
Abstract
Macropinocytosis has emerged as a scavenging pathway that cancer cells exploit to survive in a nutrient-deprived microenvironment. Tumor cells are especially reliant on glutamine for their survival, and in pancreatic ductal adenocarcinoma (PDAC) cells, glutamine deficiency can enhance the stimulation of macropinocytosis. Here, we identify the atypical protein kinase C (aPKC) enzymes, PKCζ and PKCι, as regulators of macropinocytosis. In normal epithelial cells, aPKCs associate with the scaffold proteins Par3 and Par6 to regulate cell polarity, affecting several targets, including the Par1 kinases and we find that each of these proteins is required for macropinocytosis. Mechanistically, aPKCs are regulated by EGFR signaling or by the transcription factor CREM to promote the Par3 relocation to microtubules, facilitating macropinocytosis in a dynein-dependent manner. Importantly, cell fitness impairment caused by aPKC depletion is rescued by the restoration of macropinocytosis and aPKCs support PDAC growth in vivo. Our findings enhance our understanding of the mechanistic underpinnings that control macropinocytic uptake in the context of metabolic stress.
Similar content being viewed by others
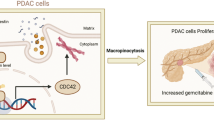

Introduction
Macropinocytosis, also known as “cellular drinking”, is a clathrin-independent endocytic pathway that non-selectively internalizes extracellular cargo into large vesicles known as macropinosomes1,2,3. In Ras-mutant cells, this uptake pathway is an amino acid supply route that allows the cancer cells to survive and proliferate despite the nutrient scarcity present in the tumor microenviroment4,5,6. By internalizing extracellular proteins, such as serum albumin, and targeting them for lysosome-dependent degradation, macropinocytosis provides a source of protein-derived amino acids. Under physiological conditions, these protein-derived amino acids can account for almost half of the intracellular and extracellular amino acid pools7. Importantly, it has been well established in multiple mouse models of cancer that macropinocytosis inhibition in vivo significantly suppresses tumor growth4,8,9,10.
Macropinocytosis in Ras-mutant cells is elevated as a response to amino acid deprivation3,5,6,11. The vast majority of pancreatic ductal adenocarcinoma (PDAC) tumors harbor a mutation in KRAS, and we previously demonstrated that macropinocytosis in PDAC cells is considerably enhanced when glutamine is limiting5. This is physiologically important since in human PDAC tumors glutamine is the most depleted amino acid relative to adjacent non-neoplastic tissue7. Glutamine is a critical nutrient in tumors as it supports various facets of cancer metabolism, including lipid and protein biosynthesis, the de novo synthesis of nucleotides, the urea cycle, glutathione production, and the biosynthesis of other amino acids12. Downstream aspects of the macropinocytosis pathway are also impacted by amino acid deprivation. For example, in KRAS-mutant mouse embryonic fibroblasts (MEFs), the lysosome-dependent degradation of macropinocytosed protein is enhanced by amino acid starvation11. This is consistent with the observation that in PDAC xenograft tumors, the highest macropinocytic rates are detected in the tumor core regions, which are depleted of glutamine and other amino acids relative to the tumor periphery5. While the functional importance of macropinocytosis in PDAC tumors is highly appreciated, the specific regulators that control macropinocytosis in the context of nutrient stress remain poorly understood.
Here, we identify PKCζ and PKCι, which constitute the family of atypical Protein Kinase C (aPKC) enzymes, as regulators of macropinocytosis. In normal epithelial cells, aPKCs are integral to a protein network that maintains cell polarity, a process disrupted in cancer cells. However, despite the loss of cell polarity, this protein network remains crucial for pro-tumorigenic activities, such as cell migration and invasion13,14. This suggests that, depending on the context, these proteins can actively contribute to cancer progression. We find that with glutamine stress, aPKCs are repurposed in PDAC cells, along with other cell polarity proteins such as Par-1a, Par3, and Par6, to drive macropinocytosis. In cell polarity, aPKCs control microtubule dynamics15,16, which are essential to maintain epithelial cells in a polarized state16,17. Interestingly, in PDAC cells, we find that the microtubule cytoskeleton and dynein-dependent microtubule transport are required for macropinocytosis and that this is associated with aPKC-dependent redistribution of Par3 to microtubules. Mechanistically, we find that glutamine deficiency primes the aPKCs for activation by EGFR. Glutamine stress can also be triggered by specifically targeting glutamine-utilizing enzymes, as is the case with 6-diazo-5-oxo-L-norleucine (DON), a glutamine antagonist that enhances macropinocytosis in PDAC cells5. Interestingly, we find that pharmacological inhibition of glutamine metabolism with DON increases the expression of the aPKCs through the transcription factor CREM. Functionally, we demonstrate that in low glutamine conditions, aPKC depletion leads to reduced proliferative capacity, a phenotype that is rescued when macropinocytosis is restored. Moreover, we elucidate that aPKCs regulate macropinocytosis in vivo and that they are essential to maintain PDAC tumor growth. Our interrogations of human PDAC clinical samples and datasets reveal that aPKCs are upregulated in tumors, contribute to patient survival, and, in a subset of tumors, are primed for activation. Altogether, our work sheds light on the mechanistic underpinnings that drive macropinocytosis in response to metabolic stress and points to the possibility of targeting cell polarity proteins in cancer.
Results
PKCζ and PKCι regulate macropinocytosis in PDAC cells
Metabolic stress caused by glutamine deprivation stimulates macropinocytosis; however, a comprehensive understanding of modulators that can control uptake in the context of nutrient stress is lacking5. In order to gain insight into the pathways that might broadly function in macropinocytosis in the context of glutamine stress, we performed a high-throughput (HT) siRNA screen using an HT-compatible macropinocytosis assay that we previously developed18,19. Both AsPC-1 and HPAF-II cells display robust stimulation of macropinocytosis upon glutamine starvation (Fig. 1a–d), but we used AsPC-1 cells for the screen since the morphology of these cells was more compatible with HT imaging. Macropinocytosis was detected through the uptake of FITC-dextran, an established marker of macropinosomes and macropinosomes were quantified by image-based high-content analysis. Using our HT assay, we performed a kinome-wide siRNA screen (siOTP; Dharmacon) to identify kinases involved in controlling macropinocytosis in cells that were exposed to glutamine stress. With this approach, we identified the atypical Protein Kinase C (aPKC) isoforms PKCζ and PKCι as modulators of glutamine-stress-induced macropinocytosis (Fig. 1e and Supplementary Fig. 1). We next validated the role of the aPKCs in macropinocytosis by performing knockdown experiments using independent siRNAs from a different library, which allowed us to mitigate potential off-target effects. For validation, in addition to AsPC-1 cells, we also used HPAF-II cells. Specific knockdown of the individual isoforms in AsPC-1 and HPAF-II cells was confirmed by western blot (Fig. 1f, g). Validating a role for each aPKC in macropinocytosis, we found that knockdown of either PKCζ or PKCι suppressed macropinocytosis in glutamine-deprived conditions (Fig. 1h–k). In addition, we also observed a significant diminishment of macropinocytosis in the same conditions when cells were treated with ACPD, a specific pharmacological inhibitor of the aPKCs20 (Fig. 1l–o).

a, b Representative fluorescent microscopy images from a FITC-dextran (green) macropinocytosis assay (a) and macropinocytosis quantification (b) in AsPC-1 cells cultured in glutamine-replete (2 mM, + Q) or glutamine-starved (0 mM, -Q) media for 24 h. Nuclei are stained with DAPI (blue). Data are shown relative to + Q. N = 3. Scale bar, 20 μm. *P(+Q vs -Q) = 0.0142. c, d Representative images (c) and quantification (d) of macropinocytosis in HPAF-II cells cultured in glutamine-replete (+ Q) or glutamine-starved (− Q) media. N = 3. Scale bar, 20 μm. **P(+Q vs -Q) = 0.0026. e Schematic depicting the steps employed for the siRNA kinome-wide screen in glutamine-stressed AsPC-1 cells. Created in BioRender. Commisso, C. (2022) BioRender.com/o37o964. f, g Immunoblots assessing PKCζ and PKCι levels in AsPC-1 (f) and HPAF-II (g) cells after transfection with non-targeting control siRNA (siCTRL) or siRNAs targeting PRKCZ (siPRKCZ#1 and siPRKCZ#2) and PRKCI (siPRKCI#1 and siPRKCI#2). β-actin was used as a loading control except for AsPC-1 cells transfected with PRKCI siRNAs, where α-tubulin was used. h, i Representative images of macropinocytosis in glutamine-starved AsPC-1 (h) and HPAF-II (i) cells transfected with the indicated siRNAs. Scale bar, 20 μm. j Quantification of macropinocytosis in glutamine-starved AsPC-1 cells transfected with the indicated siRNAs. Data are shown relative to siCTRL. N = 3. *P(siCTRL vs siPRKCZ#1) = 0.0234; *P(siCTRL vs siPRKCZ#2) = 0.0154; **P(siCTRL vs siPRKCI#1) = 0.0065; **P(siCTRL vs siPRKCI#2) = 0.0011). k Quantification of macropinocytosis in glutamine-starved HPAF-II cells transfected with the indicated siRNAs. Data are shown relative to siCTRL. N = 3. **P(siCTRL vs siPRKCZ#1) = 0.0035; *P(siCTRL vs siPRKCZ#2) = 0.0151; **P(siCTRL vs siPRKCI#1) = 0.0028; *P(siCTRL vs siPRKCI#2) = 0.0107. l, m Representative images (l) and quantification (m) of macropinocytosis in glutamine-starved AsPC-1 cells treated with vehicle (water) or ACPD at 4 μM for 72 hrs. Data are shown relative to the vehicle. N = 3. Scale bar, 20 μm. *P (Vehicle vs ACPD) = 0.0102. n, o Representative images (n) and quantification (o) of macropinocytosis in glutamine-starved HPAF-II cells treated with vehicle or ACPD at 25 μM for 72 hrs. Scale bar, 20 μm. **P (Vehicle vs ACPD) = 0.002. Statistical significance was calculated using unpaired two-tailed Student’s t test with Welch’s correction. *P < 0.05, **P < 0.01. All data are presented as the mean ± SEM. N indicates independent experiments. Source data are provided as a Source Data file.
Following incorporation into a nascent macropinosome, macropinocytic cargo is trafficked and targeted for degradation by lysosomes4. This process can be tracked using a macropinocytosed self-quenching BODIPY dye-conjugated form of bovine serum albumin (DQ-BSA), which emits a bright fluorescent signal only after undergoing lysosome-dependent proteolytic digestion4. To evaluate this maturation process, we performed a pulse-chase experiment where we incubated AsPC-1 cells cultured in either nutrient-replete or glutamine-deprived conditions with DQ-BSA for 30 min and subsequently switched them to DQ-BSA-free media for 4 h. As expected, DQ-BSA fluorescence was increased in glutamine-deprived conditions relative to nutrient-replete conditions (Supplementary Fig. 2a, b). Consistent with the aPKCs functioning in the early steps of nascent macropinosome formation, DQ-BSA degradation in glutamine-deprived conditions was significantly reduced upon PKCζ or PKCι depletion (Supplementary Fig. 2c, d). The next step in the macropinocytosis pathway entails the intracellular release of protein-derived amino acids that contribute to central carbon metabolism4,7. To assess the contribution of aPKCs to the augmentation of the intracellular amino acid pools in the context of glutamine stress, we performed a reverse labeling isotope-tracing experiment that enables specific quantitation of protein-derived amino acids produced by albumin catabolism7. Intracellular metabolites in AsPC-1 cells were labeled by culturing the cells in 13C6-glucose and 13C5-glutamine for 6 days. Cells were then transfected with control siRNAs or siRNAs targeting PKCζ or PKCι and subsequently cultured in glutamine-free media containing 13C6-glucose with or without BSA as a nutrient source for 48 h. Metabolites were extracted, and the abundance of intracellular polar metabolites was determined by gas chromatography/mass spectrometry (GC/MS) (Supplementary Fig. 2e). In control cells, we observed that addition of BSA as a nutrient source increased the levels of several unlabeled protein-derived non-essential amino acids (NEAA) and that this ability to generate these protein-derived amino acids was lost upon aPKC depletion (Supplementary Fig. 2f). Consistent with an effect on central carbon metabolism, we also observed similar results with the TCA cycle intermediates succinate, fumarate, and malate (Supplementary Fig. 2g). This indicates that when glutamine is deficient, PDAC cells rely on aPKCs to obtain nutrients derived from macropinocytic cargo, allowing them to support their oncogenic metabolic demands.
We next asked whether glutamine stress could also stimulate other endocytic pathways, such as clathrin-dependent endocytosis (CDE), and whether this was dependent on aPKCs. We analyzed CDE by monitoring the uptake of transferrin, an iron-binding protein specifically taken up by CDE21. We found that CDE was not increased in glutamine-deprived cells relative to control and that CDE was unaffected by knockdown of the aPKCs (Supplementary Fig. 3a–d). Altogether, our data indicate that the aPKCs are specifically required to control macropinocytosis in the context of glutamine stress.
PAR proteins and microtubule transport are required for glutamine stress-induced macropinocytosis
aPKCs are known to regulate apical-basal polarity in epithelial cells, which is essential for maintaining epithelial structure and integrity22,23. In cancer, the prevailing notion is that cell polarity is lost; however, different members of this protein network are known to be involved in several tumorigenic processes23,24,25. In cell polarity, aPKCs associate with the scaffold proteins Par3 and Par6, constituting the Par or cell polarity complex that controls the function and subcellular localization of multiple cell polarity-related substrates, such as Par1 kinases15,16,23,26,27,28. In our screen, in addition to the aPKCs, we also identified the microtubule-associated Par-1a kinase as a ‘hit’ (Supplementary Fig. 1). Considering the role of aPKCs and Par1 kinases in the modulation of epithelial cell polarity, we hypothesized that cell polarity proteins might be broadly required for regulating glutamine stress-driven macropinocytosis. To test this idea, we investigated the Par-1a kinase and the scaffold proteins Par3 and Par6 since these proteins form a complex with aPKC that regulates cell polarity. We found that knockdown of Par-1a, Par3, or Par6 suppressed macropinocytosis to a similar extent as aPKC depletion in glutamine-starved conditions (Fig. 2a, b and Supplementary Fig. 3e–h). These data highlight a previously unrecognized role for cell polarity proteins in the regulation of macropinocytosis.

a, b Representative images (a) and quantification (b) of macropinocytosis in glutamine-starved AsPC-1 cells transfected with the indicated siRNAs. Data are shown relative to siCTRL. N = 3. Scale bar, 20 μm **P(siCTRL vs siPAR1a#1 = 0.007); *P(siCTRL vs siPAR1a#2=0.0192); *P(siCTRL vs siPARD3#1 = 0.0368); **P (siCTRL vs siPARD3#2 = 0.0044); ***P(siCTRL vs siPARD6B#1 = 0.0004); **P(siCTRL vs siPARD6B#2 = 0.001). c, d Representative images (c) and quantification (d) of macropinocytosis in glutamine-starved AsPC-1 cells treated for 18 hrs with vehicle (water) or the microtubule inhibitors colchicine (COL, 100 ng/mL) and paclitaxel (PTX, 200 nM). Data are shown relative to the vehicle. N = 3. Scale bar, 20 μm. *P(vehicle vs COL = 0.0104); *P(vehicle vs PTX) = 0.0138). e, f Representative images (e) and quantification (f) of macropinocytosis in glutamine-starved AsPC-1 cells treated with vehicle (water) or the dynein inhibitor Ciliobrevin D (Ciliob D) for 30 minutes at 50 μM. Data are shown relative to the vehicle. N = 3. Scale bar, 20 μm. **P(vehicle vs Ciliob D = 0.0027). g Par3 protein (red) and Tubulin (green) immunostaining in AsPC-1 cells transfected with the indicated siRNAs and cultured in glutamine-replete (+) or glutamine-free (−) media. Nuclei are stained with DAPI (blue). Images are representative of N = 2. Scale 20 μm. The bottom row is higher magnification images of the boxed areas. Scale bar, 4 μm. h Quantification of the percent of Par3 protein found in subcellular puncta versus the cytoplasm in cells transfected with siCTRL (n = 42 cells/ + Q, n = 44 cells/-Q), siPRKCZ (n = 40 cells/ + Q, n = 35 cells/ − Q) or siPRKCI (n = 39 cells/ + Q, n = 38 cells/-Q). For data presentation, outliers were removed using a ROUT method test. All significant P were < 0.0001; nsP(siPRKCZ+Q vs siPRKCZ -Q) = 0.89); nsP(siPRKCI+Q vs siPRKCI -Q) = 0.291). i Immunoblot of Par3 and aPKCs proteins for the conditions described in (g). β-actin is used as a loading control. Data are representative of N = 3. j Dynein (green) and Par3 (red) protein immunostaining in AsPC-1 cells cultured in glutamine-replete (+) or free (-) media. Images are representative of N = 2. Scale bar, 10 μm. The second and fourth columns are higher magnification images of the boxed areas. Scale bar, 2 μm. Statistical significance was calculated using unpaired two-tailed Student’s t test with Welch’s correction. Ns, non-significant, *P < 0.05, **P < 0.01, ***P < 0.001. Data are presented as the mean ± SEM except for (h), where data are presented as a minimum to maximum box-and-whisker plot with the median indicated by the center line. N indicates independent experiments. Source data are provided as a Source Data file.
In the establishment of cell polarity, there is specific spatiotemporal regulation of both actin and microtubule dynamics by the Par complex17,27. While the role of actin in macropinocytosis has been extensively studied, it is not clear the extent to which microtubules contribute to macropinocytosis. There is some evidence suggesting that microtubules might be involved in the post-internalization trafficking events of mature macropinosomes29, but whether microtubules function in nascent macropinosome formation has not been examined. Since aPKCs are known to modulate microtubule dynamics17, and we detected the microtubule-associated Par-1a kinase as a macropinocytic modulator, we further examined whether microtubules could be involved in regulating macropinocytic uptake. We found that disruption of the microtubule network with either colchicine or paclitaxel, which would destabilize or stabilize microtubules, respectively, impaired glutamine stress-driven macropinocytosis (Fig. 2c, d). We next evaluated whether glutamine stress had the capacity to alter the morphology of the microtubule network. In control cells, glutamine deprivation caused the reorganization of the microtubules such that tubulin was more greatly associated with plasma membrane ruffles; however, this was not dependent on aPKCs (Supplementary Fig. 4a). These data suggested that aPKCs might be regulating macropinocytosis independent of plasma membrane ruffling. To confirm that aPKC loss does not impair membrane ruffling, we analyzed in these conditions the dynamics of the actomyosin system by co-staining the actin cytoskeleton with Myosin Light Chain 2 (MLC2), a protein required for the function of the myosin protein complex30. As expected, glutamine deprivation caused actin reorganization to induce the membrane ruffling, and this was accompanied by a preferential relocation of MLC2 to the plasma membrane (Supplementary Fig. 4b). Neither actin reorganization nor MLC2 subcellular relocation was affected in aPKC-knockdown cells, confirming that aPKCs are not required for the formation of the ruffles (Supplementary Fig. 4b). To form a nascent macropinosome, it is thought that cytoskeletal contractile forces are required to propel the macropinosome inwards, separating the macropinosome from the plasma membrane in a scission or fission process. To test this idea, we examined the function of the microtubule-associated motor protein dynein, which mediates the retrograde-directed transport of different cargoes along the microtubule network, including endocytic vesicles such as endosomes31. Dynein inhibition with ciliobrevin D nearly completely blocked macropinocytosis (Fig. 2e, f), suggesting that aPKCs could be mediating macropinosome formation through mechanisms involving the regulation of microtubule motor functions.
The cell polarity protein that is known to associate with dynein in the regulation of microtubule motor properties is the scaffold protein Par332,33. In the establishment and maintenance of cell polarity, Par3 subcellular localization is in part controlled by aPKCs that phosphorylate Par3 to promote its dissociation from the Par complex and its relocation to adherens junctions34,35,36. Considering that Par3 loss efficiently impaired macropinocytosis, we next examined whether glutamine stress was altering Par3 subcellular localization and whether any observed changes in Par3 cellular distribution were dependent on aPKC. Remarkably, we found that glutamine deprivation promoted the localization of Par3 into microtubule-associated puncta, which was dependent on aPKCs (Fig. 2g, h and Supplementary Fig. 5a, b). We found that the enhancement of Par3 protein on the microtubules was due to a redistribution of cytosolic Par3 protein to the microtubules and not due to an increase in overall Par3 protein levels (Fig. 2i and Supplementary Fig. 5c). Considering both Par3 and dynein inhibition strongly suppressed macropinocytic uptake, we next asked whether dynein could undergo a similar subcellular relocation as Par3. Dynein and Par3 were co-stained in both nutrient-replete and glutamine-deprived conditions, and we observed that both proteins undergo a similar subcellular re-distribution in glutamine stress, co-localizing at the plasma membrane and in puncta in the nuclear periphery. We did not observe dynein and Par3 co-localization in nutrient-replete conditions (Fig. 2j). Taken together, our data indicate that upon glutamine stress, aPKCs function to relocate Par3 to microtubules, where Par3 can co-operate with Dynein to regulate microtubule motor properties, facilitating macropinocytosis.
To further examine cell polarity protein signaling, we evaluated whether post-translational modification of Par-1a was affected by glutamine stress. For activation, Par-1a is phosphorylated at the threonine 211 (T211) residue located in the activation loop of the protein37. We found that phosphorylation of T211 was increased in glutamine-deprived cells, and this increase in phosphorylation was suppressed by knockdown of either PKCζ or PKCι (Supplementary Fig. 6a, b). These data indicate that glutamine stress activates Par-1a, a process that is dependent on aPKCs, reinforcing the central role of these kinases in controlling macropinocytosis promoted by metabolic stress.
Discrete modes of glutamine stress induction differentially regulate the aPKCs
6-Diazo-5-oxo-L-norleucine (DON) is a glutamine analog that broadly inhibits glutamine metabolism and drives metabolic stress that stimulates macropinocytic induction to levels comparable to glutamine deprivation5 (Supplementary Fig. 7a–d). As we observed for glutamine depletion, knockdown of the aPKCs or aPKC inhibition via ACPD blocked DON-stimulated macropinocytosis (Supplementary Fig. 7e–l). Also, DON-induced macropinocytosis was abrogated by knockdown of either Par-1a, Par3, or Par6 (Supplementary Fig. 7m–p). Since metabolic stress caused pharmacologically can often mechanistically differ from stress caused by nutrient starvation, we next investigated how these different modes of glutamine stress might impact the regulation of the aPKCs. Interestingly, we found that only the DON treatment had the capacity to increase the expression of both PKCζ and PKCι proteins and transcripts (Fig. 3a–d and Supplementary Fig. 8a, b). We next examined the phosphorylated forms of PKCζ and PKCι at the T560 and T555 residues, respectively, which are required for the full activation of these kinases38,39,40,41. We observed that only glutamine deprivation promoted an increase in the p-aPKC/aPKC ratio (Fig. 3e, f and Supplementary Fig. 8c, d). With DON treatment, although the p-aPKC/aPKC ratio was unaffected, both the phosphorylated and total forms of the proteins were enhanced (Fig. 3g, h and Supplementary Fig. 8e, f). These results suggest that DON modulates PKCζ and PKCι function by increasing their expression, while glutamine starvation primes these kinases for activation.

a, b Immunoblots assessing PKCζ and PKCι total protein levels in AsPC-1 (a) and HPAF-II (b) cells cultured for 24 hrs in glutamine-replete media treated with vehicle (water) or 2 mM DON or cultured in glutamine-free conditions. β-actin was used as a loading control. Data are representative of at least N = 3. Densitometry quantifications are presented relative to the vehicle control and normalized to β-actin. c Relative PRKCZ and PRKCI mRNA levels as assessed by RT-qPCR in AsPC-1 cells cultured in the conditions described in (a) and (b). Data are shown relative to the vehicle control. N = 3. PRKCZ: *P(Vehicle control vs DON) = 0.017); nsP(Vehicle control vs -Q) = 0. 0.7738; PRKCI: *P (Vehicle control vs DON) = 0.0233); nsP(Vehicle control vs -Q) = 0.7411. d Relative PRKCZ and PRKCI mRNA levels as assessed by RT-qPCR in HPAF-II cells cultured in the conditions described in (a) and (b). Data are shown relative to the vehicle control. N = 4. PRKCZ: *P(Vehicle control vs DON) = 0.0335); nsP(Vehicle control vs -Q) = 0.9124; PRKCI: *P(Vehicle control vs DON) = 0.0356; nsP(Vehicle control vs -Q) = 0.6503. e, f Immunoblots assessing levels of the phospho-proteins p-PKCζT560 and p-PKCιT555, and total PKCζ and PKCι protein levels in AsPC-1 (e) and HPAF-II (f) cells cultured in glutamine-replete or glutamine-free media. β-actin was used as a loading control. Densitometry quantifications are presented relative to the glutamine-replete condition and values for the phospho-forms are normalized to the total protein. Data are representative of at least N = 3. g, h Similar immunoblots as in (e) and (f) in AsPC-1 (g) and HPAF-II (h) cells cultured in glutamine-replete media treated with vehicle or DON. Densitometry quantifications are presented relative to vehicle and values for the phospho-forms are normalized to the total protein. Data are representative of N = 2. i, j Representative images (i) and quantification (j) of macropinocytosis in AsPC-1 cells transfected with the indicated siRNAs and cultured in glutamine-replete media treated with EGF at 100 nM for 10 min. Data are shown relative to siCTRL. N = 3. Scale bar, 20 μm.**P(siCTRL vs siPRKCZ) = 0.0026); *P(siCTRL vs siPRKCI) = 0.0106. Statistical significance was calculated using unpaired two-tailed Student’s t test with Welch’s correction. ns, non-significant, *P < 0.05, **P < 0.01. All data are presented as the mean ± SEM. N indicates independent experiments. Source data are provided as a Source Data file.
Glutamine deprivation promotes macropinocytosis through EGFR5; therefore, we next explored the interconnectedness between aPKC and EGFR signaling in regulating macropinocytosis upon glutamine starvation. To do this, we took advantage of the fact that stimulation with EGF ligands can mimic glutamine depletion and promote macropinocytosis5. Interestingly, we found that knockdown of either PKCζ or PKCι significantly reduced EGF-induced macropinocytosis, suggesting that the aPKCs were indeed functioning to mediate EGFR signaling to drive uptake in PDAC cells (Fig. 3i, j). Depending on the context, there are several different ways that aPKCs can regulate EGFR signaling. In mouse embryonic fibroblasts and lung epithelial cells, upon activation, aPKCs can indirectly influence EGFR signal potentiation by phosphorylating EGFR ligands, priming them for proteolytic cleavage that results in the release of soluble ligand42,43. In glioblastoma cells, aPKC phosphorylation-dependent activation is downstream of EGFR and serves to control glioblastoma progression44. In PDAC cells, we found that EGF treatment enhanced aPKC phosphorylation to similar extents as observed with glutamine deprivation (Supplementary Fig. 9a–d). Moreover, aPKC phosphorylation in glutamine-deprived conditions was EGFR-dependent as it was inhibited by erlotinib, an EGFR inhibitor (Supplementary Fig. 9e–h). Interestingly, although DON-induced macropinocytosis is aPKC-dependent, we did not observe enhanced EGFR activation in DON-treated cells, nor was DON-induced macropinocytosis suppressed by erlotinib (Supplementary Fig. 10a–d). Altogether, these data indicate that the mechanistic underpinnings of nutrient stress-driven macropinocytosis might be influenced by how the metabolic stress is achieved, but more importantly, our findings reinforce the importance of the cell polarity proteins in regulating macropinocytosis, independent of the metabolic stress source.
In glutamine deprivation, Pak kinase is downstream of the EGFR signal that drives macropinocytosis activation5,45,46. In some contexts, aPKCs are known to control Pak1 activation via phosphorylation at threonine 423 (T423)47. Since glutamine deprivation primes aPKC for activation through EGFR signaling, we asked whether, in PDAC cells aPKC can orchestrate the activation of Pak1. We found that knockdown of either PKCζ or PKCι did not prevent EGFR activation, but that Pak1 activation was inhibited when aPKCs were depleted (Supplementary Fig. 10e–h). These data indicate that in glutamine deprivation EGFR primes aPKCs for activation, thereby also activating Pak1, further confirming the essential role of aPKC in controlling nutrient stress-driven macropinocytosis.
Glutamine antagonism upregulates aPKC expression through the transcription factor CREM
Having established that priming of the aPKCs is regulated by EGFR signaling to control macropinocytosis in the context of glutamine starvation, we were next interested in examining the mechanistic underpinnings of how aPKC expression is upregulated by DON. Since we observed that DON enhanced the transcript levels of both PKCζ and PKCι, we hypothesized that this increase in expression might be controlled by a specific transcription factor. Using the ChiP Enrichment Analysis (ChEA) dataset, an application that predicts target gene-transcription factor associations by data aggregation from low and high-throughput studies48, we generated a list of 29 and 32 transcription factors (TFs) associating with the PRKCZ or PRKCI promoters, respectively (Supplementary Fig. 11a). For further analyses, we focused on nine TFs that were common to both promoters (Fig. 4a). To determine whether any of these TFs or any other potential drivers of macropinocytosis are upregulated by DON, we performed transcriptomics using RNA-Sequencing (RNASeq) in AsPC-1 cells treated with DON or vehicle control. Using this approach, we found that DON upregulated the expression of two of the nine TFs: cyclic-AMP responsive element modulator (CREM) and Rest corepressor 3 (RCOR3; Fig. 4b). As expected, we observed upregulation of PRKCZ and PRKCI in DON-treated cells, but we also found that other cell polarity transcripts including PAR1A, PARD3 and PARD6B were increased (Supplementary Fig. 11b). This further emphasizes the relevance of these proteins in the glutamine stress response.

a Predicted transcription factors binding to the PRKCZ and PRKCI promoters based on the ChEA dataset. b Top: Violin plot showing CREM and RCOR3 transcript counts in AsPC-1 cells treated with vehicle or DON (2 mM, 24 h) as assessed by RNA-seq. Bottom: Fold-change relative to vehicle and q-values for CREM and RCOR3 transcripts. c, d Representative images of macropinocytosis in DON-treated AsPC-1 (c) and HPAF-II (d) cells transfected with indicated siRNAs. Scale bar, 20 µm. e Quantification of macropinocytosis in AsPC-1 cells under the conditions described in (c). Values are relative to siCTRL. N = 4 (siCREM) or N = 3 (siRCOR3). **P(siCTRL vs siCREM) = 0.0019; nsP(siCTRL vs siRCOR3) = 0.5820. f Quantification of macropinocytosis in HPAF-II cells under the conditions described in (d). Values are relative to siCTRL. N = 3. **P(siCTRL vs siCREM) = 0.0086; nsP(siCTRL vs siRCOR3) = 0.5718. g Relative CREM mRNA levels measured by RT-qPCR in AsPC-1 cells treated with vehicle or DON (2 mM, 24 h). N = 3. **P(Vehicle vs DON) = 0.0049. h Relative CREM mRNA levels measured by RT-qPCR in HPAF-II cells in conditions described in (g). N = 3. *P(Vehicle vs DON) = 0.0314. i, j Immunoblots assessing CREM protein levels in AsPC-1 (i) and HPAF-II (j) cells cultured in glutamine-replete media containing vehicle or DON (2 mM, 24 h). β-actin was used as a loading control. Data are representative of N = 3. Densitometry quantifications are presented relative to vehicle and values are normalized to β-actin. k Relative PRKCZ and PRKCI mRNA levels assessed by RT-qPCR in the vehicle and DON-treated AsPC-1 cells transfected with indicated siRNAs. N = 3. PRKCZ: *P(siCTRL vehicle vs siCTRL DON) = 0.0153; *P(siCTRL DON vs siCREM DON) = 0.0498; nsP(siCREM vehicle vs siCREM DON) = 0.2779; PRKCI: **P(siCTRL vehicle vs siCTRL DON) = 0.002; *P(siCTRL vehicle vs siCREM DON) = 0.0479; nsP(siCREM vehicle vs siCREM DON) = 0.7431. l Immunoblots assessing aPKC total protein levels in AsPC-1 cells cultured in glutamine-replete media containing vehicle or DON. β-actin was used as a loading control. Data are representative of at least N = 3. Densitometry quantifications are presented relative to vehicle and values are normalized to β-actin. m Consensus sequence of CREM binding motif obtained from the JASPAR CORE database. n Location of the CRE consensus sequence in the PRKCZ promoter. o Fold enrichment of CREM binding to PRKCZ gene promoter in AsPC-1 cells cultured in glutamine-replete media containing vehicle or DON. N = 3. ***P(DON + IgG vs DON + CREM) = 0.0004; ***P(Vehicle + CREM vs DON + CREM) = 0.0002; nsP(Vehicle + IgG vs Vehicle + CREM) = 0.0743. p Graphical depiction of macropinocytosis regulated by aPKC isoforms during glutamine stress. Statistical significance was determined by unpaired two-tailed Student’s t test with Welch’s correction. ns, non-significant, *P < 0.05, **P < 0.01, ***P < 0.001. All data are presented as the mean ± SEM. N indicates independent experiments. Source data are provided as a Source Data file.
To elucidate whether CREM and/or RCOR3 might be involved in DON-stimulated macropinocytosis, we examined uptake in PDAC cells where these TFs were knocked down using specific siRNAs that were validated by RT-qPCR (Supplementary Fig. 11c, d). We observed that the depletion of CREM attenuated DON-stimulated macropinocytosis, but RCOR3 did not (Fig. 4c–f). Interestingly, in AsPC-1 cells, macropinocytosis driven by glutamine starvation was unaffected by the knockdown of either CREM or RCOR3 (Supplementary Fig. 11e, f). However, in HPAF-II cells, CREM depletion did block macropinocytosis caused by glutamine deprivation, suggesting possible heterogeneity in the contextual selectivity of CREM in regulating uptake (Supplementary Fig. 11g, h). Consistent with CREM playing a role in DON-mediated responses, we found that CREM transcription and protein expression were enhanced by DON (Fig. 4g–j and Supplementary Fig. 12a, b). We next deciphered whether the observed role of CREM in macropinocytosis is tied to the regulation of aPKC expression. We measured the expression of PKCζ and PKCι at the transcript and protein levels in DON-treated conditions in PDAC cells where CREM was knocked down. We determined that CREM depletion suppresses the DON-dependent increases in aPKC transcription (Fig. 4k). Equivalent results were obtained at the protein level (Fig. 4l and Supplementary Fig. 12c). We next evaluated the capability of CREM to bind to the aPKC promoters. CREM is known to recognize palindromic sequences known as cAMP Response Elements (CRE), with the consensus 5’-TGACGTCA-3’ sequence (Fig. 4m)49,50. It is also known to bind the shorter sequences TGAC and GTCA, which are known as half-CRE sites49,50. We observed that the consensus sequence was present in the PRKCZ promoter (Fig. 4n) and that the PRKCI promoter contained several different half-CRE sites. We decided to examine the binding of CREM to the PRKCZ promoter because the binding region was more straightforward to predict. We performed chromatin immunoprecipitation analysis of CREM in vehicle and DON-treated AsPC-1 cells, followed by qPCR of a PRKCZ promoter region containing the CRE consensus sequence. We observed that the binding of CREM to PRKCZ was strongly enriched in DON-treated cells relative to the vehicle, indicating the ability of CREM to directly associate with the PRKCZ promoter (Fig. 4o). All these findings indicate that the enhanced aPKC expression that we observed with glutamine stress is modulated by CREM. Moreover, these data suggest that CREM is a modulator of the metabolic adaptations, such as macropinocytosis, that occur in response to the pharmacological inhibition of glutamine metabolism. Overall, we found that different modes of glutamine stress seem to activate aPKCs differently and that multiple cell polarity proteins converge to regulate the microtubule dynamics that control macropinocytosis (Fig. 4p).
aPKCs support PDAC cell fitness through macropinocytosis
Glutamine is the most depleted amino acid in human PDAC tumors4,7; therefore, we next assessed the contributions of the aPKCs to PDAC growth and survival under low glutamine conditions. We knocked down either PKCζ or PKCι and analyzed the proliferative capacities of these cells in media with levels of glutamine that would be comparable to what is observed in PDAC tumors4,5. In AsPC-1 and HPAF-II cells, we observed that PKCζ or PKCι depletion halted proliferation under these conditions (Fig. 5a, b and Supplementary Fig. 13a, b). With high levels of glutamine, we observed that depletion of the aPKCs slowed down proliferation, indicating that these kinases can also affect the proliferative capacities of the cells in supra-physiological levels of glutamine (Supplementary Fig. 13c, d). It is well established that macropinocytosis functions to support cancer cell fitness through the uptake of extracellular albumin, which serves as a nutrient source to confer a survival advantage to cells when glutamine is limiting4,5. Since we determined that PKCζ and PKCι are integral to macropinocytic stimulation in response to glutamine stress, we hypothesized that aPKC knockdown might suppress the ability of albumin to rescue PDAC cell dependency on free glutamine. Indeed, while albumin supplementation rescued the deleterious effects of glutamine starvation in the control cells, cells with PKCζ or PKCι loss gained no significant advantage to albumin addition (Fig. 5c–f and Supplementary Fig. 13e, f). These data suggested that aPKCs function to sustain PDAC cell survival in glutamine scarcity via macropinocytosis. While the loss of the aPKCs has been shown to drive pro-apoptotic effects during carcinogenesis51, we did not observe an effect on apoptosis in our settings, as indicated by the absence of cleaved PARP, a marker of apoptosis52 (Supplementary Fig. 13g).

a Relative cell growth of AsPC-1 cells transfected with the indicated siRNAs and cultured in low glutamine (0.1 mM) media for 6 days. Cell number was assessed by Syto60 staining and values are relative to the day 0 timepoint. N = 4. *P(siCTRL vs siPRKCZ) = 0.0138; *P(siCTRL vs siPRKCI) = 0.0365. b Relative cell growth of HPAF-II cells transfected with the indicated siRNAs and cultured in low glutamine (0.05 mM) media for 6 days. Cell number was assessed by crystal violet staining and values are relative to the day 0 timepoint. N = 4.***P(siCTRL vs siPRKCZ) = 0.0003; ***P(siCTRL vs siPRKCI) = 0.0003. c, d Representative crystal violet staining (c) and cell growth quantification (d) of AsPC-1 cells cultured for 6 days with 0 mM glutamine, with and without 2% BSA. Cell number values are relative to the day 0 timepoint. N = 3. *P(siCTRL vs siCTRL+BSA) = 0.0234; *P(siCTRL +BSA vs siPRKCZ + BSA) = 0.0397; nsP(siPRKCZ vs siPRKCZ +BSA) = 0.1699; *P(siCTRL +BSA vs siPRKCI + BSA) = 0.038); nsP(siPRKCI vs siPRKCI +BSA) = 0.1246. e, f Representative crystal violet staining (e) and cell growth quantification (f) in HPAF-II cells cultured for 6 days with 0.05 mM glutamine, with and without 2% BSA. Cell number values are relative to the day 0 timepoint. N = 3. *P(siCTRL vs siCTRL+BSA) = 0.0432; **P(siCTRL +BSA vs siPRKCZ + BSA) = 0.0054; nsP(siPRKCZ vs siPRKCZ +BSA) = 0.8177; *P(siCTRL +BSA vs siPRKCI + BSA) = 0.0101; nsP(siPRKCI vs siPRKCI +BSA) = 0.321. g, h Representative images of macropinocytosis (g) and quantification (h) in AsPC-1 cells cultured for 6 days in 0.1 mM glutamine and co-transfected with indicated siRNAs and either empty vector (EV) or Myc-Pak1-WT/T423E. Values are relative to siCTRL + EV. N = 3. Scale bar, 20 μm.*P(siCTRL vs siCTRL+Pak1 WT) = 0.0333; *P(siCTRL vs siCTRL+Pak1 T423E) = 0.0225; *P(siCTRL vs siPRKCZ) = 0.0122; **P(siPRKCZ vs siPRKCZ+Pak1 WT) = 0.0096; *P(siPRKCZ vs siPRKCZ+Pak1 T423E) = 0.0451. i Quantification of cell growth for the conditions described in (g) and (h) with and without supplementation of BSA. Cell number was assessed by crystal violet after 12 days. Data are shown relative to the day 0 timepoint. N = 3. *P(siCTRL vs siCTRL +BSA) = 0.0187; nsP(siCTRL+BSA vs siCTRL+Pak1 WT+BSA) = 0.3109; nsP(siCTRL+BSA vs siCTRL+Pak1 T423E+BSA) = 0.4076; **P(siCTRL+BSA vs siPRKCZ + BSA) = 0.0071; *P(siPRKCZ+BSA vs siPRKCZ + Pak1 WT+BSA) = 0.0152; *P(siPRKCZ+BSA vs siPRKCZ + Pak1 T423E+BSA) = 0.0173; nsP(siPRKCZ vs siPRKCZ + BSA) = 0.5386; *P(siPRKCZ+Pak1 WT vs siPRKCZ + Pak1 WT+BSA) = 0.0364; *P(siPRKCZ+Pak1 T423E vs siPRKCZ + Pak1 T423E+BSA) = 0.041. Statistical analysis used unpaired two-tailed Student’s t test. Welch’s correction in (h). ns, non-significant, *P < 0.05, **P < 0.01, ***P < 0.001. All data are presented as mean ± SEM. N indicates independent experiments. Source data are provided as a Source Data file.
If the proliferative defects observed with aPKC knockdown in low glutamine conditions are attributable to a diminishment in macropinocytic stimulation, then we posited that if macropinocytosis was restored in aPKC knockdown cells, then cell survival would also be restored. As previously mentioned, downstream of EGFR, the regulation of nutrient stress-induced macropinocytosis occurs through activation of Pak5,45,46. Activation of Pak1 occurs downstream of aPKC and, in the establishment of apical-basal cell polarity, aPKC function is known to be complemented by Pak1 activity53; therefore, we sought to overexpress Pak1 to restore macropinocytosis in aPKC-depleted cells. Control and PKCζ-depleted cells were transfected with an empty vector (E.V.), myc-tagged wild-type Pak1 (Myc-Pak1-WT), or a myc-tagged constitutively active form of Pak1 (Myc-Pak1-T423E). Cells were cultured in low glutamine media, after which macropinocytosis was assessed. Confirming our previous observations, PKCζ knockdown cells transfected with E.V. displayed lower levels of macropinocytosis compared to control cells (Fig. 5g, h). Importantly, macropinocytosis in the PKCζ-depleted cells was significantly restored upon the expression of either Myc-Pak1-WT or Myc-Pak1-T423E, indicating that Pak1 can compensate for the loss of PKCζ (Fig. 5g, h). Not surprisingly, Pak1 overexpression also enhanced macropinocytosis in the control cells (Fig. 5g, h). PKCζ knockdown and Pak1 overexpression were confirmed by western blot (Supplementary Fig. 13h). After observing the ability of Pak1 to restore macropinocytosis in PKCζ knockdown cells, we asked whether in these conditions the cell viability advantage provided by albumin addition was also restored. To test this, cells were cultured in low glutamine conditions supplemented with or without albumin and cell number was determined by crystal violet staining. In line with our previous results, albumin supplementation did not rescue cell growth in PKCζ knockdown cells (Fig. 5i). We did, however, observe a significant increase in proliferation in PKCζ knockdown cells expressing either of the Pak1 proteins when the media was supplemented with albumin (Fig. 5i). Although macropinocytosis was enhanced in control cells overexpressing Pak1, no additional survival effect was observed. Altogether, these data demonstrate that the function of aPKCs in supporting PDAC cell fitness in the context of nutrient stress is attributable to their role in regulating macropinocytosis.
aPKCs regulate macropinocytosis in vivo and support tumor growth
We next examined whether aPKCs function in tumor growth control and if they regulate macropinocytosis in vivo. To knock down the aPKCs in tumors, we generated stable AsPC-1 cell lines expressing lentiviral-delivered doxycycline-inducible PKCζ or PKCι-targeting shRNAs, as well as a non-targeting shRNA control. We validated that this doxycycline (Dox) system could selectively suppress the expression of either PKCζ or PKCι (Supplementary Fig. 14a). We also confirmed that Dox-induced knockdown of either PKCζ or PKCι could inhibit macropinocytosis induced by DON, glutamine starvation or EGF stimulation (Supplementary Fig. 14b–g).
To test aPKC function in vivo, we used a heterotopic xenograft mouse model where the stable cell lines were subcutaneously implanted in athymic mice. To induce knockdown, Dox was administered ten days after tumor cell implantation and tumor volumes were calculated based on caliper measurements for approximately five weeks. We observed a significant reduction in tumor growth with either PKCζ or PKCι knockdown relative to controls (Fig. 6a–c). At termination, knockdown in the tumors was confirmed by western blot (Fig. 6d). We observed a decrease in cell proliferation in the PKCζ or PKCι knockdown tumors relative to the controls as assessed by phospho-Histone H3 (p-HistoneH3) immunostaining (Fig. 6e, f). We did not detect a difference in apoptosis within the tumors since levels of cleaved-caspase 3 (Cl-Caspase 3) staining did not change (Supplementary Fig. 15a, b), confirming our in vitro data. To evaluate intratumoral macropinocytosis we used an ex vivo macropinocytosis assay that we have previously established4,5. In these tumors, macropinocytosis is low in the periphery and predominantly stimulated within the amino acid-depleted non-peripheral regions of the tumor5. In these non-peripheral regions, we found that the macropinocytic index was significantly lower in the PKCζ or PKCι knockdown tumors relative to the control (Fig. 6g, h). Taken together, these data indicate that the aPKCs function in controlling macropinocytosis in vivo and are required for tumor growth.

a Growth curves for AsPC-1-derived heterotopic xenograft tumors expressing the indicated Doxycycline (Dox)-inducible shRNAs. Dox was provided in the drinking water and diet. A total of n = 8 tumors were measured per condition. ***P(shCTRL vs shPRKCZ)< 0.0001; ***P(shCTRL vs shPRKCI)< 0.0001. b Representative photos of the xenograft tumors at termination. c Measurement of tumor weights at termination for n = 8 tumors per condition. ***P(shCTRL vs shPRKCZ)<0.0001; ***P(shCTRL vs shPRKCI) = 0.0003. d Immunoblots assessing PKCζ and PKCι protein levels in the tumors at termination. n = 5 and n = 4 knockdown tumors for PKCζ and PKCι, respectively, were analyzed relative to two different control tumors. e Representative images of p-HistoneH3 staining in two different tumors per condition. Scale bar, 100 μm. f Quantification of p-HistoneH3 staining in tumor sections of shCTRL (n = 44 fields), shPRKCZ (n = 46 fields) and shPRKCI (n = 43 fields). Fields were quantified from n = 3 tumors for each condition. ***P(shCTRL vs shPRKCZ)< 0.0001; ***P(shCTRL vs shPRKCI)< 0.0001. g Representative images of macropinocytosis in tumors expressing the indicated shRNAs, with macropinosomes labeled with TMR-dextran (red) and tumor cells labeled with CK8 (green). Nuclei are stained with DAPI (blue). Scale bar, 20 µm. h Macropinocytosis quantification in n = 6 tumors (shCTRL and shPRKCZ) and n = 5 tumors (shPRKCI). Data are shown relative to shCTRL. ***P(shCTRL vs shPRKCZ)<0.0001; ***P(shCTRL vs shPRKCI)< 0.0001. i Immunofluorescent staining of p-PKCζT560 or total PKCζ (purple) in control tumors, with CK8 (green) delineating peripheral and non-peripheral regions. Pictures are representative of n = 4 tumors from the control group. Scale bar,100 µm. j Quantification of peripheral and non-peripheral staining for p-PKCζT560 (n = 57 fields/periphery, n = 62 fields/non-periphery) and total PKCζ (n = 50 fields/periphery, n = 51 fields/non-periphery) in control tumors. p-PKCζT560: ***P(non-periphery vs periphery)<0.0001); PKCζ: nsP(non-periphery vs periphery) = 0.1385. k Representative co-staining images of p-PKCζT560 and total PKCζ (purple) with macropinosomes (red) in peripheral and non-peripheral regions. Nuclei labeled with DAPI (blue). Scale bar, 20 µm Statistical significance was calculated using two-way ANOVA with Tukey’s multiple comparison test for (a) at day 47. For (c, f, h, and j), statistical significance was calculated using an unpaired two-tailed Student’s t test. ns, non-significant, *P < 0.05, **P < 0.01, ***P < 0.001. All data are presented as the mean ± SEM. Source data are provided as a Source Data file.
We next sought to explore the link between aPKC activation and nutrient availability in vivo. Since glutamine and other amino acids are depleted from the non-peripheral regions of the PDAC tumors5, we hypothesized that aPKC phosphorylation might be enhanced in the tumor cores relative to the periphery. Indeed, using immunofluorescence, we observed that levels of pPKCζT560 were significantly higher in the non-peripheral regions of the tumors versus the periphery (Fig. 6i, j). This differential pattern was specific to the phosphorylated form since total PKCζ was uniformly expressed throughout the tumors (Fig. 6i, j). This observation was further confirmed by immunohistochemical staining (Supplementary Fig. 15c). Importantly, the tumor cells with enhanced pPKCζT560 levels in the non-peripheral regions of the tumors also stained positive for macropinosomes (Fig. 6k). The antibodies used to detect PKCι proteins did not work well on tissue; therefore, this precluded us from analyzing PKCι in more detail in vivo. Altogether, our findings indicate that in the nutrient-depleted regions of the tumor, PKCζ is primed for activation, which in turn enhances the macropinocytic capacity of the cells.
aPKC expression is upregulated in human PDAC and correlates with poor patient prognosis
Since we observed the difference in the pPKCζT560 levels across the different tumor regions in the AsPC-1 xenografts compared to total PKCζ, we immunostained a tissue microarray containing PDAC specimens from 153 patients that underwent surgical resection. No pPKCζT560 expression was detected in 5 of the specimens, but from the other 148, 125 showed moderate to high expression (Fig. 7a). This observed heterogeneity in pPKC levels in the human tumors is consistent with our observations in the xenografts where pPKC displays intratumoral regional differences. On the other hand, and consistent with our murine data, total PKCζ levels were more homogenous throughout the different specimens (Fig. 7a). To further characterize the cell polarity protein network in PDAC, we interrogated publicly available patient datasets. We found numerous examples where expression of PRKCI was upregulated in PDAC relative to normal tissue (Fig. 7b and Supplementary Fig. 16a). Interestingly, we found similar expression correlations with the genes encoding the cell polarity proteins Par6 and Par-1a (Fig. 7c and Supplementary Fig. 16b, c). In addition, we found that elevated levels of either PRKCI, PRKCZ, PARD3 or PARD6B were correlated with poor prognosis in PDAC patients (Fig. 7d–g). Altogether, these observations suggest that the aPKC isoforms and/or PAR cell polarity proteins could be potential prognostic indicators in pancreatic cancer.

a Representative immunohistochemical staining for p-PKCζT560 and total PKCζ from clinical samples on a tissue microarray (TMA) containing 152 surgically resected human PDAC specimens. Individual patient samples stained with p-PKCζT560 were pathologically classified according to staining intensity. Images of the TMA samples from the same patient are aligned in columns. Scale bars: 200 μm (low magnification) and 50 μm (high magnification). b, c PRKCI (b) and PARD6B and PAR1a (c) transcript levels in PDAC human samples and normal pancreatic tissue (adjacent non-neoplastic tissue) from the indicated GSE datasets. Number of patient samples for each group is indicated in parentheses. GSE16515: PRKCI: ***P(normal vs tumor) < 0.0001; GSE62452: PRKCI: ***P(normal vs tumor) <0.0001; PAR1a: ***P(normal vs tumor) < 0.0001; PAR1a: **P(normal vs tumor) = 0.0075. d, e Overall survival in patients with high and low expression of PRKCI (d) and PRKCZ (e). Number of patients included in each group is indicated in parentheses. f, g Overall survival in patients with high and low expression of PARD6B (f) and PARD3 (g). Number of patients included in each group is indicated in parentheses. Statistical significance was calculated using unpaired two-tailed Student’s t test for (b, c). For (d–g), P-values were determined with the Log-rank test. **P < 0.01, ***P < 0.001. Data are presented as the mean ± SEM in (b, c).
Discussion
The precise functions of the aPKCs in regulating PDAC progression have remained elusive. In this study, we have uncovered a role for the aPKC isoforms PKCζ and PKCι in stimulating macropinocytosis in response to glutamine stress in KRAS-mutant PDAC cells, both in vitro and in vivo. Our findings are consistent with other studies that have described pro-oncogenic roles for these kinases in tumors harboring KRAS mutations, such as lung adenocarcinoma, basal cell carcinoma, ovarian carcinoma, and PDAC54,55,56,57,58,59,60,61. Previous studies have not assessed a role for the aPKCs in the metabolic stress response in PDAC, but PKCι has been shown to support PDAC progression through the Rac1-MEK-ERK1/2 signaling axis, and PKCι inhibition leads to impaired tumor growth and a reduction in metastasis and angiogenesis54. Interestingly, in a genetically engineered mouse model that is used to study the early stages of tumor initiation, animals of the genotype LSL-KrasG12D; Ptf1aCre/+, Prkci f/f still formed pancreatic intraepithelial neoplasia (PanIN) lesions, but these lesions did not progress to invasive adenocarcinoma59. Our work demonstrating that PKCι is required for tumor maintenance in established tumors reinforces its attractiveness as a potential therapeutical target in PDAC. For the PKCζ isoform, there is evidence showing that it also participates in Ras-dependent processes, such as the organization of actin cytoskeleton62. In the context of KRAS-mutant cancers, PKCζ promotes bladder cancer progression, inducing the production of reactive oxygen species, that are key mediators of the blebbishield transformation program, an emergency program that allows cancer stem cell resurrection after apoptosis63. In PDAC, PKCζ participates in cancer cell motility in association with RhoA GTPase64 and favors pancreatic cancer growth and metastasis via activation of STAT365. Thus, PKCζ has also been identified as a positive regulator of PDAC progression. What remains enigmatic is how all these different aspects of cancer biology that are modulated by the aPKCs integrate at the level of tumor progression; however, our observations are in line with these other studies that have established these kinases as pro-oncogenic regulators of tumors harboring KRAS mutations. This contrasts with other oncogenic settings where aPKCs have been postulated to be tumor suppressors66,67,68,69,70,71. Importantly, the precise contribution of each of the isoforms to various aspects of tumor initiation and progression is still poorly understood. Our data suggest that in PDAC, both isoforms independently regulate macropinocytosis since their individual specific reduction shows similar effects, but it is likely that in other settings the isoforms might have specific roles since PKCι is known to be widely expressed, whereas PKCζ shows a more restricted expression pattern72. One possibility is that the specific roles for each of the isoforms depend on their abundance in the different tissues. For example, during development, these kinases are thought to have unique functions since mice deficient in PKCι show early embryonic lethality, whereas no lethality is observed in PKCζ-deficient mice73,74. On the other hand, in normal epithelial cells where the aPKCs are both associated with the cell polarity protein network, it has been postulated that the isoforms have similar roles72,75. Future work is required to further elucidate whether there are specific roles or cooperation of the aPKC isoforms in carcinogenesis and cancer progression. Besides aPKCs, a member of the PKC family involved in macropinocytosis is PKCδ, which controls macropinocytic uptake induced by phorbol-esters and Hepatocyte Growth Factor (HGF) in macrophages76. In this study, we did not focus on this isoform because PKCδ belongs to the subgroup of ‘novel PKCs’, whose regulation is different from that of aPKCs72. The divergence among the different PKC subgroups results in distinct cellular functions and molecular readouts. Thus, regardless of the potential involvement of other PKC members in controlling nutrient stress-induced macropinocytosis, their mechanisms might be entirely distinct from those of PKCζ and PKCι. Consequently, we conclude that this study describes a specific and unique pathway for aPKCs.
In addition to the aPKCs, we have also identified the cell polarity proteins Par3, Par6, and Par-1a as regulators of macropinocytosis. Maintenance of cell polarity has been largely associated with tumor suppression mechanisms, such as preventing epithelial-to-mesenchymal transition (EMT)77. In this context, aPKCs bind to the scaffold proteins Par3 and Par6 and block EMT through phosphorylation and destabilization of Snail78. In addition, other studies have elucidated that disruption of homeostatic levels of the aPKCs leads to cell polarity loss, which contributes to tumorigenesis79,80. However, as previously mentioned, this protein network can also support the progression of cancer13,14. Indeed, pro-tumorigenic roles for these proteins have been confirmed in different models. For example, aPKC phosphorylates Par6 to promote EMT and cell migration driven by TGFβ81 and Par6 overexpression promotes breast cancer growth24. In contrast, Par3 is known to perform dual functions in tumorigenesis, such as in prostate cancer, where it displays pro-tumoral and anti-tumoral effects82,83. In PDAC, loss of Par3 has been associated with increased metastasis, which provided an antitumoral role for Par3, and this antitumoral effect is mediated through the interaction of Par3 with Tiam184, However, the requirement of Par3 that we observed in macropinocytosis and the fact that high Par3 expression associates with poor prognosis in PDAC patients suggest that Par3 can also favor PDAC progression. This suggests that Par3 can display dual functions in pancreatic cancer. Although the loss of these cell polarity proteins leads to the abrogation of macropinocytosis, it is still unclear how, in this context, this network is regulated. We observed that aPKCs are required for the relocation of Par3 protein to the microtubule network where it is known to regulate microtubule motors, which we show facilitate macropinosome formation. aPKC and Par6 bind through the PB-1 domain of aPKC, forming a complex72,81. It is conceivable that aPKC requires the association with Par6 to modulate macropinocytic uptake and that loss of Par6 might inhibit aPKC function. In cell polarity, aPKCs phosphorylate Par-1a protein at threonine 564 (T564), promoting changes in its subcellular localization15. In this study, we observed that under nutrient stress aPKCs are required for phosphorylation of Par-1a within the activation loop, which leads to the activation of the kinase, thereby modifying microtubule dynamics and macropinocytic uptake. It is unclear whether this phosphorylation is directly performed by aPKCs or if an intermediate is required. In addition, Par3 is a known target of Par185. It is conceivable that aPKCs induce the activation of Par-1a to control the subcellular redistribution of Par3, or these kinases may separately regulate the function of Par1 and Par3. Future work is required to understand how this cell polarity protein network is coordinated to promote macropinocytosis in the context of nutrient stress. Also, it is important to mention the possible link between aPKC and Pak1 in controlling macropinocytic uptake since both are required for cell polarity establishment53. As previously shown, Pak1 activation is controlled by aPKCs, which supports our data showing that overexpression of Pak1 compensates for the loss of aPKC in driving macropinocytosis. A study from our group has shown that in EGFR signaling, Pak1 can also be activated by Rac1 GTPase86, indicating that the Pak1 function can be controlled by different modulators. It is possible that in some contexts Pak1 can be regulated independently of aPKC, which would explain why Pak1 can compensate for the loss of aPKC in cell polarity establishment53.
Prior to our work, it remained unexplored whether microtubules have a role in mediating the physical formation of nascent macropinosomes, although, it had been reported that microtubules participate in the post-internalization and trafficking of mature macropinosomes29. Here, we have shown that in glutamine stress conditions microtubules are required for macropinocytic uptake, actively participating in the membrane ruffling that drives macropinosome formation. In addition, we observed that inhibition of the motor protein dynein blocks macropinocytic uptake, showing that the retrograde motor function of microtubules is essential in establishing the tension needed to pull the macropinosome inwards, and this might be in part controlled by the accumulation of Par3 in the microtubule network, a process that depends on aPKCs. Microtubules are known to be key elements in the cellular response to metabolic stress87. For example, nutrient stress promotes hyperacetylation of tubulin, and this turns on the macroautophagy machinery as a compensation mechanism in response to the lack of nutrients88. It would not be surprising then that in this context some post-translational modifications promote the rearrangement of the microtubule network to facilitate membrane ruffling, activating the macropinocytic program. Also, microtubules participate in the cellular response to metabolic stress by acting as key sensors of intracellular ATP levels87,89,90, and it has been shown that low intracellular ATP can be counterbalanced by the macropinocytic uptake of extracellular ATP91. Thus, it is possible that low intracellular ATP promotes changes in the microtubule network that would drive the macropinocytic cascade. Future work is essential to further understand how microtubules control macropinocytosis since microtubules would not only be relevant for the formation of macropinosomes but also in controlling their intracellular trafficking and positioning. A potentially analogous process is how growth factors promote the differential localization of lysosomes in the regulation of mTORC2 and Akt signaling, and how this lysosome positioning is orchestrated by the microtubule network92. Hence, it would not be surprising that microtubules control the spatial-temporal positioning of both macropinosomes and lysosomes to permit their fusion and the subsequent digestion and release of the macropinocytic cargo. Uncovering the different post-translational modifications and regulators of the microtubule cytoskeleton that are required for driving macropinocytosis and controlling macropinosome trafficking is essential for a better understanding of how this endocytic pathway is controlled and will shed light on effective methods to manipulate its activation and repression.
Methods
Experiments involving recombinant DNA and viral vectors were reviewed and approved by the SBP Institutional Biosafety Committee (approval number IBC-CC-20-004). For the in vivo experiments, the procedures were approved by the Institutional Animal Care and Use Committee of Sanford Burnham Prebys Medical Discovery Institute (SBP; approval number 20-073).
Cell culture and reagents
AsPC-1, HPAF-II, and HEK-293T cells were obtained from the American Type Culture Collection (ATCC) and were maintained in 100 units/mL penicillin/streptomycin under 5% CO2 at 37 oC and routinely tested for mycoplasma contamination using the PCR Mycoplasma Detection Kit (ABM). Cells were cultured in the following media: AsPC-1 in RPMI (Corning) supplemented with 10% fetal bovine serum (FBS), 20 mM HEPES and 1 mM sodium pyruvate and HPAF-II and HEK-293T cells in DMEM (Corning) with 10% FBS and 20 mM HEPES. For doxycycline-inducible expression cassettes, Tet System Approved FBS (#631101, Takara) was used. AsPC-1 transduced cells were maintained in 10 μg/mL puromycin (P8833, Sigma).
The following reagents were used: 6-diazo-5-oxo-L-norleucine (DON) (D1242, Sigma); 2-acetyl-1,3-cyclopentanedione (ACPD) (R426911, Sigma); Colchicine (C9754, Sigma); Paclitaxel (T7402, Sigma); Ciliobrevin D (250401, Sigma); Erlotinib (SML2156, Sigma); EGF (E9644, Sigma); Staurosporine (1285, Tocris Biosciences); Doxycycline (AC446060250, Fisher). For DON treatment, 2 mM of DON was added for 24 h in glutamine-containing serum-free media. APCD was used for 3 days at 4 μM in AsPC-1 cells and 25 μM in HPAF-II cells. The inhibitor was refreshed every 24 hours. Colchicine and paclitaxel were used at 100 ng/mL and 200 nM, respectively, for 18 h in serum-free media. Ciliobrevin D was used at 50 μM for 30 min. For erlotinib experiments, cells were treated for 2 h at 25 μM. EGF was used at 100 nM at the indicated time points or for 10 min for the macropinocytosis uptake assays. Staurosporine was used at 100 μM for 12 h. Transduced AsPC-1 cells were treated for 3 days with 2 μg/mL of doxycycline for knockdown induction.
Glutamine deprivation
Experiments evaluating macropinocytosis in glutamine deprivation were performed in the absence of serum for 24 hours unless otherwise indicated. Cells were plated in complete culture media, which was exchanged with serum-free glutamine-deprived or glutamine-containing media 2–3 days after cell seeding. For glutamine deprivation experiments, glutamine-free RPMI (Corning) or glutamine-free DMEM (Corning) was used. For Fig. 5g and h, cells were cultured in serum-containing media supplemented with 0.1 mM of glutamine for 6 days prior to macropinocytosis assessment.
siRNA kinome screen
The HT-compatible macropinocytosis assay18 was used in conjunction with a siRNA library targeting 679 kinases (siOTP-Dharmacon). Approximately 4000 AsPC-1 cells were seeded into each siRNA-containing well of 384-well plates and reverse transfected at a final 10 nM siRNA concentration. Lipo-RNAimax (ThermoFisher) was used as a transfection reagent. 48 h after transfection, media was aspirated, and wells were thoroughly washed with sterile PBS (Fisher) and incubated with serum-free glutamine-deprived RPMI media in combination with 2 mM DON for glutamine-stress induction. After 24 h, macropinocytosis was assessed by a FITC-Dextran (ThermoFisher Scientific) uptake assay. Cells were incubated with media containing 1 mg/mL FITC-Dextran for 30 minutes at 37 oC. 4 washes with cold PBS were performed, and cells were fixed with 3.7% formaldehyde for 30 minutes. 2 μg/ml of DAPI (MilliPore) was added to stain nuclei. 24 h later, imaging of the wells was performed in the Sanford Burnham Prebys Functional Genomics core using a laser-based automated HT microscope (IN Cell 1000, Opera QEHS, Celigo). 2 rounds of screening were performed and macropinocytic index was determined as previously described18. Hits showing a reduction greater than 67% were selected for further validation.
siRNA and plasmid transfection
For macropinocytosis and immunofluorescence assays, cells were transfected in 24-well plates with Lipofectamine RNAimax transfection reagent (ThermoFisher Scientific) and with the corresponding siRNAs at a final concentration of 50 nM following manufacturer’s protocol. For RT-qPCR and Immunoblot analysis, cells were transfected in 12-well and 6-well plates, respectively. Two sequential transfections were performed for each experimental sample. For macropinocytosis, immunofluorescence analysis, DQ-BSA degradation experiments, transferrin uptake assays, and 13C-labeling assays, serum-free media with the corresponding experimental components was added 24 h after the second transfection. For knockdown validation, mRNA and protein extracts were collected 48 h after the second transfection, and no serum starvation was performed. For cell-growth curves, cells were transfected in 6-cm plates and trypsinized 24 h after the second siRNA transfection and seeded in 96-well plates.
The following siRNAs (ThermoFisher Scientific) were used: non-targeting siCTRL (4390844); siPRKCZ#1 (s11128) and #2 (s11129); siPRKCI#1 (s11110) and #2 (s11111); siPAR1a#1 (s230619) and #2 (s8514); siPARD3 #1 (132893) and #2 (132894); siPARD6B #1 (225936) and #2 (225937); siRCOR3(132395). siCREM (SASI_Hs01_00111489) was purchased from Sigma.
For overexpression assays, cells were transfected in 6 cm plates 24 h after second siRNA transfection with Lipofectamine 2000 (ThermoFisher Scientific) and 2 μg of pCMV6M-Pak1-WT (Addgene, 12209), pCMV6M-Pak1-T423E (Addgene, 12208) or empty vector (Origin, pCMV6KN). 24 h after Pak1 overexpression, cells were trypsinized and seeded in 96-well plates for cell growth analysis.
For lentiviral production, HEK-293T cells were seeded in 6 cm plates, and 4.5 μg of Lentiviral vector were cotransfected with 3 μg and 1.5 μg of the packaging vectors psPAX2 and pMD2.G. Lipo2000 (ThermoFisher Scientific) was used as transfection reagent. HEK-293T cells were transfected for 48 hours prior to viral particles collection.
In vitro macropinocytosis
Cells were seeded on acid-washed glass coverslips in 24-well plates and subjected to serum starvation with or without nutrient deprivation for 24 h. For macropinocytosis in siRNA transfected cells, cells were subjected to serum starvation 24 h after the second transfection, whereas for macropinocytosis in non-transfected or transduced cell lines, serum deprivation was performed 2–3 days after cell plating. After 24 h of starvation, media was first removed from the wells, and the same media containing 1 mg/mL FITC-Dextran (ThermoFisher Scientific) was added back to the cells, and plates were incubated for 30 minat 37 oC. Thereafter, 4–5 washes with cold PBS were done, and cells were fixed in 3.7% formaldehyde for 15 min. Nuclei were next stained with 2 μg/ml of DAPI(Millipore), and coverslips were mounted on glass slides with DAKO-fluorescent media (Agilent Technologies). Images were captured and analyzed using two different approaches. As previously described, pictures were captured at 40X magnification using the EVOS FL Cell Imaging System (Thermo Fisher Scientific) and manually analyzed using ImageJ software (NIH)5. The macropinocytic index was calculated by the total particle area per cell. Alternatively, images were automatically captured at 40X magnification using the BioTek Cytation 5 (Agilent Technologies) from the Sanford Burnham Prebys Cell Imaging Core and were subjected to automated analysis using the BioTek Gen5 software19. The calculation method of the macropinocytic index was the same as described above. Both approaches have been previously compared and have been shown to provide the same results19.
DQ-BSA degradation assay
Cells were seeded on acid-wash glass coverslips in 24 well plates and subjected to serum starvation with or without nutrient deprivation for 24 h. After 24 h of starvation, media was first removed from wells, and the same media containing 0.15 mg/mL DQ-BSA Red (ThermoFisher Scientific) was added to the wells and incubated for 30 minutes at 37 oC. Cells were then fixed with 37% formaldehyde for 15 min after a 4 h chase in DQ-BSA-free media under glutamine-containing and glutamine-free conditions. After fixation, nuclei were stained with 2 μg/ml of DAPI, and coverslips were mounted on glass slides with DAKO-fluorescent media. Upon proteolytic digestion, DQ-BSA releases fragments with an excitation wavelength of 590–620 nm. Images were captured at 40X magnification using the EVOS FL Cell Imaging System and manually analyzed using the Image J Software. DQ-BSA degradation was calculated by the total particle area per cell.
13C-labeling and tracing of polar metabolites
13C-labeling was performed by culturing AsPC-1 cells in glutamine-free and glucose-free RPMI (ICN1646854, Fisher Scientific) containing 10 mmol/L 13C6-glucose (Sigma-Aldrich) and 2 mmol/L 13C5-glutamine (Sigma-Aldrich) in the presence of 10% FBS for 6 days. Cells were first plated in 100 cm plates and cultured for 3 days in labeled media. On day 3, cells were split into 6-well plates and cultured in fresh labeled media. For GC-MS, five wells were plated for each experimental condition, and two additional wells/conditions were used for cell counting. Cells were transfected with corresponding siRNAs at day 4 and day 5. 24 h after the second transfection, labeling media was removed, and cells were washed twice with sterile PBS and cultured in glutamine-free and glucose-free RPMI containing 10 mmol/L 13C6-glucose with no serum and with or without 40 mg/mL BSA (Fraction V, Millipore) for 48 h. Around 600,000-1 × 10^6 cells were quickly washed three times with prechilled PBS and extracted with 0.45 mL 50% methanol (in water) (− 20 °C) containing 20 μmol/L L-norvaline (Sigma-Aldrich, internal standard for polar metabolites) and 0.225 mL chloroform. After centrifugation, the upper methanol/water phase was collected for analysis of 13C-labeling and quantification of intracellular free amino acids and metabolites. Sample derivatization and running of GC-MS were conducted following established protocols93. Standards for unlabeled metabolites were used for quantification of the amounts of unlabeled metabolites in samples. These data were then corrected for the fraction of metabolites that were 13C-labeled, to yield total metabolite amounts. The abundance of unlabeled non-essential amino acids and TCA cycle intermediates was calculated by correcting the unlabeled amounts (nmols) by the cell number for each experimental condition, and values were expressed as nmols per million cells. Data were represented relative to each non-BSA cultured condition.
Transferrin uptake assay
Cells were seeded on acid-wash glass coverslips in 24 well plates and subjected to serum starvation with or without nutrient deprivation for 24 h. After 24 h of starvation, cells were chilled for 5 min on ice. Cell media was replaced with fresh pre-chilled serum-free media containing 50 μg/mL transferrin (89427-102, VWR) and cells were incubated for 30 min on ice. Cold media was then replaced with prewarmed media containing serum, and cells were then incubated at 37 °C for 10 min. At room temperature, cells were washed with PBS and fixed with 37% formaldehyde for 15 min. After fixation, nuclei were stained with 2 μg/ml of DAPI, and coverslips were mounted on glass slides with DAKO-fluorescent media. Images were automatically captured at 40X magnification using the BioTek Cytation 5 and subjected to automated quantification using the BioTek Gen5 software. Transferrin uptake was calculated by the total particle area per cell.
Immunoblotting
Total protein was extracted from cells or tumor tissues using RIPA buffer (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% sodium deoxycholate, 0.1% SDS, 1% Triton X-100) with protease and phosphatase inhibitors (Roche). Protein concentrations were measured using the DC Protein Assay Kit (Bio-Rad). 25–30 µg protein samples were run in SDS–PAGE followed by protein transfer using Mini Gel Tank and Mini Blot Module (Life Technologies). Immunoblotting was detected using near-infrared fluorescence (LI-COR) and the Odyssey CLx imager (LI-COR).
The following primary antibodies were used: PKCζ (CST 9368S, 1:500), PKCι (BD Transduction Laboratories 610175, 1:700), p-PKCζT560 (Abcam ab62372, 1:2000), p-PKCιT555-63 (Abcam ab5813, 1:1000), Par3 (Millipore Sigma 07-330, 1:4000), CREM (Santa Cruz sc-101530, 1:100), Myc (CST 71D10, 1:1000), p-EGFR Y1068 (CST 3777S, 1:1000), EGFR (CST 4267, 1:1000); PARP (CST 9532S, 1:1000); p-Par1 (CST 4836S; 1:600); Par-1a (CST 9311; 1:600); p-Pak (CST 2601S; 1:600); Pak1 (Abcam ab183894; 1:1000); β-actin (Sigma A1978, 1:10000); α-tubulin (Sigma T6074; 1:10000). Densitometry analysis was performed with ImageJ software (NIH).
Quantitative real-time PCR (qPCR) and RNA sequencing
Total RNA was extracted from cells with the PureLink RNA Mini Kit (Thermo Fisher Scientific). For quantitative real-time PCR (qPCR), cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). cDNA samples were diluted 1:4 in RNAse-free water. qPCR was performed in triplicates with the SYBR Premix Taq II master mix (Takara) and specific primers on the LightCycler 96 (Roche). Relative target gene expression was determined by comparing average threshold cycles (CT) with that of housekeeping genes (β-actin or RPL13) by the delta-delta CT method. The following primers were used in this study: PRKCZ-F: ATTCATGTTTTCCCGAGCAC; PRKCZ-R: TGTTAAAGCGCTTGGCTTG; PRKCI-F: CGAACAGCTCTTCACCATGA; PRKCI-R: CGTTCTGGTACACAAGGGAAC; PAR1a F: CAGTCTCCTCAC CACAAAGTGC; PAR1a-R: TGCTGGTCTGACTCCTTTTCGG; PARD3 F-ATACCAGCTGTCCCCTACAGT; PARD3-R- CTGGACTCAAAGAGCAGTCG; PARD6B-F -GGTGAAGAGCAAGTTTGGAGC; PARD6B-R TAAGCAGTGGATTGGCCGTT; RCOR3-F GTTGGGATGAGAGTCGGAGC; RCOR3-R AACATGCCAAGTGCCTGTTC; CREM-F TGATGGTGTTCAGGGACTGC; CREM-R ATGTCACCAGTGGCAGCTTG; RPL13-F GTTCGGTACCACACGAAGGT; RPL13-R TGGGGAAGAGGATGAGTTTG; β-ACTIN-F ATTGGCAATCAGCGGTTC; β-ACTIN-R CGTGGATGCCACAGGACT.
RNA-Seq was performed by the Sanford Burnham Prebys Genomics Core. AsPC-1 cells were treated with either vehicle or DON for 24 h in serum-free media. Thereafter, Poly(A) RNA was isolated using the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB), and barcoded libraries were made using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB). Libraries were pooled and single-end sequenced (1 × 75 bp) on the Illumina NextSeq 500 using the High-Output V2 Kit (Illumina). Read data were processed in BaseSpace, and reads were aligned to the Homo sapiens genome (hg19) using STAR aligner with default settings.
Chromatin immunoprecipitation (ChIP)-qPCR
For the ChIP assay, AsPC-1 cells were treated with vehicle (H2O) or DON (2 mM) for 24 h. ChIP was performed using the CUTANA™ ChIC/CUT&RUN Kit (EpiCypher) according to the manufacturer’s instructions. Vehicle and DON-treated cells were incubated overnight with 1 μg of CREM antibody (Abnova H00001390M02) or 1 μg of IgG negative-control antibody included in the CUTANA™ ChIC/CUT&RUN Kit. 500,000 cells were collected for each experimental condition. ChIP was followed by RT-qPCR validation. Fold enrichment was calculated by subtracting the average threshold cycles (CT) of the IgG negative control-incubated conditions from CREM antibody incubated samples. The following primers against PRKCZ promoter were used: F-5’ GGACTGTGCTGATGCAGAGA; R- 5’ AAAACACGGCATGGATGTGC-3’.
Immunofluorescence
Cells seeded on acid-wash coverslips were washed with cold PBS and fixed with 4% Paraformaldehyde (PFA) for 15 min at room temperature. Cells were then permeabilized with 0.2%-Triton-X-100 in PBS for 10 min and blocked in 10% Normal Goat Serum in PBS for 1 h. Thereafter, cells were stained overnight with primary antibodies for tubulin (Sigma T6074, 1:500), Par3 (Sigma HPA030443, 1:50), dynein (ThermoFisher MA1-070, 1:100) and MLC-2 (CST 8505S, 1:100). For actin staining, cells were stained overnight with Acti-stain 488 phalloidin (Cytoskeleton PHDG1-A, 1:140) diluted in blocking buffer. Three washes of PBS were performed, and cells were incubated with secondary antibodies Alexa Fluor 488 (ThermoFisher Scientific A32723, 1:1000) for tubulin and dynein and Alexa Fluor 594 (ThermoFisher Scientific A11012, 1:1000) for Par3 and MLC2 staining. Secondary antibody solutions were supplemented with 2 μg/mL of DAPI (Millipore Sigma) for nuclear staining. Finally, cells were washed three times with PBS and mounted on glass slides using fluorescent-DAKO mounting media (Agilent Technologies). Images were captured at the Sanford Burnham Prebys Cell Imaging Core, using a Zeiss LSM 710 laser scanning confocal microscope equipped with a Plan Apochromat 63X/1.4 NA oil immersion objective. Quantification of the Par3 puncta/cytosolic fraction ratios was performed with the ImageJ software (NIH). Cytosolic fraction was calculated by subtracting the intensity (mean gray value*total area) of the Par3 puncta from the total Par3 intensity (Cytosol intensity = Total Par3intensity- Puncta Par3intensity). The Percentage of Par3 puncta/cytosol was determined by dividing Par3 puncta intensity to the intensity of the cytosolic fraction ((Puncta Par3intensity/ Cytosol intensity) *100).
Tumor sections frozen in OCT compound and injected with TMR-dextran were first sectioned by the SBP Histology Core. Samples were then thawed at room temperature for 5–10 minutes and fixed with 3.7% formaldehyde for 15 min. Slides were permeabilized with 0.2%-Triton-X-100 in PBS for 10 min and blocked in 10% Normal Goat Serum and 1% BSA in PBS for 1 hour. After three washes with PBS, samples were incubated with primary antibodies for p-PKCζT560 (Abcam ab62372, 1:400); PKCζ (Abcam ab59364, 1:400); CK8 (TROMO-I, DSHB, 1:400). After three PBS washes, slides were incubated with the Alexa-Fluor 488 (ThermoFisher Scientific A11006, 1:1000) and Alexa-Fluor 647 (ThermoFisher Scientific, A32733, 1:1000) for CK8 and p-PKCζ/ PKCζ staining, respectively. Secondary antibody solutions were supplemented with 2 μg/mL of DAPI (Millipore Sigma). Pictures were captured using an EVOS M5000 Cell Imaging System (ThermoFisher Scientific) equipped with a Plan Fluorite 40x/0.75 NA air objective. The intensity of p-PKCζ and total PKCζ staining in the peripheral and non-peripheral regions of the tumor was quantified with ImageJ software (NIH). The tumor periphery was established by the presence of non-tumor cells (CK8 negative) at the edges of the tumors. For single CK8 immuno-staining, no permeabilization was performed, and the primary antibody was incubated for 4 h at the same dilution.
Cell growth assays
After 24 h of the second siRNA transfection or Pak1 overexpression, cells were trypsinized, and 5000 cells were seeded in wells of 96-well plates. 24 h after cell plating, time 0 of the growth curve was determined, and media was replaced with serum-containing media with 0 or 0.1 mM (for Pak1 rescue experiments) glutamine for AsPC-1 cells and 0.05 mM glutamine for HPAF-II cells, supplemented and non-supplemented with 2% serum-albumin (126609, Millipore). Media was refreshed every 3 days for cell growth analysis in knockdown cells, and every 5–6 days for experiments of PKCζ knockdown with Pak1 overexpression. Cell growth curves in low-glutamine in AsPC-1 cells were established at 0.1 mM glutamine conditions and at 0.05 mM glutamine for HPAF-II cells. Cell growth curves in high-glutamine were established at 4 mM glutamine for both AsPC-1 and HPAF-II cells. For BSA supplementation experiments in aPKC knockdown cells, BSA rescues were determined in AsPC-1 cells cultured in 0 mM glutamine and in 0.05 mM glutamine for HPAF-II cells. BSA rescues in AsPC-1 PKCζ knockdown cells with Pak1 overexpression were determined at 0.1 mM glutamine cell culture conditions. Cell growth in AsPC-1 and HPAF-II aPKC knockdown cells was determined on day 3 and day 6 after time 0 establishment. For BSA rescue experiments in aPKC knockdown cells, cell growth was determined at day 3 and 6 and for BSA rescues in AsPC-1 knockdown cells overexpressed with Pak1, growth was determined at day 12.
For cell growth analysis with crystal violet, media was aspirated, and cells were stained with 0.5% crystal violet for 30 minutes and thoroughly washed in tap water. When plates were dry, these were scanned, and the relative number was calculated by determining the stained area with ImageJ software (NIH). For Syto60 staining, wells were aspirated and incubated for 45 min with 1 μM Syto60 (S11342, ThermoFisher) in 0.2% Triton X-100 in PBS. Wells were washed in PBS, and plates were imaged at 700 nm on the Odyssey CLx imager (LI-COR). Relative cell number was calculated as described above. Three replicates per condition were used in each independent experiment.
Lentiviral production and transduction
Tet-pLKO-puro (Addgene plasmid #21915) constructs for doxycycline-induced shRNA expression were generated according to the manufacturer’s instructions. For constructs generation, the following oligonucleotides sequences were used: shPRKCZ#1 (TRCN0000001221); shPRKCZ#2 (TRCN0000199469); shPRKCI#1 (TRCN0000006039); shPRKCI#2 (TRCN0000006041) from the TRC database. The non-targeting shRNA (shCTRL) was previously obtained using the sequences: F 5’-CCGG CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG TTTTTG-3’ and R 5’-AATTCAAAAACCTAAGGTTAAGTCGCCCTCGCTCGAG CGAGGGCGACTTAACCTTAGG-3’94.
Lentiviral production was carried out as described above. 48 h after transfection, lentiviral particles were collected, and the medium containing viral particles was filtered with a 0.45 μm filter (Fisher). Cells were transduced with virus-containing media and 8 μg/ml Polybrene (MilliporeSigma). 48 h after transduction, media containing 10 μg/ml puromycin were added for selection. Infected cells were maintained in media supplemented with 10 μg/ml puromycin for further use.
Animal studies
Female nude mice (Foxn1nu/Foxn1nu) aged between 6 to 7 weeks old were purchased from The Jackson Laboratories. All animals were housed in sterile caging and maintained under pathogen-free conditions. The Sanford Burnham Prebys Medical Discovery Institute (SBP) IACUC policy limits tumors in mice to 2 cm3 and this was not exceeded. Mice were housed in four per cage in individually ventilated cage systems. Relative humidity in the rodent rooms is monitored and recorded daily by animal technicians but is not controlled. Generally, animal room RH (relative humidity) is within the acceptable range of 30–70% for rodents. A standard diurnal light cycle of 12 hr light: 12 hr dark was used with a rodent room thermostat temperature set point of 72 °F.
One million cells were resuspended in a 1:1 dilution of PBS and Matrigel (Corning) and subcutaneously injected in both flanks of the mice. Tumor growth was monitored using an electronic caliper and tumor volume was determined according to the V = (L × W2)/2 equation. 10 days after cell inoculation, doxycycline was supplemented in the diet (TD.01306, Envigo) and in the drinking water containing 2 mg/mL of doxycycline and 5% sucrose. Drinking water and diet were changed twice and once a week, respectively. Mice were euthanized 27–30 days after tumor cell injection, and tumors were sliced and accordingly processed for immunohistochemistry, immunofluorescence, and macropinocytosis analysis. A portion of the tissue was snap-frozen in liquid nitrogen for immunoblot analysis.
Ex vivo macropinocytosis
As previously described95,96, sections with ~ 5 mm thickness were cut from fresh tumor tissues and injected at different sites with 20 mg/mL 10-kDa TMR-Dextran (ThermoFisher Scientific) and immersed in 400–500 μL TMR-Dextran for 15 min. After quickly washing three times with PBS, cross sections were embedded in the OCT compound and snap-frozen on dry ice. Frozen sections were cut by the SBP Histology Core and subjected to CK8 immunofluorescence staining as previously explained. Images were captured at 40X magnification using the EVOS FL Cell Imaging System (ThermoFisher Scientific) and analyzed with the ImageJ software (NIH)5.
Immunohistochemistry
Xenografts were fixed in 10% formalin. Fixed tissue was embedded in paraffin and sectioned by the SBP Histology Core. Antigen retrieval was performed by microwave-heating in 10 mM sodium citrate (pH 6) and endogenous peroxidases were quenched in 3% hydrogen peroxide. After that, sections were permeabilized for 10 min with 0.2%-Triton X-100 in TBS containing 0.1% Tween-20 (TBS-T), blocked in 2% BSA, 10% goat serum in TBS-T for 1 h at room temperature and then incubated with primary antibodies diluted in 2% BSA/TBS-T overnight at 4 °C. After washes, sections were incubated with biotinylated goat anti-rabbit secondary antibody (1:1000, Vector, BA-1000-1.5) for 1.5 h at room temperature followed by incubation with the VECTASTAIN Elite ABC HRP Kit (Vector Labs) and the DAB HRP Substrate Kit (Vector Labs). Nuclear counterstaining was performed by hematoxylin staining. Images were captured with a brightfield Olympus CX-31 microscope coupled with an INFINITY camera and INFINITY capture software (Lumenera). The following primary antibodies and dilutions were used: p-Histone H3 (CST, 9701,1:200,); p-PKCζT560 (Abcam ab62372, 1:400); PKCζ (Abcam ab59364, 1:400) and cleaved caspase 3 (CC3) (CST 9664,1:1,000) For the pHis-H3 and CC3, the number of positive nuclei per image field was determined using the Fiji software (NIH).
Clinical data analysis
Immunohistochemical staining surgically resected PDAC (153 patients) was performed using p-PKCζT560 (Abcam ab62372, 1:400) and PKCζ (Abcam ab59364, 1:400) antibodies on a previously described PDAC TMA (quote PMID:20142597) that was provided by the Hirshberg Foundation funded UCLA Pancreas Bank. p-PKCζT560 staining was independently scored by a gastrointestinal pathologist (DWD) based on cytoplasmic intensity (0–3; 0-absent; 1-low; 2-moderate; 3-high) on multiple (2–3) cores for each tumor. The final IHC score for each tumor was calculated as the average intensity across its multiple cores. PKCζ staining intensity was uniformly high (3 +) across all tumor cores and, therefore, not individually scored or evaluated.
PDAC patient data for expression analysis were obtained from the following GSE datasets: GSE62452, GSE28735, GSE15471, GSE16515, and GSE183795. To analyze survival correlations, patient data was accessed through the R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl), and the “Mixed pancreatic adenocarcinoma (2022vs32)” dataset from TCGA was selected. Overall survival correlation with PRKCI, PRKCZ, PARD3, and PARD6B expression levels was assessed using the following setting: cut_off modus = scan, minimal group size = 8. Cut-off was established based on significance.
Quantification and statistical analysis
All graphs were made using GraphPad Prism software (GraphPad). Results are shown as the mean of at least three independent experiments ± standard error of the mean (SEM) unless stated otherwise. Statistical significance was determined by the unpaired two-tailed Student’s t test with Welch’s correction when appropriate, except for tumor growth analysis, that was determined by two-way ANOVA followed by the Tukey test for multiple comparisons. Where indicated, an outlier test using the ROUT method was performed. Statistical significance was confirmed both before and after the removal of the outliers. P-values less than 0.05 were considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001 and ns = not significant).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA sequencing data generated in this study have been deposited in the GEO database under accession code GSE276007. The rest of the data are within the article, supplementary information, or source data file. Source data are provided in this paper.
References
Bloomfield, G. & Kay, R. R. Uses and abuses of macropinocytosis. J. Cell Sci. 129, 2697–2705 (2016).
Recouvreux, M. V. & Commisso, C. Macropinocytosis: A metabolic adaptation to nutrient stress in cancer. Front. Endocrinol. 8, 261 (2017).
Palm, W. Metabolic functions of macropinocytosis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 374, 20180285 (2019).
Commisso, C. et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637 (2013).
Lee, S. W. et al. EGFR-Pak signaling selectively regulates glutamine deprivation-induced macropinocytosis. Dev. Cell 50, 381–392 (2019).
Lambies, G. & Commisso, C. Macropinocytosis and Cancer: From tumor stress to signaling pathways. Subcell. Biochem. 98, 15–40 (2022).
Kamphorst, J. J. et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 75, 544–553 (2015).
Zhang, Y. et al. Macropinocytosis in cancer-associated fibroblasts is dependent on CaMKK2/ARHGEF2 signaling and functions to support tumor and stromal cell fitness. Cancer Discov. 7, 1808–1825 (2021).
Kim, S. M. et al. PTEN deficiency and AMPK activation promote nutrient scavenging and anabolism in prostate cancer cells. Cancer Discov. 8, 866–883 (2018).
Jayashankar, V. & Edinger, A. L. Macropinocytosis confers resistance to therapies targeting cancer anabolism. Nat. Commun. 11, 1121 (2020).
Nofal, M., Zhang, K., Han, S. & Rabinowitz, J. D. mTOR inhibition restores amino acid balance in cells dependent on catabolism of extracellular protein. Mol. Cell 67, 936–946 (2017).
Cluntun, A. A., Lukey, M. J., Cerione, R. A. & Locasale, J. W. Glutamine metabolism in cancer: Understanding the heterogeneity. Trends Cancer 3, 169–180 (2017).
Gandalovicova, A., Vomastek, T., Rosel, D. & Brabek, J. Cell polarity signaling in the plasticity of cancer cell invasiveness. Oncotarget 7, 25022–25049 (2016).
Heikenwalder, M. & Lorentzen, A. The role of polarisation of circulating tumour cells in cancer metastasis. Cell Mol. Life Sci. 76, 3765–3781 (2019).
Hurov, J. B., Watkins, J. L. & Piwnica-Worms, H. Atypical PKC phosphorylates PAR-1 kinases to regulate localization and activity. Curr. Biol. 14, 736–741 (2004).
Wu, Y. & Griffin, E. E. Regulation of cell polarity by PAR-1/MARK Kinase. Curr. Top. Dev. Biol. 123, 365–397 (2017).
Harris, T. J. & Peifer, M. aPKC controls microtubule organization to balance adherens junction symmetry and planar polarity during development. Dev. Cell 12, 727–738 (2007).
Fennell, M., Commisso, C., Ramirez, C., Garippa, R. & Bar-Sagi, D. High-content, full genome siRNA screen for regulators of oncogenic HRAS-driven macropinocytosis. Assay Drug Dev. Technol. 13, 347–355 (2015).
Galenkamp, K. M. O., Galapate, C. M., Zhang, Y. & Commisso, C. Automated imaging and analysis for the quantification of fluorescently labeled macropinosomes. J. Vis. Exp. 174, https://doi.org/10.3791/62828 (2021).
Ratnayake, W. S., Apostolatos, A. H., Ostrov, D. A. & Acevedo-Duncan, M. Two novel atypical PKC inhibitors; ACPD and DNDA effectively mitigate cell proliferation and epithelial to mesenchymal transition of metastatic melanoma while inducing apoptosis. Int. J. Oncol. 51, 1370–1382 (2017).
Mayle, K. M., Le, A. M. & Kamei, D. T. The intracellular trafficking pathway of transferrin. Biochim. Biophys. Acta 1820, 264–281 (2012).
Hong, Y. aPKC: the Kinase that phosphorylates cell polarity. F1000Res. 7, https://doi.org/10.12688/f1000research.14427.1 (2018).
Aranda, V., Nolan, M. E. & Muthuswamy, S. K. Par complex in cancer: a regulator of normal cell polarity joins the dark side. Oncogene 27, 6878–6887 (2008).
Nolan, M. E. et al. The polarity protein Par6 induces cell proliferation and is overexpressed in breast cancer. Cancer Res. 68, 8201–8209 (2008).
Nakamura, H. et al. Expression of Par3 polarity protein correlates with poor prognosis in ovarian cancer. BMC Cancer 16, 897 (2016).
Suzuki, A. & Ohno, S. The PAR-aPKC system: lessons in polarity. J. Cell Sci. 119, 979–987 (2006).
Raman, R., Pinto, C. S. & Sonawane, M. Polarized organization of the cytoskeleton: Regulation by cell polarity proteins. J. Mol. Biol. 430, 3565–3584 (2018).
Goransson, O. et al. Regulation of the polarity kinases PAR-1/MARK by 14-3-3 interaction and phosphorylation. J. Cell Sci. 119, 4059–4070 (2006).
Williamson, C. D. & Donaldson, J. G. Arf6, JIP3, and dynein shape and mediate macropinocytosis. Mol. Biol. Cell 30, 1477–1489 (2019).
Sitbon, Y. H., Yadav, S., Kazmierczak, K. & Szczesna-Cordary, D. Insights into myosin regulatory and essential light chains: a focus on their roles in cardiac and skeletal muscle function, development and disease. J. Muscle Res. Cell Motil. 41, 313–327 (2020).
Reck-Peterson, S. L., Redwine, W. B., Vale, R. D. & Carter, A. P. Publisher Correction: The cytoplasmic dynein transport machinery and its many cargoes. Nat. Rev. Mol. Cell Biol. 19, 479 (2018).
Schmoranzer, J. et al. Par3 and dynein associate to regulate local microtubule dynamics and centrosome orientation during migration. Curr. Biol. 19, 1065–1074 (2009).
Jouette, J., Guichet, A. & Claret, S. B. Dynein-mediated transport and membrane trafficking control PAR3 polarised distribution. Elife 8, https://doi.org/10.7554/elife.40212 (2019).
Soriano, E. V. et al. aPKC Inhibition by Par3 CR3 flanking regions controls substrate access and underpins apical-junctional polarization. Dev. Cell 38, 384–398 (2016).
Morais-de-Sa, E., Mirouse, V. & St Johnston, D. aPKC phosphorylation of Bazooka defines the apical/lateral border in Drosophila epithelial cells. Cell 141, 509–523 (2010).
Walther, R. F. & Pichaud, F. Crumbs/DaPKC-dependent apical exclusion of Bazooka promotes photoreceptor polarity remodeling. Curr. Biol. 20, 1065–1074 (2010).
Lizcano, J. M. et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 23, 833–843 (2004).
Messerschmidt, A. et al. Crystal structure of the catalytic domain of human atypical protein kinase C-iota reveals interaction mode of phosphorylation site in turn motif. J. Mol. Biol. 352, 918–931 (2005).
Newton, A. C. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem. J. 370, 361–371 (2003).
Li, X. & Gao, T. mTORC2 phosphorylates protein kinase Czeta to regulate its stability and activity. EMBO Rep. 15, 191–198 (2014).
Baldwin, R. M., Parolin, D. A. & Lorimer, I. A. Regulation of glioblastoma cell invasion by PKC iota and RhoB. Oncogene 27, 3587–3595 (2008).
Dang, M. et al. Epidermal growth factor (EGF) ligand release by substrate-specific a disintegrin and metalloproteases (ADAMs) involves different protein kinase C (PKC) isoenzymes depending on the stimulus. J. Biol. Chem. 286, 17704–17713 (2011).
Geribaldi-Doldan, N., Gomez-Oliva, R., Dominguez-Garcia, S., Nunez-Abades, P. & Castro, C. Protein Kinase C: Targets to regenerate brain injuries? Front. Cell Dev. Biol. 7, 39 (2019).
Kusne, Y. et al. Targeting aPKC disables oncogenic signaling by both the EGFR and the proinflammatory cytokine TNFalpha in glioblastoma. Sci. Signal. 7, ra75 (2014).
Redelman-Sidi, G., Iyer, G., Solit, D. B. & Glickman, M. S. Oncogenic activation of Pak1-dependent pathway of macropinocytosis determines BCG entry into bladder cancer cells. Cancer Res. 73, 1156–1167 (2013).
Amstutz, B. et al. Subversion of CtBP1-controlled macropinocytosis by human adenovirus serotype 3. EMBO J. 27, 956–969 (2008).
Islam, S. M. A., Patel, R. & Acevedo-Duncan, M. Protein Kinase C-zeta stimulates colorectal cancer cell carcinogenesis via PKC-zeta/Rac1/Pak1/beta-Catenin signaling cascade. Biochim. Biophys. Acta Mol. Cell Res. 1865, 650–664 (2018).
Lachmann, A. et al. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444 (2010).
Rauen, T., Hedrich, C. M., Tenbrock, K. & Tsokos, G. C. cAMP responsive element modulator: a critical regulator of cytokine production. Trends Mol. Med. 19, 262–269 (2013).
Martianov, I. et al. Cell-specific occupancy of an extended repertoire of CREM and CREB binding loci in male germ cells. BMC Genomics 11, 530 (2010).
Vorhagen, S. et al. Shared and independent functions of aPKClambda and Par3 in skin tumorigenesis. Oncogene 37, 5136–5146 (2018).
Belmokhtar, C. A., Hillion, J. & Segal-Bendirdjian, E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 20, 3354–3362 (2001).
Aguilar-Aragon, M. et al. Pak1 kinase maintains apical membrane identity in epithelia. Cell Rep. 22, 1639–1646 (2018).
Scotti, M. L., Bamlet, W. R., Smyrk, T. C., Fields, A. P. & Murray, N. R. Protein kinase Ciota is required for pancreatic cancer cell transformed growth and tumorigenesis. Cancer Res. 70, 2064–2074 (2010).
Scotti, M. L. et al. Protein kinase C iota regulates pancreatic acinar-to-ductal metaplasia. PLoS ONE 7, e30509 (2012).
Ali, S. A., Justilien, V., Jamieson, L., Murray, N. R. & Fields, A. P. Protein kinase ciota drives a NOTCH3-dependent stem-like phenotype in mutant KRAS lung adenocarcinoma. Cancer Cell 29, 367–378 (2016).
Yin, N. et al. Protein kinase ciota and Wnt/beta-catenin signaling: Alternative pathways to Kras/Trp53-driven lung adenocarcinoma. Cancer Cell 36, 692–693 (2019).
Yin, N. et al. Protein kinase Ciota mediates immunosuppression in lung adenocarcinoma. Sci. Transl. Med. 14, eabq5931 (2022).
Inman, K. S. et al. Prkci regulates autophagy and pancreatic tumorigenesis in mice. Cancers 14, https://doi.org/10.3390/cancers14030796 (2022).
Atwood, S. X., Li, M., Lee, A., Tang, J. Y. & Oro, A. E. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature 494, 484–488 (2013).
Wang, Y., Hill, K. S. & Fields, A. P. PKCiota maintains a tumor-initiating cell phenotype that is required for ovarian tumorigenesis. Mol. Cancer Res. 11, 1624–1635 (2013).
Uberall, F. et al. Evidence that atypical protein kinase C-lambda and atypical protein kinase C-zeta participate in Ras-mediated reorganization of the F-actin cytoskeleton. J. Cell Biol. 144, 413–425 (1999).
Jinesh, G. G., Taoka, R., Zhang, Q., Gorantla, S. & Kamat, A. M. Novel PKC-zeta to p47 phox interaction is necessary for transformation from blebbishields. Sci. Rep. 6, 23965 (2016).
Della Peruta, M., Giagulli, C., Laudanna, C., Scarpa, A. & Sorio, C. RHOA and PRKCZ control different aspects of cell motility in pancreatic cancer metastatic clones. Mol. Cancer 9, 61 (2010).
Butler, A. M. et al. Protein kinase C zeta regulates human pancreatic cancer cell transformed growth and invasion through a STAT3-dependent mechanism. PLoS ONE 8, e72061 (2013).
Galvez, A. S. et al. Protein kinase Czeta represses the interleukin-6 promoter and impairs tumorigenesis in vivo. Mol. Cell Biol. 29, 104–115 (2009).
Ma, L. et al. Control of nutrient stress-induced metabolic reprogramming by PKCzeta in tumorigenesis. Cell 152, 599–611 (2013).
Llado, V. et al. Repression of intestinal stem cell function and tumorigenesis through direct phosphorylation of beta-Catenin and Yap by PKCzeta. Cell Rep. 10, 740–754 (2015).
Kim, J. Y. et al. c-Myc phosphorylation by PKCzeta represses prostate tumorigenesis. Proc. Natl. Acad. Sci. USA 110, 6418–6423 (2013).
Nakanishi, Y. et al. Control of paneth cell fate, intestinal inflammation, and tumorigenesis by PKClambda/iota. Cell Rep. 16, 3297–3310 (2016).
Nakanishi, Y. et al. Simultaneous loss of both atypical protein kinase C genes in the intestinal epithelium drives serrated intestinal cancer by impairing immunosurveillance. Immunity 49, 1132–1147 (2018).
Reina-Campos, M., Diaz-Meco, M. T. & Moscat, J. The dual roles of the atypical protein kinase Cs in cancer. Cancer Cell 36, 218–235 (2019).
Soloff, R. S., Katayama, C., Lin, M. Y., Feramisco, J. R. & Hedrick, S. M. Targeted deletion of protein kinase C lambda reveals a distribution of functions between the two atypical protein kinase C isoforms. J. Immunol. 173, 3250–3260 (2004).
Leitges, M. et al. Targeted disruption of the zetaPKC gene results in the impairment of the NF-kappaB pathway. Mol. Cell 8, 771–780 (2001).
Rodriguez-Boulan, E. & Macara, I. G. Organization and execution of the epithelial polarity programme. Nat. Rev. Mol. Cell Biol. 15, 225–242 (2014).
Singla, B. et al. PKCdelta-Mediated Nox2 activation promotes fluid-phase pinocytosis of antigens by immature dendritic cells. Front. Immunol. 9, 537 (2018).
Lee, M. & Vasioukhin, V. Cell polarity and cancer–cell and tissue polarity as a non-canonical tumor suppressor. J. Cell Sci. 121, 1141–1150 (2008).
Jung, H. Y. et al. Apical-basal polarity inhibits epithelial-mesenchymal transition and tumour metastasis by PAR-complex-mediated SNAI1 degradation. Nat. Cell Biol. 21, 359–371 (2019).
Durgan, J., Kaji, N., Jin, D. & Hall, A. Par6B and atypical PKC regulate mitotic spindle orientation during epithelial morphogenesis. J. Biol. Chem. 286, 12461–12474 (2011).
Linch, M. et al. Regulation of polarized morphogenesis by protein kinase C iota in oncogenic epithelial spheroids. Carcinogenesis 35, 396–406 (2014).
Gunaratne, A., Thai, B. L. & Di Guglielmo, G. M. Atypical protein kinase C phosphorylates Par6 and facilitates transforming growth factor beta-induced epithelial-to-mesenchymal transition. Mol. Cell Biol. 33, 874–886 (2013).
Zhou, P. J. et al. Elevated expression of Par3 promotes prostate cancer metastasis by forming a Par3/aPKC/KIBRA complex and inactivating the hippo pathway. J. Exp. Clin. Cancer Res. 36, 139 (2017).
Zhou, P. J. et al. Loss of Par3 promotes prostatic tumorigenesis by enhancing cell growth and changing cell division modes. Oncogene 38, 2192–2205 (2019).
Guo, X. et al. Par3 regulates invasion of pancreatic cancer cells via interaction with Tiam1. Clin. Exp. Med. 16, 357–365 (2016).
Benton, R. & St Johnston, D. Drosophila PAR-1 and 14-3-3 inhibit Bazooka/PAR-3 to establish complementary cortical domains in polarized cells. Cell 115, 691–704 (2003).
Lee, S. W. & Commisso, C. Rac1 and EGFR cooperate to activate Pak in response to nutrient stress. Biochem. Biophys. Res. Commun. 533, 437–441 (2020).
Parker, A. L., Kavallaris, M. & McCarroll, J. A. Microtubules and their role in cellular stress in cancer. Front. Oncol. 4, 153 (2014).
Geeraert, C. et al. Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J. Biol. Chem. 285, 24184–24194 (2010).
Bershadsky, A. D. & Gelfand, V. I. ATP-dependent regulation of cytoplasmic microtubule disassembly. Proc. Natl. Acad. Sci. USA 78, 3610–3613 (1981).
Infante, A. S., Stein, M. S., Zhai, Y., Borisy, G. G. & Gundersen, G. G. Detyrosinated (Glu) microtubules are stabilized by an ATP-sensitive plus-end cap. J. Cell Sci. 113, 3907–3919 (2000).
Qian, Y. et al. Extracellular ATP is internalized by macropinocytosis and induces intracellular ATP increase and drug resistance in cancer cells. Cancer Lett. 351, 242–251 (2014).
Jia, R. & Bonifacino, J. S. Lysosome positioning influences mTORC2 and AKT signaling. Mol. Cell 75, 26–38 (2019).
Ratnikov, B. et al. Glutamate and asparagine cataplerosis underlie glutamine addiction in melanoma. Oncotarget 6, 7379–7389 (2015).
Galenkamp, K. M. O. et al. Golgi acidification by NHE7 regulates cytosolic pH homeostasis in pancreatic cancer cells. Cancer Discov. 10, 822–835 (2020).
Lee, S. W., Alas, B. & Commisso, C. Detection and quantification of macropinosomes in pancreatic tumors. Methods Mol. Biol. 1882, 171–181 (2019).
Commisso, C., Flinn, R. J. & Bar-Sagi, D. Determining the macropinocytic index of cells through a quantitative image-based assay. Nat. Protoc. 9, 182–192 (2014).
Acknowledgements
We thank all members of the Commisso laboratory for their helpful comments, discussions, and scientific advice. This work was supported by NIH grants R01CA254806 and R01CA207189 to C.C. Sanford Burnham Prebys Medical Discovery Institute core services are supported by NCI Cancer Center Support grant P30CA030199. DAS is supported by NCI Research Specialist award R50 CA283813.
Author information
Authors and Affiliations
Contributions
G.L. and C.C. designed the study and wrote the manuscript. G.L. performed most of the experimental work and prepared all the figures. S.-W.L. and P.A.-B. performed the siRNA kinome-wide screen. S.-W.L. performed validation for the hits from the siRNA screen. K.D.-P. performed the immunohistochemical staining of the TMA. S.M. assisted with the immunohistochemical staining of the tumor xenograft sections. C.-M.G. performed the transferrin uptake assays. A.D. and A.-J.D. assisted with the ChIP studies. D.-A.S. supervised the isotope-tracing assays, performed the GC-MS study, and analyzed the data. D.W.D. provided the TMA samples and scored the stained TMAs. C.C. supervised the study and obtained funding. All authors reviewed, edited, or commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
C.C. is an inventor on a U.S. patent titled “Cancer diagnostics, therapeutics, and drug discovery associated with macropinocytosis,” Patent number: US-11209420-B2. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lambies, G., Lee, SW., Duong-Polk, K. et al. Cell polarity proteins promote macropinocytosis in response to metabolic stress. Nat Commun 15, 10541 (2024). https://doi.org/10.1038/s41467-024-54788-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54788-9
This article is cited by
-
Targeting macropinocytosis for cancer therapy
Nature Reviews Cancer (2025)
