
Abstract
Two or more catalysts conducting multistep reactions in the same reactor, concurrent tandem catalysis, could enable (bio)pharmaceutical and fine chemical manufacturing to become much more sustainable. Herein we report that co-immobilization of metal nanoparticles and a biocatalytic system within a synthetic covalent organic framework capsule, COFcap-2, functions like an artificial cell in that, whereas the catalysts are trapped within 300-400 nm cavities, substrates/products can ingress/egress through ca. 2 nm windows. The COFcap-2 reactor is first coated onto an electrode surface and then used to prepare eleven homochiral amines using dinitrogen as a feedstock. The amines, including drug product intermediates and active pharmaceutical ingredient, are prepared in >99% enantiomeric excess under ambient conditions in water. Importantly, the COFcap-2 system is recycled 15 times with retention of performance, addressing the relative instability and poor recyclability of enzymes that has hindered their broad implementation for energy-efficient, low waste production of chemicals and (bio)pharmaceuticals.
Similar content being viewed by others
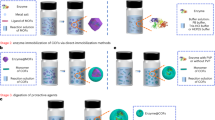
Introduction
Dinitrogen (N2) is the most abundant atmospheric gas and the primary commodity for nitrogenated industrial and natural products1. With respect to industrial production, fixation and conversion of N2 is conducted by the Haber-Bosch process, a process introduced in 1905 that, because it requires high-purity N2/H2 gas and extreme conditions, consumes 1–2% of global energy production2. Emerging techniques, especially electrocatalytic nitrogen reduction reactions (NRRs) using chemocatalysts, promise a green and more sustainable alternative3,4. The primary N2 fixation product, ammonia (NH3)5, is also a chemical commodity that is utilized directly or to produce value-added products such as chiral amines that serve as active pharmaceutical ingredients (e.g., (R)-sitagliptin6, (R)-ramatroban7, or (S)-rivastigmine8). Chemocatalytic approaches for chiral amine production (hydrogenation of Schiff bases, diastereoisomeric crystallization or nucleophilic addition9,10) are typically heterogeneous and often suffer from poor enantioselectivity and/or product yield11. Biocatalytic approaches (enzymes) are inherently greener as they offer better control (chemo-, regio-, and enantioselectivity) under milder reaction conditions. Unfortunately, the use of enzymes in homogenous systems renders recycling difficult and enzymes are relatively fragile outside physiological conditions. Whereas the potential utility of multi-step processes driven by multiple catalysts, concurrent tandem catalysis, is well and long recognized12, implementation has been hindered by inability to recycle the catalysts and mutual interference of catalysts and substrates. Minteer’s group13 developed a homogeneous N2 fixation system to realize N2 conversion to chiral amines by a multi-enzyme approach involving nitrogenase, diaphorase, L-alanine dehydrogenase, and ω-Transaminase. However, homogenous catalytic systems are difficult to recycle, and nitrogenase is environmentally sensitive and costly. Immobilization techniques offer potential for improving the performance of tandem chemocatalytic and/or biocatalytic systems14. Indeed, hydrogels15 and metal-organic frameworks (MOFs)16 have been used to immobilize enzymes for bioelectrocatalysis reactions although they have not yet been demonstrated on tandem catalytic systems involving biocatalysts. It is therefore desirable to develop new immobilization techniques that enable tandem catalytic reactors that enable chemocatalysts, electrocatalysts and/or biocatalysts to work in synergy. Herein, we introduce such an approach by using mild conditions to co-immobilize metal nanoparticles and an enzyme cascade system within a nanoscale (COF) capsule (COFcap-2) coated onto an electrode surface. The resulting tandem catalytic reactor was demonstrated to efficiently convert N2 to chiral amines at least as well as its homogenous counterpart and, because of its heterogenous nature, it was readily recycled by water washing.
In principle, rigid porous capsules with nanoscale dimensions and appropriate functionality should be able to mimic cell membranes/walls and enable co-immobilization of multiple catalysts17,18,19. COFs are synthetic materials, usually with 2D structures, that are inherently modular and offer mesoporous features20. We recently introduced COFcaps prepared in a three-step approach by exploiting metal–organic frameworks (MOFs) as sacrificial templates. Whereas the first generation (Gen 1) COFcap, COFcap-1, can immobilize enzymes there are limitations: the MOF templates can compress the enzymes and reduce their activity; only MOF particles with regular polyhedral shapes were effective as templates21; the range of catalysts that can be encapsulated is limited. A related strategy using prefabricated COFs encounters the same handicaps22,23,24,25 as physisorption of enzymes is size limited and tends to result in pore blockage, leading to slow flux rates for substrates and products26,27. Here we introduce a Gen 2 COFcap, COFcap-2, which was prepared by a PEG-driven emulsion templating method, PEG polymers promoting the liquid-liquid phase separation (LLPS) to form stable droplets, that resulted in 300–400 nm COFcaps (Fig. 1) suitable for one-pot integration of multiple enzymes (ω-transaminase (ω-TA), glutamate dehydrogenase (GluDH), glucose dehydrogenase (GCDH)) and chemocatalysts (nanoparticle, NAD+) (Fig. 1). The presence of. As detailed below and in Fig. 1, in a second step, COFcap-2 was coated onto a carbon paper electrode and investigated as a concurrent tandem catalytic reactor for nitrogen fixation and chiral amine production.

The PEG-driven emulsion templating approach to prepare COFcap-2 enabled immobilization of multiple catalysts and subsequent conversion of N2 to chiral amines. SEM image of COFcap-2 on the upper right.
Results and discussion
Preparation and characterization of COFcap-2
Preparation of COFcap-2 was conducting by reacting three COF monomers, DTH (2,5-diethoxyterephthalohydrazide) and TB (1,3,5-triformylbenzene), with an additive, DTH-400 (Mw = 400 g/mol), in an emulsion formed by water and chloroform with scandium trifluoromethanesulfonate (Sc(OTf)3) as a catalyst (Fig. 2b). The ratio of DTH-400:DTH:TB was varied (0:30:20; 1:29:20; 3:27:20; 5:25:20; 10:20:20; 20:10:20; 30:0:20) and profoundly impacted the reaction outcome. Transmission electron microscopy (TEM), scanning electron microscopy (SEM), and dynamic light scattering (DLS) were used to characterize the morphology and particle size distribution of the COFcap-2 products. Figure 2a and Supplementary Fig. 1 reveal that the 0:30:20 reaction (pure COF-42) did not produce capsules. Using the PEGylated polymer additive, DTH-400, afforded monodisperse high-quality COFcap-2 (hollow spherical morphology and smooth shell surfaces). This is consistent with our previous work in which DTH-400 improved the processability, mechanical strength, and flexibility of COF membranes28. DTH-400 also serves as an emulsion stabilizer to facilitate the formation of COF capsules. The PEG chains would form interlayer or intralayer chain entanglement with an increase in the contents of DTH-400. COFcap-2 was identified by powder X-ray diffraction (PXRD). Among the ratios studied, 0:30:20, 1:29:20, 3:27:20; 5:25:20, 10:20:20 matched the characteristic PXRD peaks of COF-42 (2θ = 3.4°, 7.0°, and 26.9°, Fig. 2c)28,29. N2 sorption studies of COFcap-2 (DTH-400:DTH:TB = 0:30:20, 1:29:20, 3:27:20, 5:25:20, 10:20:20) at 77 K revealed BET surface areas of 1051 m2/g, 825 m2/g, 581 m2/g, 466 m2/g, and 115 m2/g, respectively (Fig. 2d). Pore size distribution as determined using the DFT method revealed intrinsic pores of 2.2 nm (COF-42) as well as a new pore of 1.2 nm which increased with the percentage of DTH-400 (Supplementary Fig. 2). COFcap-2 (DTH-400:DTH:TB = 20:10:20 and 30:0:20) was observed to be amorphous with a broad peak centered at 2θ ≈ 22° and low BET surface area (Supplementary Fig. 3). Fourier transform infrared spectra (FT-IR) showed enhancement of stretching modes at 2867 cm−1 in COFcap-2 which we attribute to PEGylated DTH-400 (Supplementary Fig. 4). The shell thickness of COFcap-2 was adjusted from 80 nm to 10 nm by increasing the percentage of DTH-400 (Fig. 2e). We also found that the ratio of chloroform to water affected capsule size from 190 ± 30 nm to 825 ± 45 nm (TEM, Fig. 2f; DLS, Supplementary Fig. 5). Reaction conditions (e.g., reaction time) were further optimized as detailed in SI, crystallinity being studied by PXRD (Supplementary Fig. 6), capsule size by TEM (Supplementary Fig. 7).

a TEM images of COFcap-2 prepared with different reagent ratios (from left to right, DTH-400:DTH:TB = 0:30:20, 1:29:20, 3:27:20, 5:25:20, 10:20:20, 20:10:20, 30:0:20). b Synthesis of COFcap-2. c, d PXRD patterns and N2 sorption isotherms of COFcap-2 prepared with different ratios. e The shell thickness of COFcap-2 with different ratios. f The size and corresponding TEM images of COFcap-2 with different volumes of water/trichloromethane (mL/mL): 0.5/7.5, 1.0/7.5, 2.0/7.5, 3.0/7.5. Scale bar: 200 nm. The error bar of the sample refers to the standard deviation of three groups prepared at different times (n = 3). Source data are provided as a Source Data file.
Preparation and characterization of enzyme◎COFcap-2
Lipase (Aspergillus niger lipase) was selected to test enzyme encapsulation by COFcap-2 (enzyme◎COFcap-2) during the synthesis of COFcap-2. 3:27:20 (DTH-400:DTH:TB) was used to produce lipase◎COFcap-2 (Fig. 3a and Supplementary Fig. 8). The porosity and pore size of lipase◎COFcap-2 matched COFcap-2 (Supplementary Fig. 9), confocal microscopy indicating that lipase was present inside COFcap-2 (Fig. 3c and Supplementary Fig. 10). The different sizes of COFcap-2 formed by adjusting synthesis conditions influenced enzyme immobilization and enzymatic activity. The COFcap-2 with a diameter of 396 ± 30 nm possessed better relative activity and is large enough to immobilize multiple catalysts (Supplementary Fig. 11a and b). We investigated if lipase influenced the synthesis of lipase◎COFcap-2. Tracking PXRD peaks of lipase◎COFcap-2, we found that the crystallinity gradually decreased with increase in lipase (Supplementary Fig. 11c–f). Thus, load efficiency and crystallinity can be used as an indicator to judge whether enzyme◎COFcap-2 indeed clathrates lipase. Moreover, lipase◎COFcap-2 (3:27:20) offered good protection under various conditions and better recyclability than lipase◎COFcap-2 without DTH-400 (Supplementary Fig. 12), highlighting the benefit of mixed polymeric building blocks.

a PXRD patterns of lipase◎COFcap-2 with different ratios: 0:30:20, 1:29:20, 3:27:20, 5:25:20, 10:20:20. b The L-glutamic acid yield rate of GluDH◎COFcap-2 and 2-amino-4-phenylbutane yield rate of ω-TA◎COFcap-2. The error bar of the sample refers to the standard deviation of three groups prepared at different times (n = 3). c 3D view of CLSM image of FITC-tagged lipase◎COFcap-2. d SIM image of GCDH&GluDH&ω-TA◎COFcap-2. Scale bar: 500 nm. Source data are provided as a Source Data file.
We next investigated encapsulation and reactivity of the enzymes GluDH, GCDH, and ω-TA separately or together within COFcap-2. For GluDH, GCDH and ω-TA, load amounts of 0.10 g g−1, 0.10 g g−1 and 0.19 g g−1, respectively, afforded optimal activities of 74%, 77% and 78% vs. the respective free enzymes (Supplementary Figs. 13–15). GluDH◎COFcap-2 produced L-glutamic acid (0.067 mM mgenzyme−1 h−1) and ω-TA◎COFcap-2 generated 2-amino-4-phenylbutane (0.08 mM mgenzyme−1 h−1). GluDH:ω-TA = 1.2:1.0 was selected for optimal efficiency (Fig. 3b). Since GluDH often requires a stoichiometric amount of coenzyme, we also encapsulated NAD+. X-ray photoelectron spectroscopy (XPS) supported the presence of NAD+ in NAD+◎COFcap-2 (Supplementary Fig. 16 and 17), with low leakage of NAD+ (1.32%) after 5 hours (Supplementary Fig. 18). To test for cascade reactions in COFcap-2, we co-encapsulated GluDH, GCDH, and ω-TA into COFcap-2. A super-resolution imaging system (SIM) suggested uniform dispersion of GluDH, GCDH, and ω-TA in GCDH&GluDH&ω-TA◎COFcap-2 (Fig. 3d). The N2 Thermo gravimetric analysis (TGA) curve also indicated the presence of enzymes in COFcap-2 (Supplementary Fig. 19).
N2 fixation inside COFcap-2
We studied conversion of N2 to ammonia to test our COFcap-2 reactor (Fig. 4a). Three enzymes (ω-TA, GluDH, and GCDH), a chemocatalyst (Au nanoparticle, Au NPs) and coenzyme NAD+ were co-immobilized by COFcap-2. The Au NPs as a chemocatalysts can be used for the electrochemical N2 reduction and ammonia generation3,30. Au NPs were pre-synthesized and then added with DTH-400, DTH and TB to form Au◎COFcap-2. TEM images reveal Au NPs of diameter 80 ± 15 nm (Supplementary Fig. 20). TEM images and energy-dispersive spectroscopy (EDS) elemental mappings of Au◎COFcap-2 indicate that Au NPs had indeed been encapsulated (Fig. 4b). The Au content in Au◎COFcap-2 was determined to be 5.2 wt% using atomic absorption spectroscopy (AAS). Electrochemical activity toward the NRR was assessed in a neutral liquid-based electrochemical system at room temperature and atmospheric pressure. Electrodes were prepared by coating carbon paper with a paste of Au◎COFcap-2 before being air dried (Supplementary Fig. 21). Linear sweep voltammetry (LSV) curves of Au◎COFcap-2 were measured in an Ar- or N2-saturated environment, revealing a change from −0.2 to −0.6 V vs. reversible hydrogen electrode (RHE) in the N2-saturated solution, indicative of NRR activity in this potential range (Supplementary Fig. 22). Chronoamperometry (CA) indicated that the highest NH3 yield, 40.5 μg h–1 mg–1, was at −0.40 V versus RHE (Nessler’s reagent method). The greatest faradaic efficiency of 42% was obtained at −0.30 V vs. RHE (Fig. 4c and Supplementary Fig. 23). 15N isotopic labeling was used to confirm the nitrogen source for NH3 (Fig. 4d). NH3 productivity was determined by the Nessler’s reagent method, indophenol blue approach, 1H NMR (14N2) and 1H NMR (15N2) spectroscopy (Supplementary Figs. 24–28). Moreover, no N2H4 by-product was observed in the electrolyte after NRR (Supplementary Fig. 29). We also explored whether other nanoparticles (Fe2O3 and Cu) could be used as NRR catalysts. Fe2O3◎COFcap-2 and Cu◎COFcap-2 were prepared with 3.1 wt% Fe and 3.5 wt% Cu, respectively, and their NH3 productivity and faradaic efficiency were determined to be 47% and 34% of that of Au◎COFcap-2 (Supplementary Figs. 30–32). Meanwhile, the XPS data illustrated that oxidation states of nanoparticles have not changed pre- and post-catalysis (Supplementary Figs. 33 and 34).

a Schematic illustration of N2 transformation to chiral amines by combining NRR and enzyme cascade reactions. The amine was provided by Au nanoparticle induced NRR, and then GluDH regenerated L-glutamic acid from α-ketoglutarate and ammonia with the regenerated NAD+ as the coenzyme that was recycled by GCDH. Next, ω-TA transferred the amino group from glutamic acid to ketone substrates and produced the desired chiral amine products. b TEM image and EDS elemental mappings of Au◎COFcap-2 indicate the C+Au, C, and Au elemental distributions. Scale bar: 200 nm. c Rates of ammonia production and Faradaic efficiencies of Au◎COFcap-2 at various potentials. The error bar of the sample refers to the standard deviation of three groups prepared at different times (n = 3). d 1H NMR spectra of standard NH4Cl samples and post-electrolytes using 15N2 and 14N2. e, The 2-amino-4-phenylbutane yield rate of catalysts◎COFcap-2. f The 2-amino-4-phenylbutane yield rate of catalysts◎COFcap-2x (x = a, b, c representing the GluDH + GCDH + ω-TA amount, a = 0.15 + 0.15 + 0.125 mg; b = 0.30 + 0.30 + 0.25 mg; c = 0.6 + 0.6 + 0.5 mg). The error bar of the sample refers to the standard deviation of three groups prepared at different times (n = 3). g Liquid chromatograph-mass spectrometer (LCMS) spectra of producing 2-amino-4-phenylbutane using 15N2 and 14N2. h, Performance of recycled catalysts◎COFcap-2 over 15 cycles. The error bar of the sample refers to the standard deviation of three groups prepared at different times (n = 3). Source data are provided as a Source Data file.
Our success with NRR for converting N2 to NH3 prompted us to also study an enzyme cascade reaction. Co-encapsulated GluDH, GCDH, ω-TA, Au NPs and NAD+ in COFcap-2 were found to convert N2 into chiral amines on the surface of an electrode as prepared above in an H-shaped dual-chamber reactor. The yield of 2-amino-4-phenylbutane was 105 μM mg−1 after 24 h, the highest productivity being 62 μM h−1 (Fig. 4e). We also prepared chiral amines in the presence of 15N-labeled N2 and liquid chromatography-mass spectrometry (LCMS) analysis revealed formation of 15N-labeled 2-amino-4-phenylbutane (Fig. 4g). As shown in Supplementary Fig. 35, the catalysts◎COFcap-2 reactor was ineffective in the absence of even one of these co-catalysts. Furthermore, catalysts◎COFcap-2x with various amounts of GluDH + GCDH + ω-TA were studied (x = a = 0.15 + 0.15 + 0.125 mg; x = b = 0.30 + 0.30 + 0.25 mg; x = c = 0.6 + 0.6 + 0.5 mg) to evaluate the influence of collision efficiency on catalytic performance. The best performance was exhibited by catalysts◎COFcap-2b (Fig. 4f). It has been reported previously that higher concentrations of enzymes can cause repeated collisions and reduce overall performance31. Meanwhile, the product yield of catalysts◎COFcap-2b is significantly higher than that of corresponding free systems (Supplementary Fig. 36). The catalysts◎COFcap-2 with different molar ratio (GluDH:GCDH:ω-TA = 0.6:0.6:1.0, 1.2:1.2:1.0, 2.4:2.4:1.0) were studied the efficiency of the reaction pathway. The catalysts◎COFcap-2 (1.2:1.2:1.0) shown the similar yield rate with 2.4:2.4:1.0, much higher than 0.6:0.6:1.0, because these three enzymes in catalysts◎COFcap-2 (1.2:1.2:1.0) can be well matched in long-path catalysis (Supplementary Fig. 37). Importantly, the heterogeneous nature of catalysts◎COFcap-2 enabled facile recovery and recycling; after 15 cycles productivity of >60 % was retained (Fig. 4h). Meanwhile, catalysts◎COFcap-2 maintained good stereoselectivity, enzyme activity and NH3 yield, and there was almost no enzyme leakage after 15 cycles (Supplementary Fig. 38). Catalysts◎COFcap-2 retained crystallinity and morphology (Supplementary Figs. 39 and 40).
To test the scope of this tandem catalyst system, eleven prochiral ketones were studied as example substrates (Table 1, Supplementary Figs. 41–51 and Supplementary Table 1), seven of which had been previously studied with a homogeneous cascade biocatalytic system13. The catalysts◎COFcap-2 reactor exhibited high productivity and excellent stereoselectivity (>99% eep) to afford the corresponding (R)-amines (Supplementary Figs. 52–62). Notably, the productivity of catalysts◎COFcap-2 matched or surpassed that of the previously reported homogeneous biocatalyst system. Moreover, we studied four more substrates (Table 1), three pharmaceutical intermediates and one active pharmaceutical ingredient. (R)-1b, with a productivity = 62 μM h−1, is a precursor of the antihypertensive dilevalol32,33. (R)-2b (productivity = 43 μM h−1) has the same backbone of the antiarrhythmic, antimyotonic, analgesic drug (R)-mexiletine. The antimyotonic, analgesic drug (R)-mexiletin34 and (R)-11b (productivity = 30 μM h−1) is the active ingredient of Januvia®, a blockbuster drug product for treatment of type 2 diabetes35.
A key feature of COFcap-2 is that, when compared to COFcap-1, pure COFs, and MOFs, all of which can encapsulate biocatalysts, its internal diameter is much larger. This enables it to accommodate multiple catalysts in such a way that it in effect mimics a homogenous system. In essence, the cavity is large enough to prevent conformational compression of enzymes, which can reduce the performance of biocatalysts. These features also distinguish COFcap-2 from porous solids such as activated carbon, MCM-41, and MIL-100 (Cr), all of which possess pores that are too small to accommodate such catalysts (Supplementary Figs. 63 and 64). Moreover, catalysts◎COFcap-2 surpassed the performance of other in-situ immobilization platforms. Indeed, the productivity of 2-amino-4-phenylbutane was enhanced by a factor of >4.5 vs. catalysts@ZIF-90, >3.5 vs. catalysts@COFcap-1 and >8.8 vs. catalysts@NKCOF-98 (Supplementary Figs. 65–68). We assert that this is because the previous approaches restrict enzyme freedom and rapid mass transfer. In addition, since catalysts◎COFcap-2 was coated to an electrode surface, it could be recycled just by washing with water, avoiding the need for filtration to recycle the enzyme system.
In summary, we detail a Gen 2 COF-based capsules, COFcap-2, with dimensions (250-300 nm internal diameter) and features (mesopores) to facilitate encapsulate multiple chemo and biocatalysts. The resulting system, catalysts◎COFcap-2, serves as a nanoreactor for tandem catalysis exemplified by fixation of N2 into homochiral amines under mild conditions by combining NRR with an enzyme cascade process. Catalysts◎COFcap-2 therefore combines the advantages of biocatalysts and chemocatalysts, including activity better than a related homogenous system, and enantioselectivity, with the recyclability of heterogeneous catalysts. The scope of this particular catalysts◎COFcap-2 system was demonstrated on 11 substrates, 7 of which had been previously studied with a homogeneous biocatalytic cascade system, another of which is a precursor to a blockbuster drug molecule.
Whereas catalysts◎COFcap-2 demonstrated with fixation of N2 and formation of chiral amines, the mild in situ encapsulation process makes COFcap-2 a versatile platform for clathration of multiple catalysts, including enzymes, in confined space that is large enough to mimic solution. Further, the mesoporous functionality enables ingress of substrates and egress of products while the larger catalyst species is clathrated. The technique reported herein, tandem catalysis involving a cascade biocatalysis system and electrocatalysts, afforded products of relevance to pharmaceutical manufacturing and addresses the limitations of existing immobilization approaches as the observed catalytic performance at least matches that of solution and recycling is facile. The COFcap-2 reactor is not inherently limited to the catalysts studied herein and so could offer a new paradigm for utility of tandem catalysis systems to enable sustainable manufacturing in various fields, including energy, pharmaceuticals, fine chemicals and biotechnology.
Methods
Synthesis of COFcap-2 and lipase◎COFcap-2 with different ratios of DTH-400:DTH:TB
The DTH-400:DTH (0.03 mmol) with the different ratios (0:30, 1:29, 3:27, 5:25, 10:20, 20:10, 30:0) and water/chloroform mixture (V/V = 1.0/7.5 mL/mL) was added into a 50 mL bottle, and stirred at > 600 rpm for 10 min. Then, TB (3.3 mg, 0.02 mmol) was ultrasonic dissolved in 1.0 mL chloroform and 1.0 mL acetonitrile. The TB solution and Sc(OTf)3 aqueous solution (70 μL, 1.0 g/mL) were added to the above mixture by drops and stirred at room temperature for 50 min. At the end of the reaction, the solid was precipitated with 15 mL methanol, and the faint yellow solid was obtained by filtration.
For the preparation of lipase◎COFcap-2, DTH-400:DTH (0.03 mmol) with the different ratios, lipase (6.0 mg) and water/chloroform mixture (V/V = 1.0/7.5 mL/mL) was added into a 50 mL bottle and stirred at > 600 rpm for 10 min. Then, TB (3.3 mg, 0.02 mmol) was ultrasonic dissolved in 1.0 mL chloroform and 0.1 mL acetonitrile. The TB solution and Sc(OTf)3 aqueous solution (10 μL, 1.0 g/mL) were added to the above mixture by drops and stirred at room temperature for 50 min. The product was collected by centrifugation at 6000 × g for 10 min, washed three times with DI water, and dried at room temperature. After the encapsulation and washing process, the supernatant of each operation was collected and the concentration of protein was quantified by following the standard protocol of the Bradford assay, before which the calibration curve of lipase was fitted. The loading efficiency (LE %) and the loading capacity (LC, g·g−1) were calculated as follows:
Synthesis of Au nanoparticles (Au NPs)
The Au NPs were synthesized on the basis of the reported methods36. Au NPs were synthesized by adding 2.4 g of PVP (MW of 40k) in 15 mL of DMF solvent with 22.5 μL of 1 M stock solution of NaOH. Further, 24 μL of 169 mM stock solution of HAuCl4 was added to the uniformly mixed solution at room temperature for 4 h. Then, Au NPs were centrifuged at 6000 × g for 15 min and washed with deionized water three times. Finally, the Au NPs were dispersed with 1.0 mL of water.
Synthesis of Au◎COFcap-2
The DTH-400:DTH (3:27, 0.03 mmol), the dispersion of Au NPs (300 μL), and water/chloroform mixture (V/V = 1.0/7.5 mL/mL) were added into a 50 mL bottle and stirred at > 600 rpm for 10 min. Then, TB (3.3 mg, 0.02 mmol) was ultrasonic dissolved in 1.0 mL chloroform and 0.1 mL acetonitrile. The TB solution and Sc(OTf)3 aqueous solution (10 μL, 1.0 g/mL) were added to the above mixture by drops and stirred at room temperature for 50 min. The product was collected by centrifugation at 6000 × g for 10 min, washed three times with DI water, and dried at room temperature.
Synthesis of catalyst◎COFcap-2x (x = a, b, c)
The DTH-400:DTH (3:27, 0.03 mmol), the dispersion of Au NPs (300 uL), NAD+ (5.0 mg), GluDH, GCDH, ω-TA (x representing the GluDH + GCDH + ω-TA amount, a = 0.15 + 0.15 + 0.125 mg; b = 0.30 + 0.30 + 0.25 mg; c = 0.6 + 0.6 + 0.5 mg), and water/chloroform mixture (V/V = 1.0/7.5 mL/mL) were added into a 50 mL bottle, and stirred at > 600 rpm for 10 min. Then, TB (3.3 mg, 0.02 mmol) was ultrasonic dissolved in 1.0 mL chloroform and 0.1 mL acetonitrile. The TB solution and Sc(OTf)3 aqueous solution (10 μL, 1.0 g/mL) were added to the above mixture by drops and stirred at room temperature for 50 min. The product was collected by centrifugation at 6000 × g for 10 min, washed three times with DI water, and dried at room temperature.
Expression and purification of ω-TA
A mutant (R)-selective ω-transaminase ATA-117 originated from the Arthrobacter genus and was constructed by Savile (Genbank accession No. JA717225.1). The expression construct for the variant of ATA-117 was designed in pET28a with BamHI and XhoI restriction sites (pET28a_ATA-117) and using E. coli BL21(DE)3 as host. Expression and purification of the variant of ATA-117 were performed as described by Savile et al.6. By ultrasonic spallation, the recombinant E. coli BL21 (pET28a_ATA-117) was lysed with PBS buffer (0.1 mM sodium phosphate, 0.5 mM NaCl, pH 7.0). To obtain pure enzymes, a linear imidazole gradient (20-300 mM) in the same PBS buffer would be used to elute the proteins. The protein concentration of the purified ATA-117 was measured by the Bradford method with bovine serum albumin as the standard. With sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrospray mass spectrometry, the protein samples were analyzed (Supplementary Figs. 69 and 70).
Calculation of ammonia yield rate and faradaic efficiency
The faradaic efficiency from −0.2 to −0.6 V vs. reversible hydrogen electrode (RHE) for NRR was defined as the amount of electric charge used for synthesizing NH3 divided by the total charge passed through the electrodes during electrolysis. The total amount of NH3 was measured by colorimetric methods. Assuming three electrons were required to produce one NH3 molecule, the Faradaic efficiency could be calculated as:
The rate of ammonia formation (\({{{\rm{R}}}}_{{{{\rm{NH}}}}_{3}}\)) was calculated by using the following equation:
where F is the Faraday constant, \({c}_{{{{\rm{NH}}}}_{3}}\) is the measured ammonia concentration, V is the volume of electrolyte for NRR, Q is the quantity of applied electricity, t is the electro-reaction time, and mcat. is the mass of the catalyst.
Electrochemical NRR measurements
Electrochemical measurements were taken on the electrochemical station using a three-electrode system. Au◎COFcap-2, Fe2O3◎COFcap-2, or Cu◎COFcap-2 (2.0 mg) were used to modify the working electrode, while Ag/AgCl (saturated KCl) and Pt plate worked as the reference and counter electrodes, respectively. And the electrolyte is 0.1 M Na2SO4 water solution. Before each NRR process, the electrochemical system was first bubbled continuously with N2 gas for at least 1 h. And then, the gas was maintained throughout the electrochemical reaction. Potentiostatic measurements were carried out at a series of applied potentials, including –0.2, –0.3, –0.4, –0.5, and –0.6 V versus Ag/AgCl for 2 h at room temperature at 95% iR compensation.
The synthesis and analysis of generated 15N-labeled chiral amine
The catalytic system in vitro synthesis of chiral amine chemicals was performed in an H-shaped dual-chamber bioelectrochemical reactor at a constant potential of −0.4 V vs Ag/AgCl at 30 °C. The catalyst◎COFcap-2 (5.0 mg) capsule was used as the working electrode. Electrodes were prepared by coating carbon paper with a paste of catalyst◎COFcap-2 with water (containing 0.2 wt% Nafion) before being air dried. The headspace of the reactor was filled with high-purity Ar and then filled with high-purity 15N2. The electrolyte is 30 mL Na2SO4 (0.1 M) water solution containing 1 mM PLP, 5 mM glucose, 1 mM 2-ketoglutaric acid, 1 mM ketone substrate, and 1% DMSO. Potentiostatic measurement was carried out at −0.4 V vs Ag/AgCl for 24 hours at room temperature. The reaction mixture was extracted with ethyl acetate. The organic layers were separated by centrifugation and finally dried over Na2SO4. The enantiomeric excess of the corresponding amine was determined by LCMS analysis.
Data availability
All data are available in the main text or the supplementary materials, and from corresponding authors upon request. Source data are provided with this paper.
References
Jia, H.-P. & Quadrelli, E. A. Mechanistic aspects of dinitrogen cleavage and hydrogenation to produce ammonia in catalysis and organometallic chemistry: relevance of metal hydride bonds and dihydrogen. Chem. Soc. Rev. 43, 547–564 (2014).
Martín, A. J., Shinagawa, T. & Pérez-Ramírez, J. Electrocatalytic reduction of nitrogen: from haber-bosch to ammonia artificial leaf. Chem 5, 263–283 (2019).
He, H. et al. Metal–organic framework supported Au nanoparticles with organosilicone coating for high-efficiency electrocatalytic N2 reduction to NH3. Appl. Catal. B: Environ. 302, 120840 (2022).
Liu, S. et al. Proton-filtering covalent organic frameworks with superior nitrogen penetration flux promote ambient ammonia synthesis. Nat. Catal. 4, 322–331 (2021).
Foster, S. L. et al. Catalysts for nitrogen reduction to ammonia. Nat. Catal. 1, 490–500 (2018).
Savile, C. K. et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 329, 305–309 (2010).
Busto, E., Simon, R. C., Grischek, B., Gotor-Fernández, V. & Kroutil, W. Cutting short the asymmetric synthesis of the ramatroban precursor by employing ω-transaminases. Adv. Synth. Catal. 356, 1937–1942 (2014).
Fuchs, M., Koszelewski, D., Tauber, K., Kroutil, W. & Faber, K. Chemoenzymatic asymmetric total synthesis of (S)-Rivastigmine using omega-transaminases. Chem. Commun. 46, 5500–5502 (2010).
Breuer, M. et al. Industrial methods for the production of optically active intermediates. Angew. Chem. Int. Ed. 43, 788–824 (2004).
Ghislieri, D. & Turner, N. J. Biocatalytic approaches to the synthesis of enantiomerically pure chiral amines. Top. Catal. 57, 284–300 (2013).
Kelly, S. A. et al. Application of omega-transaminases in the pharmaceutical industry. Chem. Rev. 118, 349–367 (2018).
Wasilke, J.-C., Obrey, S. J., Baker, R. T. & Bazan, G. C. Concurrent tandem catalysis. Chem. Rev. 105, 1001–1020 (2005).
Chen, H. et al. Upgraded Bioelectrocatalytic N2 Fixation: From N2 to Chiral Amine Intermediates. J. Am. Chem. Soc. 141, 4963–4971 (2019).
McFadden, K. M., Kays, L. G., Boucher, D. G. & Minteer, S. D. Bioelectrocatalysis for synthetic applications: Utilities and challenges. Curr. Opin. Electrochem. 44, 101458 (2024).
Radomski, J., Vieira, L. & Sieber, V. Bioelectrochemical synthesis of gluconate by glucose oxidase immobilized in a ferrocene based redox hydrogel. Bioelectrochemistry 151, 108398 (2023).
Yang, X.-G. et al. Enhanced Activity of Enzyme Immobilized on Hydrophobic ZIF-8 Modified by Ni2+ Ions. Angew. Chem. Int. Ed. 62, e202216699 (2023).
Huo, J., Aguilera-Sigalat, J., El-Hankari, S. & Bradshaw, D. Magnetic MOF microreactors for recyclable size-selective biocatalysis. Chem. Sci. 6, 1938–1943 (2015).
Borodina, T. et al. Controlled Release of DNA from Self-Degrading Microcapsules. Macromol. Rapid Commun. 28, 1894–1899 (2007).
Wang, X., Feng, J., Bai, Y., Zhang, Q. & Yin, Y. Synthesis, properties, and applications of hollow micro-/nanostructures. Chem. Rev. 116, 10983–11060 (2016).
Oliveira, F. L. et al. Enzyme immobilization in covalent organic frameworks: strategies and applications in biocatalysis. Chempluschem 85, 2051–2066 (2020).
Li, M. et al. Fabricating covalent organic framework capsules with commodious microenvironment for enzymes. J. Am. Chem. Soc. 142, 6675–6681 (2020).
Liang, J. et al. Insight into bioactivity of in-situ trapped enzyme-covalent-organic frameworks. Angew. Chem. Int. Ed. 62, e202303001 (2023).
Zheng, Y. et al. Green and scalable fabrication of high-performance biocatalysts using covalent organic frameworks as enzyme carriers. Angew. Chem. Int. Ed. 61, e202208744 (2022).
Chao, H. et al. Template-free in situ encapsulation of enzymes in hollow covalent organic framework capsules for the electrochemical analysis of biomarkers. ACS Appl. Mater. Inter. 14, 20641–20651 (2022).
Kandambeth, S. et al. Self-templated chemically stable hollow spherical covalent organic framework. Nat. Commun. 6, 6786 (2015).
Paul, S. et al. Hierarchical covalent organic framework-foam for multi-enzyme tandem catalysis. Chem. Sci. 14, 6643–6653 (2023).
Tang, Y. et al. Fabrication of hollow covalent-organic framework microspheres via emulsion-interfacial strategy to enhance laccase immobilization for tetracycline degradation. Chem. Eng. J. 421, 129743 (2021).
Wang, Z. et al. PolyCOFs: A new class of freestanding responsive covalent organic framework membranes with high mechanical performance. ACS Cent. Sci. 5, 1352–1359 (2019).
Uribe-Romo, F. J., Doonan, C. J., Furukawa, H., Oisaki, K. & Yaghi, O. M. Crystalline covalent organic frameworks with hydrazone linkages. J. Am. Chem. Soc. 133, 11478–11481 (2011).
Sun, H. et al. Porous β-FeOOH nanotube stabilizing Au single atom for high-efficiency nitrogen fixation. Nano Res. 15, 3026–3033 (2022).
Mao, D. et al. DNA nanomachine with multitentacles for integral processing of nanoparticles and its application in biosensing. ACS Appl. Bio Mater. 3, 2940–2947 (2020).
Ye, L. J. et al. Engineering of amine dehydrogenase for asymmetric reductive amination of ketone by evolving rhodococcus phenylalanine dehydrogenase. ACS Catal. 5, 1119–1122 (2015).
Sanfilippo, C., Paternò, A. A. & Patti, A. Resolution of racemic amines via lipase-catalyzed benzoylation: Chemoenzymatic synthesis of the pharmacologically active isomers of labetalol. Mol. Catal. 449, 79–84 (2018).
Cavalluzzi, M. M. et al. Synthesis of (R)-, (S)-, and (RS)-hydroxymethylmexiletine, one of the major metabolites of mexiletine. Tetrahedron.: Asymmetry 18, 2409–2417 (2007).
Desai, A. A. Sitagliptin manufacture: a compelling tale of green chemistry, process intensification, and industrial asymmetric catalysis. Angew. Chem. Int. Ed. 50, 1974–1976 (2011).
Deng, X. et al. Yolk–Shell Structured Au Nanostar@Metal–organic framework for synergistic chemo-photothermal therapy in the second near-infrared window. Nano Lett. 19, 6772–6780 (2019).
Acknowledgements
We appreciate the financial support from the National Key Research and Development Program of China (2021YFC2102100), National Natural Science Foundation of China (22371136), Research Ireland through the Synthesis and Solid State Pharmaceutical Centre (grant 12/RC/2275_P2) and Haihe Laboratory of Synthetic Biology (22HHSWSS00008).
Author information
Authors and Affiliations
Contributions
Q.Q.Z. developed the electrocatalytic/biocatalytic system and performed most of the experiments. Q.Q.Z., J.Y.Y., Z.F.W., T.W., J.J.L assisted in synthetic experiments. S.Q., S.B.G, Y.L.Z. performed the TEM, HRTEM, and Confocal characterization. Q.Q.Z. and P.J.D. wrote the original draft, with input from all authors. J.Z.Z., Y.C., M.J.Z., P.C., Q.Q.Z. reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Rahul Banerjee and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, Q., Dong, P., Yu, J. et al. COFcap2, a recyclable tandem catalysis reactor for nitrogen fixation and conversion to chiral amines. Nat Commun 16, 992 (2025). https://doi.org/10.1038/s41467-025-56214-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56214-0
