
Abstract
Enhancers serve as pivotal regulators of gene expression throughout various biological processes by interacting with transcription factors (TFs). While transcription factor binding sites (TFBSs) are widely acknowledged as key determinants of TF binding and enhancer activity, the significant role of their surrounding context sequences remains to be quantitatively characterized. Here we propose the concept of transcription factor binding unit (TFBU) to modularly model enhancers by quantifying the impact of context sequences surrounding TFBSs using deep learning models. Based on this concept, we develop DeepTFBU, a comprehensive toolkit for enhancer design. We demonstrate that designing TFBS context sequences can significantly modulate enhancer activities and produce cell type-specific responses. DeepTFBU is also highly efficient in the de novo design of enhancers containing multiple TFBSs. Furthermore, DeepTFBU enables flexible decoupling and optimization of generalized enhancers. We prove that TFBU is a crucial concept, and DeepTFBU is highly effective for rational enhancer design.
Similar content being viewed by others

Introduction
Precise regulation of gene expression is essential in complex biological processes such as development and differentiation1,2. Central to this regulation is the binding of transcription factors (TFs) to enhancers3,4. Deciphering the activity of enhancers5,6 provides insight into the fundamental principles of gene regulation7 and facilitates the design of synthetic enhancers for manipulating gene expression in genetic engineering and gene therapy applications8,9. Conventionally, the transcription factor binding sites (TFBSs), particularly TF binding motifs that usually range from 5 to 20 base pairs, are acknowledged as key determinants of TF binding to enhancers10,11,12. Studies have shown that arranging multiple TFBSs within a DNA sequence can improve its function as an enhancer13,14,15. The orientation and sequential order of TFBSs also significantly influence enhancer activity16. Leveraging these properties, manipulating TFBS arrangements and combinations on DNA sequences is widely used in designing synthetic enhancers17,18.
However, DNA sequences with identical TF binding motifs can exhibit different TF binding behaviors across the genome12. This variability emphasizes the critical role of the context sequences surrounding these motifs in determining TF binding efficacy and enhancer activity19,20,21,22,23,24. For example, short tandem repeats within these context sequences may influence TF binding by directly interacting with TFs21 and acting as DNA antennae to attract them21,23,25. Intrinsically disordered regions (IDRs) of TFs help recognize certain context sequences to facilitate TF binding26. Factors such as the local DNA shape22,27,28,29, the presence of weak binding sites30,31,32, and other TFBSs within the context sequences22,33 may also collectively influence TF binding. Our recent research demonstrated that enhancer activity can be significantly improved by designing sequences between TFBSs to fit the common characteristics of strong enhancers34. These findings collectively highlight the limitation of models that focus solely on TF binding motifs30,35,36. However, unlike TF binding motifs, which can be explicitly described and visualized, the complex features within the context sequences make it difficult to assess their impact on TF binding and enhancer activity. Therefore, there is an urgent need for a quantitative method to characterize the impact of context sequences on TF binding and enhancer activity across different TFs and cell types. This would facilitate the understanding of gene regulation and the rational design of full-length synthetic enhancers with desired functions.
To address these challenges, we proposed the concept of the transcription factor binding unit (TFBU) for modularized enhancer modeling and design. The TFBU concept integrates the effect of the core TFBS and its surrounding context sequences (TFBS-context). This integration enables the quantitative evaluation of a long DNA sequence’s potential to bind targeted TF and drive transcription (enhancer activity) in specific cell types. Using deep learning models, we identified key patterns in the TFBS-context from chromatin immunoprecipitation sequencing (ChIP-seq) data that influence TF binding. These models facilitate the rational design of synthetic enhancers with desired functions, expanding the design scope for specific TFs from core TFBSs to entire enhancer sequences. Based on the TFBS-context models, we developed a series of enhancer design methods and integrated them into a toolkit named DeepTFBU.
We measured the enhancer activity of over 36,000 sequences designed by DeepTFBU to validate our enhancer modeling and design strategy under various scenarios. Our results demonstrated that the sequence patterns of functional TFBS-contexts can be specific to TFs and cell types. Designing the context sequence within a TFBU can significantly modulate the enhancer activity for most TFs (82.9%, 97/117). For a single TFBU, such design can achieve an average enhancer activity increase of over 20-fold without introducing other obvious TFBSs. It can also produce cell type-specific enhancer activity up to 60-fold. Moreover, the TFBU-based strategy can be extended to the de novo design of enhancers containing multiple TFBSs. We showed that designing context sequences can improve the activity of typical synthetic enhancers containing tandem repeats of TFBSs by over 30-fold. Finally, we proposed a flexible and generalized framework for enhancer optimization by decoupling the enhancer effect into individual TFBUs. Using this framework, we elevated the activity of the cytomegalovirus (CMV) enhancer by 60% with only a few mutations. This demonstrates that enhancer sequences can be modularized into TFBUs and redesigned with TFBUs in an intuitively understandable and modularized bottom-up manner. All these results proved that the TFBU is a crucial concept for modeling and designing enhancers and DeepTFBU is a powerful toolkit for rational enhancer design.
Results
Overview of DeepTFBU
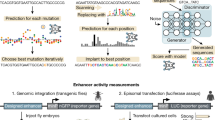
We proposed the concept of TFBU to quantify the capability of DNA sequences to be bound with TFs and the subsequent impact on enhancer activity. This concept forms the foundational support for the DeepTFBU toolkit. In this work, the typical length of TFBUs was set to be 168 base pairs, based on the restriction of massively parallel reporter assays (MPRA) experiment and the model performance (see Section “Methods”). Each TFBU consists of two parts: the core TFBS and its context sequence denoted as TFBS-context (Fig. 1a). To evaluate the binding effect of a TFBU, we separately quantified the matching scores of both parts for a certain TF. The matching score of the core TFBS is directly calculated using a position probability matrix (PPM, see Section “Methods”). Unlike the core TFBS, the TFBS-context includes complex and implicit features that cannot be directly quantified. To address this, we employed deep learning models to extract the TF-specific preferences for TFBS-context in the human genome, thus obtaining the TFBS-context matching score.

a Illustration of the TFBU. A TFBU is defined as a fragment of DNA sequence that consists of two parts: the core TFBS and the TFBS-context. b Illustration of selecting training samples for the TFBS-context model. Both positive and negative samples were from accessible genome regions with high motif PPM matching scores. The GC content distribution and the histone modification state were balanced between positive and negative samples. Then the core TFBS in these DNA fragments were masked to form TFBS-context datasets for the deep learning model. c Optimizing TFBS-contexts by genetic algorithm with the guidance of the TFBS-context model. The TFBS-contexts deep learning model is TF specific, and was separately trained for each TF. d Illustrations of evaluating the TFBU’s function. The TFBUs were inserted into the plasmid as enhancers to validate their enhancer activity. e Illustrations of tasks based on the concept of TFBU.
The deep learning model takes one-hot encoded TFBS-context sequences as input and processes them through several layers. First, 1D convolution layers followed by 1D pooling capture local sequence patterns. These features are then passed through a bidirectional LSTM layer, which captures long-range dependencies in both directions along the sequence. Finally, Dense Blocks integrate these features and output the TFBS-context matching score (Fig. 1c, Supplementary Fig. 1). This architecture enables the model to capture both local and global features of the TFBS-context, enabling us to quantitatively evaluate whether certain TFBS-context sequences facilitate TF binding and consequently impact enhancer activity.
In training the TFBS-context deep learning model, we utilized ChIP-seq data as the primary information source. ChIP-seq data directly reflect the binding states of a certain TF across the genome, thereby indicating the binding preference of this TF. We obtained ChIP-seq data for 198 TFs in the HepG2 cell line from the ENCODE database37. To identify sequence patterns conducive to TF binding, we selected positive samples within the peaks identified by ChIP-seq and negative samples outside these peaks. We created a TFBS-context dataset for each TF by constructing candidate samples and balancing properties between positive and negative samples (Fig. 1b, see Section “Methods”). We trained 198 TF-specific TFBS-context models to capture the binding preference and quantitatively evaluate the TFBS-context score for the corresponding TF. The evaluation on the test set showed the binding preference patterns of each TF were stably captured by the model (with the area under curve (AUC) values significantly higher than 0.5, one-tailed t-test p-value < 0.05, mean AUC values ranging from 0.5611 to 0.9595, Supplementary Data 1).
Since TF binding preference impacts enhancer activity12, TFBUs can serve as an elementary feature for enhancer design. We implemented this by combining TFBS-context models with a genetic algorithm (Fig. 1c). For a given DNA sequence segment, the TFBS-context model assigns a matching score. This score forms the basis for a task-specific evaluation metric. At each optimization step, the genetic algorithm introduces mutations and recombination into the sequences. The TFBS-context model then calculates the evaluation metric for each sequence, selecting those with higher scores to proceed to the next round. Through successive cycles of mutation, recombination, and selection, the algorithm converges on a set of optimized sequences, which serve as designed enhancers. This algorithm is flexible and can accommodate various optimization objectives. Based on this algorithm, we developed a series of enhancer design strategies and integrated them into the DeepTFBU toolkit (see Section “Methods”). To validate our design methods and to avoid the complexities inherent in the genome, we constructed the designed sequence into plasmids for enhancer activity measurement (Fig. 1d).
In the following sections, we will evaluate the function of TFBUs in enhancer modeling and design using DeepTFBU (Fig. 1e). First, we designed enhancers with a single TFBU and demonstrated that designing TFBS-context can modulate enhancer activity for most TFs. Next, we successfully designed cell type-specific enhancers by introducing cell type-specific TFBS-context models. We then designed enhancers containing multiple TFBSs by applying joint optimization of TFBUs with the TFBS-context model. Finally, we deciphered the enhancer function using a flexible framework based on TFBS-context model and optimized the existing strong enhancer with this framework. All these designs were validated with biological experiments.
Manipulating TFBU sequences can significantly regulate enhancer activity
To measure the effect of TFBUs on enhancer activity, we conducted an MPRA experiment. We chose ELF1, HNF1A, and HNF4A, known as key regulators in the HepG2 cells16,38, to validate the effect of the core TFBS and the TFBS-context separately. To validate the effect of the core TFBS, we selected mutated core TFBS sequences for each TF and inserted them into various TFBS-contexts (see Section “Methods”). Consistent with previous reports, the enhancer activity was correlated with the core TFBS match score. We observed a significant correlation between the mean enhancer activity and the core TFBS match score in the logarithmic scale. (Pearson correlation coefficients (PCCs): 0.7410 for ELF1, 0.9360 for HNF1A, 0.7899 for HNF4A, Supplementary Fig. 2a). Besides, the relative ranking of these different core TFBSs tended to be similar with different TFBS-contexts (Supplementary Fig. 2b). This suggests that the effect of the core TFBS on enhancer activity is generally consistent with different context sequences.
To determine whether designing TFBS-context can impact enhancer activity, we generated a series of high-scoring TFBS-context sequences for the selected three TFs using the TFBS-context model (see Section “Methods”). As controls, we also selected two genome region sequences previously validated to have no enhancer activity in the HepG2 cell line13,16. Employing the same core TFBS (the consensus motif sequence), the measured enhancer activities of the optimized TFBS-context for ELF1 and HNF1A were significantly higher than those of the control sequences (fold change: 3.31 for ELF1, 3.30 for HNF1A, one-tailed t-test p-value < 0.05, Fig. 2a). To further investigate the relationship between TFBS-context score and the enhancer activity, we generated a range of TFBS-context sequences and selected TFBS-context sequences from the genome with varying scores (see Section “Methods”). The method for designing TFBS-context with aimed scores is incorporated as the SigTFBU module in DeepTFBU). The MPRA experiment results showed a strong correlation between the mean enhancer activity and the TFBS-context scores of sequences (PCC: 0.8091/0.8444 for ELF1 generated/genome TFBS-context, 0.9670/0.9344 for HNF1A generated/genome TFBS-context, 0.9699 for HNF4A genome TFBS-context, Fig. 2b, d). We noticed that the generated TFBS-context sequences for HNF4A with the highest scores collapsed into repeated fragments of the HNF4A TFBS, resulting in a notable decrease in enhancer activity. After filtering out sequences with all obvious TFBSs in TFBS-context (see Section “Methods”), the optimized HNF4A TFBS-context showed significantly higher activity compared to the control sequences (fold change: 2.87, one-tailed t-test p-value < 0.05, Fig. 2a), and the PCC between the mean enhancer activity and the scores of generated TFBS-contexts for HNF4A also significantly improved (PCC from 0.6781 to 0.9626, Fig. 2c). Notably, the enhancer activity of optimized TFBS-contexts for ELF1 and HNF1A was not significantly affected after filtering out sequences with obvious TFBSs (Fig. 2a), and the PCC between mean enhancer activity and TFBS-context score was also consistent (from 0.8091 to 0.8136 for ELF1, from 0.9670 to 0.9632 for HNF1A). These results demonstrated that designing TFBS-context can significantly modulate the enhancer activity, and the function of TFBS-contexts could be achieved without the presence of other obvious TFBSs.

a The enhancer activity measured from the MPRA experiment. The control denotes control backbone sequences, with/without a diagonal shadow in control bars denotes the control backbone is inserted/not inserted with a core TFBS; the optimized denotes optimized TFBS-context inserted with a core TFBS; without other TFBS means the optimized TFBS-contexts with other obvious TFBS in them were filtered out; negative control denotes negative control sequences from genome, including control backbone sequences not inserted with a core TFBS. Each sample in optimized groups is the average of three biological replicates for a TFBU. Each point in control groups denotes one biological replicate for the control sample. The box plots in a show the median (center line), interquartile range between Q1 and Q3 (box limits), minimum and maximum values within 1.5 times the IQR from Q1 and Q3 (whiskers), and outliers (hollow circles). b–d The enhancer activity (y-axis) of TFBUs (TFBS-context with consensus motif as core TFBS) with different predicted values from models (x-axis). Each point denotes the average of three biological replicates for a TFBU. The model-generated TFBS-contexts are shown in b, c and genome TFBS-contexts are shown in d. The gray points denote TFBS-contexts with obvious TFBSs in them and were ignored when calculating the PCC in (c). Two outliers with extremely large measured enhancer activity were removed in (d). e Top: the illustration of points in the volcano plot. Bottom left: The volcano plot for the enhancer activity of optimized TFBS-context against the control backbone. Each point denotes the result for a TF. Bottom right: The volcano plot after filtering out TFBS-context with obvious TFBS in them. Error bars in a–d denote mean ± s.d. of corresponding groups. Source data are provided as a Source Data file.
To understand the key DNA features contributing to the enhancer activity of TFBUs, we conducted further analysis focusing on the patterns in TFBS-context, including the TFBSs of other TFs, the DNA shape patterns, the count of weak binding sites of the core TF, and the DNA secondary structure features. Since it is reported that the TFBSs of HNF4A and FOXA2 tend to show co-occurrence in natural functional regulatory elements39, we first analyzed the FOXA2 binding sites in the TFBS-context of HNF4A. In the HNF4A’s TFBS-contexts with the top 10% enhancer activity, we observed significantly higher FOXA2 TFBS matching scores compared to the bottom 10% (independent one-tailed t-test for the max matching score, p-value < 0.05, Supplementary Fig. 3a top). This observed trend aligned with the model predicted value (independent one-tailed t-test for max matching score, p-value < 0.05, Supplementary Fig. 3a middle and bottom), indicating that the model extracted such synergistic effect accumulated in the process of evolution, which is involved in the TFBS-context containing other TFBSs. Subsequently, we extended the analysis to encompass the TFBS signal of all TFs within the TFBS-contexts containing obvious TFBSs (see Section “Methods”). Visualization of the TFBS signal in TFBS-contexts revealed distinct patterns between those TFBS-contexts with high and low enhancer activity (Supplementary Fig. 3b). Several TFs with varying TFBS matching scores have been previously reported to interact with the core TF of the corresponding TFBU40,41 (e.g., the KLF family with ELF1, NR2F2 with HNF4A; Supplementary Table 1). Furthermore, it is reported that the DNA shape of the context sequence surrounding the TFBS and the presence of weak binding sites may influence TF binding22,28,29,30,31,32. Therefore, we analyzed these two features within the TFBS-context sequences. The result showed both the DNA shape feature (Supplementary Fig. 3c) and the count of weak binding sites of the core TF (independent two-tailed t-test p-value < 0.05, Supplementary Fig. 3d. see Section “Methods”) exhibited differences between TFBS-contexts with high and low enhancer activity. We also analyzed the R-loop features and non-B motif features of the DNA sequences (see Section “Methods”). We found that, for ELF1 and HNF1A, the probability of R-loop formation in high enhancer activity TFBS-contexts is significantly lower than in low enhancer activity TFBS-contexts (independent two-tailed t-test p-value < 0.05). HNF4A exhibits an opposite trend, but the effect is not as significant as the other two TFs (see Supplementary Data 2). In the analysis of non-B motifs within the DNA sequences, we observed that in ELF1, the proportion of TFBS-contexts containing G-quadruplex motifs is significantly higher in those with low enhancer activity, while in HNF4A, the proportion of TFBS-contexts containing short tandem repeat motifs is higher in those with high enhancer activity (two-tailed Fisher’s exact test p-value < 0.05). These results collectively indicated that explicit features like TFBS composition, along with known and unknown implicit or weak features such as DNA shape patterns, the count of weak binding sites of the core TF and DNA structure features, may all contribute to the enhancer activity of the entire TFBU.
The effect of TFBS-context on enhancer activity is significant for most TFs
We have demonstrated that designing TFBS-context can significantly modulate enhancer activity for the selected three TFs. Consequently, we investigated whether such effects could be generalized to other human TFs. We first tested if high enhancer activity TFBS-contexts are exchangeable across different TFs. We selected several high-scoring TFBS-contexts and exchanged their core TFBSs to form TFBUs (see Section “Methods”). The experiment results revealed that the TFBS-contexts yielding strong enhancer activity varied among different TFs (Spearman’s rank correlation coefficient: 0.6880 between ELF1 and HNF1A, 0.7221 between ELF1 and HNF4A, 0.6743 between HNF1A and HNF4A, Supplementary Fig. 4). Given that enhancer activity generated by TFBS-context appears TF-specific, we then aimed to verify if the influence of TFBS-context on enhance activity is universal for TFs. We designed another MPRA library containing TFBS-contexts of more TFs. From 198 TFs with available ChIP-seq data of HepG2 cells in the ENCODE database, we selected 118 TFs by removing TFs with similar binding motifs. For these 118 selected TFs, we generated high-scoring TFBS-context sequences with the TFBS-context model and aimed to compare their enhancer activity with the control sequences13,16 as described in the above section (see Section “Methods”).
The MPRA experiment results indicated that the enhancer activity generated by high-scoring TFBS-context for 105 out of 117 TFs (one TF missed when constructing the library) exhibited a significant increase compared to the control sequences (89.74%, one-tailed t-test p-value < 0.05, fold changes range from 1.16 to 22.03, Fig. 2e bottom left). These results demonstrated that the enhancer activity of TFBU can be significantly modulated by designing TFBS-context for most TFs, and the TFBS-context model successfully captured the patterns conducive to transcription for these TFs.
We then tested whether TFBS-context could still function without the presence of other obvious TFBSs. After filtering out TFBS-contexts containing other obvious TFBSs (FIMO42 adjusted p-value threshold 1e-4, see Section “Methods”), the enhancer activity increased significantly for 55 out of 68 TFs (80.88%, one-tailed t-test p-value < 0.05, fold changes range from 1.16 to 20.95, Fig. 2e bottom right). We also filtered out TFBS-contexts containing other TFBSs with a stricter criterion (FIMO adjusted p-value threshold 2e-3, see Section “Methods”), the result showed the enhancer activity increased significantly for 72 out of 90 TFs after the stricter filtering (80.00%, one-tailed t-test p-value < 0.05, fold changes range from 1.14 to 5.10, the range of samples being statistically analyzed has been expanded due to the insufficient number of samples, see Section “Methods”). This reaffirmed that the enhancer function of TFBUs could be achieved without the presence of other obvious TFBSs within the TFBS-context. For some TFs, their generated TFBS-contexts all contained other TFBSs. This may be due to that other TFBSs exist near their core TFBSs in the natural genome, and such features were captured by the deep learning model.
The TFBS-context can generate cell type-specific enhancer activity
After verifying the TF-specific effect of TFBU, we investigated whether the same TFBU had different effects across cell types, thereby facilitating the design of cell type-specific enhancers. To compare the binding preference of TF in different cell types, we trained additional TFBS-context models using ChIP-seq data obtained from the K562 cell line. Using the previously examined ELF1 as an example, we observed a significant correlation between the TFBS-context scores derived from the model trained on K562 data and the model trained on HepG2 data (PCC: 0.7259, Supplementary Fig. 5). However, notable distinctions were also observed in the preferred TFBS-context sequences between these two cell lines, which were effectively captured and modeled by our TFBS-context models. This contrasts with the core TFBS, which usually exhibits no significant differences between cell types. Consequently, we leveraged the discrepancies in ChIP-seq data across different cell types for a given TF to design TFBS-contexts with cell type-specific enhancer activity.
To assess the cell type-specificity of a particular TF, three models were built to evaluate TFBS-contexts. The first and second models were TFBS-context models trained on HepG2 and K562 data, respectively, to evaluate the functionality of TFBS-context in specific cell types. The third model was the HepG2 vs K562 model, which was designed to evaluate whether the TFBS-context met the requirements for cell type specificity. To generate cell type-specific TFBS-contexts, the genetic algorithm was applied by utilizing the combination of outputs from these three independent models as the loss function (see Section “Methods”, Fig. 3a). The method for designing cell type-specific enhancers is incorporated as the CSpTFBU module in DeepTFBU.

a Illustration of measuring TFBS-context’s cell type-specificity with deep learning models. b Measured enhancer activity of TFBUs in different cell lines. The generated denotes the designed TFBS-context with the models; genome denotes TFBS-context selected from the genome sequence by the models; -SC denotes the core TFBSs were scrambled. The fold value is the mean activity fold change across cell types. The p-values are based on a paired one-tailed t-test, where ns represents not significant. c The fold change of the enhancer activity of TFBUs across cell types. The yellow bar denotes the TFBU’s core TFBSs were scrambled. The fold value denotes the mean effect on the fold change caused by scrambling the core TFBS. The p-values are based on a paired one-tailed t-test, where ns represents not significant. d The fold change of the enhancer activity across cell types before/after filtering out TFBUs with obvious TFBSs in their TFBS-context. The fold value was calculated with the mean value of each group. The box plots in b–d show the median (center line), interquartile range between Q1 and Q3 (box limits), minimum and maximum values within 1.5 times the IQR from Q1 and Q3 (whiskers), and outliers (hollow circles). Each point in b–d denotes the average of three biological replicates. Source data are provided as a Source Data file.
To evaluate the cell type-specific enhancer activity achieved through our design method, we selected four TFs with high activity in HepG2 or K562 cell lines according to the model performance and the MPRA experiment result. Initially, for each TF, we designed 10 cell type-specific TFBS-contexts for a certain cell type and selected 10 cell type-specific TFBS-contexts from the genome with models. These TFBS-contexts were all inserted with the consensus motif as core TFBS to form TFBUs. We examined their enhancer activity in both types of cells using flow cytometry (see Section “Methods”). The experiment result showed both the generated and selected TFBS-contexts exhibited significant cell type-specific enhancer activity in the expected cell line for all four TFs (paired one-tailed t-test p-value < 0.05, mean fold changes range from 7.63 to 27.43, max fold change of single TFBS range from 27.51 to 80.29, Fig. 3b, c).
Subsequently, to ensure the cell type-specific enhancer activity of TFBUs is achieved by manipulating the flanking sequences of the core TFBS rather than generating functional sequences independent of the core TFBS, we scrambled the core TFBS of each TFBU (see Section “Methods”). This resulted in a noticeable decrease in both enhancer activity levels and fold change (fold change decrease range from 2.18 to 19.94, Fig. 3b, c), indicating that the cell type-specific enhancer activity of TFBUs depends on the presence of the corresponding core TFBS rather than solely on TFBS-context. Additionally, to ensure the cell type-specificity was not generated by simply adding other TFBSs, we filtered out TFBUs containing other obvious TFBSs in their TFBS-context (FIMO adjusted p-value threshold 1e-4, see Section “Methods”), and the cell type-specific enhancer activity was not significantly influenced (independent one-tailed t-test p-value > 0.1, Fig. 3d). We then filtered out TFBUs containing TFBSs in their TFBS-context with the stricter criterion (FIMO adjusted p-value threshold 2e-3, see Section “Methods”). Among all TFBUs tested for cell type-specificity, only five samples were retained after the stricter filtering, but they still showed cell type-specificity (fold changes range from 3.70 to 71.21). All these results demonstrated that TFBUs can generate cell type-specific enhancer activity by designing proper TFBS-contexts, and the cell type-specificity of TFBUs can be achieved without the presence of obvious TFBSs in their TFBS-context.
Joint optimization of TFBUs enables the de novo design of enhancers containing multiple TFBSs
In the preceding sections, we demonstrated the significant impact of a single TFBU on enhancer activity. Here, we aimed to elevate the enhancer activity of DNA sequences containing multiple core TFBS using the TFBS-context model. Enhancer sequences typically contain several potential TFBS. These binding sites, along with their respective TFBS-contexts, may overlap with each other. This means that the same sequence segment may be shared by multiple TFBUs and will occupy different positions within different TFBUs. To maximize overall enhancer activity, sequence design should optimize all TFBUs simultaneously. Therefore, each position should be evaluated based on the scores of all overlapping TFBS-contexts, ensuring compatibility among key TFBSs and elevating total enhancer activity. We explored whether multiple TFBUs within the DNA sequence could be individually addressed yet jointly optimized to generate DNA sequences with enhanced enhancer activity (Fig. 4a, b).

a Illustration of jointly designing multiple TFBU. b Illustration of enhancer sequences in different groups. c The measured enhancer activity of different groups. Error bars denote mean ± s.d. for three biological replicates. d The measured enhancer activity distribution. The fold value was calculated with the mean enhancer activity of each group. The dashed line denotes the enhancer activity of the baseline. The p-values are based on a one-tailed t-test, where ns represents not significant. The box plots show the median (center line), interquartile range between Q1 and Q3 (box limits), minimum and maximum values within 1.5 times the IQR from Q1 and Q3 (whiskers), and outliers (hollow circles). Each point denotes the average of three biological replicates. Source data are provided as a Source Data file.
Previous work reported that stronger enhancer activity could be achieved by arranging multiple TFBSs within a DNA sequence13,14,15. Further studies extended such strategy into more specialized applications43 and proposed certain interval sequences for the design of synthetic enhancers44. Here we adopted the interval sequence between tandem repeats of TFBSs proposed in previous work43,44 as the baseline (Fig. 4b top). We fixed eight core TFBSs on the given sequence and optimized the TFBS-context of these TFs while keeping the core TFBSs unchanged. Through the implementation of joint optimization (see Section “Methods”), we obtained optimized enhancers containing multiple high-scoring TFBUs (Fig. 4b middle). Specifically, we selected four TFs exhibiting significantly improved predicted enhancer activity for optimization. The flow cytometry results showed that for 3 out of the 4 selected TFs, the mean enhancer activity of the context-optimized sequences was significantly higher than that of the baseline sequence (one-tailed t-test p-value < 0.05, mean optimized fold change: 13.01 for GATA2, 2.20 for ONECUT2, 2.01 for SIX1, Fig. 4c, d). Moreover, there were consistently context-optimized sequences exhibiting higher enhancer activity compared to the baseline sequence (fold change for highest optimized enhancer: 43.16 for GATA1, 3.68 for ONECUT2, 5.79 for SIX1, 1.71 for TEAD3, Fig. 4c).
Next, we asked whether stronger enhancer activity could be achieved with fewer core TFBSs and thus a larger context sequence design space (Fig. 4b bottom). We reduced the number of fixed core TFBSs by half and conducted optimization on the enhancers. Remarkably, for all four selected TFs, the context-optimized sequences with only half the number of core TFBSs exhibited significantly higher mean enhancer activity compared to the baseline sequence (one-tailed t-test p-value < 0.05, mean optimized fold change: 31.42 for GATA2, 3.50 for ONECUT2, 3.48 for SIX1, 1.98 for TEAD3, fold change for highest optimized enhancer: 109.38 for GATA1, 7.34 for ONECUT2, 6.28 for SIX1, 3.59 for TEAD3, Fig. 4c, d). Additionally, they tended to show higher enhancer activity compared to sequences with more core TFBSs (fold change of mean enhancer activity: 2.41 for GATA1, 1.59 for ONECUT2, 1.73 for SIX1, 3.24 for TEAD3, Fig. 4d). These observations suggested that the enhancer activity can be elevated by simultaneously designing the TFBS-context of multiple core TFBSs, and higher enhancer activity could be achieved with a proper balance between the number of core TFBSs and the design space. The method for the de novo design of enhancers containing multiple TFBSs is incorporated as the DeMulTFBU module in DeepTFBU.
TFBU-based modeling promises a flexible framework for enhancer decoupling and optimization
In the generalized form, enhancers typically contain multiple TFBSs of various TFs, and enhancer sequences lengths may vary considerably. In practical applications, modeling and optimizing enhancers of varying lengths present significant challenges. Enhancer activity data obtained through high-throughput strategies such as MPRA typically have constraints on sequence lengths, making it difficult to accurately predict and optimize the activity of longer enhancers with the currently available datasets. To fit this generalized scenario, we decoupled enhancers into basic units with TFBUs and developed a flexible framework for modeling and optimizing enhancers beyond the length constraint.
We first generated TFBU-based features to decouple enhancer functions. As discussed earlier, both the matching score of core TFBS and the score of TFBS-context contribute to the enhancer effect of a TFBU. Therefore, we used these scores to form the basic feature of enhancers for estimating enhancer activity (Fig. 5a). For a given enhancer sequence, we identified the position with the highest core TFBS matching score for each of the 198 TFs with available ChIP-seq data and extracted their corresponding TFBS-context. By multiplying the core TFBS score with the corresponding TFBS-context score, we obtained the overall TFBUs score. The TFBU-based feature of this given enhancer sequence can be represented by concatenating all TFs’ core TFBS score, TFBS-context score, and the overall TFBUs score.

a Illustration of decoupling the enhancer effect into individual TFBU effects. b The performance of modeling the enhancer activity with different features. The dashed lines denote modeling enhancers with natural language processing models. Error bars denote mean ± s.d. for ten computational replicates. c The measured enhancer activity of optimized CMV enhancers. Error bars denote mean ± s.d. for three biological replicates. d Comparison of enhancer activity between optimized CMV enhancers with one nucleic acid base mutation and more mutations for higher predicted score. The p-value is based on a one-tailed t-test. The box plots show the median (center line), interquartile range between Q1 and Q3 (box limits), and minimum and maximum values (whiskers). Each point denotes the average of three biological replicates. e The importance of each nucleic acid base in CMV enhancers evaluated by the model and important regions as key TFBSs. f The one nucleic acid base mutation caused the highest enhancer activity increase strengthened the core motif signal of RFX5. g The mutation caused a high enhancer activity increase during the greedy optimization strengthened the core motif signal of NFIB. Source data are provided as a Source Data file.
To evaluate the efficacy of our decoupling strategy, we extracted the TFBU-based feature of DNA sequences in the MPRA library containing 118 TFs’ TFBUs and trained a linear regression model to predict the enhancer activity of DNA sequences. Results on the independent test set showed that the performance of the linear regression model using TFBU-based features is even better than that of typical deep learning models using original sequences as input34,45 (PCC: 0.7942 ± 0.0002 Fig. 5b). We also used samples for a specific TF as the independent test set to evaluate the model’s performance on unseen TFs. The results demonstrated considerable variability in the performance across different TFs (Supplementary Fig. 6). The linear regression model using TFBU-based features still outperformed the deep learning model that used original sequences as input. This indicates that the enhancer sequence can be decoupled into basic units made up of TFBUs effectively. Together, all TFBS-context models and the linear model form a comprehensive TFBU-based framework for enhancer modeling and optimization, which is incorporated as the ModOptTFBU module in DeepTFBU.
To validate the function of our TFBU-based framework in enhancer optimization, we conducted experiments to optimize the CMV enhancer by introducing mutations guided by the framework46. Initially, we visualized the contribution of each nucleotide base of the CMV enhancer as determined by the framework. The results revealed that the framework could identify several significant motifs on the original CMV enhancer (Fig. 5e). Subsequently, we selected the top 10 mutations predicted to elevate the enhancer activity and constructed corresponding mutant enhancers for experimental evaluation. The mean enhancer activity of 8 mutant enhancers was found to be higher than that of the original enhancer (with the highest enhancer activity elevated to 1.41-fold, Fig. 5c). Analysis of the framework-learned features indicated that the mutant enhancer with the highest mean enhancer activity primarily increased the activity by enhancing the core TFBS score of the key TF RFX547 in HepG2, which is also validated by visualizing the contribution of each nucleotide base (Fig. 5f). Additionally, we also observed one mutation successfully increased enhancer activity mainly by enhancing the TFBS-context score of HNF4A. Finally, we introduced more mutations to achieve higher predicted scores greedily with up to 15 mutations and tested 10 optimized CMV enhancers with experiments. The activity of the optimized CMV enhancers showed a significant improvement (independent one-tailed t-test p-value < 0.05, with the highest enhancer activity elevated to 1.59-fold, Fig. 5c, d). We also observed that the score of typical core TFBSs of key TFs in HepG2, such as NFIB48 (Fig. 5g), contributed to the increase in enhancer activity by taking both the effect of core TFBS and TFBS-context into account. All these results demonstrated that the enhancer activity can be improved with the framework based on the TFBU effects.
Discussion
In this work, we proposed the concept of TFBU to modularly model and design enhancers. We successfully quantified the TF-specific and cell type-specific functions of TFBS-context using deep learning models. Our results indicated that designing TFBS-context can regulate enhancer activity for most TFs, and the TFBU-based methods enable enhancer design for various situations. These findings demonstrated that the TFBU can serve as a fundamental concept for enhancer modeling and design. We effectively decoupled enhancer sequences into basic units composed of TFBUs, providing a perspective for understanding transcription initiation. Based on the TFBU feature, we can develop a quantified system to describe and comprehend the fundamental code of life.
The length of TFBU was set to be 168 bp in this study, which was jointly determined by the model’s performance and the constraints of MPRA experiments. Theoretically, longer sequences can provide more information. However, excessively long sequences may introduce noises unfavorable to sequence design and synthesis. As shown in Supplementary Fig. 7, the 168 bp length appears to contain substantial relevant information for predicting enhancer activity without introducing significant noise associated with excessively long sequences. However, this does not imply that 168 bp is the optimal length. The optimal TFBU length is difficult to determine in practice, and it may also vary for different TFs. Our explorations of various lengths for TF binding predictions reveal that the elbow points differ among TFs, with a range from 100 bp to 300 bp as shown in Supplementary Fig. 8. We conclude that while the 168 bp length is effective for most TFs, the optimal TFBU length remains to be determined through further systematic studies.
We demonstrated that properly designed TFBUs could generate significant cell type-specific transcription activity. The divergence in cofactors or post-translational modification of TFs across cell types may be the source of such specificity49. We applied the TFBS-context model to analyze an enhancer with liver cancer cell-specific activity, which was validated in vivo50. The result showed that for the key TF HNF4G in hepatocytes, its TFBS-context score in HepG2 cells was nearly maximal (0.9998), while in healthy liver cells, the score was below 0.6. This indicates the potential of TFBU for in vivo applications. We also verified that joint optimization of TFBUs is effective in the de novo design of enhancers containing multiple TFBSs. These approaches can be combined for rational enhancer design in complicated and crucial tasks, such as constructing artificial gene circuits and precision therapies, thereby demonstrating strong potential for practical applications.
When designing enhancers containing multiple TFBSs, we found that simply increasing the number of TFBSs does not necessarily result in stronger enhancer activity. There is a trade-off between the number of TFBSs and the design space within the same sequence length. Furthermore, for different TFs, whether to introduce adjacent TFBS within their TFBS-context may also vary. For some TFs, their TFBS-context tends to include homologous or synergistic TFBS. This design strategy is more suitable for these TFs. We observed that, for certain TFs, their high-scoring TFBS-contexts frequently contain TFBSs that resemble those of their own (see Section “Methods”, Supplementary Data 3). This suggests that for these TFs, the presence of similar binding sites around their TFBS increases the likelihood of binding to the DNA sequence. On the other hand, the experimental results showed that, for certain TFBSs, there is a tendency for other distinct TFBSs to appear in their TFBS-context. These observations suggest the potential for interactions between these TFs in vivo, and indicate that the TFBS-context model has the potential to capture the synergistic functions of different TFs.
We successfully decoupled and optimized the existing strong enhancer using our TFBU-based framework. This framework is not restricted by a specific sequence length, and as TFBU data accumulates, the efficiency and quality of such optimization will continue to improve. This decoupling and optimization is designed for the general form of enhancers. For applications with strong prior knowledge, such as when there are multiple TFBSs for a particular TF on an enhancer, although the information of these binding sites will be reflected in the TFBS-context scores, the sequence optimization effect may not be as good as the joint optimization of multiple TFBS-contexts mentioned above. Therefore, when there are several known crucial TFBSs on an enhancer sequence, using the joint optimization of multiple TFBU would be more appropriate. Users can choose the proper strategy in the DeepTFBU toolkit according to the specific situation for aimed enhancer design.
During transcription, the binding of TFs is crucial12. By evaluating the binding signal across the genome, we made the explicit and implicit factors in TFBS-context measurable. When searching for binding sites, TFs typically undergo a process involving three-dimensional diffusion and one-dimensional search, during which non-specific binding occurs before specific binding51. Some studies suggest that when TFs search for potential binding sites, certain context sequences could attract TFs to relatively proximal regions or prolong the time TFs spent in corresponding regions23, resulting in different binding states with the same core motif. Additionally, TFs may also show the tendency of non-specific binding with certain context sequences before specifically binding to the core motif52. Proper TFBS-context may increase the binding probability of certain TF by prolonging the one-dimensional diffusion when searching for specific binding sites or facilitating the non-specific binding of TF. Our experiment results suggest that weak binding sites in the TFBS-context can affect the function of TFBU. It is conceivable that weak binding sites resembling the core TFBS may potentially exhibit synergistic effects that facilitate TF binding, and weak binding sites associated with other TFs may also enhance TF binding through various interactions. The explicit and implicit factors on the DNA sequence were captured and integrated by TFBS-context models, thus facilitating quantitative evaluation of TFBS-context and modularized rational enhancer design.
The process of gene regulation may involve the synergistic action of multiple binding events, and TFs may cooperate through direct or indirect mechanisms to interact with the enhancer3. The concept of TFBU is suitable for modeling these patterns. By estimating the matching score of each TF to the enhancer sequence, the modularized features offered by TFBU can effectively model the function of enhancers. Previous work employed machine learning and deep learning algorithms to predict TF binding and model enhancers using features like k-mers, TFBS, and biophysical information53,54,55, or by establishing end-to-end models with DNA sequences24,45,56,57,58,59. In contrast, our work introduces the idea of modularizing enhancer with functional units, and the core TFBSs and their context sequences were modeled separately. This concept evaluates the functional units for each TF, and the sequences surrounding TFBSs are flexibly reused in the evaluation for different TFs. Additionally, this approach does not impose strict requirements on the length of enhancer sequences, making it a flexible approach that can leverage existing enhancer data for different designing tasks.
In our previous work34, we successfully elevated the enhancer activity through designing sequences between TFBSs by leveraging the common characteristics of natural enhancers extracted with a deep learning model DeepSEED. This approach is applicable with the sequence data providing the general pattern information of target gene regulatory elements. In this work, we emphasized the influence of TF-specific and cell type-specific preferences for the context sequences surrounding the core TFBS. This enables detailed and precise modeling of the sequences beyond the core TFBSs and can serve as generalized fundamental features of enhancers with the support of numerous publicly available data. This method also enables the precise design of gene regulatory elements tailored to specific TFs and cell types. In future research, by leveraging more information on TFs and TFBU properties, we could construct a more detailed and comprehensive TFBU system, thereby facilitating more precise DNA sequence modeling and regulatory elements designing based on TFBU.
Methods
Cell lines
The cell lines used in this study were the liver cancer cell line HepG2 and the chronic myelogenous leukemia cell line K562 obtained from the Pricella Life Science&Technology. The cells HepG2 and K562 were maintained in DMEM (GIBCO, 11965118) and IMEM (GIBCO, 31980030) respectively, supplemented with 10% FBS (GIBCO, 16000-044), 1× NEAA (GIBCO, 11140050), and 0.5× penicillin–streptomycin (Solarbio, P1400) at 37 °C and 5% CO2. Sex was not considered in this study.
MPRA library design
The TFBU sequences in the MPRA library are all in the form of TFBS-context + core TFBS. There are two typical sources of TFBS-context: one is the TFBS-context generated by the genetic algorithm with the guidance of the deep learning model, and the other source is the TFBS-context from the genome. For TFBS-context generated by the genetic algorithm, there are already masked regions and the core TFBS can be directly inserted into the masked regions to form the TFBU sequence. For TFBS-context from the genome, when inserting core TFBSs into the genome sequences, the center part of the genome sequences was replaced by the core TFBS to form TFBU sequences.
The MPRA library of the three functional TFs (ELF1, HNF1A, HNF4A) in the HepG2 cell line contains the following sequences: (19288 in total, Supplementary Data 4).
For each TF, 2000 TFBS-contexts with different TFBS-context scores were generated with the genetic algorithm. 4000 genome TFBS-contexts with different TFBS-context scores were selected from the TFBS-context samples. The consensus motif sequence was inserted into these TFBS-contexts as the core TFBS to form TFBUs. (6000*3 TFs=18000).
To evaluate the effect of the core TFBS, for each TF, 10 mutated core TFBS sequences with the most occurrences in the ChIP-seq data were inserted into 25 TFBS-contexts with different scores as the core TFBS. (10*25*3 TFs = 750).
To validate whether the preferred TFBS-context is the same across different TFs, for each TF pair, 80 TFBS-contexts were inserted with the consensus motif sequences of the two TFs as core TFBSs. (80*2*3 TF pairs - 14 repeated=466).
Two genome region sequences previously validated to have no enhancer activity in the HepG2 cell line13,16,60,61 were selected as control backbone sequences. The consensus motif sequence was inserted into these control backbone sequences as core TFBS to form the control samples. (2*3 TFs = 6). And 66 control sequences were added to this library (including 3 additional negative control sequences13,62).
The MPRA library of the 118 TFs contains the following sequences: (18,000 in total, Supplementary Data 5).
For each TF, 20 genome TFBS-contexts were selected from the positive TFBS-context samples, and 20 genome TFBS-contexts were selected from the negative TFBS-context samples. 110 TFBS-contexts with different TFBS-context scores were generated with the genetic algorithm. (150*118 TFs =17700).
Two genome region sequences previously validated to have no enhancer activity in the HepG2 cell line13,16 were selected as control backbone sequences. The consensus motif sequence was inserted into these control backbone sequences as core TFBS to form the control samples. (2*118 TFs = 236). And 64 control sequences were added to this library.
MPRA library construction and cloning
MPRA plasmid libraries were generated as previously described63. Two libraries oligos were synthesized by TWIST Bioscience as 198 bp in length containing 168 bp of designed sequence and 15 bp of adapter sequence at both 5′ and 3′ end. Oligos were cloned into the pMPRA1 plasmid (Addgene ID no. 49349) to measure the activity of designed sequences. The plasmid and primer sequences used in this study are detailed in Supplementary Data 6.
The 20 bp barcodes were added by performing 6 emulsion PCR reactions by DNA emulsion and purification Kit (EURX, E3600). Each reaction was a 50 μL water phase in the volume containing approximately 108 ~ 109 copies of oligo, 25 μL NEBNext® Ultra™ II Q5® Master Mix (NEB, M0544S), 2 μM Library_F and Barcode_R primers, and 0.01 mg/ml of the acetylated BSA (Aladdin Scientific, A397958). The emulsion preparation process follows the manufacturer’s protocol. The 350 μL of Emulsion mixture was equally divided into 4 PCR tubes and cycled with the following conditions; 95 °C for 2 min, 20 cycles (95 °C for 10 s, 67 °C for 30 s, 72 °C for 15 s), 72 °C for 5 min. The PCR reaction was cleaned up using the attached purification kit, then concentrated with Zymo Clean and Concentrator Kit (Zymo research, D4014), eluting into 10 μL of Ultra-pure H2O.
To create pMPRA1-oligo library, barcoded oligos were inserted into sfiI digested pMPRA1 by Gibson assembly (NEB, E2621) using 1.1 μg of barcoded oligos and 1 μg of digested pMPRA1 in a 60 μL reaction incubated for 60 min at 50 °C. The reaction was cleaned with Zymo Clean and Concentrator Kit and elution in 10 μL of Ultra-pure H2O. All of the ligated vector was then transformed into 100 μL of NEB10-beta (Biomed, BC401-01) by electroporation. The estimated transformation efficiency is ~3 × 106 CFU, each oligo has ~200 barcodes on average.
The miniP-EYFP cloned into the pMPRA1-oligo to create the final library. pMPRA1-oligo plasmid was digested by AsiSI (NEB, R0630). miniP-EYFP was cloned from the plasmids with NEBNext® Ultra™ II Q5® Master Mix (NEB, M0544S) for 35 cycles using primers EYFP_F and EYFP_R at 1 μM. 6.6 μg of miniP-EYFP fragments and 2 μg of digested pMPRA1-oligo plasmid in a 100 μL Gibson assembly reaction for 90 min at 50 °C. The reaction was cleaned with Zymo Clean and Concentrator Kit and elution in 40 μL of Ultra-pure H2O. To remove the remaining uncut plasmids, the 40 U of AsiSI, 20 U of RecBCD (NEB, M0345), 20 μL 10 mM ATP, and 20 μL 1× rCutSmart Buffer were incubated in a 200 μl reaction for 4 h at 37 °C. The product was concentrated by Zymo Clean and Concentrator Kit (Zymo research, D4014) and eluted into 10 μL of Ultra-pure H2O. All of the ligated vector was then transformed into 100 μL of NEB10-beta (Biomed, BC401-01) by electroporation. The estimated transformation efficiency is >107 CFU.
MPRA transfections, RNA extraction and cDNA synthesis
In transfection experiments, approximately 6.5 × 106 HepG2 cells were plated in 15 cm cell culture plates (NEST Biotechnology, 715001) and grown to 60-70% cell density for ∼24 h. On the day of transfection, media was replaced with 30 mL fresh complete DMEM followed by transfection with 87.5 μL of Lipofectamine 3000 (Invitrogen, L3000150) and 35 μg of DNA using the manufacturer’s protocol. Cells were incubated with transfection reagents for 24 h to RNA Extraction. Three separate transfection replicates were performed.
Total RNA was extracted from cells using Qiagen RNeasy Midi Kit (Qiagen, 75144) following the manufacturer’s protocol including the on-column DNase digestion. A second DNase treatment was performed on the purified RNA using 10 μL of Turbo DNase (Life Technologies, AM2238) in 500 μL of total volume for 1 h at 37 °C. Subsequent wash steps concentrated isolated RNA with Qiagen Minelute Cleanup Kit (Qiagen, 74204) and elution in a total of 50 μL of RNA-free H2O. Next, 25 μL of isolated RNA was converted to cDNA with Transcript II one-step gDNA Removal and cDNA Synthesis SuperMix (TransGen Biotech, AH311) and a primer specific to the 3’UTR (MPRA_v3_Amp2Sc_R) in 120 μL reaction following the protocol. The cDNA reaction mixture was cleaned with Zymo Clean and Concentrator Kit (Zymo research, D4014) and elution in 30 μL of RNA-free H2O.
Sequencing preparation
To determine oligo/barcode combinations within the MPRA pool, the 4 separate amplifications with 200 ng of pMPRA1-oligo in a 100 μL NEBNext® Ultra™ II Q5® Master Mix (NEB, M0544S) PCR reaction containing 0.5 μM of MPRA_v3_Amp2Sa_Illumina_F and Illumina_Universal_Adapter_R primers under the following conditions: 98°C for 30 s, 6 cycles (98 °C for 10 s, 65 °C for 20 s, 72 °C for 30 sec), 72 °C for 2 min. The reaction was cleaned with Zymo Clean and Concentrator Kit and elution in 30 μL of Ultra-pure H2O. Sequencing indexes were then attached by Illumina_Multiplex_F and Illumina_Universal_Adapter_R primers. This process utilized 20 μL of the eluted product, and the reaction conditions were adjusted, reducing the number of cycles to 5. All of the reactions were run on a 1% agarose gel and then purified by a Multifunctional DNA purification Kit (Aidlab, DR0303).
The condition of sequencing preparation of the pMPRA1-oligo-miniP-EYFP plasmid same as the pMPRA1-oligo except the MPRA_v3_Illumina_EYFP_F and Illumina_Universal_Adapter_R primers used in the first PCR reaction.
The 10 μL of cDNA from three separate transfection replicates were amplified with NEBNext® Ultra™ II Q5® Master Mix (NEB, M0544S) using primers MPRA_v3_Illumina_EYFP_F and Illumina_Universal_Adapter_R at 0.5 μM in 50 μL reaction with the following conditions: 98 °C for 30 s, 12 cycles (98 °C for 10 s, 65 °C for 20 s, 72 °C for 30 s), 72 °C for 2 min. The reaction was cleaned with Zymo Clean and Concentrator Kit and elution in 40 μL of Ultra-pure H2O. Sequencing indexes were then attached by Illumina_Multiplex_F and Illumina_Universal_Adapter_R primers, with all of the eluted product in 100 μL reaction under the following conditions: 98 °C for 30 s, 6 cycles (98 °C for 10 s, 69 °C for 20 s, 72 °C for 30 s), 72 °C for 2 min. The reaction was cleaned with Zymo Clean and Concentrator Kit and elution in 20 μL of Ultra-pure H2O for final sequencing.
All of the samples were sequenced using 2 × 150 bp chemistry on Illumina NovaSeq instruments through the sequencing service by Azenta company.
Flow cytometry and data analysis
HepG2 and K562 cells were transfected using Lipofectamine 3000 (Invitrogen, L3000150) following the manufacturer’s guidelines. For each transfection experiment, approximately 1.9 × 105 cells were suspended in 1 mL of complete medium and plated into each well of a 12-well plate (NEST Biotechnology, 712002). The cells were then incubated at 37 °C and 5% CO2 for approximately 24 h to achieve around 70% confluency. On the day of transfection, the existing medium was replaced with 1 mL of fresh culture medium. The transfection mix for each well was prepared using 3 μL of Lipofectamine 3000 (Invitrogen, L3000150) and 1.5 μg of plasmid DNA carrying the candidate enhancer, adhering strictly to the protocol provided by the manufacturer.
Cells were harvested to assess the fluorescence expression of candidate enhancers. For HepG2 cells, the adherent cells were trypsinized using 0.25% trypsin-EDTA (GIBCO, 25200072). The suspended cells were then transferred to a 15 mL centrifuge tube and centrifuged at 300 × g for 5 min at room temperature to pellet the cells. For K562 cells, the suspended cells were directly centrifuged at 300 × g for 5 min at room temperature to pellet the cells. Then, the cells were washed with 1 mL of DPBS (GIBCO, 14190144). This washing step was repeated once to remove any residual trypsin and serum. The cells were resuspended in DPBS to a total volume of 300 µl, ready for analysis.
For flow cytometry analysis, the resuspended cells were analyzed using LSRFortessa flow cytometer (BD Biosciences). The instrument settings for fluorescent protein detection were as follows: excitation at 488 nm, emission filtered through a 530/30 filter, and photomultiplier tube (PMT) voltage set at 200 V for EYFP measurement. For each sample, approximately 1 × 105 cell events were collected. The raw data were processed using FlowJo (Version 10.8.1) for downstream analysis. For data analysis, raw data were filtered through the FSC and SSC to get the single cells (Supplementary Fig. 9). The EYFP mean was normalized by the CMV enhancer-promoter as the activity of the designed sequences (CMV enhancer-promoter as 100). Three independent biological replicates were performed for each sequence. All of the flow cytometry results are shown in Supplementary Data 7.
MPRA data processing
The enhancer activity of DNA sequences in the MPRA library is obtained by calculating the ratio of the corresponding relative abundance of RNA to the corresponding relative abundance of DNA64. For each barcode, the relative abundance in DNA-seq and RNA-seq is quantified as the normalized read counts with Eq. (1):
The barcodes mapped to more than one enhancer are filtered out. For a certain DNA sequence, the normalized read counts of all barcodes mapped to it are used to calculate its enhancer activity with Eq. (2):
The enhancers with less than three barcodes and enhancers whose summed normalized RNA counts or DNA counts is less than 0.1 were filtered out to reduce the noise. The mean enhancer activity of the biological replicates is calculated as the final enhancer activity. The correlation of different replicates is shown in Supplementary Fig. 10. All of the MPRA results are shown in Supplementary Data 8, 9.
Core TFBS matching score calculation
The matrix information of core motifs is from the JASPAR database65,66. For each TF, the position frequency matrix (PFM) is firstly transformed into the original \({{{{\rm{PPM}}}}}_{0}\) (in the size of \({L}_{0}\times 4\), where \({L}_{0}\) denotes the original length of the motif). Then the information content of each position in \({{{{\rm{PPM}}}}}_{0}\) is calculated by:
where \(N=4\)(denotes 4 kinds of nucleic acid bases), \({p}_{n}\) is the probability of the \(n\)-th nucleic acid base at this position. To get more solid information and increase the sequence designing space, we choose part of \({{{{\rm{PPM}}}}}_{0}\) from the first position with information content larger than 0.3 to the last position with information content larger than 0.3 as the final \({{{\rm{PPM}}}}\) (in the size of \(L\times 4\)) that we use in this work. The threshold of 0.3 is adjustable and can be increased or decreased according to specific application requirements. Alternatively, users can also choose not to set a threshold and use the entire \({{{{\rm{PPM}}}}}_{0}\) directly.
With a given \({{{\rm{PPM}}}}\) of a TF and a DNA segment \(C\) of length \(L\), the core TFBS matching score of this TF is calculated as:
where \({{{{\rm{PPM}}}}}_{i}\) denotes the probability vector of \({{{\rm{PPM}}}}\) at the position \(i\), \({C}_{i}\) denotes the nucleic acid base at position \(i\).
When searching for the position with max matching score of a given TF on a sequence, the \({S}_{{{{\rm{core}}}}}\) of all possible DNA segments of length \(L\) form this DNA sequence and its reverse complement sequence are calculated, and the max one is selected as the max matching score and the corresponding DNA segment is selected as the core TFBS.
For TFs with multiple PFMs in the JASPAR database, we prioritized selecting the latest version of the PFM. Additionally, we used the motif enrichment tool STREME67 to perform motif enrichment analysis on the ChIP-seq data we used. If a PFM significantly deviates from the latest version, we replace it with a PFM that more closely matches the motifs enriched from the ChIP-seq data we used. The PFM matrix IDs used in this work can be found in Supplementary Data 10. Furthermore, since the ChIP-seq data used in this work are all specific to a particular TF, we did not utilize the PFMs for Homo-/Hetero protein complexes from the JASPAR database in this research.
Preparation of dataset for deep learning model
To train the deep learning models, we constructed separate datasets for each TF using data from the ENCODE37,68,69 database. For each TF, we generated positive samples, derived from ChIP-seq data, and negative samples, derived from ATAC-seq and ChIP-seq data. We used the bed narrow peak files in this work.
In generating positive samples, for each peak identified in the ChIP-seq data, we used the PPM corresponding to the TF to find the location within the peak with the highest matching score, marking it as the core TFBS. If multiple positions yield the same highest score, the position closest to the summit of the peak is chosen as the core TFBS. The context sequence of the selected core TFBS is taken as a positive sample of the TFBS-context.
Negative samples were generated from ATAC-seq data by selecting peaks from regions that do not overlap with any peaks in the ChIP-seq data. The same method used to define positive samples (using the PPM and context sequence extraction) was applied to derive the potential negative TFBS-context samples from these ATAC-seq peaks.
To prevent the model from focusing on simple features, we balanced the dataset for GC content and histone modification states. First, the GC content was calculated for each sample. Additionally, samples were labeled based on their histone modification states for H3K4me3 and H3K4me1. Negative samples were selected to ensure that the distribution of GC content and the number of samples with specific histone modification states matched between positive and negative sets.
For the dataset of cell type-specific models, we followed a similar procedure to derive positive samples of both cell types from the ChIP-seq data. Then the positive samples of both cell types were balanced by GC content and histone modification states to form the dataset of the cell type-specific model.
The TFBU information
The length of TFBU is set to be 168 bp in this work, this is mainly decided by the MPRA experimental restriction. We tried to model the experiment result with different TFBU lengths. The result showed that longer TFBUs will provide more information, but the length of 168 could cover most information. The model’s performance shows a saturation trend before the TFBU length of 168 (Supplementary Fig. 7).
We use a DenseNet-LSTM-based model to predict whether a TFBS-context sequence is favorable for TF binding, and it’s set to be a binary classification model. The TFBU is transformed into one-hot encoding, with the core TFBS masked by Ns (which is \([{{\mathrm{0,0,0,0}}}]\) in one-hot encoding). The core TFBS is in the middle of TFBU. When the length of core TFBS is singular, the TFBS-context part on the 5’ side of core TFBS will contain 1 more nucleic acid base than the TFBS-context part on the 3’ side of core TFBS.
Sequence design and optimization
The genetic algorithm is applied for sequence generation. In the genetic algorithm, the maximum population size is set to 2000. Each iteration selects the top 30% of the parent samples as elite parents, and will generate new samples equivalent to 30% of the current population size. The algorithm runs for up to 300 iterations, a number experimentally validated to ensure convergence for our design tasks.
In each iteration, half of the new samples are derived from elite parents, and the other half from non-elite parents. For each parent sample, random mutation and recombination occur with equal probability. In mutation, a random base is selected from the parent sample to mutate. In recombination, the parent sequence is recombined with another randomly selected sequence from the current samples pool, with each position in the new sequence equally likely to come from either parent. Sequences with top evaluation scores, based on the evaluation metric, are selected for the next round of iterations.
The TFBS-context models trained with data from ChIP-seq experiments are binary classification models with an output range from \([{{\mathrm{0,1}}}]\). When optimizing sequences with a certain aim, the genetic algorithm is applied to the initial sequence by introducing mutation and reorganization in the region except for the fixed core TFBS. To avoid the bias introduced by the randomness during model training, the mean score of 10 models trained with the same data is used in this work to evaluate the score of a given sequence. The sequence segment same as the core TFBS was avoided from appearing in the TFBS-context. For different tasks, the evaluation metric is different, but the aim is always set to maximize the corresponding metric like:
When optimizing the TFBS-context of a given TF, the evaluation metric is the score of this TFBS-context as:
The difference between the GC ratios of the generated sequences and the mean GC ratio of the training data should be less than 10%. The Levenshtein distance between each pair of sequences finally selected should be equal to or larger than 50.
When optimizing the sequence with multiple core TFBS, the evaluation metric is the sum of scores of all the TFBS-context of these core TFBSs as:
The lower bound of Levenshtein distance in this task is set to be 5 in the situation with 8 core TFBSs and set to be 10 in the situation with 4 core TFBSs. The interval sequence segments between core TFBSs were avoided to be all the same to facilitate the construction of the optimized sequences.
When optimizing the cell type-specific TFBS-context, there are results from three models that should be taken in the metric. The score of the HepG2 model denotes the score that the TFBS-context is functional in the HepG2 cell line. The score of the K562 model denotes the score that the TFBS-context is functional in the K562 cell line. The score of the specific model denotes the relative specificity of the TFBS-context, and is set to be 1 as HepG2-specific, 0 as K562-specific. The metric for HepG2-specific TFBS-contexts is:
The metric for K562-specific TFBS-contexts is:
The upper bound of the GC ratio difference between generated sequences and the training data is set to be 10%. The lower bound of Levenshtein distance in this task is set to be 30.
The strategy for designing cell type-specific TFBS-contexts can also be generalized to more general scenarios. For cases involving three or more cell types, the cell types where expression is desired are denoted as active_types (number of active_types denoted as n), and the cell types where expression is undesired are denoted as mute_types (number of mute_types denoted as m). Using the method described above, a TFBS-context model is trained for each cell type, as well as a specificity model between active_types and mute_types. The evaluation metric is defined using the following formula:
By applying this metric with the genetic algorithm, it is possible to design enhancers with specificity in multi-cell-type contexts.
Features of TFBS in TFBS-context
The motif analyzing tool FIMO42 is applied to search for the TFBS in the TFBS-context sequence against the reference of human TF motif matrixes from JASPAR database. The p-value threshold is set to be 1e-4 as default (adjusted for multiple motif comparisons using the Benjamini–Hochberg method70). For each of the three key TFs, we selected the top/bottom 100 sequences with the highest/lowest enhancer activity from the sequences whose TFBS-context contains TFBS to represent TFBS-context with high/low enhancer activity. The TFBS matching scores for TFs whose PFMs are available in JASPAR database were calculated to form the feature vector. The feature vectors were visualized with the dimension reduction algorithm UMAP71.
In the analysis of high-scoring TFBS-contexts designed for 118 TFs, we used the TOMTOM72 tool to compare other obvious TFBSs found within the TFBS-contexts against the core TFBS. We applied TOMTOM’s default threshold, considering matched TFBSs as similar binding sites to the core TFBS. We then increased the recognition threshold to 0.95, and any TFBSs that still failed to match under this threshold were classified as distinct binding sites from the core TFBS. The detailed results are listed in Supplementary Data 3.
Identification of TFBS in TFBS-context
To assess whether the TFBS-context can still function in the absence of obvious TFBS, we applied the same method described in the previous section to search for obvious TFBS within the TFBS-context. Sequences that did not contain identifiable TFBS after this search were considered to be TFBS-context without obvious TFBS.
On the basis of the aforementioned, we also determined a more stringent criterion for filtering out TFBSs. We increased the default p-value threshold of FIMO by 20 times (from 1e-4 to 2e-3), hoping to use this more relaxed threshold to filter out more potential TFBSs. At this threshold, the blank control sequences, previously reported as lacking enhancer activity and used as blank backbone for TFBS effect, were all detected to contain potential match sites. Additionally, we randomly extracted 168 bp DNA sequences from each accessible region in HepG2 cells that did not exhibit ChIP-seq binding signals. At this threshold, more than 88% of these randomly extracted sequences were found to contain potential match sites. These results suggest that even in randomly extracted non-binding regions, this threshold can identify potential binding sites in the majority of sequences. We adopted this threshold as a more stringent criterion to filter out TFBSs, believing that this criterion can filter out most of TFBSs.
Since TFBSs are considered a crucial component of TFBU functionality in designing TFBUs, we didn’t directly filter out TFBSs during the design process. This resulted in many sequences optimized for 118 TFs contain TFBS under the stricter criterion. In the analysis of these optimized results under the stricter criterion, we expanded the statistical scope and included all TFBU samples with a TFBS-context score greater than 0.5. This ensured that as many TFs as possible had at least 10 samples for statistical evaluation.
DNA shape feature of TFBS-context
Previous studies have reported that the DNA shape surrounding TFBS may influence the binding of TFs22,28,29. Therefore, we analyzed the patterns of DNA shape, hoping to see if there is a difference in the DNA shape patterns between TFBS-context sequences with high enhancer activity and those with low enhancer activity.
Since the TFBS in the TFBS-context is a relatively strong feature to DNA shape, to avoid being masked by the feature of TFBS, the feature of DNA shape is generated with TFBS-contexts that don’t contain obvious TFBS. The TFBS-contexts with top/bottom 10% enhancer activity that don’t contain obvious TFBS are selected to generate the feature of DNA shape.
The minor groove width (MGW), roll, propeller twist (ProT), and helix twist (HelT) were calculated as the feature of DNA shape73. The feature vectors composed of these features were encoded by the unsupervised learning model based on contrastive learning and visualized with the dimension reduction algorithm UMAP. Since DNA shape is a high-dimensional feature that cannot be directly visualized for comparison, UMAP can help us intuitively visualize whether the DNA shape of TFBS-contexts with high enhancer activity and low enhancer activity have different distributions in the latent space.
The definition of weak binding sites
Weak binding sites are defined based on the p-value of the matching degree between a DNA sequence and the corresponding PPM. For a given TF and its PPM, we use the FIMO tool to search for potential match sites within the sequences. The potential match sites with p-values greater than 0.0001 and less than 0.001 are defined as weak binding sites. Here, we only consider the PPM of the core TFBS used in generating TFBS-contexts, without taking into account redundant PPMs for the same TF (specific matrix IDs can be found in Supplementary Data 10). The TFBS-contexts with top/bottom 10% enhancer activity are selected to count the number of weak binding sites.
DNA structure features of TFBS-context
We further analyzed the DNA structure features of the TFBS-contexts which don’t contain obvious TFBS. In the analysis of DNA structure, similar to the analysis of DNA shape, we selected samples with top/bottom 10% enhancer activity for each TF. We first utilized an algorithm called R-looper74 to calculate the probability of R-loop formation within these TFBS-contexts. The parameter for the size of the superhelical domain was set to auto, and we used the other default parameters of R-looper for the calculations. R-looper can provide the probability of forming an R-loop at each base in a sequence. For each sequence, we selected the maximum value as the probability of forming an R-loop in that sequence. After calculating the probabilities for all sequences, we compared those from the top/bottom 10% enhancer activity sequences.
Next, we employed the alignment tool provided by Non-B DB75 to identify non-B motifs within these sequences. For each of the seven types of non-B motifs, we applied Fisher’s exact test to assess whether there was a significant difference in the proportion of sequences containing that type of non-B motif between TFBS-contexts with top/bottom 10% enhancer activity.
Scrambling a TFBS
In designing cell type-specific TFBUs, we scrambled the core TFBS as a control group. The scrambling is realized by introducing mutations at several positions within a TFBS, ensuring that the mutated TFBS no longer matches any TFBS motifs in the JASPAR database while introducing as few mutations as possible. We used the TOMTOM tool to evaluate whether the scrambled sequences still matched any other known TFBS motifs. To maximize the distinction between the scrambled sequences and other TFBS motifs, we set the TOMTOM recognition threshold to 0.95. The core TFBS sequences before/after the scramble are as follows: BHLHE40: TCACGTGA/TCAGTTAA; GABPA: ACCGGAAGAG/ACCGGCCAAG; GATA2: CTTATCT/CTTCGCT; SP1: CCCCGCCCCC/CCAAGACCCC.
Sequence in the task of multiple TFBU optimization
The interval sequence between multiple TFs proposed by Tim Lu is applied in this task to form the sequence in the control group. The form of the sequence is:
TFBS-AGA-TFBS-TCG-TFBS-GAC-TFBS-CTA-TFBS-ACT-TFBS-TGC-TFBS-GTA-TFBS-G.
Here TFBS is the consensus sequence of the corresponding motif. One thing should be noticed is that in our method, the positions with information content less than 0.3 at both sides of the motif are taken as a part of TFBS-context, which means that the interval sequence that we optimize may be a little longer than 3 bp. To validate whether stronger enhancer activity could be achieved with fewer core TFBSs, we set another group where the 2nd, 4th, 6th, and 8th TFBSs are not taken as core TFBS as are changeable during the optimization.
Decoupling enhancers into individual TFBUs
The maximum core TFBS matching score may occur at any position within the sequence. If the position is not at the middle of our experimental TFBU, the context sequence of the selected core TFBS would not be covered with the origin TFBU. To address this issue, we appended the sequence connected to TFBU on the plasmid at both ends when getting the context sequence of the selected core TFBS. When a TF’s highest TFBS matching score occurs at multiple positions on a sequence, only the position with the highest TFBS-context score is considered.
Statistics
Statistics were calculated using Scipy76. Detailed statistics information was provided in the Supplementary Data 2 for statistic information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the enhancer activities data generated in this work are provided in Supplementary Data 7–9. The list of data from ENCODE used in this work is provided in Supplementary Data 11. The TFBS-context deep learning model weights for the 198 TFs are available in the ZENODO database via https://doi.org/10.5281/zenodo.1093182577. The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (GSA)78 in National Genomics Data Center79, China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences under accession codes: GSA: CRA015817, GSA-Human: HRA007140. The minimal dataset supporting the results and analysis in this study has been deposited in the figshare database [https://doi.org/10.6084/m9.figshare.26354725]80. Source data are provided with this paper.
Code availability
Detailed codes and the required files for the DeepTFBU toolkit and related analyses are publicly available and has been deposited in Github at https://github.com/WangLabTHU/DeepTFBU, under Apache-2.0 license. The specific version of the code associated with this publication is archived in the Zenodo repository and is accessible via https://doi.org/10.5281/zenodo.1455232181.
References
Maston, G. A., Evans, S. K. & Green, M. R. Transcriptional regulatory elements in the human genome. Annu. Rev. Genom. Hum. Genet. 7, 29–59 (2006).
Chatterjee, S. & Ahituv, N. Gene regulatory elements, major drivers of human disease. Annu. Rev. Genom. Hum. Genet. 18, 45–63 (2017).
Spitz, F. & Furlong, E. E. M. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 13, 613–626 (2012).
Stormo, G. D. & Zhao, Y. Determining the specificity of protein–DNA interactions. Nat. Rev. Genet. 11, 751–760 (2010).
Gerland, U., Moroz, J. D. & Hwa, T. Physical constraints and functional characteristics of transcription factor–DNA interaction. Proc. Natl Acad. Sci. USA 99, 12015–12020 (2002).
Gheorghe, M. et al. A map of direct TF–DNA interactions in the human genome. Nucleic Acids Res. 47, e21–e21 (2019).
Hajheidari, M. & Huang, S. C. Elucidating the biology of transcription factor–DNA interaction for accurate identification of cis-regulatory elements. Curr. Opin. Plant Biol. 68, 102232 (2022).
Khalil, A. S. & Collins, J. J. Synthetic biology: applications come of age. Nat. Rev. Genet. 11, 367–379 (2010).
Ruder, W. C., Lu, T. & Collins, J. J. Synthetic biology moving into the clinic. Science 333, 1248–1252 (2011).
Jolma, A. et al. DNA-binding specificities of human transcription factors. Cell 152, 327–339 (2013).
Lambert, S. A. et al. The human transcription factors. Cell 172, 650–665 (2018).
Grossman, S. R. et al. Systematic dissection of genomic features determining transcription factor binding and enhancer function. Proc. Natl Acad. Sci. USA. 114, E1291–E1300 (2017).
Smith, R. P. et al. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat. Genet. 45, 1021–1028 (2013).
Sharon, E. et al. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat. Biotechnol. 30, 521–530 (2012).
Levo, M. & Segal, E. In pursuit of design principles of regulatory sequences. Nat. Rev. Genet. 15, 453–468 (2014).
Georgakopoulos-Soares, I. et al. Transcription factor binding site orientation and order are major drivers of gene regulatory activity. Nat. Commun. 14, 2333 (2023).
Weingarten-Gabbay, S. et al. Systematic interrogation of human promoters. Genome Res. 29, 171–183 (2019).
Liu, X. et al. De novo design of programmable inducible promoters. Nucleic Acids Res. 47, 10452–10463 (2019).
Levo, M. et al. Unraveling determinants of transcription factor binding outside the core binding site. Genome Res. 25, 1018–1029 (2015).
Ghoshdastidar, D. & Bansal, M. Flexibility of flanking DNA is a key determinant of transcription factor affinity for the core motif. Biophys. J. 121, 3987–4000 (2022).
Horton, C. A. et al. Short tandem repeats bind transcription factors to tune eukaryotic gene expression. Science 381, eadd1250 (2023).
Dror, I., Golan, T., Levy, C., Rohs, R. & Mandel-Gutfreund, Y. A widespread role of the motif environment in transcription factor binding across diverse protein families. Genome Res. 25, 1268–1280 (2015).
Dror, I., Rohs, R. & Mandel‐Gutfreund, Y. How motif environment influences transcription factor search dynamics: Finding a needle in a haystack. BioEssays 38, 605–612 (2016).
De Almeida, B. P., Reiter, F., Pagani, M. & Stark, A. DeepSTARR predicts enhancer activity from DNA sequence and enables the de novo design of synthetic enhancers. Nat. Genet. 54, 613–624 (2022).
Castellanos, M., Mothi, N. & Muñoz, V. Eukaryotic transcription factors can track and control their target genes using DNA antennas. Nat. Commun. 11, 540 (2020).
Brodsky, S. et al. Intrinsically disordered regions direct transcription factor in vivo binding specificity. Mol. Cell 79, 459–471.e4 (2020).
Rohs, R. et al. The role of DNA shape in protein–DNA recognition. Nature 461, 1248–1253 (2009).
Gordân, R. et al. Genomic regions flanking E-Box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep. 3, 1093–1104 (2013).
Samee, Md. A. H., Bruneau, B. G. & Pollard, K. S. A de novo shape motif discovery algorithm reveals preferences of transcription factors for DNA shape beyond sequence motifs. Cell Syst. 8, 27–42.e6 (2019).
Tanay, A. Extensive low-affinity transcriptional interactions in the yeast genome. Genome Res. 16, 962–972 (2006).
Farley, E. K. et al. Suboptimization of developmental enhancers. Science 350, 325–328 (2015).
Crocker, J. et al. Low affinity binding site clusters confer hox specificity and regulatory robustness. Cell 160, 191–203 (2015).
Morgunova, E. & Taipale, J. Structural perspective of cooperative transcription factor binding. Curr. Opin. Struct. Biol. 47, 1–8 (2017).
Zhang, P. et al. Deep flanking sequence engineering for efficient promoter design using DeepSEED. Nat. Commun. 14, 6309 (2023).
Avsec, Ž. et al. Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet. 53, 354–366 (2021).
Slattery, M. et al. Absence of a simple code: how transcription factors read the genome. Trends Biochem. Sci. 39, 381–399 (2014).
The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Ernst, J. et al. Genome-scale high-resolution mapping of activating and repressive nucleotides in regulatory regions. Nat. Biotechnol. 34, 1180–1190 (2016).
Alder, O. et al. Hippo signaling influences HNF4A and FOXA2 enhancer switching during hepatocyte differentiation. Cell Rep. 9, 261–271 (2014).
Göös, H. et al. Human transcription factor protein interaction networks. Nat. Commun. 13, 766 (2022).
Chen, Y.-R., Huang, H.-C. & Lin, C.-C. Regulatory feedback loops bridge the human gene regulatory network and regulate carcinogenesis. Briefings Bioinform. 20, 976–984 (2019).
Grant, C. E., Bailey, T. L. & Noble, W. S. FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011).
Wu, M.-R. et al. A high-throughput screening and computation platform for identifying synthetic promoters with enhanced cell-state specificity (SPECS). Nat. Commun. 10, 2880 (2019).
Lu, T. K., Nissim, L. & Wu, M.-R. Synthetic promoters. US11718860B2 (2018).
Vaishnav, E. D. et al. The evolution, evolvability and engineering of gene regulatory DNA. Nature. https://doi.org/10.1038/s41586-022-04506-6 (2022).
Taskiran, I. I. et al. Cell-type-directed design of synthetic enhancers. Nature 626, 212–220 (2024).
Zhao, Y. et al. The transcription factor RFX5 is a transcriptional activator of the TPP1 gene in hepatocellular carcinoma. Oncol. Rep. 37, 289–296 (2017).
Zhang, Q. et al. p53-induced microRNA-1246 inhibits the cell growth of human hepatocellular carcinoma cells by targeting NFIB. Oncol. Rep. 33, 1335–1341 (2015).
Arvey, A., Agius, P., Noble, W. S. & Leslie, C. Sequence and chromatin determinants of cell-type–specific transcription factor binding. Genome Res. 22, 1723–1734 (2012).
Huang, H. et al. Oncolytic adenovirus programmed by synthetic gene circuit for cancer immunotherapy. Nat. Commun. 10, 4801 (2019).
Redding, S. & Greene, E. C. How do proteins locate specific targets in DNA? Chem. Phys. Lett. 570, 1–11 (2013).
Kalodimos, C. G. et al. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science 305, 386–389 (2004).
Ghandi, M., Lee, D., Mohammad-Noori, M. & Beer, M. A. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput. Biol. 10, e1003711 (2014).
Zhang, Q., Shen, Z. & Huang, D.-S. Modeling in-vivo protein-DNA binding by combining multiple-instance learning with a hybrid deep neural network. Sci. Rep. 9, 8484 (2019).
Tareen, A. & Kinney, J. B. Biophysical Models of Cis-Regulation as Interpretable Neural Networks. https://doi.org/10.1101/835942 (2019).
Alipanahi, B., Delong, A., Weirauch, M. T. & Frey, B. J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 33, 831–838 (2015).
Zeng, H., Edwards, M. D., Liu, G. & Gifford, D. K. Convolutional neural network architectures for predicting DNA–protein binding. Bioinformatics 32, i121–i127 (2016).
Quang, D. & Xie, X. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 44, e107 (2016).
Zheng, A. et al. Deep neural networks identify sequence context features predictive of transcription factor binding. Nat. Mach. Intell. 3, 172–180 (2021).
Visel, A., Minovitsky, S., Dubchak, I. & Pennacchio, L. A. VISTA enhancer Browser-a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–D92 (2007).
Donoghue, M., Ernst, H., Wentworth, B., Nadal-Ginard, B. & Rosenthal, N. A muscle-specific enhancer is located at the 3’ end of the myosin light-chain 1/3 gene locus. Genes Dev. 2, 1779–1790 (1988).
Koesterich, J. et al. Characterization of de novo promoter variants in autism spectrum disorder with massively parallel reporter assays. IJMS 24, 3509 (2023).
Tewhey, R. et al. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell 165, 1519–1529 (2016).
Yu, T. C. et al. Multiplexed characterization of rationally designed promoter architectures deconstructs combinatorial logic for IPTG-inducible systems. Nat. Commun. 12, 325 (2021).
Castro-Mondragon, J. A. et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50, D165–D173 (2022).
Rauluseviciute, I. et al. JASPAR 2024: 20th anniversary of the open-access database of transcription factor binding profiles. Nucleic Acids Res. gkad1059 https://doi.org/10.1093/nar/gkad1059 (2023).
Bailey, T. L. STREME: accurate and versatile sequence motif discovery. Bioinformatics 37, 2834–2840 (2021).
Luo, Y. et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 48, D882–D889 (2020).
Hitz, B. C. et al. The ENCODE uniform analysis pipelines. https://doi.org/10.1101/2023.04.04.535623 (2023).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 57, 289–300 (1995).
McInnes, L., Healy, J. & Melville, J. UMAP: Uniform manifold approximation and projection for dimension reduction. https://doi.org/10.48550/ARXIV.1802.03426 (2018).
Gupta, S., Stamatoyannopoulos, J. A., Bailey, T. L. & Noble, W. S. Quantifying similarity between motifs. Genome Biol. 8, R24 (2007).
Zhou, T. et al. DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res. 41, W56–W62 (2013).
Stolz, R. et al. Interplay between DNA sequence and negative superhelicity drives R-loop structures. Proc. Natl. Acad. Sci. USA 116, 6260–6269 (2019).
Cer, R. Z. et al. Non-B DB v2.0: a database of predicted non-B DNA-forming motifs and its associated tools. Nucleic Acids Res. 41, D94–D100 (2012).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 17, 261–272 (2020).
Li, J. et al. The deep learning model weights for the TFBU in HepG2. Zenodo, https://doi.org/10.5281/zenodo.10931825 (2024).
Chen, T. et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics Proteomics Bioinformatics 19, 578–583 (2021).
CNCB-NGDC Members and Partners. et al. Database resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 50, D27–D38 (2022).
Li, J. et al. Modeling and designing enhancers by introducing and harnessing transcription factor binding units. figshare. Dataset. https://doi.org/10.6084/m9.figshare.26354725 (2024).
Li, J. et al. Modeling and designing enhancers by introducing and harnessing transcription factor binding units. Zenodo, https://doi.org/10.5281/zenodo.14552321 (2024).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 62250007, 62225307 to X.W.W.), National Key R&D Program of China (Nos. 2020YFA0906900, 2023YFF1204500 to X.W.W.), and a grant from the Guoqiang Institute, Tsinghua University (2021GQG1023 to X.W.W.).
Author information
Authors and Affiliations
Contributions
J.Q.L., P.C.Z., X.W.W. conceived this study. J.Q.L. developed the algorithm, designed and implemented the computational experiments. P.C.Z. developed the experimental system, designed and implemented the biological experiments. X.W.W. designed some experiments and supervised the study. J.Q.L., P.C.Z., X.X., L.W., L.Y.L. and X.W.W. wrote and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Qiang Lyu, Mikhail Spivakov and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, J., Zhang, P., Xi, X. et al. Modeling and designing enhancers by introducing and harnessing transcription factor binding units. Nat Commun 16, 1469 (2025). https://doi.org/10.1038/s41467-025-56749-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56749-2
