research and Innovation Program, H2020 FORECEE, under Grant Agreement No. 634570 (M.W.), the European Union’s Horizon 2020 European Research Council Program, H2020 BRCA-ERC under Grant Agreement No. 742432 (M.W.) as well as the Land Tirol (EUTOPS) and the charity, The Eve Appeal (https://eveappeal.org.uk/) (M.W.). We are grateful to the participants of the FORECEE and BRCA-ERC studies and the management team, research nurses, interviewers, research assistants and other staff who have taken part in gathering the data for this study.
Author information
Authors and Affiliations
Contributions
C.M.S.H.: conceptualization, formal analysis, data curation, writing—original draft, writing—review and editing, visualization, and supervision. B.T.: formal analysis, data curation, writing—original draft, writing—review and editing, and visualization. A.J. and I.E.: investigation, data curation, writing—review and editing. L.B., M.Z., D.C., N.H., N.C., resources. N.P., writing—review and editing. M.W. conceptualization, supervision, project administration, funding acquisition, and writing—review and editing.
Corresponding author
Ethics declarations
Competing interests
C.M.S.H. and M.W. are shareholders of Sola Diagnostics GmbH, which holds an exclusive license to the intellectual property that protects the commercialization of cervical sample DNA methylation-based tests (WID-qtBC, WID-BC). At the time of the publication of this manuscript, a UK patent application is pending for work described in this manuscript. A.J. and I.E. are named inventors on a patent of the WID-BC test. N.C. has reported fees for advisory board membership for AstraZeneca, Clovis Oncology, Eisai, GSK, Immunogen, Mersana, MSD/Merck, Nuvation Bio, Oncxerna, Pieris, Roche, Novocure; fees as an invited speaker for AstraZeneca, Clovis Oncology, GSK, MSD/Merck, Eisai, and institutional research grants from AstraZeneca, Roche, GSK. She has also reported non-remunerated activities as a member of the ESMO Guidelines Steering Committee and chair of the Scientific Committee of ACTO (Alleanza contro il tumore ovarico). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jacob Kresovich, Xiaoqi Zheng and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Herzog, C.M.S., Theeuwes, B., Jones, A. et al. Systems epigenetic approach towards non-invasive breast cancer detection. Nat Commun 16, 3082 (2025). https://doi.org/10.1038/s41467-024-53696-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53696-2
This article is cited by
-
Genetic Aspects of Breast Cancer: an Update
Current Breast Cancer Reports (2026)
-
Functionally enriched epigenetic clocks reveal tissue-specific discordant aging patterns in individuals with cancer
Communications Medicine (2025)
