
Abstract
Medulloblastoma is the most frequent malignant primary brain tumor in children. Despite recent advances in integrated genomics, the prognosis in children with high-risk medulloblastoma remains devastating, and new tumor-specific therapeutic approaches are needed. Here, we present an atlas of naturally presented T cell antigens in medulloblastoma. We map the human leukocyte antigen (HLA)-presented peptidomes of 28 tumors and perform comparative immunopeptidome profiling against an in-house benign database. Medulloblastoma is shown to be a rich source of tumor-associated antigens, naturally presented on HLA class I and II molecules. Remarkably, most tumor-associated peptides and proteins are subgroup-specific, whereas shared presentation among all subgroups of medulloblastoma (WNT, SHH, Group 3 and Group 4) is rare. Functional testing of top-ranking novel candidate antigens demonstrates the induction of peptide-specific T cell responses, supporting their potential for T cell immunotherapy. This study is an in-depth mapping of naturally presented T cell antigens in medulloblastoma. Integration of immunopeptidomics, transcriptomics, and epigenetic data leads to the identification of a large set of actionable targets that can be further used for the translation into the clinical setting by facilitating the informed design of immunotherapeutic approaches to children with medulloblastoma.
Similar content being viewed by others
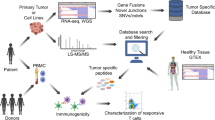


Introduction
Central nervous system (CNS) tumors remain the leading cause of cancer-related mortality in childhood1,2,3. Medulloblastoma is a highly aggressive pediatric primitive neuroectodermal tumor and the most frequent malignant CNS tumor in children—accounting for 25 % of all intracranial neoplasms4.
Over the past two decades, large-scale genomic efforts have profoundly advanced our understanding of medulloblastoma and helped disentangle the molecular basis of medulloblastoma, including biologically and clinically relevant intertumoral heterogeneity5,6. Integrating molecular subgroups into the WHO classification highlights the importance of molecular risk stratification in patients with medulloblastoma and calls for new tumor subtype-specific therapeutic approaches7,8,9. Non-specific, “one-fits-all” treatment modalities like chemotherapy and craniospinal irradiation seem not targeted and adapted enough for a biologically heterogeneous group of tumors with distinct tumor response patterns among the subgroups. Immunotherapy is a highly attractive therapy modality to optimize the targeting of tumor cells while sparing the developing, vulnerable brain tissue in children.
One prerequisite for T cell-based immunotherapy is the presentation of tumor-associated peptides by human leukocyte antigens (HLA). A major impediment in developing meaningful immunotherapeutic approaches against medulloblastoma is the knowledge gap that concerns the natural antigenic landscape of this tumor entity.
Here, we show the T cell antigen landscape of medulloblastoma. An integrative multi-omics antigen discovery pipeline, with comparative profiling against an in-house benign immunopeptidome database10, enables the identification of a set of T-cell targets exclusively presented on native tumor tissue. Top-ranking candidate peptides undergo further validation in functional T cell assays to investigate their potential for T cell-based immunotherapy.
Results
Patient characteristics
This study is based on an in-depth immunopeptidome analysis and candidate peptide selection derived from integrative multi-omics and functional immunogenicity testing (Fig. 1a). We provide a T cell antigen atlas for medulloblastoma (n = 28), including a set of novel medulloblastoma-associated antigens naturally presented on HLA class I and II molecules including in vitro immunogenicity testing for top-ranking candidates.

a Workflow outlining the three-phase antigen discovery strategy with an in-depth immunopeptidome analysis, candidate peptide selection, and functional immunogenicity testing of selected candidate peptides. Created in BioRender. Neidert, M. (2021) BioRender.com/e22b172. b A swimmer survivable plot shows each patient’s clinical course and treatment modalities; each patient is represented by one bar. GTR gross total resection, STR subtotal resection, AWD alive with disease, AWND alive with no disease. c t-SNE analysis shows DNA methylation clustering of the patient cohort (n = 28) with distinct methylation patterns according to the subgroups (WNT, SHH, Group 3, Group 4). (Used data set for the reference cohort:). d Bar plots depict the major chromosomal changes based on DNA methylation profiles for each subgroup. e HLA class I and II protein levels were assessed on medulloblastoma samples by immunohistochemistry, and nuclei were counterstained with hematoxylin; one patient of each subgroup (Pat. 4 / WNT, Pat. 9 / SHH, Pat. 18 / Group 3, Pat. 25 / Group 4) is shown exemplarily. The IHC staining was performed once. Source data are provided as a Source Data file.
We included 28 patients with a median age at diagnosis of 8 years (range: 17 months to 37 years). The female-to-male ratio was 2:3. Data on the clinical course and treatment are shown in Fig. 1b. Molecular subgroup annotation of the 28 medulloblastoma samples is shown in Supplementary Table 1. Individual patient and sample characteristics, including HLA typing and available sample mass for HLA-IP, are listed in Supplementary Table 2. Preoperative MR images are shown in Supplementary Fig. 1.
DNA methylation profiling defines the major molecular subgroups of medulloblastoma
DNA-methylation profiles were obtained from all tumor samples. Based on molecular classification using the Heidelberg Brain Tumor Methylation Classifier the 28 patient samples were classified as follows: WNT (n = 4), SHH (n = 9), Group 3 (n = 7), Group 4 (n = 8) based on the most confident classifier prediction (Fig. 1c)11. Bar plots of subgroup specific chromosomal gains and losses are depicted in Fig. 1d; CNV for each tumor sample are depicted in Supplementary Fig. 2.
Mapping the antigenic landscape of medulloblastoma yields a broad set of naturally presented HLA class I and II peptides
Molecular HLA expression is a prerequisite for immunopeptidome profiling and was assessed via immunohistochemistry. Robust membrane staining was detected for both HLA class I and II molecules and an example is shown for each molecular subgroup (Fig. 1e; Pat. 4/WNT, Pat. 9/SHH, Pat. 18/Group 3, Pat. 25/Group 4).
HLA class I- and II-bound peptides were extracted from 28 snap-frozen tumor samples. The study cohort comprised 60 distinct HLA class I allotypes covering 99.95% of the world population, whereby 92.08% of all individuals are expected to be positive for at least three allotypes (Supplementary Fig. 3). The most frequent allotypes among medulloblastoma patients were HLA-A*02:01 (39%), -A*03:01 (32%), and -C*06:02 (32%) (Supplementary Table 3).
A median of 2016 [88–5867] HLA class I peptides and 1347 [400–4164] HLA class II-presented peptides per sample were identified. HLA class I peptide eluates exhibited a purity of at least 70%. None of the samples were censored for low peptide yield or low percentage of HLA class I ligands. The total number of unique HLA class I and II peptides, as well as the purity (fraction of peptides with predicted HLA binding) of HLA class I peptide eluates, are shown for each sample in Fig. 2a. The length distribution of HLA class I peptides peaked at 9 amino acids length (Fig. 2b), whereas HLA class II-presented peptides were typically 13- to 18-mers (Fig. 2c). Except for HLA class I ligands eluted from snap frozen tissue, the correlation between sample mass and the number of yielded peptides was poor (Supplementary Fig. 4).

a HLA class I and II peptide yield of all 28 samples. Calculated purities refer to the proportion of HLA class I peptides annotated to an HLA allotype of the respective patient in silico by peptide binding prediction. b Length distribution of HLA class I and II peptides (each line represents one sample) shows that 71% of HLA class I peptides were 9-mers, whereas 67% of HLA class II-presented peptides had a length between 13 and 18 AA (c). Saturation analysis for identifying HLA class I proteins (d) and peptides (e) identified 91% and 75% of the estimated maximum attainable amount of distinct source proteins and peptides, respectively. f Comparative profiling of medulloblastoma’s HLA class I proteins versus an in-house benign database. Each bar in this waterfall plot (x-axis) represents a single source protein. In contrast, the frequency of positive HLA peptidomes is shown on the y-axis, separately for medulloblastoma (n = 28) and benign samples without testes (n = 418 covering 29 different human tissues). Source proteins detected on a maximum of one non-CNS-related tissue were designated as medulloblastoma-associated, whereby n = 77 were identified on at least two tumors (enlarged view on the left). The number of all source proteins detected is shown on the right. g Bar plot showing the set of 15 medulloblastoma-associated proteins naturally presented on 11–29% of tumors. h Comparative profiling of HLA class I peptides presented on medulloblastoma versus an in-house benign database. Peptides were designated as medulloblastoma-associated when detected on a maximum of one non-CNS-related tissue, whereby n = 1963 were identified on at least two tumors (enlarged view on the left). The number of all distinct HLA class I peptides is illustrated by the Venn diagram on the right. i The entirety of identified candidate peptides covers 98.64% of the world population. The percentage of individuals with a specific number of matching peptides is indicated by bar charts (left y-axis). The line diagram (associated with the right y-axis) shows the cumulative percentage of population coverage. Waterfall plots (F and H) were generated in three steps, 1. descending sorting by detection frequency (multiplied with −1) on normal samples, whereby a single detection on non-CNS tissues is treated equally to 0 detections, 2. descending sorting by detection frequency on tumors, 3. descending sorting by ratio of absolute detection numbers on tumors/absolute detection numbers on tumors + normal samples. As in the step before, a single detection on non-CNS tissues is treated equally to 0 detections. Source data are provided as a Source Data file.
Tumor association is defined according to the immunological pivotal level of the natural immunopeptidome
The overall poor correlation between the abundance of precursors (RNA and protein) with HLA peptides for individual genes/proteins limits in silico predictions of HLA peptides based on RNA sequencing or protein data and warrants direct analysis of immunopeptidomes for antigen discovery 12,13,14,15. Therefore, we performed an in-depth immunopeptidome analysis to identify targets by comparative profiling of patient samples in an in-house benign database comprising 30 distinct primary human organs (n = 418 HLA class I peptidome datasets including among others the HLA Ligand Atlas; www.hla-ligand-atlas.org. The term medulloblastoma-associated was assigned to peptides and corresponding source proteins that were not identified in CNS tissues (i.e. brain, cerebellum, and spinal cord) and for which a maximum of one non-CNS-related sample was positive in the benign database. As additional criterion to select targets for cancer immunotherapies, RNA expression data acquired across a large set of benign human tissues and deposited in the GTEx database (www.gtexportal.org) was reported for every candidate protein’s gene. Source proteins of detected HLA peptides with absent or low gene expression in normal tissues - defined as less than ten transcripts per million (TPM)—were considered potential candidates.
Mapping the antigenic landscape of naturally presented HLA class I peptides reveals medulloblastoma-associated antigens
HLA ligandome profiling of medulloblastoma tumor samples (n = 28) identified 9821 distinct source proteins, represented by HLA class I peptides on native tumor tissue (91% of the estimated maximum attainable amount of distinct source proteins, Fig. 2d). Although a considerable overlap between the HLA class I peptidome of medulloblastoma and the in-house benign database was observed, peptides of 77 proteins were exclusively presented on at least two medulloblastoma samples (Fig. 2f). Upon further manual curation, including quality control of underlying peptides for HLA motifs and subtracting peptides mapping to several source proteins, a set of 15 medulloblastoma-associated proteins and corresponding peptides naturally presented on 11–29% of tumors was created. Among these, SPINK8, WNT5A, and SHISA9 were the most frequent (Fig. 2g). SPINK8 was presented across SHH, Group 3, and Group 4, WNT5A was presented in WNT, SHH, and Group 4, and SHISA9 was found in Group 3 and Group 4 tumor samples. SLC22A1 was only presented in Groups 3 and 4. None of the medulloblastoma-associated HLA class I source proteins were found in all subgroups highlighting the need for molecular subgroup-specific antigen discovery. Peptide sequences and their HLA restriction, a listing of positive patient samples, and the GTEx profile of the corresponding source protein can be retrieved from the Supplementary Table. 4. Notably, none of the medulloblastoma-associated HLA class I proteins exhibited a cancer-testis antigen (CTA)-like gene expression profile.
Analysis of medulloblastoma-associated HLA class I-presented peptides yielded 32,199 distinct HLA class I ligands, representing 75% of the estimated maximum attainable coverage (Fig. 2e). Subtraction of the in-house benign database revealed 1963 medulloblastoma-associated peptides on at least two tumors (Fig. 2h). A set of 34 peptides derived from 38 proteins and presented on 18–29% of tumors was defined after manual curation. Among these, the most frequent HLA class I peptides are derived from GFAP, HNRNPK, SNX14, AGRN, DNMT3A, KIF1A, and GABRG2. Considering the different subgroups, four peptides proved to be pan-medulloblastoma targets and were presented among all subgroups. Two HLA class I peptides were exclusively part of the immunopeptidome of the WNT and SHH subgroups, whereas 2 HLA class I peptides were only presented among Group 3 and Group 4. One HLA class I peptide was presented solely in the SHH subgroup (5 samples). Peptide sequences of medulloblastoma-associated HLA class I peptides and their HLA restriction, a listing of positive patients, and the corresponding source protein are listed in Supplementary Table 5.
Combining the list of peptides derived from HLA class I-presented medulloblastoma-associated proteins (Supplementary Table 4) with that of HLA class I peptides designated as medulloblastoma-associated (Supplementary Table 5) yielded n = 66 candidate target peptides for cancer immunotherapy. Taking the HLA allotype restriction of all targets into account, these cover 98.64% of the world population (Fig. 2i), whereby an average of 14 peptides are expected to match per patient. The population coverage on a per-country basis is shown in Supplementary Fig. 5.
Mapping the HLA class II ligandome identifies medulloblastoma-associated proteins and peptides
HLA class II peptidome analyses of medulloblastoma identified 4922 distinct source proteins giving rise to HLA class II-restricted peptides (71% of the estimated maximum attainable number of distinct proteins, Fig. 3a). Despite the large overlap with benign samples, 60 proteins were exclusively presented by at least two tumors (Fig. 3c). Following manual curation of the underlying peptides for peptide length and the presence of length variants and multi-mapping to several source proteins, a set of ten medulloblastoma-associated proteins and corresponding peptides naturally presented on 11–36% of tumors was created. The most frequent were IGFBPL1, CBX4, ESCO1, SYCP3, and CCDC59 (Fig. 3d). None of the medulloblastoma-associated HLA class II proteins were identified in all subgroups. Six of ten tumor-associated proteins were presented on HLA class II in adult and childhood medulloblastomas (IGFBPL1, CBX4, ESCO1, CCDC59, PCDH20, CGGBP1), although the present study population included only two adults. Peptide sequences, a listing of positive patients, and the GTEx profile of the corresponding source protein can be retrieved from Supplementary Table 4. One of these tumor-associated HLA class II proteins (SYCP3) exhibited a CTA-like expression profile, with the related SYCP1 listed in the CTDatabase16.

Saturation analysis showed that 71% and 66% of the estimated maximum attainable amount of distinct HLA class II-presented proteins (a) and peptides (b) had been identified, respectively. c Comparative profiling of the HLA class II peptidome of medulloblastoma versus an in-house benign database revealed medulloblastoma-associated source proteins (n = 60), which were identified on at least two neoplasms (enlarged view on the left). Each bar in this waterfall plot (x-axis) represents a single source protein. In contrast, the y-axis depicts the frequency of positive immunopeptidomes for medulloblastoma (n = 28) and benign samples without testes (n = 364 covering 30 different human tissues). The Venn diagram on the right compares the overlap of HLA class II peptides between tumors and benign samples (d). A set of 10 tumor-associated proteins naturally presented on 11–36% of medulloblastomas was defined and is depicted in the bar plot. e Comparative profiling of HLA class II peptides on medulloblastoma identified n = 11,613 medulloblastoma-associated peptides, whose source proteins were subjected to hotspot analysis. The Venn diagram on the left illustrates the total number of HLA class II-restricted peptides. Waterfall plots (c,e) were generated in three steps, 1. descending sorting by detection frequency (multiplied with −1) on normal samples, whereby a single detection on non-CNS tissues is treated equally to 0 detections, 2. descending sorting by detection frequency on tumors, 3. descending sorting by ratio of absolute detection numbers on tumors/absolute detection numbers on tumors + normal samples. As in the step before, a single detection on non-CNS tissues is treated equally to 0 detections. Source data are provided as a Source Data file.
On the peptide level, 25,076 distinct HLA class II-presented peptides were eluted from medulloblastomas (n = 28), obtaining 66% of the estimated maximum attainable coverage (Fig. 3b). Given that comparative profiling cannot reflect length variants and common core sequences, we aimed at grouping peptides sharing a core sequence. Thus, after comparative profiling, all proteins represented by at least one tumor-associated HLA class II-presented peptide were subjected to a “hotspot” analysis (Fig. 3e)17. Medulloblastoma-associated HLA class II presentation hotspots were defined to have a minimum length of eight AA and to be covered by peptides identified in at least five patients, while not having matching sequences in benign samples. This generated a set of 11 proteins harboring regions uniquely presented on tumor tissue, with peptide-specific frequencies reaching up to 43% of positive immunopeptidomes. Two hotspot targets were presented within the immunopeptidome of all medulloblastoma subgroups (WNT, SHH, Group 3, and Group 4). Peptide sequences, a listing of positive patients, and the corresponding source protein are provided in Supplementary Table 6.
A comparative analysis between medulloblastoma-associated HLA class I- (n = 15) and II-represented source proteins (n = 10), as well as medulloblastoma-associated HLA class I- (n = 38 source proteins) and II-restricted peptides (n = 11 source proteins), revealed an essentially unique antigenic repertoire inherent to HLA class I and II peptidomes. Notably, the medulloblastoma-associated source proteins IGFBPL1, INSM1, and INSM2 yielded both HLA class I- and II-presented candidate target peptides. Gene expression levels in the healthy developing brain of medulloblastoma-associated targets were assessed using the “BrainSpan: Atlas of the developing brain” (https://brainspan.org/rnaseq/search/index.html). Heatmaps as an overview (Supplementary Fig. 6 a–d) show overall low gene expression of targets in the surrounding developing brain, detailed and absolute gene expression data can be obtained as a supplement (Supplementary Data 1–4).
Gene ontology and network analysis show enrichment of pan-cancer pediatric and medulloblastoma gene sets
Functional annotation based on gene ontology (GO) terms revealed distinct cellular compartments, molecular functions, and biological processes for the genes of HLA ligand source proteins presented in HLA class I compared to HLA class II (Supplementary Figs. 7 and 8). Furthermore, comparison of the collection Network of Cancer Genes18 with the underlying genes of HLA class I ligand source proteins, the two top-ranking enriched gene sets were “pan-cancer pediatric” and “medulloblastoma” (Fig. 4a; Supplementary Data 5), pointing to the biological relevance of detected candidate targets. The involved genes and the overlap between enriched terms are displayed in a Gene-Concept Network (CNET) plot (Fig. 4b).

a Bar plot of the enriched gene set (The Network of Cancer Genes) based on the gene list of all HLA class I peptides. The y-axis contains the identified gene sets that were available in the Network of Cancer Genes catalog (https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1612-0). The counts on the x-axis show the number of genes that are overlapping with the identified gene sets from the catalog. The colors are used to annotate the significance of each gene set, ranging from blue to red (p-value 0.025–0.005, respectively). The p-adjusted method of Benjamini-Hochberg was used to correct for multiple comparison with a p-value cut-off of 0.05 to determine the enriched gene set. b CNET plot of enriched genes based on the same list, however, shows the top 5 enriched gene sets (indicated with a yellow dot), genes are indicated in gray and the lines represent the presence within a gene set. c Identification of established TAAs, CTAs, and medulloblastoma-associated proteins across the present immunopeptidome dataset. (Left column) HLA class I peptides derived from 116 naturally presented TAAs and CTAs were identified, and 16 were represented by medulloblastoma-associated peptides on at least two tumors (highlighted with orange diamonds). The frequency of positive immunopeptidomes was assessed based on HLA class I peptides for tumor samples, whereby benign hits were reported independent of HLA binding probabilities of the underlying peptide identifications. (Right column) HLA class II peptides derived from 85 naturally presented TAAs and CTAs were identified, of which eight yielded tumor-associated peptides attaining a presentation frequency of ≥7% (highlighted with orange diamonds). While peptides mapping to multiple source proteins were considered to calculate the frequency of positive immunopeptidomes, these were excluded from reporting the representation by medulloblastoma-associated peptides. CTAs and TAAs exclusively identified on benign samples were not listed. Source data are provided as a Source Data file.
Established CTAs and TAAs show a high identification rate but overall low presentation frequencies
Considering a total number of 366 selected CTA and tumor-associated antigens (TAA),19,20,21, as well as 16 proteins reported to be associated with medulloblastoma (n = 15 overlapped with the general list of CTAs and TAAs), the present immunopeptidome dataset acquired from medulloblastoma, was screened for previously published tumor antigens. Of these, n = 117 and n = 84 were represented by HLA class I and HLA class II-presented peptides, respectively. Although identification rates were comparatively high, CTAs and TAAs were presented at low frequency, especially those exclusively identified on medulloblastoma. While none of the HLA class I-presented TAAs and CTAs, fulfilling the aforementioned criteria to be designated as tumor-associated antigens, were identified on more than one sample, 16 of them were represented by medulloblastoma-associated HLA class I peptides on at least two and a maximum of four tumors. In HLA class II, 13 proteins were exclusively identified in the peptidome of medulloblastomas, with only RAD51 and GPC2 found in two tumors. Eight further CTAs and TAAs were represented by tumor-associated HLA class II-presented peptides in 7–14% of samples (Fig. 4c, Supplementary Table 7).
Transcriptomic profiling of medulloblastomas reveals subgroup-specific differential expression of target antigens and characterizes the immune microenvironment
Bulk RNA sequencing of the entire cohort revealed that samples cluster according to their DNA methylation subgroups (Fig. 5a), thus orthogonally validating the subgroup annotation and highlighting the quality of the transcriptomics data. A multi-omics approach linking the HLA ligandome level to the transcriptomics data integrated differential gene expression of the top-ranking target antigens on the peptide (Fig. 5b) and source protein level (Fig. 5c). Subgroup-specific differential gene expression was found for a large portion of target antigens except for HLA class II source proteins. Individual comparisons of subgroups regarding the integration of HLA ligandome and transcriptome are depicted as circos and volcano plots (Supplementary Fig. 9a, b). Deconvolution of cell composition based on gene expression reveals roughly equal proportions of lymphocytes and myeloid cells (Fig. 5d) in the immune cell population of the tumor microenvironment. Dedicated lymphoid (Fig. 5e) and myeloid (Fig. 5f) panels are based on deconvolution of transcriptomics data. Cytotoxic CD8 + T cells were enriched in group 4. Alle subgroups showed frequent gene expression signatures matching to M0 polarized macrophages.

a Principal component analysis of bulk RNA sequencing data sets. Supervised heatmap using HLA peptide (b) and protein (c) information as input. Protein symbols were converted to gene symbols to match the gene symbols in the transcriptomics data. Transcript abundance (normalized) is indicated in the heatmap using the blue-red scale (blue for low and red for high abundance). Metadata if the samples (subgroup and sex) can be distinguished using different colors. Furthermore, the significance column indicates whether a gene symbol was found during the differentially expression analysis of the transcriptomics data in subgroup comparisons (see Supplementary Fig. 9). Relative frequencies of lymphocytes and myeloid cells (d), as well as a dedicated lymphoid (e) and myeloid (f) panel, are based on deconvolution of transcriptomics data. The deconvolution is performed with published data, which shows an accurate observation of the identified cell types in the dataset99,100. Source data are provided as a Source Data file.
Functional testing of medulloblastoma-associated candidate peptides revealed antigen-specific immune recognition
Based on frequency, the top 4 HLA-A*02-restricted medulloblastoma-associated candidate peptides were synthesized and subjected to functional immunogenicity testing. These peptides were derived from the source proteins chemokine-like receptor 1 (CMKKLR1), Wnt-5a (WNT5A), doublecortin-like kinase 1/2 (DCLK1/2), and BarH-like 1 homeobox protein (BARHL1). The frequencies of these candidates among patients (HLA-A*02) were 29%, 50%, 7%, and 21%, respectively. Peptide-specific T cell recognition was shown for CMKKLR1, WNT5A, and DCLK1/2, via priming of non-autologous naïve CD8+ T cells followed by HLA tetramer staining (Fig. 6). These functional immunogenicity data shows that de novo T cell responses can be triggered in healthy donors and that T cell receptors with specificity for our top-ranking candidates exist. Thus, T cell priming identifies these antigens as prime targets for immunotherapy.

a Workflow of non-autologous CD8 + T cell priming with artificial antigen-presenting cells. T cell responses were defined via tetramer staining and were considered positive when a 3-fold increase compared to the negative control and >1% of tetramer-positive cells among CD8 + T cells was observed. Created in BioRender. Neidert, M. (2021) BioRender.com/w57d406. b Left column (negative control): medulloblastoma-specific tetramer staining of CD8 + T cells primed with an unrelated, HLA-matched peptide. Right column: Positive tetramer staining of CD8 + T cells primed with DCLK1/287-96, WNT5A20-28, and CMKKLR181-90 demonstrating a significant antigen-specific response. Source data are provided as a Source Data file.
Discussion
We provide an in-depth atlas of naturally presented T cell antigens by mapping the HLA ligandome of 28 medulloblastomas (WNT (n = 4); SHH (n = 9); Group 3 (n = 7); Group 4 (n = 8)). This atlas provides a set of actionable targets that can be translated into the clinical setting for devising new therapeutic approaches in patients with medulloblastoma. Functional testing of a subset of these novel candidates has demonstrated the induction of antigen-specific T cell responses pointing out their potential for T cell-based immunotherapy.
HLA class I antigen-processing machinery component defects (e.g., HLA-A, -B, and -C, calnexin, β2m, LMP2, LMP7) have previously been reported in various cancer types at high frequency, including medulloblastoma22. However, medulloblastoma tumor samples yielded considerable numbers of HLA class I- and II-presented peptides, with gene enrichment analysis pointing at tumor cells as the origin of HLA ligands. In addition, we observed robust immunostaining of tumor cells for both HLA class I and II (Fig. 1e). In total, 60 distinct HLA class I allotypes were identified, covering 99.95% of the world population, with 92.08% of all individuals expected to be positive for at least three allotypes. Nevertheless, changes in the antigen processing and presenting machinery in tumor cells—extensively reviewed by Leone et al.23—could be one explanation for a commonly observed phenomenon: HLA-ligands can be tumor-exclusive on the peptide level, while other peptides deriving from the same source protein are also found on normal tissues24.
We found medulloblastoma to be a rich source of candidate targets for cancer immunotherapy: 15 HLA class I and 10 HLA class II frequently presented medulloblastoma-associated proteins were identified. Among these, IGFBPL1 (36%), SPINK8 (29%), WNT5A (25%), CBX4 (21%), ESCO1 (21%), SHISA9 (18%), SYCP3 (18%), and CCDC59 (18%) were the most frequent. IGFBPL1 belongs to the neurotrophic insulin-like growth factor axis, which is linked to cell proliferation and inhibition of apoptosis—dysregulation of the IGF-IR systems promotes growth in medulloblastoma, ependymoma, meningioma, and glioma, via anti-apoptotic and mitogenic peptides25,26,27. High levels of several members of the insulin-like growth factor binding protein family have been associated with a poor prognosis in medulloblastomas28. IGFBPL1-derived peptides were found in patients with SHH, Group 3 and Group 4 medulloblastoma (Supplementary Table 4). Peptides derived from SPINK8, a serine peptidase inhibitor, were identified in 29 % of our tumor samples and found among SHH, Group 3, and Group 4 tumors. SPINK8 is not expressed above background levels (10 TPM) in benign human tissues and is considered an unfavorable prognostic marker in breast cancer 29,30,31.
WNT5A-derived HLA peptides were found in WNT, SHH, and Group 4. WNT5A is a member of the oncogenic WNT protein family, renowned for tumor-associated elevated expression28,32. Another candidate protein, CBX4, exerts an oncogenic function by activating Notch1 signaling33. Moreover, CBX4-mediated stimulation activates the oncogenic DNA repair protein BMI-1, which is under investigation for targeted therapy in pediatric diffuse intrinsic pontine glioma34,35,36,37. Besides promoting cell cycle progression, cancer cell migration, and metastasis, CBX4 is also angiogenic by promoting VEGF expression and governing the hypoxia-inducible factor 1-α38. The protein SYCP3 not only exhibited a CTA-like expression profile23, but is also described as a testis-specific protein regulating sister chromosome segregation during meiosis with aberrant expression in malignant cells (including brain tumor)39,40,41. Since the related gene SYCP1 is listed in the CTDatabase16 it appears that SYCP3 represents a novel CTA. Natural presentation of HLA class II molecules and the absence of HLA ligands in our normal tissue database orthogonally validate the CTA-like properties on the immunologically pivotal level of the immunopeptidome in addition to transcriptomic data.
Overlapping medulloblastoma-associated HLA class I- and II-presented proteins (n = 10), as well as tumor-associated HLA class I- and II-restricted peptides, revealed an essentially unique antigenic repertoire inherent to HLA class I and II peptidomes. Specifically, only three candidate proteins (IGFBPL1, INSM1, INSM2) yielded both HLA class I- and II-presented target peptides. This finding underlines the importance of investigating the peptidome of both HLA classes for comprehensive target discovery approaches. The importance of mapping not only HLA class I peptides but also the HLA class II peptidome is based on data on the crucial role of HLA class II antigens and CD4+ helper T cells in clinical anti-tumor responses42,43,44. In addition, including HLA class II-restricted antigens in multi-epitope T cell approaches containing both HLA class I- and HLA class II antigens is likely to induce and boost synergistic CD8+ and CD4+ anti-tumor responses.
Before further translational use, we encourage everyone to critically revise and validate candidate antigens, as additional data sets on HLA ligands and precursor data on the DNA and RNA level will become available. The extent of tumor-association should always be assessed on the most recently available data set at any given time point.
We found a small proportion of pan-medulloblastoma candidate peptide targets, which were coincidently eluted from WNT, SHH, Group 3, and Group 4 tumors (n = 4 HLA class I peptide targets, n = 4 HLA class II peptide targets). In contrast, none of the candidate proteins proved to be pan-medulloblastoma. This allows the conclusion that medulloblastoma subgroups exhibit a common T cell antigen signature on the peptide level, but major differences prevail in the T cell signature among the subgroups on the protein level.
Our multi-omics integration of HLA ligandome and transcriptomics data showed both medulloblastoma subgroup-specific gene expression of target antigens (e.g. WNT5A) and pan-medulloblastoma expression of targets (e.g., CMKKLR1). Combining the list of medulloblastoma-associated proteins and peptides presented on HLA class I (n = 66 candidate target peptides for cancer immunotherapy), these achieve a global population coverage of 98.64%, whereby an average of 14 peptides are expected to match per patient.
CTA are frequently re-expressed genes in tumors whose expression is typically restricted to germ cells located in immunoprivileged sites. CTAs are considered promising candidates for cancer immunotherapy45. Established CTAs and TAAs often fail to fulfill the requirements of frequent and tumor-associated presentation as being either anecdotally detected in very few tumors or broadly identified on both malignant and benign tissues. Accordingly, these data did not contribute to defining candidate targets for medulloblastoma immunotherapy.
The importance of investigating HLA ligandomes from human healthy and diseased tissues has been well recognized46,47,48. Our antigen discovery approach is based on the comparison of tumor datasets to a large database of benign human tissues, mainly derived from autopsies. A major rationale for this approach is the poor correlation of HLA ligands with their precursors (mRNA transcripts and proteins)15,49,50 limiting approaches based on in silico HLA-binding predictions in combination with transcriptomics and proteomics data alone51. Furthermore, morphologically normal tissue adjacent to the tumor (normal tissues adjacent to tumor, NATs) were used as an alternative source of benign tissue in cancer research. However, NATs represent not truly benign tissue but an intermediate state between healthy and disease52. Additionally, for brain tumors such as medulloblastoma, surgical resection of NATs is ethically not feasible.
The medulloblastoma and normal tissue datasets were acquired on the same LC-MS setup using the same methods, thus allowing for the best possible data consistency and comparability. The fact that a few of the medulloblastoma-associated peptides described in this manuscript were recently identified on normal tissues during a re-analysis using a different much more sensitive LC-MS setup53 suggests that these peptides are present in only low abundance on these normal tissues compared to medulloblastoma. Future antigen discovery approaches could therefore also investigate differences in absolute antigen abundance, for instance by using targeted mass spectrometry.
The ideal control for this medulloblastoma T cell antigen cohort would be a large set of HLA ligandome data based on normal tissue samples from developing brain at various ages. For obvious reasons, these samples are impossible to obtain and we have used publicly available transcriptome data of “BrainSpan: Atlas of the developing brain” to show low expression of targets of the healthy developing brain.
Next-generation sequencing techniques and the clinical success of immune checkpoint inhibitors have shifted much attention to T cell antigens derived from somatic missense mutations (neoepitopes). Neoepitopes are, in theory, tumor-specific and immunogenic due to their “foreign” sequence—recent studies showed promising clinical results in tumor entities other than medulloblastoma44,54,55. Our antigen discovery pipeline is not designed to detect neoepitopes because the immunopeptidome data (LC-MS/MS) acquired in this study was processed against a reference proteome. However, this approach enables the sensitive detection of non-mutated antigens derived from “self”, which are well established in tumor immunology19,56 and proven targets of tumor-associated T cell responses 50,57. In addition, the tumor mutational burden (TMB) in medulloblastoma is very low, even lower than in glioblastoma58. Even in tumors with a high TMB, such as melanoma, HLA-presented peptides derived from mutated sequences are rarely detected, making it impossible in medulloblastoma as well59,60. In general, the number of mutated tumor antigens that elicit frequent and effective anti-tumor immune responses appears to be relatively small, even in the setting of immune checkpoint inhibition. One reason for a relatively low prevalence of neoantigens in tumors with a high TMB might be the clonal selection of antigen loss variants by immunoediting61. Taken together, this discrepancy highlights the importance of exploiting and including the group of non-mutated antigens in designing meaningful anti-tumor T cell approaches. In the GAPVAC trial, which combined HLA ligandome profiling and next generation sequencing (NGS) in glioblastoma patients, a median of 36 somatic, non-synonymous mutations were detected by NGS however none of the 643 mutations could be confirmed by mass spectrometry62. This is in line with previously reported rates of below 1% for the transition of genetic mutations into HLA ligands in other tumor entities63,64.
Immunophenotyping in medulloblastoma by deconvolution of next-generation sequencing techniques has been previously performed, for instance using deconvolution of DNA methylation profiling data by Grabovska et al.65 or based on transcriptomic (as in our case) data by Bockmayr et al.66. The latter found immune cell infiltration signatures that are in line with our findings, for instance the pronounced infiltration of cytotoxic lymphocytes in subgroup 4.
We acknowledge as a limitation that we focused the functional immunogenicity assays on the top-ranking medulloblastoma-associated candidate peptides that are HLA-A*02:01-restricted. HLA-A*02 was not only the most frequent HLA class I allotype in our medulloblastoma cohort, but it also has a pronounced high allele frequency across the world67. HLA-A2 ligands have a much higher likelihood of being “top-ranking” in this selection process due to the high frequency of HLA-A2 in the general population. Thus, the functionally validated targets in this study cover a large number of medulloblastoma patients that might benefit from T cell-based approaches.
In this study, we have mapped the antigenic landscape of medulloblastoma in an unprecedented depth, unveiling a novel set of canonical, non-mutated, tumor-associated peptides and proteins with natural and frequent presentation on HLA class I and II molecules. This comprehensive antigen atlas facilitates the development of antigen-specific immunotherapies such as vaccination with mRNA, peptides, peptide-pulsed dendritic cells (DC), or the adoptive transfer of antigen-specific T cells.
Methods
Ethical compliance statement
This study was conducted in accordance with all relevant ethical regulations. All analyses were performed according to the guidelines of the local review boards (Kantonale Ethikkommission Zürich/KEK-ZH-Nr. 2015-0163; Ethik-Kommission der Ärztekammer Hamburg/PV4904; authorized vote-free usage of residual tissue collected for other purposes in Sankt Augustin and Würzburg). Written informed consent was obtained from each patient and their legal representative, as well as from patients and healthy donors that contributed to the benign immunopeptidome database in accordance with the local ethical requirements and the Declaration of Helsinki. No participant compensation was given.
Patients and blood samples
Newly diagnosed medulloblastoma samples were collected at the Department of Neurosurgery of the University Hospital Zürich, Asklepios Hospital Sankt Augustin, University Medical Center Hamburg-Eppendorf and the University Children’s Hospital Würzburg. Tumor tissue was snap-frozen in liquid nitrogen, Bambanker medium, or Tissue-Tek O.C.T. during surgery and stored at −80 °C until use. Peripheral blood mononuclear cells (PBMC) of HLA-typed healthy donors were provided by the Swiss Red Cross Blood Transfusion Services Zurich and Chur, Switzerland.
Clinical and radiological data
Patient characteristics (sex and age at diagnosis) and tumor- and treatment-related data (tumor location and extension, extent of resection, recurrence, and further treatment) were retrieved from hospital records. Assigned sex of participants was used and matched with the chromosomal sex based on copy number variation (CNV) plots in all cases (Supplementary Table. 2). The extent of the initial resection was assessed by magnetic resonance imaging within 72 h after surgery.
Immunohistochemistry
A representative paraffin block was selected for four patients (Pat. No 4/9/18/25). One patient was taken from each subgroup to exemplify the expression of HLA class I and HLA class II in each subgroup (WNT, SHH, Group 3, Group 4) by immunohistochemistry. Unstained 3-μm-thick slides of formalin-fixed paraffin-embedded tissues were obtained and submitted for immunostaining. The following primary antibodies were used: MHC-I, clone EPR1394Y Abcam Limited, and MHC-II, clone M5/114 Laboratory of Prof. Zinkernagel. External positive and negative controls were used for all antibodies. The IHC staining was performed once. Typically, IHC is only repeated if an unexpected result occurs or if the on-slide control staining is negative.
HLA typing
Sequence-based typing was performed for all samples by sequencing exons 1–8 for HLA-I alleles and exons 2–6 for HLA-II alleles (Histogenetics, Ossining, NY)68.
HLA ligand immunopurification (IP)
HLA-I and HLA-II molecules were isolated from snap-frozen tissue using standard immunoaffinity chromatography. CNBr-activated sepharose (GE Healthcare, Little Chalfont, UK) was cross-linked with either pan-HLA-I-specific antibody W6/32 (ATCC HB-95)68, and HLA-DR-specific antibody L243 (ATCC HB-55)69, or Tü3970 specific for HLA-DP, -DQ, -DR (produced in-house, Department of Immunology, University of Tübingen) at a ratio of 40 mg per 1 mg of antibody with 0.5 M NaCl/0.1 M NaHCO3 at pH 8.3. To precipitate HLA-peptide complexes, at least 1 mg antibody (W6/32 for HLA-I or Tü39 and L243 mixed 1:1 for HLA-II) was employed, which was increased to 1 mg per 1 g of tissue for samples >1 g. Tissue homogenization was performed in lysis buffer consisting of CHAPS (PanReac AppliChem, Darmstadt, Germany) and one cOmplete protease inhibitor cocktail tablet (Roche, Basel, Switzerland) in PBS. As far as possible, Tissue-Tek O.C.T. and Bambanker medium were cut off from embedded samples before HLA-IP and RNA sequencing while keeping specimens frozen during this step. After mechanical disruption using a scalpel and a Potter-Elvehjem tissue homogenizer, samples were incubated for 60 min on a shaker at 4 °C. To further disrupt cell membranes, samples underwent five cycles of pulsed sonification, each lasting 20 s, followed by another 1 h incubation period on the shaker at 4 °C. 2× concentrated lysis buffer (used only during chopping tissue finely using forceps and scalpel) consisted of 33 ml PBS/0.4 g CHAPS (PanReac AppliChem, Darmstadt (Germany))/1 protease inhibitor cocktail tablet (cOmplete/Roche, Basel (Switzerland)). In all subsequent steps, 1x lysis buffer (2x lysis buffer diluted 1:1 with PBS) was used. Overall lysis lasted two hours (plus shorter time periods to process all samples of the same batch), 1 h after mechanical disruption of tissue (initially using scalpel and forceps followed by treatment with a Potter-Elvehjem tissue homogenizer), and 1 h after pulsed sonication. The lysate was cleared by centrifugation for 1 h at 3,100 × g and 4 °C for 1 h (repeated once for the supernatant of tissues ≥1 g) and by sterile filtration employing 5.0 μm low protein binding filter units (Merck Millipore). For each sample, two columns were placed one above the other, so that flow through the first one, containing HLA class I-specific antibodies, drained directly into the second column equipped with HLA class II-specific antibodies. Lysates were passed cyclically through the columns overnight at 4 °C. After washing the affinity columns for 30 min with PBS and for 1 h with water, peptide elution was performed by four successive incubations on a shaker with 100 µl 0.2% TFA per 1 mg antibody. Peptides were separated from the HLA molecules by ultrafiltration using 3 kDa and 10 kDa Amicon filter units (Merck Millipore) for HLA-I and HLA-II, respectively. By lyophilization, the eluate volume was subsequently reduced to approximately 30 µl.
Finally, peptide solutions (only performed for Pat. 6/7/14/19/25/26/27) were desalted by reversed-phase liquid chromatography using ZipTipc18 Pipette tips (Merck Millipore, 0.6 µl bed volume) and using 32.5% ACN/0.2% TFA as eluent. The hydrophobic solvent content was removed by vacuum centrifugation, 1% ACN / 0.05% TFA was added to a final volume of 35 µl, and samples were stored at −80 °C until LC-MS/MS. Samples not cleaned up using ZipTipc18 Pipette tips were directly subjected to vacuum centrifugation following lyophilization (referred to as direct injection).
LC-MS/MS and data analysis
The purified peptides were analyzed by nanoflow high-performance liquid chromatography (Dionex Ultimate 3000 Series liquid chromatography system) and tandem mass spectrometry. Four technical replicates were acquired for each tissue and HLA class, consuming a 5 µl sample and lasting 130 min each. Peptide solutions were loaded with 1% ACN/0.05% TFA on a 75 µm × 2 cm Acclaim PepMap RSLC column (Thermo Fisher Scientific) at a flow rate of 4 µl/min for 10 min at 50 °C. Subsequently, peptides were gradually eluted from the separation column (Acclaim C18 PepMap RSLC column 50 µm × 25 cm, Thermo Fisher Scientific) with a linear gradient from 3% to 40% solvent B (0.15% FA/80% ACN) at a flow rate of 300 nl/min (Orbitrap Fusion Lumos) over 90 min. Between minutes 101 and 106, the column was washed with solvent B, which mounted up to 95%.
Pre-fractionated peptides eluting from the separation column were introduced into an online-coupled tandem mass spectrometer (Orbitrap Fusion Lumos, Thermo Fisher Scientific) equipped with a nanospray ion source and operated in data-dependent acquisition (DDA) mode. Resolution for MS1 was set to 120,000, whereas MS2 data were acquired at a resolution of 30,000. This applied to both HLA class I (400-650 m/z, positive charge states 2–3) and HLA class II peptides (400–1000 m/z, positive charge states 2–5). For data processing, precursor mass tolerance was set to ±5 ppm and fragment mass tolerance was set to ±0.02 Da.
Fragment spectra were searched against the Swiss-Prot release from September 27th, 2013 (20,279 reviewed protein sequences) using the SEQUEST71 search algorithm embedded in Proteome Discoverer 1.4.1.14 (Thermo Fisher Scientific) with methionine oxidation as the only dynamic modification. Peptide length was set to 8–12 (HLA class I) or 8–25 amino acids (HLA class II), respectively. The false discovery rate estimated by the Percolator algorithm (2.04) was set to ≤5%. HLA class I datasets were deconvoluted using a stand-alone version of NetMHCpan-4.072,73 (percentile rank score ≤2%) and an enhanced version of SYFPEITHI74 (SYFPEITHI score ≥60%).
Benign database of HLA ligands
Our in-house benign database comprised 30 distinct primary human organs (n = 418 HLA class I peptidome datasets including among others the HLA Ligand Atlas; www.hla-ligand-atlas.org10. Tissue is mainly based on autopsies and surgeries of persons that have not been diagnosed with any malignancy. This approach was previously described and is accepted as a surrogate source of benign tissue52,75. This definition of “benign tissue” is also in agreement with the definition used by the Genotype-Tissue Expression Consortium,75,76 which transcriptomic data of benign tissues originating from autopsies.
RNA sequencing
RNA and genomic DNA were isolated from fresh frozen tumor tissue (≥20 mg) using the AllPrep DNA/RNA Mini Kit (Qiagen). RNA sequencing was performed for all 28 tumors at CeGaT GmbH, Tübingen. Following quality control for RNA integrity, sequencing libraries were prepared from 78 ng RNA using the KAPA RNA HyperPrep Kit with RiboErase (HMR; Roche), whereby ribosomal RNA was depleted. Paired-end sequencing at 100 bp read length was performed on a NovaSeq 6000 sequencing system (Illumina). Sequencing reads were demultiplexed employing the Illumina bcl2fastq 2.20, and untrimmed FASTQ12-formatted files were uploaded to qPortal13 (Quantitative Biology Center, University of Tübingen) for further evaluation.
Software, statistical analysis, and online tools
Transcriptomic reads were quantified using the Nextflow-based nf-core pipeline rnaseq v1.4.277 with the standard parameters. The quality of the FASTQ files was determined with FastQC, followed by adapter trimming with Trim Galore v0.6.412. Trimmed reads were used as input for the STAR v2.6.113 aligner to map reads to the reference genome GRCh37. RNA-seq data quality control was performed with RSeQC v3.0.114 and read counting of the features (e.g., genes) with featureCounts v1.6.478. Ultimately, an aggregation of the quality control for the RNAseq analysis was performed with MultiQC v1.779.
The nf-core rnaseq pipeline contained the raw read count table resulting from featureCounts. This content was processed with the R package DESeq2 v1.34.080 and used for the differential expression analysis. Transcripts with an absolute log fold change >2 and an FDR < 0.01 were differentially expressed.
Gene ontology analysis was dependent on the R packages clusterprofiler v4.2.281 and DOSE v3.20.182. Gene identifiers were converted to Entrez IDs using org.Hs.eg.db v3.14.083. A GO enrichment analysis was performed to retrieve the biological processes (BP), cellular components (CC), and molecular functions (MF) with a 0.05 p-value cut-off. Additionally, we performed ontology using disease84, Network of cancer genes18, Disease Gene Network (DisGeNET)85, and KEGG86,87,88 annotation references.
A supervised heatmap was created with pheatmap v.1.0.1289 using peptide and protein information as input. Protein symbols were converted to gene symbols (biomaRt v2.50.390,91) to match the gene symbols in the transcriptomics data. Furthermore, differentially expressed genes that matched protein names were highlighted in the annotation column. Graphs were produced in Rstudio92 with R version 4.1.393 (2022-03-10), mainly using the R package ggplot2 v3.3.694.
DNA methylation-based clustering and copy number variation plots (CNVs)
Tumor DNA was extracted from fresh-frozen tissue samples using the Qiagen AllPrep DNA/RNA Mini Kit (see above). Genome-wide DNA methylation profiling was performed using Infinium HumanMethylation EPIC Kit according to the manufacturer’s instructions (Illumina, San Diego, CA, USA)95. DNA methylation-based classification was performed using the Heidelberg Brain Tumor Methylation Classifier v12 (www.molecularneuropathology.org)11. The minfi Bioconductor package v.1.24.0 was used to calculate raw signal intensities from IDAT files, normalization was performed for each sample individually and batch effects were corrected using the removeBatchEffect function of the limma package v3.34.596,97. Both methylated and unmethylated signals were corrected individually. Subsequently, beta values were calculated, CpG probes for downstream analysis selected and unsupervised nonlinear dimension reduction was calculated using the 1-variance weighted Pearson correlation between the samples to construct a distant matrix11. This matrix was used as input for t-SNE visualization11. Copy number variation analysis was performed using the conumee package v1.12.0 and copy number changes were evaluated by inspecting the respective plots11.
Peptide synthesis
Peptides for functional T cell experiments were synthesized by INTAVIS Peptide services (Tübingen, Germany) using standard solid-phase Fmoc chemistry and quality control was performed by MALDI-MS and RP HPLC.
CD8+ T cell priming using aAPC
Priming of enriched human-derived CD8+ T cells was performed using artificial antigen-presenting cells (aAPC) as previously described98. On day 1, PBMC were isolated from healthy donor buffy coats via density gradient centrifugation. Buffy coats of healthy donors were provided by the Blood Transfusion Service, Swiss Red Cross, Schlieren, Switzerland. CD8+ T cells were MACS-enriched by positive selection at day 2 (Miltenyi Biotec, Bergisch Gladbach, Germany). In vitro priming and stimulation of CD8+ T cells with aAPC loaded with peptide-carrying MHC monomers (Tetramer Shop ApS, Denmark) and anti-CD28 were initiated on day three. Specifically, 106 cells were incubated with 8 × 105 aAPC in a 96 well plate in 200 µl IMDM containing 25 mM HEPES (Thermo Fisher Scientific) supplemented with 10% heat-inactivated human AB plasma (Sigma-Aldrich), 2 mM L-glutamine, 50U/ml penicillin, 50 µg/ml streptomycin. Together with aAPC, 5 ng/ml human IL-12 p70 (R&D System) was added. After 3 days of incubation at 37 °C, half of the medium was exchanged with fresh medium supplemented with 40 U/ml human IL-2, and cells were incubated for four days before the following stimulation. This priming cycle was repeated three times in total and followed by tetramer staining on day 28.
Tetramer staining
To quantify the frequency of peptide-specific CD8+ T cells, staining with PerCP anti-human CD8a antibody (Biolegend, HIT8a clone, cat.300921) and peptide-loaded PE-conjugated tetramers (Tetramer Shop ApS, Denmark) was performed after 28 days of stimulation and priming. Samples were analyzed on a BD FACSverse (BD Biosciences). A testing peptide was considered immunogenic when more than 1% of tetramer-positive cells among the CD8+ T cells were detected and showed a threefold increase compared with the corresponding negative control. As a negative control staining, PE-conjugated tetramers were loaded with a naïve peptide to which the cells had not been previously exposed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA sequencing data generated in this study has been deposited in the NCBY’s Gene Expression Ominibus (GEO) database under accession code GSE238195 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE238195. The DNA methylation data has also been deposited in the GEO database under accession code GSE250586 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE250586. The mass spectrometry generated in this study has been deposited in the Proteomics Identifications Database (PRIDE) under accession code PXD043770 https://www.ebi.ac.uk/pride/archive/projects/PXD043770.
The complete peptide list (HLA class I and HLA class II) of the entire cohort is available in the supplementary materials (Supplementary Data 6, 7). All other data are available in the article and its Supplementary files or from the corresponding author upon request. Source data are provided with this paper.
References
Pui, C. H., Gajjar, A. J., Kane, J. R., Qaddoumi, I. A. & Pappo, A. S. Challenging issues in pediatric oncology. Nat. Rev. Clin. Oncol. 8, 540–549 (2011).
Salloum, R. et al. Late morbidity and mortality among medulloblastoma survivors diagnosed across three decades: A report from the Childhood Cancer Survivor Study. J. Clin. Oncol. 37. https://doi.org/10.1200/JCO.18.00969 (2019).
Siegel, R. L., Miller K. D., Jemal A. Cancer statistics, 2019. CA Cancer J. Clin. 69. https://doi.org/10.3322/caac.21551 (2019).
Northcott, P. et al. Medulloblastomics: the end of the beginning. Nat. Rev. 12, 818–834 (2012).
Kool, M. et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 123, 473–484 (2012).
Northcott, P. A. et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 29, 1408–1414 (2011).
Ramaswamy, V. et al. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol. 131, 821–831 (2016).
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 23. https://doi.org/10.1093/neuonc/noab106 (2021).
Hovestadt, V. et al. Medulloblastomics revisited: biological and clinical insights from thousands of patients. Nat. Rev. Cancer. 20. https://doi.org/10.1038/s41568-019-0223-8 (2020).
Marcu, A. et al. HLA Ligand Atlas: a benign reference of HLA-presented peptides to improve T-cell-based cancer immunotherapy. J. Immunother. Cancer 9. https://doi.org/10.1136/jitc-2020-002071 (2021).
Capper, D. et al. DNA methylation-based classification of central nervous system tumours. Nature. 555. https://doi.org/10.1038/nature26000 (2018).
Andrews, S. et al. https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 29. https://doi.org/10.1093/bioinformatics/bts635 (2013).
Wang, L., Wang S., Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics. 28. https://doi.org/10.1093/bioinformatics/bts356 (2012).
Weinzierl, A. O. et al. Distorted relation between mRNA copy number and corresponding major histocompatibility complex ligand density on the cell surface. Mol. Cell. Proteom. 6. https://doi.org/10.1074/mcp.M600310-MCP200 (2007).
Almeida, L. G. et al. CTdatabase: a knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res. 371. https://doi.org/10.1093/nar/gkn673 (2009).
Bilich, T. et al. The HLA ligandome landscape of chronic myeloid leukemia delineates novel T-cell epitopes for immunotherapy. Blood. 133. https://doi.org/10.1182/blood-2018-07-866830 (2019).
Repana, D. et al. The Network of Cancer Genes (NCG): a comprehensive catalogue of known and candidate cancer genes from cancer sequencing screens. Genome Biol. 20. https://doi.org/10.1186/s13059-018-1612-0 (2019).
Vigneron, N., Stroobant, V., van den Eynde, B. J., van der Bruggen, P. Database of T cell-defined human tumor antigens: the 2013 update. Cancer Immun. 13 (2013).
Gjerstorff, M. F., Andersen, M. H., Ditzel, H. J. Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget. 6. https://doi.org/10.18632/oncotarget.4694 (2015).
Cheever, M. A. et al. The prioritization of cancer antigens: a National Cancer Institute pilot project for the acceleration of translational research. Clin. Cancer Res. 15. https://doi.org/10.1158/1078-0432.CCR-09-0737 (2009).
Raffaghello, L. et al. Expression and functional analysis of human leukocyte antigen class I antigen-processing machinery in medulloblastoma. Cancer Res. 67. https://doi.org/10.1158/0008-5472.CAN-06-4735 (2007).
Leone, P. et al MHC class I antigen processing and presenting machinery: Organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 105. https://doi.org/10.1093/jnci/djt184 (2013).
Kowalewski, D. J. et al. HLA ligandome analysis identifies the underlying specificities of spontaneous antileukemia immune responses in chronic lymphocytic leukemia (CLL). Proc. Natl. Acad. Sci. USA. 112. https://doi.org/10.1073/pnas.1416389112 (2015).
Russo, V. C., Gluckman, P. D., Feldman, E. L., Werther, G. A. The insulin-like growth factor system and its pleiotropic functions in brain. Endocr. Rev. 26. https://doi.org/10.1210/er.2004-0024 (2005).
Wang, J. Y. et al. Activation of the IGF-IR system contributes to malignant growth of human and mouse medulloblastomas. Oncogene. 20. https://doi.org/10.1038/sj.onc.1204532 (2001).
Zumkeller, W. & Westphal, M. The IGF/IGFBP system in CNS malignancy. Mol. Pathol. 54, 227–229 (2001).
de Bont, J. M. et al. Various components of the insulin-like growth factor system in tumor tissue, cerebrospinal fluid and peripheral blood of pediatric medulloblastoma and ependymoma patients. Int. J. Cancer. 123. https://doi.org/10.1002/ijc.23558 (2008).
Aguet, F. et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 369(6509). https://doi.org/10.1126/SCIENCE.AAZ1776 (2020)
Jeong, J. et al. Expressional and functional analyses of epididymal SPINKs in mice. Gene Expression Patterns. 31. https://doi.org/10.1016/j.gep.2018.12.001 (2019).
Jalkanen, J., Kotimäki, M., Huhtaniemi, I., Poutanen, M. Novel epididymal protease inhibitors with Kazal or WAP family domain. Biochem. Biophys. Res. Commun. 349. https://doi.org/10.1016/j.bbrc.2006.08.023 (2006).
Howng, S. L. et al. Differential expression of Wnt genes, β-catenin and E-cadherin in human brain tumors. Cancer Lett. 183. https://doi.org/10.1016/S0304-3835(02)00085-X (2002).
Zeng, J. S. et al. CBX4 exhibits oncogenic activities in breast cancer via Notch1 signaling. Int. J. Biochem. Cell Biol. 95. https://doi.org/10.1016/j.biocel.2017.12.006 (2018).
Qiu, M. et al. MicroRNA-200c suppresses cell growth and metastasis by targeting Bmi-1 and E2F3 in renal cancer cells. Exp. Ther. Med. 13. https://doi.org/10.3892/etm.2017.4147 (2017).
Ismail, I. H. et al. CBX4-mediated SUMO modification regulates BMI1 recruitment at sites of DNA damage. Nucleic Acids Res. 40. https://doi.org/10.1093/nar/gks222 (2012).
Hu, C. et al. CBX4 promotes the proliferation and metastasis via regulating BMI-1 in lung cancer. J. Cell Mol. Med. 24. https://doi.org/10.1111/jcmm.14771 (2020).
Kumar, S. S. et al. BMI-1 is a potential therapeutic target in diffuse intrinsic pontine glioma. Oncotarget 8. https://doi.org/10.18632/oncotarget.18002 (2017).
Li, J. et al. Cbx4 governs HIF-1α to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell. 25. https://doi.org/10.1016/j.ccr.2013.12.008 (2014).
Aarabi, M. et al. Testicular expression of synaptonemal complex protein 3 (SYCP3) messenger ribonucleic acid in 110 patients with nonobstructive azoospermia. Fertil. Steril. 86. https://doi.org/10.1016/j.fertnstert.2005.12.070 (2006).
Parra, M. T. et al. Involvement of the cohesin Rad21 and SCP3 in monoporal attachment of sister kinetochores during mouse meiosis I. J. Cell Sci. 117. https://doi.org/10.1242/jcs.00947 (2004).
Mobasheri, M. B. et al. Expression of two testis-specific genes, TSGA10 and SYCP3, in different cancers regarding to their pathological features. Cancer Detect. Prev. 31. https://doi.org/10.1016/j.cdp.2007.05.002 (2007).
Kreiter, S. et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520. https://doi.org/10.1038/nature14426 (2015).
Schumacher, T. et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 512. https://doi.org/10.1038/nature13387 (2014).
Tran, E. et al. Cancer immunotherapy based on mutation-specific CD4 + T cells in a patient with epithelial cancer. Science 344(6184). https://doi.org/10.1126/science.1251102 (2014)
Whitehurst, A. W. Cause and consequence of cancer/testis antigen activation in cancer. Annu. Rev. Pharmacol. Toxicol. 54. https://doi.org/10.1146/annurev-pharmtox-011112-140326 (2014).
Caron, E., Aebersold R., Banaei-Esfahani A., Chong C., Bassani-Sternberg M. A Case for a Human Immuno-Peptidome Project Consortium. In: Immunity. 47. https://doi.org/10.1016/j.immuni.2017.07.010 (2017).
Vizcaíno, J. A. et al. The human immunopeptidome project: A roadmap to predict and treat immune diseases. Mol. Cell. Proteom. 19. https://doi.org/10.1074/mcp.R119.001743 (2020).
Faridi, P., Purcell, A. W., Croft, N. P. In Immunopeptidomics We Need a Sniper Instead of a Shotgun. Proteomics. 18. https://doi.org/10.1002/pmic.201700464 (2018).
Fortier, M. H. et al. The MHC class I peptide repertoire is molded by the transcriptome. J. Exp. Med. 205. https://doi.org/10.1084/jem.20071985 (2008).
Schuster, H. et al. The immunopeptidomic landscape of ovarian carcinomas. Proc. Natl. Acad. Sci. USA. 114. https://doi.org/10.1073/pnas.1707658114 (2017).
Boegel, S., Castle, J. C., Kodysh, J., O’Donnell, T., Rubinsteyn, A. Bioinformatic methods for cancer neoantigen prediction. In: Progress in Molecular Biology and Translational Science. 164.; Academic Press, 2019. https://doi.org/10.1016/bs.pmbts.2019.06.016
Aran, D. et al. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat. Commun. 8. https://doi.org/10.1038/s41467-017-01027-z (2017).
Hoenisch, Gravel N. et al. TOFIMS mass spectrometry-based immunopeptidomics refines tumor antigen identification. Nat. Commun. 14. https://doi.org/10.1038/s41467-023-42692-7 (2023).
Robbins, P. F. et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 19. https://doi.org/10.1038/nm.3161 (2013).
van Rooij, N. et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. 31. https://doi.org/10.1200/JCO.2012.47.7521 (2013).
van der Bruggen, P. et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 254(5038). https://doi.org/10.1126/science.1840703 (1991).
Kowalewski, D., Stevanovic, S. Biochemical large-scale identification of MHC class I ligands. Methods Mol. Biol. 960. https://doi.org/10.1007/978-1-62703-218-6 (2013).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature. 500. https://doi.org/10.1038/nature12477 (2013).
Andersen, R. S. et al. Dissection of T-cell antigen specificity in human melanoma. Cancer Res. 72. https://doi.org/10.1158/0008-5472.CAN-11-2614 (2012).
Kvistborg, P. et al. TIL therapy broadens the tumor-reactive CD8 + T cell compartment in melanoma patients. Oncoimmunology. 1. https://doi.org/10.4161/onci.18851 (2012).
Verdegaal, E. M. E. et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature. 536. https://doi.org/10.1038/nature18945 (2016).
Hilf, N. et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 565. https://doi.org/10.1038/s41586-018-0810-y (2019).
Yadav, M. et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 515. https://doi.org/10.1038/nature14001 (2014).
Kalaora, S. et al. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget. 7. https://doi.org/10.18632/oncotarget.6960 (2016).
Grabovska, Y. et al. Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat. Commun. 11. https://doi.org/10.1038/s41467-020-18070-y (2020).
Bockmayr, M. et al. Subgroup-specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology. 7. https://doi.org/10.1080/2162402X.2018.1462430 (2018).
González-Galarza, F. F. et al. Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 43. https://doi.org/10.1093/nar/gku1166 (2015).
Barnstable, C. J. et al. Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens-new tools for genetic analysis. Cell. 14, 9–20 (1978).
Shackelford, D. A., Lampson, L. A., Strominger, J. L. Separation of three class II antigens from a homozygous human B cell line. J. Immunol. 130 (1983)
Ødum, N., et al. Homotypic aggregation of human cell lines by HLA class II-, class Ia- and HLA-G-specific monoclonal antibodies. Eur. J. Immunol. 21. https://doi.org/10.1002/eji.1830210921 (1991).
Eng, J. K., McCormack, A. L., Yates, J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5. https://doi.org/10.1016/1044-0305(94)80016-2 (1994).
Hoof, I. et al. NetMHCpan, a method for MHC class i binding prediction beyond humans. Immunogenetics. 61. https://doi.org/10.1007/s00251-008-0341-z (2009).
Jurtz, V. et al. NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 199. https://doi.org/10.4049/jimmunol.1700893 (2017).
Rammensee, H. G., Bachmann, J., Emmerich, N. P. N., Bachor, O. A., Stevanović, S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 50. https://doi.org/10.1007/s002510050595 (1999).
Melé, M. et al. The human transcriptome across tissues and individuals. Science. 348(6235). https://doi.org/10.1126/science.aaa0355 (2015).
Consortium TGte, A. K. G., et al. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 348(6235) (2015)
Ewels, P., Hammarén, R., Peltzer, A., Moreno, D., Garcia, M. https://zenodo.org/record/3503887.
Liao, Y., Smyth, G. K., Shi, W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 30. https://doi.org/10.1093/bioinformatics/btt656 (2014).
Ewels, P., Magnusson, M., Lundin, S., Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32. https://doi.org/10.1093/bioinformatics/btw354 (2016).
Love, M. I., Huber, W., Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2. https://doi.org/10.1016/j.xinn.2021.100141 (2021).
Yu, G., Wang, L. G., Yan, G. R., He, Q. Y. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics. 31. https://doi.org/10.1093/bioinformatics/btu684 (2015).
Carlson, M., Falcon, S., Pages, H., Li, N. org. Hs. eg. db: Genome Wide Annotation for Human. R package version. 3(2) (2019).
Schriml, L. M., et al. Disease ontology: A backbone for disease semantic integration. Nucleic Acids Res. 40. https://doi.org/10.1093/nar/gkr972 (2012).
Piñero, J., Saüch, J., Sanz, F., Furlong, L. I. The DisGeNET cytoscape app: Exploring and visualizing disease genomics data. Comput. Struct. Biotechnol. J. 19. https://doi.org/10.1016/j.csbj.2021.05.015 (2021).
Kanehisa, M., Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M., Tanabe, M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 49. https://doi.org/10.1093/nar/gkaa970 (2021).
KoldeR. 2012
Durinck, S. et al. BioMart and bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 21. https://doi.org/10.1093/bioinformatics/bti525 (2005).
Durinck, S., Spellman, P. T., Birney, E., Huber, W. Mapping identifiers for the integration of genomic datasets with the R/ Bioconductor package biomaRt. Nat. Protoc. 4. https://doi.org/10.1038/nprot.2009.97 (2009).
RStudio Team. RStudio: Integrated Development for R. RStudio, PBC. Published online 2020.
R. Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Published online 2022.
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag, ISBN 978-3-319-24277-4. Published online 2016.
Pajtler, K. W. et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. 27. https://doi.org/10.1016/j.ccell.2015.04.002 (2015).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 30. https://doi.org/10.1093/bioinformatics/btu049 (2014).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43. https://doi.org/10.1093/nar/gkv007 (2015).
Walter, S. et al. Cutting edge: predetermined avidity of human CD8 T cells expanded on calibrated MHC/ANTI-cd28-coated microspheres. J. Immunol. 171. https://doi.org/10.4049/jimmunol.171.10.4974 (2003).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 12. https://doi.org/10.1038/nmeth.3337 (2015).
Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37. https://doi.org/10.1038/s41587-019-0114-2 (2019).
Acknowledgements
This study was supported by the Filling-The-Gap Program of the University of Zurich (J.V., MCN), the Béatrice Ederer-Weber Foundation (MCN), the Prof. Dr. Max Cloëtta Foundation (J.V.), the EANS Research Grant (JV), the Clinical Research Priority Program (CRPP) of the University of Zurich for the CRPP ImmunoCure (PR, MS, MiW) and the Detas foundation (MiW), the German Academic Scholarship Foundation (Studienstiftung des Deutschen Volkes) (DRG), the non-profit organization Ein Kiwi gegen Krebs (DRG), the ERC grant 339842 (HGR), the Deutsche Forschungsgemeinschaft under Germany’s Excellence Strategy (Grant EXC2180 390900677) (MD, JW), the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, Grant WA 4608/1-2) (J.W.), German Cancer Consortium (DKTK) (J.W.), Wilhelm Sander Stiftung (Grant 2016.177.2) (J.W.) and Else Kroener Fresenius Stiftung (2022_EKSE.79) (J.W.).
We acknowledge support from the Open Access Publication Fund of the University of Zürich.
Author information
Authors and Affiliations
Contributions
J.V., L.K.F., G.M., Mi.W., M.C.N.—designed the overall study. J.V., L.K.F., G.M., M.M., K.L., M.W., M.E., A.R., M.M.J., H.R., R.B.C., B.M.F., N.K., O.B., L.R., Mi.W., M.C.N.—provided tumor tissue, cancer cells, buffy coats and participated in designing experiments. J.V., L.K.F., G.M., M.D., D.R.G., D.J.K., F.S.—performed and analyzed experiments (e.g. immunopeptidome experiments, T cell priming). M.D., D.R.G., M.S.,. S.N., M.G., F.S., M.C.N.—performed bioinformatic analysis. J.V., L.K.F.,. G.M., M.D., M.M., D.R.G., D.J.K., A.B.P., A.M.Z., K.B., K.K., Man.S.; T.W., H.G.W., P.R., Mi.G., H.G.R., E.J.R. F.S., J.S.W., Mi.W., M.C.N.—interpreted the data and contributed to writing the manuscript. J.V., L.K.F., G.M., M.M., D.R.G., J.S., D.J.K., A.B.P.,. A.M.Z., K.L., M.W., M.E., M.M.J., A.R., H.R., K.B., R.B.C., B.M.F., K.K., M.S., S.N., M.G., Man.S., T.W., H.G.W., P.R., Mi.G., N.K., O.B., L.R., H.G.R., E.J.R., J.S.W., Mi.W., M.C.N.—participated in logistics, and reviewed the manuscript. Mi.W., M.C.N.—supervised this study. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
L.K.F. and D.J.K. are employees, and H.G.R. is a shareholder of Immatics Biotechnologies GmbH. H.G.R. is also a shareholder of Curevac AG and ViferaXS GmbH. K.K. is currently an employee of Roche Diagnostics International Ltd and holds non-voting equity securities in F. Hoffmann-La Roche Ltd. G.M. is currently an employee of Roche Diagnostics International Ltd. M.M.J. received honoraria from Integra. T.W. has received honoraria for advisory board participation from Philogen and a research grant from Cellis. P.R. has received honoraria for lectures or advisory board participation from Bristol-Myers Squibb, Boehringer Ingelheim, Debiopharm, Merck Sharp and Dohme, Novocure, QED, and Roche and research support from Merck Sharp and Dohme and Novocure. FS received honoraria from Bayer and Illumina. F.S. is Co-founder and shareholder of Heildeberg Epignostix GmbH. Mi.W. has received research grants from Quercis and Versameb, and honoraria for lectures or advisory board participation or consulting from Novartis, Novocure, Orbus, and Philogen. M.C.N. has received a research grant from Novocure, honoraria for consulting, or lectures from WISE and MSD and Osteopore. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Velz, J., Freudenmann, L.K., Medici, G. et al. Mapping naturally presented T cell antigens in medulloblastoma based on integrative multi-omics. Nat Commun 16, 1364 (2025). https://doi.org/10.1038/s41467-025-56268-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56268-0
