
Abstract
Biophysical cues play a crucial role in T cell biology, yet their implications in adoptive T cell therapy (ACT) remain largely unknown. Here, we investigate the effect of electrical stimuli on CD8+ T cells using a charged substrate composed of electroactive nanocomposites with tunable surface charge intensities. Electrical stimuli enhance the persistence and tumor-suppressive efficacy of transferred T cells, with effects dependent on substrate charge. Single-cell RNA-sequencing analysis unveils a decrease in virtual memory T (Tvm) cells and an increase in proliferative potential T (Tpp) cells, which exhibit superior antitumor activity and metabolic adaptations relative to those treated with uncharged substrate. ATAC-seq profiling demonstrates heightened accessibility at upstream binding sites for EGR1, a transcription factor critical for Tpp cell differentiation. Mechanistically, the charged substrate disrupts ionic TCR-lipid interactions, amplifies TCR signaling, and activates EGR1, thereby impeding Tvm polarization during ex vivo culture. Our findings thus highlight the importance of extracellular electrical stimuli in shaping T cell fate, offering potential for optimizing ACT for therapeutic applications.
Similar content being viewed by others


Introduction
Adoptive cellular transfer (ACT) has emerged as a promising therapeutic approach in oncology, enabling the precise targeting and elimination of cancer cells. However, the efficacy of ACT in solid tumors is often compromised by impaired CD8+ T cell functions, inadequate activation, and limited lifespan of tumor-reactive T cells1,2,3. The TCR-CD3 complex mediates the recognition of tumor antigens, initiating downstream signaling essential for a robust immune response. Yet, the immunosuppressive tumor microenvironment can inhibit CD8+ T cell activation and cytotoxicity, facilitating tumor immune evasion4,5,6. Unlike the immune checkpoint inhibitors, ACT relies on the ex vivo activation and expansion of T cells, including chimeric antigen receptor (CAR) T cells. Accumulating evidence indicates that this phase can influence the therapeutic efficacy of tumor-specific CD8+ T cells against cancer7,8,9. Hence, optimizing culture conditions and elucidating the underlying mechanisms are critical for broadening the clinical utility of ACT immunotherapy.
T cell activation, proliferation, and differentiation into effector and memory T cells are orchestrated by a combination of extrinsic and intrinsic signaling cascades, essential for combating malignancies and establishing durable protection10,11. Following T cell receptor (TCR) engagement, antigen recognition is reinforced by the formation of TCR clusters at the cell membrane12,13. This leads to the establishment of an immune synapse, which activates a series of transcription factors (TF), including early growth response 1 (EGR1), nuclear factor of activated T cells (NFAT), and activator protein 1 (AP-1), driving the transcription of genes crucial for T cell effector function14,15. These TFs also regulate chromatin accessibility, thereby shaping T cell function and fate determination16,17. Identifying the molecular regulators that modulate TF activity and guide T cell specification is crucial for enhancing T cell-mediated antitumor responses.
Among these TFs, EGR1 plays a pivotal role in CD8+ T cell responses. EGR1 is rapidly induced in T cells upon antigen receptor cross-linking or TCR stimulation, serving as a key mediator linking early TCR signaling to downstream gene expression14,18. Studies have demonstrated the importance of EGR1 in various T cell processes, including thymic development and the regulation of key cytokines such as IL-2 and TNF, as well as its crucial role in the clonal expansion of virus-specific CD8+ T cells in a lymphocytic choriomeningitis virus (LCMV) model19,20,21. Utilizing piezoelectric membrane materials, we demonstrate the role of electrical stimulation in accelerating bone regeneration and combating bacteria22,23. Building on this work, our study reveals that treatment with a charged substrate enhances EGR1 expression and increases chromatin accessibility at EGR1 binding sites in CD8+ T cells, driving proliferation and polarization. Importantly, we find that EGR1 activation is antigen-dependent, underscoring its role in shaping CD8+ T cell responses under different immune contexts.
In most clinical settings, autologous CD8+ T cells are genetically modified to express transgenic antigen receptors, followed by a brief period of in vitro activation and expansion before being reinfused into the patient. Efforts to improve T cell functionality typically focus on biological or chemical interventions24,25,26,27. While biochemical factors influencing T cell regulation have been extensively studied, the role of biophysical cues, such as shear stress, solid stress, and matrix stiffness, is increasingly recognized as critical in CD8+ T cell activation8,28,29,30,31. These physical characteristics can independently or synergistically impact the tumor microenvironment and the efficacy of CAR-T cells. Moreover, membrane potential (MP) depolarization in T cells within tumor microenvironments or inflamed tissues has been shown to affect receptor mobility, while characteristic oscillations at immunological synapses and electrostatic interactions in TCR signaling regulation have been observed32,33,34,35,36. However, the mechanisms by which electrical signals are converted into biochemical signaling within T cells and whether external electrical stimulation can be used to modulate T cell activity in cancer therapy, remain elusive.
In this study, we demonstrate that external electrical stimulation during ex vivo culture refines CD8+ T cell differentiation, enhancing the efficacy of ACT or CAR-T therapy in vivo. By augmenting TCR signaling transduction, electrical stimulation reduces the polarization of virtual memory T cells and promotes the expansion of CD8+ T cells with higher proliferative potential, key factors in the antitumor effects of ACT. Our findings highlight the critical role of extracellular electrical stimuli in shaping CD8+ T cell fate, providing a foundational strategy for harnessing proliferative potential T cells in cancer therapy.
Results
Electrical modulation of T cell response through ferroelectric nanocomposite membranes
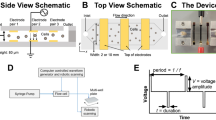
Changes in cell membrane potential (MP) are implicated in the physiological processes of various immune cells, such as B cells, macrophages, and neutrophils37,38,39,40. However, the impact of altering MP on the biological functions of T cells remains unclear. Using the anionic membrane potential-sensitive fluorescence probe DiBAC4(3), we observed depolarization of plasma MP during T cell activation (Fig. 1a). To investigate the effect of bioelectrical signals on T cell responses, we designed a substrate electrical stimulation platform using a ferroelectric nanocomposite membrane as a modulator of T cell MP (Supplementary Fig. 1a). Initially, the ferroelectric nanocomposite membrane was fabricated by incorporating BaTiO3 nanoparticles into poly(vinylidene fluoridetrifluoroethylene) (P(VDF-TrFE)) matrix. By adjusting parameters of annealing and corona poling, we achieved tunable regulation of electrical magnitude of the nanocomposite membrane, referred to as non-charged (NC, 55 °C, no poling), low-charged (LC, 55 °C, poling), mid-charged (MC, 90 °C, poling) and high-charged (HC, 120 °C, poling) nanocomposite membranes, respectively (Supplementary Fig. 1b–e). Surface potential measurements using Kelvin probe force microscopy (KPFM) revealed homogeneous distribution and substantial increases from LC to HC groups (Supplementary Fig. 1b, c). In addition, residual polarization and piezoelectric coefficient (d33) increased with increasing annealing temperature (Supplementary Fig. 1 d, e). Further surface structure analysis confirmed uniform dispersion of BaTiO3 nanoparticles within the P(VDF-TrFE) matrix (Supplementary Fig. 1f), with no significant difference in surface roughness observed between nanocomposite membranes with different charge parameters (Supplementary Fig. 1g, h). To investigate whether electrical stimuli from the charged substrate can affect T cell MP, OT-I cells were cultured on the nanocomposite membranes and activated by OVA peptide for 6 h or antibodies against CD3/CD28 for 24 h. Using membrane voltage-reporting dye and whole-cell patch-clamp recording, we observed enhanced T cell membrane depolarization with increasing surface charge of the nanocomposite membrane (Fig. 1b–d and Supplementary Fig. 2a). Importantly, biocompatibility evaluation demonstrated minimal cytotoxicity of the electroactive nanocomposite membranes (Supplementary Fig. 2b–d). Collectively, we fabricated a series of BaTiO3/P(VDF-TrFE) nanocomposite membranes with varying surface electrical potential to investigate the effects of extracellular electrical stimulation on T cells, independent of other surface properties.

a Flow cytometric analysis of T cell membrane potential. OT-I cells were isolated and activated with OVA257–264 peptides (2 μg/mL) for 6 h followed by DiBAC4(3) staining (n = 5 biological replicates, mean ± sem, ****P < 0.0001, two-tailed unpaired Student’s t test). b Graphic illustration of the study design for evaluating T cell activation. OT-I cells were isolated and cultured on the nanocomposite membranes with varying surface charges, and activated by OVA257–264 peptides (2 μg/mL) for 6 h. NC, non-charged nanocomposite membranes; LC, low-charged nanocomposite membranes; MC, mid-charged nanocomposite membranes; HC, high-charged nanocomposite membranes. c Flow cytometric analysis of T cell membrane potential. OT-I cells were isolated and cultured on the nanocomposite membranes with varying surface charges and treated with OVA peptides (2 μg/mL) for 6 h, followed by DiBAC4(3) staining (n = 6 biological replicates, mean ± sem, *, compared with NC; &, compared with LC; #: compared with MC; ***P = 0.0002, ****P < 0.0001, & & & &P < 0.0001, ####P < 0.0001, two-tailed unpaired Student’s t test). d Whole-cell patch-clamp recording of T cell membrane potential. OT-I cells were isolated and activated with OVA257–264 peptides (2 μg/mL) for 6 h, followed by electrophysiological recording (n = 4 biological replicates, mean ± sem, ***P = 0.0002, two-tailed unpaired Student’s t test). e In vitro cytotoxicity assay of OVA expressing LLC cells by OT-I transgenic T cells at decreased E:T ratios. E, effector cells; T, target cells (n = 4 biological replicates, mean ± sem, *, compared with NC; &, compared with LC; #: compared with MC; *P = 0.0169, **P = 0.0047 (E:T = 1:1), **P = 0.0030 (E:T = 0.5:1), ****P < 0.0001, & &P = 0.0023, & & & &P < 0.0001, ###P = 0.0006, ####P < 0.0001, one-way ANOVA). f A schematic illustration of the adoptive T cell therapy. Briefly, NOD-SCID mice were inoculated subcutaneously with LLC-OVA cells (2 × 106) on day 0. OT-I naïve T cells, isolated from lymph nodes and spleen of OT-I mice, were cultured on the nanocomposite membranes with varying surface charges, activated with OVA peptides (2 μg/mL) for 2 days, and expanded with IL-2 (10 U/mL) for another 4 days. OT-I cells (3 × 106) were intravenously injected into tumor-bearing mice on day 7 post-tumor inoculation. g Macroscopic evaluation of tumors from mice intravenously injected with OT-I cells (n = 6 mice). h Tumor volume of mice intravenously injected with OT-I cells was monitored over time (n = 6 mice, mean ± sem, *, compared with NC; &, compared with LC; ns, not significant (P > 0.05), ****P < 0.0001, && &P = 0.0002, & & & & P < 0.0001, one-way ANOVA). i Quantity of tumor-infiltrating immune cells in mice intravenously injected with OT-I cells 21 days after implantation (n = 6 mice, mean ± sem, *, compared with NC; &, compared with LC; #, compared with MC; ns, not significant (P > 0.05), *P = 0.0371, **P = 0.0043, ****P < 0.0001, & &P = 0.0050, #P = 0.0423, one-way ANOVA). j Flow cytometric analysis of the frequency of CD8+ T cells in tumors from mice intravenously injected with OT-I cells 21 days after implantation (n = 6 mice, mean ± sem, *, compared with NC; &, compared with LC; #, compared with MC; ns, not significant (P > 0.05), *P = 0.0162, ***P = 0.0004, ****P < 0.0001, & & & &P < 0.0001, ##P = 0.0019, one-way ANOVA). k Flow cytometric analysis of the frequency of CD8+ T cells in draining lymph node (dLN) from mice intravenously injected with OT-I cells 21 days after implantation (n = 6 mice, mean ± sem, *, compared with NC; &, compared with LC; #, compared with MC; ns, not significant (P > 0.05), *P = 0.0447, ***P = 0.0001, ****P < 0.0001, & & &P = 0.0001, #P = 0.0462, one-way ANOVA). l Flow cytometric analysis of the expression of TNF in tumor-infiltrated OT-I cells (n = 6 mice, mean ± sem, *, compared with NC; &, compared with LC; ns, not significant (P > 0.05), *P = 0.0326, **P = 0.0012, ****P < 0.0001, &P = 0.0126, one-way ANOVA). m Flow cytometric analysis of the expression of IFNγ in tumor-infiltrated OT-I cells (n = 6 mice, mean ± sem, *, compared with NC; &, compared with LC; ns, not significant (P > 0.05), *P = 0.0299, &P = 0.0365 (MC vs. LC), &P = 0.0156 (HC vs. LC), one-way ANOVA). n Flow cytometric analysis of the expression of GZMB in tumor-infiltrated OT-I cells (n = 6 mice, mean ± sem, *, compared with NC; &, compared with LC; #, compared with MC; ns, not significant (P > 0.05), ****P < 0.0001, & & & &P < 0.0001, ##P = 0.0090, one-way ANOVA). o Graphic illustration of the study design for investigating the status of tumor-infiltrated OT-I cells. Briefly, NOD-SCID mice were inoculated subcutaneously with LLC-OVA cells (2 × 106) on day 0. OT-I naïve T cells, isolated from lymph nodes and spleen of OT-I mice, were cultured on the nanocomposite membranes with varying surface charges, activated with OVA peptides (2 μg/mL) for 2 days, and expanded with IL-2 (10 U/mL) for another 4 days. OT-I cells (3 × 106) were intravenously injected into tumor-bearing mice on day 7 post-tumor inoculation. Tumor-infiltrated OT-I cells were isolated on day 21 and subjected to SMART-sequencing. p, q Tumor-infiltrated OT-I cells were isolated and subjected to SMART-seq. GSEA of genes expressed in the HC group and NC group. ES, enrichment score; NES, normalized enrichment score. Data are representative of two (d, e) independent experiments. Source data are provided as a Source Data file.
We next evaluated the cytolytic function of T cells through in vitro cytotoxicity assay and observed a significant decrease in target cell viability when co-cultured with electrically stimulated T cells (Fig. 1e). To confirm these findings in vivo, we isolated naïve OT-I cells and cultured them on nanocomposite membranes with varying surface charge. After activation and proliferation induction by OVA peptide and IL2, respectively, OT-I cells were adoptively transferred to mice bearing LLC-OVA tumors (Fig. 1f). As shown in Fig. 1g, h, T cells induced by electrical stimuli from charged substrates exhibited superior tumor control, with efficacy dependent on the magnitude of electrical charge. In addition, compared to the blank group, the NC substrate hardly affected the cytotoxic function of T cells (Fig. 1g, h). Furthermore, increasing amounts and percentages of CD8+ T cells were isolated from tumor-infiltrating lymphocytes (TIL) and draining lymph nodes (dLN) in a dose-dependent manner (Fig. 1i–k and Supplementary Fig. 3a). Moreover, we detected the status of tumor-infiltrated T cells 24 h post-infusion and found that the proportion of infiltrating T cells was comparable between the two groups (Supplementary Fig. 3b). However, Ki-67 expression was significantly higher in the HC group (Supplementary Fig. 3c), indicating that the charged substrate enhanced T cell survival and proliferation, contributing to a more sustained T cell presence at later stages, rather than an initial increase in infiltration.
In addition to T cell accumulation, we observed enhanced expression of effector molecules such as TNF, IFNγ, and GZMB in tumor-infiltrating OT-I cells (Fig. 1l–n). To further elucidate the molecular mechanisms, we applied SMART-seq to examine the status of tumor-infiltrating CD8+ T lymphocytes previously cultured on ferroelectric nanocomposite membranes (Fig. 1o). Gene set enrichment analysis revealed selective enrichment of genes related to “effector CD8 T cell” and “T cell receptor signaling pathway” in T cells cultured on HC nanocomposite membranes (Fig. 1p, q). Subsequent flow cytometry analysis also showed the protein levels of CD247 and CD28 were elevated on T cells infiltrated in tumors following HC treatment, which was consistent with transcriptomic data (Supplementary Fig. 3d, e). We also performed an enrichment analysis to examine the functional characteristics of the upregulated genes in the HC group. As shown in Supplementary Fig. 3f, HC treatment led to the upregulation of genes associated with TCR signaling, T cell proliferation, and IFNγ production, compared to the NC group. Accordingly, genes associated with the BioCarta pathways “T cytotoxic cell surface molecules” and “CTL mediated immune response against target cells” were overrepresented in the significantly upregulated genes in HC group (Supplementary Fig. 3g). The above findings thus demonstrate that electrical stimulation enhances the antitumor effects of T cells both in vitro and in vivo.
Enhanced antitumor efficacy of CAR-T therapy through charged substrate treatment
Adoptive cell therapy with chimeric antigen receptor (CAR) immunotherapy holds great promise for cancer treatment. Here, we investigate whether charged substrate treatment can augment the antitumor efficacy of CAR-T therapy. We utilized anti-CD19 CAR-T cells cultured on ferroelectric nanocomposite membranes or without membrane culturing (blank control), which were subsequently administered into a subcutaneous tumorigenic model (Fig. 2a). We first assessed CAR expression levels by flow cytometry and found that the CAR expression levels were consistent between NC and HC conditions (Supplementary Fig. 4a). Remarkably, tumor growth of Raji leukemia cells was markedly restricted in mice receiving CAR-T cells from the high-charged (HC) group (Fig. 2b, c). As expected, the tumor volume in the blank group was the same as that in the NC group, indicating that no charged membrane culturing hardly affects T cell function (Fig. 2b, c). Consistent with these findings, a higher abundance of effector T cells was observed in tumors from mice treated with T cells cultured on charged substrates (Fig. 2d–f). Furthermore, intravenous infusion of CAR-T cells from the HC group resulted in substantial tumor suppression and significantly prolonged survival in an intravenous xenograft tumor model (Fig. 2g–i). Notably, analysis of immune cells in peripheral blood revealed greater numbers of CAR-T cells in the HC group compared to the NC group (Fig. 2j), with similar trends observed in liver and spleen samples (Fig. 2k and Supplementary Fig. 4b). These findings collectively indicate that electrical stimulation provided by charged substrate treatment sustains CAR-T cell viability and enhances the efficacy of CAR-T cell therapy against cancer.

a A schematic illustration of the adoptive CAR-T cell therapy. Briefly, NSG mice were inoculated subcutaneously with Raji (5 × 105) on day 0. T cells were cultured on the nanocomposite membranes with varying surface charges or without membrane culturing (blank) and activated by anti-CD3/anti-CD28 mAb-coated beads. After transduced with CD19 CAR, T cells were expanded with IL2 and intravenously injected into tumor-bearing mice on day 7 post-tumor inoculation. b Macroscopic evaluation of tumors from mice intravenously injected with CD19 CAR-T cells (n = 6 mice). c Tumor volume of mice intravenously injected with CD19 CAR-T cells was monitored over time (n = 6 mice, mean ± sem, ns, not significant (P > 0.05), ****P < 0.0001, one-way ANOVA). d Flow cytometric analysis of the expression of IFNγ in CAR-T cells (n = 6 mice, mean ± sem, ns, not significant (P > 0.05), ***P = 0.0009, one-way ANOVA). e Flow cytometric analysis of the expression of TNF in CAR-T cells (n = 6 mice, mean ± sem, ns, not significant (P > 0.05), ****P < 0.0001, one-way ANOVA). f Flow cytometric analysis of the expression of GZMB in CAR-T cells (n = 6 mice, mean ± sem, ns, not significant (P > 0.05), ***P = 0.0003, one-way ANOVA). g A schematic illustration of the adoptive CAR-T cell therapy. Briefly, NSG mice were intravenously injected with luciferase-expressing Raji cells (5 × 105) on day 0. T cells were cultured on the nanocomposite membranes with varying surface charges and activated by anti-CD3/anti-CD28 mAb-coated beads. After transduced with CD19 CAR, T cells were expanded with IL2 and intravenously injected into tumor-bearing mice on day 7 post-tumor inoculation. h NSG mice were intravenously injected with 5 × 105 luciferase-expressing Raji cells on day 0 and 2 × 106 CAR T cells were intravenously injected on day 7. Tumor cells were measured by bioluminescence imaging every week (n = 5 mice). i Survival analysis of NSG mice intravenously injected with CD19 CAR-T cells (n = 5 mice, *P = 0.0133, **P = 0.0018, log-rank (Mantel-Cox) test). j Flow cytometric analysis of the frequency of CAR-T cells (GFP+ cells) in peripheral blood mononuclear cells (PBMC) from NSG mice intravenously injected with CD19 CAR-T cells (n = 6 mice, mean ± sem, ***P = 0.0004, two-tailed unpaired Student’s t test). k Representative multiplex immunohistochemistry staining images of T cells in the liver from mice intravenously injected with CAR-T cells. Green, CD3; red, EPCAM; blue, DAPI (the scale bars represent 100 μm). Source data are provided as a Source Data file.
Charged substrate treatment enhances T cell mediated antitumor immunity in tumor rechallenge model
To assess the secondary immune response, we conducted secondary tumor challenge experiments. In a subcutaneous tumor model, T cells cultured on a charged substrate were reinfused to kill the primary tumor, and tumors were re-implanted following the excision of initial tumors. Specifically, OT-I cells expanded on HC and NC membranes were transferred into tumor-bearing NOD-SCID mice. After 21 days, initial tumors were excised, and new tumors were implanted in the contralateral axilla to evaluate secondary activation responses. Our results showed that T cells cultured with the high-charged membranes retained a strong secondary response, demonstrating preserved memory function despite an enhanced effector phenotype (Fig. 3a–c). We also observed increased infiltration of CD8+ T cells in the tumors, suggesting superior tumor-homing capabilities of these OT-I cells (Fig. 3d).

a A schematic illustration of the study designed to assess the OT-I cell-mediated secondary immune response. Briefly, NOD-SCID mice were inoculated subcutaneously with LLC-OVA cells (2 × 106) on day 0. OT-I naïve T cells, isolated from lymph nodes and spleen of OT-I mice, were cultured on the nanocomposite membranes with varying surface charges, activated with OVA peptides (2 μg/mL) for 2 days, and expanded with IL-2 (10 U/mL) for another 4 days. OT-I cells (3 × 106) were intravenously injected into tumor-bearing mice on day 7 post-tumor inoculation. 21 days later, the subcutaneous tumor was excised and tumor cells (5 × 106) were re-implanted in the opposite axillary region. b Macroscopic evaluation of second tumors (n = 8 mice). c Tumor volume of the secondary inoculation was monitored over time (n = 8 mice, mean ± sem, ****P < 0.0001, two-tailed unpaired Student’s t test). d Flow cytometric analysis of the frequency of CD8+ T cells in second tumors (n = 8 mice, mean ± sem, ***P = 0.0002, two-tailed unpaired Student’s t test). e A schematic illustration of the study designed to assess the CAR-T cell-mediated secondary immune response. Briefly, NSG mice were intravenously injected with luciferase-expressing Raji cells (5 × 105) on day 0. T cells were cultured on the nanocomposite membranes with varying surface charges and activated by anti-CD3/anti-CD28 mAb-coated beads. After transduced with CD19 CAR, T cells were expanded with IL2 and intravenously injected into tumor-bearing mice on day 7 post-tumor inoculation. After 30 days, tumor-free mice were selected and Raji cells (2 × 106) were re-injected into the mice via the tail vein. Naive mice refer to mice that have not previously been inoculated with tumors. f Tumor cells were measured by bioluminescence imaging at indicated times (n = 4 mice). Source data are provided as a Source Data file.
Furthermore, we also performed the CAR-T tumor model. As shown in Fig. 3e, f, mice that received CAR-T therapy and achieved complete tumor clearance displayed significantly reduced susceptibility to tumor relapse compared to untreated controls. These findings highlight that electrical culture amplified effector cells while preserving the critical tumor reactivity of memory T cells, a key factor for long-term therapeutic success.
Charged substrate treatment shapes the balance between Tvm and Tpp
To elucidate the mechanism underlying the enhancement of T cell-mediated antitumor immune responses by charged substrate treatment, we cultured OT-I cells on the nanocomposite membranes and induced their activation and expansion with OVA peptide and IL2, respectively, and conducted single-cell RNA sequencing (scRNA-seq) using the 10 × Genomics platform (Fig. 4a). We identified five distinct T cell subpopulations-proliferative potential T cells (Tpp), virtual memory T cells (Tvm), central memory T cells (Tcm), effector memory T cells (Tem), and effector T cells (Teff)—based on the unique expression patterns of canonical marker genes from the transcriptomic data (Fig. 4b and Supplementary Fig. 5a, b). As shown in Fig. 4c and Supplementary Fig. 5c, the proportion of Tvm (CD44+CD122+CD5+CCL5+Ly49-) significantly decreased upon electrical stimulation. Consistent with previous findings characterizing Tvm as exhibiting a memory-like phenotype without foreign antigen, genes associated with TCR signaling were selectively suppressed in Tvm compared to Tpp or Tem (Fig. 4d and Supplementary Fig. 5d). Furthermore, ligand-receptor mapping revealed cell-cell communication patterns, indicating that the association of Tvm with other T cell types was partially inhibited by electrical stimulation (Fig. 4e and Supplementary Fig. 5e). We also compared the differentially expressed genes (DEG) of Tvm cells under NC and HC culture conditions (Supplementary Fig. 5f) and performed GO enrichment analysis to examine the functional characteristics of the upregulated DEGs (Supplementary Fig. 5g). As shown in Supplementary Fig. 5g, HC treatment led to the upregulation of genes associated with glucose metabolism and the cell cycle, compared to Tvm cells under NC condition. These findings indicate that HC treatment not only suppresses the quantity of Tvm cells but also alters their functional properties.

a Graphic illustration of the study design for evaluating T cell function. OT-I cells were stimulated with OVA peptides (2 μg/mL) for 2 days and IL-2 (10 U/mL) for 4 days. b T cells were induced activation and expansion, followed by 10 × single-cell RNA-sequencing (scRNA-seq). The t-SNE plot shows the single T cells cultured on NC and HC nanocomposite membranes of 5 distinct clusters, including proliferative potential T cells (Tpp), virtual memory T cells (Tvm), central memory T cells (Tcm), effector memory T cells (Tem) and effector T cells (Teff). c Proportions of the 5 different clusters of T cells cultured on the NC and HC nanocomposite membranes. d GSEA of genes expressed in Tpp and Tvm. ES, enrichment score; NES, normalized enrichment score. e Network depicting interaction weights/strength between various cell types. The edge width is proportional to the indicated strength of statistically significant detected interactions. f Volcano plot analysis of gene expression in Tpp and Tvm cells. Red, genes upregulated in Tpp cells. Blue, genes downregulated in Tpp cells. Statistical significance was assessed by the Wilcoxon Rank-Sum test. g Expression of proliferation-associated genes in single T cells cultured on NC and HC nanocomposite membranes. h Genes that were significantly upregulated in Tpp cells cultured on HC nanocomposite membranes, compared with Tpp cells cultured on NC nanocomposite membranes, were analyzed using the GO database. Statistical significance was assessed by a one-sided hypergeometric test. i GSEA of genes expressed in Tpp from the HC group and NC group. ES, enrichment score; NES, normalized enrichment score. j OT-I cells were isolated and cultured on the nanocomposite membranes. T cells were treated with OVA peptides for 2 days and IL2 for 4 days, and proliferation was determined by CFSE dilution assay. k Flow cytometric analysis of the expression of Ki-67 in OT-I cells cultured on the nanocomposite membranes with varying surface charges. OVA peptides (2 μg/mL) and IL-2 (10 U/mL) were used to stimulate T cell activation and ensuing proliferation (n = 4 biological replicates, mean ± sem, ****P < 0.0001, two-tailed unpaired Student’s t test). l Flow cytometric analysis of the expression of CD25 in OT-I cells cultured on the nanocomposite membranes with varying surface charges. OVA peptides (2 μg/mL) and IL-2 (10 U/mL) were used to stimulate T cell activation and ensuing proliferation (n = 4 biological replicates, mean ± sem, ****P < 0.0001, two-tailed unpaired Student’s t test). m Flow cytometric analysis of the expression of phospho-STAT5 in OT-I cells cultured on the nanocomposite membranes with varying surface charges. OVA peptides (2 μg/mL) and IL-2 (10 U/mL) were used to stimulate T cell activation and ensuing proliferation (n = 4 biological replicates, mean ± sem, ****P < 0.0001, two-tailed unpaired Student’s t test). Data are representative of two (k, l, m) independent experiments. Source data are provided as a Source Data file.
In contrast to the reduction in Tvm, the proportion of Tpp, which highly expressed genes associated with cell proliferation such as Mki67 and Top2a, was significantly increased in the HC group (Fig. 4f, g and Supplementary Fig. 6a, b). Analysis of DEGs between the HC and NC groups revealed a global upregulation of gene sets implicated in the IL2-STAT5 as well as glycolysis signaling pathways, which are essential for T cell proliferation (Fig. 4h, i and Supplementary Fig. 6c, d). To further validate the stimulatory effects of charged substrate treatment on T cell proliferation, we performed flow cytometry analysis and the results demonstrated a significant upregulation in the expression of proliferation-associated markers and the proliferative potential of T cells in the HC group (Fig. 4j–m). In addition, identical levels of memory-associated transcription factor TCF7 were detected in both NC-treated T cells and HC-treated T cells (Supplementary Fig. 6e, f), further indicating preserved memory function.
Tpp cells exhibit robust antitumor immunity upon charged substrate treatment
To investigate the role of Tpp cells in adoptive cell therapy (ACT) against cancer, we cultured OT-I cells on HC nanocomposite membranes and isolated CFSEhigh (CFSEhi) and CFSElow (CFSElo) cells (Fig. 5a). RNA sequencing analysis revealed preferential expression of genes associated with T cell proliferation, cell cycle, and amino acid metabolism in CFSElo T cells (Fig. 5b, c and Supplementary Fig. 7a, b). Integration of RNA sequencing and 10 × single-cell RNA sequencing data indicated that CFSElo T cells shared similar characteristics with cluster Tpp cells (Supplementary Fig. 7c, d). Subsequently, CFSEhi and CFSElo OT-I cells were separately adoptively transferred into mice bearing LLC-OVA tumors, and tumor growth was monitored. Notably, the growth of tumors was significantly restricted in mice transferred with CFSElo T cells (Fig. 5d, e). Moreover, the CFSElo group exhibited increased infiltration of tumor-infiltrating immune cells and a higher percentage of CD8+ T cells compared to the CFSEhi group (Fig. 5f, g). Furthermore, infiltrated T cells in the CFSElo group displayed significantly higher expression of effector molecules compared to the CFSEhi group (Fig. 5h–k). Overall, our findings demonstrate that the induction of Tpp cells significantly contributes to enhanced antitumor immunity following charged substrate treatment.

a Graphic illustration of the study design for the investigation the role of proliferative T cells in antitumor immunity. OT-I cells were cultured on HC nanocomposite membranes. OVA peptides (2 μg/mL) and IL-2 (10 U/mL) were used to stimulate T cell activation and ensuing proliferation. CFSElo and CFSEhi cells were isolated by BD FACS Aria II flow cytometer and subjected to RNA-sequencing. b Volcano plot analysis of gene expression in CFSElo and CFSEhi T cells. Red, genes upregulated in CFSElo T cells. Blue, genes downregulated in CFSElo T cells. Statistical significance was assessed by the Wald test. c Genes that were significantly upregulated in CFSElo T cells were analyzed using GO database. Statistical significance was assessed by oa ne-sided hypergeometric test. d Macroscopic evaluation of tumors from mice intravenously injected with CFSElo and CFSEhi T cells (n = 5 mice). e Tumor volume of mice intravenously injected with CFSElo and CFSEhi T cells cultured on HC nanocomposite membrane was monitored over time (n = 5 mice, mean ± sem, ***P = 0.0005 (Day 13), ***P = 0.0008 (Day 15), ***P = 0.0003 (Day 19), ****P < 0.0001, two-tailed unpaired Student’s t test). f Quantity of tumor-infiltrating immune cells in mice intravenously injected with CFSElo and CFSEhi T cells (n = 5 mice, mean ± sem, **P = 0.0055, two-tailed unpaired Student’s t test). g Flow cytometric analysis of the frequency of CD8+ T cells in tumors from mice intravenously injected with CFSElo and CFSEhi T cells (n = 5 mice, mean ± sem, ****P < 0.0001, two-tailed unpaired Student’s t test). h Flow cytometric analysis of the expression of IFNγ in tumor-infiltrated OT-I cells (n = 5 mice, mean ± sem, **P = 0.0042, two-tailed unpaired Student’s t test). i Flow cytometric analysis of the expression of TNF in tumor-infiltrated OT-I cells (n = 5 mice, mean ± sem, **P = 0.0013, two-tailed unpaired Student’s t test). j Flow cytometric analysis of the expression of GZMB in tumor-infiltrated OT-I cells (n = 5 mice, mean ± sem, ***P = 0.0001, two-tailed unpaired Student’s t test). k Flow cytometric analysis of the expression of KLRG1 in tumor-infiltrated OT-I cells (n = 5 mice, mean ± sem, ****P < 0.0001, two-tailed unpaired Student’s t-test). Source data are provided as a Source Data file.
Charged substrate treatment modulates metabolism in Tpp cells
In line with the robust proliferative activity, genes associated with cellular metabolism pathways were enriched in Tpp cells from the HC group compared to those from the NC group, indicating the necessity to meet the metabolic demands of T cell proliferation (Fig. 6a, b). Subsequent RNA-sequencing and quantitative PCR analyses further validated these findings, revealing enhanced mitochondrial respiration in HC group (Fig. 6c–h). To gain deeper insights into alterations in metabolic pathways, untargeted metabolomic analysis based on LC/MS acquisition was conducted to identify metabolites in T cells with or without electrical stimulation. Principal component analysis (PCA) illustrated notable differences between the extracts from the two groups (Fig. 6i). Notably, metabolites enriched in the HC group were associated with “Glycolysis,” “Oxidation of fatty acids” and “amino acid metabolism” (Fig. 6j, k). We subsequently assessed the impact of electrical stimulation on T cell metabolism by measuring the oxygen consumption rate (OCR). As expected, T cells cultured on a charged substrate exhibited significantly higher OCR and spare respiratory capacity (SRC) (Fig. 6l–n). Furthermore, we observed that the proliferative potential induced by electrical stimulation from the charged substrate hardly led to replicative stress and genome instability (Fig. 6o). Overall, our findings highlight the role of electrical stimulation by the charged substrate in enhancing T cell metabolic fitness and promoting the polarization of Tpp cells.

a OT-I cells were isolated and cultured on the nanocomposite membranes with varying surface charges. T cells were treated with OVA peptides for 2 days and IL2 for 4 days. Genes upregulated in the HC group were analyzed with GO terms. Statistical significance was assessed by a one-sided hypergeometric test. b OT-I cells were isolated and cultured on the nanocomposite membranes with varying surface charges. T cells were treated with OVA peptides for 2 days and IL2 for 4 days. Genes upregulated in the HC group were analyzed with metabolism pathways. c–h RT–qPCR analysis of indicated gene expression levels in T cells cultured on the nanocomposite membranes with varying surface charges. Relative expression of the genes was normalized by Actb (n = 4 biological replicates, mean ± sem, **P = 0.0012 (Ndufb2), **P = 0.0027 (Ndufaf4), **P = 0.0013 (Ndufa5), ***P = 0.0005 (Cox7a1), ***P = 0.0008 (Ndufa12), ****P < 0.0001, two-tailed unpaired Student’s t test). i T cells were stimulated with OVA peptides (2 μg/mL) for 2 days and IL-2 (10 U/mL) for 4 days, and cell extracts were subjected to LC/MS analysis. PCA analysis of metabolites within T cells cultured on NC and HC nanocomposite membranes. j Heatmap plot showing the top metabolites in T cells cultured on nanocomposite membranes. T cells were stimulated with OVA peptides (2 μg/mL) for 2 days and IL-2 (10 U/mL) for 4 days, and cell extracts were subjected to LC/MS analysis. k T cells were stimulated with OVA peptides (2 μg/mL) for 2 days and IL-2 (10 U/mL) for 4 days, and cell extracts were subjected to LC/MS analysis. Metabolites that were significantly upregulated in T cells cultured on HC nanocomposite membranes, compared with T cells cultured on NC nanocomposite membranes, were analyzed with the KEGG pathway. Statistical significance was assessed by a one-sided hypergeometric test. l–n OT-I cells were isolated and cultured on the nanocomposite membranes with varying surface charges. Metabolic characteristics of T cells were evaluated by OCR and SRC levels. Oligomycin (500 nM), carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) (1 μM), and antimycin A & rotenone (500 nM) were used (n = 6 biological replicates, mean ± sem, ****P < 0.0001, two-tailed unpaired Student’s t test). o OT-I cells were isolated and cultured on the nanocomposite membranes with varying surface charges. T cells were treated with OVA peptides for 2 days and IL2 for 4 days. Flow cytometric analysis of the expression of γ-H2AX in T cells was performed. (n = 3 biological replicates, mean ± sem, ns, not significant (P > 0.05), two-tailed unpaired Student’s t test). Data are representative of two (c–h, o) independent experiments. Source data are provided as a Source Data file.
Direct current stimulation enhances T cell mediated antitumor immunity
To determine whether other forms of electrical stimulation could similarly enhance T cell responses, we applied direct current (DC) stimulation to T cells. Consistent with previous findings, DC stimulation suppressed tumor growth and enhanced the efficacy of adoptive T cell therapy (Supplementary Fig. 8a, b). In addition, infiltrated T cells in both the HC and DC groups expressed higher levels of effector molecules compared to the NC group (Supplementary Fig. 8c–e). As observed with ferroelectric nanocomposite membranes, in vitro experiment showed that DC stimulation potentiated T cell proliferation marker, Ki-67 (Supplementary Fig. 8f). Subsequent quantitative PCR revealed that genes related to mitochondrial respiration were upregulated with DC stimulation (Supplementary Fig. 8g–l). Moreover, T cells with DC stimulation exhibited significantly higher oxygen consumption rate (OCR) and spare respiratory capacity (SRC) (Supplementary Fig. 8m–o).
EGR1 acts as the core downstream target of T cells upon charged substrate treatment
To elucidate the underlying mechanism driving the polarization of Tpp cells upon charged substrate treatment, we employed transposase-accessible chromatin sequencing (ATAC-seq) to map open chromatin regions. Notably, ATAC-seq peaks were prominently enriched at gene regulatory regions, with electrical stimulation markedly enhancing the overall ATAC-seq signals at transcription start sites (TSS) (Supplementary Fig. 9a, b). Analysis of the differential accessibility of gene loci revealed a global increase in chromatin accessibility induced by electrical stimulation (Fig. 7a). Consistent with transcriptome data, the majority of open chromatin regions or ATAC peaks identified were associated with genes implicated in T cell activation and proliferation signatures (Fig. 7b and Supplementary Fig. 9c). Given that transcription factor (TF) binding plays a pivotal role in transcriptional regulation, we conducted ATAC-seq and TF motif integrative analysis, identifying early growth response factor 1 (EGR1) as the top-ranked TF (Fig. 7c). Noteworthy, representative EGR1 binding sites were observed in the promoter regions of key genes (Fig. 7d). Interestingly, charged substrate electrical stimulation and DC stimulation elicited EGR1 expression (Fig. 7e and Supplementary Fig. 9d). To decipher the functional relevance of EGR1 in T cell responsiveness to electrical stimulation, we generated Egr1 knockout mice using CRISPR-Cas9 technology (Supplementary Fig. 10a, b), and confirmed the absence of off-target effects through PCR and sequencing (Supplementary Fig. 10c). In addition, we assessed the impact of Egr1 deficiency on T cell development by examining the size of various immune organs, including the thymus, spleen, inguinal lymph nodes, and mesenteric lymph nodes. We also compared the percentage of CD4+ T cells/CD8 + T cells in these organs. As shown in Supplementary Fig. 11a–h, under quiescent conditions, the absence of Egr1 hardly affects the development of immune organs or the host immune response. Compared with wild-type (WT) T cells, loss of EGR1 abrogated T cell activation (Fig. 7f), effector function (Fig. 7g, h), and proliferation (Fig. 7i and Supplementary Fig. 11i, j) induced by electrical stimulation. Furthermore, EGR1 deletion undermined the electro-responsive T cell-mediated antitumor immunity, as evidenced by gross tissue evaluation, tumor growth curve, and quantity and function of infiltrated lymphocytes (Fig. 7j–n and Supplementary Fig. 11k–m). We also utilized a lentiviral system to overexpress the transcription factor EGR1 in T cells (Supplementary Fig. 12a). Following this, we activated and expanded the T cells under both NC and HC culture conditions. As shown in Supplementary Fig. 12b, overexpression of EGR1 significantly promoted T cell activation (indicated by increased CD25+CD69+ and CD25-CD69+ populations) under NC culture conditions. As expected, HC treatment further enhanced this effect. Similarly, EGR1 overexpression significantly increased IFNγ expression in T cells under both culture conditions (Supplementary Fig. 12c). Lastly, we assessed T cell proliferation using Ki67 staining. As shown in Supplementary Fig. 12d, HC treatment facilitated the EGR1-mediated enhancement of T cell proliferation. Collectively, these findings underscore the essential role of the transcription factor EGR1 in mediating T cell responsiveness to electrical stimulation.

a OT-I cells were isolated and cultured on the nanocomposite membranes with varying surface charges and treated with OVA peptides (2 μg/mL) for 24 h, followed by ATAC-seq. Volcano plot analysis of pairwise comparison of ATAC-seq density between HC and NC OT-I cells was shown. Statistical significance was assessed by the Wald test. b Genes upregulated in the HC group were analyzed with GO terms. Statistical significance was assessed by a one-sided hypergeometric test. c A schematic illustration of the identification of transcription factor EGR1. The sequence motifs identified from the peaks were analyzed and predicted binding transcription factors. d Representative EGR1 binding sites in the promoter region of key genes were displayed. e Immunoblot analysis of protein expression level of EGR1 in T cells cultured on the nanocomposite membranes with varying surface charges in the presence or absence of OVA peptides. f Flow cytometric analysis of the frequencies of CD25−CD69− cells, CD25−CD69+ cells, and CD25+CD69+ cells in OT-I cells cultured on the nanocomposite membranes with varying surface charges in the presence or absence of EGR1. OVA peptides (2 μg/mL) were used to stimulate T cell activation (n = 4 biological replicates, mean ± sem, *P = 0.0353, ****P < 0.0001, two-tailed unpaired Student’s t test). g Flow cytometric analysis of the expression of IFNγ in OT-I cells cultured on the nanocomposite membranes with varying surface charges in the presence or absence of EGR1. OVA peptides (2 μg/mL) were used to stimulate T cell activation (n = 4 biological replicates, mean ± sem, ns, not significant (P > 0.05), ****P < 0.0001, two-tailed unpaired Student’s t test). h Flow cytometric analysis of the expression of TNF in OT-I cells cultured on the nanocomposite membranes with varying surface charges in the presence or absence of EGR1. OVA peptides (2 μg/mL) were used to stimulate T cell activation (n = 4 biological replicates, mean ± sem, *P = 0.0115, ****P < 0.0001, two-tailed unpaired Student’s t test). i Flow cytometric analysis of the expression of Ki-67 in OT-I cells cultured on the nanocomposite membranes with varying surface charges in the presence or absence of EGR1. OVA peptides (2 μg/mL) and IL2 were used to stimulate T cell activation and expansion (n = 4 biological replicates, mean ± sem, ns, not significant (P > 0.05), ***P = 0.0001, two-tailed unpaired Student’s t-test). j NOD-SCID mice were inoculated subcutaneously with LLC-OVA cells (2 × 106) on day 0. OT-I cells with or without EGR1 were cultured on the nanocomposite membranes with varying surface charges, activated with OVA peptides (2 μg/mL) for 2 days, and expanded with IL-2 (10 U/mL) for another 4 days. OT-I cells (3 × 106) were intravenously injected into tumor-bearing mice on day 7 post-tumor inoculation. Macroscopic evaluation of tumors from mice intravenously injected with WT or Egr1─/─ OT-I cells 21 days post-implantation (n = 5 mice). k Tumor volume of mice intravenously injected with WT or Egr1─/─ OT-I cells was monitored over time (n = 5 mice, mean ± sem, *P = 0.0350, ****P < 0.0001, two-tailed unpaired Student’s ttest). l Flow cytometric analysis of the frequency of CD8+ T cells in tumors from mice intravenously injected with WT or Egr1─/─ OT-I cells (n = 5 mice, mean ± sem, ns, not significant (P > 0.05), ****P < 0.0001, two-tailed unpaired Student’s t test). m Flow cytometric analysis of the frequency of CD8+ T cells in dLN from mice intravenously injected with WT or Egr1─/─ OT-I cells (n = 5 mice, mean ± sem, *P = 0.0182, ****P < 0.0001, two-tailed unpaired Student’s t test). n Flow cytometric analysis of the expression of GZMB in tumor-infiltrated WT or Egr1─/─ OT-I cells (n = 5 mice, mean ± sem, ns, not significant (P > 0.05), **P = 0.0082, two-tailed unpaired Student’s t test). Data are representative of two (e–i) independent experiments. Source data are provided as a Source Data file.
Charged substrate treatment disrupts ionic TCR-lipid interaction and amplifies TCR signaling
In addition to T cell proliferation, we examined T cell activation and effector functions in the presence of a charged substrate. Our findings revealed a significant increase in the percentage of activated (CD25+CD69+) T cells upon charged substrate treatment (Fig. 8a), accompanied by enhanced production of effector cytokines, such as IFNγ (Fig. 8b). Similar observations were made in murine primary T cells stimulated with antibodies against CD3 and CD28 (Supplementary Fig. 13a–d). These findings suggest that the HC nanocomposite membrane did not enhance OVA peptide loading onto MHC class I molecules but facilitated T cell activation. Consistently, transcriptomic analysis unveiled upregulation of genes associated with GO terms like “T cell activation”, “T cell proliferation” and “negative regulation of apoptotic process” in the HC group (Supplementary Fig. 13e), with a more pronounced enrichment of genes related to the “T cell antigen receptor TCR signaling pathway” compared to the NC group (Fig. 8c). Enhanced CD28 protein expression and slight upregulation of ERK1/2 protein level were detected 24 h after T cell activation (Supplementary Fig. 13f). Calcium ion influx is one of the key signaling events following T cell receptor (TCR) stimulation. Calcium influx assays using Fluo-4 AM as an indicator of intracellular calcium levels showed that T cells cultured on HC membranes exhibited a higher calcium influx compared to those cultured on NC membranes (Supplementary Fig. 13g).

a Flow cytometric analysis of the frequencies of CD25−CD69− cells, CD25−CD69+ cells, and CD25+CD69+ cells in OT-I cells cultured on the nanocomposite membranes with varying surface charges. OVA peptides (2 μg/mL) were used to stimulate T cell activation. Blank, cells cultured on a standard dish without nanocomposite membrane (n = 3 biological replicates, mean ± sem, *P = 0.04 (CD25−CD69− cells), *P = 0.0266 (CD25+CD69+ cells), **P = 0.0038, ***P = 0.0007, ****P < 0.0001, one-way ANOVA). b Flow cytometric analysis of the expression of IFNγ in OT-I cells cultured on the nanocomposite membranes with varying surface charges. OVA peptides (2 μg/mL) were used to stimulate T cell activation (n = 3 biological replicates, mean ± sem, *, compared with NC; &, compared with LC; #: compared with MC, ***P = 0.0003, ****P < 0.0001, &P = 0.0160, & & & &P < 0.0001, #P = 0.0132, one-way ANOVA). c OT-I cells were isolated and cultured on the nanocomposite membranes with varying surface charges. Cells were treated with OVA peptides (2 μg/mL) for 6 hours and subjected to RNA-seq. GSEA of genes expressed in the HC group and NC group. ES, enrichment score; NES, normalized enrichment score. d OT-I cells were isolated and cultured on the nanocomposite membranes with varying surface charges and treated with OVA peptides (2 μg/mL) for 6 h. T cells were collected and subjected to Western Blot assay to assess the indicated protein expression. e–g Immunological synapse (IS) formation between PE·Cy7 labeled OT-I cells and GFP expressing LLC-OVA cells was detected by Imaging flow cytometry. OT-I cells and LLC-OVA cells were mixed (1:1) on the nanocomposite membranes with varying surface charges for 30 min. Cells were collected and fixed, permeabilized, and then stained with TRITC-labeled Phalloidin, followed by Imaging flow cytometry analysis. Statistical analysis was performed using the Ideas software (v6.0) (n = 4 biological replicates, mean ± sem, **P = 0.0048, two-tailed unpaired Student’s t test) (f) (n = 15 cells, mean ± sem, **P = 0.0013, two-tailed unpaired Student’s t test) (g). h–j The membrane binding of the cytoplasmic domain of the CD3ε chain was measured by dequenching FRET. The FRET donor was the mTFP1 fused to the C terminus of the CD3ε and the FRET acceptor was R18 dye inserted into the plasma membrane. The short linker constructs, KIR2DL3-3 amino acid (3 AA)-mTFP1 and KIR2DL3-50 amino acid (50 AA)-mTFP1 served as the positive control and negative control, respectively. Jurkat cells with different constructs were cultured on the nanocomposite membranes with varying surface charges and treated with or without anti-CD3/anti-CD28 mAb-coated beads for 30 minutes. BP, before photobleaching; AP, after photobleaching. The scale bars represent 10 μm. k The FRET efficiency was calculated as [DonorAfter - DonorBefore]/[DonorAfter] (n = 15 cells, mean ± sem, ****P < 0.0001, one-way ANOVA). Data are representative of two (a, b, d, f) independent experiments. Source data are provided as a Source Data file.
We also conducted experiments to evaluate activation induced cell death (AICD) and overall T cell expansion. As shown in Supplementary Fig. 13h, T cells cultured with the charged membrane exhibited higher proliferation compared to controls on day 6 post-activation. In addition, there was a reduction in the percentage of apoptotic T cells (7AAD+) in the charged membrane condition (Supplementary Fig. 13i). We also assessed Fas and FasL expression and observed a transient increase in FasL expression on day 2 in T cells cultured with the charged membrane (Supplementary Fig. 13j), which we believe is related to the degree of T cell activation. Importantly, no significant differences were observed at other time points (Supplementary Fig. 13j, k), indicating that charged substrate treatment hardly induced AICD of T cells. Notably, treatment with Phorbol 12-myristate 13-acetate (PMA) and ionomycin, mimicking T cell activation via protein kinase C stimulation and Ca2+ influx, resulted in comparable levels of T cell activation irrespective of the presence of charged substrate (Supplementary Fig. 14a), further indicating the synergistic effects of electrical stimulation and TCR signaling.
Given the pivotal role of TCR signaling in T cell proliferation and Tvm cell differentiation, we hypothesize that the regulatory effect of charged substrate treatment on T cell differentiation is achieved through modulating the transduction of the TCR signaling pathway. Utilizing Western Blot and Imaging Cytometry assays, we observed potentiated TCR signaling and enhanced immunological synapse formation in the HC group and DC group compared to the NC group (Fig. 8d–g and Supplementary Fig. 14b). In addition, enhanced activation of TCR signaling was observed in the HC group compared to the NC group upon OVA peptide treatment for 30 min, which confirmed the potentiating effect of the charged substrate on anti-TCR stimulation early after TCR engagement (Supplementary Fig. 14c). Electrostatic interactions are known to regulate immune receptor signaling in T cells, including CD3ε/ζ and CD2841,42,43,44. Hence, we employed a fluorescence resonance energy transfer (FRET)-based experiment to measure the distance between the cytoplasmic domain of CD3ε and the plasma membrane43,45. T cells stimulated with magnetic beads conjugated with monoclonal antibodies against CD3/CD28 showed a significant decline in FRET efficiency of CD3ε-mTFP1 in the HC group, indicating dissociation of the cytoplasmic domain of CD3ε from the plasma membrane upon TCR engagement (Fig. 8h–k and Supplementary Fig. 14d).
We further introduced mutations into the basic amino acids of the CD3ε tail, replacing them with neutral amino acids to disrupt the tail’s responsiveness to charge changes. We then assessed the impact of these mutations using FRET assay. As shown in Supplementary Fig. 14e, f, i, T cells stimulated with magnetic beads conjugated with monoclonal antibodies against CD3/CD28 showed a significant decline in FRET efficiency of CD3ε-mTFP1 in the HC group. This decline indicates dissociation of the cytoplasmic domain of CD3ε from the plasma membrane upon TCR engagement. Importantly, when the basic amino acids in the CD3ε tail were mutated to neutral amino acids, this effect was abolished (Supplementary Fig. 14g–i). These results suggest that the dynamics of the CD3ε tail, which are influenced by charge changes, play a crucial role in the observed TCR signaling effects. Together, our findings reveal that electrical stimulation disrupts ionic TCR-lipid interaction and enhances TCR signaling, leading to a decrease in virtual memory T cells and an increase in proliferative potential T cells. We also assessed the phosphorylation levels of CAR ITAMs and the results show a significant increase in CAR phosphorylation in T cells treated with charged substrates (Supplementary Fig. 14j). However, electrical stimulation by charged substrate had minimal impact on TCR internalization, suggesting that electrical stimulation do not significantly influence TCR turnover processes (Supplementary Fig. 14k).
Considering interspecies differences between humans and mice, we isolated CD8+ T lymphocytes from the peripheral blood of patients undergoing physical examination and induced their activation. Electrical stimulation via charged substrate promoted the expression of cytotoxic effector molecules (Supplementary Fig. 15a–c) and genes associated with proliferation (Supplementary Fig. 15d, e) in human-derived T cells, suggesting the potential clinical utility of extracellular electrical stimulation in enhancing adoptive T cell-based antitumor therapy.
Together, our study demonstrate charged substrate treatment promotes T cell proliferation and inhibits Tvm polarization, which in turn enhances T cell mediated cancer immunotherapy (Supplementary Fig. 15f).
Discussion
The efficacy of adoptive transfer of antigen-specific T cells against solid tumors remains limited with challenges including sub-optimal T cell expansion and waning functionality post-transfer, leading to potential relapse in patients, even in hematological contexts where CAR-T therapies have shown promise46,47. In this study, we report that extracellular electrical stimulation disrupts the TCR-lipid interaction and augments TCR signaling transduction by charged substrate treatment. Beyond T cell activation, this treatment promotes tumor-reactive T cell proliferation while restraining the emergence of tumor-neglecting T cells or virtual memory T cells, thereby optimizing T cell-based immunotherapy against cancer. We propose that bioelectrical stimuli represent a promising avenue for modulating T cell activity to enhance the efficacy of ACT therapy.
Emerging evidence highlights the electro-responsiveness of immune cells. For instance, electrical stimulation has been shown to promote cytokine-induced macrophage polarization48 and enhance macrophage phagocytic uptake function49. In addition, studies using electrotaxis-on-chip (ETOC) platforms demonstrate that neutrophils migrate in response to both electrical and chemotactic gradients50. In our study, we find that charged substrates modify the surface charge of T cells, promoting the dissociation of TCR tails from acidic lipids on the inner membrane, thereby enhancing T cell activation. This process is distinct from electroporation, which uses electrical pulses to create temporary pores in the cell membrane, allowing for the delivery of nucleic acids or protein into cells51,52. While electroporation has been instrumental in engineering CAR-T cells, combining electrical stimulation with nanotechnology has recently emerged as an effective strategy to enhance CAR gene delivery. This method has demonstrated superior delivery efficiency compared to viral transduction, producing CAR-T cells with strong antitumor activity and the ability to suppress the growth of lymphoma Raji cells both in vitro and in vivo53. Moreover, electroporation-based non-viral CAR-T cells have been shown to generate a higher proportion of memory T cells, which have demonstrated impressive clinical outcomes. Importantly, non-viral approaches avoid the risks associated with viral transduction, such as random DNA integration, which can lead to genomic instability or unintended mutations and in rare cases, tumorigenesis54.
Nevertheless, T cell electro-immunology is still in its infancy, and understanding how T cells respond to varying electrical cues could lead to not only novel molecular insights in T cell electro-biology but also fruitful discoveries to facilitate emerging T cell-based therapies. Here, we demonstrate that electrical stimulation by ferroelectric nanocomposite membrane enhances TCR signal transduction, which in turn promotes transcription of cytotoxic effectors and T cell proliferative potential. In this process, nuclear transcription factor EGR1 is shown to be essential for T cell electro-responsiveness. Our study thus delineates the processes and mechanisms of electro-transduction from extracellular stimuli to the cell membrane, cytoplasm, and nucleus of T cells. Our findings build upon previous research suggesting that repetitive stimulation with tumor antigens in vitro can partially restore the responsiveness of virtual memory-like bystander T cells towards tumors, implying that enhancing TCR signaling activation strength may reverse Tvm cell differentiation55. Expanding upon this, our study reveals that in vitro expansion of naïve T cells induces various T cell differentiation stages, including Tvm cells, accompanied by alterations in T cell membrane potential. By engineering a charged substrate capable of modifying T cell membrane potential, we demonstrate that treatment with this substrate enhances depolarization of the membrane of activated T cells. This not only facilitates TCR signaling transduction, thereby augmenting T cell activation, but also limits the differentiation potential of naïve T cells toward Tvm cells.
Interestingly, our findings show a discrepancy with previous studies56,57. We have observed that traditional in vitro T cell expansion leads to the generation of Tvm cells, which express high levels of CD127 and CD122. These Tvm cells compete with Tcm cells for IL-7 and IL-15, limiting the formation of Tcm cells. However, in T cells treated with charged substrates, we found that the generation of Tvm cells was significantly suppressed, which in turn supports the survival of memory T cells in vivo, thereby enhancing response to secondary immune challenges. As a result, T cells treated with the charged substrate exhibit heightened tumor cytotoxicity and targeted elimination capability. This suggests that reversing or impeding Tvm cell differentiation holds promise for enhancing the efficacy of ACT therapy in tumor treatment.
Given the challenges associated with CAR T cell therapy, particularly regarding the persistence of adoptively transferred cells, strategies focusing on the generation and maintenance of long-lasting T cells are imperative3. Manipulating the metabolic characteristics of T cells and CAR-T cells emerges as a promising approach to enhance their antitumor functions3,58. Here, we identify a population of T cells with proliferative potential induced by electrical stimulation. Through modulation by the transcription factor EGR1 in response to electrical stimuli, these cells exhibit enhanced metabolic fitness and prolonged longevity. Thus, electrical stimulation represents a potent strategy for regulating immune functionality and T cell longevity post-reinfusion in adoptive transfer therapy.
Although different structures exit in intracellular domains of CAR and TCR, CAR with polymeric charged particles (PCP) or CD3ζ expressed on the T cell surface also have electrostatic interactions with negatively charged phospholipids in the cell membrane. In addition, incorporating PCPs into CARs with the 4-1BB co-stimulation domain can optimize tonic signaling and promote CAR-T cell persistence33, highlighting the importance of charge density in enhancing CAR signaling and improving T cell performance. Moreover, altered membrane binding of endogenous CD3ε could interact with LCK, thus contributing to greater phosphorylation of CAR ITAMs and the observed phenotype induced by the charged substrate.
In addition to utilizing charged substrates, we also explore the use of DC to stimulate T cells. Our results demonstrate that both electrical stimulation via charged substrates and DC treatment effectively suppress tumor growth and enhance the efficacy of adoptive T cell therapy. Mechanistic analysis reveals that both high-charged substrates and DC stimulation activate TCR signaling and facilitate the expression of EGR1, highlighting the crucial role of electrical cues in modulating T cell responses. While both methods induce similar short-term effects, the charged nanocomposite membranes offer a distinct advantage in terms of long-term stability. The persistent electric field generated by the charged substrates likely exerts a more sustained influence on T cell polarization, potentially through the regulation of chromatin accessibility. This enduring impact may enhance the longevity and efficacy of the immune response compared to DC stimulation, which is typically more transient in nature. As such, while both electrical approaches show promising therapeutic potential, further studies are required to better understand the precise mechanisms at play and to explore the long-term advantages of each method, particularly in terms of their effects on immune cell differentiation and memory formation. These insights could pave the way for optimizing T cell-based therapies, ensuring both robust and durable antitumor responses.
In summary, we present an electrical stimulation-based strategy for T cell-mediated immunotherapy, whereby modulation of T cell fate is achieved. By activating and sustaining tumor-reactive T cells, we demonstrate the potential of electrical stimuli to prime EGR1-mediated cytotoxic effectors and support their long-term persistence. Importantly, these findings are applicable to both mouse and human-derived T lymphocytes, highlighting the translational potential of extracellular electrical stimulation in ACT or CAR-T immunotherapy.
Methods
Mice
Egr1─/─ mice (C57BL/6 J background) were generated by CRISPR-Cas9-mediated gene editing by ourselves. The sequence (GAGGGCAGCGGCGGTAATAG) of the murine Egr1 gene was targeted. All animals were housed and maintained under specific pathogen-free conditions. All sex- and age-matched animal experiments were performed in accordance with protocols approved by the Ethics Committee of Peking University Health Science Center (approved number PUIRB-LA2022609). NOD-SCID mice (NOD.CB17-Prkdcscid/NcrCrl, Cat. 406) and NSG mice (NOD.Cg-PrkdcscidIl2rgtm1Sug/JicCrl, Cat. 408) were purchased from Vital River Laboratory Animal Technology. OT-I mice (The Jackson Laboratory, C57BL/6-Tg (TcraTcrb) 1100Mjb/J, Cat. 003831) were originally provided by Dr. Hong Tang (Institute Pasteur of Shanghai Chinese Academy of Sciences).
Patients and specimens
Human peripheral blood was collected from patients undergoing physical examination. All procedures were carried out under the approval of the Ethics Committee of Peking University School and Hospital of Stomatology (approved number PKUSSIRB-202385029), and informed consent was obtained from all participants (in accordance with the Helsinki Declaration).
Adoptive T cell therapy
LLC (ATCC, CRL-1642)-OVA cells were produced as previously described14. Male NOD-SCID mice (6–8 weeks old) were inoculated subcutaneously with LLC-OVA cells (2 × 106) on day 0. OT-I naïve T cells, isolated from lymph nodes and spleen of OT-I mice, were activated with OVA257–264 peptides (Invivogen, vac-sin) (2 μg/mL) for 2 days and expanded with IL-2 (PeproTech, 212-12) (10 U/mL) for another 4 days. OT-I cells (3 × 106) were intravenously injected into tumor-bearing mice on day 7 post-tumor inoculation. Tumors were monitored every other day and tumor volume was calculated as (length × width2)/2. Tumor volume was monitored every other day and tumor-bearing mice were euthanized when tumor size exceeded 2000 mm3.
CAR-T therapy
Raji cells (CCL-86) were purchased from ATCC. Luciferase-expressing Raji cells were generated by stably transducing a lentiviral vector encoding firefly luciferase. 5 × 105 Raji cells were inoculated subcutaneously or intravenously into male NSG mice (6–8 weeks old). CAR19 was generated using the single-chain variable fragment (scFv)-targeting domain derived from FMC-63 mouse hybridoma and linked to CD8 transmembrane and hinge domain, followed by 4-1BB (CD137) co-stimulatory domain and CD3ζ signaling domain. Activated murine CD8+ T cells were transduced with retroviral vector encoding CD19 CAR and EGFP tag, and the EGFP-positive cells were sorted by a BD FACS Aria II flow cytometer. After in vitro expansion with IL2 (PeproTech, 212-12), CAR-T cells (2 × 106) were intravenously injected into tumor-bearing mice. Tumors were monitored every week and tumor regression was determined by bioluminescent imaging using a Xenogen IVIS-200 instrument (PerkinElmer). Images were analyzed using Living Image, v.4.1, software (PerkinElmer). The mice were monitored every day. Euthanasia was performed if the animals exhibited a rapid weight loss of 20%, or if they lost mobility, were unable to eat or drink, or showed severe behavioral abnormalities and clinical symptoms. The mice were euthanized by excessive amounts of CO2.
Tumor re-challenge model
To assess the OT-I cell-mediated secondary immune response, male NOD-SCID mice (6–8 weeks old) were inoculated subcutaneously with LLC-OVA cells (2 × 106) on day 0. OT-I naïve T cells, isolated from lymph nodes and spleen of OT-I mice, were cultured on the nanocomposite membranes with varying surface charges, activated with OVA peptides (Invivogen, vac-sin) (2 μg/mL) for 2 days, and expanded with IL-2 (PeproTech, 212-12) (10 U/mL) for another 4 days. OT-I cells (3 × 106) were intravenously injected into tumor-bearing mice on day 7 post-tumor inoculation. 21 days later, the subcutaneous tumor was excised and tumor cells (5 × 106) were re-implanted in the opposite axillary region. Tumor volume was monitored every other day and tumor-bearing mice were euthanized when tumor size exceeded 2000 mm3.
To assess the CAR-T cell-mediated secondary immune response, male NSG mice (6–8 weeks old) were intravenously injected with luciferase-expressing Raji cells (5 × 105) on day 0. T cells were cultured on the nanocomposite membranes with varying surface charges and activated by anti-CD3/anti-CD28 mAb-coated beads (Thermo Fisher Scientific, 11452D). After transduced with CD19 CAR, T cells were expanded with IL2 (PeproTech, 212-12) and intravenously injected into tumor-bearing mice on day 7 post-tumor inoculation. After 30 days, tumor-free mice were selected, and Raji cells (2 × 106) were re-injected into the mice vipeopa the tail vein. Naive mice refer to mice that have not previously been inoculated with tumors.
The mice were monitored every day. Euthanasia was performed if the animals exhibited a rapid weight loss of 20%, or if they lost mobility, were unable to eat or drink, or showed severe behavioral abnormalities and clinical symptoms. The mice were euthanized by excessive amounts of CO2.
In vitro cytotoxicity assay
OT-I naïve T cells were isolated and activated with OVA257–264 peptides (Invivogen, vac-sin) (2 μg/mL) for 24 h. LLC-OVA cells were produced as previously described14. OT-I cells were then counted as effectors and added to target cells (OVA expressing LLC cells (ATCC, CRL-1642)) (E:T) in ratios ranging from 1:1 to 0.25:1 and co-cultivated for 12 h at 37 °C. Cells were washed by PBS for 3 times, and the viability of tumor cells was assessed by Cell Counting Kit-8 (Dojindo, CK04) according to the manufacturer’s protocol.
Fabrication of the ferroelectric nanocomposite membranes
The BaTiO3/P(VDF-TrFE) nanocomposite membranes were fabricated based on our previously reported protocol23. Briefly, BaTiO3 nanoparticles were dispersed ultrasonically in dopamine hydrochloride aqueous solution to modify the nanoparticles. Subsequently, dopamine-modified nanoparticles with 5 vol% and P(VDF-TrFE) co-polymer (70/30 mol% VDF/TrFE) powders were dispersed in N, N-dimethylformamide (DMF) to form a stable mixture. The mixture was then cast into nanocomposite membranes of approximately 30 μm thickness and subjected to heating for solvent vaporization. For polarization, the nanocomposite membranes were annealed at 90 °C and 120 °C respectively for 30 min and then treated under an electric field strength of 1 kV/mm by corona poling at room temperature for 30 min, thus obtaining nanocomposite membranes that are non-charged (NC, 55 °C, no poling), low-charged (LC, 55 °C, poling), mid-charged (MC, 90 °C annealing, poling) and high-charged (HC, 120 °C annealing, poling).
Characterization of the ferroelectric nanocomposite membranes
For ferroelectric property characterization of the BaTiO3/P(VDF-TrFE) nanocomposite membranes, the surface potential distribution was characterized by Kelvin probe force microscopy (KPFM, USA). The polarization-electric field (P-E) loop was analyzed using a commercial ferroelectric analyzer (TF1000, Germany). The piezoelectric coefficient (d33) was detected by a piezoelectric coefficient meter (ZJ-3AN, IACAS). The surface morphology of the nanocomposite membranes after annealing and polarization was examined by field emission scanning electron microscopy (FE-SEM, S-4800, HITACHI). The surface roughness of the membranes was characterized by atomic force microscopy (AFM, Bruker).
Immunoblot analysis
Murine T cells were lysed by lysis buffer containing 10% (v/v) glycerol, 0.5% (v/v) NP-40, 150 mM NaCl and 0.1 mM EDTA with protease inhibitor cocktail (Roche). The cell lysates were then quantified and subjected to SDS-Page.
Preparation of immune cells
For isolation of tumor-infiltrated lymphocytes59, the tumor tissues were cut into small pieces and digested by digestion solution containing 0.5 mg/mL collagenase D (Roche, 11088866001) and 0.1 mg/mL DNase (Sigma, DN25) at 37 °C. After passed through filters, samples were centrifuged using a Percoll gradient fractionation method.
PBMCs from patients were isolated by Ficoll density gradient centrifugation60. CD8+ T cells were separated from PBMCs by magnetic cell sorting (MACS) using CD8 microbeads (Miltenyi Biotec, 130-098-194). T cells were stimulated with anti-CD3/anti-CD28 mAb-coated Human T-Expander beads (Thermo Fisher Scientific, 11141D) and cultured in RPMI1640 medium supplemented with IL2 (PeproTech, 200-02).
Flow cytometry
To analyze cell surface maker expression, the cells were incubated with specific antibodies for 30 min at room temperature. To perform intracellular marker staining, cells were treated with a protein transport inhibitor cocktail (eBioscience, 00-4980-03) for 5 h, then fixed and permeabilized, followed by staining with specific antibodies. To detect the expression of Ki-67, cells were fixed and permeabilized, followed by staining with specific antibodies. To detect expression of phosphorylated STAT5, cells were fixed with 2% (w/v) paraformaldehyde and then permeabilized in 90% ice-cold methanol, followed by two washes with PBS. Cells were then followed by incubation with antibody against phospho-STAT5 (eBioscience, SRBCZX, 12-9010-42) for 1 h at room temperature. To detect T cell membrane potential, cells were collected and stained with 5 μM DiBAC4(3) (Solarbio, D9800) for 20 min at 37 °C, followed by flow cytometry analysis. To detect T cell proliferation, isolated T cells were stained with 1 μM CFSE (Thermo Fisher Scientific, 65-0850-84) for 10 min at room temperature. Cells were then induced to activation and proliferation and collected at indicated times, followed by flow cytometry analysis. Antibodies information is supplied in Supplementary Data 1.
SMART-seq
For cDNA library preparation, tumor-infiltrated CD8+ T cells were sorted by a BD FACS Aria Sorp flow cytometer and placed directly into a lysis buffer. Total cellular mRNAs were then reverse transcripted to cDNAs and amplified according to the protocol of Single Cell Full Length mRNA-Amplification Kit (Vazyme, N712). The sequencing libraries were established using TruePrep DNA Library Prep Kit V2 for Illumina (Vazyme, TD501). The library quality was assessed using the Agilent 2100 Bioanalyzer system, and sequencing was performed on the Illumina Hiseq platform with paired-end 150 bp reads. The SMART-seq reads were aligned to the mm10 assembly of the mouse genome using HISAT2 and quantified by RSEM. Thereafter, confirmatively expressed genes were used for downstream analysis. For GSEA, differentially expressed genes were analyzed using applications from Broad Institute 21 with default parameters.
RNA-seq
To study the role of charged substrate in the regulation of T cell function, OT-I cells were cultured on different nanocomposite membranes and treated with OVA peptides for 6 h. To study the role of T cell proliferation in antitumor immunity, OT-I cells cultured on HC nanocomposite membrane were stimulated with OVA peptides for 2 days and IL2 for 4 days, and CFSElo and CFSEhi cells were isolated by BD FACS Aria II flow cytometer. Total RNA was extracted using TRIzol reagent and then purified using poly-T oligo-attached magnetic beads. RNA-seq libraries were constructed using NEBNext UltraTM RNA 24 Library Prep Kit for Illumina (NEB, E7530) and sequenced on an Illumina platform with 125 bp/150 bp paired-end reads. According to the manufacturer’s instructions, clean data were obtained by removing reads containing adapter and poly-N as well as low-quality reads from the raw data. Clean reads were mapped with the reference genome Hisat2 (version 2.0.5) based on the gene model annotation file. Fragments per kilobase per million mapped reads (FPKM) of each gene were calculated based on the length of the gene and the reads count mapped to this gene. Gene sets from RNA-seq data were analyzed for overlap with curated datasets in the MSigDB database.
ATAC-seq
OT-I cells were cultured on the nanocomposite membranes and treated with OVA peptides for 24 h. The unfixed nuclei of these cells were tagged with tn5 transposase using TruePrep DNA Library Prep Kit V2 for Illumina (Vazyme, TD501), and the resulting library fragments were generated by 16 PCR cycles and sequenced on an Illumina NovaSeq 6000 instrument. After adapters were removed, the reads were aligned to the mouse reference genome (mm10) and were annotated to the nearest gene TSSs using esATAC. All duplicated reads, mitochondrial reads, and low-quality reads were removed. Visualization of peak distribution along genomic regions of genes of interest was performed with the Integrative Genomics Viewer (IGV).
Single cell RNA-sequencing
20,000 cultured cells were barcoded and pooled using a 10 × Genomics device. The samples were prepared following the manufacturer’s protocol and sequenced on an Illumina NextSeq sequencer. To produce a raw unique molecular identifier (UMI) count matrix, the raw data were aligned and quantified using the Cell Ranger Single-Cell Software Suite (version 5.0.0, 10 × Genomics) against the mm10 reference genome. The matrix was converted into a Seurat object by the R package Seurat software (version 3.2.3). For quality control, cells with over 15% mitochondrial-derived UMI counts were removed, and the remaining single cells were subjected to downstream analyses, separately. The UMI count matrix was normalized with the SCTransform function. Then, we performed integration on two datasets. In this process, potential anchors were created with the FindIntegrationAnchors function of Seurat using the top 3000 variable genes, and the IntegrateData function was used to integrate data and create a new matrix, in which the potential batch effect was regressed out. To reduce the dimensionality of the scRNA-Seq dataset, principal component analysis (PCA) was performed. The top 40 PCs were used to perform the downstream analysis with the Elbowplot function. Subsequently, uniform manifold approximation and projection (UMAP) were performed. The Seurat FindClusters function was used to divide all cells into several clusters with a resolution set at 0.5, and the Seurat FindAllMarkers function was performed to identify preferentially expressed genes within clusters. The Seurat FindMarkers function was used to identify differentially expressed genes between the two samples in each group. The R package dplyr software (version 1.0.3) was used to count the proportion of each cell type. Primers used for quantitative real-time PCR and sequencing are included in Supplementary Table 1 and Supplementary Table 2.
Downmodulation of cell-surface TCR–CD3 upon TCR engagement
TCR turnover detection was performed according to a previous study61. Cell-surface CD3ε molecules were labeled with FITC conjugated anti-CD3ε Abs (BioLegend, 145-2C11, 100305), and Jurkat cells were activated with anti-CD3/CD28 mAb-coated beads (Thermo Fisher Scientific, 11141D) at 37 °C for the indicated periods of time. After the incubation, the Abs bound to the cell-surface CD3ε were stripped with an acidic buffer, and then the amount of intracellular FITC-conjugated anti-CD3ε Abs was measured by flow cytometry.
Multiplex immunohistochemistry (mIHC)
Tumor tissues were fixed with formaldehyde, embedded within paraffin, and then sectioned at 4 μm thickness. The sections were dehydrated with graded concentrations of ethanol, and endogenous peroxidase activity was blocked with 3% (v/v) hydrogen peroxide in methanol. Then, antigen retrieval was carried out in a microwave oven. The sections were then washed with PBS for three times, blocked with 5% (w/v) BSA for 30 minutes, followed by incubation with primary antibodies overnight at 4 °C. Then, the sections were washed with PBS and incubated with the secondary antibodies for 30 min at room temperature. Citrate buffer was used to elute the primary and secondary antibodies thoroughly by heating the samples using a microwave oven. Antigens were labeled by distinct fluorophores and the cell nuclei were finally stained with DAPI. Images were captured under a confocal laser scanning microscope (Leica).
Detection of immunological synapse formation
For the immunological synapse formation experiment, PE·Cy7 labeled OT-I cells were isolated by BD FACS Aria II flow cytometer and mixed with GFP expressing LLC-OVA cells (1:1) on the nanocomposite membranes with varying surface charges for 30 min. Cells were collected and fixed, permeabilized, and then stained with TRITC-labeled Phalloidin (Solarbio, CA1610), followed by Imaging flow cytometry analysis (FlowSight, Merk Millipore).
Live cell FRET
Investigation of the membrane binding of the cytoplasmic domain of CD3ε in live T cells was performed as previously described43,45. The construct of a human CD3ε chain with a fused C-terminal mTFP1 was constructed. The constructs with flexible linkers of 3 and 50 amino acid residues of the repeat sequence Gly-Gly-Ser (GGS) between the transmembrane domain of KIR2DL3 and mTFP1 were used as a negative control or positive control, respectively. Jurkat cells stably expressing the above constructs were obtained by lentiviral infection and Flow cytometry sorting. Jurkat cells were cultured on the nanocomposite membranes with varying surface charges and stimulated with magnetic beads covalently conjugated with monoclonal antibodies against CD3/CD28 for 30 min. Cells were transfer to a glass-bottom dish, and the cell membrane was stained with 2.5 μg/mL octadecyl rhodamine B (R18) (MCE, HY-W127703) for 10 min. The FRET efficiency between mTFP1 (donor) and R18 (acceptor) was measured by the dequenching method.
The mTFP1 was excited with the 488 nm laser and R18 was excited with 561 nm laser. When performing the photobleaching, we illuminated the chosen cell with 50% power of a 561 nm laser for 3 frames, which could cause more than 95% photobleaching efficiency. The FRET efficiency was calculated as [DonorAfter - DonorBefore]/[DonorAfter].
Metabolite extraction and detection
To extract metabolites of T cells, the cell pellet was re-suspended with 500 μL methanol (prechilled to − 80 °C) and froze in liquid nitrogen. These quenched cells were then thawed and vortexed for 30 s. After centrifugation at 800 g at 4 °C for 1 min, the supernatant was transferred to a fresh tube and lyophilized under vacuum. The dried samples were reconstituted in 80% (v/v) methanol and incubated at 4 °C for 15 min. The samples were then centrifuged at 12,000 × g at 4 °C for 20 min and the supernatant was used for LC/MS analysis.
Metabolic assays
For estimation of T cell metabolic characteristics, OT-I cells were cultured on the nanocomposite membranes and treated with OVA peptides (Invivogen, vac-sin) for 2 days and IL2 (PeproTech, 212-12) for 4 days, followed by mitochondrial stress tests (SeaHorse Bioscience, 03015-100), performed as per the manufacturer’s specifications. 20,000 cells per well in XF base medium supplemented with 10 mM glucose, 1 mM sodium pyruvate, and 2 mM glutamine were then placed into a SeaHorse XF Extracellular 96 Flux Analyzer to measure their oxygen consumption rate (OCR). Oligomycin (500 nM), carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) (1 μM), and antimycin A & rotenone (500 nM) were used to treat the cells.
Electrophysiological recordings
OT-I cells were cultured on the nanocomposite membranes, treated with OVA peptides (Invivogen, vac-sin) for 12 h, and incubated on poly-L-lysine (Solarbio, P2100)-coated coverslips for adhesion. Membrane currents were measured in the whole-cell configuration of the patch clamp technique. Membrane currents were corrected for liquid junction potentials.
Statistical analysis
Statistical analysis was performed using the Prism GraphPad software v8.0. Statistical significance was determined with the unpaired Student’s t test (two-tailed), one-way ANOVA, the log-rank (Mantel-Cox) test, one-sided hypergeometric test, Wilcoxon Rank-Sum test, or Wald test. P < 0.05 was considered significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The scRNA-seq data that supported the findings in this study has been deposited in the GEO database with the accession code GSE243388. The SMART-seq data that supported the findings in this study has been deposited in the SRA database with the accession code PRJNA958611. The ATAC-seq data has been deposited in the SRA database with the accession code PRJNA958476. The RNA-seq data has been deposited in the SRA database with the accession code PRJNA958467. All data are included in the Supplementary Information or available from the authors, as are unique reagents used in this Article. The raw numbers for charts and graphs are available in the Source Data file. Source data are provided in this paper.
References
Labanieh, L., Majzner, R. G. & Mackall, C. L. Programming CAR-T cells to kill cancer. Nat. Biomed. Eng. 2, 377–391 (2018).
Gumber, D. & Wang, L. D. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine 77, 103941 (2022).
Liu, Y. et al. Strategies to enhance CAR-T persistence. Biomark. Res. 10, 86 (2022).
Cheng, J. et al. Cancer-cell-derived fumarate suppresses the anti-tumor capacity of CD8(+) T cells in the tumor microenvironment. Cell Metab. 35, 961–978 (2023).
Liu, G., Rui, W., Zhao, X. & Lin, X. Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cell Mol. Immunol. 18, 1085–1095 (2021).
Tang, L. et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat. Biotechnol. 36, 707–716 (2018).
Watanabe, N., Mo, F. & McKenna, M. K. Impact of Manufacturing Procedures on CAR T Cell Functionality. Front. Immunol. 13, 876339 (2022).
Adu-Berchie, K. et al. Generation of functionally distinct T-cell populations by altering the viscoelasticity of their extracellular matrix. Nat. Biomed. Eng. 7, 1374–1391 (2023).
Dickinson, M. J. et al. A novel autologous CAR-T therapy, YTB323, with preserved T-cell stemness shows enhanced CAR T-cell efficacy in preclinical and early clinical development. Cancer Discov. 13, 1982–1997 (2023).
Kishton, R. J., Sukumar, M. & Restifo, N. P. Metabolic regulation of T Cell longevity and function in tumor immunotherapy. Cell Metab. 26, 94–109 (2017).
Irvine, D. J., Maus, M. V., Mooney, D. J. & Wong, W. W. The future of engineered immune cell therapies. Science 378, 853–858 (2022).
Friedl, P. & Storim, J. Diversity in immune-cell interactions: states and functions of the immunological synapse. Trends Cell Biol. 14, 557–567 (2004).
Demetriou, P. et al. A dynamic CD2-rich compartment at the outer edge of the immunological synapse boosts and integrates signals. Nat. Immunol. 21, 1232–1243 (2020).
Dan, L. et al. The phosphatase PAC1 acts as a T cell suppressor and attenuates host antitumor immunity. Nat. Immunol. 21, 287–297 (2020).
Heim, L. et al. NFATc1 Promotes antitumoral effector functions and memory CD8(+) T-cell differentiation during non-small cell lung cancer development. Cancer Res. 78, 3619–3633 (2018).
Belk, J. A. et al. Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell 40, 768–786 (2022).
Yukawa, M. et al. AP-1 activity induced by co-stimulation is required for chromatin opening during T cell activation. J. Exp. Med. 217, e20182009 (2020).
Xi, H. & Kersh, G. J. Induction of the early growth response gene 1 promoter by TCR agonists and partial agonists: ligand potency is related to sustained phosphorylation of extracellular signal-related kinase substrates. J. Immunol. 170, 315–324 (2003).
Miyazaki, T. Two distinct steps during thymocyte maturation from CD4-CD8- to CD4+CD8+ distinguished in the early growth response (Egr)-1 transgenic mice with a recombinase-activating gene-deficient background. J. Exp. Med. 186, 877–885 (1997).
Du, N. et al. EGR2 is critical for peripheral naive T-cell differentiation and the T-cell response to influenza. Proc. Natl. Acad. Sci. USA 111, 16484–16489 (2014).
Singh, A., Svaren, J., Grayson, J. & Suresh, M. CD8 T cell responses to lymphocytic choriomeningitis virus in early growth response gene 1-deficient mice. J. Immunol. 173, 3855–3862 (2004).
Zhang, X. et al. Nanocomposite Membranes Enhance Bone Regeneration Through Restoring Physiological Electric Microenvironment. ACS Nano 10, 7279–7286 (2016).
Song, J. et al. Manipulation of surface electrical charge on nanocomposite membranes confers wide spectrum bactericidal effects and promotes tissue regeneration. Adv. Funct. Mater. 34, https://doi.org/10.1002/adfm.202314024 (2024).
Zheng, Y., Nandakumar, K. S. & Cheng, K. Optimization of CAR-T cell-based therapies using small-molecule-based safety switches. J. Med. Chem. 64, 9577–9591 (2021).
Ma, S. et al. Identification of a small-molecule Tim-3 inhibitor to potentiate T cell-mediated antitumor immunotherapy in preclinical mouse models. Sci. Transl. Med. 15, eadg6752 (2023).
Mo, F. et al. An engineered IL-2 partial agonist promotes CD8(+) T cell stemness. Nature 597, 544–548 (2021).
Verma, V. et al. MEK inhibition reprograms CD8(+) T lymphocytes into memory stem cells with potent antitumor effects. Nat. Immunol. 22, 53–66 (2021).
Hope, J. M. et al. Fluid shear stress enhances T cell activation through Piezo1. BMC Biol. 20, 61 (2022).
Hickey, J. W. et al. Engineering an artificial T-cell stimulating matrix for immunotherapy. Adv. Mater. 31, e1807359 (2019).
Joseph, J. P., Gugulothu, S. B., Nandi, D. & Chatterjee, K. Mechanical properties affect primary T cell activation in 3D bioprinted hydrogels. ACS Macro Lett. 12, 1085–1093 (2023).
O’Connor, R. S. et al. Substrate rigidity regulates human T cell activation and proliferation. J. Immunol. 189, 1330–1339 (2012).
Nagy, E. et al. Membrane potential distinctly modulates mobility and signaling of IL-2 and IL-15 receptors in T cells. Biophys. J. 114, 2473–2482 (2018).
Chen, J. et al. Tuning charge density of chimeric antigen receptor optimizes tonic signaling and CAR-T cell fitness. Cell Res. 33, 341–354 (2023).
DeFord-Watts, L. M. et al. The CD3 zeta subunit contains a phosphoinositide-binding motif that is required for the stable accumulation of TCR-CD3 complex at the immunological synapse. J. Immunol. 186, 6839–6847 (2011).
Wang, H., Huang, Y. & Xu, C. Charging CAR by electrostatic power. Immunol. Rev. 320, 138–146 (2023).
Xu, C. et al. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell 135, 702–713 (2008).
Hulsmans, M. et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 169, 510–522 (2017).
Yu, W. et al. Kir2.1-mediated membrane potential promotes nutrient acquisition and inflammation through regulation of nutrient transporters. Nat. Commun. 13, 3544 (2022).
Chacon-Cruz, E., Oelberg, D. G., Davis, P. & Buescher, E. S. Membrane depolarization and depletion of intracellular calcium stores are associated with delay of apoptosis in human neutrophils. J. Leukoc. Biol. 64, 759–766 (1998).
Lobner, M., Leslie, R. G., Prodinger, W. M. & Nielsen, C. H. Spontaneous complement activation on human B cells results in localized membrane depolarization and the clustering of complement receptor type 2 and C3 fragments. Immunology 128, e661–e669 (2009).
Dobbins, J. et al. Binding of the cytoplasmic domain of CD28 to the plasma membrane inhibits Lck recruitment and signaling. Sci. Signal 9, ra75 (2016).
Guo, X. et al. Lipid-dependent conformational dynamics underlie the functional versatility of T-cell receptor. Cell Res. 27, 505–525 (2017).
Li, L. et al. Ionic CD3-Lck interaction regulates the initiation of T-cell receptor signaling. Proc. Natl. Acad. Sci. USA 114, E5891–E5899 (2017).
Deford-Watts, L. M. et al. The cytoplasmic tail of the T cell receptor CD3 epsilon subunit contains a phospholipid-binding motif that regulates T cell functions. J. Immunol. 183, 1055–1064 (2009).
Shi, X. et al. Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids. Nature 493, 111–115 (2013).
Huang, R. et al. Recent advances in CAR-T cell engineering. J. Hematol. Oncol. 13, 86 (2020).
Pietrobon, V. et al. Improving CAR T-Cell Persistence. Int. J. Mol. Sci. 22, 10828 (2021).
Gu, J. et al. Effects of electrical stimulation on cytokine-induced macrophage polarization. J. Tissue Eng. Regen. Med. 16, 448–459 (2022).
Hoare, J. I., Rajnicek, A. M., McCaig, C. D., Barker, R. N. & Wilson, H. M. Electric fields are novel determinants of human macrophage functions. J. Leukoc. Biol. 99, 1141–1151 (2016).
Moarefian, M., Davalos, R. V., Burton, M. D. & Jones, C. N. Electrotaxis-on-Chip to Quantify Neutrophil Migration Towards Electrochemical Gradients. Front. Immunol. 12, 674727 (2021).
An, J. et al. Enhancement of the viability of T cells electroporated with DNA via osmotic dampening of the DNA-sensing cGAS-STING pathway. Nat. Biomed. Eng. 8, 149–164 (2024).
Koh, S., Shimasaki, N. & Bertoletti, A. Redirecting T cell specificity using T cell receptor messenger RNA electroporation. Methods Mol. Biol. 1428, 285–296 (2016).
Shokouhi, A. R. et al. Engineering efficient CAR-T cells via electroactive nanoinjection. Adv. Mater. 35, e2304122 (2023).
Zhang, J. et al. Non-viral, specifically targeted CAR-T cells achieve high safety and efficacy in B-NHL. Nature 609, 369–374 (2022).
Seok, J., Cho, S. D., Seo, S. J. & Park, S. H. Roles of virtual memory T cells in diseases. Immune Netw. 23, e11 (2023).
Ren, Z. et al. Transient hydroxycholesterol treatment restrains TCR signaling to promote long-term immunity. Cell Chem. Biol. 31, 920–931.e926 (2024).
Zhang, Q. et al. Akt inhibition at the initial stage of CAR-T preparation enhances the CAR-positive expression rate, memory phenotype and in vivo efficacy. Am. J. Cancer Res. 9, 2379–2396 (2019).
Huang, Y. et al. Rewiring mitochondrial metabolism to counteract exhaustion of CAR-T cells. J. Hematol. Oncol. 15, 38 (2022).
Song, J. et al. The deubiquitinase OTUD1 enhances iron transport and potentiates host antitumor immunity. EMBO Rep. 22, e51162 (2021).
Song, J. et al. Loss of RPA1 Impairs Peripheral T Cell Homeostasis and Exacerbates Inflammatory Damage through Triggering T Cell Necroptosis. Adv. Sci. 10, e2206344 (2023).
Ge, Y., Paisie, T. K., Chen, S. & Concannon, P. UBASH3A Regulates the Synthesis and Dynamics of TCR-CD3 Complexes. J. Immunol. 203, 2827–2836 (2019).
Acknowledgements
This work was supported by the National Key Research and Development Program of China 2022YFA1207304 (to X.D.) and 2021YFB3800800 (to Xuehui Z.), the National Natural Science Foundation of China 82221003 (to X.D.), 81991505 (to X.D.), U22A20160 (to X.D.), 52273258 (to Xuehui Z.), U24A20372 (to D.L.), 82171826 (to D.L.) and 82301076 (to J.S.), the China Postdoctoral Science Foundation 2022M720008 (to J. S.), the Beijing Natural Science Foundation L222017 (to D.L.), National Program for Multidisciplinary Cooperative Treatment on Major Diseases PKUSSNMP-202002 (to X.D.), Beijing Selecting and Advance Plan for Medical Science and Technology Innovation Achievements Transformation YC202301QX0137 (to Xuehui Z.) and Central Government Guidance Funding for Local Science and Technology Projects in Shanxi Province YDZJSX2024D075 (to Xuehui Z.).
Author information
Authors and Affiliations
Contributions
X.D., D.L., and Xuehui Z. conceived the study. J.S., Y.L., and L. L. performed most of the experiments and analyzed the data. X.H., Z.L., Q.C., Y.G., and Xiaona Z. performed the animal experiments. Y. M. provided human samples. Xin Z. and F. Y. performed bioinformatics analysis. J.S., D.L., X.D., B.C.H., and Xuehui Z. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Nicolai van Oers, Govind Srimathveeravalli, and Chenqi Xu for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, J., Lu, Y., Liu, L. et al. Charged substrate treatment enhances T cell mediated cancer immunotherapy. Nat Commun 16, 1585 (2025). https://doi.org/10.1038/s41467-025-56858-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56858-y
