
Abstract
Photo-oxidation of methane (CH4) using hydrogen peroxide (H2O2) synthesized in situ from air and water under sunlight offers an attractive route for producing green methanol while storing intermittent solar energy. However, in commonly used aqueous-phase systems, photocatalysis efficiency is severely limited due to the ultralow availability of CH4 gas and H2O2 intermediate at the flooded interface. Here, we report an atomically modified metal-organic framework (MOF) membrane nanoreactor that promotes direct CH4 photo-oxidation to methanol at the gas-solid interface in a reticular open framework. We show that the domino synergy between colocalized single-atom palladium and iron on MOF nodes enables efficient generation and in situ utilization of H2O2 in the absence of liquid water, thus circumventing H2O2 dilution. Meanwhile, the “breathable” MOF membrane, optimized by solar-driven interfacial water management, provides high-flux channels to facilitate efficient gas diffusion and rapid methanol desorption and transfer. As a result, we demonstrate over 210 hours of continuous photosynthesis of 0.25 M methanol with unity selectivity, achieving an exceptional methanol productivity of 14.4 millimoles per gram of catalyst per hour.
Similar content being viewed by others
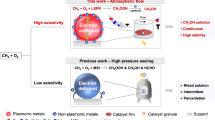


Introduction
Methanol (CH3OH) is an efficient energy carrier and a vital platform molecule for synthesizing key bulk chemicals such as olefins and aromatics1,2,3,4,5,6,7,8. The conventional route to its industrial synthesis involves reforming methane (CH4) into syngas under high temperatures and pressures, an expensive and energy-intensive process5,9. Direct oxidation of CH4 into CH3OH would be more attractive but confronts significant challenges, including the high C–H bond dissociation energy of CH4 and the over-oxidation of CH3OH product9,10,11,12. Tremendous efforts have been devoted to surmounting these hurdles for decades, among which hydrogen peroxide (H2O2) promotes the highly selective oxidation of CH4 to CH3OH by triggering a radical reaction pathway7,13,14. Recently, photocatalytic tandem methane oxidation (PTMO) has attracted increasing attention for producing H2O2 in situ via the photocatalytic reaction of air and water instead of using the expensive H2O2 synthesis through the substituted anthraquinone route1,2.
However, current PTMO processes for methanol production require operation in aqueous-phase systems, where liquid water serves as an essential transporter for both the hydrophilic H2O2 intermediate and methanol product1,2,5. The flooded catalytic interface inevitably leads to sluggish gas transfer and inefficient H2O2 utilization. On one hand, CH4 can scarcely dissolve in water under standard conditions15. This significantly constrains the diffusion of CH4 gas through liquid water to the catalyst’s surfaces3,4. On the other hand, the key H2O2 intermediates generated at the liquid-solid interface via photocatalytic oxygen reduction (POR) are immediately diluted by water, causing a relatively low H2O2 concentration proximate to the CH4 oxidation centres. This significantly dampens the kinetics of the subsequent H2O2-mediated methane oxidation (HMO) process16,17. In addition, methanol generated in aqueous-phase PTMO systems is mixed with substantial liquid water, severely hindering subsequent concentration and separation processes required to obtain pure liquid chemical solutions in practical applications.
Proceeding PTMO at a gas-solid interface can theoretically avoid water flooding, thereby boosting the diffusion of gaseous reactants, eradicating the dilution of H2O2 intermediate, and enabling the direct collection of high-concentration methanol by condensing its vapor. However, without liquid water as the carrier, the hindered transfer of hydrophilic H2O2 intermediates from POR to allopatric HMO sites has thus drastically limited gas-phase PTMO to produce methanol1,18. In addition, to prevent overoxidation, an ideal gas-solid PTMO system should enable the continuous sweeping of the catalyst layer with high-throughput water vapor to promote the desorption and rapid transfer of methanol products9,19. But water vapor will inevitably stagnate and condense on the gas-solid interface, causing a dramatic decline in gas-solid mass transfer efficiency20,21. Thus, to achieve gas-phase PTMO, it is essential not only to promote high-flux gas diffusion but also to enable two crucial mass transfer processes at the gas-solid interface: (i) the effective transport and utilization of hydrophilic H2O2 intermediate and (ii) the rapid expulsion of interfacial adsorption water.
Here, we present a metal-organic framework (MOF) confined reticular gas-solid catalytic interface design that, by simultaneously regulating the spatial intimacy of cascade catalytic sites and the microenvironment of MOF pores, enables efficient gas-phase PTMO to produce high-concentration methanol at high activity and unity selectivity continuously (Fig. 1a). We show that the colocalized single-atomic palladium (Pd1) and iron (Fe1) on MOF nodes, which serve as the POR and HMO sites, respectively, can synergistically trigger the H2O2-mediated PTMO at the gas-solid interface. The in situ generated H2O2 on Pd1 can be utilized locally by adjacent Fe1 species to catalyze methane oxidation, circumventing the need for liquid water as a transport medium and thus avoiding H2O2 dilution (Fig. 1a). Meanwhile, sunlight-derived low-grade heat can effectively expel the interfacial adsorption water within MOF pores, maintaining efficient gas-solid mass transfer while promoting methanol desorption and transfer (Fig. 1b). These methanol products, therefore, can be rapidly and continuously pushed out of the MOF membrane and collected in its high-concentration form through a simple cold condensation process.

a Schematic illustration for photocatalytic tandem oxidation of methane into methanol under sunlight irradiation. b Upon exposure to sunlight, the photothermally induced low-grade heat from the carbon-based GDL substrate can effectively expel the interfacial adsorption water within the MOF pores, facilitating water vapor diffusion while minimizing water vapor retention and condensation at the gas-solid catalytic interface, thus maintaining an ultrahigh efficiency in gas-solid mass transfer.
Results and discussion
Molecular dynamics simulations
We began by modeling the catalytic interface in different PTMO scenarios and employing molecular dynamics (MD) simulations to explore how the mass transmission efficiency of CH4, O2, and in situ generated H2O2 would be promoted by regulating the interfacial microenvironment (Supplementary Figs. 1, 2). Among various candidates, we chose a reticular chemistry-based MOF photocatalyst, namely NH2-UiO-66 (aUiO, Supplementary Fig. 3), as the base material to fabricate the reticular PTMO interface because of its high photoactivity, suitable band gap, high porosity and, especially, its plentiful pore environment around the active MOF nodes that provide easy access for gaseous reactants22,23,24,25,26. In these simulations, gas-phase CH4, O2, and water, or liquid water containing dissolved CH4 and O2, are introduced continuously across the MOF (NH2-UiO-66) membrane to represent the reactant’s mass transfer process in gas-solid and liquid-solid systems, respectively. In addition, MD simulations do not depict the formation process of the H2O2 intermediate. Seeking to compare the mass transfer behaviors of the H2O2 intermediate in both gas-solid and liquid-solid systems, we randomly inserted several H2O2 molecules near the Zr–O nodes within the MOF pores to simulate the in situ generated H2O2. MD simulations suggest that by breaking gas solubility limitations, the local concentrations of CH4 and O2 molecules can be dramatically enhanced at the gas-solid interface within the MOF membrane (Fig. 2a, b and Supplementary Figs. 4, 5). Additionally, the H2O2 intermediates generated at the water-flooded liquid-solid interface are rapidly diluted by water and consequently migrate away from the MOF matrix (Fig. 2c, Supplementary Figs. 6, 7). In sharp contrast, at the gas-solid interface, H2O2 molecules mainly remain near the Zr–O nodes of aUiO and rather than being diluted by liquid water (Fig. 2d, e), thereby maintaining a significantly high H2O2 availability surrounding the CH4 oxidation centres.

a Mass density profiles of CH4 in gas-solid and liquid-solid systems along the gas diffusion (gas-solid system) or water flow (liquid-solid system) direction under 160 ps simulation. In the gas-solid system, gas-phase CH4, O2, and water were continuously introduced across the MOF (NH2-UiO-66) membrane. In the liquid-solid system, liquid water containing dissolved CH4 and O2 molecules flowed through the MOF membrane. b Mass density profiles of O2 in gas-solid and liquid-solid systems along the gas diffusion (gas-solid system) or water flow (liquid-solid system) direction under 160 ps simulation. Two-dimensional mass density distribution maps of the H2O2 molecules under different simulation times for the liquid-solid system (c), as well as the gas-solid systems at ambient (d) and elevated (e) temperatures. Several hydrogen peroxide molecules are placed near the Zr-O nodes inside the MOF pores to simulate the in situ generated H2O2. Snapshots of the water molecule distribution in the gas-solid system at ambient (f) and elevated (g) temperatures under 40 and 199 ps simulation. Water molecules are highlighted as yellow sticks.
However, with prolonged operation of the gas-solid PTMO system under ambient temperature (25 oC), water molecules accumulate increasingly within the aUiO pores through the hydrogen-bond interaction (Fig. 2f, Supplementary Fig. 8)27. This results in forming a water adsorption layer at the initial gas-solid photocatalysis interface, creating a barrier to gas diffusion and locally diluting the H2O2 intermediate (Fig. 2d). Given that low-grade heat can facilitate the regeneration of aUiO28, we proceeded to explore the gas-solid mass transfer behavior at an elevated temperature (75 oC). Our findings indicate no significant increase in water molecule density within the MOF pores throughout the simulation (Fig. 2g, Supplementary Fig. 9). This suggests that increasing the environmental temperature effectively suppresses water molecule aggregation within MOF27, thus maintaining a clear gas-solid catalysis interface free from water vapor retention and condensation. As a result, greater availability of CH4, O2, and in situ generated H2O2 at the gas-solid catalytic interface can be facilitated at an elevated temperature compared to ambient conditions (Fig. 2a, b and e), thereby significantly enhancing the reaction probability for PTMO.
Co-functionalization of aUiO matrix with cooperative POR and HMO sites
We developed a two-step strategy to introduce POR and HMO sites into the aUiO matrix. We first chose Pd species to functionalize the Zr–O nodes as POR sites due to their remarkable capabilities to catalyze the hydrogenation of O2 in thermocatalysis29. By creating defects on the Zr–O nodes of aUiO30,31, atomically dispersed Pd species (0.78 wt.%) were successfully grafted onto the nodes to form the Pd1/aUiO catalyst, as revealed by energy-dispersive X-ray spectroscopy (EDS), high-angle annular dark-field scanning TEM (HAADF-STEM) and extended X-ray absorption fine structure (EXAFS) analyses (Supplementary Fig. 10). Notably, the Pd1/aUiO (0.78 wt.%) catalyst is at least two orders of magnitude more active than bare aUiO for photocatalytic H2O2 evolution (Supplementary Figs. 11–13), verifying the crucial role of Pd1 species in catalyzing POR. However, a further increase in the loading of Pd (to 0.83 wt.%) resulted in the partial conglomeration of Pd atoms (Supplementary Fig. 14), which in turn decreased POR activity, as revealed by density functional theory (DFT) calculations and photocatalytic tests (Supplementary Figs. 12 and 15). This suggests there were no sufficient bonding sites on Zr–O nodes for additionally anchoring atomically dispersed metal species.
Inspired by natural nonheme iron enzymes that use oxidizers (i.e., dioxygen) to generate high-spin Fe(IV) = O species for various oxygenation reactions32, we further sought to develop mononuclear Fe (Fe1) species bonded to Zr–O nodes to act as the HMO centres. Given the ultrasmall size (less than 1 nm) and isolated distribution of the Zr–O nodes in aUiO33, we proposed that confining Pd1 and Fe1 simultaneously onto MOF nodes could lead to the colocalization of POR and HMO sites. In this configuration, the Fe1 sites would be oxidized into Fe(IV) = O active species by H2O2 generated by the adjacent Pd1 sites, thereby efficiently catalyzing methane oxidation.
To test this hypothesis, additional defect sites were introduced near the Pd1 on Zr–O nodes to serve as bonding sites for Fe1 using a simple hydrogen (H2) treatment strategy. We demonstrated that the H2 could dissociate on Pd1 sites to generate highly active atomic hydrogen species34,35, which would diffuse into and interact with the Zr–O nodes to create oxygen defect sites in the vicinity of the Pd1 (Supplementary Figs. 16, 17). Electron spin resonance (ESR) spectroscopy, along with Zr K-edge EXAFS analyses, revealed a substantial increase in the density of oxygen defects on the Zr–O nodes of hydrogen-treated Pd1/aUiO catalyst [Pd1/aUiO(H)] (Supplementary Fig. 17), thereby rendering these nodes conducive for hosting robustly bonded and isolated Fe atoms.
Following this, we further integrated Fe species into Pd1/aUiO(H) through a photo-deposition and annealing process and synthesized Pd1FeOx/aUiO(H) photocatalyst (Supplementary Fig. 18). EDS mappings and HAADF-STEM images of Pd1FeOx/aUiO(H) (with 0.89 wt.% Fe loading) revealed no nanometer-sized particles, indicating that the Pd and Fe species were highly dispersed in the MOF matrix (Fig. 3a–c). Pd K-edge EXAFS results exhibited the only existence of Pd–O configuration in Pd1FeOx/aUiO(H) (Fig. 3d), verifying the atomic dispersion of Pd species. Notably, the Fe K-edge EXAFS spectrum indicated the coexisting Fe–O and Fe–Fe paths in the Pd1FeOx/aUiO(H) catalyst (Fig. 3e). Combining with the 57Fe Mössbauer spectra (Fig. 3f), we propose that the Fe1 species and small iron oxide clusters may coexist in the Pd1FeOx/aUiO(H)13,36.

a HAADF-STEM image and corresponding EDS mappings for Pd1FeOx/aUiO(H) sample. AC-HAADF-STEM images for (b) Pd1FeOx/aUiO(H) and (c) Pd1Fe1/aUiO(H), indicating the high dispersion of Pd and Fe species in the MOF matrix. d Pd K-edge FT-EXAFS spectra of Pd1FeOx/aUiO(H) and Pd1Fe1/aUiO(H), with Pd foil and PdO as references. e Fe K-edge FT-EXAFS spectra of Pd1FeOx/aUiO(H) and Pd1Fe1/aUiO(H), with Fe foil and Fe2O3 as references. f 57Fe Mössbauer spectra of Pd1FeOx/aUiO(H) and Pd1Fe1/aUiO(H) samples. g N2 adsorption-desorption isotherms (at 77 K) for Pd1FeOx/aUiO(H) and Pd1Fe1/aUiO(H). h Pd K-edge XANES spectra of Pd1FeOx/aUiO(H), Pd1Fe1/aUiO, and Pd1/aUiO(H) catalysts, indicating the electronic structure of Pd1 was perturbed by the incorporated Fe1 species. i Pd K-edge XANES spectra of Pd1Fe1/aUiO(H) catalysts with different mass loading of Fe1 species.
To further distinguish between Fe1 and oxide clusters, we used a dilute sulphuric acid solution to selectively leach the iron oxide clusters from the Pd1FeOx/aUiO(H) and obtained Pd1Fe1/aUiO(H) catalyst. The N2 sorption isotherms showed a significant increase in both surface areas and pore volumes after acid treatment (Fig. 3g and Supplementary Table 1), suggesting that iron oxide clusters were embedded inside the MOF pores. In addition, the surface area and pore volume of Pd1Fe1/aUiO(H) closely match those of the Pd1/aUiO(H), further indicating that the acid treatment without impacting the MOF structure (Supplementary Table 1). The Fe loading decreased to 0.37 wt.%, leaving only the atomically dispersed Fe (Fig. 3c). Fe K-edge EXAFS analyses and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) revealed that the Fe1 bound to the MOF by −O/−OHx ligands, capping the defect sites on the Zr–O nodes (Fig. 3e, Supplementary Fig. 19)30,31. The significant decrease in peak intensity of −O/OHx groups in DRIFTS spectra following Fe1 species incorporation suggests these Fe1 were confined on the defect sites created by Pd1-catalyzed hydrogenation and close to Pd1 (Supplementary Fig. 19). Additionally, the presence of Fe1 on Zr–O nodes perturbed the electronic structure of Pd1 (Fig. 3h). As the density of Fe1 increases, the disturbance to the electronic structure of Pd1 becomes increasingly pronounced, further validating the proximity of Pd1 and Fe1 sites on the Zr–O nodes (Fig. 3i). Additionally, when the hole scavenger was introduced during the photo-deposition process, it resulted in the exclusive integration of atomically dispersed Fe species into aUiO(H), with iron oxide clusters being notably absent in the resultant sample (Supplementary Fig. 20). Hence, the Fe1 and Pd1 in Pd1FeOx/aUiO(H) were colocalized on the Zr–O nodes, while the iron oxide clusters formed by photo-oxidation of iron ions existed in the pores of aUiO(H). These iron oxide clusters can act as hole acceptors to promote the separation of photogenerated carriers, thereby effectively boosting the PTMO process (as will be discussed in detail later)37.
Boosting PTMO at the MOF-confined reticular gas-solid interface
We then assembled the as-prepared Pd1FeOx/aUiO(H) particles onto the gas-permeable polytetrafluoroethylene (PTFE) substrates and assessed their PTMO performance in the continuous-flow gas-solid system (by using a gas-flow cell), utilizing a humidified CH4/O2 mixture (v/v = 1/1) as feed gas (Supplementary Figs. 21–23). As shown in Supplementary Fig. 24, H2O2 was detected solely in the Pd1/aUiO(H) membrane following a 1-hour photocatalytic reaction, suggesting a blocking in CH4 oxidation. Efficient PTMO was stimulated at the gas-solid interface after introducing Fe species into Pd1/aUiO(H) (Supplementary Figs. 25, 26). When the Fe loading increased, the CH3OH productivity and selectivity followed a volcanic curve, rising from 0.59 mmol g–1 h–1 and 50.4% to 4.37 mmol g–1 h–1 and 83.6%, respectively (Supplementary Fig. 25). We therefore screened out the optimal Fe loading as 0.89 wt.%.
Seeking to shed more light on the reactions that play a role in the PTMO process, we evaluated the CH4 photo-oxidation performance of only 0.89 wt.% Fe modified aUiO (FeOx/aUiO, which exhibits a consistent distribution of Fe species with the Pd1FeOx/aUiO(H) catalyst but absence Pd1 species) in the gas-solid system (Supplementary Fig. 27). It was discovered that the FeOx/aUiO membrane was nearly inert for CH4 photo-oxidation by supplying a humidified CH4/O2 mixture (v/v = 1/1) as the reactant (Supplementary Fig. 28). However, when we introduced H2O2 by water vapor into the reaction system, it triggered the CH4 oxidation immediately (Supplementary Fig. 28). Therefore, we suggested that the gas-solid CH4 photo-oxidation process on Pd1FeOx/aUiO(H) membrane proceeds via the tandem steps of (i) H2O2 production on Pd1 sites and (ii) H2O2-mediated CH4 oxidation on Fe species5. These findings also confirmed that the colocalization of POR (Pd1) and HMO (Fe1) centres could enable effective utilization of in situ generated H2O2 at the gas-solid interface for CH4 oxidation.
Seeking to assess the promotion effect of interfacial engineering on PTMO, we compared CH3OH productivities in gas-solid and traditional liquid-solid (by dispersing MOF photocatalyst powders in liquid water) systems. As shown in Fig. 4a, using Pd1FeOx/aUiO(H) as the catalyst, the CH3OH productivity in the gas-solid system far surpassed that of the liquid-solid system. With a feed gas flow rate set at 150 standard cubic centimeters per minute (sccm), we observed an impressive CH3OH yield of 4.37 mmol g–1 h–1 on the Pd1FeOx/aUiO(H) membrane, an 8-fold increase compared to the optimized liquid-solid system.

a Comparison of the CH3OH yields (left axis) and selectivity (right axis) on Pd1FeOx/aUiO(H) membrane nanoreactor (gas-solid system) and particles (liquid-solid system) under different flow rates of feed gas. Reaction conditions: 50 mg of MOF catalyst, 1 hour, 25 oC, feed gas with 50% CH4/50% O2, 1200 revolutions per minute (liquid-solid system), and under simulated sunlight irradiation (AM 1.5 G). b Time-on-line water uptake (right axis) and amounts of residual CH3OH and H2O2 (left axis) within the MOF membrane. Reaction condition: 150 sccm humidified feed gas, 25 oC, 1 hour. c Water uptake (right axis) and amounts of residual CH3OH and H2O2 (left axis) within the MOF membrane under different system temperatures. Reaction condition: 150 sccm humidified feed gas, 1 hour. d Comparison of the CH3OH productivity (left axis) and selectivity (right axis) on Pd1FeOx/aUiO(H) membrane (gas-solid system) and particles (liquid-solid system) under different temperatures. e Long-term performance test of the GDL-equipped Pd1FeOx/aUiO(H) membrane under the optimized reaction condition. f Photo-oxidation of CH4 to CH3OH by air and tap water under natural solar light. The error bars and bands correspond to the standard deviation of three independent measurements.
To deeply understand how the reticular gas-solid interface design promotes PTMO, we compared the H2O2 evolution performance of Pd1/aUiO(H) and Pd1FeOx/aUiO(H) samples at different catalytic interfaces (Supplementary Fig. 29). Remarkably, shifting from the liquid-solid to the gas-solid system increased H2O2 productivity on the Pd1/aUiO(H) catalyst by 4.7 times, indicating that interfacial engineering enhanced O2 mass transfer and thereby boosted POR. However, when using the Pd1FeOx/aUiO(H) catalyst, the gain factor of H2O2 productivity from the liquid-solid to gas-solid system fell to only 1.3 (Supplementary Fig. 29). This suggests that the gas-solid catalytic interface not only promoted H2O2 generation on Pd1 sites but also enhanced its utilization efficiency by Fe species. Firstly, compared to the water-flooded liquid-solid interface, the reticular gas-solid interface design can boost high-flux gas diffusion via interconnected MOF pores, enabling exceedingly high local concentrations of gaseous reactants near the metal active sites. Secondly, the colocalized POR and HMO sites on MOF nodes facilitate the in-situ utilization of the H2O2 intermediate at the gas-solid interface, thereby preventing H2O2 dilution and markedly enhancing its utilization efficiency.
Optimizing gas-solid PTMO via interfacial water management
Although high PTMO performance was achieved, the Pd1FeOx/aUiO(H) membrane nanoreactor has yet to reach its optimized operating state due to the rapid deterioration of the interfacial microenvironment during the catalytic process. As shown in Supplementary Fig. 30, within an hour of continuous operation, the CH3OH activity and selectivity declined notably. Residual CH3OH was verified to remain trapped within the MOF membrane (Fig. 4b, Supplementary Fig. 31). This hindered the effective desorption of the product, leading to the overoxidation of methanol to formaldehyde (Supplementary Fig. 32). In addition, the accumulation of H2O2 intermediate on the catalytic interface, instead of directly taking part in PTMO as expected, was also detected (Fig. 4b). We speculated that these issues were due to interface water invasion. As shown in Fig. 4b, under ambient temperature (25 oC), water vapor in the humidified feed gas could be trapped in the pores of MOF during the PTMO process, causing water condensation on the gas-solid interface (Supplementary Fig. 33). As soon as this occurred, the H2O2 intermediate experienced dilution, while simultaneously, the diffusion pathways for gaseous reactants (i.e., CH4 and O2) and CH3OH vapor became obstructed; thus, the PTMO efficiency quickly dropped (Supplementary Figs. 34, 35).
To further optimize the PTMO performance, we sought to suppress the water accumulation in the pores of MOF by accelerating the rapid water desorption during the catalytic process. Guiding by the MD simulations, we turned our attention to optimizing the operation temperature of the gas-solid PTMO system. We observed a drastic decrease in water uptake as we raised the system temperature above 55 oC (Fig. 4c, Supplementary Fig. 36). Thus, increasing the temperature of the MOF membrane nanoreactor could promote water vapor diffusion through MOF pores while minimizing water retention and condensation at the gas-solid catalytic interface (Supplementary Figs. 37). This, in turn, remarkably enhanced the efficiency of gaseous reactant transfer, H2O2 intermediate utilization, and methanol desorption (Supplementary Figs. 36 and 37)9,19. Moreover, the elevated reaction temperature further facilitates the efficient transfer of desorbed methanol from the MOF membrane in vapor form, thereby effectively suppressing overoxidation (Supplementary Fig. 38). It is well known that increasing the reaction temperature can also promote PTMO by accelerating reaction kinetics. Thus, we further assessed the contribution of interfacial water management in promoting PTMO. As shown in Fig. 4d, by improving the reaction temperature from 25 to 75 oC, the gain factor of CH3OH yields on the gas-solid interface ( ~ 3.3) was much higher than that in the liquid-solid system ( ~ 1.7). This finding confirms that the significantly enhanced PTMO performance at the gas-solid interface, following a rise in system temperature, mainly resulted from substantially improved gas-solid mass transfer. As a result, under an elevated temperature of 75 oC, the Pd1FeOx/aUiO(H) membrane achieved significantly improved CH3OH productivity and selectivity of up to 0.73 mmol h–1 (14.6 mmol g–1 h–1) and ~100% (Fig. 4d, Supplementary Fig. 39), demonstrating an ultrahigh solar-to-chemical (STC) energy conversion efficiency of up to 1.2% (Supplementary Fig. 40). Carbon source for methanol production were also studied in the presence of isotopic labeled 13CH4 (Supplementary Fig. 41), where the signal of mass spectra (MS) at m/z = 33 was ascribed to 13CH3OH, confirming that CH4 was the carbon source for oxygenates production.
Based on our findings above, we developed a photothermal-conversion-layer (carbon-based gas diffusion layer, GDL) equipped hybrid MOF membrane to use infrared light from the solar spectrum to heat the Pd1FeOx/aUiO(H) layer to ~75 oC directly, eliminating all extra energy consumption for maintaining the membrane temperature (Fig. 4e and Supplementary Figs. 42, 43). In a 210-hour continuous test, the hybrid Pd1FeOx/aUiO(H) membrane exhibited an average STC conversion efficiency of 1.17% with an impressive CH3OH selectivity of ~100% (Fig. 4e). Nearly 98% of the generated CH3OH could be collected in real-time by an easy cold condensation process, resulting in a total of 0.6 litres (L) of 0.25 M high-concentration CH3OH product, corresponding to an average productivity of 14.4 mmol g–1 h–1 (Fig. 4e, Supplementary Fig. 44). This significantly exceeds the productivity of advanced photocatalysts reported under similar reaction conditions (Supplementary Table 3). In addition, the Pd1FeOx/aUiO(H) membrane demonstrated a constant PTMO selectivity over 210 hours and only exhibited a slight decay in methanol yield, mainly owing to the slight collapse in the MOF structure (Fig. 4e; Supplementary Fig. 45 and Table 5).
Our reticular gas-solid catalytic interface design also exhibits tremendous practical potential for the continuous conversion of CH4 to CH3OH under natural solar light by utilizing air as an oxidant and naturally available water as the proton source. On the one hand, the reticular gas-solid interface promotes efficient gas transfer, facilitating the enrichment of gaseous reactants with initially low concentrations. As shown in Supplementary Fig. 46, the gas-solid system displayed a 38-fold increase in CH3OH productivity compared to the liquid-solid system when the feed gas comprised low concentration CH4 and synthetic air (CH4/O2/N2, 17/17/66). On the other hand, operating PTMO on a gas-solid interface effectively circumvented the corroding and poisoning from impure liquid water toward photocatalysts (Supplementary Fig. 47)38. As a result, during a 24-cycle test under outdoor natural sunlight irradiation (on North Campus at the Heibei University, Baoding City, Hebei Province, China), our MOF membrane-based gas-solid system achieved an average CH3OH productivity of 11.8 mmol g–1 h–1, with high CH3OH selectivity of ~100% when utilizing air as the oxidant and tap water as the proton source (Fig. 4f).
Domino synergy between the colocalized Pd1 and Fe1 at the gas-solid interface
The efficient utilization of the H2O2 intermediate at the gas-solid interface hinges upon the colocalization of Pd1 and Fe1 sites on Zr–O nodes, which is the key to enabling an effective domino cascade between POR and HMO process. To verify this inference, we synthesized an additional Fe porphyrin (Fe-TCCP) decorated Pd1/aUiO(H) catalyst [Pd1Fe(TCCP)/aUiO(H)] and probed the effects of spatial proximity between the Pd1 and Fe1 sites on the efficiency of H2O2 intermediate utilization and CH3OH generation (Supplementary Fig. 48)39. In Pd1Fe(TCCP)/aUiO(H), the Fe1 sites displayed a similar electronic structure and intrinsic HMO reactivity as those in the Pd1Fe1/aUiO(H) catalyst (Supplementary Fig. 49); however, these Fe1 sites were located at the cage of the MOF and not co-anchored on the Zr–O nodes with Pd139. Notably, in the gas-solid system (75 oC), the residue of the H2O2 intermediate was scarcely detectable in the Pd1Fe1/aUiO(H) membrane (Fig. 5a), indicating that the H2O2 generated on Pd1 sites could be efficiently utilized by adjacent Fe1 to trigger PTMO. Contrarily, the Pd1Fe1(TCCP)/aUiO(H) membrane demonstrated inefficient utilization for in situ generated H2O2 and sluggish PTMO reactivity, primarily caused by the obstructed H2O2 transfer from Pd1 to allopatric Fe1 sites, given the absence of liquid water as the transporter.

a CH3OH yields (right axis) and amounts of residual H2O2 (left axis) in the Pd1Fe1/aUiO(H) and Pd1Fe(TCCP)/aUiO(H) membrane. Reaction conditions: 50 mg of MOF catalyst, 1 hour, 150 sccm humidified feed gas, and under simulated sunlight irradiation (AM 1.5 G). b Operando Fe K-edge XANES spectra of Pd1Fe1/aUiO(H) and Fe1/aUiO catalysts under PTMO conditions. c In situ EPR spectra of DMPO radical for observing reactive methyl and hydroxyl radicals on different photocatalysts under simulated sunlight irradiation (AM 1.5 G). d Isotope-labeled in situ ATR–FTIR spectra of Pd1FeOx/aUiO(H) and Pd1Fe1/aUiO catalysts under PTMO conditions. The absorption peaks at 886.3 and 853.3 cm–1 to be assigned to FeIV = 16O and FeIV = 18O species, respectively42. In situ ATR-FTIR spectra of Pd1Fe1/aUiO(H) catalyst under PTMO conditions by using humidified CH4/O2 (e) and CH4/Ar (f) as feed gas. The error bars correspond to the standard deviation of three independent measurements.
Seeking to understand the domino synergy between Pd1 and Fe1 sites at the gas-solid interface, we first investigated the activation mechanism of CH4 molecules on Fe1 sites. X-ray absorption near-edge structure (XANES) analysis was used to examine the electronic structure of Fe1 species during the PTMO process. Besides Pd1Fe1/aUiO(H), we also prepared the Fe1/aUiO catalyst featuring only Fe1 decoration (0.39 wt.%) for comparative study (Supplementary Fig. 50). Ex situ XANES analyses revealed the oxidation state of the Fe1 in both the Pd1Fe1/aUiO(H) and Fe1/aUiO catalysts to be close to +3 valence (Supplementary Fig. 51). However, when treated with H2O2 solutions, both compounds exhibited enhanced white line intensities in their XANES spectra, suggesting the oxidation of Fe1 by H2O2 (Supplementary Fig. 52). Notably, the in situ XANES spectra revealed a noticeable increase in the valence state of Fe1 in Pd1Fe1/aUiO(H) during the gas-phase PTMO process, similar to that of Fe1 in H2O2-treated Pd1Fe1/aUiO(H) (Fig. 5b). In contrast, the valence state of Fe1 sites in Fe1/aUiO remained unchanged, with only a marginal increase observed in the Pd1Fe(TCCP)/aUiO(H) catalyst (Fig. 5b and Supplementary Fig. 53). Combined with the in situ EPR spectroscopy (Fig. 5c), we propose that the H2O2 generated on the Pd1 could directly oxidize the colocalized Fe1 sites during the PTMO process. This leads to the release of hydroxyl radicals ( ∙ OH) while generating high-valence Fe1 sites that act as the CH4 activation centres for facilitating the dehydrogenation of CH4 into methyl radicals ( ∙ CH3).
In situ attenuated total reflectance–Fourier transform infrared reflection spectroscopy (ATR–FTIR) was then carried out to characterize further the HMO process on Fe1 sites. Upon light irradiation, the Fe1 sites in Pd1Fe1/aUiO(H) were immediately oxidized to the high-valence FeIV = O species by the in situ generated H2O2, as verified by isotope-labeled in situ ATR–FTIR experiments (Fig. 5d, Supplementary Fig. 54)40,41. In addition, the CH3 deformation vibrational mode at ~1471 cm−1 appears and enhances gradually on the Pd1Fe1/aUiO(H) with increasing illumination time when using a humidified CH4/O2 mixture as feed gas, indicating the prompt activation of CH4 to methyl radicals ( ∙ CH3) on the FeIV = O sites (Fig. 5e)42,43,44. Meanwhile, the peak at ~1091 cm−1, corresponding to the C–O stretching modes of CH3OH, also gradually enhances with prolonged illumination time, confirming the efficient coupling of ∙CH3 and ∙OH45. However, no analogous peak (at ~1471 and ~1091 cm−1) can be observed on the Pd1Fe1/aUiO(H) when using a humidified CH4/Ar mixture as feed gas (Fig. 5f), suggesting that the in situ generated H2O2 via POR process was necessary for triggering the evolution of Fe1 sites from FeIII–OH to active FeIV = O species.
We also assessed the function of iron oxide clusters within the Pd1FeOx/aUiO(H) catalyst by studying the evolution of these species during the PTMO. Under light irradiation, the typical CH3 deformation vibrational mode at 1470 cm−1 was observed on the Pd1FeOx/aUiO(H) catalyst under humidified CH4/O2 and CH4/Ar conditions (Supplementary Fig. 55). However, almost no CH3OH molecular could be detected in the CH4/Ar condition (Supplementary Fig. 55a), implying that the coupling reaction between ∙CH3 and ∙OH was impeded due to the absence of the H2O2 initiator (Supplementary Fig. 56). Isotope-labeled in situ ATR–FTIR experiments further revealed that the iron oxide clusters in the Pd1FeOx/aUiO(H) catalyst could effectively capture photogenerated holes and be oxidized into FeIV = O species during the PTMO (Fig. 5d, Supplementary Figs. 57 and 58)40,41. Consequently, throughout the PTMO process, the iron oxide clusters serve dual roles: inhibiting electron-hole recombination while providing more FeIV = O centers to accelerate CH4 activation (Supplementary Fig. 59).
Following our research, we propose that the CH4 activation in the PTMO process relies on FeIV = O species, while the evolution of methanol depends on subsequent radical reactions. Initially, photons excite electrons from MOF ligands (2-aminoterephthalic acid) to the Zr–O nodes, promoting the POR on Pd1 sites and leaving holes in the ligands. The Fe1 anchored on the –O nodes are then oxidized to FeIV = O active species by the H2O2 generated on the vicinal Pd1 sites, during which hydroxyl radicals are released46,47. Meanwhile, the iron oxide clusters can effectively capture photoholes and be oxidized to FeIV = O species. Subsequently, CH4 molecules are adsorbed onto the resulting FeIV = O centers, followed by the homolytic cleavage of a C–H bond, forming FeIII–OH species and methyl radicals48. The methyl and hydroxyl radicals then react to generate CH3OH molecules. Finally, the photo-generated H2O2 and holes reoxidize the FeIII–OH species to FeIV = O, setting this cycle anew (Supplementary Fig. 60).
In summary, we have developed a proof-of-concept MOF membrane nanoreactor by assembling MOF crystals (NH2-UiO-66) decorated by sterically colocalized single-atom palladium (Pd1) and iron (Fe1) onto a gas diffusion layer. We show that the PTMO could be boosted at the gas-solid interface by maximizing the spatial intimacy of the photocatalytic H2O2 generation (Pd1) and CH4 oxidation (Fe1) sites, thereby effectively circumventing H2O2 dilution. Moreover, photothermal effect-driven interfacial water management enables the simultaneous effective desorption of hydrophilic CH3OH and high-flux diffusion of hydrophobic CH4 within the MOF membrane nanoreactor. Consequently, this MOF membrane nanoreactor demonstrates unprecedented catalytic activity and stability for directly converting methane to high-concentration methanol under simulated sunlight irradiation. Moreover, our MOF membrane nanoreactor represents a photocatalytic platform with both scientific value and practical application potential for a wide range of three-phase photocatalytic reactions. The powerful toolbox in single-atom catalysis and MOF chemistry endows us with almost infinite possibilities to tune the chemical compositions and pore microenvironments of MOF, enabling the design of specialized MOF membrane nanoreactors optimized for diverse three-phase tandem catalytic reactions and facilitating the investigation of these reactions’ mechanisms at an atomic level. The facile preparation of hybrid MOF membranes, the easy scale-up of such photocatalysis devices, and the minimized usage of moisture (instead of freshwater) make them even more suitable for applications in regions with limited access to pure water and abundant solar energy.
Methods
Catalyst synthesis and preparation
Preparation of aUiO particles
In a typical synthesis process, 10.5 mg of ZrCl4 was dissolved in 5 mL of anhydrous N, N-dimethylformamide (DMF) in a 20 mL glass vial. Simultaneously, another 14.5 mg of NH2-BDC was similarly dissolved in 5 mL of anhydrous DMF in a separate vial. The two solutions were combined, and 1.3 mL of acetic acid was added. The mixture solution is shaken vigorously and then subjected to 12 h of heating at a stable temperature of 120 °C. Subsequently, light yellow powders were collected through centrifugation (7000 xg) for 5 min. The collected powders were then washed serially: five times with DMF over 48 h and five times with water over 36 h. After a comprehensive cleaning process, the powders were dried overnight under a dynamic vacuum to yield light yellow powders. In the final stage, these as-prepared powders were activated under high dynamic vacuum and temperature to produce aUiO. Typically, 200 mg of the as-prepared powders were placed into a quartz crucible and accommodated in a vacuum oven. The oven was kept at 100 °C for 8 h, after which the temperature was gradually raised to 185 °C and sustained for another 28 h. When these conditions were met, the resulting product was a brownish-yellow powder obtained after the final cooling to room temperature.
Preparation of Pd1/aUiO (0.78 wt.%)
In a typical synthesis process, 80 mg of aUiO powders were first dispersed fully in 60 mL of ultrapure water. This is followed by the slow injection of 1.25 mL of Na2PdCl4 aqueous solution of 5 mM concentration at a rate of 0.5 mL min–1 using an injection pump. This dispersion was then stirred for 10 h under the dark condition. Following this, the products were obtained by centrifugation (7000 xg) for 5 min, and they were subsequently rinsed twice with ultrapure water. Post-drying in a vacuum oven at 80 °C, these products were placed into a quartz boat and then heated in a tubular oven under high-purity Ar protection at 200 °C for 1 h.
Preparation of Pd1/aUiO(H)
Typically, the Pd1/aUiO(H) was prepared by treating 200 mg of as-prepared Pd1/aUiO powders in H2 (150 mL min–1) gas flow at 80 °C for 10 min, which was performed in an alumina crucible located at the center of a glass tube.
Preparation of Pd1FeOx/aUiO(H)
Fe species was introduced into Pd1/aUiO(H) by a photo-deposition and annealing process. Typically, 50 mg of Pd1/aUiO(H) powders were fully dispersed in 50 mL of ultrapure water. Then, 1.8 mL of FeCl3 aqueous solution (5 mM) was slowly injected (0.5 mL min–1) into the above dispersion with an injection pump. The mixture was then stirred in the dark for 3 h. Then, the solution was irradiated with a 300 W Xe-lamp (Beijing China Education Au-light Co., Ltd.) for 30 min. After that, the products were collected via centrifugation (8000 rpm, 5 min) and washed with ultrapure water (two times). After drying under 80 °C, the resultant powders are heated in a tubular oven under high-purity Ar protection at 200 °C for 1 h and then stored under high-purity Ar protection. From ICP-AES, the Fe amount of the as-synthesized Pd1FeOx/aUiO(H) was analyzed to be 0.89 wt.%. In this work, we controlled the Fe loadings by regulating the concentration of the precursor solution. For instance, to synthesize Pd1FeOx/aUiO(H) catalysts with Fe loadings of 0.23, 0.45, 0.67, 0.89, 1.06 wt.%, we injected 0.45, 0.9, 1.35, 1.8, and 2.15 mL of a 5 mM FeCl3 aqueous solution as the precursor.
Preparation of Pd1Fe1/aUiO(H)
Typically, the Pd1Fe1/aUiO(H) was prepared by treating the as-prepared Pd1FeOx/aUiO(H) powders in 0.5 M H2SO4 solution for 1 h. The resulting samples were collected by centrifugation, repeatedly washed with ultrapure water, and dried in the vacuum oven under 80 °C. From ICP-AES, the Fe amount of the Pd1Fe1/aUiO(H) was analyzed to be 0.37 wt.%. Pd1Fe1/aUiO(H) catalysts with different Fe mass loading were synthesized using a consistent acid pickling method, with the sole alteration being the adjusted Fe content in Pd1FeOx/aUiO(H).
Preparation of FeOx/aUiO and Fe1/aUiO
To prepare FeOx/aUiO, we thoroughly dispersed 50 mg of aUiO powders in 50 mL of ultrapure water. Then, 1.8 mL of a FeCl3 aqueous solution (5 mM) was slowly introduced into the dispersion at a rate of 0.5 mL min–1 using an injection pump. The mixture was stirred at room temperature for 3 h. Following that, the solution undergoes a 30-minute irradiation process under a 300 W Xe-lamp (Beijing China Education Au-light Co., Ltd.). After the irradiation, the resultants are gathered through a 5-minute centrifugation (7000 xg) and then washed twice with ultrapure water. After drying in the vacuum oven under 80 °C, the resultant powders are heated in a tubular oven under high-purity Ar protection at 200 °C for 1 h and then stored under high-purity Ar protection. Using ICP-AES, we establish that the Fe amount in the FeOx/aUiO(H) is around 0.89 wt.%. The Fe1/aUiO catalyst was prepared by treating the as-prepared FeOx/aUiO(H) powders in 0.5 M H2SO4 solution for 1 h. The resulting samples were collected by centrifugation, repeatedly washed with ultrapure water, and dried in the vacuum oven under 80 °C. From ICP-AES, the Fe amount of the Fe1/aUiO was analyzed to be 0.39 wt.%.
Preparation of Pd1Fe(TCCP)/aUiO(H)
The Pd1Fe(TCCP)/aUiO(H) catalyst was prepared using a solvothermal method based on previously reported work48. Typically, 10.5 mg of ZrCl4 was dissolved in 5 mL of anhydrous DMF in a 20 mL glass vial. Simultaneously, another 14.5 mg of NH2-BDC and 0.8 mg Fe-TCPP were similarly dissolved in 5 mL of anhydrous DMF in a separate vial. Following this, the two solutions were combined, and 1.3 mL of acetic acid was added to the mixture. The combined solution was shaken vigorously and then subjected to 12 h of heating at a stable temperature of 120 °C. After a comprehensive cleaning and activation process, 80 mg of as-prepared powders are dispersed fully in 60 mL of ultrapure water. This was followed by the slow injection of 1.25 mL of Na2PdCl4 aqueous solution of 5 mM concentration at a rate of 0.5 mL min–1 using an injection pump. The dispersion was then stirred for 10 h in the dark. Following this, the products were obtained by centrifugation (7000 xg) for 5 min, and they were subsequently rinsed twice with ultrapure water. Post-drying in a vacuum oven at 80 °C, these products are placed into a quartz boat and then heated in a tubular oven under high-purity Ar protection at 200 °C for 1 h. Finally, the resulting powders were treated in 150 sccm H2/Ar (10 vol% H2) gas flow at 80 °C for 10 min to obtain Pd1Fe(TCCP)/aUiO(H) catalyst.
Fabrication of hybrid MOF membranes
Typically, 50 mg of MOF photocatalyst powders [Pd1FeOx/aUiO(H), Pd1Fe1/aUiO(H), Pd1Fe(TCCP)/aUiO(H) or Fe1/aUiO] were first dispersed in a mixture of isopropanol, ultrapure water, and Nafion solution (150 μL of Nafion solution in 10 mL of 7:1 isopropanol: water mixture). The mixture was then sonicated for 30 min to produce MOF inks. Then, the inks were deposited onto one side of commercially available polytetrafluoroethylene (PTFE) films or carbon-based gas diffusion layer (GDL) substrate (Freudenberg, Germany) to fabricate the hybrid MOF membranes through vacuum filtration. After that, the as-prepared membranes were dried in Ar and activated in a high dynamic vacuum at 80 °C.
PTMO in the conventional liquid-solid system
The PTMO experiments were carried out in an outer irradiation-type photo-reactor (Pyrex glass). A 50 mg portion of catalyst and 30 mL H2O were added into a 50 mL photo-reactor equipped with a cross-shaped magnetic stirrer bar. The system was placed 3 cm in front of an Xe lamp. Before the reaction, the reaction system was purged by the CH4 and O2 (v/v = 1/1, 150 mL min−1) for at least 30 min to reach equilibrium. After that, the reactor was irradiated by a Xe lamp with an AM 1.5 G filter (Beijing China Education Au-light Co., Ltd) for a desired reaction time under stirring.
The liquid products (e.g., CH3OH, HCOOH) were quantified with NMR. Quantitative analysis was based on the linear relationship between CH3OH or HCOOH concentration and relative area versus dimethyl sulfoxide (DMSO) as an internal standard. Typically, 0.5 mL of the resultant solution was extracted from the reactor and mixed with 100 µL of D2O (99.9 at.% D, Sigma-Aldrich) containing 0.5 μL of dimethyl sulfoxide (99.9%, Sigma-Aldrich) as an internal standard. HCHO was measured by the colorimetric method based on the reaction between acetylacetone and HCHO in the presence of acetic acid and ammonium acetate49. The H2O2 concentration was measured by a cerium sulfate Ce(SO4)2 titration method based on the mechanism that the yellow solution of Ce4+ could be reduced to colourless Ce3+ by H2O2 according to the following equation:
\(2{{\rm{Ce}}}^{4+}+{{\rm{H}}}_{2}{{\rm{O}}}_{2}\to 2{{\rm{Ce}}}^{3+}+{{\rm{O}}}_{2}+2{{\rm{H}}}^{+}\)
Thus, the concentration of H2O2 could be obtained by measuring the concentration of Ce4+ (UV-visible absorption at the wavelength of 316 nm)50.
The gaseous products (e.g., CO2) were quantified by online gas chromatography equipped with a methanizer and flame ionization detector (FID). The concentration of CO2 is often too low to be detected by the insensitive thermal conductivity detectors; an FID with a methanizer is highly recommended to make a reliable quantification and avoid over-estimation of the selectivity to target products.
PTMO in the continuous-flow gas-solid system
The PTMO tests of the hybrid MOF membranes in the gas-solid system were carried out in a homemade outer irradiation-type gas-flow cell connected to a closed gas-circulation system (Supplementary Fig. 22). Typically, the as-prepared membrane was transferred to the middle of the reactor. Then, a stainless ring was installed to fix the membrane. The reactor was then sealed using a rubber O-ring and a stainless cap. The MOF membrane underwent irradiation via a Xe lamp (with AM 1.5 G filter) introduced through the cap’s quartz window. The temperature of the catalyst bed could be monitored by the temperature probe with a thermocouple at the bottom. The gas mixture (CH4/O2 v/v = 1/1) purged the reaction system for at least 0.5 h to reach equilibrium prior to assessing the photocatalytic activity. During the photocatalysis process, humidified CH4 and O2 mixture gas (v/v = 1/1) was supplied to the reactor with tunable flow rates so that the CH4, O2, and H2O molecules could continuously pass across the membranes. The humidified feed gas-carried liquid products (e.g., CH3OH, HCHO) were collected by a cold trap and then quantified with NMR and colorimetric methods. The gas-phase products (e.g., CO2) were quantified by online gas chromatography (Shimadzu GC-2014C). In order to detect the residual CH3OH and H2O2 within the MOF membrane, the membrane was sufficiently washed with ultrapure water after the PTMO process to ensure the efficient collection of the residual products. Then, NMR and cerium sulfate titration were used to measure the concentrations of CH3OH and H2O2 in the aforesaid aqueous solution, respectively. For the isotope labeling 18O2 experiment, 18O2 was used as the reactant, and the product was analyzed by a GC–mass spectrometry instrument (GC 8890-MS 5977).
The STC energy conversion efficiency in our gas-solid system was assessed under AM 1.5 G simulated sunlight conditions. During the PTMO process, the HMO reaction at Fe1 sites occurs spontaneously without photocatalysis process; therefore, we estimated the STC energy conversion efficiency via the POR process. Theoretically, producing one equivalent of CH3OH requires expending one equivalent of photo-generated H2O2. Consequently, we can calculate the STC energy conversion efficiency using the formula below:
where 117000 J mol−1 is the Gibbs free energy for H2O2 generation.
Characterization
XRD measurements of the obtained catalyst powders were performed on a Stoe STADI-P instrument using Mo Kα1 radiation. XPS measurements were performed by a Thermo VG ESCALAB-250 system with Al-Kα and Mg-Kα sources operated at 15 kV. The binding energies referred to the C 1 s peak (284.8 eV) from adventitious carbon. UV-vis diffuse reflectance data were recorded in a Shimadzu Solid Spec-3700 spectrophotometer in the 200 − 800 nm wavelength range. The one-dimensional 1H spectra were recorded on Bruker ARX-400 spectrometer. N2 sorption isotherms were measured at 77 K on a Quantachrome ASiQMVH002-5 absorption apparatus. Before tests, the samples were pre-activated at 120 °C for 12 h. The pore size distributions were estimated by the DFT method from a N2 sorption experiment at 77 K.
The metal loading in our catalysts was determined by an ICP-AES spectrometer (Model Optima 2000, PerkinElmer). A series of solutions for the measurements were prepared by dissolving 20 mg of samples in 4 mL of aqua regia (75 vol.% HCl and 25 vol.% HNO3). The solution was left overnight to allow complete dissolution. The resultant solution was diluted to 50 mL with deionized water in a volumetric flask and then analysed using ICP-AES.
The high-angle annular dark-field scanning TEM (HAADF-STEM) images and STEM-EDX elemental mapping were obtained on a Tecnai G2 F30 (FEI) microscope at 300 kV. The samples were prepared by dropping water/ethanol dispersion of samples onto ultrathin carbon film and immediately evaporating the solvent. The HAADF-STEM images were obtained on a JEM-ARM200F transmission electron microscopy working at 200 kV, equipped with a probe spherical aberration corrector. The SEM images and EDX elemental mapping were acquired from JEOL S-4800 and Zeiss Supra 55 scanning electron microscopes.
The Pd K-edge and Fe K-edge XAFS spectra were obtained at beamline BL14W1 of the Shanghai Synchrotron Radiation Facility (SSRF) and beamline BL01C1 of the National Synchrotron Radiation Research Centre (NSRRC). The samples were measured in fluorescence mode using a solid-state detector to collect the data. Athena and Artemis codes were used to extract the data and fit the profiles. For the X-ray absorption near edge structure (XANES) part, the experimental absorption coefficients as a function of energies μ(E) were processed by background subtraction and normalization procedures and reported as “normalized absorption” for all the measured samples and standard references. For the extended X-ray absorption fine structure (EXAFS) part, the Fourier transformed (FT) data in R space were analyzed by applying different models for M–O, M–Cl, and M–M (M = Pd, Fe) contributions. The passive electron factors, S02, were determined by fitting the experimental data on metal foils and fixing the coordination number (CN) of M-M, and then fixed for further analysis of the measured samples. The parameters describing the electronic properties (e.g., correction to the photoelectron energy origin, E0) and local structure environment, including CN, bond distance (R), and Debye-Waller factor around the absorbing atoms, could vary during the fit process. During operando experiments, the photocatalyst was irradiated with a 450 nm diode emission laser, with an irradiation power of 300 mW cm−2 over a spot with a diameter of 0.5 cm2.
The in-situ ATR–FTIR experiments were conducted using a Thermo Fisher Nicolet IS50 II FTIR spectrometer with an internal reflection element of ZnSe crystal integrated with a photocatalytic in situ photochemical IR accessory. One milligram powder catalyst sample was spread on the surface of the internal reflection element. After sample loading, the system was degassed for 30 min under an Ar atmosphere to remove the absorbates. Subsequently, a humidified CH4 and O2 mixture (v/v = 1/1, 60 mL min−1) or humidified CH4 (60 mL min−1) was introduced into the chamber for 10 min. For isotope-labelled in-situ ATR–FTIR experiments, 18O2 and D218O were used to replace O2 and H2O as oxidant and proton sources, respectively. The system was then exposed to light irradiation, and the spectra were collected at different irradiation times. The in situ EPR was conducted using a Bruker EMX PLUS spectrometer with a 300 W Xe lamp.
MD simulation method and model
MD simulation was performed using large-scale parallel Atomic/Molecular simulation software (LAMMPS23)49,50. In the process of MD simulation, the all atom (OPLS-AA) force field which is a classical force field widely used in molecular dynamics simulation of liquid systems is used to describe the interaction between atoms51. It is designed to provide accurate intermolecular interactions, especially in liquid environments. The cell structure was constructed with PACKMOL software and the water molecule model was set as TIP3P52,53. The box length is a = 28.9676, b = 28.9676, and c = 120.4832 angstrom, respectively. The graphite-carbon acts as a wall of atoms placed on both sides of the model to block the passage of molecules. Twenty hydrogen peroxide (H2O2) molecules were randomly placed in the MOF pore and the methane, oxygen, and water molecules were placed on top of the MOF in quantities according to the actual experimental conditions (20 CH4 molecules, 640 water molecules, and 20 O2 molecules in the liquid-solid system; 320 CH4 molecules, 200 water molecules, and 320 O2 molecules in the gas-solid system). Prior to MD simulation, the energy of the initial model was minimized by using the steepest descent algorithm. Then, under the NPT ensemble (T = 298 K, and P = 101 Kpa), the temperature was controlled by the Nose-Hoover temperature controller, and the structural relaxation of 100 ps (with a time step of 0.1 fs) was performed to bring the model to equilibrium. In the MD simulation process, the NVT ensemble (T = 298 and 348 K) and a total timestep of 200 ps MD simulation with 0.1 fs time step was carried out.
The molecular force field is composed of both bonded and nonbonded interactions. Nonbonded interactions include van der Waals (vdW) forces and electrostatic interactions, which are described by Eqs. 1 and 2, respectively54.
In the equation, qi and qj are atomic charges, rij is the distance between atoms, σ is the atomic diameter, and ε is the atomic energy parameter. For different kinds of atoms, the geometric mix rules were adopted for vdW interactions, which follows Eq. 3. The cutoff distance of vdW and electronic interactions was set to 1.2 nm, and the pppm method was employed to calculate long-range electrostatic interactions.
In all the MD simulations, the motion of atoms was described by classical Newton’s equation, which was solved using the velocity-Verlet algorithm. The calculated results were visualized and graphed using OVITO software.
Density functional theory (DFT) calculation
The spin-polarized density functional theory (DFT) calculations have been conducted on Vienna ab-initio simulation package (VASP) to study the properties of prepared materials55,56,57. The Projector augmented wave method with a cutoff energy of 450 eV accompanied by Perdew-Burke-Ernzerhof functional has been used in the DFT calculations58. DFT-D3 method has been used to correct the influence of van der Waals interactions59. One unit structure containing a Zr6O4(OH)4 cluster and twelve terephthalic acid ligands from the UiO-66 MOF has been used. Then, one terephthalic acid ligand was replaced by one Pd atom with two hydroxyl ligands and one water ligand to build the Pd1/aUiO model. The carbon atoms in the Pd1/aUiO model have been fixed to keep the crystal structure of the origin MOF. To build the Pd-111 model, four layers of the Pd (111) facet have been cleaved, and a vacuum of 20 Å has been put on the surface to break the periodicity of the z direction. Half of the bottom layers have been fixed to simulate the bulk phase. All models have been fully relaxed with the energy convergence criterion of 10-5 eV and the force convergence criterion of 0.02 eV/Å, respectively. Only the Γ point has been used in the K-point mesh. The adsorption energy (Eads) has been calculated using formula 4,
The Etotal, Esubstrate, and Eadsorbate represent the energy of adsorption structure, substrate, and adsorbate, respectively. The free energies have been calculated using the following formula 5,
The G, EDFT, ZPE, and TS represent the free energy, energy from DFT calculations, zero point energy, and entropic contributions, respectively.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Schematic diagrams are provided for Fig. 1 in the paper. Source data for Figs. 2–5 are included with this paper. Additional data are available from the corresponding author upon request Source data are provided with this paper.
References
An, B. et al. Direct photo-oxidation of methane to methanol over a mono-iron hydroxyl site. Nat. Mater. 21, 932–938 (2022).
Feng, N. et al. Efficient and selective photocatalytic CH4 conversion to CH3OH with O2 by controlling overoxidation on TiO2. Nat. Commun. 12, 4652 (2021).
Cui, X. et al. Room-temperature methane conversion by graphene-confined single iron atoms. Chem 4, 1902–1910 (2018).
García de Arquer, F. P. et al. CO2 electrolysis to multicarbon products at activities greater than 1 A cm–2. Science 367, 661–666 (2020).
Jin, Z. et al. Hydrophobic zeolite modification for in situ peroxide formation in methane oxidation to methanol. Science 367, 193–197 (2020).
Li, X., Wang, C. & Tang, J. Methane transformation by photocatalysis. Nat. Rev. Mater. 7, 617–632 (2022).
Agarwal, N. et al. Aqueous Au-Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions. Science 358, 223–227 (2017).
Periana, R. A. et al. Platinum catalysts for the high-yield oxidation of methane to a methanol derivative. Science 280, 560–564 (1998).
Liu, Z. et al. Water-promoted interfacial pathways in methane oxidation to methanol on a CeO2-Cu2O catalyst. Science 368, 513–517 (2020).
Song, H., Meng, X., Wang, Z.-J., Liu, H. & Ye, J. H. Solar-energy-mediated methane conversion. Joule 3, 1606–1636 (2019).
Schwach, P., Pan, X. & Bao, X. Direct conversion of methane to value-added chemicals over heterogeneous catalysts: challenges and prospects. Chem. Rev. 117, 8497–8520 (2017).
Wu, X. et al. Atomic-scale Pd on 2D titania sheets for selective oxidation of methane to methanol. ACS Catal. 22, 14038–14046 (2021).
Xie, J. et al. Highly selective oxidation of methane to methanol at ambient conditions by titanium dioxide-supported iron species. Nat. Catal. 1, 889–896 (2018).
Xing, Y. et al. Fe/Fe3C boosts H2O2 utilization for methane conversion overwhelming O2 generation. Angew. Chem. Int. Ed. 60, 8889–8895 (2021).
Guo, G. J. & Zhang, Z. Open questions on methane hydrate nucleation. Commun. Chem. 4, 102 (2021).
Zhang, S.-K. et al. Interfacial electrochemical-chemical reaction coupling for efficient olefin oxidation to glycols. Joule 7, 1887–1091 (2023).
Meng, X. et al. Hierarchical triphase diffusion photoelectrodes for photoelectrochemical gas/liquid flow conversion. Nat. Commun. 14, 2643 (2023).
Li, Y. et al. Constructing solid-gas interfacial fenton reaction over Alkalinized-C3N4 photocatalyst to achieve apparent quantum yield of 49% at 420 nm. J. Am. Chem. Soc. 138, 13289–13297 (2016).
Sushkevich, V. L., Palagin, D., Ranocchiari, M. & Bokhoven, J. A. V. Selective anaerobic oxidation of methane enables direct synthesis of methanol. Science 356, 523–527 (2017).
Yang, Y. et al. Self-flooding behaviors on the fuel cell catalyst surface: an in situ mechanism investigation. Energy Environ. Sci. 16, 491–501 (2023).
Suguro, T. et al. A hygroscopic nano-membrane coating achieves efficient vapor-fed photocatalytic water splitting. Nat. Commun. 13, 5698 (2022).
Yaghi, O. M., Kalmutzki, M. J. & Diercks, C. S. Introduction to reticular chemistry: metal-organic frameworks and covalent organic frameworks. Wiley-VCH. Weinheim (2019).
Zhang, Q. et al. Covalent organic framework–based porous ionomers for high-performance fuel cells. Science 378, 181–186 (2022).
Hao, Y.-C. et al. Metal-organic framework membranes with single-atomic centers for photocatalytic CO2 and O2 reduction. Nat. Commun. 12, 2682 (2021).
Sun, D. et al. Studies on Photocatalytic CO2 Reduction over NH2-UiO-66(Zr) and its derivatives: towards a better understanding of photocatalysis on metal-organic frameworks. Chem. Eur. J. 19, 14279–14285 (2013).
Furukawa, H., Cordova, K. E., O’Keeffe, M. & Yaghi, O. M. The chemistry and applications of metal-organic frameworks. Science 341, 1230444–1230456 (2013).
Kim, H. et al. Water harvesting from air with metal-organic frameworks powered by natural sunlight. Science 356, 430–434 (2017).
Lu, F.-F., Gu, X.-W., Wu, E., Liu, B. & Qian, G. Systematic evaluation of water adsorption in isoreticular UiO-type metal–organic frameworks. J. Mater. Chem. A. 11, 1246–1255 (2023).
Yu, S. M. et al. High activity and selectivity of single palladium atom for oxygen hydrogenation to H2O2. Nat. Commun. 51, 4737 (2022).
Ma, X. et al. Modulating Coordination Environment of single-atom catalysts and their proximity to photosensitive unit for boosting MOF photocatalysis. J. Am. Chem. Soc. 143, 12220–12229 (2021).
Abdel-Mageed, A. M. et al. Highly active and stable single-atom Cu catalysts supported by a metal–organic framework. J. Am. Chem. Soc. 141, 5201–5210 (2019).
Nam, W., Lee, Y. M. & Fukuzumi, S. Tuning reactivity and mechanism in oxidation reactions by mononuclear nonheme iron(IV)-oxo complexes. Acc Chem Res 47, 1146–1154 (2014).
Kalmutzki, M. J., Hanikel, N. & Yaghi, O. M. Secondary building units as the turning point in the development of the reticular chemistry of MOFs. Sci. Adv. 4, eaat9180 (2018).
Wang, S. et al. H2-reduced phosphomolybdate promotes room-temperature aerobic oxidation of methane to methanol. Nat. Catal. 6, 895–905 (2023).
Xu, Y. et al. Pd-catalyzed instant hydrogenation of TiO2 with enhanced photocatalytic performance. Energy Environ. Sci. 9, 2410–2417 (2016).
Li, J. et al. Identification of durable and non-durable FeNx sites in Fe–N–C materials for proton exchange membrane fuel cells. Nat. Catal. 4, 10–19 (2021).
Qi, Y. et al. Unraveling of cocatalysts photodeposited selectively on facets of BiVO4 to boost solar water splitting. Nat. Commun. 13, 484 (2022).
Lee, W. H. et al. Floatable photocatalytic hydrogel nanocomposites for large-scale solar hydrogen production. Nat. Nanotech. 18, 754–762 (2023).
Sui, J. et al. Bioinspired microenvironment modulation of metal-organic framework-based catalysts for selective methane oxidation. Sci. Bull. 68, 1886–1893 (2023).
Zandi, O. & Hamann, T. W. Determination of photoelectrochemical water oxidation intermediates on haematite electrode surfaces using operando infrared spectroscopy. Nat. Chem. 8, 778–783 (2016).
Zhao, Y. et al. α-Fe2O3 as a versatile and efficient oxygen atom transfer catalyst in combination with H2O as the oxygen source. Nat. Catal. 4, 684–691 (2021).
Song, S. et al. A selective Au-ZnO/TiO2 hybrid photocatalyst for oxidative coupling of methane to ethane with dioxygen. Nat. Catal. 4, 1032–1042 (2021).
Li, X. et al. PdCu nanoalloy decorated photocatalysts for efficient and selective oxidative coupling of methane in flow reactors. Nat. Commun. 14, 6343 (2023).
Li, X. et al. Efficient hole abstraction for highly selective oxidative coupling of methane by Au-sputtered TiO2 photocatalysts. Nat. Energy 8, 1013–1022 (2023).
Luo, L. et al. Water enables mild oxidation of methane to methanol on gold single-atom catalysts. Nat. Commun. 12, 1218 (2021).
Fang, Y. et al. Realizing methanol synthesis from CO and water via the synergistic effect of Cu0/Cu+ over Cu/ZrO2 catalyst. J. Energy Chem. 93, 126–134 (2024).
Zhao, H. B. et al. The role of Cu1–O3 species in single-atom Cu/ZrO2 catalyst for CO2 hydrogenation. Nat. Catal. 5, 818–831 (2022).
Cheng, Q. et al. Maximizing active Fe species in ZSM-5 zeolite using organic-template-free synthesis for efficient selective methane oxidation. J. Am. Chem. Soc. 145, 5888–5898 (2023).
Martínez, L., Andrade, R., Birgin, E. G. & Martínez, J. M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157–2164 (2009).
Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 117, 1–19 (1995).
Jorgensen, W. L. & Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110, 1657–1666 (1988).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Schott-Verdugo, S. & Gohlke, H. PACKMOL-Memgen: A simple-to-use, generalized workflow for membrane-protein-lipid-bilayer system building. J. Chem. Inf. Model. 59, 2522–2528 (2019).
Yao, A. & Liu, J. T. Microporous polyarylate membranes based on 3D phenolphthalein for molecular sieving. Sci. Adv. 10, eado7687 (2024).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Tu, H. Y., Hou, H. S. & Ji, X. B. Carbon dots from alcohol molecules: principles and the reaction mechanism. Chem. Sci. 14, 12194 (2023).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (Nos. U23A20139, 22405069, 52371220, 22322101, and 22375017), the National Key Research and Development Program of China (No. 2020YFB1506300), the Beijing Institute of Technology Research Fund Program, the Natural Science Foundation of Hebei Province (Nos. 2023HBQZYCXY001, B2024201096, B2023204034, B2023201107, B2022201090, B2021201074, B2021201034, and F2021203097), the Hebei Provincial Department of Science and Technology (No. 216Z4303G), and the Hebei Education Department (No. BJ2025011).
Author information
Authors and Affiliations
Contributions
Y.C.H., B.W., and J.H.Y. designed the research. Y.C.H. and L.W.C. performed the catalyst preparation, characterization, photocatalytic tests, and data analysis. H.D.L., W.F.N., X.J.G., J.N.L., H.Z.H., C.S., C.C.L., S.B.N., L.J.G., Y.G.L., and S.F.W. assisted with the material synthesis, characterizations, and catalysis measurements. Y.C.H., L.W.C., A.X.Y., B.W., and J.H.Y. wrote the paper. B.W. and J.H.Y. supervised the research. All authors discussed the results and assisted during manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Li-Hua Gan, Pengfei Zhu and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hao, Y., Chen, L., Liu, H. et al. Continuous photo-oxidation of methane to methanol at an atomically tailored reticular gas-solid interface. Nat Commun 16, 747 (2025). https://doi.org/10.1038/s41467-025-56180-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56180-7
