
Abstract
Targeted protein delivery with nanocarriers holds significant potential to enhance therapeutic outcomes by precisely directing proteins to specific organs or tissues. However, the complex interactions between nanocarriers and the biological environment pose considerable challenges in designing effective targeted delivery vehicles. In this study, we address this challenge by leveraging DNA-barcoded high-throughput screening. We construct a nanocapsule library via in-situ polymerization, incorporating various monomers to create nanocapsules with unique surface properties. In vitro and in vivo screening, using female mice, identify nanocapsules with high cell association and different biodistribution. Our investigation into kidney-enriched nanocapsules highlights the crucial role of polymer composition in biodistribution, demonstrating the potential of surface engineering for precise control over nanoparticle distribution. The kidney-enriched nanocapsule successfully delivers catalase, showcasing its therapeutic potential in mitigating cisplatin-induced acute kidney injury. Overall, our study presents an approach for identifying protein delivery vehicles, with the capacity to broaden the application of proteins as therapeutic agents or research tools.
Similar content being viewed by others

Introduction
Targeted delivery has long been the holy grail for nanomedicine researchers as it offers numerous advantages regarding drug efficacy and reduced side effects1,2,3,4. Although pursued for many years, the development of targeted drug delivery systems is still hampered by the limited knowledge of the complicated interactions between nanoparticles and the human body5,6,7. Once injected, nanocarriers encounter different biological barriers, interact with various biological molecules, and transport through many organs. The size, charge, shape, surface chemistry, stiffness, and chemical composition of the delivery vehicles may affect their biodistribution, cellular uptake, and overall therapeutic efficacy. Given the complexity of nano-bio interactions, optimizing a delivery carrier for a specific organ or tissue remains a significant challenge.
High-throughput screening has long been a powerful means to seek optimal solutions in complicated systems. It has been widely applied in drug discovery8, formulation optimization9,10,11, and target identification12,13,14. However, when it comes to screening nanocarriers for organ-specific drug delivery, screening throughput has been limited by the number of animals and the time required to perform in vivo studies. DNA barcoding technology offers a promising solution to the bottleneck in nanocarrier screening for targeted delivery. By incorporating unique barcode sequences into nanocarriers, researchers can create a barcoded nanocapsule library that allows for simultaneous screening of multiple nanocarriers in vivo. Scientists, including James E. Dahlman, Michael J. Mitchell, and Daniel G. Anderson have successfully demonstrated the capacity of barcode-facilitated in vivo screening in identifying targeted nanocarriers15,16,17,18,19. However, current studies mainly focus on optimizing the composition of lipid nanoparticles for nucleic acid delivery. Strategies for targeted protein delivery are still lacking.
Targeted protein delivery is important for both pharmaceutical applications and scientific research. Protein therapeutics hold great promise for treating a wide range of diseases, thanks to their high specificity and essential role in modulating biological processes20,21. However, systematic administration of protein therapeutics often causes on-target side effects due to the multitudinous function of proteins22,23,24. Therefore, the development of nanocarriers for targeted protein delivery is crucial to achieve effective and safe therapy. Furthermore, targeted protein delivery holds significance in scientific research due to its pivotal role in potential applications like gene editing25,26,27,28,29. Previously, we developed a nanoencapsulation technology that enables sustained protein delivery30,31,32,33. In brief, one-step in situ polymerization is performed on the protein surface to produce protein nanocapsules with the protein embedded in a polymeric network, protecting the protein from degradation and prolonging its release. This technology has shown promise in delivering various proteins, such as enzymes, cytokines, and growth factors, to target tissues and organs.
In this work, we aim to develop a modified approach that enables high-throughput synthesis of a combinatorial nanocapsule library containing protein nanocapsules with high stability and diverse surface properties. Aided by the DNA barcoding technology, we can profile the tropism of nanoparticles in a high-throughput manner, thereby enabling a comprehensive understanding of the impact of surface properties on nanoparticle tropism. With streptavidin as the core protein, we synthesize a nanocapsule library with biotin-labeled DNA barcodes (Fig. 1a). The nanocapsule library comprises 143 nanocapsules with varied surface properties, synthesized through the polymerization of various combinations of monomers. In vitro screening identifies nanocapsules that efficiently bind to cell membranes, demonstrating the feasibility of screening the nanocapsule library. Subsequent in vivo screening enables us to identify nanocapsules with different tropism. We investigate the compositional factors influencing the accumulation of nanocapsules in the kidneys. Our findings indicate that the polymer composition, rather than size or surface charge, plays a more significant role in determining the kidney accumulation potential of these nanocapsules. We then use the kidney-enriched nanocapsule to deliver catalase to the kidney, effectively alleviating the cisplatin-induced acute kidney injury. Together, we demonstrate that surface engineering is a feasible way to manipulate the biodistribution of nanoparticles.

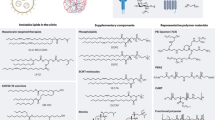
a The schematic illustration of the synthesis, screening, and barcode reading. The illustration was created in BioRender. Wang, L. (2025) https://BioRender.com/a96u763. b The design of the DNA barcodes. c Monomers used for the synthesis of the nanocapsule library. The monomers are classified into five categories: hydrophilic neutral monomers (W), hydrophobic neutral monomers (O), positively charged monomers (P), negatively charged monomers (N), and functional monomers (F). d The predicted pKa, log D (pH 7.4), and charge (pH 7.4) of the monomers. MMA does not possess pKa value. MPTA does not possess the pKa or logD values. e The monomer molar ratio added to the polymerization reactions for nanocapsule syntheses. Source data are provided as a Source Data file.
Results
The construction of DNA-barcoded nanocapsule library
To study the performance of multiple nanocapsules simultaneously, we rationally designed a series of DNA barcodes (Fig. 1b). Each DNA barcode has a 20-bp forward and a 20-bp reverse universal primer site, making it possible for PCR amplification. The 10 nucleotide “barcode” was situated in the center of the 50 nucleotide DNA sequence. We have selected 96 out of the 410 possible DNA combinations for nanocapsule barcoding (Supplementary Table 1). To facilitate the connection of the DNA barcodes to the streptavidin nanocapsules, a click reaction was used to conjugate biotin to the 5’ end of the barcode with a photocleavable (PC) linker (Fig. 1a)34,35. A fluoresceinamine is attached to the 3’ end of the barcode to enable affinity-based enrichment of the barcode during barcode extraction.
To allow high-throughput nanocapsule synthesis and ensure the stability of the nanocapsules during screening, we first developed a modified nanocapsule synthesis procedure that utilizes photo-initiated in situ polymerization (Fig. 1a). Streptavidin (SA) was selected as the core protein for the library to allow biotinylated DNA barcodes to attach. To ensure consistent reaction conditions across the entire library, we chose photopolymerization to allow precise control over the polymerization duration. Photo initiator eosin 5-isothiocyanate (EITC) was conjugated to streptavidin to yield protein macroinitiators (SA-eosin) for subsequent in situ polymerization. Eosin fluorescence observed in the SDS-PAGE (Supplementary Fig. 1a) indicated the successful conjugation of EITC to SA.
To enhance the diversity of surface properties of nanocapsules, we selected polymerization monomers based on their structural characteristics, chemical properties, and commercial availability. We then selected a total of 19 distinct monomers, which were divided into five categories (Fig. 1c), i.e., hydrophilic neutral monomers (W), hydrophobic monomers (O), cationic monomers (P), anionic monomers (N), and functional monomers (F). With distinctive charges, pKa values, and logDs (Fig. 1d), these monomers are suitable for developing a nanocarrier library with diverse surface properties. For each nanocapsule in the library, two or three monomers were selected as monomers in the polymerization reaction (Fig. 1e and Supplementary Table 1). To ensure good solubility of the nanocapsules, one or two hydrophilic monomers were selected as the major component forming each nanocapsule. Additional charged or functional monomer was included in some of the nanocapsules to increase the library diversity. The monomers, together with the crosslinker (N, N-methylene bisacrylamide, BIS), were added to the SA-eosin solution, with a molecular ratio of monomers:BIS:protein = 40000:4000:1. Subsequently, the photo-initiated polymerization was carried out using a green light (530 nm) emitted by a light emitting diode (LED) lamp for 5 min.
Following the synthesis of streptavidin nanocapsules (nSA), we performed agarose gel electrophoresis to determine the surface charges of nanocapsules (Supplementary Fig. 1b), as the electrophoretic mobility of electroosmotic flow is minimal in agarose gel36. Out of the 192 nanocapsules we constructed, 49 of them are excluded from the final libraries due to precipitation/gelation formation or unwanted surface charge. The rest 143 nanocapsules were divided into two libraries according to their surface charge. Finally, we have 69 nanocapsules in the cationic library (Library Z) and 74 nanocapsules in the anionic library (Library F). We randomly selected 10 nSAs from the libraries to characterize their sizes. Dynamic light scattering (DLS) measurement indicated that all 10 nanocapsules had mean hydrodynamic diameters of 15–49 nm (Fig. 2a and Supplementary Fig. 1c), consistent with our previous observations30,31,32,33. Together, these results demonstrated that we have successfully constructed a cationic and an anionic nanocapsule library.

a The size distribution of selected nSAs. b The agarose gel electrophoresis images of nSA-Z25 in complex with biotin-DNA oligomers with different lengths at a 1:1 mol ratio. The nSA-Z25 was visualized with Typhoon RGB (Cy2 channel). DNA was stained with gel red and imaged using the Cy3 channel. This experiment was repeated independently three times with similar results. c The size distributions of nSA-Z25 and nSA-Z25-DNA complex. d The ζ potentials of nSA-Z25 and nSA-Z25-DNA complex. e The schematic illustration of the method to enrich 3’-FAM-labeled DNA barcodes with anti-FITC magnetic beads (Created in BioRender. Wang, L. (2025) https://BioRender.com/a96u763). f The enrichment efficiency of biotin-DNA at different concentrations with anti-FITC magnetic beads. Data are presented as mean with individual data points. n = 2. g The enrichment efficiency of nSA-biotin-DNA complex at 40 nM concentration in PBS buffer or 293 T cell lysate using anti-FITC magnetic beads. Data are presented as mean with individual data points. n = 2. Source data are provided as a Source Data file.
With the nSA in hand, we next established a workflow for the incorporation and extraction of DNA barcodes. First, we tested the capability of nSA to bind biotinylated DNA oligomers. At a 1:1 molar ratio, the SA nanocapsules capture the DNA oligomers (10–60 bp) with a nearly quantitative yield, as demonstrated in the cases of nSA-Z25, nSA-Z01, and nSA-F24 (Fig. 2b and Supplementary Fig. 2a–c). Therefore, the designed 50-bp DNA barcode can be efficiently incorporated into the nanocapsules. Furthermore, DNA barcode incorporation did not alter the particle sizes or surface ζ potentials of nSA-Z25, nSA-Z01, and nSA-F24 (Fig. 2c, d and Supplementary Fig. 2d, e).
To facilitate the dissociation of DNA barcodes from the nSA-DNA complex during barcode extraction, we designed a photo-cleavable (PC) linker between biotin and DNA barcode (Supplementary Fig. 3a). The biotinylated barcodes were synthesized from Biotin-DBCO and DNA-N3 via a click reaction (Supplementary Fig. 3b). After the exploration of different stoichiometric ratios (Supplementary Fig. 3c), a Biotin-DBCO/DNA-N3 ratio of 20:1 was used for conjugation. Mass spectrometry (MS) confirmed the successful construction of the biotinylated DNA barcode (Supplementary Fig. 3d, e). We then exposed the biotinylated DNA barcode to 30 and 60 seconds of UV light to assess the susceptibility of the photo-cleavable (PC) linker to ultraviolet (UV) radiation. This exposure led to successful cleavage of the linker, which was confirmed by MS (Supplementary Fig. 3f–h). HPLC analysis revealed changes in the polarity of DNA barcodes following conjugation with a photocleavable (PC) linker (Supplementary Fig. 3i, j). The photocleavage kinetics of biotinylated DNA barcodes linked with PC linker demonstrated a gradual increase in the accumulation of photocleavage products over time (Supplementary Fig. 3j). We also estimated the kinetics of DNA barcode release from SA-DNA complex upon UV irradiation. Most of the barcodes can be released from the complex within two minutes (Supplementary Fig. 3k). Similarly, the DNA barcodes were successfully released from the nSA-DNA complex upon UV irradiation (Supplementary Fig. 3l).
The released DNA barcodes can be extracted with anti-FITC magnetic beads (Fig. 2e). To investigate the extraction efficiency, real-time quantitative PCR (qPCR) was performed before and after barcode extraction. Results showed 95.76% recovery when the barcode was extracted from 10 nM DNA-FAM (Fig. 2f). However, when the concentration of DNA-FAM dropped to 1 nM and 100 pM, the recovery dramatically decreased to 21.14% and 1.56%, respectively. The anti-FITC magnetic beads were also used to extract the barcode from the 40 nM nSA-Z28-PC-DNA complex after UV exposure. The recovery was 75.06% in PBS and 71.56% in HEK-293T cell lysate (Fig. 2g).
Once we established the protocols for barcode incorporation and extraction, we pooled the barcoded nanocapsules to form Libray Z (cationic) and Library F (anionic). The barcodes were then extracted and sequenced. The results indicate that all barcodes in the libraries could be detected, although with varying efficiency (Supplementary Fig. 4a, b). Replicated studies found consistent barcode percentages in sequencing results, showing that the barcode extraction and sequencing workflow produced reliable and reproducible results. Taken together, we established DNA-barcoded nanocapsule libraries and developed a workflow to read the barcodes.
In vitro screening enabled the discovery of cell-adhesive nanocapsules
To demonstrate the potential of DNA-barcode-aided screening, we conducted the screening of library Z on four cell lines: Ramos, Jurkat, HeLa, and DC2.4 cells. Ramos and Jurkat cells are malignant lymphocytes that are hard to transfect37,38. HeLa cell is a widely used cancer cell line, while DC2.4 is a murine dendritic cell line. To obtain nanoparticles that interact with these cells, we first incubated Library Z with the cells for 24 h. The cells were then washed and lysed. Subsequently, the DNA barcodes were extracted and amplified. Electrophoresis indicated the successful extraction and amplification of the barcodes (Supplementary Fig. 5a). Next-generation sequencing allows us to plot the enrichment factor of each DNA barcode relative to the original nanoparticle library in Fig. 3a–d. In DC2.4 cells and HeLa cells, the enrichment factors of the particles in the library ranged from 0.5 to 2.2 (Fig. 3a, b), indicating most nanoparticles interacted with these cells with similar capability. This is consistent with previous observations that cationic nanoparticles can bind to and be internalized by these cells. In contrast, incubation with Ramos and Jurkat cells resulted in more biased enrichment within the library. On these two cell lines, nSA-Z38, nSA-Z40, and nSA-Z91 nanoparticles exhibited the highest enrichment factors (Fig. 3c, d), indicating the three nanoparticles had a strong association with Ramos and Jurkat cells. We then choose the three nanoparticles to further study their interactions with the cells.

a–d The enrichment factor of each nanoparticle after incubation with (a) HeLa cell, (b) DC2.4 cell, (c) Ramos cell, and (d) Jurkat cell. Data are presented as mean with range. n = 2. e Flow cytometry analysis of HeLa, DC2.4, Ramos, and Jurkat cells after the treatment with 430 nM fluorescently labeled nSA-Z28 (P2W5), nSA-Z38 (P3N3), nSA-Z40 (P3N4), nSA-Z91 (F3P3N4) and nSA-P3N1 for 24 h. f Confocal microscope images of HeLa cells after treatment with nSA-Z38 at 37 °C for different periods. This experiment was repeated independently three times with similar results. Scale bar = 10 μm. White arrows were used for colocalization analysis (Supplementary Fig. 5b). g Surface-bound and internalized nSA-Z38 in red blood cells after incubation at 37 °C for 1 h. Data are presented as mean ± s.d. n = 3. h The Surface-bound and internalized nSA-Z38 in mouse peripheral blood mononuclear cells (PBMCs) at different concentrations (172 nM, 86 nM, and 43 nM). Data are presented as mean ± s.d. n = 3. The n represents biologically independent replicates (g, h). Source data are provided as a Source Data file.
To confirm the binding of nSA-Z38, nSA-Z40, and nSA-Z91 on different cells, we assessed the quantity of cell-bound particles with flow cytometry. Moderately enriched nanoparticle nSA-Z28 and poorly enriched nanoparticle nSA-Z01 were chosen for comparison. Flow cytometry measurements revealed that, Z38, Z40, and Z91 have stronger binding on cells than nSA-Z28 and nSA-Z01, consistent with the sequencing results (Fig. 3e). In particular, nSA-Z38 and nSA-Z40 exhibited the strongest binding on cells. Interestingly, nSA-Z38, Z40, and Z91 share the same cationic monomer [(3-methacrylamidopropyl)trimethylammonium chloride (MPTA, P3)], but each contains a different anionic monomer. Following this principle, we then synthesized nSA-P3N1 nanoparticle, which was not included in the original nanoparticle library, and detected its cell binding or endocytosis capability. As expected, nSA-P3N1 showed strong cell binding or endocytosis (Fig. 3e). These observations demonstrated that from the library, we could select nanoparticles that strongly bind to cells. These nanoparticles were constituted of the co-polymer of MPTA and an anionic monomer.
We then investigated whether these copolymers could be internalized by the cells. We chose nSA-Z38 to represent the MPTA-anionic copolymeric nanocapsules. Confocal microscopy images reveal that nSA-Z38 rapidly attached to the cell membrane and accumulated in HeLa cells with the extension of the incubation (Fig. 3f). The internalized nanocapsules partially colocalized with the lysosome probe LysoTracker (Supplementary Fig. 5b), indicating that the nanocapsules were internalized by cells via endocytosis. We next evaluated the total and internalized nSA-Z38 in Jurkat cells. In agreement with HeLa cells, Jurkat cells showed both surface-bound and internalized nSA-Z38 after 1 h incubation (Supplementary Fig. 5c). Moreover, the internalization of nSA-Z38 was inhibited following treatment with NaN3 or incubation at low temperature (0 °C), suggesting the involvement of endocytosis. To investigate the endocytosis pathway of nSA-Z38, we co-incubate the cells with different endocytosis inhibitors. Intriguingly, only cytochalasin D induced a slight decrease in the internalized nSA-Z38 level (Supplementary Fig. 5c).
To understand the endocytosis mechanism of nSA-Z38, we used pHrodo Red as a tracer to monitor the nanoparticle endocytosis. pHrodo Red demonstrates minimal extracellular fluorescence but displays red fluorescence specifically within endosomes/lysosomes (Supplementary Fig. 5d). After conjugation with pHrodo, the nSA-Z38 nanocapsules were exposed to HeLa or Jurkat cells. The cells had been pre-treated with a variety of endocytosis inhibitors to analyze changes in endocytosis using FACS (Supplementary Fig. 5e–h). The macropinocytosis inhibitor, cytochalasin, notably reduced the entry of Z38 into both HeLa and Jurkat cells, indicating micropinocytosis is the major pathway of Z38 endocytosis. In contrast, clathrin-mediated endocytosis (chlorpromazine, sucrose, and chloroquine) only had some contribution in HeLa cells but not Jurkat cells. The insensitivity to the methyl-β-cyclodextrin (M-β-CD) inhibitor indicated that Z38 did not enter cells via caveolae-mediated endocytosis (Supplementary Fig. 5e–h). Collectively, our findings indicated macropinocytosis is the major pathway of Z38 endocytosis.
We further explored the interaction of nSA-Z38 with blood cells. nSA-Z38 exhibited dose-dependent binding to mouse red blood cells (RBCs) (Fig. 3g). RBCs lack endocytosis mechanism39. Consistently, we observed no nSA-Z38 internalization in RBCs. The association kinetics of nSA-Z38 with RBCs were rapid, reaching a plateau within 30 min (Supplementary Fig. 5i). Next, we detected the interaction of nSA-Z38 with mouse peripheral blood mononuclear cells (PBMCs) (Fig. 3h). Similar to Jurkat, PBMCs showed strong binding to nSA-Z38 and moderate endocytosis. Taken together, nSA-Z38 strongly interacted with RBCs and PBMCs and can be internalized by PBMCs.
In vivo nanoparticle screening identified organ-specific nanocapsules
Once we demonstrated that barcode-aided screening can be used to identify cell-adhesive nanoparticles, we next investigated whether the nanoparticle library could be used to screen for organ-specific nanoparticles in vivo. Twenty-four hours after the pooled barcoded nanoparticle libraries Z and F were intravenously administrated to mice, the main organs (heart, liver, spleen, lung, and kidney) were dissected and lysed. DNA barcodes from the tissue lysate were extracted and amplified. Agarose gel electrophoresis indicates the successful extraction of the barcodes (Supplementary Fig. 6a). Next-generation sequencing was performed to measure the abundance of each barcode. The enrichment factor of each DNA barcode was calculated (Fig. 4a, b), with the top enriched nanocapsules identified and listed in Fig. 4c and Supplementary Fig. 6b. Among the top-enriched nanocapsules, nSA-Z95 and nSA-Z93 did not exhibit good organ specificity in sequencing results. Consequently, we selected the top two nanocapsules for the kidney (nSA-Z20 and nSA-Z40), the top two for the lung (nSA-Z31 and nSA-Z91), and the top two liver-accumulated nanocapsules (nSA-Z38 and nSA-Z69) for further validation. After individual nanocapsule preparation, electrophoresis and DLS were performed. The SA nanocapsules exhibited expected surface charges and size distributions (Supplementary Fig. 7a). Upon injection into mice, fluorescent imaging revealed high kidney enrichment for nSA-Z20 and nSA-Z40, lung accumulation for nSA-Z91 despite wide distribution in the liver, spleen, and kidney, and selective liver accumulation for nSA-Z38 and nSA-Z69 (Fig. 4d). Similar validation with nanocapsules from Library F (nSA-F18, nSA-F69, nSA-F77, nSA-F80, and nSA-F83, Supplementary Fig. 7b) demonstrated consistent biodistribution with sequencing data (Supplementary Fig. 7c). Overall, in vivo screening enables the identification of organ-specific nanocarriers for protein delivery.

a, b The DNA barcode enrichment factors in the heart, liver, spleen, lung, and kidney from mice 24 h after the tail vein injection of the nanocapsule combination libraries (n = 2). c The enrichment factors of the Top 5 enriched nanoparticles in each organ from Library Z. Data are presented as mean with individual data points. n = 2. The n represents biologically independent replicates. d Representative ex vivo fluorescent images of major organs from mice 24 h after tail-vein injection with nSA-Z38, nSA-Z69, nSA-Z31, nSA-Z91, nSA-Z20, nSA-Z40, and streptavidin (SA) (100 μL, 8.6 μM) labeled with biotin-Cy5. Source data are provided as a Source Data file.
The kidney accumulation potentials of nanocapsules are influenced by the polymer composition rather than size or surface charge
Due to their exceptional kidney accumulation, carriers Z20 and Z40 were selected for further investigation in kidney-targeted protein delivery. Initially, we replaced SA in the nanocapsules with bovine serum albumin (BSA) to synthesize BSA nanocapsules (nBSA) using comparable protocols and feed monomer ratio (Fig. 5a). Subsequently, the nBSAs were labeled with Alexa Fluor 647 (AF647) for fluorescent imaging. The MALDI-TOF results revealed that one BSA protein was modified with an average of 4.28 EITC and 1.00 AF647 dye (Supplementary Fig. 8a–c). The kidney distribution patterns of nBSA-Z20 and nBSA-Z40 closely resembled those of nSA-Z20 and nSA-Z40 (Fig. 5b), suggesting the nanocarrier identified from the nSA libraries could be readily applied for the delivery of other proteins.

a The monomers and feed ratios for the syntheses of nBSA-Z20 and nBSA-Z40. b The fluorescence images of major organs 24 h after the injection of nBSA-Z20 or nBSA-Z40 labeled with Alexa Fluor 647 (AF647). c Fluorescence microscopy images of kidney tissue co-stained with anti-ITGA8 antibody after nBSA-Z20 and nBSA-Z40 injection. This experiment was repeated independently three times with similar results. Scale bar = 50 μm. White arrows were used for colocalization analysis (d, e). d, e The colocalization analyses of mesangial cells (highly expressed ITGA8) and nBSA-Z20 (d) or nBSA-Z40 (e) (fluorescence images were shown in Fig. 5c). f, g Biodistribution of nNluc-Z20 (f) and nNluc-Z40 (g) tested by bioluminescence. Data are presented as mean ± s.d. and were analyzed by one-way ANOVA with Tukey’s multiple comparisons test. n = 3. h The integrated radiance, sizes, and ζ potentials of the major organs 24 h after the injection of AF647-labeled BSA, nBSA-Z20, nBSA-Z40, nBSA-Z30, nBSA-P1N4, nBSA-Z23, nBSA-Z39, or nBSA-N1P3. Sizes and ζ potentials are presented as mean ± s.d. n = 3. The n represents biologically independent replicates (f–h). Source data are provided as a Source Data file.
Fluorescence microscope images showed effective accumulation of both nBSA-Z20 and nBSA-Z40 in the kidney (Supplementary Fig. 8d–g), primarily localized in the glomeruli (Supplementary Fig. 8h). We then co-stained the kidney tissue section with an antibody against ITGA8, a protein highly expressed on glomerular mesangial cells (Fig. 5c). The results showed co-localization of nBSA-Z20 and nBSA-Z40 with ITGA8, further confirming their targeting ability to the glomeruli. Colocalization analysis revealed a strong association between nBSA-Z20 or nBSA-Z40 and mesangial cells (Fig. 5d, e), indicating their potential for targeted protein delivery to these cells.
We further quantify the biodistribution of Z20 and Z40 utilizing NanoLuciferase (Nluc) as the core protein. We synthesized nNluc-Z20 and nNluc-Z40 with the same monomer ratio employed for nSA-Z20 and nSA-Z40. nNluc-Z30, Nluc protein, and PBS were served as control groups. Nluc nanocapsules or Nluc protein (26 μM, 100 μL) were intravenously injected into mice (n = 3). After a 24-hour post-injection period, mice were anesthetized with avertin and perfused with 50 mL of 0.9% NaCl through the heart to eliminate blood from the systemic circulation. The heart, liver, spleen, lung, and kidney were harvested, segmented, and weighed. 0.1 g of each tissue was homogenized with a low-temperature grinder until completely disrupted. Through bioluminescence assays, it was observed that the Nluc concentrations were significantly higher in the kidney for nNluc-Z20 and nNluc-Z40 when contrasted with other organs (Fig. 5f, g). In contrast, control nanocapsules nNluc-Z30 and unmodified Nluc demonstrated equivalent Nluc levels in both the kidney and liver (Supplementary Fig. 8i–k).
To investigate the primary compositional factors influencing the renal accumulation of Z20 and Z40 nanocapsules, a set of control nanocapsules (nBSA-Z30, nBSA-P1N4, nBSA-Z23, nBSA-Z39, nBSA-N1P3) was synthesized for comparative analysis with nBSA-Z20 and nBSA-Z40. Once the nanocapsules were prepared, labeled with AF647, and purified, they were introduced into mice via tail vein injection. Fluorescence imaging was performed to visualize the nanocapsules accumulated in the major organs of these mice (Fig. 5h and Supplementary Fig. 8l). nBSA-Z20 was synthesized by copolymerization of acrylamide (AAM) and N-(3-aminopropyl) methacrylamide (APM). When (3-methacrylamidopropyl)trimethylammonium chloride (MPTA) was used to substitute APM in the polymerization, we obtained nBSA-Z30, which showed significantly decreased kidney accumulation. On the other hand, when AAM was replaced by either an anionic monomer (2-carboxyethyl acrylate, CA) or a zwitterionic monomer (2-methacryloyloxyethyl phosphorylcholine, MPC), the nanocapsules still exhibited high kidney accumulation, as illustrated by nBSA-P1N4 and nBSA-Z23. These observations indicate that the APM constituent in the Z20 carrier is critical for nanocapsule kidney accumulation. For the nBSA-Z40 system, two control nanocapsules were synthesized, one with 2-(Dimethylamino) ethyl methacrylate (DMAEMA) replacing the cationic monomer MPTA, and the other with 2-Methyl-2-(acryloylamino)propane-1-sulfonic acid sodium salt (AMPS) substituting the anionic monomer CA. Interestingly, both control nanocapsules showed decreased kidney accumulation, indicating that CA and MPTA were both essential for kidney accumulation. Together, we demonstrated that the surface groups had a great impact on the kidney accumulation of nanocapsules.
Once we had determined the compositional determinants of kidney accumulation, we next studied whether the kidney accumulation was correlated with the size or the ζ potential of nanocapsules. The sizes of nBSA-Z20, nBSA-Z40, and the control nanocapsules (nBSA-Z30, nBSA-P1N4, nBSA-Z23, nBSA-Z39, nBSA-N1P3) ranged from 18 nm to 50 nm, surpassing the size of BSA (Fig. 5h). The absence of a noticeable link between size and kidney accumulation implies that differences in size are not the primary factor influencing the kidney enrichment of these nanocapsules. The ζ potentials of the nanocapsules ranged from − 2 mV to 10 mV, a considerable increase compared to native BSA. Nonetheless, the absence of a discernable relationship between ζ potential and kidney accumulation implies that ζ potential does not play a pivotal role in determining the kidney accumulation of these nanocapsules. While the particles showed differences in sizes and ζ potentials, these variances were constrained within the range of 18 nm to 50 nm and − 2 mV to 10 mV. Hence, the surface groups significantly influence the kidney accumulation of nanocapsules, despite variations in size and surface charge.
Delivery of catalase to kidney effectively alleviated cisplatin-induced acute kidney injury
We then assessed Z40 as a carrier for targeted protein drug delivery to the kidney, focusing on addressing cisplatin-induced acute kidney injury (AKI), a condition lacking effective treatment and posing a significant public health challenge40. AKI is linked to the overproduction of reactive oxygen species (ROS), leading to apoptosis, necroptosis, inflammation, and kidney damage41,42,43. To scavenge ROS, we selected the antioxidant enzyme catalase (CAT) as a potential therapeutic protein for AKI treatment. The kidney-targeting catalase (nCAT-Z40) was synthesized, with CAT serving as the core protein encapsulated within MPTA-CA co-polymer (Supplementary Fig. 9a). Considering the catalase has a high molecular weight, we increased the monomer-to-protein molar ratio to ensure complete protein encapsulation (Supplementary Table 2). Dynamic light scattering and ζ potential measurements demonstrated the successful construction of the nCAT-Z40 nanocapsule (Supplementary Fig. 9b, c). In addition, the resulting nCAT-Z40 exhibited comparable enzymatic activity as catalase (Supplementary Fig. 9d), indicating the mild polymerization procedure did not reduce catalase activity.
We then validated the biodistribution of nCAT-Z40. While catalase is primarily distributed in the liver, nCAT-Z40 showed efficient accumulation in kidneys, consistent with nSA-Z40 and nBSA-Z40 (Fig. 6a). The kidney enrichment was also evidenced by both the comparative analysis with other organs (Supplementary Fig. 9e) and the histological examination of tissue samples (Supplementary Fig. 9f). Further examination of the microscopic images revealed that nCAT-Z40 accumulated in the glomeruli (Fig. 6b and Supplementary Fig. 9f). CD31 staining indicated the presence of nCAT-Z40 in the interstitium between peritubular capillaries and renal tubules, in addition to its accumulation in the glomeruli (Supplementary Fig. 9g). Following nCAT-Z40 injection, examination via hematoxylin and eosin (H&E) staining showed no apparent morphological variations in the principal organs (heart, liver, spleen, lung, and kidney), indicating low toxicity (Supplementary Fig. 9f).

a Representative ex vivo fluorescence images of major organs from mice 24 h after tail-vein injection of nCAT-Z40 and catalase (CAT) labeled with Alexa Fluor 647. b Immunofluorescence images of kidney sections 24 h after the mice were injected with nCAT-Z40 and CAT labeled with Alexa Fluor 647. The tissue sections were stained for cell nuclei (blue) and nephrin (green). This experiment was repeated independently three times with similar results. Scale bar = 10 µm. c The dosing scheme for cisplatin-induced acute kidney injury (AKI) model and nCAT-Z40 treatment (Created in BioRender. Wang, L. (2025) https://BioRender.com/a96u763). d Body weights of mice on Day 3 in the cisplatin-induced AKI model. e Blood serum levels of blood urea nitrogen (BUN) in healthy mice and AKI mice treated with PBS, CAT, or nCAT-Z40. f Paller score of kidney tissue in healthy mice and AKI mice treated with PBS, CAT, or nCAT-Z40. g Dihydroethidium (DHE) fluorescence intensities in kidney sections on Day 3 of PBS, CAT, or nCAT-Z40 treatment of cisplatin-induced AKI. h Fluorescence images of DAPI and DHE-stained kidney tissues from healthy mice and AKI mice treated with PBS, CAT, or nCAT-Z40. This experiment was repeated independently two times with similar results. Scale bar = 20 μm. d–g Data are presented as mean ± s.d. and were analyzed by one-way ANOVA with Tukey’s multiple comparisons test. n = 5. The n represents biologically independent replicates (d–g). Source data are provided as a Source Data file.
We then assessed the therapeutic efficacy of nCAT-Z40 in a cisplatin-induced acute kidney injury model. Mice received pre-treatment of cisplatin (20 mg/kg s.c.) 2 h prior to the administration of nCAT-Z40, CAT, or PBS. The mice were euthanized 72 h after the cisplatin treatment (Fig. 6c). Cisplatin-treated mice had increased blood urea nitrogen (BUN) and serum creatinine levels, as well as elevated ROS level, indicating its nephrotoxicity (Fig. 6d–h and supplementary Fig. 10a). The nCAT-Z40 treatment mitigated the decrease in body weight, suggesting reduced nephrotoxicity (Fig. 6d). In addition, cisplatin-induced BUN and creatinine elevation were rescued by nCAT-Z40 treatment (Fig. 6e and supplementary Fig. 10a). Periodic acid-Schiff (PAS) and H&E staining of the kidney revealed extensive injury in the cisplatin group, while nCAT-Z40 treatment showed alleviated necrosis and preserved brush borders in most proximal tubules (supplementary Fig. 10b). Following Paller’s method44,45, the renal tubular injury score was analyzed. Compared with the PBS and CAT, nCAT-Z40 more effectively alleviated the cisplatin-induced kidney injury (Fig. 6f). As ROS production was reported to be the key mediator of cisplatin-induced nephrotoxicity, we then performed Dihydroethidium (DHE) staining of kidney tissues to quantify ROS. Results revealed a substantial increase in ROS production in the cisplatin-treated group, while the nCAT-Z40 treatment alleviated ROS increase caused by cisplatin (Fig. 6g, h). Therefore, the delivery of catalase to the kidney reduced the ROS generation in cisplatin-induced acute kidney injury, thereby decreasing the cisplatin-associated nephrotoxicity.
Discussion
In conclusion, our study pursued a high-throughput screening approach using barcoded DNA technology to discover organ-specific protein delivery nanocarriers. Through the utilization of diversified monomer combinations in an in situ polymerization process, two nanocarrier libraries were established, giving rise to nanocapsules with a broad spectrum of surface properties. By conducting in vitro and in vivo screenings, we have demonstrated that DNA-barcoded screening facilitates the identification of both cell-adhesive and organ-specific nanocapsules. Further investigations into the kidney-specific nanocapsules indicated that the polymer composition, rather than size or surface charge, significantly influenced the accumulation of nanocapsules in the kidneys. The kidney-enriched nanocapsule was then successfully employed for the delivery of catalase, demonstrating its therapeutic potential in alleviating cisplatin-induced acute kidney injury. Overall, our findings highlight the significance of surface engineering in tailoring nanocarriers for precise control over biodistribution, representing a significant step forward in the quest for effective and safe protein delivery.
To further extend our work in clinical applications, several key challenges need to be addressed in the future. Firstly, although photocatalysis faces some challenges in scalability in the traditional reactors, current progress in continuous flow reactors largely tackled this challenge. In addition, continuous flow reactors have shown to be more efficient and cost-effective compared to batch reactors, making them a promising option for large-scale production of nanocapsules. Secondly, large animal studies should be conducted to explore whether the biodistribution and therapeutic efficacy of the kidney-enriched nanocapsules are consistent with the results obtained from small animal models. Finally, the potential immunogenicity of the nanocapsules and their long-term safety profile also need to be thoroughly evaluated before clinical translation. Overall, our study provides a promising platform for the development of targeted protein delivery systems with potential applications in a variety of diseases and disorders.
From our structure-activity studies, we found that nanocapsules containing APM or CA/MPTA could accumulate in the kidney. In addition, the correlation between kidney accumulation and size/zeta potential is weak. We speculate that protein corona composition around the nanocapsules may play an important role in determining the nanoparticle-organ interaction. Previous evidence has shown that the protein corona formed on nanoparticles in biological fluids can greatly influence their pharmacokinetics and biodistribution46,47,48. Therefore, further investigation into the protein corona composition and its effects on the biodistribution of our nanocapsules is necessary for a better understanding of their in vivo behavior and potential clinical applications.
Methods
Ethical statement
All animal protocols used in this study are approved by the IACUC (Institutional Animal Care and Use Committee) of Tsinghua University (Protocol # 17-DJJ1) and performed in accordance with the guidelines of the IACUC. The laboratory animal facility of Tsinghua University has been accredited by AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care International).
Materials
Monomers including acrylic acid (AA; 147230), acrylamide (AAM; A9099), 2-Hydroxyethyl acrylate(HA; 292818), methyl methacrylate (MMA; M55909), 2-Methacryloyloxyethyl phosphorylcholine(MPC; 730114), N-[Tris(hydroxymethyl)methyl]acrylamide(NTA; 364959), Poly(ethylene glycol) methyl ether acrylate(PEG; 454990), N-(3-Aminopropyl)methacrylamide hydrochloride(APM; 731099), 2-Methyl-2-(acryloylamino)propane-1-sulfonic acid sodium salt(AMPS; 655821), (3-methacrylamidopropyl)trimethylammonium chloride(MPTA; 280658), 3-(Acrylamido) phenylboronic acid (APB; 71465), glycosyloxyethl methacrylate solution(GEMA; 659576), 2-N-Morpholinoethyl methacrylate(MEMA; 729833), 2-(Dimethylamino) ethyl methacrylate (DMAEMA; 234907), 2-Carboxyethyl acrylate (CA; 552348) and crosslinker N,N-methylene bisacrylamide(BIS; M1533) were purchased from Sigma-Aldrich. 3-[Dimethyl-[2-(2-methylprop-2-enoyloxy)ethyl]azaniumyl]propane-1-sulfonate (SBMA; A011473) was purchased from Energy Chemical. 1-Vinyl-2-pyrrolidinone (NVP; 177747) was purchased from J&K Scientific. 1-Vinylimidazole (VDL; V820454) was purchased from Macklin Biochemical Technology Co., Ltd (Shanghai, China). Bovine serum albumin (BSA; 9048-46-8) was purchased from Solarbio Life Science. Catalase (CAT; E3289) was purchased from Sigma-Aldrich. Roswell Park Memorial Institute 1640(RIPM1640; C11875093) and Dulbecco’s modified Eagle’s medium (DMEM; C11995073) were purchased from Gibco. Fetal bovine serum (FBS; 900-108) was obtained from Gemini. Recombinant Anti-CD31 antibody (Mouse mAb) (GB15063-100) and Anti-Nephrin Mouse mAb (GB12343-100) was obtained from Wuhan Servicebio Technology CO., LTD. Cell dishes and cell culture plates were obtained from NEST.
Animals and cells
Female C57BL/6 mice (6–8 weeks) were purchased from Vital River Laboratory Animal Center (Beijing, China). The mice were kept under SPF conditions in the animal facilities at Tsinghua University that are regularly checked for standard pathogens per general husbandry protocols. The mice were housed at Tsinghua University Animal Facilities, where they were kept under specific pathogen-free conditions with a 12 h light/dark cycle. They had access to sterile pellet food and water ad libitum and were maintained at 22–26 °C, 30–70% relative humidity. For each experiment, mice were randomly allocated by blinded investigators to each group. The mice used for all experiments were naive. No drug tests and prior vaccinations were done.
DC2.4 cells (murine dendritic cell line) were gifts from Haidong Tang’s lab and cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin, and 50 μM β-Mercaptoethanol at 37 °C in 5% CO2 atmosphere. Jurkat cells were gifts from Gelin Wang’s lab and cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin at 37 °C in 5% CO2 atmosphere. Ramos cells (1101HUM-PUMC000048) and HeLa cells (1101HUM-PUMC000011) were purchased from the Cell Resource Center, Peking Union Medical College (PCRC). Ramos cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS 1% penicillin/streptomycin at 37 °C in 5% CO2 atmosphere. HeLa cells were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated FBS (Gemini) and 1% penicillin/streptomycin (Gibco) at 37 °C in 5% CO2 atmosphere.
DNA barcode synthesis and biotin labeling
The DNA barcode with modification of 5’-N3 and 3’-FAM was synthesized from Sangon Biotech Co., Ltd (Shanghai). The 5’ N3-labeled DNA barcode was then reacted with photocleavable Biotin-DBCO (Confluore, BBBD-16) in 0.5 × PBS buffer at 37 °C for 16 h. Afterward, the reaction mixture was subjected to the treatment with 10 M ammonium acetate at a 3:1 volume ratio, followed by precipitation using a 1:1 volume ratio of cold ethanol and isopropanol. The resulting precipitate was then pelleted and subsequently washed thrice with cold 75% ethanol. The molecular weight of the biotinylated DNA oligomer was characterized with ESI mass spectrometry (n = 1).
For mass spectrometry analysis, DNA oligomers were diluted to concentrations within the range of 5–10 µM, and a total of 25 µL of this solution was loaded for mass spectrometric analysis. The analysis was conducted using an LTQ XL linear ion trap mass spectrometer from Thermo Scientific, USA. The instrumental parameters were set as follows: the ionization spray voltage was set at 4.5 kV, the nebulizer temperature was set at 350 °C, and the ion mode was configured for negative ion detection. The raw mass spectrometry data were processed using Xcalibur software for data acquisition, followed by in-depth analysis with ProMass for Xcalibur software.
pKa, logD, and charge prediction
To obtain the pKa value of monomers, we employed Schrödinger’s Epik 7 for the prediction of small molecule pKa values. Initially, the structures of monomers were drawn using ChemDraw software and exported as sdf files using Chem3D. Subsequently, the files containing the monomer structures were imported into Schrödinger. LigPrep was then employed to prepare the structures, focusing on generating stereoisomers and performing energy minimization. Epik 7 panel was then utilized to predict pKa values within specified pH ranges (pH 7.4 ± 1.0). The resultant pKa data were exported individually for further analysis. And the logD of the monomers under the condition of pH 7.4 was gained from the database of the ChemSpider website (https://www.chemspider.com/). For the monomers that can dissociate protons, the charge of the monomer under the condition of pH 7.4 was calculated from the following formula (1). While monomers can absorb protons, the charge of the monomer under the condition of pH 7.4 was calculated from the following formula (2).
For the monomer APMS, SMP, AA, CA, AAM, HA, NTA, APB, GEMA:
For the monomer APM, DMAEMA, MPC, SBMA, NVP, VDL, and MEMA:
There is no corresponding pKa and charge value for compounds in the form of salts in the monomer library, such as MPTA. The monomer MMA does not possess a pKa value.
Synthesis and characterization of nanoparticles
SA, BSA, catalase (CAT), or NanoLuciferase (Nluc) was first conjugated with eosin-5-isothiocyanate (Aladdin, E131264), a type II photoinitiator at 1:20 equivalent in 50 mM Na2CO3-NaHCO3 buffer (pH 8.5) for 3 h at room temperature. After conjugation, the reaction solution was dialyzed against PBS with a 10 kDa molecular weight cut-off (Thermo Scientific, 88245) overnight or ultrafiltrated (10 kDa cut-off) to remove residual small molecules, yielding SA-eosin, CAT-eosin, Nluc-eosin, or BSA-eosin. Monomers and crosslinker BIS were prepared in 20% (w/v) stock freshly. The nanoparticle synthesis reaction was carried out in PBS buffer containing 0.5 mg/mL SA-eosin, BSA-eosin, CAT-eosin, or Nluc-eosin and defined amounts of monomers and crosslinkers (Supplementary Tables 1 and 2). TEMED (N, N, N’, N’-Tetramethylethylenediamine) (TEMED: protein = 160000:1, molar ratio) was used as a polymerization catalyst. The polymerization reaction was initiated with a 530 nm LED lamp (12 W/cm2) for 5 min. Subsequently, the polymerization solution was then dialyzed against PBS to remove the remaining monomers.
For Agarose gel electrophoresis, 5 μg nanoparticles or protein were loaded to 1% agarose gel, and electrophoresis was performed for 20 min with a voltage of 160 V. The gel can be visualized with Typhoon (General Electric Company) with 488 nm as the excitation wavelength.
For nanoparticles’ size and ζ potential characterizations, dynamic light scattering (DLS) was performed using ZetaSizer (Malvern Instruments). Briefly, nanoparticles were first filtered through a 0.22 μm filter. Size distribution was measured using disposable solvent-resistant micro cuvettes (Malvern, ZEN0040) and ζ potential was measured using disposable folded capillary cells (Malvern, DTS1070) at 25 °C.
The purification efficiency of FAM-labeled barcodes
To evaluate the barcode purification efficiency using anti-FITC beads, DNA-FAM (AGACGATGCACGAGCCGTCACGACCGGCTGAAATACGATCGAGTCGAGTCA with 3’ FAM modification) was added to HEK-293T lysate (2 mg/mL, extract using Cytoplasmic Protein Extraction Kit, Beyotime, P0027) containing 0.5 mg/mL heparin sodium salt (Solarbio, H8060) with or without 0.1% triton X-100. The final concentrations of DAN-FAM in the mixtures were 100 pM, 1 nM, or 10 nM. After 20 min shaking, 20 μL anti-FITC magnetic beads (Biomagbeads, 10 mg/mL) were added to the mixture for additional 3 h shaking. The beads were then collected with magnets (Thermo, DynaMa-2), washed thrice with PBS, and then eluted with 100 μL elution buffer (5.5 M urea, 1 M NaCl, 1% Triton X-100, pH 7). The eluent was then purified with Oligo Clean & Concentrator (Zymo Research, D4060) to yield the DNA barcodes. The concentration of recovered DNA-FAM was quantified using quantified using real-time quantitative PCR (qPCR). The forward and reverse primers were AGACGATGCACGAGC and TGACTCGACTCGATCGTA, respectively.
To test the total enrichment efficiency on nSA-Z28-PC-DNA after UV cleavage and anti-FITC beads enrichment, 200 μL nSA-Z28-PC-DNA (40 nM) in PBS buffer or 293 T lysate under UV light exposure for 5 min and the enriched using the above method and quantified using qPCR to calculate the enrichment efficiency.
DNA barcode extraction and purification
To enrich the DNA barcode from cell or tissue lysate, a 10 mg/mL solution of heparin sodium was added to the cell lysate or tissue lysate (1/20, v/v), and it was shaken completely for 1 h at room temperature. Subsequently, UV light (150 mW/cm2, 365 nm) was irradiated for 5 min, and then 1/10 (v/v) of anti-FITC magnetic beads (Biomagbeads, 10 mg/mL) were added and allowed to bind at room temperature for 3 h. The beads were then enriched using a magnetic stand (Thermo, DynaMa-2) and washed twice with a PBS solution. Finally, 100 μL of elution buffer (5.5 M urea, 1 M NaCl, 1% Triton X-100, pH 7) was added to elute the DNA barcode. The eluted DNA barcode was purified using Oligo Clean & Concentrator (Zymo Research, D4060).
DNA Barcode amplification and next-generation sequencing of DNA barcodes
The adapters for next-generation sequencing were added to the barcodes during PCR amplification. The PCR reactions were prepared as follows: 25 μL Q5 High-Fidelity 2 × Master Mix (NEB), the 2 μL extracted barcodes; 2.5 μL forward primer (AATGATACGGCGACCACCGAGATCTACAC- I7, 10 μM) and reverse primer (I5-ACACTCTTTCCCTACGACGACGCTCTTCCGATCT-AAATCGATGTGGTCAGGAAG, 10 μM) were mixed, where index code I5 and I7 are 8-base sequences used to distinguish between different template sources (Supplementary Table 3). DEPC water was added to make to total volume of 18 μL. The PCR reaction was conducted under the following conditions: initial denaturation at 98 °C for 10 s, followed by annealing at 63 °C for 30 s, and extension at 72 °C for 1 min, for a total of 20 cycles. Subsequently, the amplified DNA was obtained at 72 °C for 2 min, analyzed through agarose gel electrophoresis, and purified using the Zymoclean Gel DNA Recovery Kit (Zymo Research, D4007). The amplified and purified DNA barcodes with unique index codes I5 and I7 to distinguish sample sources were pooled together and then sequenced using the NovaSeq PE 150 system to generate 1 Gb reads for each sample.
The sequencing results were processed by utilizing the central 10 base pairs of DNA barcodes extracted from the sequences to differentiate between nanocapsules. Subsequently, the barcodes were counted49. The original nanocapsule libraries were also extracted, amplified, and sequenced with the same procedure as described above, serving as the baseline. The enrichment factor was calculated as the ratio of barcode percentages after and before the screening (with Z01 in the heart as an example):
The percentage of a nanoparticle in library was calculated using the following formula (4), with Nanoparticle Z01 from library Z as an example).
Cell and tissue lysis
In a 6-well plate, Ramos, Jurkat, and DC2.4 cells were seeded at a density of 1.5 × 106 cells/mL, while HeLa cells were seeded at 5 × 105 cells/mL one night before the experiment. On the day of the experiment, library Z was added to the cell culture to reach a final concentration of 215 nM, and the cells were incubated for 24 h at 37 °C. After that, the cells were collected at 300 × g for 5 min and washed thrice using PBS. HeLa cells were subjected to trypsin digestion before collection. Subsequently, the cell pellet was resuspended in 200 μL hypotonic buffer (10 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, 0.1% Triton X-100, 2 mg/mL ABESF, pH 7.9) and incubated on ice for 15 min. Finally, the mixture was centrifuged for 5 min at 16,000 × g at 4 °C, and the supernatant was collected.
Mice (C57BL/6 J, 6–8 weeks) were injected with nanoparticle library Z or F (100 μL, 8.6 μΜ) via the tail vein. 24 h after the injection, mice were anesthetized with avertin and subsequently perfused with 50 mL of 0.9% NaCl through the heart to remove blood from the body. Subsequently, the main organs, including the heart, liver, spleen, lung, and kidney, were dissected and quick-frozen in liquid nitrogen to prevent degradation. The tissue was disrupted using a CryoPREP Tissue Disruption Systems (Covaris), followed by the addition of hypotonic buffer (10 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, pH 7.9) to resuspend the tissue. The suspension was further homogenized by ultrasound (S220 Focused-ultrasonicator, Covaris) to obtain the final tissue lysate.
Flow cytometric analysis
In a 24-well plate, Ramos, Jurkat, HeLa, and DC2.4 cells were seeded at a density of 106 cells/mL one night before the experiment. On the day of the experiment, nanoparticles (nSA-Z01, nSA-Z28, nSA-Z38, nSA-Z40, nSA-Z91, and nSA-P3N3) were added to the medium to a final concentration of 430 nM to allow a 24-hour incubation at 37 °C. After that, the cells were collected by centrifugation at 300 × g for 5 min and washed thrice using PBS. The cells were resuspended and analyzed with a BD LSRFortessa Flow Cytometer. The FITC channel was used to analyze the fluorescence from the eosin on the nanocapsules. Data were analyzed with FlowJo Software (Version 10).
Endocytosis inhibition
Jurkat or HeLa cells were pre-treated with inhibitors targeting endocytic pathways at specified concentrations for 30 min at 37 °C. The inhibitors used were sucrose (450 μM), chloroquine (50 μM), methyl-β-cyclodextrin (M-β-CD, 10 μg/mL), cytochalasin D (5 μM), amiloride (400 μM), and Sodium azide (NaN3, 10 mM). For the 4 °C control group, cells were pre-incubated under 4 °C for 1 h. Subsequently, nSA-Z38 or nSA-Z38 conjugated with pHrodo Red succinimidyl ester (Invitrogen, P36600, 4 eq) was added to the cells and further incubated at 37 °C for 1 h. The cells were then rinsed twice with pre-chilled PBS and placed on ice before being analyzed using flow cytometry (BD LSRFortessa) to quantify the cellular uptake of nanocapsules. To differentiate internalized nanocapsules from those remaining on the cell surface, cell surface fluorescence was quenched with a 0.1% (w/v) trypan blue solution, and the fluorescence signal was measured using flow cytometry to determine the internalized amount. The surface-bound nanocapsules were calculated by subtracting the internalized amount from the total amount. FlowJo software was utilized for processing and analyzing the data.
Confocal microscopy
HeLa cells were seeded into a 4-chamber confocal dish (In Vitro Scientific, D35C4-20-1.5-N) precoated with poly-L-lysine solution (Sigma-Aldrich, P4707) and then incubated overnight at 37 °C to allow cell adhesion. Nanoparticle Z38 was added into the HeLa cell to a final concentration of 86 nM. Upon incubation at 37 °C for varying durations, the cells were subjected to three rounds of cold PBS washing. Subsequent staining involved LysoTracker Red (Invitrogen, L7528, 1:10,000 dilution) for 1 h and Hoechst 33342 (Sigma-Aldrich, B2261, 5 μg/mL) for 15 min at 37 °C. Following PBS rinsing, imaging was performed using a confocal microscope (Zeiss 880) with a UPLSAPO 63 × oil-immersion objective lens (Olympus). Colocalization analysis was conducted with ZEN software (version 3.4).
RBC and PBMC isolation
Mice aged 6–8 weeks (C57BL/6 J) were anesthetized for blood collection into EDTA-coated tubes. The blood was then diluted with 1640 medium on ice and combined with lymphocyte isolate LSM (GE, 17-1440-02) along the tube wall. Centrifugation at 600 × g for 30 min with zero acceleration yielded red blood cells at the bottom and white blood cells at the interface. The cells were carefully collected and washed twice with PBS to yield RBCs and PBMCs for further analysis.
Biodistribution of nanoparticles
The nanocapsules were first fluorescently labeled for biodistribution studies. nSAs were labeled with Cy5 by mixing nSA and Biotin-Cy5 (AAT Bioquest, Cat. # 3100, 4 eq) at room temperature for 4 h. nBSA and nCAT were labeled with Alexa Fluor 647 (Invitrogen, a20006, 4 eq) at room temperature for 2 h. The labeled nanoparticles were purified by ultrafiltration (10 kDa cut-off molecular weight). Then nanoparticles were sterilized by filtering through a 0.22 μm filter before tail-vein injection into mice 100 μL 8.6 μM nSA or 100 μL 2 μM nCAT per mouse. Twenty-four hours after the injection, mice were anesthetized with avertin and subsequently perfused with 50 mL of 0.9% NaCl through the heart to remove blood from the body. Subsequently, the major organs (heart, liver, spleen, lung, and kidney) were excised and imaged on black cardboard using IVIS Spectrum (Perkin).
Fluorescent microscope images of tissue sections and fluorescence semi-quantitation
Heart, liver, spleen, lung, and kidney tissues were excised from mice 24 h after the tail-vein injection of Alexa Fluor 647-labeled nanoparticles. The tissues were then embedded in O.C.T. medium, frozen, and sliced into 8 μm cryosections. The sections were subsequently washed twice with PBS, fixed in 95% ethanol for 30 s, and counter-stained with DAPI for nuclei. Imaging was performed using a high-throughput slide scanner, AXIO Scan Z1 (Zeiss). Quantification of nanoparticle fluorescence across different tissues was performed with ZEN software.
Expression and purification of Nluc protein
Nluc protein was expressed in Escherichia coli BL21 (DE3) containing plasmid pET28a-Nluc-His6 with C-terminal His-tag. The bacteria were grown in LB medium with 100 mg/mL kanamycin at 37 °C until the bacteria optical density (OD 600) reached 1.0 when isopropyl-β-D-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM to induce protein expression. The culture was allowed to grow 16 h at 22 °C with constant shaking (220 rpm). After that, the bacteria were harvested by centrifugation (2000 × g, 4 °C, 20 min). The pellet was resuspended in 30 mL buffer A (50 mM Tris-HCl, 300 mM NaCl, 2 mM dithiothreitol, 2 mM EDTA, pH 8.0) and lysed by a high-pressure homogenizer. The cell debris and insoluble proteins were removed by centrifugation (12,000 × g, 4 °C, 30 min), followed by the addition of 1 mL Ni-NTA resin (Qiagen, Germany) into the cleared cell lysate. After a binding period of 3 h at 4 °C, the resin was loaded in a gravity column, washed with buffer A, buffer A contained 10 mM imidazole, and buffer A contained 20 mM imidazole, and then eluted with buffer A contained 250 mM imidazole. The eluted protein was buffer-exchanged into PBS by dialysis. The endotoxin in the Nluc protein was removed by using Triton X-114.
Bioluminescence of Nluc and nNluc
Nanocapsules or Nluc protein (26 μM, 100 μL) were administrated by tail-vein injection into mice (n = 3), and after 24 h after the injection, mice were anesthetized with avertin and subsequently perfused with 50 mL 0.9% NaCl through the heart to remove blood from the body. Subsequently, the major organs (heart, liver, spleen, lung, and kidney) were excised and cut to small pieces. Each organ was weighted 0.1 g tissue and added 1250 μL NP-40 Lysis Buffer containing 1 mM AEBSF and then homogenized with a low-temperature grinding machine until fully cracked. The lysate was then centrifuged by 12,000 × g for 5 min. Then, 5 μL supernatant or Nluc standard was mix with 95 μL reaction buffer (100 mM MES, 150 mM KCl, pH 6.3, 1 mM DTT, 50 nM Furimazine) to test the bioluminescence.
Cisplatin-induced acute kidney injury model
Eight-week-old male C57BL/6 J mice were intraperitoneally injected with cisplatin (HY-17394, MCE) at a dose of 20 mg/kg to induce acute kidney injury. Two hours post-cisplatin administration, various agents were administered to the mice. The PBS group received sterile PBS (100 μL), the CAT group received CAT protein (100 μL, 2 μM), and the nCAT-Z40 group received nCAT-Z40 nanoparticles (100 μL, 2 μM). After three days, the mice were euthanized. The serum and kidneys were collected for further analysis.
Immunofluorescence staining of Nephrin, ITGA8, or CD31
The frozen paraformaldehyde-fixed kidney slides were blocked in PBS with 5% BSA and 0.3% Triton X-100. The section was then labeled in a humidified chamber overnight at 4 °C with Nephrin (Wuhan Servicebio Technology Co., Ltd., GB12343-100), ITGA8 (Proteintech, 30714-1-AP, lot # 00145891) or CD31 (Wuhan Servicebio Technology Co., Ltd., GB15063-100) antibody. After three times washing with PBS, the sections were exposed to Cy3-labeled secondary antibody, and the nuclei were counterstained with DAPI. The slides were imaged using a high throughput slide scanner AXIO Scan Z1 (Zeiss).
DHE staining for ROS level in tissue
Fresh kidney tissue from mice was excised and promptly embedded in an O.C.T. medium, followed by freezing and slicing into 8 μm cryosections for DHE staining to assess superoxide levels. Specifically, the frozen tissue slides were treated with a 10 μM DHE fluorescent probe (YEASEN, 50102ES02) for 30 min at room temperature, then washed thrice with PBS to eliminate excess probe. Nuclei were counterstained with DAPI, and imaging was performed using a high-throughput slide scanner, AXIO Scan Z1 (Zeiss), to visualize superoxide.
Renal function restoration analysis and blood biochemistry analysis
Following a 3-day treatment with nCAT-Z40, CAT, and PBS, mice were euthanized, and blood was sampled via the retro-orbital veins. CRE and BUN levels in the collected blood samples were measured by Wuhan Servicebio Technology CO., LTD.
H&E, PAS staining and Paller scores
Hematoxylin and eosin (H&E) staining was conducted on paraformaldehyde-fixed tissue sections of the heart, liver, spleen, lung, and kidney by Wuhan Service Technology Co., Ltd. to assess organ-specific toxicity. Kidney tissue sections were subjected to Periodic acid-Schiff (PAS) staining to evaluate the extent of cisplatin-induced damage, following the manufacturer’s protocols (KeyGEN BioTECH, Nanjing, China). The stained sections were imaged using the Pannoramic SCAN slide scanner (3DHISTECH Ltd.). Assessment of at least eight randomly chosen fields included evaluation of tubular lysis and dilation, necrosis, cast formation, loss of brush border, glomerular atrophy, and the presence of sloughed cells. To evaluate the Paller score, five fields of kidney H&E results were randomly selected, and under high magnification, 10 renal tubular regions were randomly selected in each field of view. The average of the score of 10 tubulars regions was taken as the tissue score for this visual field. The scoring criteria were Paller’s method44. The standard is described as follow: normal renal tubules (0 point), obviously dilated renal tubules (1 point), flat or swollen cells are scored (1 point), renal brush border membrane injury (1 point), cell debris (2 points), tubular type (2 points), cell shedding and necrosis in the lumen of renal tubules but without tubular type and cell debris (1 point)45.
Statistics and reproducibility
All the experiments described in the current study were performed two or three times with similar results. The data presented in the manuscript are representative results. The data (mean ± s.d.) are plotted and analyzed using GraphPad Prism software (9.4.1). The ordinary one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test or Dunnet’s multiple comparisons test was employed statistical difference calculation. Normalization was conducted with respect to proper experimental controls as mentioned in the corresponding figure captions. Statistical significance levels are labeled in the figures. No statistical method was used to predetermine the sample size. The sample size was determined by the type of study. Typically, screenings were performed in duplicates. In vitro studies were performed in triplicates. In vivo imaging studies were performed in duplicates. Animal disease models were performed with 5 replicates. No data were excluded from the analyses. The experiments were randomized. The Investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The next-generation sequence data generated in this study have been deposited in the Sequence Read Archive (SRA) database of the National Center of Biotechnology Information (NCBI) under BioProject number PRJNA1138090. Source data are provided in this paper.
Code availability
The code used in this study is available on GitHub (https://github.com/LuyaoWang2024/Barcoded-Screening-Identifies-Nanocarriers-for-Targeted-Protein-Delivery-to-Kidney.git), and Zenodo49.
References
Dilliard, S. A. & Siegwart, D. J. Passive, active and endogenous organ-targeted lipid and polymer nanoparticles for delivery of genetic drugs. Nat. Rev. Mater. 8, 282–300 (2023).
Kim, J., Eygeris, Y., Ryals, R. C., Jozic, A. & Sahay, G. Strategies for non-viral vectors targeting organs beyond the liver. Nat. Nanotechnol. 19, 428–447 (2023).
Liu, J., Zhou, Y., Lyu, Q., Yao, X. & Wang, W. Targeted protein delivery based on stimuli-triggered nanomedicine. Exploration 4, 20230025 (2024).
Su, H., Rong, G., Li, L. & Cheng, Y. Subcellular targeting strategies for protein and peptide delivery. Adv. Drug. Deliver. Rev. 212, https://doi.org/10.1016/j.addr.2024.115387 (2024).
Cabral, H., Li, J., Miyata, K. & Kataoka, K. Controlling the biodistribution and clearance of nanomedicines. Nat. Rev. Bioeng. 2, 214–232 (2024).
Liu, J., Guo, M. & Chen, C. Nano-bio interactions: A major principle in the dynamic biological processes of nano-assemblies. Adv. Drug. Deliv. Rev. 186, 114318 (2022).
Poon, W., Kingston, B. R., Ouyang, B., Ngo, W. & Chan, W. C. W. A framework for designing delivery systems. Nat. Nanotechnol. 15, 819–829 (2020).
Macarron, R. et al. Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discov. 10, 188–195 (2011).
Mann, J. L. et al. An ultrafast insulin formulation enabled by high-throughput screening of engineered polymeric excipients. Sci. Transl. Med. 12, https://doi.org/10.1126/scitranslmed.aba6676 (2020).
Chai, Q., Shih, J., Weldon, C., Phan, S. & Jones, B. E. Development of a high-throughput solubility screening assay for use in antibody discovery. MAbs 11, 747–756 (2019).
Mitragotri, S., Burke, P. A. & Langer, R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat. Rev. Drug Discov. 13, 655–672 (2014).
Schenone, M., Dancik, V., Wagner, B. K. & Clemons, P. A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 9, 232–240 (2013).
Vincent, F. et al. Phenotypic drug discovery: recent successes, lessons learned and new directions. Nat. Rev. Drug Discov. 21, 899–914 (2022).
Mitchell, D. C. et al. A proteome-wide atlas of drug mechanism of action. Nat. Biotechnol. 41, 845–857 (2023).
Xue, L. et al. High-throughput barcoding of nanoparticles identifies cationic, degradable lipid-like materials for mRNA delivery to the lungs in female preclinical models. Nat. Commun. 15, 1884 (2024).
Dahlman, J. E. et al. Barcoded nanoparticles for high throughput in vivo discovery of targeted therapeutics. Proc. Natl. Acad. Sci. USA 114, 2060–2065 (2017).
Sago, C. D. et al. Nanoparticles that deliver RNA to bone marrow identified by in vivo directed evolution. J. Am. Chem. Soc. 140, 17095–17105 (2018).
Rhym, L. H., Manan, R. S., Koller, A., Stephanie, G. & Anderson, D. G. Peptide-encoding mRNA barcodes for the high-throughput in vivo screening of libraries of lipid nanoparticles for mRNA delivery. Nat. Biomed. Eng. 7, 901–910 (2023).
Sago, C. D. et al. High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc. Natl. Acad. Sci. USA 115, E9944–E9952 (2018).
Ebrahimi, S. B. & Samanta, D. Engineering protein-based therapeutics through structural and chemical design. Nat. Commun. 14, 2411 (2023).
Leader, B., Baca, Q. J. & Golan, D. E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 7, 21–39 (2008).
Postow, M. A., Sidlow, R. & Hellmann, M. D. Immune-rlated adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 378, 158–168 (2018).
Bernard, G. R. et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N. Engl. J. Med. 344, 699–709 (2001).
Suntharalingam, G. et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 355, 1018–1028 (2006).
An, M. R. et al. Engineered virus-like particles for transient delivery of prime editor ribonucleoprotein complexes in vivo. Nat. Biotechnol. 42, 1526–1537 (2024).
Kreitz, J. et al. Programmable protein delivery with a bacterial contractile injection system. Nature 616, 357–364 (2023).
Wei, T., Cheng, Q., Min, Y. L., Olson, E. N. & Siegwart, D. J. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 11, 3232 (2020).
Zhang, S., Shen, J., Li, D. & Cheng, Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics 11, 614–648 (2021).
Zuris, J. A. et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 33, 73–80 (2015).
Yan, M. et al. A novel intracellular protein delivery platform based on single-protein nanocapsules. Nat. Nanotechnol. 5, 48–53 (2010).
Liu, Y. et al. Biomimetic enzyme nanocomplexes and their use as antidotes and preventive measures for alcohol intoxication. Nat. Nanotechnol. 8, 187–192 (2013).
Tian, H. J. et al. Growth-factor nanocapsules that enable tunable controlled release for bone regeneration. Acs Nano 10, 7362–7369 (2016).
Jia, X. Q. et al. A Biodegradable antigen nanocapsule promotes anti-tumor immunity via the cGAS-STING pathway. Adv. Funct. Mater. 33, 2212085 (2023).
Choi, S. K. Photonanotechnology for Therapeutics and Imaging. (Elsevier, Amsterdam, 2020).
Wegner, S. V., Senturk, O. I. & Spatz, J. P. Photocleavable linker for the patterning of bioactive molecules. Sci. Rep. 5, 18309 (2015).
Stellwagen, N. C. & Stellwagen, E. Effect of the matrix on DNA electrophoretic mobility. J. Chromatogr. A. 1216, 1917–1929 (2009).
Brenner, M. K. Will T-cell therapy for cancer ever be a standard of care? Cancer Gene Ther. 19, 818–821 (2012).
Ebert, O. et al. Lymphocyte apoptosis: induction by gene transfer techniques. Gene Ther. 4, 296–302 (1997).
Gao, X., Yue, T., Tian, F., Liu, Z. & Zhang, X. Erythrocyte membrane skeleton inhibits nanoparticle endocytosis. Aip. Adv. 7, https://doi.org/10.1063/1.4985052 (2017).
Bao, Y., Yuan, Y., Chen, J. & Lin, W. Kidney disease models: tools to identify mechanisms and potential therapeutic targets. Zool. Res. 39, 72–86 (2018).
Fu, Z. et al. HIF−1 alpha-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox. Biol. 36, 101671 (2020).
Lan, T. et al. Dual-responsive curcumin-loaded nanoparticles for the treatment of cisplatin-induced acute kidney injury. Biomacromolecules 23, 5253–5266 (2022).
Zhao, M. et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 11, 1845–1863 (2021).
Paller, M. S., Neumann, T. V., Patten, M. & Knobloch, E. Reactive oxygen species and rat renal epithelial-cells during hypoxia and reoxygenation. Kidney Int. 40, 1041–1049 (1991).
Liu, W. F. et al. Caspase-1 inhibitor reduces pyroptosis induced by brain death in kidney. Front. Surg. 8, 760989 (2021).
Mahmoudi, M. et al. The protein corona from nanomedicine to environmental science. Nat. Rev. Mater. 8, 422–438 (2023).
Ngo, W. et al. Identifying cell receptors for the nanoparticle protein corona using genome screens. Nat. Chem. Biol. 18, 1023–1031 (2022).
Miao, Y. Q. et al. Regulating protein corona on nanovesicles by glycosylated polyhydroxy polymer modification for efficient drug delivery. Nat. Commun. 15, 1159 (2024).
Wang, L. et al. Code used in the Study: Barcoded Screening Identifies Nanocarriers for Targeted Protein Delivery to Kidney. Zenodo. https://doi.org/10.5281/zenodo.14565684 (2024).
Acknowledgements
This work was supported by the National Key R&D Program of China (2017YFA0207900, J.D.) and the Tsinghua-Peking Joint Center for Life Sciences (CLS) Program (J.D.). The authors thank Zhenqin Wang (Tsinghua University) for assisting with the gel analysis of nSA and DNA complex and Bin Yu (Core Facility, Center of Biomedical Analysis, Tsinghua University) for technical support with flow cytometry analysis. The authors also thank Yue Sun and Jingjing Wang (Center of Biomedical Analysis, Tsinghua University) for assistance with using the slide scanner and Longfeng Zhang (Laboratory Animal Resources Center, Tsinghua University) for assistance with IVIS Spectrum.
Author information
Authors and Affiliations
Contributions
J.D. and L.W. conceived and designed the experiments. L.W. performed the experiments and analyzed the data. W.Z. performed the kidney accumulation experiments of nBSA-Z20, nBSA-Z40, nNluc-Z20, nNluc-Z40, and other selected nanocapsules. W.Z. performed the FACS experiment of the internalized nSA-Z38 modified with pHrodo and kidney tissue section staining for ITGA8. H.C., P.Z., M.W., Y.Z., Y.D., X.J., and T.L. performed the animal anatomy experiments. H. C. assisted in constructing the mouse model of cisplatin-induced acute kidney injury and drug treatment experiment design. R.Z. and Y.D. assisted with the DNA barcode modification. P.Z. assisted in the prediction of small molecule pKa values using Schrödinger’s Epik 7. H.J. assisted with the confocal experiments. L.W., W.Z., and J.D. analyzed the data. B.T. assisted in the analysis of the NGS sequence result of the in vivo screening. B.D. and Y.P. assisted in the analysis of kidney targeting results. J.D. and L.W. co-wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Yamin Li, Weiqiang Lin, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, L., Zhou, W., Chen, H. et al. Barcoded screening identifies nanocarriers for protein delivery to kidney. Nat Commun 16, 899 (2025). https://doi.org/10.1038/s41467-025-56257-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56257-3
This article is cited by
-
Spatial fluorescence barcode by transiently luminescent DNA beads
Nature Communications (2025)
