
Abstract
Endometrial cancers are characterized by frequent alterations in the PI3K-AKT-mTor, IGF1 and DNA repair signaling pathways. Concomitant inhibition of these pathways was warranted. ENDOLA phase I/II trial (NCT02755844) was designed to assess the safety/efficacy of the triplet combination of the PARP inhibitor olaparib, metronomic cyclophosphamide (50 mg daily), and PI3K-AKT-mTor inhibitor metformin (1500 mg daily) in women with recurrent endometrial carcinomas. Olaparib dose-escalation (100-300 mg twice-a-day (bid)) was used to determine the recommended-phase II-trial-dose (RP2D, primary endpoint), followed by an expansion cohort to determine the non-progression rate at 10 weeks (NPR-10w, secondary endpoint). 31 patients were treated. Olaparib RP2D was defined as 300 mg bid. The tolerability was acceptable, and grade 3-4 adverse events (51% patients) were mainly hematological. The NPR-10w was 61.5%, and the median progression-free survival (mPFS) was 5.2 months. In a post-hoc analysis, when explored by molecular subtypes/alterations, longer PFS were observed in patients with tumors characterized by a non-specific-molecular-profile (NSMP, n = 4; mPFS, 9.1 months), and by both TP53 altered & high number of large genomic alterations (LGA ≥ 8)(n = 10, mPFS, 8.6 months)). The analyses about kinetics of circulating biomarkers and pharmacodynamic effects are not reported here. In total, the benefit/toxicity ratio of the all-oral olaparib/cyclophosphamide/metformin regimen was favorable in heavily pretreated patients with recurrent endometrial cancer.
Similar content being viewed by others

Introduction
Endometrial cancer is the fourth most common malignancy among women in the United States, with an estimated incidence of 67,880 cases/year1. For advanced/metastatic disease, the standard first-line treatment relies on hormone therapy or on the carboplatin-paclitaxel regimen combined with an anti-PD1 mononoclonal antibody2,3, while pembrolizumab + lenvatinib combination is a standard therapeutic option in recurrent setting in patients who were not previously treated with an immune checkpoint inhibitor (ICI)4,5. There is an unmet medical need for new therapeutic innovative options in patients with recurrent advanced/metastatic disease beyond ICIs.
From a molecular point of view, endometrial carcinomas are usually classified into 4 subtypes: 1) rare POLE ultra-mutated diseases; 2) tumors characterized by a defective mismatch repair (dMMR) and/or microsatellite instability (MSI-high); 3) TP53 altered diseases; and the remaining 4) Non-specific molecular profile (NSMP)6. In addition, homologous recombination deficiency (HRD) was reported in up to 28.5% of endometrial cancers7,8. Moreover, frequent alterations of the PI3K-AKT-mTor pathway have been reported in all endometrial cancer molecular subtypes9 PARP inhibitors were shown to enhance cytotoxicity in combination with DNA alkylating agents like cyclophosphamide10. All these data supported the development of an innovative triplet regimen with olaparib, metronomic cyclophosphamide, and metformin.
Poly(ADP-ribose)polymerase (PARP) inhibitors were found to be effective in patients with different HRD-positive tumors11,12,13,14,15,16. Therefore, we assessed the utility of the PARP inhibitor olaparib in patients with advanced endometrial carcinomas. Furthermore, the UTOLA phase II trial recently showed that patients with endometrial tumors with high levels of genome instability as reflected by higher number of large genomic alterations (LGA), a typical marker of serous-like tumors, derived a deeper benefit from olaparib17. To increase the efficacy of olaparib, we combined it with the PI3K-mTor inhibitor metformin. Metformin was thought to exert anticancer effects through several mechanisms of action. First, it is known to interact with PI3K-AKT-mTor signaling pathway by inhibiting AKT activation18. Moreover, metformin was expected to reduce IGF1 circulating levels, thereby inhibiting IGF1R up-regulation commonly reported in endometrial carcinomas19. Furthermore, the PI3K signaling pathway has been reported to maintain homologous recombination steady state, to stabilize and to preserve double-strand break repair by interacting with the homologous recombination complex20. The suppression of PI3K function was shown to impair homologous recombination20. Several pre-clinical studies suggested that the sensitivity of cancer cells to PARP inhibitors, including endometrial carcinoma cells was increased by PTEN deficiency21,22,23,24,25.
The combination of olaparib and metformin was completed with metronomic cyclophosphamide. Endometrial carcinomas are sensitive to alkylating agents, such as platinum compounds or cyclophosphamide26, and PARP inhibitors have been shown to enhance cytotoxicity in combination with DNA alkylating agents27. A major feature of HRD is the susceptibility to platinum and other DNA-damaging agents27.
As a consequence, we assumed that the combination of metronomic cyclophosphamide would increase the anti-proliferative effects of olaparib, in addition to its anti-cancer effects. Altogether, the PARP inhibitor, cyclophosphamide, and metformin combination were expected to induce a synergistic therapeutic effect in recurrent advanced/metastatic endometrial cancer beyond their own activity.
To explore this hypothesis, the ENDOLA phase I/II trial was designed to assess the safety of this triplet combination, defining the olaparib recommended phase II trial dose (RP2D), and investigating the preliminary efficacy. Here, we present the outcomes about the tolerability of the triplet combination, the RP2D, and the efficacy outcomes in the whole population, and in molecularly pre-defined subgroups.
Results
Patient characteristics and exposure
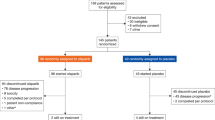
Thirty-five patients were enrolled in this study between September 2016 and October 2019. Four patients, who did not meet the inclusion criteria, did not receive the study treatment and were considered screen-failed. Consequently, the intent-to-treat population comprised 31 patients, including 17 in Phase I and 14 in Phase II (Fig. 1).

RP2D: recommended-phase II-trial-dose of olaparib.
The median patient age was 69 years (Range: 46–80, Inter-Quartile-Range: 62–72, Table 1). Most patients had an endometrioid carcinoma (58.1%) or a serous carcinoma (35.5%), while two patients had a carcinosarcoma (6.5%). Most patients had previously received ≥4 lines of treatment (54.8%), including 29.1% who had received ≥6 lines. Of note, 6.5% had previously received an ICI, 6.5% received an anti-VEGF treatment, and 16.1% received an endocrine therapy.
Olaparib dose-escalation in the phase I part
Of the 17 patients enrolled from September 2016 to August 2018 and assessable for the primary endpoint, two patients were included in the 100 mg dose-level group, four patients in the 150 mg group, two in the 200 mg group, three in the 250 mg group, and six in the 300 mg group (olaparib given on a twice-a-day basis). Dose-escalation was performed up to the maximal dose level. No DLT, the primary safety outcome, was observed. The RP2D (primary endpoint) was therefore defined as the dose level 4: olaparib 300 mg twice-a-day with metronomic cyclophosphamide (50 mg/day) and metformin (500 mg three-times-a-day).
Safety and tolerability
The combination of these three drugs was generally well tolerated (Table 2). Regarding secondary outcomes, all patients experienced at least one adverse event. However, these toxicities were mainly grade 1 or 2. The most frequent treatment-related adverse events of all grades included anemia (71.0%), fatigue (61.3%), nausea (54.8%), lymphocytopenia (38.7%), diarrhea (32.3%), neutropenia (32.3%) and thrombocytopenia (29.0%). Treatment-related grade ≥3 adverse events were reported in 16 patients (51.6%), and were mostly biological side effects. The most frequent were lymphocytopenia (32.3%), anemia (16.1%), neutropenia (16.1%), fatigue (12.9%), and thrombocytopenia (6.5%). Treatment-related grade 4 events included three cases of lymphocytopenia, and one case of neutropenia. No treatment-related deaths occurred. Overall, five patients (16.1%) experienced at least one treatment-related serious adverse event (SAE), including one patient experiencing two SAEs (three lymphocytopenia, two fatigue, and one neutropenia). Eighteen patients (58.0%) required dose reduction or discontinuation (three patients), mostly owing to cytopenias.
Efficacy
Twenty-six patients were assessable for the non-progression rate at 10 weeks (NPR-10w, secondary outcome) (Fig. 1). The NPR-10w was 61.5% (90% CI 0.44–0.77). There was negligible difference between the patients treated at the RP2D (n = 10/16, 62.5%, 90% CI 0.39–0.82), or those treated at a lower dose (n = 6/10, 60.0%, 90% CI 0.30-0.85). Median follow up was 5.1 months (IQR 3.0–8.1). Regarding other secondary outcomes, among the 26 assessable patients, the median PFS was 5.2 months (95% CI 5.1–8.8) (Fig. 2A). The patients with an endometrioid carcinoma had a longer PFS (7.6 months, 95% CI 5.2–10.6), than those with a serous carcinoma (5.1 months, 95% CI 3.8-Not Assessable) (Fig. 2B). The small number of carcinosarcomas did not allow for a relevant assessment of the efficacy, although one patient experienced a complete response. The best radiological response was assessable in 24 patients. The ORR was 20.8% (95% CI 7.1–42.1), and the disease control rate was 66.7% (95% CI 44.7–84.4) (Fig. 3). Overall survival data, although not mature, is above 80% at 6 months (Supplementary Fig. 3).

A Progression-free survival (PFS) in patients assessable for efficacy. B Progression-free survival according to the histological subtypes. Mean values are represented with their 95% confidence interval. NA not assessable.

Percentage change in tumor size from baseline by best overall response (n = 24 evaluable patients).
The secondary endpoints about the pharmacodynamic effects and kinetics of serum tumor markers are still pending, and are therefore not presented in this article.
Exploratory efficacy analyses according to the molecular subtypes
Post-hoc exploratory analyses were performed to explore the efficacy of the triplet regimen according to the molecular subtypes in the ITT population. The molecular subtype was explored in 20 patients (64.5%): POLE (0%), dMMR/MSI-high (n = 3, 15.0%); NSMP (n = 4, 20.0%); and TP53 altered (n = 13, 65.0%). The LGA status was assessed in 23 patients (74.2%). The tumors of 15 patients were LGA-low (LGA score ≤7, 65.2%), whereas those of 8 patients were LGA-high (LGA score ≥8; 34.8%). No tumor exceeded LGA > 20 that is the threshold used in ovarian carcinomas to defined an HRD tumor. As expected, all patients with TP53 wild-type tumors were LGA-low, while the only BRCA2 mutated tumor had a high LGA score of 15.
The lowest efficacy outcomes were observed in patients with dMMR/MSI-high tumors (NPR-10w, 18.2%; median PFS, 5.2 months, 95% CI 3.2 – NA). These numbers were NPR-10w 54.5% and median PFS 5.2 months (95% CI 2.9 – NA) in patients with TP53 altered tumors, and 27.3% and 9.1 months (95% CI 5.1 – NA) in those with NSMP tumors, respectively (Fig. 4A). No clear impact of the LGA score on efficacy was found when considered alone: LGA-low: NPR-10w, 63.6%, median PFS, 5.1 months (95% CI 3.2 – NA); LGA-high: NPR-10w, 36.4%, median PFS, 5.3 months (95%CI 3.6 – NA) (Supplementary Fig. 2). However, when LGA and TP53 were combined, as already performed in the UTOLA study17 a worse median PFS was observed in patients with TP53 altered & LGA-low (n = 5, 3.2 months, 95%CI 2.3 – NA), and a better median PFS was observed in patients with TP53 altered & LGA-high (n = 10, 8.6 months, 95% CI 3.6 – NA, Fig. 4B).

A Progression-free survival according to the molecular subtypes. B Progression-free survival depending on TP53 status and homologous recombination deficiency status explored by the large genomic alterations (LGA) score. NMSP nonspecific molecular subtype, MSI microsatellite instable, dMMR mismatch repair deficient. NA Not Assessable.
Discussion
ENDOLA phase I/II trial was designed as a proof-of-concept study meant to determine whether the concurrent blockage of the main pathways dysregulated in endometrial cancers, using the triplet olaparib + metformin + metronomic cyclophosphamide, would exert synergistic activity, and would be well tolerated.
The enrolled population was representative of the real-life population of patients with recurrent advanced/metastatic endometrial carcinomas, including a median age of 69 years [inter-quartile-range 62–72] which is consistent with an elderly women population, and a high number of previous treatment lines (54.8% of patients with ≥4 lines, supplementary table 2).
The all-oral triplet combination was clinically well tolerated, enabling the olaparib maximum dose-level to be reached without any DLT. The RP2D was defined as 300 mg olaparib twice-a-day, 50 mg daily cyclophosphamide and 500 mg metformin three-times-a-day. The observed adverse events were mainly grade 1–2, while grade 3 were mostly cytopenias and considered manageable. They were consistent with the expectations based on the limited literature data28. In terms of efficacy, the preliminary results of ENDOLA trial suggest that the triplet regimen is active, given the heavily pretreated population. The efficacy endpoint was reached (61.5 % non-progression rate at 10 weeks) and the median PFS was 5.2 months, including 7.6 months for endometrioid carcinomas, and 5.1 months for serous carcinoma.
Altogether, the benefit/toxicity ratio of the triplet regimen in a heavily pre-treated population of patients with all types of endometrial carcinoma appears promising compared to other innovative treatments reported in the literature, although this remains a small study. The median PFS observed with pembrolizumab + lenvatinib in the KEYNOTE-775 was higher (median PFS 7.2 months), but the patients were treated only in second-line setting, and 88.9% of patients experienced grade 3–4 adverse events. In hormone receptor-positive populations, the median PFS observed in patients treated with vistusertib + anastrozole in 1st or 2nd line setting was 5.2 months in VICTORIA trial, with 41% severe adverse events (versus 13% for anastrozole)29 and 8.3 months with letrozole + palbociclib in PALEO trial, with significant hematological toxicity (42% grade 3-4 neutropenia)30. Similarly, Soliman et al tested the interest of everolimus + letrozole and metformin in patient with recurrent endometrial carcinoma in 2nd or 3rd line, and reported a 50% NPR-16w (27/54)31
The outcomes of four randomized trials that tested maintenance with PARP inhibitors after platinum-based chemotherapy in patients with advanced endometrial carcinomas were recently reported. The UTOLA phase II trial that investigated olaparib as a maintenance treatment after first-line platinum chemotherapy was negative in the whole population, but a trend in favor of olaparib was found in patients with higher levels of LGA (median PFS, 5.4 vs 3.6 months, HR = 0.59, 95% CI 0.35–0.99), especially in those with both LGA-high & TP53 alteration (median PFS, 5.3 vs 3.5 months, HR = 0.54, 95% CI 0.30–0.99)17. On the other hand, a second randomized phase II trial suggested that maintenance treatment with rucaparib could potentially increase the PFS compared to placebo with an impressive median PFS of 28.1 months versus 8.7 months (HR 0.45 95%CI 0.26–0.80, p = 0.005)32. The DUO-E phase III trial assessed the utility of adding durvalumab to standard chemotherapy, followed by maintenance with olaparib and durvalumab in first-line treatment. A relevant 15.1-month PFS was reported. The subgroup analysis suggested that the benefit from olaparib added to durvalumab + chemotherapy was restricted to patients with pMMR tumors (15.0 vs 9.9 months, HR = 0.76, 95% CI 0.59–0.99)2. Similarly, the RUBY part 2 trial recently reported favorable outcomes with the addition of dostarlimab to chemotherapy followed by maintenance with dostarlimab + niraparib in the same population of patients (14.3 vs 8.3 months, HR = 0.63, 95% CI 0.44–0.91)33.
In this context, it was rational to explore the efficacy of the olaparib-based triplet regimen, assessed according to the main molecular alteration patterns in ENDOLA (available for 64.5% patients). Of note, our cohort was enriched in patient with TP53 alterations, a biomarker that is usually associated with worse outcome. The distribution across the different subtypes (POLE, 0%; dMMR/MSI-high, 15%; NSMP, 20%; TP53 altered, 65%) was closer to those observed in UTOLA trial (POLE, 1%; dMMR/MSI-high, 12%; NSMP, 35%; TP53 altered, 53%)17, than in those reported in RUBY trial (POLE, 1%; dMMR/MSI-high, 23%; NSMP, 54%; TP53 altered, 22%)34. The efficacy of olaparib combined with metronomic cyclophosphamide and metformin was limited in patients with dMMR/MSI-high tumors, acknowledging that only 6.5% patients previously received immunotherapy. It was higher with mPFS > 8.5 months in patients with NSMP tumors, as well as those with TP53 altered and LGA-high tumors, which is consistent with the outcomes reported in the UTOLA trial17.
These outcomes are promising when compared to other trials which tested the efficacy of combinations based on PARP and immune-checkpoint-inhibitors, such as niraparib + dostarlimab (median PFS 2.4 months, 95% CI 1.6–3.735,), olaparib + durvalumab (median PFS 3.4 months, 95% CI 2.8–6.236,), and talazoparib + avelumab (median PFS 3.6 months, 95% CI 2.4–5.437,).
The results presented here should be interpreted with caution, as ENDOLA trial has several limitations. First, it was a small single-arm trial with a limited number of selected patients treated in highly specialized centers, and was therefore prone to selection biases likely to reduce the applicability of the results to the general population. When the trial was designed in 2015, the therapeutic utility of characterizing the molecular subtypes (based on POLE, dMMR/MSI-high, and TP53 alterations) was still unclear and poorly integrated into the disease management routine. This explains why these biomarkers were not prospectively collected in the trial dataset, and were available for only 65% of patients. Nevertheless, the BRCA and LGA status, assessed centrally using biobanked tumor samples, was available for 74% of the enrolled patients. Interestingly, despite their exploratory nature and the small number of patient in each subgroup, our outcomes were consistent with those reported by Joly et al. in the UTOLA trial regarding the maximum benefit in patients with TP53 altered & LGA-high diseases17. A limitation of ENDOLA trial is that the design did not allow to discriminate the respective effects of each drug, especially those of metformin which actual anti-cancer activity has been questioned in several clinical trials since ENDOLA trial was designed38. Finally, only a limited number of patients had prior treatment with immunotherapy, whilst we need data on the efficacy of targeted agents following treatment with immune checkpoint inhibitors.
In summary, ENDOLA shows promising signs of efficacy/toxicity in a representative population of heavily pretreated patients with recurrent endometrial carcinomas (including elderly patients). It therefore generates data for a future trial testing an all-oral therapeutic option based on a PARP inhibitor and metronomic cyclophosphamide warranting further investigation, especially in recurrent settings after immunotherapy. Indeed, there is a strong need for alternative therapeutic options beyond immune checkpoint inhibitors in patients with endometrial carcinomas.
Methods
Ethical conduct of research
The trial was conducted in accordance with the Declaration of Helsinki and Good Clinical Practices guidelines of the International Council for Harmonization. The protocol, along with seven amendments, was validated by French health authorities (ANSM, 151657A-12) and the central ethics committee (CPP Sud-Est 6 n°AU1238). The investigators declare that they have obtained appropriate institutional review board approval and have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. All the patients provided signed written informed consent.
Study design and treatments
Patient selection
This academic non-randomized, open-label, multi-parameter phase I/II trial (NCT02755844, www.clinicaltrial.gov) was funded by the French National Cancer Institute, sponsored by Lyon University Hospital, and conducted in 6 early phase trial centers of the French gynecology-oncology collaborative group GINECO. The trial protocol is available in the Supplementary Note.
The main key inclusion criteria comprised women with histologically and/or cytologically documented endometrial carcinoma (type I or type II), recurrent after platinum-based chemotherapy, age ≥18 years and <81 years; life expectancy\(\ge \) 16 weeks; Eastern Cooperative Oncology Group (ECOG) performance status (PS) ≤ 1, RECIST 1.1 measurable disease; archival tumor tissue available, or otherwise tumor lesion biopsy feasible; adequate renal, liver and bone marrow function measured within 28 days prior to administration of study treatment i.e., absolute neutrophil count ≥1.5 × 109/L, platelet count ≥100 × 109/L and hemoglobin ≥9.0 g/dL and no blood transfusions in the past 28 days.
The exclusion criteria included prior therapy with PARP inhibitors; use of systemic therapy during the last 2 weeks; treatment with another investigational agent; concomitant 2nd cancer; myelodysplastic syndrome; acute myeloid leukemia; concomitant use of known strong CYP3A4 inhibitor; persistent toxicity (adverse event grade ≥2 caused by previous cancer therapy; illness incompatible with metformin or cyclophosphamide treatment; previous treatment and/or allergy/hypersensitivity to cyclophosphamide; illness that does not allow oral medication; Female patients who are pregnant or lactating or of childbearing potential without a medically acceptable method of birth control; active infection to HIV, hepatitis B or C, or have other forms of hepatitis or cirrhosis; symptomatic uncontrolled brain metastases; major surgery planned within 14 days of starting study treatment; patients must have recovered from any effects of any previous major surgery; resting ECG with QTc >470 ms on 2 or more time points within a 24 h period or family history of long QT syndrome; concomitant treatment with vitamin K antagonists; patients under guardianship.
Objectives and endpoints
The primary objective of the phase I part of the study was to determine the safety of the combination of olaparib, cyclophosphamide, and metformin using NCI-CTAE v.4 criteria, along with the RP2D). The phase II part was designed to evaluate the efficacy of the combination in terms of the non-progression rate at 10 weeks (NPR-10w), among other secondary outcomes like the best radiological response to assess overall response rate (ORR); progression-free survival (PFS); and safety throughout the study. The NPR-10w was chosen as the efficacy endpoint to align with the timing of the first imaging assessment, which occurred 10 weeks after the start of treatment, taking into account that the first cycle lasted 6 weeks.
Phase I part
A Bayesian continual reassessment method (CRM) was used to guide the olaparib dose-escalation into six predefined dose-levels (two patients by level, Supplementary Table 1). At the end of the phase I part, the RP2D was defined as the highest dose-level at which ≤20% of patients would experience dose-limiting toxicities (DLT) with ≥4 patients treated at this dose-level. DLTs were defined as the occurrence of any of the following events during the first treatment cycle, which were judged by the investigator(s) to be at least possibly related to either drug (olaparib and/or cyclophosphamide): (i) Grade 4 neutropenia absolute neutrophil count (ANC) < 0.5 × 109/l, for 7 or more consecutive days, (ii) Grade ≥3 febrile neutropenia: ANC < 1.0 × 109/l and fever ≥38.5 °C, (iii) Grade 3 thrombocytopenia: absolute platelets count <50 × 109/l or thrombocytopenic bleeding, (iv) Grade ≥3 non-hematologic toxicity despite adequate medical intervention excluding toxicities that do not pose a safety risk (e.g., alopecia, asymptomatic hypophosphatemia), as judged by the investigator.
The phase I part was planned to stop when: (i) 20 patients evaluable for DLT would have been included; or (ii) in the case of 10 consecutive patients would have been treated at the same dose-level upon CRM guidance; or (iii) in the case of excessive number of DLTs observed at the lowest dose-level.
During cycle 1 only, the three drugs were gradually introduced during run-in periods: (i) olaparib alone from day 1 to day 7 (week 1), (ii) olaparib + metronomic cyclophosphamide from day 8 to day 14 (week 2), (iii) olaparib + metronomic cyclophosphamide + metformin on day 15, and onwards (week 3–6), to understand potential clinical and biological interactions between the three drugs (Supplementary Fig. 1). Subsequent cycles, based on the continuous administration of the three drugs, were administered on 4-week cycles.
Phase II part
Additional patients were then enrolled at the RP2D (defined during the phase I part) to assess the efficacy in terms of NPR-10w using a Simon minimax 2- 2-stage design. Treatment would be considered interesting if the NPR-10w was ≥50% (Hypothesis H1), and not interesting if it was ≤20% (Hypothesis H0). The 20% and 50% thresholds for NPR-10w were based on the outcomes of previous studies with chemotherapy and endocrine treatments in the second-line setting, with non-progression rates reported between 39% and 45% (before the publication of KEYNOTE-775)39,40,41. This design required a maximum of 17 patients, with a one-sided alpha risk of 10% and power of 90%.
NPR-10w calculation was based on the number of patients with stable disease (SD), partial response (PR) and complete response (CR) defined according to RECIST v.1.1, after 10 weeks of treatment. During the first stage of the Simon design, a maximum of 10 patients assessable for tumor response rate had to be enrolled at RP2D. If less than three successes were observed among the 10 first patients assessable, the trial would be closed for non-efficacy. On the other end, if at least three successes were observed within the first 10 assessable patients at RP2D, the recruitment would continue in the second stage of the design for a total of 17 patients (including at least 4 patients treated at RP2D during the phase I part). If less than six successes were observed, the treatment would be considered as not worthy, and the trial would be closed for non-efficacy. However, if at least six successes were observed the treatment would be considered as worthy for further development. No efficacy hypothesis was established based on the molecular subtypes.
Safety evaluation
Clinical examination, urine dipsticks, ECG and biological tests were performed at baseline, after 1 week, and then every other week during cycle 1, and then at each cycle. Adverse events were recorded and graded using CTCAE v4.0. Patients were evaluated for DLTs during the first (6 weeks) cycle. DLTs are defined in the Supplementary Data.
Efficacy evaluation
Tumors were measured using computed tomography and/or magnetic resonance imaging scans at inclusion, at 10 weeks (at the end of cycle 2), and then every 2 cycles (8 weeks). The best tumor response was assessed using RECIST (v1.1).
Molecular subtype characterization
The mismatch repair status of tumor cells (deficient MMR (dMMR/MSI-High); or proficient MMR (pMMR/MSI-Negative)), along with TP53 alteration (mutated/abnormal TP53, vs. wild-type/normal) and POLE mutations were defined locally on archival tissue, according to the local standard of care.
In addition, tumor samples were centrally reviewed to assess BRCA mutational status and genome profiles as potential complementary prognostic factors for efficacy. High level of copy number alterations may serve as an additional predictor of olaparib benefit, beyond histopathological classification. Translational analyses previously conducted on samples from patients enrolled in the randomized phase 2 UTOLA trial indicated that those with both TP53 alterations and high levels of large genomic alterations (LGA) derived the greatest benefit from olaparib maintenance therapy17.
Tumor cell content was systematically evaluated by a pathologist on H&E-stained slides. Only samples with tumor cell content ≥25% were analyzed for BRCA1/2 and LGA statuses. Ideally, 100 ng (minimum 20 ng) of FFPE-derived DNA was used to construct the libraries with the Agilent SureSelect XT-HS library prep kit (Agilent Technologies, Santa Clara, CA). The pooled pre-capture and capture libraries were sequenced using a NovaSeq 6000 (Illumina Inc., San Diego, CA). The number of LGAs (large genomic alterations, defined as gains or losses >9 Mb) was determined using the shallow Whole Genome Sequencing (sWGS) methodology via the shallowHRDv2 bioinformatics pipeline based on pre-capture library preparation with a mean coverage of ~1x42. Tumors were categorized as LGA-low (≤7 LGA events per genome) or LGA-high (≥8 LGA events per genome), assumed to better discriminate serous-like and endometroid-like carcinomas, as previously applied for UTOLA trial.
For BRCA1/2 sequencing, we prepared capture libraries using the Agilent SureSelect CD Curie CGP panel. Bioinformatic analysis was conducted as previously described43. Variant analysis focused on BRCA1 and BRCA2 genes. We applied a minimum allelic fraction threshold of 10% and a minimum read depth of 100. Only pathogenic or likely pathogenic mutations (including nonsense mutations such as frameshift deletions/insertions, stopgain, and splicing alterations) were reported.
Statistical analyses and design
Progression-free survival (PFS), defined as the time from randomization to progression according to RECIST 1.1 criteria or death (whichever came first) was evaluated over the entire follow-up period, using the Kaplan-Meier method to estimate the median survival time, along with its 95% confidence interval. Confidence intervals for proportions were calculated using the Clopper–Pearson exact method.
Statistical analyses were performed with SAS software (version 9.4) and with R (version 4.0.3).
Reporting summary
Further information on research design is available in Nature Portfolio Reporting Summary linked to this article.
Data availability
ENDOLA trial data may be shared on request according to the Lyon University Hospital rules, based on the following principles: no data should be released that would compromise an ongoing trial or study; there must be a strong scientific or other legitimate rationale for the data to be used for the requested purpose; investigators who have invested time and effort into developing a trial or study should have a period of exclusivity in which to pursue their aims with the data, before key trial data are made available to other researchers; the resources required to process requests should not be underestimated, particularly successful requests that lead to preparing data for release, thus adequate resources must be available to comply in a timely manner or at all, and the scientific aims of the study must justify the use of such resources; and data exchange complies with Information Governance and Data Security Policies in all the relevant countries. If the request fits with these rules, the data may be available within a 3 month time window after sponsor approval, for a maximum 6 month period. Researchers wishing to access data from the ENDOLA should contact sylvie.bin@chu-lyon.fr in the first instance. All remaining data can be found in the article, Supplementary and Source Data files. Source data are provided with this paper.
References
Endometrial Cancer | Uterine Cancer | American Cancer Society n.d. https://www.cancer.org/cancer/types/endometrial-cancer.html (accessed 11 June 2024).
Westin, S. N. et al. Durvalumab plus carboplatin/paclitaxel followed by maintenance durvalumab with or without olaparib as first-line treatment for advanced endometrial cancer: the phase III DUO-E trial. J. Clin. Oncol. 42, 283 (2024).
Mirza, M. R. et al. Dostarlimab for primary advanced or recurrent endometrial cancer. N. Engl. J. Med. 388, 2145–2158 (2023).
Makker, V. et al. Lenvatinib plus pembrolizumab for advanced endometrial cancer. N. Engl. J. Med. 386, 437–448 (2022).
Halla, F. N. P.-C. K. Emerging treatment options for advanced or recurrent endometrial cancer. J. Adv. Pract. Oncol. 13, 45–59 (2022).
Kommoss, S. et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann. Oncol. 29, 1180–1188 (2018).
Siedel, J. H. et al. Clinical significance of homologous recombination deficiency score testing in endometrial Cancer. Gynecol. Oncol. 160, 777–785 (2021).
De Jonge, M. M. et al. Frequent homologous recombination deficiency in high-grade endometrial carcinomas. Clin. Cancer Res. 25, 1087–1097 (2019).
Getz, G. et al. Integrated genomic characterization of endometrial carcinoma. Nature 497, 7447 (2013).
Donawho C. K., et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 13. https://doi.org/10.1158/1078-0432.CCR-06-3039 (2007).
Kaye, S. B. et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J. Clin. Oncol. 30, 372–379 (2012).
Kaufman, B. et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 33, 244–250 (2015).
Fong, P. C. et al. Inhibition of poly(ADP-Ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 361, 123–134 (2009).
Lee, J. M., Ledermann, J. A. & Kohn, E. C. PARP inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann. Oncol. 25, 32–40 (2014).
Mhawech-Fauceglia, P. et al. Expression of DNA repair proteins in endometrial cancer predicts disease outcome. Gynecol. Oncol. 132, 593–598 (2014).
Konstantinopoulos, P. A. et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J. Clin. Oncol. 28, 3555–3561 (2010).
Joly Lobbedez, F. et al. LBA42 Olaparib vs placebo as maintenance therapy after platinum-based chemotherapy in advanced/metastatic endometrial cancer patients: The GINECO randomized phase IIb UTOLA trial. Ann. Oncol. 34, S1283–S1284 (2023).
Ferreira G. D., et al. Metformin modulates PI3K and GLUT4 expression and Akt/PKB phosphorylation in human endometrial stromal cells after stimulation with androgen and insulin. Eur. J. Obstetr. Gynecol. Reprod. Biol. 175. https://doi.org/10.1016/j.ejogrb.2014.01.009 (2014).
Bruchim, I., Sarfstein, R. & Werner, H. The IGF hormonal network in endometrial cancer: functions, regulation, and targeting approaches. Front Endocrinol. (Lausanne) 5, 76 (2014).
De, P. et al. Doubling down on the PI3k-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia 16, 43–72 (2014).
Reinbolt, R. E. & Hays, J. L. The role of PARP inhibitors in the treatment of gynecologic malignancies. Front. Oncol. 3, 237 (2013).
Mendes-Pereira, A. M. et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 1, 315–322 (2009).
Dedes, K. J. et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci. Transl. Med. 2, 53ra75 (2010).
Janzen, D. Y. et al. Low levels of circulating estrogen sensitize PTEN-Null endometrial tumors to PARP inhibition in vivo. Mol. Cancer Ther. 12, 2917–2928 (2013).
Miyasaka, A. et al. Anti-tumor activity of olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor, in cultured endometrial carcinoma cells. BMC Cancer 14, 179 (2014).
Aoki, Y. et al. Adjuvant chemotherapy as treatment of high-risk stage I and II endometrial cancer. Gynecol. Oncol. 94, 333–339 (2004).
O’Sullivan, C. C., Moon, D. H., Kohn, E. C. & Lee, J. M. Beyond breast and ovarian cancers: PARP inhibitors for BRCA mutation-associated and BRCA-like solid tumors. Front. Oncol. 4, 42 (2014).
Lee, C. K. et al. Phase 1 trial of olaparib and oral cyclophosphamide in BRCA breast cancer, recurrent BRCA ovarian cancer, non-BRCA triple-negative breast cancer, and non-BRCA ovarian cancer. Br. J. Cancer 120, 279–285 (2019).
Heudel, P. et al. Safety and efficacy of the mTOR inhibitor, vistusertib, combined with anastrozole in patients with hormone receptor−positive recurrent or metastatic endometrial cancer. JAMA Oncol. 8, 1–9 (2022).
Mirza M. R., et al. Palbociclib versus placebo in combination with letrozole for patients with advanced or recurrent endometrial cancer: the NSGO ENGOT-EN3/PALEO trial. J. Clin. Oncol. 35. https://doi.org/10.1200/jco.2017.35.15_suppl.tps5612 (2017).
Soliman, P. T. et al. Everolimus, letrozole, and metformin in women with advanced or recurrent endometrioid endometrial cancer: a multi-center, single-arm, phase II study. Clin. Cancer Res. 26, 581–587 (2020).
Corr B., et al. A phase II, randomized, double-blind study of the use of rucaparib vs placebo maintenance therapy in metastatic and recurrent endometrial cancer. J. Clin. Oncol. 41:TPS5626–TPS5626 (2023).
Dostarlimab-Based Combinations in Advanced Endometrial Cancer—The ASCO Post n.d. https://ascopost.com/news/march-2024/dostarlimab-based-combinations-in-advanced-endometrial-cancer/ (accessed 11 June 2024).
Mirza, M. R. et al. 740MO - Dostarlimab + chemotherapy for the treatment of primary advanced or recurrent endometrial cancer (pA/rEC): Analysis of progression free survival (PFS) and overall survival (OS) outcomes by molecular classification in the ENGOT-EN6-NSGO/GOG-3031/RUBY trial. Ann. Oncol. 34, S507–S542 (2023).
Madariaga, A. et al. Clinical outcome and biomarker assessments of a multi-centre phase II trial assessing niraparib with or without dostarlimab in recurrent endometrial carcinoma. Nat. Commun. 14, 1452 (2023).
Post, C. C. B. et al. Efficacy and safety of durvalumab with olaparib in metastatic or recurrent endometrial cancer (phase II DOMEC trial). Gynecol. Oncol. 165, 223–229 (2022).
Konstantinopoulos, P. A. et al. Evaluation of treatment with talazoparib and avelumab in patients with recurrent mismatch repair proficient endometrial cancer. JAMA Oncol. 8, 1317–1322 (2022).
Lord, S. R. & Harris, A. L. Is it still worth pursuing the repurposing of metformin as a cancer therapeutic? Br. J. Cancer 128, 958–966 (2023).
Lindemann, K. et al. Examestane in advanced or recurrent endometrial carcinoma: a prospective phase II study by the Nordic Society of Gynecologic Oncology (NSGO). BMC Cancer 14, 68 (2014).
Muggia, F. M., Blessing, J. A., Sorosky, J. & Reid, G. C. Phase II trial of the pegylated liposomal doxorubicin in previously treated metastatic endometrial cancer: a gynecologic oncology group study. J. Clin. Oncol. 20, 2360–2364 (2002).
Fleming G. F. Second-line therapy for endometrial cancer: the need for better options. J. Clin. Oncol. 33. https://doi.org/10.1200/JCO.2015.61.7225 (2015).
Eeckhoutte, A. et al. ShallowHRD: detection of homologous recombination deficiency from shallow whole genome sequencing. Bioinformatics 36, 3888–3889 (2020).
Callens, C. et al. Shallow whole genome sequencing approach to detect homologous recombination deficiency in the PAOLA-1/ENGOT-OV25 phase-III trial. Oncogene 42, 3556–3563 (2023).
Acknowledgements
This trial was supported by the French National Cancer Institute (Institut National du Cancer) within the CLIP2 program. Olaparib was provided by Astra-Zeneca, in partnership with The French National Cancer Institute (Institut National du Cancer, Grant # INCa_9211). Lyon University Hospital (Hospices Civils de Lyon, Lyon, France), was the sponsor of the trial and therefore endorsed the following rules: designing the study, securing funding, ensuring regulatory compliance, overseeing data collection and analysis, and safeguarding participant safety and trial integrity.
Author information
Authors and Affiliations
Contributions
Conceptualization: B.Y. Methodology: M.B., M.A., B.Y. Software: M.B. Validation: M.P., S.B., M.B., M.R., B.Y. Formal analysis: M.P., M.R., B.Y. Investigation: A.L., P.F., C.A., F.J., A.B., C.C., V.S., G.F., M.R., B.Y. Resources: S.B., L.V., M.R. Data curation: M.P., S.B., M.B., M.R., B.Y. Writing original draft: M.P., M.R., B.Y. Writing review and editing: M.P., A.L., P.F., C.A., F.J., S.B., M.B., A.B., C.C., L.V., M.A., V.S., G.F., M.R., B.Y. Visualization: M.P., M.B. Supervision: B.Y. Project administration: S.B., L.V., B.Y. Funding acquisition: B.Y.
Corresponding author
Ethics declarations
Competing interests
Florence Joly declares links of interest with AstraZeneca for expertise and the development of UTOLA trial. Gilles Freyer declares links of interest with AstraZeneca for expertise and consulting. Manuel Rodrigues reports personal fees for serving as an advisor for AstraZeneca; travel support from AstraZeneca; funds to his institution to support research from Merck Sharp & Dohme, Janssen-Cilag, Daiichi-Sankyo. Celine Callens is a co-inventor of the ShallowHRDv2 method (International Patent Application number PCT/EP2024/061709, filed on April 28th, 2024, published on October 31st, 2024 under the International publication number WO2024/223927 and entitled « METHODS FOR DIAGNOSING A HOMOLOGOUS RECOMBINATION DEFICIENCY IN HUMAN TUMORS »). Benoit You has links of interest with Astra-Zeneca for consulting, expertise and travel expenses Philippe Follana has links of interest with Astra-Zeneca for lectures, presentations, manuscript writing or educational events. Alexandra Leary has links of interest with Astra-Zeneca for travel to congress, fees for participation to boards, and funding for trials given to its institution. The other authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript, apart from those disclosed. No writing assistance was used in the production of this manuscript.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Piffoux, M., Leary, A., Follana, P. et al. Olaparib combined to metronomic cyclophosphamide and metformin in women with recurrent advanced/metastatic endometrial cancer: the ENDOLA phase I/II trial. Nat Commun 16, 1821 (2025). https://doi.org/10.1038/s41467-025-56914-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56914-7
