
Abstract
In the spore-forming bacterium Bacillus subtilis transcription and translation are uncoupled and the translational machinery is located at the cell poles. During sporulation, the cell undergoes morphological changes including asymmetric division and chromosome translocation into the forespore. However, the fate of translational machinery during sporulation has not been described. Here, using microscopy and mass spectrometry, we show the localisation of ribosomes during sporulation in wild type and mutant Bacillus subtilis. We demonstrate that ribosomes are associated with the asymmetric septum, a functionally important organelle in the cell’s developmental control, and that SpoIIDMP-driven peptidoglycan rearrangement is crucial for ribosomes packing into the forespore. We also show that the SpoIIIA-SpoIIQ ‘feeding-tube’ channel is not required for ribosome translocation. Our results demonstrate that translation and translational machinery are temporally and spatially organised in B. subtilis during sporulation and that the forespore ‘inherits’ ribosomes from the mother cell. We propose that the movement of ribosomes in the cell may be mediated by the bacterial homologs of cytoskeletal proteins and that the cues for asymmetric division localisation may be translation-dependent. We anticipate our findings to elicit more sophisticated structural and mechanistic studies of ribosome organisation during bacterial cell development.
Similar content being viewed by others

Introduction
The view of a bacterial cell as a disorganised collection of macromolecules enclosed by a rigid cell wall is slowly but surely going into obscurity1. The number of proteins having discrete subcellular addresses in the bacterial cell has been increasing with examples including cell division proteins FtsZ and the Min system (involved in cell division site selection)2,3, or chromosome translocases FtsK and SpoIIIE in Escherichia coli and Bacillus subtilis, respectively4,5. Interestingly, the translational machinery itself has been somewhat neglected in this type of studies, however, not due to a lack of subcellular location. In vegetative cells of B. subtilis, the ribosomes were shown to be localised at the cell poles, away from the centrally located nucleoid and transcriptional machinery6,7. The functional consequences of this separation were recently described by Johnson et al.8 in their seminal paper showing that transcription and translation are uncoupled in B. subtilis.
In response to nutrient limitation, vegetative cells of B. subtilis can undergo a developmental change to form dormant and resistant spores. Sporulation is a tightly regulated and highly trackable process, both temporally and spatially. The hallmark of sporulation is formation of an asymmetric septum which divides the cell into two compartments with differing fates: small forespore and a larger mother cell. In brief, sporulation consists of precisely regulated positioning of the asymmetric septum, chromosome translocation, engulfment of the forespore by the mother cell, spore coat and cortex development and finally, mother cell lysis and spore release (Fig. 1a)9,10,11,12. In this sequence of morphological events, two processes receive considerable attention—rearrangements of the cell membrane and peptidoglycan and chromosome translocation13,14,15,16. However, although the translational machinery including ribosomes and translation factors constitutes ~20% of the cell volume17, little is known about its localisation and dynamics during sporulation, and only recently the ribosomes became objects of detailed observations in the sporulating B. subtilis18.

a Schematic of the sporulation process in B. subtilis. A vegetative cell undergoes asymmetric septation, followed by chromosome translocation into the forespore. Next, the mother cell engulfs the forespore which then develops spore cortex and coat. A mature spore is then released as a result of mother cell autolysis. B. subtilis WT expressing GFP-tagged ribosomes, RpsB-GFP (b) or GFP-tagged RNA polymerase β' subunit, RpoC-GFP (d). Cells were stained with DAPI to visualise the chromosome and FM4-64, a membrane stain, to track the asymmetric septation and sporulation progress. Blue arrowhead points to the localisation of the chromosome (DAPI) and green arrowheads point to the localisation of ribosomes (GFP). Samples were taken before sporulation induction (T0) and every hour of the sporulation process (T1–T5). Scale bar is 2 µm. Example cells and plots of mean GFP fluorescence intensity (green) across the cells show subcellular localisation of ribosomes (c) or RNA polymerase (e) during sporulation in relation to the chromosome (blue) and cell membrane and asymmetric septum (red). The cell lengths were normalised and fluorescence was measured along a line drawn across the cell long axis. The cells are oriented so that the spore is located to the left. The number of cells (n) analysed for each strain and time point is as follows: RpsB-GFP (T0, n = 109; T1, n = 110; T2, n = 225; T3, n = 246; T4, n = 116; T5, n = 138); RpoC-GFP (T0, n = 116; T1, n = 205; T2, n = 163; T3, n = 119; T4, n = 84; T5, n = 99). Scale bar is 1 µm. f Microscopic images of B. subtilis WT cells treated with OPP and stained with Alexa 488 and DAPI, illustrating active translation during 5 h of sporulation (T0–T5). White arrowheads point to the location of selected forespores. Scale bar is 2 µm. g Plots of the mean OPP-Alexa 488 fluorescence intensity (green) across the cell showing localisation and intensity of active translation during sporulation in relation to the chromosome localisation (blue). The cell lengths were normalised and fluorescence was measured along a line drawn across the cell long axis. The number of analysed cells (n) is as follows (T0, n = 186; T1, n = 609; T2, n = 435; T3, n = 346; T4, n = 338; T5, n = 422). Scale bar is 1 µm. Error bars represent ±SD (standard deviation) of the fluorescence intensities. Source data are provided as a Source Data file.
In this work, we used a combination of microscopic assays to monitor positioning of the ribosomes during sporulation in B. subtilis. We showed that localisation of the ribosomes correlates with the active translation sites and that the asymmetric septum plays an important role in the spatial organisation of ribosomes. We also showed that the ribosomes enter the forespore in a sequential manner, after the chromosome translocation and this is dependent on septal peptidoglycan rearrangements.
Results
Ribosome packing into the forespore is sequential
Using fluorescence microscopy, we observed distribution of ribosomes during sporulation in B. subtilis in 1-h intervals, beginning at T0 (logarithmic growth, immediately prior to sporulation induction) to T5 (5 h post sporulation induction). We tagged the small ribosomal protein RpsB (S2) with GFP to monitor the position of ribosomes in relation to the asymmetric septum and the chromosome, stained with FM4-64 and DAPI, respectively (Fig. 1b, c). At T0, mid-exponential phase, the GFP signal is localised throughout the cell, with a slight increase at the cell poles. This pattern becomes more pronounced 1 h post sporulation induction, where both poles are enriched in ribosomes. However, at T2 when the asymmetric septum is being formed at ~20th percentile of the cell’s length, we observed a drop in GFP signal in favour of DAPI at the cell pole where the forespore is developing. Approximately 3 h post sporulation induction, membrane migration is mostly complete, based on the increase in FM4-64 signal at the cell pole where the spore is (Fig. 1b, c, Supplementary Fig. 1), and this is accompanied by chromosome translocation resulting in forespore inflation to ~30% of the mother cell’s length. Interestingly, we observed that during asymmetric septation and chromosome translocation (T2–T3), the fluorescence signal corresponding to ribosomes at the cell pole (T1) decreases, suggesting that less ribosomes are detected in the developing forespore. It appears that the ribosomes gather and wait at the asymmetric septum at the mother cell side and during or very shortly after the engulfment by the mother cell, the ribosomes are translocated into the spore (T4). To further investigate whether such sequential packing of the developing spore is characteristic of sporulation and applies also to transcriptional machinery, we observed the localisation of GFP-tagged β' subunit of RNA polymerase (RpoC) during sporulation. We show that the RpoC colocalizes with the chromosome during asymmetric septation and membrane migration (T2 and T3), and is not excluded from the developing forespore prior to engulfment (T4) (Fig. 1d, e). The SpoIIIE channel strips the translocating chromosome of the DNA-binding proteins including RNA polymerase. However, as the chromosome segregation begins before asymmetric septation, a proportion of RNA polymerase is present at the cell pole prior to SpoIIIE channel formation. The increase in fluorescence intensity in the forespore results from expression of rpoC-gfp from the native locus, which is present in the forespore compartment at the time of polar septation19. This is supported by the detection of de novo RNA synthesis in the forespore at T2 and T3, measured by incorporation and fluorescent tagging of the uridine analogue, 5-ethynyluridine (EU)20 (Supplementary Fig. 2). The initial expression of RpoC-GFP in the early forespore implies, however, the presence of translating ribosomes in this compartment. We investigated this using an Alexa 488 labelled alkyne analogue of puromycin (O-propargyl-puromycin (OPP)). Puromycin terminates translation by mimicking an aminoacyl-tRNA and binding to the nascent polypeptide chain. Fluorescently labelled nascent chains allow for visualisation and relative quantification of the incorporated OPP, or in other words, of the active translation21. As shown previously in Iwanska et al.22, in the early time points translation localises to the cell poles, away from the chromosome (Fig. 1f, g), which is consistent with the uncoupled transcription–translation in B. subtilis discussed in the seminal paper by Johnson et al. 8. At T2, around the time of asymmetric division, translation declines significantly which correlates with the loss of polar localisation of the ribosomes. Once the asymmetric septation is complete, translation takes place mostly at the septum on the mother cell side, and then inside the spore (T4). It should be noted, however, that past T4 the fluorescence signal from the spore is limited due to, most probably, spore impermeability and the results presented in Fig. 1f, g regarding late sporulation (T5) are more representative of the mother cell. Low level of translation in the forespore prior to engulfment is consistent with our observation that there is a small proportion of ribosomes present in the forming forespore and in fact, most of the ribosomes are localised at the septum on the mother cell side. Around the time of engulfment, however, ribosomes are translocated into the forespore. We also followed these dynamic subcellular changes in the ribosome and RNA polymerase localisation using time-lapse microscopy (Supplementary Videos 1 and 2, Supplementary Figs. 3 and 4). Based on the above observations we propose that the ribosomes are packed into the forespore sequentially, after the chromosome translocation, and that this shift is associated with membrane migration or cell wall remodelling during engulfment.
Mass spectrometry indicates direct interaction of the ribosome with components of the protein machinery required for engulfment
We applied tandem mass tag-mass spectrometry (TMT-MS) analysis to determine which proteins may interact with the ribosome at the site of asymmetric septation. We collected the cells 3 h post sporulation induction and performed digitonin-based lysis on DSP (dithiobis(succinimidyl propionate)) cross-linked and non-cross-linked cultures, in duplicates. Digitonin-based lysis allowed for membrane solubilisation and enrichment of the lysates with membrane interacting and/or bound proteins. We used flash-frozen and pulverised cells (according to Kopik et al.23) as a control of the lysis method, as well as cells harvested in the logarithmic phase as a control for the developmental stage (sporulating vs non-sporulating cells). We separated the 70S ribosome and disome-containing fractions using sucrose gradient centrifugation (Fig. 2a, Supplementary Fig. 5). We used close to physiological salt concentration and did not prepurify the ribosomes using sucrose cushion to allow for co-migration of even transiently associated factors. Although we are aware this may result in high background, including for example, large multiprotein complexes like flagellum (Fli, Flh, Flg and Hag proteins)24, we did intend to cast a wide net in our search. The TMT-MS analysis revealed 641 proteins, including 55 ribosomal proteins and their paralogs (Supplementary Data 1). We performed PCA analysis of log2 normalised reporter intensities for all samples, revealing adequate grouping of data based on preservation of the native ribosomes (lysis-dependent, PC1) and growth stage (sporulating vs logarithmic, PC2) (Supplementary Fig. 5). We compared the consecutive cross-linked (DSP) vs non-cross-linked (D) samples lysed with digitonin and identified 204, 174, 111 and 82 enriched proteins in the consecutive cross-linked fractions which we categorised into 11 functional groups (Fig. 2b, Supplementary Data 1). We also compared both experimental groups with the control of the lysis method and the monosomal fractions of cells in sporulating vs logarithmic growth phases (Supplementary Fig. 6). The biggest functional group of proteins enriched in cross-linked samples was broadly defined metabolism, mostly due to the polar and/or membrane-associated localisation of proteins within this group. The second and third next functional groups were sporulation and cell envelope and cell division proteins. In the DSP cross-linked samples, we also observed enrichment of translation factors including IF-2 (infB), IF-3 (infC), EF-G (fusA), EF-Tu (tufA), EF-P (efp) and RRF (frr) indicating that we have collected actively translating ribosomes. We observed enrichment of the cell wall and membrane-associated proteins (cell envelope and cell division group), such as components of the Min system, bacterial cytoskeletal homologues (MreB, MreC, Mbl, DivIVA, FtsH) and regulatory proteins (FtsE-FtsX, EzrA, GpsB). Within the sporulation group, we identified proteins related to spore coat and cortex assembly (SpoIVA, SpoVR, SpoVS, CotE, CotO, SafA, CwlD), mother cell and forespore-associated penicillin-binding proteins (DacF, SpoVD) and specifically, components of the feeding tube channel (SpoIIIAG, SpoIIIAH, SpoIIQ and GerM) and septal peptidoglycan degradation module (SpoIIP and SpoIIB) (Fig. 2b). Since both of these protein complexes are essential for engulfment and constitute a potential gateway for the ribosomes to enter the spore, we further investigated whether either one or both may have a role in translocation of the ribosomes into the developing spore.

a Polysome profiles of the DSP cross-linked (DSP) and non-cross-linked (D) samples of sporulating B. subtilis cells lysed with digitonin. Fractions analysed by MS, in both DSP and D conditions, are highlighted in grey: monosome (fr1), monosomal shoulder (fr2) and disome (fr 3 and 4). Positions of the ribosomal subunits (30S, 50S), monosome (70S) and polysomes are indicated. b Volcano plots illustrating the mass spectrometry proteomics data for cross-linked vs non-cross-linked samples (DSP vs D), for the consecutive fractions 1–4. The x-axis shows the calculated Log2 fold change and p values are represented on the y-axis as −Log10(p value) (Student’s T-test, two-sided; no adjustments for multiple comparisons). The vertical and horizontal dotted lines show the cut-off of Log2(Fold Change) = ± 1 (Fold Change = ± 2), and for p value = 0.05, respectively. The enriched proteins are categorised into 11 functional groups, colour-coded accordingly to the legend shown in bottom left. Protein components of the feeding tube channel (SpoIIIAH-AG, SpoIIQ and GerM) and peptidoglycan degradation module (SpoIIP and SpoIIB) are labelled. Source data are provided as a Source Data file.
Deletion of the components of SpoIIIA-SpoIIQ complex does not influence ribosome localisation
The SpoIIIA proteins, encoded in the single operon spoIIIA (SpoIIIAA-SpoIIIAH), together with SpoIIQ and GerM required for assembly, form a multimeric channel connecting the mother cell and the forespore. The channel was proposed to constitute a hybrid secretion-like transport machinery and function as a feeding tube. Indeed, the SpoIIIA-SpoIIQ channel was shown to maintain forespore physiology and mediate small-molecule intercellular transport25,26,27. Deletion strains of any component of the complex produce small forespores with membrane invaginations and limited transcription during late sporulation (expression of σG regulon), generally resulting in defective spore formation28. SpoIIIAH and SpoIIQ were also identified as having additional role in membrane migration during engulfment, forming a zipper-like structure29. Because of this dual function, we investigated ribosome localisation during sporulation in ΔspoIIIAH and ΔspoIIQ strains in half-hour intervals, between 2 and 5 h post sporulation induction, using GFP-tagged ribosomes as above. Both strains formed asymmetric septa between 2 and 2.5 h post sporulation induction, maintaining the ribosome localisation to the mother cell side of the septum, similarly to WT cells, and failed to conclude spore engulfment by T3, as expected. However, in the later stages of sporulation (T3–T3:30) we detected the GFP signal at the forespore membrane vicinity, and by T4 the ribosomes were localised inside the forespore (Fig. 3a–c, Supplementary Fig. 7a, b). This implies that the Q-AH complex may aid in, but is not essential for the ribosome translocation into the spore of B. subtilis. We also investigated localisation of the transcriptional machinery and active translation in ΔspoIIQ strain (Supplementary Fig. 8). Interestingly, we found that although both transcriptional and translational machineries are present in the forespore of the mutant strain, there is little fluorescence signal corresponding to the active translation in the spore and the strongest OPP-Alexa 488 signal is localised to the asymmetric septum in late sporulation (T4 and T5). This is most probably a direct consequence of limited transcription in the spore described by others25,30, however, not due to the lack of relevant protein machineries. The feeding tube channel has been shown to transport small molecules including arginine30, however, we also investigated the effect of deletion of the ring-forming complex of the channel31, ΔspoIIIAG, on the ribosome localisation. The mutant showed the expected small spores phenotype, and the ribosomal translocation into the forespore was not affected suggesting that the feeding tube channel does not take part in ribosome translocation into the developing spore (Supplementary Fig. 9).

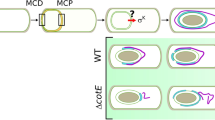
a Selected cells of the ΔspoIIIAH, ΔspoIIQ and ΔspoIID strains with fluorescently tagged RpsB-GFP and RpoC-GFP proteins. Cells were stained with DAPI to visualise the chromosome and FM4-64, a membrane stain, to track the asymmetric septation and sporulation progress. Samples were taken every 30 min starting 2 h post sporulation induction (T2) up to 5 h post sporulation induction (T5). Scale bar is 1 µm. b Plots of the mean fluorescence intensity illustrating the localisation of ribosomes (RpsB-GFP) or RNA polymerase (RpoC-GFP), in relation to the asymmetric septum (FM4-64, red) and chromosome (DAPI, blue) in ΔspoIIIAH, ΔspoIIQ and ΔspoIID strains during sporulation (T2–T5). The cell lengths were normalised and fluorescence was measured along a line drawn across the cell long axis. The number of cells (n) analysed for each strain and time point is as follows: ΔspoIIIAH RpsB-GFP (T2, n = 507; T2:30, n = 364; T3, n = 532; T3:30, n = 425; T4, n = 560; T4:30, n = 505; T5, n = 477); ΔspoIIQ RpsB-GFP (T2, n = 718; T2:30, n = 684; T3, n = 351; T3:30, n = 356; T4, n = 512; T4:30, n = 456; T5, n = 393); ΔspoIID RpsB-GFP (T2, n = 273; T2:30, n = 391; T3, n = 338; T3:30, n = 269; T4, n = 182; T4:30, n = 159; T5, n = 198); and ΔspoIID RpoC-GFP (T2, n = 445; T2:30, n = 450; T3, n = 434; T3:30, n = 437; T4, n = 262; T4:30, n = 238; T5, n = 331). c Cartoon representation of the ribosome and RNA polymerase localisation (green) in the sporulating B. subtilis cell 4 h post sporulation induction (post engulfment, T4) in the ΔspoIIIAH, ΔspoIIQ and ΔspoIID strains. d Microscopic images of selected ΔspoIID cells treated with OPP and stained with Alexa 488 and DAPI, illustrating active translation during sporulation (T0–T5). Scale bar is 1 µm. Below, plots of the mean OPP-Alexa 488 fluorescence intensity (green) across the ΔspoIID cells showing localisation and intensity of active translation during sporulation in relation to the chromosome localisation (DAPI, blue). The number of cells (n) analysed for each time point is as follows: (T0, n = 353; T1, n = 358; T2, n = 609; T3, n = 345; T4, n = 415; T5, n = 457). The cell lengths were normalised and fluorescence was measured along a line drawn across the cell long axis. Cells exhibiting the bulged membrane phenotype were selected for the analysis. Error bars represent ±SD (standard deviation) of the fluorescence intensities. Source data are provided as a Source Data file.
Peptidoglycan remodelling by the SpoIIDMP is crucial for ribosome translocation into the spore
The SpoIIDMP complex localises at the leading edge of the mother cell membrane during engulfment and partially degrades peptidoglycan at the asymmetric septum allowing for membrane migration and new peptidoglycan synthesis. The SpoIIP and SpoIID are cell wall hydrolases and SpoIIM localises the complex to the septum. In B. subtilis, all three proteins are essential for engulfment and deletion mutants demonstrate developmental blockage during sporulation after asymmetric septation and produce bulged septal membranes12,32,33,34. We, therefore, investigated localisation of the translational machinery (GFP-tagged ribosomes) in the ΔspoIID strain between 2 and 5 h post sporulation induction. We observed asymmetric septation in the ΔspoIID strain between T2 and T3, followed by bulging of the septum (Fig. 3a, b, Supplementary Fig. 7c). Similar to WT, in the ΔspoIID strain ribosomes are also mostly excluded from the forespore and localised at the mother cell side of the asymmetric septum (T2:30–T3). However, unlike in WT, we did not record enrichment of the GFP signal inside the forespore at the later time points, suggesting that translocation of the ribosomes into the developing spore depends on peptidoglycan rearrangement. To test whether this applies also to transcriptional machinery, we observed the localisation of RpoC-GFP and recorded fluorescence signal in the forespore post asymmetric septation (Fig. 3a–c, Supplementary Fig. 7d). We then investigated translational activity of the ΔspoIID strain and showed that after the asymmetric division, there was negligible level of active translation in the forespores of the mutant strain (Fig. 3d, Supplementary Fig. 7e). We also investigated this in the ΔspoIIM and ΔspoIIP mutants and obtained similar results (Supplementary Fig. 10). In the light of SpoIIIE protein stripping activity during chromosome translocation discussed above, a decrease of OPP-Alexa 488 fluorescence intensity in the forespore at T3 (Fig. 3d) together with the presence of RNA polymerase in the forespore led us to further investigate the ribosomal localisation during sporulation. We used structured illumination microscopy (Lattice SIM) to observe the localisation of ribosomes (RpsB-GFP) in the WT and ΔspoIID strains between 1 and 4 h post sporulation induction in 30 min intervals, as well as localisation of the RNA polymerase (RpoC-GFP) in WT as a control (Fig. 4). We show that during asymmetric septation a small proportion of ribosomes is associated with the forespore membrane. In the WT strain, as the chromosome translocation concludes and as the peptidoglycan rearrangement driving spore engulfment progresses, the fluorescent signal in the spore membrane increases suggesting relocation of the ribosomes into the spore (T2–T3). However, this is not the case for the ΔspoIID strain in which blockage of the peptidoglycan degradation-driven engulfment prevents ribosome translocation into the spore. As the bulged, newly synthesised membrane is devoid of the GFP signal, this implies that the cell possibly uses existing mother cell membrane proteins to anchor and transport ribosomes. This may also suggest that de novo ribosome biosynthesis in the developing spore is very limited if not null. Levels of RNA polymerase on the other hand are only briefly decreased in the forespore, at the time of chromosome translocation by the SpoIIIE (T2), and the fluorescence intensity and distribution quickly become similar in both mother cell and the forespore in WT.

Lattice SIM images showing representative cells of WT (a) and ΔspoIID (b) strains with GFP-tagged ribosomes and WT strain with GFP-tagged RNA polymerase (c) during sporulation from T1 (1 h post sporulation induction) to T4 (4 h post sporulation induction). Images in (a, b) were reconstructed using SIM2 reconstruction algorithm and images in (c) were reconstructed using SIM reconstruction algorithm. Images processed in ImageJ using Fire LUT represent fluorescence signal intensity (blue—low; white—high; colour scale in the range of 0–256 [arbitrary unit]). Scale bar is 1 µm.
Discussion
In this work, we propose a model of ribosomes translocation into the developing spore in B. subtilis (Fig. 5). Contrary to the current model, we propose that the ribosomes are not already present in the forespore and displaced to the forespore periphery during chromosome translocation18. Rather, the translocation of ribosomes into the forespore is temporally separate from and subsequent to chromosome translocation and the forespore inherits the mother cell’s ribosomes. Our data strongly suggest that ribosomes are restricted from the developing forespore rather than trapped in the divided cytoplasm. The ribosomes move away from the pole where the asymmetric septation will take place, and only a small proportion of membrane-associated ribosomes is included in the developing forespore. The majority of the ribosomes then wait at the asymmetric septum at the mother cell side where they remain translationally active. This supports the view of the asymmetric septum as a bacterial organelle serving as a hub in the cell’s developmental control, proposed by Shapiro and Losick35. Peptidoglycan rearrangement by SpoIIDMP is crucial for ribosome packing into the spore after the chromosome translocation and we hypothesise that this transfer is mediated by mother cell membrane or cell wall proteins, possibly via the septal pore, after SpoIIIE loss of focal localisation and before the spore peptidoglycan synthesis. Once the chromosome becomes dehydrated, ribosomes are released from the spore membrane and move towards the centrally located chromosome. The proposed model implies two things—(I) ribosomes can be transported to different subcellular locations during development and the cell most likely employs homologues of cytoskeletal proteins for this; (II) the cues for localisation of asymmetric septation may be translational and also based on the sites of ribosome location. Whether membrane targeting and membrane association of the ribosomes is via signal recognition particles, short nascent peptides36 or by direct binding with the membrane or cell wall components (perhaps lipid II mediated?), remains to be answered.

Cartoon model of the sporulation process in B. subtilis WT and ΔspoIID mutant illustrating chromosome and ribosomes translocation into the developing spore. Chromosome is depicted in grey, RpsB/ribosomes in green and RpoC/RNA polymerase in yellow.
We postulate that the SpoIIDMP role in ribosomes translocation into the forespore can be underlined by the evolutionary conservation of these proteins in the sporulation gene set of Firmicutes37 and can suggest that ribosome transport may be widespread and universal in spore-forming bacteria. Since we did not observe ribosome biogenesis factors in the MS data nor the GFP signal in the bulged membrane of ΔspoIID, this might suggest that ribosomes are not synthesised de novo in the forespore and are inherited from the mother cell. Also, the ribosomal transport from the mother cell to the developing spore appears to be in fact required for successful sporulation. SpoIIIA-SpoIIQ feeding tube channel is another example of a conserved complex essential for engulfment in sporulating bacteria. Although our data show that it is not necessary for ribosome transport into the forespore, we do not exclude the possibility that it may play an indirect role.
Collectively, our results contribute to the changing idea of the bacterial cell as a spatially organised system and place the translational machinery in the context of dynamic subcellular localisation during sporulation.
Methods
Strains and growth conditions
B. subtilis 168 strain was used as a wild type and the remaining strains are derivatives thereof, as listed in Supplementary Table 1a. The strains were grown overnight in LB medium at 30 °C with shaking then diluted to OD600 = 0.1 in CH medium38 and grown until OD600 reached 0.5–0.6, at 37 °C with shaking. Sporulation was induced by medium exchange to sporulation medium as described by Sterlini and Mandelstam39, except the cells were harvested by filtration and filters were transferred into the culture flasks. The sporulation medium was supplemented with 3% v/v of the culture in CH medium at OD600 = 0.5–0.6 to promote sporulation.
Strains construction
The primers and plasmids used in this study are listed in Supplementary Tables 1b and c, respectively.
B. subtilis single deletion strains were purchased from Bacillus Genetic Stock Center (BGSC, www.bgsc.org)40.
For strains with the GFP tag, the RpsB ribosomal protein or β' subunit of RNA polymerase, RpoC, was tagged with GFP at the C termini. The fusion was performed by a double cross-over and integrated into the chromosome in the native locus. The tag was introduced by transforming the competent B. subtilis cells41 with a linear DNA construct prepared by overlap PCR method42. The ~5.5 kb DNA construct consisted of two genomic regions 2 kb upstream and downstream of the STOP codon of rpsB or rpoC genes amplified from the genomic DNA of B. subtilis using dedicated UPFOR/UPREV and DOWNFOR/DOWNREV primers, and GFP tag and spectinomycin resistance cassette were amplified from pSHP2 plasmid with MID1FOR/MID2REV primers introducing appropriate flanking regions (Supplementary Tables 1b and c). The STOP codons of rpsB and rpoC were omitted. The transformants were selected on nutrient agar plates with spectinomycin and the results of transformations were verified by PCR and visually (expression of GFP). WT strains with GFP-tagged ribosomes and RNA polymerase demonstrated normal morphology and growth in CH medium at 37 °C compared to WT.
Confocal microscopy
Confocal microscopy was carried out with an inverted confocal system Nikon C1. Images were taken with Plan-Apo VC 100×/1.40 oil immersion objective. GFP/Alexa 488 were excited at 488 nm, DAPI at 408 nm, FM4-64 at 543 nm. The fluorescence signals were collected using filter sets: 515/30 nm for GFP/Alexa 488, 480/40 nm for DAPI and 610LP nm for FM4-64. Imaging of specimens having more than one fluorophore was performed in a sequential scan mode to prevent bleed-through of signal. B. subtilis strains were grown and sporulation was induced as described above. Aliquots of the cultures were sampled every hour for 6 h into sporulation, beginning at time T0—prior to sporulation induction. Cells were immobilised on 1% agarose pads prepared with 1.0 × 1.0-cm GeneFrames (Thermo Fisher Scientific). Unless stated otherwise, the cells were visualised with FM4-64 dye at a final concentration of 10 μg ml−1 and 4′,6-diamidino-2-phenylindole (DAPI) dye at a final concentration of 5 μg ml−1. For translation visualisation, cells were incubated with OPP using Click-iT® Plus OPP Alexa Fluor® 488 Protein Synthesis Assay Kit (Invitrogen) according to the manufacturer’s guidelines. Briefly, cells we incubated with 13 μM OPP for 30 min at 37 °C with shaking. Cells were then fixed with 3.7% formaldehyde and permeabilized with 0.1% Triton X-100. Fluorescent labelling was performed for 30 min with Alexa Fluor 488 reaction cocktail and then, cells were stained with DAPI.
Time-lapse microfluidics microscopy (TLMM)
TLMM experiments were performed using a Delta Vision Elite inverted microscope equipped with a 100×/1.4 Oil objective. B. subtilis strains were grown and sporulation was induced as described above. Immediately after sporulation induction cells were loaded into the ONIX observation chamber (B04A-03-5PK CellASIC ONIX plates for bacteria cells) and further cultured in sporulation medium with addition of 0.57–3% of the rich medium. Images were recorded at 5 min intervals for 6 h (35 ms exposure time, 10% transmittance on transmitted light channel, 15 ms exposure time and 10% of transmittance on EGFP channel). The temperature during the experiment was maintained at 37 °C.
Lattice SIM imaging
Strains were grown and sporulation was induced as described above. Cultures were sampled hourly beginning with T1 (1 h post sporulation induction), to T4 (4 h post sporulation induction). Lattice SIM Imaging was performed using an Elyra 7 (Zeiss) inverted microscope equipped with an sCMOS 4.2 CL HS camera and an alpha Plan-Apochromat 100×/1.46 Oil DIC M27 objective with an Optovar 1.6× magnification changer. Fluorescence was excited with 488 nm laser (100 mW), the signals were observed through multiple beam splitter (405/488/561/641 nm) and laser block filters (405/488/561/641 nm). Cells were immobilised on 1% agarose pads (1.0 × 1.0-cm GeneFrames; Thermo Fisher Scientific). Cells were illuminated with 488 nm laser with 0.9% intensity and 150 ms exposure time and with 0.5% intensity and 100 ms exposure time for respectively RpsB-GFP and RpoC-GFP fusions. Each Z-plane was illuminated in Lattice-SIM mode comprising 13 phases. Image reconstruction was performed with the ZEN 3.0 SR software (Zeiss) using SIM2 reconstruction algorithm with standard parameters. Images were viewed and analysed in ImageJ and the middle planes were selected for visualisation.
Microscopy data analysis
Images were analysed using ImageJ43 and graphs were prepared in R44. For fluorescence localisation measurements, a 1p line was drawn across the cells (region of interest, ROI). The cell lengths were normalised to 100% and the fluorescence intensities across the line were recorded and visualised. The number of cells used to calculate the mean fluorescence for each time point was as follows: for WT expressing RpsB-GFP (Fig. 1c), 109, 110, 225, 246, 116 and 138 cells were analysed at T0, T1, T2, T3, T4 and T5, respectively. For WT expressing RpoC-GFP (Fig. 1e), 116, 205, 163, 119, 84 and 99 cells were measured at T0, T1, T2, T3, T4 and T5, respectively. In the case of the translation visualisation experiments (Fig. 1g), 186, 609, 435, 346, 338 and 422 cells were analysed at T0, T1, T2, T3, T4 and T5, respectively. For the ΔspoIIIAH mutant expressing RpsB-GFP (Fig. 3b), 507, 364, 532, 425, 560, 505 and 477 cells were analysed at T2, T2:30, T3, T3:30, T4, T4:30 and T5, respectively. For the ΔspoIIQ mutant expressing RpsB-GFP (Fig. 3b), 718, 684, 351, 356, 512, 456 and 393 cells were quantified at T2, T2:30, T3, T3:30, T4, T4:30 and T5, respectively. Similarly, for the ΔspoIID mutant expressing RpsB-GFP (Fig. 3b), 273, 391, 338, 269, 182, 159 and 198 cells were analysed at T2, T2:30, T3, T3:30, T4, T4:30 and T5, respectively. For the ΔspoIID mutant expressing RpoC-GFP (Fig. 3b), 445, 450, 434, 437, 262, 238 and 331 cells were examined at the same respective time points. In the translation visualisation experiment for the ΔspoIID (Fig. 3d), 353, 358, 609, 345, 415 and 457 cells were analysed at T0, T1, T2, T3, T4 and T5, respectively. To enhance the visualisation of the green channel intensity (OPP and EU), the minimum and maximum values of the green channel histogram were scaled to a range of 0–2500 using ImageJ ‘Brightness/Contrast…’ option. The example cells were individually selected and cropped to create isolated images. These images were then rotated to a standardised orientation in GIMP.
Ribosome purification and mass spectrometry
Ribosomes were purified from B. subtilis WT cultures harvested 3 h post sporulation induction, in duplicates. Before harvesting, cultures were treated with 0.3 mM chloramphenicol. Cross-linking was performed using 2.5 mM DSP for 30 s, followed by quenching with 50 mM Tris for 30 s. The cross-linked cultures were collected by centrifugation, 10 min, 2500 × g, 4 °C. Otherwise, cells were collected by filtration and flash-frozen in liquid nitrogen. In case of detergent-based lysis, frozen cells were incubated in lysis buffer (20 mM Tris pH 7.6, 10 mM MgAcet, 150 mM KAcet and 1 mM chloramphenicol), with 1% digitonin, 1 mg/ml lysozyme and 100 U/ml Dnase I for 30 min at 30 °C. Lysis by grinding was performed with mortar and pestle with addition of aluminium oxide, in lysis buffer without digitonin (20 mM TRIS pH 7.6, 10 mM MgAcet, 150 mM KAcet, 0.4% TRITON X-100, 1 mM chloramphenicol, 100 U/ml Dnase I). As a control, cultures in logarithmic growth phase were also harvested. These were grown in CH medium at 37 °C with shaking and harvested when OD600 = 0.8. Lysed cells were then centrifuged and debris-free lysates were collected. Concentration of RNA was measured spectrophotometrically. Linear sucrose gradients (5%–30% w/v, 12.5 ml) were prepared in polysome buffer (20 mM Tris, 10 mM MgAcet, 5 mM KAcet,100 mM NH4Cl) with 0.1% addition of digitonin for the samples lysed by detergent-based lysis, in polypropylene ultracentrifuge tubes. The same amounts of samples (700 µg of total RNA) were layered on top of the sucrose gradients and centrifuged in a Thermo Scientific Sorval WX Ultra Series ultracentrifuge for 3 h at 96,589 × g (average g-force), 4 °C using a Thermo Scientific Th-641 swinging bucket rotor. Polysome profiles were recorded at 254 nm and gradients were fractionated into 500 μl fractions using BioComp’s Piston Gradient Fractionator™. Polysome profiles were analysed and fractions corresponding to the monosome were sent for mass spectrometry analysis.
The collected samples were subjected to chloroform/methanol precipitation and the resulting protein pellets were washed with methanol. The pellets were air-dried for 10 min and resuspended in 100 mM HEPES pH 8.0. TCEP (final conc. 10 mM) and chloroacetamide (final conc. 40 mM). Trypsin was added in a 1:100 enzyme-to-protein ratio and incubated overnight at 37 °C. The digestion was terminated by the addition of trifluoroacetic acid (TFA, final conc. 1%). The resulting peptides were labelled using an on-(StageTip) column TMT labelling protocol45. Peptides were eluted with 60 μl 60% acetonitrile/0.1% formic acid in water. Equal volumes of each sample were pooled into two TMT12plex samples and dried using a SpeedVac concentrator. Peptides were then loaded on StageTip columns and washed with 0.1% TEA/5%ACN, eluted with 0.1% TEA/50%ACN, and dried using a SpeedVac concentrator. Prior to LC-MS measurement, the peptide fractions were dissolved in 0.1% TFA, and 2% acetonitrile in water. Chromatographic separation was performed on an Easy-Spray Acclaim PepMap column 50 cm long × 75 µm inner diameter (Thermo Fisher Scientific) at 55 °C by applying 180 min acetonitrile gradients in 0.1% aqueous formic acid at a flow rate of 300 nl/min. An UltiMate 3000 nano-LC system was coupled to a Q Exactive HF-X mass spectrometer via an easy-spray source (all Thermo Fisher Scientific). The Q Exactive HF-X was operated in TMT mode with survey scans acquired at a resolution of 60,000 at m/z 200. Up to 15 of the most abundant isotope patterns with charges 2–5 from the survey scan were selected with an isolation window of 0.7 m/z and fragmented by higher-energy collision dissociation with normalised collision energies of 32, while the dynamic exclusion was set to 35 s. The maximum ion injection times for the survey scan and the MS/MS scans (acquired with a resolution of 45,000 at m/z 200) were 50 and 96 ms, respectively. The ion target value for MS was set to 3e6 and for MS/MS to 1e5, and the minimum AGC target was set to 1e3. Data were processed with MaxQuant v. 1.6.17.046, and peptides were identified from the MS/MS spectra searched against a modified Uniprot Bacillus Subtilis reference proteome (UP000001570 was extended by four entries: protein RpsN1 sequence MAKKSMIAKQQRTPKFKVQEYTRCERCGRPHSVIRKFKLCRICFRELAYKGQIPGVKKASW; protein RpsN2 sequence MAKKSKVAKELKRQQLVEQYAGIRRELKEKGDYEALSKLPRDSAPGRLHN RCMVTGRPRAYMRKFKMSRIAFRELAHKGQIPGVKKASW; protein RpmG2 sequence MRKKITLACKTCGNRNYTTMKSSASAAERLEVKKYCSTCNSHTAHLETK; protein RpmG3 sequence MRVNVTLACTETGDRNYITTKNKRTNPDRLELKKYSPRLKKYTLHRETK) using the built-in Andromeda search engine. Reporter ion MS2-based quantification was applied with reporter mass tolerance = 0.003 Da and min. reporter PIF = 0.75. Normalisation was selected, weighted ratio to all TMT channels. Cysteine carbamidomethylation was set as a fixed modification and methionine oxidation as well as glutamine/asparagine deamination were set as variable modifications. For in silico digests of the reference proteome, cleavages of arginine or lysine followed by any amino acid were allowed (trypsin/P), and up to two missed cleavages were allowed. The false discovery rate was set to 0.01 for peptides, proteins, and sites. A match between runs was enabled. Other parameters were used as pre-set in the software. Unique and razor peptides were used for quantification enabling protein grouping (razor peptides are the peptides uniquely assigned to protein groups and not to individual proteins). Reporter intensity corrected values (values) for protein groups were loaded into Perseus v. 1.6.10.047. Standard filtering steps were applied to clean up the dataset: reverse (matched to decoy database), only identified by site, and potential contaminant (from a list of commonly occurring contaminants included in MaxQuant) protein groups were removed. The values were log2 transformed and proteins groups with at least one valid value across all 11 sample groups were kept. The values were then normalised to the ribosomal protein content of each sample (within each sample a median value of 54 detected ribosomal proteins was subtracted from the values of non-ribosomal proteins). Log2 fold changes were calculated for: DSP fr1-4 vs. D fr1-4 (cross-linked vs. non-cross-linked samples lysed with digitonin), DSP fr1-4 and D fr1-4 vs. monosomal fraction P (control for lysis method), and sporulating sample P vs. logarithmic sample Log fr1-2 (control for growth stage). PCA plot was generated in R. Volcano plots were generated using the EnhancedVolcano package48 in R. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium49 via the PRIDE50 partner repository with the dataset identifier PXD047497.
Statistics and reproducibility
The mass spectrometry experiments were performed in biological duplicates. The microscopic observations using confocal microscopy were performed in biological triplicates or duplicates and data from a single replicate was presented in this manuscript. No data were excluded from the analyses. No statistical method was used to predetermine sample size. Randomisation is not applicable in this study. All samples were analysed and processed in the same manner thus, no blinding was required.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry proteomics data generated in this study have been deposited in the ProteomeXchange Consortium49 via the PRIDE50 partner repository under accession code PXD047497. The microscopic measurements and log2FoldChange values for the protein mass spectrometry data generated in this study, for all data presented in graphs within the Figures are provided in the Source Data file. The calculated log2FoldChange values for the protein mass spectrometry data generated in this study are provided in the Supplementary Data 1 file. Source data are provided with this paper.
References
Rudner, D. Z. & Losick, R. Protein subcellular localization in bacteria. Cold Spring Harb. Perspect. Biol. 2, a000307 (2010).
Whitley, K. D. et al. FtsZ treadmilling is essential for Z-ring condensation and septal constriction initiation in Bacillus subtilis cell division. Nat. Commun. 12, 1–13 (2021).
Feddersen, H., Würthner, L., Frey, E. & Bramkamp, M. Dynamics of the bacillus subtilis min system. MBio 12, 10–1128 (2021).
Chan, H., Mohamed, A. M. T., Grainge, I. & Rodrigues, C. D. A. FtsK and SpoIIIE, coordinators of chromosome segregation and envelope remodeling in bacteria. Trends Microbiol. 30, 480–494 (2022).
Bath, J., Wu, L. J., Errington, J. & Wang, J. C. Role of Bacillus subtilis SpoIIIE in DNA transport across the mother cell-prespore division septum. Science 290, 995–997 (2000).
Lewis, P. J., Thaker, S. D. & Errington, J. Compartmentalization of transcription and translation in Bacillus subtilis. EMBO J. 19, 710 (2000).
Mascarenhas, J., Weber, M. H. W. & Graumann, P. L. Specific polar localization of ribosomes in Bacillus subtilis depends on active transcription. EMBO Rep. 2, 685 (2001).
Johnson, G. E., Lalanne, J. B., Peters, M. L. & Li, G. W. Functionally uncoupled transcription–translation in Bacillus subtilis. Nature 585, 124–128 (2020).
Errington, J. Regulation of endospore formation in Bacillus subtilis. Nat. Rev. Microbiol. 1, 117–126 (2003).
Piggot, P. J. & Hilbert, D. W. Sporulation of Bacillus subtilis. Curr. Opin. Microbiol. 7, 579–586 (2004).
Higgins, D. & Dworkin, J. Recent progress in Bacillus subtilis sporulation. FEMS Microbiol. Rev. 36, 131–148 (2012).
Khanna, K., Lopez-Garrido, J. & Pogliano, K. Shaping an endospore: architectural transformations during Bacillus subtilis Sporulation. Annu. Rev. Microbiol. 74, 361–386 (2020).
Fleming, T. C. et al. Dynamic SpoIIIE assembly mediates septal membrane fission during Bacillus subtilis sporulation. Genes Dev. 24, 1160–1172 (2010).
Khanna, K. et al. The molecular architecture of engulfment during Bacillus subtilis sporulation. eLife 8, e45257 (2019).
Mohamed, A. et al. Chromosome segregation and peptidoglycan remodeling are coordinated at a highly stabilized septal pore to maintain bacterial spore development. Dev. Cell 56, 36–51.e5 (2021).
Cramer, K., Reinhardt, S. C. M., Auer, A., Shin, J. Y. & Jungmann, R. Comparing divisome organization between vegetative and sporulating Bacillus subtilis at the nanoscale using DNA-PAINT. Sci. Adv. 10, eadk5847 (2024).
Bosdriesz, E., Molenaar, D., Teusink, B. & Bruggeman, F. J. How fast-growing bacteria robustly tune their ribosome concentration to approximate growth-rate maximization. FEBS J. 282, 2029–2044 (2015).
Lopez-Garrido, J. et al. Chromosome translocation inflates Bacillus forespores and impacts cellular morphology. Cell 172, 758–770.e14 (2018).
Marquis, K. A. et al. SpoIIIE strips proteins off the DNA during chromosome translocation. Genes Dev. 22, 1786–1795 (2008).
Jao, C. Y. & Salic, A. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc. Natl. Acad. Sci. USA 105, 15779–15784 (2008).
Liu, J., Xu, Y., Stoleru, D. & Salic, A. Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc. Natl. Acad. Sci. USA 109, 413–418 (2012).
Iwańska, O. et al. Translation in Bacillus subtilis is spatially and temporally coordinated during sporulation. Nat. Commun. 15, 1–13 (2024).
Kopik, N., Chrobak, O., Latoch, P., Kovalenko, M. & Starosta, A. L. RIBO-seq in bacteria: a sample collection and library preparation protocol for NGS sequencing. J. Vis. Exp. 2021, e62544 (2021).
Maki-Yonekura, S., Yonekura, K. & Namba, K. Conformational change of flagellin for polymorphic supercoiling of the flagellar filament. Nat. Struct. Mol. Biol. 17, 417–422 (2010).
Doan, T. et al. Novel secretion apparatus maintains spore integrity and developmental gene expression in Bacillus subtilis. PLOS Genet. 5, e1000566 (2009).
Zeytuni, N. & Strynadka, N. C. J. A hybrid secretion system facilitates bacterial sporulation: a structural perspective. Microbiol. Spectr. 7, 10–1128 (2019).
Riley, E. P., Schwarz, C., Derman, A. I. & Lopez-Garrido, J. Milestones in Bacillus subtilis sporulation research. Microb. Cell 8, 1 (2020).
Morlot, C. & Rodrigues, C. D. A. The new kid on the block: a specialized secretion system during bacterial sporulation. Trends Microbiol. 26, 663–676 (2018).
Rubio, A. & Pogliano, K. Septal localization of forespore membrane proteins during engulfment in Bacillus subtilis. EMBO J. 23, 1636–1646 (2004).
Riley, E. P., Lopez-Garrido, J., Sugie, J., Liu, R. B. & Pogliano, K. Metabolic differentiation and intercellular nurturing underpin bacterial endospore formation. Sci. Adv. 7, 6385–6407 (2021).
Zeytuni, N. et al. Near-atomic resolution cryoelectron microscopy structure of the 30-fold homooligomeric SpoIIIAG channel essential to spore formation in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 114, E7073–E7081 (2017).
Morlot, C., Uehara, T., Marquis, K. A., Bernhardt, T. G. & Rudner, D. Z. A highly coordinated cell wall degradation machine governs spore morphogenesis in Bacillus subtilis. Genes Dev. 24, 411–422 (2010).
Gutierrez, J., Smith, R. & Pogliano, K. SpoIID-mediated peptidoglycan degradation is required throughout engulfment during Bacillus subtilis sporulation. J. Bacteriol. 192, 3174–3186 (2010).
Ojkic, N., López-Garrido, J., Pogliano, K. & Endres, R. G. Cell-wall remodeling drives engulfment during Bacillus subtilis sporulation. eLife 5, e18657 (2016).
Shapiro, L. & Losick, R. Dynamic spatial regulation in the bacterial cell. Cell 100, 89–98 (2000).
Bornemann, T., Jöckel, J., Rodnina, M. V. & Wintermeyer, W. Signal sequence–independent membrane targeting of ribosomes containing short nascent peptides within the exit tunnel. Nat. Struct. Mol. Biol. 15, 494–499 (2008).
Galperin, M. Y., Yutin, N., Wolf, Y. I., Alvarez, R. V. & Koonin, E. V. Conservation and evolution of the sporulation gene set in diverse members of the firmicutes. J. Bacteriol. 204, e00079-22 (2022).
Sharpe, M. E., Hauser, P. M., Sharpe, R. G. & Errington, J. Bacillus subtilis cell cycle as studied by fluorescence microscopy: constancy of cell length at initiation of DNA replication and evidence for active nucleoid partitioning. J. Bacteriol. 180, 547–555 (1998).
Sterlini, J. M. & Mandelstam, J. Commitment to sporulation in Bacillus subtilis and its relationship to development of actinomycin resistance. Biochem. J. 113, 29–37 (1969).
Koo, B. M. et al. Construction and analysis of two genome-scale deletion libraries for Bacillus subtilis. Cell Syst. 4, 291–305.e7 (2017).
Jenkinson, H. F. Altered arrangement of proteins in the spore coat of a germination mutant of Bacillus subtilis. J. Gen. Microbiol. 129, 1945–1958 (1983).
Forloni, M., Liu, A. Y. & Wajapeyee, N. Creating insertions or deletions using overlap extension polymerase chain reaction (PCR) mutagenesis. Cold Spring Harb. Protoc. 2018, pdb.prot097758 (2018).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
R Core Team. R: a language and environment for statistical computing. https://www.R-project.org (2024).
Myers, S. A. et al. Streamlined protocol for deep proteomic profiling of FAC-sorted cells and its application to freshly isolated murine immune cells*. Mol. Cell. Proteom. 18, 995a–1009a (2019).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Blighe, K., Rana, S. & Lewis, M. EnhancedVolcano: publication-ready volcano plots with enhanced colouring and labeling. R package version 1.22.0. at https://github.com/kevinblighe/EnhancedVolcano (2024).
Deutsch, E. W. et al. The ProteomeXchange consortium at 10 years: 2023 update. Nucleic Acids Res. 51, D1539–D1548 (2023).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Acknowledgements
European Molecular Biology Organization Installation Grant 3914, A.L.S. Foundation for Polish Science FIRST TEAM grant POIR.04.04.00-00-3E9C/17-00, A.L.S.
Author information
Authors and Affiliations
Contributions
Conceptualisation: A.L.S., J.Z.C. Investigation: O.I., M.K., M.L., J.H., R.S., A.G. Visualisation: P.L. Funding acquisition: A.L.S. Writing—original draft: O.I. Writing—review & editing: A.L.S., P.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Iwańska, O., Latoch, P., Kovalenko, M. et al. Ribosomes translocation into the spore of Bacillus subtilis is highly organised and requires peptidoglycan rearrangements. Nat Commun 16, 354 (2025). https://doi.org/10.1038/s41467-024-55196-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-55196-9
This article is cited by
-
Modulation of the Sporulation Dynamics in the Plant-Probiotic Bacillus velezensis 83 via Carbon and Quorum-Sensing Metabolites
Probiotics and Antimicrobial Proteins (2025)
