
Abstract
The electrical double layer (EDL) at metal oxide-electrolyte interfaces critically affects fundamental processes in water splitting, batteries, and corrosion. However, limitations in the microscopic-level understanding of the EDL have been a major bottleneck in controlling these interfacial processes. Herein, we use ab initio-based machine learning potential simulations incorporating long-range electrostatics to unravel the molecular-scale picture of the EDL at the prototypical anatase TiO2-electrolyte interface under various pH conditions. Our large-scale simulations, capable of capturing interfacial water dissociation/recombination reactions and electrolytic proton transport, provide unprecedented insights into the detailed structure of the EDL. Moreover, the larger capacitance of the EDL under basic relative to acidic conditions, originating from the higher affinity of the cations for the oxide surface, is found to give rise to distinct charging mechanisms on negative and positive surfaces. Our results are validated by the agreement between the computed EDL capacitance and experimental data.
Similar content being viewed by others


Introduction
When a metal oxide is interfaced with an aqueous electrolyte with a pH different from its pH point of zero proton charge (pHPZC), a net charge forms on the surface1,2,3: at pH <pHPZC, the surface adsorbs protons from the electrolyte and becomes positively charged, while at pH > pHPZC it adsorbs hydroxide ions and becomes negatively charged. Electrolyte ions of opposite charges are then drawn closer to the interface to balance the surface charge. This electrostatic attraction, competing with thermal fluctuations and (de)solvation thermochemistry, leads to inhomogeneous ion distributions near the interface. Additionally, adjacent water molecules reorient in response to the surface and ionic charges. The combination of the charged surface, adjacent ions, and neighboring water molecules constitutes what is known as the electrical double layer (EDL)1,2,3. The EDL governs the chemical reactivity and physical properties of the interface and is crucial in diverse environmental, biological, colloidal, and electrochemical processes4,5,6,7.
The EDL has often been described based on the Gouy-Chapman-Stern (GCS) model, which assumes a uniformly charged, laterally homogeneous surface and treats water as a homogeneous dielectric continuum3,8. However, as the understanding of solid-electrolyte interfaces has advanced through theory and experiment, it has become apparent that the properties of the first few interfacial water layers depend on the surface structure and deviate considerably from those of bulk water9, a clear indication of the limitations of the GCS mean-field description3,4,5. To overcome these limitations, GCS has been combined with surface complexation models that account for discrete active surface sites10,11,12,13. These approaches have been useful for the interpretation of several experimental data but could not explicitly deal with key features such as the water ions originating from the pH of the solution. Moreover, growing experimental evidence underscores the inadequacy of classical mean-field descriptions in the presence of dynamic events such as proton/electron transfer and chemical reactions3,4,6,14. There is thus an urgent need for a microscopic understanding of the EDL that goes beyond mean-field theory and semi-empirical descriptions.
Investigations of the EDL at oxide-electrolyte interfaces are notoriously challenging. Experimentally, although surface-sensitive techniques are becoming increasingly available to investigate the structure and dynamics of solid-liquid interfaces3,15,16, the EDL’s microscopic properties are still difficult to probe. Theoretically, force field simulations and multiscale modeling have been widely used and provided important insights8,15,17,18, but are generally not accurate enough for describing reactive processes such as water dissociation and proton transfer at aqueous interfaces19,20,21. In contrast, density functional theory (DFT)-based ab initio molecular dynamics (AIMD) can in principle provide more accurate information on both atomistic details of the EDL and on macroscopic observables19,20,21,22. However, the computational cost of AIMD simulations makes them practical only for system sizes (hundreds of atoms) and simulation times (tens of picoseconds) that are often insufficient to characterize the EDL and pH-dependent surface chemistry21,22,23. For example, AIMD provided estimates of Helmholtz capacitances at oxide-electrolyte interfaces19,24,25, but could not predict equilibrium ion distributions in the EDL, for which longer simulation times and larger cells would be needed.
In the past few years, machine-learned potentials (MLPs)26,27,28,29,30 have emerged as a viable approach to enable large-scale ab initio-level simulations of bulk systems, interfaces and reactive processes21,23,31,32,33,34. Of such MLP methods, the Deep Potential (DP) scheme27,28 has been successfully applied to model bulk aqueous electrolytes31 and water-oxide interfaces21,23,32,33,34. Moreover, this scheme has recently been extended to include long-range electrostatic interactions35, a component missing in standard MLPs but found to be important for simulating the acid-base chemistry in water36.
In this work, we use the deep potential long-range (DPLR) method35 to conduct large-scale molecular dynamics simulations of the anatase TiO2 (101) surface in contact with electrolyte solutions at various pHs. TiO2 is a typical functional oxide and one of the most widely used materials in (photo-)electrochemistry37,38,39. Our simulations provide a comprehensive molecular-scale picture of the EDL at TiO2 interfaces including the occurrence of distinct microscopic surface charging mechanisms at low and high pHs. We also developed a method to calculate the electrostatic potential profile with ab initio DFT accuracy that enabled us to evaluate the interfacial capacitance due to the adsorption of protons or hydroxide ions and the corresponding counterions, a macroscopic property measurable from experiments that is highly reflective of the EDL’s nature19,24,25,40. The computed capacitances agree with experimental results40, underscoring the reliability of our ab initio-based, machine-learned description of the EDL.
Results and discussion
Structure of the EDL
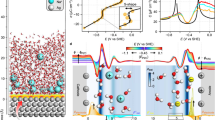
To characterize the EDL, we performed DPLR molecular dynamics simulations on periodically repeated systems consisting of a five-layer anatase (101) slab with a (\(30.7{\text{\AA}} \times 33.9{\text{\AA}}\)) surface cell in contact with a 67 Å thick aqueous electrolyte (Fig. 1a). In the following discussion we focus on three experimentally relevant solutions40, namely neutral 0.4 M NaCl (NaCl(aq), composed of 2376 H2O molecules and 18 NaCl), acidic 0.4 M NaCl + 0.2 M HCl (with 10 HCl added to the neutral solution), and basic 0.4 M NaCl + 0.2 M NaOH (with 10 NaOH added to the neutral solution). In the electrolyte, NaCl serves as a background salt, and adding 0.2 M NaOH or HCl changes the pH of the initial configuration from a nominal value of 7 to 13.3 or 0.7. Additional results for TiO2 interfaces with electrolytes containing higher concentrations of NaOH and HCl are reported in Supplementary Section 10 and Fig. 3c. The magnified view of the interface (Fig. 1b) shows that the TiO2 surface exposes five-fold coordinated titanium (Ti5c) and two-fold coordinated oxygen (O2c) atoms (108 Ti5c and 108 O2c sites in total, considering the two surfaces of the slab). The undercoordinated Ti5c atoms act as (Lewis) acid sites for the adsorption of water or OH− ions, while the O2c atoms act as Brønsted bases which can accept a hydrogen bond or a H+ from water. Water dissociation thus results in protons on O2c sites (bridging hydroxyls) and OH− groups on Ti5c sites (terminal hydroxyls), as depicted in Fig. 1b.

a Supercell of the anatase (101)-electrolyte interface employed in the DPLR simulations. The TiO2 regions are shaded in gray, while the electrolyte region is shaded in blue. b Magnified view of the interface showing surface undercoordinated Ti5c and O2c sites and adsorbed H2O, H+ and OH− at these sites. For visual clarity, only relevant atoms are shown. c Surface charge density \(\sigma=e({{{\rm{N}}}}_{{{\rm{H}}}+}-{{{\rm{N}}}}_{{{\rm{OH}}}-})/S\), where S is the surface area, as a function of simulation time for anatase (101) in contact with three different types of electrolytes. All results and error bars (shaded areas) were derived from simulations using two independent DPLR models. Source data are provided as a Source Data file.
For the TiO2-NaCl(aq) interface, the simulations show an equal number of adsorbed H+ (\({{{\rm{N}}}}_{{{\rm{H}}}+}\)) and OH– (\({{{\rm{N}}}}_{{{\rm{OH}}}-}\)) species, originating from the dissociation of adsorbed water molecules. Consequently, the surface charge density (black line in Fig. 1c) is close to zero. This suggests that the pH of the neutral NaCl(aq) is approximately equal to the pHPZC of anatase, a result consistent with the experimental pHPZC of anatase being around 6 ( ± 1)13,41,42. To support this inference, we performed enhanced sampling DPLR simulations to explicitly evaluate the pHPZC of the anatase (101)-neat water interface (Supplementary Section 4). We focused on neat water in this simulation because pHpzc is defined to reflect only the protonation reactions43, unaffected by the presence of salt ions. We obtained a pHPZC of 7.0\(\pm 0.1\), a value within the experimental range.
Interfaces of TiO2 with acidic and basic solutions were generated starting from an equilibrated configuration of TiO2-NaCl(aq), and subsequently adding 0.2 M HCl or NaOH at random positions within the electrolyte solution. All additional H+ (or OH−) ions were gradually adsorbed on the surface within 3 ns, leading to a positively (or negatively) charged surface (Fig. 1c). Averaging over 3-10 ns, we obtained surface charge densities \({{\sigma }_{a}=7.69\pm 0.04\mu C/{cm}}^{2}\) and\(\,{{\sigma }_{b}=-7.54\pm 0.13\mu C/{cm}}^{2}\) at the interfaces with the acidic and basic electrolyte, respectively. In experiments13, such values of \({\sigma }_{a}\) and \({\sigma }_{b}\) equilibrate with bulk solutions that have pHs of ≈ 4.4 and 7.4, respectively. For our simulation cell size (2376 water molecules), a pH value of 4.4 (or 7.4) corresponds to a negligible amount of ~\(2\times {10}^{-3}\) H+ (or \(1\times {10}^{-5}\) OH−) ions in the electrolyte solution. This is consistent with the fact that there are no H+ or OH− ions left in the bulk region of our electrolyte solution.
Upon surface charging, an EDL forms to compensate for the surface charge1,2,3. In the GCS model, the electrolyte ions form discrete (Helmholtz) planes, with the outer Helmholtz plane (OHP) separating the Stern layer from a diffuse layer where the ions follow the Poisson-Boltzmann distribution (Fig. 2a). The counter-ion and co-ion densities converge to an equal value in the EDL’s tail, indicating that the electrolyte recovers its bulk-like behavior. Within the Stern layer, the inner Helmholtz plane (IHP) is formed by ions that are specifically adsorbed at the surface and lack a complete hydration shell (Fig. 2a).

a Schematic of the Gouy-Chapman-Stern (GCS) model of the electric double layer (EDL): the outer Helmholtz plane (OHP) separates the EDL into Stern and diffuse layers. Within the Stern layer, the inner Helmholtz plane (IHP) is defined by the distance at which ions specifically adsorb. b Ion density distributions as functions of distance \(\Delta z=z-{z}_{{{\rm{surface}}}}\) from the solid surface, obtained from DPLR simulations of anatase (101) in contact with different electrolytes; the position of the solid surface, \({z}_{{{\rm{surface}}}}\), corresponds to the average position of the O2c sites. Besides the IHP predicted by the GCS model, these simulations reveal two additional ionic density peaks within the Stern layer, which we define as the intermediate Helmholtz planes (IMHPs). The inset in b amplifies the small IMHP1 peak in the neutral system. The computed ion densities are averaged over the two interfaces in the supercell. All results and error bars were derived from simulations using two independent DPLR models. c–e Illustrative snapshots of the microscopic structures of IHP, IMHP1, and IMHP2. Color code: purple (Na+), green (Cl-), white (H), light red (O in water or water ions), dark red (O in TiO2), and gray (Ti). Lines between ions and neighboring oxygens indicate distances smaller than the ionic hydration shell radius. Dashed blue lines indicate hydrogen bonds between water and surface O2c. For visualization purposes, only the most relevant atoms are shown. Source data are provided as a Source Data file.
The electrolyte ion distributions obtained from our simulations are shown in Fig. 2b. The structure of the diffuse layer agrees with that given by the GCS model, i.e., the Cl⁻/Na⁺ density decreases/increases with increasing distance from the OHP until the two densities become identical within the error bars of the simulation. However, the ion distribution within the Stern layer is more complex than suggested by the GCS model and by previous force field simulations15,18. Specifically, within the Stern layer, we identify not just a single ionic peak, but three distinct peaks: a first Na⁺ peak at 1.5 Å from the surface, followed by a Cl⁻ peak at 3.4 Å of low intensity due to the repulsion between the Cl⁻ and the electronegative O2c atoms, and another Na⁺ peak at 4.2 Å. The Na⁺ ions forming the first peak are, on average, coordinated to two surface O2C atoms (Fig. 2c), consistent with previous DFT results13. The distance, \({r}_{{{\rm{Na}}}-{{\rm{O}}}2{{\rm{c}}}}\), between Na⁺ and O2C fluctuates around 2.3 Å (Supplementary Fig. 17), a value similar to the distance between Na+ ions and water molecules in their first hydration shell in bulk solution. With the O2C atoms substituting hydration water molecules, these Na⁺ ions exhibit incomplete hydration shells (Fig. 2c) and can thus be identified as inner-sphere surface complexes15,18 and the corresponding peak as the IHP. Instead, the Cl⁻ ions forming the second peak (Fig. 2 d) and the Na⁺ ions contributing to the third peak (Fig. 2e) exhibit complete hydration shells. While this characteristic is consistent with the definition of outer-sphere surface complexes44, these ions do not satisfy the OHP criterion that their interactions with the surface involve only long-range electrostatic forces45. As shown in Fig. 2d and Fig. 2e, some water molecules in the hydration shell of these ions are either adsorbed on Ti5c atoms or form strong hydrogen bonds with surface O2c atoms. Since these surface water molecules have a diffusion coefficient 1~2 orders of magnitude smaller than that of bulk water46, these ions can be considered as semi-adsorbed on the surface via surface water molecules. Consequently, these two ionic peaks cannot be classified as either IHP or OHP. In this work, we designate the corresponding layers as intermediate Helmholtz planes (IMHPs), since their peaks fall between the IHP and the OHP. We also find that the ions within the Stern layer are not fixed but can exchange with other ions of the same type in the electrolyte. Specifically, Na+ ions in the IHP have a residence time on the order of nanoseconds, while ions in IMHPs have a residence time of tens to hundreds of picoseconds (Supplementary Information Section 5).
In contrast to the GCS prediction that the EDL only forms when a net (electronic or protonic) surface charge is present, our results show that the charge-neutral TiO2-NaCl(aq) interface also exhibits an EDL (Fig. 2b). This occurs because, even when the overall surface charge is zero, local charges remain imbalanced. Specifically, the outermost oxide surface layer is composed of electronegative O2c atoms, which favor the adsorption of Na+ ions and, in combination with the water molecules adsorbed or hydrogen bonded to the surface, determine the ionic peak positions (Fig. 2c-e). Therefore, altering the surface charge density modifies the peak intensities of the ion distribution but does not appreciably change their relative positions (Fig. 2b). In particular, regardless of the surface charge, anions dominate at the OHP and in the diffuse layer to compensate the Na+ ions in the IMHP2 − another important feature missed by the GCS model.
The water molecules within the EDL exhibit distribution and orientation patterns which, as the ion distributions, are mostly determined by the surface structure. Our DPLR simulations show that the water distribution and orientation undergo subtle changes in response to both the surface charge and the adjacent ions (Supplementary Section 6).
Differential capacitance
The microscopic structure of the EDL determines the differential capacitance C of the interface, which is composed of contributions from the capacitance of the Stern layer (\({C}_{H}\), also called Helmholtz capacitance) and the diffuse layer (\({C}_{D}\))47, with \(\frac{1}{C}=\frac{1}{{C}_{H}}+\frac{1}{{C}_{D}}\). While \({C}_{H}\) has been studied previously by AIMD simulations19,24,25, the calculation of C with ab initio accuracy has been a long-standing challenge due to the long simulation times and large simulation cells needed to equilibrate the EDL. Interestingly, experiments found that, at equivalent magnitudes of charge densities, negatively charged oxide interfaces have a higher capacitance than their positively charged counterparts for metal oxides such as anatase40, rutile40, and zinc oxide48. This is in contrast to the symmetric capacitance predicted by the GCS model, suggesting that the EDL at these oxide surfaces can screen negative surface charges more effectively than positive ones. However, the underlying reason for this phenomenon remains a topic of active debate. Some studies attributed the larger capacitance under basic conditions to the high affinity of cations to the oxide12,40. In contrast, recent AIMD studies proposed that the difference is due to the distinct properties of water under basic and acidic conditions24,25,49. For example, Ref 24. attributed it to the larger interfacial water fluctuations under basic conditions, Ref 25. explained it in terms of the orientation of chemisorbed water, and Ref 49. attributed it to the water adsorption energy.
To obtain insight into the observed asymmetry of C, we calculated this quantity with ab initio accuracy using our DPLR method. Specifically, starting from the definition of C as the first-order derivative of the surface charge density σ with respect to the electrostatic potential drop ψ, \(C=\frac{{{\rm{d}}}\sigma }{{{\rm{d}}}\psi }\), we used the finite difference expression \(C\approx \frac{\Delta \sigma }{\Delta \psi }\), where ∆ represents the deviation of a charged interface from a neutral reference interface. While σ is readily available in simulations (Fig. 1c), ψ depends not only on the distribution of the ions but also on that of valence electrons, which is generally not available in simulations based on force fields. In our DPLR approach35, however, ψ can be explicitly calculated (see “Methods” for details). The average potential profile calculated in this way for the TiO2-NaCl(aq) interface is displayed by the green line in Fig. 3a. Within the TiO2 region, the potential exhibits pronounced oscillations, while the homogeneous and isotropic nature of the liquid electrolyte results in a more uniform profile. Macroscopic averaging50 of \(\phi (z)\) yields the black line in Fig. 3a, from which the potential drop ψ, defined as the potential difference between solid and liquid phases, is extracted. At this point, the capacitance can be calculated. Taking the neutral TiO2-NaCl(aq) interface as the reference and using the results in Fig. 1c, we have \(\Delta {\sigma }_{b}={-7.54\pm 0.13\mu C/{cm}}^{2}\) for the TiO2-NaCl+NaOH basic solution interface, and \(\Delta {{\sigma }_{a}=7.69\pm 0.04\mu C/{cm}}^{2}\) for the TiO2-NaCl+HCl acidic solution interface. By comparing the macroscopically averaged50 electrostatic potential at the acidic and basic solution interfaces with the neutral reference (Fig. 3b), we determine the potential drop differences \(\Delta {\psi }_{a}=131.6\pm 10.6{mV}\) and \(\Delta {\psi }_{b}=-78.3\pm 11.2{mV},\) respectively. Consequently, the differential capacitances are \({C}_{a}=58.4\pm 4.7\mu F/{{cm}}^{2}\) and \({C}_{b}=96.2\pm 13.8\mu F/{{cm}}^{2}\) under acidic and basic conditions, respectively. As shown in Fig. 3c, our results (which include the computed capacitances at higher HCl and NaOH concentrations reported in Supplementary Section 10) reproduce the experimental trends40,51 quite satisfactorily. In particular, our calculated ratio of \({C}_{b}/{C}_{a}=1.6\pm 0.3\) agrees well with previous DFT19,24 and experimental40 results showing \({C}_{b}/{C}_{a}\approx 1.5\) at similar oxide/electrolyte interfaces.

a Representative snapshot from our DPLR simulation of the anatase TiO2-NaCl solution interface (top) and plane-averaged electrostatic potential \(\phi\) along the \(z\)-direction, before and after macroscopic (macro-) average (bottom). The potential drop \(\psi\) is determined from the macro-averaged potential difference between the bulk solid and bulk liquid regions. b Macro-averaged electrostatic potential at the TiO2 interfaces with three different types of electrolytes. All curves are aligned to zero \(z=0\). The magnified view in the inset shows the potential drop differences between the negatively (positively) charged surface and the neutral surface, denoted as \(\Delta {\psi }_{b}\) (\(\Delta {\psi }_{a}\)), which is obtained by further averaging the macro-averaged electrostatic potentials over 31.8 \(\mathring{\rm A}\) < z < 51.8 \(\mathring{\rm A}\). All results and error bars were derived from simulations using two independent DPLR models. c Comparison between our computed interfacial capacitance (star symbols) and experiments for rutile TiO2-aqueous NaNO3 interfaces (solid lines)40[,51. Different colors of the experimental curves represent different concentrations of the background NaNO3 salt. Source data are provided as a Source Data file.
Based on our simulations, the larger capacitance observed under basic conditions in comparison to acidic ones can be understood as follows. For the negative (positive) surface, the positive Na+ (negative Cl−) ions are drawn towards the surface to screen the surface charge. However, the abilities of Na+ and Cl− to screen the surface charge differ. Figure 2b shows that Na+ can approach the surface more closely than Cl− ions because the outermost layer of TiO2 surface is composed of electronegative O2c atoms. This allows Na+ to screen the surface charge more effectively than Cl–. In more detail, when the system is negatively charged under basic conditions, the affinity of Na+ ions to the electronegative surface O2c atoms draws them close to the surface, effectively screening the surface OH− ions. This effect is evidenced by Fig. 2b, which shows a significant amplification of the Na+ peak intensity when transitioning from a neutral to a basic system. In contrast, when the system is positively charged under acidic conditions, a substantial proportion of O2c atoms (82% in our acidic system) do not bind H+ ions (Fig. 2d). These electronegative O2c atoms repel Cl- ions, preventing them from moving close to the surface to screen the surface H+ ions. Consequently, the transition from a neutral to an acidic system induces a modest increase in the Cl− peak intensity as shown in Fig. 2b. This leads to a smaller absolute value of \(\Delta {\psi }_{b}\) than \(\Delta {\psi }_{a}\), and consequently a larger \({C}_{b}\) than \({C}_{a}\). Compared to the significant changes of ion distributions at different pHs, the water distribution and orientation undergo smaller changes (Supplementary Section 6). Therefore, our simulations agree with the suggestion that the larger capacitance under basic conditions is primarily due to the higher affinity of cations to the oxide. Differences between our findings and previous AIMD simulations19,24 are likely related to the higher ion concentrations used in the latter, their short equilibration times, and their focus on the Helmholtz part only of the differential capacitance.
Microscopic surface charging mechanism
The distinct interfacial capacitances resulting from the higher affinity of cations to the oxide surface, compared to anions, are found to give rise to distinct microscopic charging mechanisms on negative and positive surfaces. As shown in Fig. 4a, for the charge-neutral TiO2-NaCl(aq) interface, \(14\pm 2\%\) water molecules adsorbed at Ti5c sites are dissociated into adsorbed H+ at O2c and OH− at Ti5c, with zero net surface charge. Starting with an equilibrated TiO2-NaCl(aq) configuration, when we add bases or acids into the electrolyte, the OH− or H3O+ ions have two potential pathways to be adsorbed at the surface. For an electrolyte OH– (Fig. 4b), one pathway is to recombine with a surface H+ ion at the O2c site to form a water molecule in the liquid, which decreases the surface’s H+ population. Another pathway is to recombine with an H+ from an adsorbed H2O, which results in an additional OH− ion adsorbed at the surface Ti5c site. Similarly, an electrolyte H+ can either recombine with a surface terminal OH− into a water molecule or occupy an empty O2c site (Fig. 4c). Although both pathways result in the same net surface charge σ, the total number of surface charges, NSC = NH+ + NOH−, is different. The first pathway reduces NSC, whereas the second increases it.

Time evolution of the surface H+ and OH− coverage on TiO2 interfaces with (a) neutral, (b) basic, and (c) acidic solutions. The legends list the surface ion coverages averaged between 3-10 ns. Schematics in (a) show molecularly absorbed H2O at Ti5C sites and an H2O dissociated into adsorbed H+ at O2c and OH− at Ti5C sites. Schematics in (b and c) illustrate two potential pathways an additional water ion might take upon adsorption to the surface. Specifically, in b, a solvated OH− can either recombine with an adsorbed H+ to form an H2O molecule (pathway 1) or replace an adsorbed H2O molecule (pathway 2). Similarly, in (c) a solvated H+ can either recombine with an adsorbed OH− to form an H2O molecule (pathway 1) or get adsorbed at a O2c site (pathway 2). Pathway 2 is preferred in (b) while pathway 1 is preferred in (c) as discussed in the text. Source data are provided as a Source Data file.
The averaged ion coverages in Fig. 4 suggest that OH– ions prefer the second pathway (increasing NSC), whereas protons slightly prefer the first (decreasing NSC). As a result, if we define the water dissociation fraction as the smaller value between the fraction of surface H⁺ ions and the fraction of surface OH⁻ ions, the average water dissociation fraction decreases significantly (to ~9%) with the acidic electrolyte whereas it remains approximately the same as that of the neutral interface under basic conditions, consistent with the increase of water dissociation fraction with the pH reported in a previous AIMD study25. These trends can be related to the different capacitances under basic and acidic conditions. The large capacitance under basic conditions indicates a strong ability of the EDL to screen the OH− surface charge. This is evidenced by the high number density of Na+ ions surrounding an OH− surface ion, as shown in Fig. 5a. Consequently, the surface can accommodate more OH− ions in-plane, making the second pathway favorable under basic condition. In contrast, the small capacitance under acidic conditions reflects a weak ability of the EDL to screen the H+ surface charge. This is demonstrated by the low probability of Cl- ions surrounding an H+ surface ion, as shown in Fig. 5b. As a result, the surface cannot accommodate more H+ ions in-plane, making the first pathway favorable under acidic conditions. The fact that negatively charged surfaces allow a larger density of surface hydroxyl groups is important in photocatalysis because these groups can trap photo-generated holes and form hydroxyl radicals38,52, which are key intermediates of many photo-oxidation reactions. The photooxidation of water is indeed known to be faster at high pH53.

Top view of the anatase TiO2 surface, with x and y corresponding to the [\(\bar{1}01\)] and [010] directions, respectively. The contours represent the 2D projected number densities of (a) the IHP Na+ ion around a surface OH− ion for the TiO2-NaCl(aq)+NaOH(aq) interface, and (b) the IMHP1 Cl- ion around a surface H+ ion for the TiO2-NaCl(aq)+HCl(aq) interface. Source data are provided as a Source Data file.
The ab initio-level molecular-scale picture of the EDL at the TiO2-electrolyte interface provided by our simulations exhibits Stern layer features that are not included in the GCS model, particularly intermediate Helmholtz planes associated with outer-sphere complexes of electrolyte ions. The shorter adsorption distance of positive vs. negative ions results in a larger capacitance at high pH, in good agreement with experiment. The different capacitances at low and high pHs are associated with distinct microscopic surface charging mechanisms, a feature that can help optimize photo(electro)catalytic processes. These results underscore the utility of machine learning-enabled, ab initio-quality simulations to characterize the specific chemistry and inhomogeneity of (photo)electrochemical interfaces and pave the way to further studies including the presence of external fields.
Methods
DFT calculations
Static DFT calculations were conducted using the strongly constrained and appropriately normed (SCAN)54 exchange-correlation functional as implemented in the Quantum ESPRESSO55 package. The SCAN functional54 has been found to well describe TiO2 interfaces56 and electrolyte solutions31,57,58 in previous studies. Valence electron-ion interactions were described by Optimized Norm-Conserving Vanderbilt (ONCV)59 pseudopotentials. Electron wavefunctions were expanded in plane waves using a cutoff energy of 150 Ry. A total energy convergence threshold of 1 × 10 Ry was adopted. Because of the large size of our supercells, the Brillouin zone sampling was restricted to the Gamma point. Following each self-consistent calculation, maximally localized Wannier functions (MLWFs)60 were determined using the Wannier90 code61. Each MLWF was associated with its closest Ti, O, Na, or Cl atom, resulting in each of these atoms carrying four doubly occupied MLWFs. From the MLWFs, the coordinates of the Wannier Centroids (WCs) relative to their corresponding atoms were obtained by computing the average position of the Wannier centers associated with each given atom35.
Deep neural network (DNN) models
The training dataset for the DNN models was collected through an active machine-learning approach28. This dataset comprehensively spans the configurational space of bulk anatase TiO2, water, and various aqueous electrolyte solutions (NaCl, NaOH, HCl, NaCl + NaOH, and NaCl + HCl solutions), as well as anatase (101) interfaces with each of these liquids (see Supplementary Table 1). The exploration spanned temperatures of 200−800 K (systems 1–11) or 300−400 K (systems 12-15), under conditions of either a pressure of 1 bar or a constant volume corresponding to experimental densities. The final dataset comprises 30,103 configurations in total. The DPLR and Deep Wannier (DW)62 DNN models were trained on this training dataset using the DeePMD-kit package63. For both DPLR and DW, two independent models were generated using different initial random parameters. The two models were used to run independent simulations from which average properties and corresponding error bars were derived.
To evaluate the performance of our DNN models, we generated a testing dataset of configurations not included in the training. This was achieved by conducting deep potential long-range (DPLR) molecular dynamics (MD) simulations on the following four representative systems.
-
The anatase (101)-liquid water interface, comprising 60 TiO2 units and 82 H2O molecules.
-
The anatase (101)-NaCl(aq) solution interface, comprising 60 TiO2 units, 82 H2O molecules, and 1 NaCl ion pair.
-
The anatase (101) -NaCl(aq)+NaOH(aq) solution interface, comprising 60 TiO2 units, 80 H2O molecules, 1 NaCl ion pair, and 1 NaOH ion pair.
-
The anatase (101)-NaCl(aq)+ HCl(aq) solution interface, comprising 60 TiO2 units, 81 H2O, 1 NaCl ion pair, and 1 HCl ion pair.
In the above, the 60 TiO2 units correspond to a five-layer (1 × 3) anatase (101) slab. For each system, we conducted 5 ns DPLR MD simulations within the canonical ensemble at 330 K. The initial 1 ns of each simulation was discarded for equilibration purposes. From the subsequent 4 ns, 50 configurations were uniformly extracted from each trajectory, resulting in a total of 200 configurations for the testing dataset.
DPLR method
The DPLR method35 assumes that the potential energy surface has short- and long-range contributions. The short-range contribution is represented as in the standard deep potential model64, while the long-range contribution is approximated by the electrostatic energy of a system of spherical Gaussian charges associated with the ions (nuclei + core electrons) and the valence electrons. We calculate the electrostatic energy of the Gaussians via the particle-particle-particle-mesh method65 for evaluating Ewald sums. The location, charge magnitude, and spatial spread of the Gaussians are determined as follows.
-
Location: Ionic Gaussians are centered at the atomic sites, and electronic Gaussians are centered at the maximally localized Wannier centers66. For computational simplicity, Wannier centers associated with the same atom are combined into a single Wannier Centroid (WC)35, located at the instantaneous average position of these Wannier centers. The locations of WCs are predicted by the DW DNN model62.
-
Charge magnitude: The magnitudes of the ionic charges are \({+Z}_{V}e\), with \({Z}_{V}\) being the number of their valence electrons. The pseudopotential applied in this work treats the 3 s23p6 3d24s2 electrons of Ti, 1 s electron of H, 2 s22p4 electrons of O, 2 s22p6 3 s1 electrons of Na, and 3s23p5 electrons of Cl as valence electrons explicitly. Consequently, \({Z}_{V}\) equals 12, 1, 6, 9, and 7 for Ti, H, O, Na, and Cl, respectively. For electrons, each Wannier center carries a charge of \(-2e\). Given that each Ti, O, Na, and Cl ion has four Wannier centers, their WCs carry a charge of \(-8e\). The DFT calculations show that the average distance of Ti’s WCs from the Ti ions is significantly smaller (0.002 Å, averaged over \(x\), \(y\), and \(z\) directions) compared to that of O’s WCs from O ions (0.026 Å, averaged over \(x\), \(y\), and \(z\) directions). This suggests a negligible contribution of the polarization of Ti’s WCs to the overall electrostatic energy. Therefore, for computational efficiency, our DPLR model further simplifies the representation of Ti ions: instead of accounting for a \(+12e\) charge on the Ti ion counterbalanced by a \(-8e\) charge from its WCs, we simplify our DPLR model by omitting the WCs of Ti and treating each Ti ion as a \(+4e\) charge. The WCs of O, Na+, and Cl- are all treated explicitly.
-
Spatial spread: The long-range electrostatic contribution up to dipole terms is independent of the spread parameter, which can then be chosen to ensure charge neutrality in the bulk of the solution as well as good numerical representability by a DNN of the short-range contribution. In our DPLR, the spatial spread of the Gaussians for both ions and electrons is (2\({\beta \left)\right. }^{-1},\) where \(\beta\) is an adjustable parameter. In the limit of \(\beta \to 0\), the Gaussian width is infinite and the DPLR model reduces to the standard short-range DP (hereafter denoted as DPSR) model. Conversely, as \(\beta \to \infty\), the Gaussian charges become point-like, leading to singular potentials incompatible with DNN representations35. To determine an optimal \(\beta\) for our system, we conducted systematic training of DPLR models across a range of \(\beta\) values: 0.0, 0.1, 0.2, 0.3, and 0.4 \({{{\text{{\AA}}}}}^{-1}\). The performance analysis, depicted in Supplementary Fig. 1, reveals that, for both the training and testing datasets, \(\beta=0.1{{{\text{{\AA}}}}}^{-1}\) minimizes the root mean square error (RMSE) in the prediction of energies and forces in comparison to the DFT calculations. We thus adopted \(\beta=0.1{{\text{\AA}} }^{-1}\) for our DPLR simulations.
Supplementary Fig. 1 also shows that the RMSE for the testing dataset is lower than that for the training dataset. This is because the training dataset includes higher temperature (up to 800 K) configurations compared to the 330 K temperature used for the testing dataset. At higher temperatures, the magnitudes of forces and energies are larger, resulting in larger absolute errors. Given that the production runs of our work were conducted at 330 K, the errors at 330 K are more representative and indicative of the model’s performance under our specific conditions of interest.
DPSR fails to properly describe oxide-electrolyte interfaces, because, in the absence of long-range interactions, charge accumulation at the interface due to surface charging and/or EDL formation may result in charge neutrality violation in the bulk of an electrolytic solution. DPLR remedies this deficiency because the electrostatic energy penalizes bulk charging (Supplementary Section 3). In the absence of dissolved ions, as in the case of the anatase (101) interface with neat water, DPLR gives results that agree with DPSR (Supplementary Section 2).
DPLR MD simulations
The large-scale DPLR MD simulations presented in the main manuscript were conducted on model systems consisting of a five-layer (\(3\times 9\)) anatase (101) slab (540 TiO2 units) in contact with a 67 \({\text{\AA}}\) thick layer of aqueous electrolyte within a periodically repeated supercell of size \(30.7\,{\text{\AA}} \times 33.9\,{\text{\AA}} \times 83.4\,{\text{\AA}}\) along the three orthogonal directions \([\bar{1}01],\,[010]{{\rm{and}}}[101]\) of the anatase crystal lattice. All simulations were conducted in the canonical ensemble for 10 ns with a temperature of 330 K. The 30 K elevation is to partially compensate for the overestimation of the melting temperature of ice by the SCAN functional and describe a liquid with diffusivity close to that of water at standard conditions67. The TiO2 interfaces with the acidic and basic solutions were initially simulated at 400 K for 0.9 ns to accelerate equilibration and subsequently cooled to the target temperature of 330 K. In all simulations, hydrogen was replaced by deuterium to allow the use of a larger simulation time step of 0.5 fs. The ion and water densities presented in this work were averaged over the period from 3 to 10 ns. To check the convergence of ion and water densities with respect to simulation time, we plotted the ion and water density averaged over the periods from 3 to 6.5 ns and from 6.5 to 10 ns, respectively, in Supplementary Figs. 2 and 3. The results averaged over 3 to 6.5 ns agree well with those averaged over 6.5 to 10 ns, indicating the 10 ns simulation is well converged.
Electrostatic potential calculation
The electrostatic potential drop, \(\psi\), is an average property that requires knowledge of the plane-averaged density profile of the electrons along \(z\), which can be estimated accurately from the positions of the Wannier centroids, provided by the DW31 neural network model, and from the average spread of the associated electron distributions, provided by DFT calculations on smaller systems. The density profile of the total charge (ions + valence electrons) is obtained by adding the plane-averaged ion density profile to the valence electron density profile. Then, \(\phi (z)\), the electrostatic potential profile, is calculated by solving a one-dimensional Poisson’s equation. As shown in Supplementary Section 7, this procedure is remarkably accurate when compared to DFT calculations on reference systems.
Data availability
The complete DFT training datasets and the deep potential models generated in this study have been deposited in the Figshare database (https://figshare.com/s/3c4e7eb02ee64fd8b3bf)68. Source data are provided as a Source Data file. Source data are provided with this paper.
Code availability
Deep potential long-range molecular dynamics simulations were conducted using the DeePMD-kit package (https://github.com/deepmodeling/deepmd-kit) in conjunction with LAMMPS. The code used to generate the plots shown in the main text is available from the corresponding author upon request.
References
Brown, G. E. et al. Metal oxide surfaces and their interactions with aqueous solutions and microbial organisms. Chem. Rev. 99, 77–174 (1999).
Bourikas, K., Kordulis, C. & Lycourghiotis, A. Titanium dioxide (anatase and rutile): surface chemistry, liquid-solid interface chemistry, and scientific synthesis of supported catalysts. Chem. Rev. 114, 9754–9823 (2014).
Gonella, G. et al. Water at charged interfaces. Nat. Rev. Chem. 5, 466–485 (2021).
Rehl, B. et al. Water structure in the electrical double layer and the contributions to the total interfacial potential at different surface charge densities. J. Am. Chem. Soc. 144, 16338–16349 (2022).
Lee, S. S., Koishi, A., Bourg, I. C. & Fenter, P. Ion correlations drive charge overscreening and heterogeneous nucleation at solid–aqueous electrolyte interfaces. Proc. Natl Acad. Sci. 118, e2105154118 (2021).
Shin, S.-J. et al. On the importance of the electric double layer structure in aqueous electrocatalysis. Nat. Commun. 13, 174 (2022).
Li, J.-Q., Meng, L., Sprik, M. & Cheng, J. Thermodynamic Investigation of proton/electron interplay on the pourbaix diagram at the TiO2/electrolyte interface. J. Phys. Chem. C. 124, 19003–19014 (2020).
Becker, M. et al. Multiscale modeling of aqueous electric double layers. Chem. Rev. 124, 1–26 (2023).
Björneholm, O. et al. Water at Interfaces. Chem. Rev. 116, 7698–7726 (2016).
Lutzenkirchen, J. Surface Complexation Modelling. (Elsevier, 2006).
Hiemstra, T., Van Riemsdijk, W. H. & Bolt, G. H. Multisite proton adsorption modeling at the solid/solution interface of (hydr)oxides: A new approach: I. Model description and evaluation of intrinsic reaction constants. J. Colloid Interface Sci. 133, 91–104 (1989).
Bourikas, K., Hiemstra, T. & Van Riemsdijk, W. H. Ion pair formation and primary charging behavior of titanium oxide (Anatase and Rutile). Langmuir 17, 749–756 (2001).
Ridley, M. K., Machesky, M. L. & Kubicki, J. D. Anatase nanoparticle surface reactivity in NaCl media: a CD–MUSIC model interpretation of combined experimental and density functional theory studies. Langmuir 29, 8572–8583 (2013).
Boettcher, S. W. & Surendranath, Y. Heterogeneous electrocatalysis goes chemical. Nat. Catal. 4, 4–5 (2021).
Zhang, Z. et al. Ion Adsorption at the rutile−water interface: linking molecular and macroscopic properties. Langmuir 20, 4954–4969 (2004).
Brown, M. A. et al. Determination of surface potential and electrical double-layer structure at the aqueous electrolyte-nanoparticle interface. Phys. Rev. X 6, 011007 (2016).
Předota, M. et al. Electric double layer at the rutile (110) surface. 1. structure of surfaces and interfacial water from molecular dynamics by use of ab initio potentials. J. Phys. Chem. B 108, 12049–12060 (2004).
Předota, M., Zhang, Z., Fenter, P., Wesolowski, D. J. & Cummings, P. T. Electric double layer at the rutile (110) surface. 2. adsorption of ions from molecular dynamics and X-ray experiments. J. Phys. Chem. B 108, 12061–12072 (2004).
Cheng, J. & Sprik, M. The electric double layer at a rutile TiO2 water interface modelled using density functional theory based molecular dynamics simulation. J. Phys.: Condens. Matter 26, 244108 (2014).
Le, J.-B., Yang, X.-H., Zhuang, Y.-B., Jia, M. & Cheng, J. Recent progress toward Ab Initio modeling of electrocatalysis. J. Phys. Chem. Lett. 12, 8924–8931 (2021).
Schran, C. et al. Machine learning potentials for complex aqueous systems made simple. Proc. Natl Acad. Sci. USA 118, e2110077118 (2021).
Groß, A. Challenges for ab initio molecular dynamics simulations of electrochemical interfaces. Curr. Opin. Electrochem. 40, 101345 (2023).
Calegari Andrade, M. F., Ko, H.-Y., Zhang, L., Car, R. & Selloni, A. Free energy of proton transfer at the water–TiO2 interface from ab initio deep potential molecular dynamics. Chem. Sci. 11, 2335–2341 (2020).
Zhang, C., Hutter, J. & Sprik, M. Coupling of surface chemistry and electric double layer at TiO2 electrochemical interfaces. J. Phys. Chem. Lett. 10, 3871–3876 (2019).
Jia, M., Zhang, C. & Cheng, J. Origin of asymmetric electric double layers at electrified oxide/electrolyte interfaces. J. Phys. Chem. Lett. 12, 4616–4622 (2021).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Zhang, L., Han, J., Wang, H., Car, R. & E, W. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Phys. Rev. Lett. 120, 143001 (2018).
Zhang, L., Lin, D.-Y., Wang, H., Car, R. & E, W. Active learning of uniformly accurate interatomic potentials for materials simulation. Phys. Rev. Materials 3, 023804 (2019).
Behler, J. Four generations of high-dimensional neural network potentials. Chem. Rev. 121, 10037–10072 (2021).
Unke, O. T. et al. Machine Learning Force Fields. Chem. Rev. 121, 10142–10186 (2021).
Zhang, C., Yue, S., Panagiotopoulos, A. Z., Klein, M. L. & Wu, X. Why dissolving salt in water decreases its dielectric permittivity. Phys. Rev. Lett. 131, 076801 (2023).
Quaranta, V., Behler, J. & Hellström, M. Structure and dynamics of the liquid–water/zinc-oxide interface from machine learning potential simulations. J. Phys. Chem. C. 123, 1293–1304 (2019).
Wen, B., Calegari Andrade, M. F., Liu, L.-M. & Selloni, A. Water dissociation at the wate-rutile TiO2(110) interface from ab-initio-based deep neural network simulations. Proc. Natl Acad. Sci. 120, e2212250120 (2023).
Zeng, Z. et al. Mechanistic insight on water dissociation on pristine low-index TiO2 surfaces from machine learning molecular dynamics simulations. Nat. Commun. 14, 6131 (2023).
Zhang, L. et al. A deep potential model with long-range electrostatic interactions. J. Chem. Phys. 156, 124107 (2022).
Calegari Andrade, M., Car, R. & Selloni, A. Probing the self-ionization of liquid water with ab initio deep potential molecular dynamics. Proc. Natl Acad. Sci. 120, e2302468120 (2023).
Linsebigler, A. L., Lu, G. & Yates, J. T. Photocatalysis on TiO2 surfaces: principles, mechanisms, and selected results. Chem. Rev. 95, 735–758 (1995).
Schneider, J. et al. Understanding TiO2 photocatalysis: mechanisms and materials. Chem. Rev. 114, 9919–9986 (2014).
Guo, Q., Zhou, C. Y., Ma, Z. B. & Yang, X. M. Fundamentals of TiO2 photocatalysis: concepts, mechanisms, and challenges. Adv. Mater. 31, e190199 (2019).
Be´rube´, Y. G. & de Bruyn, P. L. Adsorption at the rutile-solution interface: II. model of the electrochemical double layer. J. Colloid Interface Sci. 28, 92–105 (1968).
Bourikas, K., Vakros, J., Kordulis, C. & Lycourghiotis, A. Potentiometric mass titrations: experimental and theoretical establishment of a new technique for determining the point of zero charge (PZC) of Metal (Hydr)Oxides. J. Phys. Chem. B 107, 9441–9451 (2003).
Kosmulski, M. Isoelectric points and points of zero charge of metal (hydr)oxides: 50years after Parks’ review. Adv. Colloid Interface Sci. 238, 1–61 (2016).
Sverjensky, D. A. Prediction of surface charge on oxides in salt solutions: revisions for 1:1 (M+L−) electrolytes. Geochimica et. Cosmochimica Acta 69, 225–257 (2005).
Lee, S. S., Fenter, P., Nagy, K. L. & Sturchio, N. C. Changes in adsorption free energy and speciation during competitive adsorption between monovalent cations at the muscovite (001)-water interface. Geochimica et. Cosmochimica Acta 123, 416–426 (2013).
Bard, A. J., Faulkner, L. R. & White, H. S. Electrochemical Methods: Fundamentals and Applications. (Wiley, 2022).
Calegari Andrade, M. F., Ko, H. Y., Car, R. & Selloni, A. Structure, polarization, and sum frequency generation spectrum of interfacial water on anatase TiO2. J. Phys. Chem. Lett. 9, 6716–6721 (2018).
Wu, J. Understanding the electric double-layer structure, capacitance, and charging dynamics. Chem. Rev. 122, 10821–10859 (2022).
Blok, L. & de Bruyn, P. L. The ionic double layer at the ZnOsolution interface: III. comparison of calculated and experimental differential capacities. J. Colloid Interface Sci. 32, 533–538 (1970).
Jia, M., Wang, J., Liu, Q., Yang, X. & Zhang, C. https://doi.org/10.26434/chemrxiv-2024-bs1rr (2024).
Baldereschi, A., Baroni, S. & Resta, R. Band offsets in lattice-matched heterojunctions: a model and first-principles calculations for GaAs/AlAs. Phys. Rev. Lett. 61, 734–737 (1988).
Bérubé, Y. G. & de Bruyn, P. L. Adsorption at the rutile-solution interface: I. thermodynamic and experimental study. J. Colloid Interface Sci. 27, 305–318 (1968).
Anpo, M., Shima, T. & Kubokawa, Y. Esr and photoluminescence evidence for the photocatalytic formation of hydroxyl radicals on small TiO2 particles. Chem. Lett. 14, 1799–1802 (1985).
Imanishi, A., Okamura, T., Ohashi, N., Nakamura, R. & Nakato, Y. Mechanism of water photooxidation reaction at atomically flat TiO2 (Rutile) (110) and (100) surfaces: dependence on solution pH. J. Am. Chem. Soc. 129, 11569–11578 (2007).
Sun, J., Ruzsinszky, A. & Perdew, J. P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 115, 036402 (2015).
Giannozzi, P. et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys.: Condens. Matter 29, 465901 (2017).
Calegari Andrade, M. F., Ko, H.-Y., Car, R. & Selloni, A. Structure, polarization, and sum frequency generation spectrum of interfacial water on anatase TiO 2. J. Phys. Chem. Lett. 9, 6716–6721 (2018).
Zhang, C., Yue, S., Panagiotopoulos, A. Z., Klein, M. L. & Wu, X. Dissolving salt is not equivalent to applying a pressure on water. Nat. Commun. 13, 822 (2022).
Liu, R. et al. Structural and dynamic properties of solvated hydroxide and hydronium ions in water from ab initio modeling. J. Chem. Phys. 157, 024503 (2022).
Hamann, D. R. Optimized norm-conserving Vanderbilt pseudopotentials. Phys. Rev. B 88, 085117 (2013).
Marzari, N. & Vanderbilt, D. Maximally localized generalized Wannier functions for composite energy bands. Phys. Rev. B 56, 12847–12865 (1997).
Pizzi, G. et al. Wannier90 as a community code: new features and applications. J. Phys.: Condens. Matter 32, 165902 (2020).
Zhang, L. et al. Deep neural network for the dielectric response of insulators. Phys. Rev. B 102, 041121 (2020).
Wang, H., Zhang, L., Han, J. & E, W. DeePMD-kit: a deep learning package for many-body potential energy representation and molecular dynamics. Computer Phys. Commun. 228, 178–184 (2018).
Zhang, L. et al. in Advances in Neural Information Processing Systems 31 (eds S. Bengio et al.) 4436–4446 (Curran Associates, Inc., 2018).
Eastwood, J. W., Hockney, R. W. & Lawrence, D. N. P. 3M3DP - THE 3-DIMENSIONAL PERIODIC PARTICLE-PARTICLE-PARTICLE-MESH PROGRAM. Computer Phys. Commun. 19, 215–261 (1980).
Marzari, N., Mostofi, A. A., Yates, J. R., Souza, I. & Vanderbilt, D. Maximally localized Wannier functions: theory and applications. Rev. Mod. Phys. 84, 1419–1475 (2012).
Piaggi, P. M., Panagiotopoulos, A. Z., Debenedetti, P. G. & Car, R. Phase equilibrium of water with hexagonal and cubic ice using the SCAN functional. J. Chem. Theory Comput. 17, 3065–3077 (2021).
Zhang, C. et al. Data from “molecular-scale insights into the electrical double layer at oxide-electrolyte interfaces”. figshare. Dataset https://doi.org/10.6084/m9.figshare.25461061.v2 (2024).
Acknowledgements
This work was conducted within the “Chemistry in Solution and at Interfaces” (CSI) Center funded by the USA Department of Energy under Award DE-SC0019394 (C.Z., M.F.C.A., Z.K.G., A.S.R., Y.L., P.M.P., X.W., R.C., and A.S.), with further support from DoE BES, CSGB Division under Award DESC0007347. The work at the Lawrence Livermore National Laboratory was performed under the auspices of the U.S. Department of Energy under Contract DE-AC52-07NA27344 (M.F.C.A.). This research used computational resources from the National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy Office of Science User Facility located at Lawrence Berkeley National Laboratory, operated under Contract No. DE-AC02-05CH11231, and from the Oak Ridge Leadership Computing Facility at the Oak Ridge National Laboratory, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC05-00OR22725. We also acknowledge computational resources from the Princeton Research Computing resources at Princeton University which is consortium of groups led by the Princeton Institute for Computational Science and Engineering (PICSciE) and Office of Information Technology’s Research Computing. We thank Michiel Sprik for insightful discussions and comments on our manuscript.
Author information
Authors and Affiliations
Contributions
A.S. and R.C. designed the project. C.Z. and M.F.C.A. carried out the simulations and performed the analysis. C.Z., A.S., and R.C. wrote the manuscript. M.F.C.A. and Z.K.G. reviewed the manuscript. A.S.R., Y.L., P.M.P., and X.W. contributed fruitful discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Chao Zhang, Jun Cheng, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, C., Calegari Andrade, M.F., Goldsmith, Z.K. et al. Molecular-scale insights into the electrical double layer at oxide-electrolyte interfaces. Nat Commun 15, 10270 (2024). https://doi.org/10.1038/s41467-024-54631-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54631-1
This article is cited by
-
Comparative study of the effect of strong and weak proton donor and coagulation baths on the properties of millimeter-sized spherical porous ZrO2 supports prepared via chitosan gelation
Journal of Sol-Gel Science and Technology (2026)
-
Multiscale Theoretical Calculations Empower Robust Electric Double Layer Toward Highly Reversible Zinc Anode
Nano-Micro Letters (2026)
-
Machine learning of charges and long-range interactions from energies and forces
Nature Communications (2025)
-
Deep learning potential simulations of Pt(211)/water interface at potential of zero charge
Ionics (2025)
-
Computational Modeling of Solid–Liquid Interfaces: From Ab Initio Methods to Machine-Learning Potentials
Journal of the Indian Institute of Science (2025)
