
Abstract
Although most cancer deaths are caused by metastasis, there are no effective therapeutic approaches. This study describes the efficacy of a short synthetic mRNA (s-mRNA) designed by the sequence of non-vesicular extracellular IL1β-mRNA found in the pre-metastatic lung of tumor-bearing mice. The administration of s-mRNA inhibits murine lung metastasis by inducing the innate and adaptive immune systems. s-mRNA binds to ZC3H12D, an RNA-binding protein on natural killer cells and cytotoxic T lymphocytes. The ZC3H12D-s-mRNA complex translocated to the nucleus without being involved in translation. This process induces cytolytic activity and cell death in cancer cells without inducing a cytokine storm, and immune cells retain their antitumor activity. Although the antitumor activity of cytotoxic lymphocytes declines as the disease progresses in cancer patients, s-mRNA induces sustained high killing capacities of natural killer cells and cytotoxic T lymphocytes from colon cancer patients. Therefore, s-mRNA could be a breakthrough solution to prevent metastasis.
Similar content being viewed by others


Introduction
Cancer is a significant health concern due to its increasing incidence among people younger than 50 and its association with longevity1. Although treatments for more than 200 different types of primary cancer2 have improved in the last decade, metastasis remains a threat to the prognosis of cancer2,3. Metastasis is very complicated; the interplay between cancer cells, immune cells, and tissue that metastasizes contributes to metastasis, and metastasis may differ from cancer to cancer and from tissue to tissue4,5. Studies have shown that cancer cells create a microenvironment that facilitates metastasis to target organs before metastasis occurs in mouse models and cancer patients6,7,8,9. At this stage, a small population of antimetastatic immune cells exists10,11,12; however, the majority of immune cells become guardians of cancer cells7. Therefore, to prevent metastasis, it is practical to intervene in immune cells and promote their ability to combat cancer cells. Natural killer (NK) cells and cytotoxic T lymphocytes (CTLs) are potential targets of intervention due to their strong direct tumor-killing ability in metastasis13. They are key players in both innate and adaptive immunity, and their therapeutic value for cancer treatment has been demonstrated in several ways14,15,16.
Extracellular mRNAs that are not enclosed in a vesicle play an important role17 in premetastatic organs8,18. A detailed study revealed that full-length IL1β-mRNA (full-IL1β-mRNA) is present in premetastatic lungs and is incorporated into NK cells in a manner dependent on the surface receptor ZC3H12D19 to enhance their antimetastatic ability. The molecular mechanism of external mRNA signaling is not fully understood, but the IL1β-mRNA-ZC3H12D complex is transferred to the nucleus without being used for protein translation. In a mouse model study, the transfer of NK cells through adoptive cell transfer (ACT) combined with in vitro stimulation by full-IL1β-mRNA significantly suppressed metastasis17. This finding suggests that the administration of full-IL1β-mRNA is highly effective in preventing metastasis. However, full-IL1β-mRNA was degraded in the blood because serum contains RNase (Supplementary Fig. 1a).
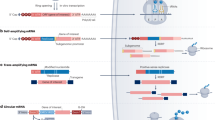
This study successfully demonstrates that a short IL1β-mRNA with a core immunostimulatory sequence activates not only NK cells but also CTLs, increasing their tumoricidal ability (Fig. 1a, summary). In addition, this synthetic mRNA can be administered intravenously to provide sustainable prevention of metastasis. The chemical modifications introduces in the mRNA protect it from degradation by RNase, and thus, it retains its abilities for an extended period.

a Summary of metastasis inhibition by systemic s-mRNA-primed NK cells and CTLs. b Detection of core functional sIL1β-mRNA in humans and mice. c Structural formulas of RNase-resistant synthetic nucleotides. d Binding analysis of mRNAs by EMSA. Lane 1, FITC-mouse IL1β-mRNA (50-mer) alone; lane 2, FITC-mouse IL1β-mRNA (50-mer) + ZC36 protein; lane 3, FITC-mouse IL1β-mRNA (50-mer) + ZC36 protein + non-labeled unrelated RNA (50-mer); lane 4, FITC-mouse IL1β-mRNA (50-mer) + ZC36 protein + non-labeled mouse IL1β-mRNA (50-mer); lane 5, FITC-sIL1β-mRNA_OMe_AE alone; lane 6, FITC-sIL1β-mRNA_OMe_AE + ZC36 protein; lane 7, FITC-sIL1β-mRNA_OMe_AE + ZC36 protein + non-labeled unrelated RNA (50-mer); lane 8, FITC-IL1β-mRNA_OMe_AE + ZC36 protein + non-labeled mouse IL1β-mRNA (50-mer). e Migration analyses of ZC3H12D-overexpressing 786-O cells using various synthetic modified IL1β-mRNA. N = 3 independent experimental replicates for IL1β-mRNA, IL1β-mRNA_OMe, IL1β-mRNA_S, IL1β-mRNA_AE, IL1β-mRNA_OMe_AE, and poly A-mRNA_OMe_AE and N = 2 independent experimental replicates for β-actin-mRNA_OMe_AE. One-way ANOVA with Bonferroni correction. Mean ± SEM. f, g FITC-labeled sIL1β-mRNA was incubated with 20% mouse and human serum. sIL1β-mRNA_OMe_AE was more resistant than nonmodified sIL1β-mRNA. Representative electrophoresis gel image (f) and its quantification (g). h Trace scheme of s-mRNA in vivo. Blood and lungs were examined after tail vein injection of FITC-s-mRNA into mice stimulated with TCM. i Detection of fluorescein in mouse serum at 30 min or 48 hr after tail vein injection of FITC-sIL1β-mRNA_OMe_AE (right, serum). The left panels show positive controls after a mixture of PBS or FITC-sIL1β-mRNA_OMe_AE and serum in vitro. j Representative images of FITC-s-mRNA near metastatic PKH26-labeled E0771 cells in the lungs taken 24 hr after injection with either PBS or FITC-sIL1β-mRNA_OMe_AE. Scale bar: 50 μm, 10 μm. Electropherogram of the retrieved mRNA from the lungs 30 min after tail vein injection of FITC-s-mRNA. 0, FITC-sIL1β-mRNA_OMe_AE; 1 and 2, mRNA from the lung without FITC-s-mRNA injection (two mice); 3 and 4, mRNA from the lung with FITC-sIL1β-mRNA_OMe_AE injection (two mice). k Quantification of FITC-s-mRNA signals around E0771 cells in the lungs. The distance between the FITC signal and the metastatic tumor cell was set at 20 μm. N = 30 areas from 3 mice. One-way ANOVA with Bonferroni correction. Mean ± SEM. Source data are provided as a Source Data file.
Results
Synthetic RNA sequences and characteristics
To determine the functional region of IL1β-mRNA as a non-vesicular extracellular signaling molecule, we first focused on the 3’ untranslated region (UTR). This is because the ZC3H12D receptor protein recognizes this region17. We used a migration assay to screen the functional region. The 130-mer region of the 3’UTR of IL1β-mRNA was selected based on a comparison of mouse and human IL1β-mRNA sequences, and this region retained a biological activity equivalent to that of the full-IL1β-mRNA (Supplementary Fig. 1b). This region induced migration in ZC3H12D-overexpressing 786-O cells. Further sequence alignment revealed that the potential functional region is an AU-rich element containing a 50-mer (Fig. 1b and Supplementary Fig. 2 and 3a). Before preparing the in vivo-grade 50-mer fragment (synthetic IL1β-mRNA, sIL1β-mRNA), we tested several modifications to confirm that the 50-mer was RNase resistant (Fig. 1c). The binding ability of 50-mer sIL1β-mRNA to the receptor protein was then confirmed by electrophoretic mobility shift assay (EMSA) using recombinant ZC3H12D protein (ZC36) (Fig. 1d). Modifications of 4’-aminoethyl-2’-F20 in the ribose group at both ends of uracil and 2’-O-methyl21 for other nucleotides (referred to as _OMe_AE) increased the migration activity to levels as high as those of unmodified RNA (Fig. 1e). However, the migration activity of RNA with phosphorothioate modifications (_S)22 was as low as that of the negative control, sPoly A-mRNA_OMe_AE and sβ-actin-mRNA_OMe_AE. This indicates that sIL1β-mRNA_OMe_AE retains the biological activity of native IL1β-mRNA (Fig. 1e). The concentration dependency of the migration activity induced by sIL1β-mRNA_OMe_AE showed a typical bell-shaped pattern, allowing us to set the optimal concentration as 10 ng/mL for subsequent in vitro research. It was reported that single-stranded RNA (ssRNA) is recognized by intracellular TLR723. To investigate whether TLR 7 is involved in the s-mRNA-mediated migration, we first established a ZC3H12D-overexpressing RAW264.7 cell line because TLR7 is expressed in the cells. Then, TLR7 knockdown was carried out in the ZC3H12D+ cells (Supplementary Fig. 3b). Our migration assay data showed that sIL1β-mRNA_OMe_AE induced both control and TLR7 siRNAs-treated groups at the same level (Supplementary Fig. 3c). To confirm the stability of sIL1β-mRNA_OMe_AE, synthetic RNAs were incubated with mouse or human serum. The unmodified RNAs were rapidly degraded by endogenous RNases, but the sIL1β-mRNA_OMe_AEs remained intact for 48 hr (Fig. 1f, g, and Supplementary Fig. 4a–d). This indicates that s-mRNA are suitable for in vivo research.
To locate the s-mRNA, we introduced a fluorescein isothiocyanate (FITC) label at the 5’ end before conducting the metastasis study. It is well established that a primary tumor can remotely create a premetastatic microenvironment that is favorable for metastasis7. We replicated this microenvironment in a healthy mouse model by intraperitoneally injecting tumor-conditioned medium (TCM) (Fig. 1h). TCM is expected to contain many signaling factors24 released from tumor cells that facilitate tumor metastasis. We observed resistance of FITC-s-mRNA in 100% concentrations of mouse serum in vitro (Fig. 1i, left). However, FITC-s-mRNA was present in the serum 30 min after intravenous injection but not 48 hr after intravenous injection (Fig. 1i, in vivo), implying that the s-mRNA was excreted from the body or absorbed in the tissues within 48 hr. Next, we investigated the localization of FITC-s-mRNA to determine the relationship between the premetastatic microenvironment and FITC-s-mRNA (Fig. 1j gel picture and Supplementary Fig. 4e). To visualize the premetastatic microenvironment, we injected fluorescent-labeled breast cancer cells (E0771 cells) into mice because these cancer cells tend to settle within 20 μm of the premetastatic sites9. FITC-s-mRNA was injected for 1 h after PKH26-labeled E0771 cells were injected. After 24 hr, the lungs exhibited clear recruitment of FITC signals near the PKH26-positive tumor cells (Fig. 1j, k).
sIL1β-mRNA uptake and tumoricidal ability in vitro
It has been demonstrated that full-IL1β-mRNA can induce tumoricidal activity when incorporated into NK cells17. Regarding ZC3H12D, two ZC3H12D-derived splicing variants encoding potential tumor suppressors, P58TFL and P36TFL, were first identified25. It was reported that ZC3H12D is preferentially expressed on normal lymphoid tissues. In addition, the tumor-bearing situation influenced the recruitment of ZC3H12D to the cell surface17. Initially, we examined the expression of ZC3H12D in NK and T cells derived from healthy human peripheral blood mononuclear cells (PBMCs) and mouse splenocytes. Fluorescence-activated cell sorting (FACS) and immunohistochemistry (IHC) revealed that ZC3H12D was expressed at homeostatic levels in these cells (Supplementary Figs. 5 and 6a–c).
We tested whether s-mRNAs are incorporated into human NK or T cells (Fig. 2a, b, and Supplementary Fig. 7). Our FACS analysis revealed the uptake of FITC-sIL1β-mRNA_OMe_AE, but not of FITC-sPoly A-mRNA_OMe_AE, by CD56+CD3- NK and CD3+ T cells (Fig. 2a, b). Similarly, mouse NK and T cells also captured FITC-sIL1β-mRNA_OMe_AE but not FITC-sPoly A-mRNA_OMe_AE (Fig. 2c). This uptake occurred in a ZC3H12D-dependent manner, as shown by the use of Zc3h12 d knockout (KO) mice26 (Fig. 2d). Fluorescence microscopy confirmed the transport of s-mRNA into the nucleus (Fig. 2e, f). Then, we assessed the s-mRNA-mediated immune cell response. Previous studies have reported phenotypic and functional differences between the CD56dim and CD56bright subpopulations27. Therefore, we assessed these subpopulations independently. The production of interferon-γ (IFNγ) in CD56bright cells stimulated with sIL1β-mRNA_OMe_AE was as high as that in CD56bright cells stimulated with the IL-12 protein, which is classically known as an immune cell stimulator28,29 (Supplementary Fig. 8a, b). In contrast, CD56dim cells did not show any induction of IFNγ production (Supplementary Fig. 8c). Regarding the T-cell response, IFNγ induction by sIL1β-mRNA_OMe_AE in CD3+ T cells was detected at a similar level to that of the IL-12 protein (Supplementary Fig. 8d, e). In addition, sIL1β-mRNA_OMe_AE or IL-12 protein stimulation increased the cellular growth rate of CD56bright cells (Supplementary Fig. 9a, b).

a Histogram after the uptake of FITC-sIL1β-mRNA_OMe_AE in human CD56+CD3- NK and CD3+ T cells derived from PBMCs using flow cytometry. FITC-sPoly A-mRNA_OMe_AE (50-mer) was used as a control. b Quantification of the number of cells that were taken up by s-mRNA-FITC. N = 5 individuals. One-way ANOVA with Bonferroni correction. Mean ± SEM. c, d Quantification of FITC-sIL1β-mRNA_OMe_AE uptake in NK1.1+ NK, CD4+ T and CD8+ T cells from splenocytes of wild-type (WT) (c) and Zc3h12d knockout (KO) mice (d) by FACS. N = 3 mice. One-way ANOVA with Bonferroni correction. Mean ± SEM. e Representative confocal microscopy images of FITC-s-mRNA relocation in the nucleus of human CD56dimCD3- NK cells at 1 h after the addition of 10 ng/ml FITC-sIL1β-mRNA_OMe_AE. 10 stacked images. Scale bar: 10 μm. f Quantitative analyses of nuclear FITC signals in human NK cells after treatment with FITC-s-mRNA. N = 13 cells from 3 individuals, none; N = 37 cells from 3 individuals, sIL1β-mRNA_OMe_AE; N = 44 cells from 3 individuals, sPoly A-mRNA_OMe_AE. One-way ANOVA with Bonferroni correction. Mean ± SEM. g Schematic of the tumoricidal assay for NK cells and T cells (see ‘Methods’ for details). h Representative Zombie+ cells indicate cells killed by sIL1β-mRNA_OMe_AE-activated CD56dimCD3- NK cells. i–k Quantitative analysis of killed tumor cells, DLD-1 (i), Caco-2 (j), and MDA-MB-231 (k) cells. Scale bar: 20 μm. N = 3 independent experimental replicates. One-way ANOVA with Bonferroni correction. Mean ± SEM. l Representative Zombie+ dead tumor cells killed by sIL1β-mRNA_OMe_AE-activated CD8+ T cells. m Quantitative analysis of tumor cells killed by s-mRNA- or protein-stimulated CD8+ T cells cocultured with (left) or without dendritic cells (DCs) (right). Scale bar: 20 μm. N = 3 independent experimental replicates. One-way ANOVA with Bonferroni correction. Mean ± SEM. n Strategy for the tumoricidal assay using s-mRNA-primed CD56dimCD3- NK cells or CD8+ T cells after multiple rounds of stimulation (see ‘Methods’ for details). o–q Quantitative analysis of tumor cells killed by CD56dimCD3- NK cells after single (o), two (p), or three (q) stimulation events. N = 3 independent experimental replicates. One-way ANOVA with Bonferroni correction. Mean ± SEM. r–t Quantitative analysis of tumor cells killed by CD8+ T cells after single (r), two (s), or three (t) stimulations. N = 3 independent experimental replicates. One-way ANOVA with Bonferroni correction. Mean ± SEM. Source data are provided as a Source Data file.
To evaluate the tumoricidal activities of human NK and T cells in vitro, we examined whether sIL1β-mRNA_OMe_AE-primed NK cells can kill colon cancer cells (CaCO2, DLD-1) and breast cancer cells (MDA-MB-231). The tumoricidal effect was found to be influenced by the ratio of immune cells to tumor cells30. An equal ratio of immune cells to tumor cells was chosen for in vitro experiments. This is because subsequent in vivo metastasis experiments (Fig. 3) assume that the numbers of tumor cells injected in mice and NK cells in the blood are approximately the same. The tumoricidal activity of CD56dim NK cells was enhanced by sIL1β-mRNA_OMe_AE to the same extent as the IL-12 protein (Fig. 2g–k), whereas that of CD56bright NK cells was not (Supplementary Fig. 9c). In addition, sIL1β-mRNA_OMe_AE enhanced the cytotoxic activity of CD8+ T cells in the presence of dendritic cells (DCs) stimulated with tumor cell lysate (Fig. 2g, l and m). Notably, sIL1β-mRNA_OMe_AE-induced CD8+ T-cell activation was lost when DCs were absent (Fig. 2m). In contrast, the addition of sIL1β-mRNA_OMe_AE did not enhance the cytotoxic activity of CD4+ T cells (Supplementary Fig. 9d–f). We also investigated the effect of repetitive stimulation by sIL1β-mRNA_OMe_AE, as it is well known that repetitive stimulation can cause immune cells to be refractory to stimulation (Fig. 2n). When used for single stimulation, sIL1β-mRNA_OMe_AE, the IL1β-protein, and the IL-12 protein similarly enhanced NK cell tumoricidal activities (Fig. 2o). However, in the second and third rounds, the stimulatory power of the IL1β-protein and IL-12 proteins gradually decreased, and sIL1β-mRNA_OMe_AE retained its enhancing effect (Fig. 2p, q). Regarding CD8+ T-cell-mediated cytotoxic ability, sIL1β-mRNA_OMe_AE showed a booster effect after a series of stimulations (Fig. 2r–t). These data indicate that sIL1β-mRNA_OMe_AE was able to maintain the activated state of NK cells and CTLs without exhaustion, which other classic stimulators cannot achieve.

a Schematic of the antimetastatic experiment using E0771 and MC-38 cells. Tumor-bearing mice were treated with intravenous (iv) injection of s-mRNA or PBS, followed by iv injection of PKH26-labeled tumor cells. b Representative image of the primary tumor taken on day 7 after mammary fat pad (mfp) injection of E0771 cells. The white-lined squares indicate 1 cm. c Volumes of primary tumors treated with s-mRNA or PBS on day 7. N = 5 mice, PBS; N = 5 mice, sPoly A-mRNA_OMe_AE; N = 4 mice, 1 μg of sIL1β-mRNA_OMe_AE; N = 4 mice, 10 μg of sIL1β-mRNA_OMe_AE. One-way ANOVA with Bonferroni correction. Mean ± SEM. d Representative images of PKH26-labeled E0771 cells in the lungs on day 7. e Number of E0771 cells in the lungs of mice treated with s-mRNA or PBS. N = 5 mice, PBS; N = 5 mice, sPoly A-mRNA_OMe_AE; N = 4 mice, 1 μg of sIL1β-mRNA_OMe_AE; N = 4 mice, 10 μg of sIL1β-mRNA_OMe_AE. One-way ANOVA with Bonferroni correction. Mean ± SEM. f Number of E0771 cells in the lungs of mice treated with PBS, 1 μg of sIL1β-mRNA_OMe_AE, or 1 μg of full-length IL1β-mRNA. N = 5 mice. One-way ANOVA with Bonferroni correction. Mean ± SEM. g Scheme for the antimetastasis assay, similar to Fig. 3a, in WT and Zc3h12d KO mice. h Number of metastatic cells in the lung of E0771-bearing WT and Zc3h12d KO mice treated with sIL1β-mRNA_OMe_AE or PBS. N = 5 mice. One-way ANOVA with Bonferroni correction. Mean ± SEM. i, j Metastasis of MC-38 cells. Number of metastatic cells in the lung (j) after treatment with sIL1β-mRNA_OMe_AE or PBS. The primary tumor volume is also shown (i). N = 5 mice, PBS; N = 6 mice, sIL1β-mRNA_OMe_AE. Student’s two-sided t test. Mean ± SEM. k Schematic of an antimetastatic experiment using sIL1β-mRNA_OMe_AE in mice with NK cell depletion (l–n), T-cell depletion (o–r), and NK/T-cell double depletion (s) in combination with anti-ASGM-1 or/and anti-CD4/CD8a antibodies. l, m Validation of NK cell depletion among lymphocytes in the blood (l) and lungs (m). N = 5 mice, PBS; N = 6 mice, Anti-ASGM1. Student’s two-sided t test. Mean ± SEM. n Number of metastatic E0771 cells in the lungs of NK cell-depleted mice. N = 5 mice, PBS; N = 6 mice, Anti-ASGM1. Student’s two-sided t test. Mean ± SEM. o, p Analyses of the percentages of CD4+ T and CD8a+ T cells among TCRβ+ lymphocytes in the blood (o) and lung (p). N = 5 mice. Student’s two-sided t test. Mean ± SEM. q Number of metastatic E0771 cells in T-cell-depleted lungs. N = 5 mice, PBS; N = 6 mice, Anti-CD4/CD8a. Student’s two-sided t test. Mean ± SEM. r Number of metastatic E0771 cells in T-cell-depleted lungs. N = 5 mice. One-way ANOVA with Bonferroni correction. Mean ± SEM. s Number of metastatic E0771 cells in the lungs of both PBS-treated control mice and NK/T double-depleted mice. N = 5 mice. Student’s two-sided t test. Mean ± SEM. Source data are provided as a Source Data file.
Antimetastatic ability of systemic administration of sIL1β-mRNA
To assess the effectiveness of systemic administration of sIL1β-mRNA_OMe_AE, we conducted a metastasis inhibition assay (Fig. 3a). We used tumor cell lines, such as E0771 or MC-38 (a colon cancer cell line), to induce the premetastatic phase in tumor-bearing mice. At this stage, the lungs do not contain metastatic cells, despite the presence of a primary tumor at a distant site. E0771 or MC-38 cells were injected into the mammary fat pad (mfp) or subcutaneously (sc), respectively. The s-mRNA were then intravenously (iv) administered. To mimic the metastatic phase, PKH26-labeled tumor cells were additionally iv injected, and the lungs were examined (Fig. 3a). In this study, we used a three-dose regimen because it was more effective than a single administration at suppressing metastasis (Supplementary Fig. 10a–d). Furthermore, multiple administrations are expected to have a booster effect (Fig. 2q and t). Considering the short half-life of s-mRNA (< 48 hr) in the serum (Fig. 1g and i), continuous antimetastatic treatment might be necessary during primary tumor growth. Mammary fat pad implantation of E0771 cells resulted in the formation of a tumor nodule (~ 5 mm) within one week (Fig. 3b), and s-mRNA treatment did not affect the size of the tumor (Fig. 3c). However, this treatment reduced the number of metastatic cells in the lungs (Fig. 3d) compared to that in the control groups (Fig. 3e). To ensure consistency, we used a 1 μg injection/dose of s-mRNA for one mouse in subsequent experiments, as this dose was found to have the same antimetastatic effect as a 10 μg injection/dose (Fig. 3e). We also tested unmodified full-IL1β-mRNA in this assay and found that injection of the full-IL1β-mRNA had no significant effect on inhibiting metastasis (Fig. 3f). Furthermore, the data indicated that Zc3h12d KO mice had more metastatic cells than did WT mice in this assay (Fig. 3g, h), suggesting a ZC3H12D-dependent effect. To confirm the antimetastatic effect of s-mRNA, we repeated the experiment using a different cell line, MC-38. Similarly, treatment with s-mRNA did not affect the size of the primary tumor (Fig. 3i and Supplementary Fig. 10e) but did reduce the number of lung metastases (Fig. 3j). The data suggest that the antimetastatic effect of s-mRNA treatment is effective in the lung environment, as it does not appear to be effective in the initial colonization of tumor cells in the liver (Supplementary Fig. 10f).
The assay was also performed under immune cell depletion conditions (Fig. 3k). To eliminate NK cells, anti-ASGM1 (5 μl)31 was injected, and the loss of NK cells in the blood and lungs was confirmed on day 7 (Fig. 3l, m, and Supplementary Fig. 11a). The results of the assay indicated that the effect of s-mRNA was reversed by NK cell depletion (Fig. 3n). To eliminate T cells, we injected CD4 and/or CD8a antibodies (both 250 μg)32 into mice (Fig. 3o, p, and Supplementary Fig. 11b). The s-mRNA effect was also reversed when CD8a+ T-cell depletion was applied (Fig. 3q and r). When both depletions were combined, the number of metastases more than doubled (Fig. 3s and Supplementary Fig. 11c and d). The antimetastatic effect of s-mRNA was mainly observed on NK and CD8+ T cells.
Antimetastatic effects of sIL1β-mRNA with no noticeable side effects
Firstly, to consider the s-mRNA-mediated side effect, we examined the location of ZC3H12D+ cells. ZC3H12D was mainly expressed on CD45+ leukocytes in the lung, liver, kidney, and in the primary tumors of tumor-bearing mice (Supplementary Fig. 12a). In contrast, ZC3H12D-positive cells were rare in lung, liver, and kidney tissues of non-tumor-bearing mice. Next, we investigated the side effects of systemic administration of s-mRNA by examining the serum, lung, liver, kidney, and spleen 6 hr after the final injection of s-mRNA or controls (Fig. 4a). In this study, we used IL-12 protein injection as a reference. IL-12 potentially exerts antitumor effects, but it can also cause toxicity and systemic induction of immunosuppressive signals33. Based on published data, the antitumor effect of intraperitoneal injection of the IL-12 protein (1 μg) for 5 consecutive days34 appears to be comparable to that of the three-dose s-mRNA treatment.

a Schematic of screening for immunogenic side effects. The s-mRNA and control PBS groups received intravenous injections 3 times. The IL-12 group received intraperitoneal injections for 5 consecutive days. All groups were dissected 6 hr after the final injections, and their organs and blood were examined. b HE staining of the lung, liver, kidney, and spleen. The necrotic area in the livers of mice treated with IL-12 was increased. Scale bar: 20 µm, 50 μm. c Comparison of blood biochemical values among the 3 groups. N = 5 mice, PBS; N = 6 mice, sIL1β-mRNA_OMe_AE; N = 4 mice, IL-12. One-way ANOVA with Bonferroni correction for multiple comparisons. Mean ± SEM. d Comparison of blood cytokine values among the 3 groups. N = 5 mice, PBS; N = 6 mice, sIL1β-mRNA_OMe_AE; N = 4 mice, IL-12. One-way ANOVA with Bonferroni correction for multiple comparisons. Mean ± SEM. e Comparison of IFNα values among the 4 groups by the ELISA assay. N = 6 mice. One-way ANOVA with Bonferroni correction for multiple comparisons. Mean ± SEM. Source data are provided as a Source Data file.
Histologically, hematoxylin-eosin (HE) staining did not reveal any significant changes, such as inflammation, increased apoptotic bodies, or necrosis, in the organs of the s-mRNA and PBS groups (Fig. 4b). However, the IL-12 group exhibited spotty necrosis in the liver and neutrophil infiltration in the splenic red pulp (Fig. 4b). Furthermore, the IL-12 group exhibited elevated serum AST and ALT levels compared to those in the s-mRNA and PBS groups (Fig. 4c). In addition, blood cytokine levels were quantified 6 hr after s-mRNA injection to investigate the potential cytokine storm35 induced by IFNγ and/or TNFα (Fig. 4d). The levels of IFNγ and/or TNFα in the IL-12 group were greater than those in the s-mRNA and PBS groups, with the latter two groups exhibiting similar levels. In the ELISA assay, culture medium derived from mouse splenocytes treated with s-mRNA did not increase IFNα (Fig. 4e). Furthermore, we performed the same examination 48 hr after the final injection of s-mRNA or controls and obtained similar results (Supplementary Figs. 12b, 12c, 13a, 13b, and 14). In addition, we investigated the potential side effects of continuous administration for one month (Supplementary Fig. 15a) in anticipation of future regular administration to human patients. The results showed no remarkable damage to organs or abnormal blood indicator levels (Supplementary Figs. 15b–d and 16).
Long-term observation models and xenograft model
To assess the long-term effect of s-mRNA on lung metastasis, we excised the lungs 14 days after the tail vein injection of tumor cells (Fig. 5a) to count the metastatic nodules. To simulate a clinical scenario in which only a few metastatic nodules are present in the lungs of cancer patients, we optimized the number of tumor cells used for injection (Supplementary Fig. 17a–d). Thus, we determined empirically that an injection of 1 × 105 tumor cells yielded optimal results without any apparent changes in body or lung weight (Fig. 5b, c). In this study, compared with control treatment, s-mRNA treatment significantly reduced tumor metastasis (Fig. 5d, e). Ki-67 staining of the lungs, which enables the detection of micrometastasis, also demonstrated the antitumor effect of s-mRNA treatment (Fig. 5f, g).

a Scheme of preventive metastatic nodule assay. TCM-stimulated mice were pre-treated with s-mRNAs or PBS and then injected intravenously with metastatic tumor cells. The mice were dissected after 2 weeks. b Weights of the body (left) and lungs (right) among three groups. N = 6 mice per group. One-way ANOVA with Bonferroni correction. Mean ± SEM. c Representative photo of lungs with metastatic nodules (red circles). The white lines indicate 1 cm squares. d, e Blockade of metastasis through the use of sIL1β-mRNA. Photos of the lungs were taken 14 days after E0771 injection for each treatment (d). The green arrows show metastatic nodules. Some nodules are present on the dorsal surface. Number of metastatic nodules (e). N = 6 mice per group. One-way ANOVA with Bonferroni correction. Mean ± SEM. f, g IHC analysis of metastatic cells in the lungs using Ki-67 staining. Representative images of Ki-67 signals (f). Scale bar: 50 μm. Number of the Ki-67-positive cells (g). N = 6 mice per group. One-way ANOVA with Bonferroni correction. Mean ± SEM. h Scheme of the orthotopic model using E0771 cells. Primary tumors were removed 14 days after initial orthotopic implantation, and mice were treated with s-mRNAs or PBS. Lung metastases were counted 3 weeks after removal of the primary tumor. i Representative image of the lung with metastatic nodules (red circle). White lines indicate 1 cm squares. j Number of metastatic nodules. N = 5 mice per group. Student’s two-sided t test. Mean ± SEM. k Scheme of the long-term survival model. Primary tumors were removed 14 days after initial orthotopic implantation, and mice were treated with s-mRNAs or PBS. l Representative image of the lung with metastatic nodules (yellow arrows). White lines indicate 1 cm squares. m Survival data were plotted on a Kaplan-Meier survival curve and statistical significance was calculated using the log-rank test. N = 10 mice, PBS; N = 8 mice, sIL1β-mRNA_OMe_AE. n Scheme of the xenograft model using NOD/Shi-scid, IL-2RγKO (NOG) mouse, and MDA-MB-231 cells. CD56+CD3- NK cells or CD8+ T cells from a healthy donor were intravenously injected into TCM-stimulated mice, followed by intravenous injection of PKH26-labeled tumor cells. o Number of MDA-MB-231 cells in the lungs of mice treated with s-mRNA or PBS. N = 5 mice. One-way ANOVA with Bonferroni correction. Mean ± SEM. Source data are provided as a Source Data file.
We also evaluated the effect of s-mRNA in a spontaneous metastasis model using orthotopic transplantation (Fig. 5h). Since E0771 tumor cells do not spontaneously metastasize unless a primary tumor is removed, primary tumor resection was used in this assay36. Then, s-mRNAs were injected intravenously during the first week. We counted metastatic foci in the lungs 21 days after primary tumor resection. The assay result clearly showed that s-mRNA treatment prevented lung metastasis (Fig. 5i, j). Based on this result, a survival assay system was designed (Fig. 5k), and our result showed that the s-mRNA treatment group had a better prognosis than the control group (Fig. 5l and m).
Finally, we performed a xenograft model using human cell lines to better mimic clinical conditions (Fig. 5n). This assay used NOD/Shi-scid, IL-2RγKO (NOG) mouse, which lacks NK and T cells and has a high engraftment rate of human cells. The results showed that s-mRNA treatment reduced lung metastasis after intravenous injection of human cancer cells (Fig. 5o). Thus, these assay data indicate that s-mRNA treatment may be effective in suppressing lung metastasis in humans.
Nuclear response of NK and CTL cells to sIL1β-mRNA
This investigation revealed that s-mRNA is an antimetastatic drug with no inflammatory side effects. To understand the underlying mechanisms of s-mRNA, we conducted molecular-level research. We applied FITC-sIL1β-mRNA to human PBMCs and separated FITC + CD56+CD3- NK cells or FITC + CD3+ T cells using a cell sorter 30 min or 6 hr after administration because IL1β-mRNA uptake began at 30 min (Supplementary Fig. 18a). Total RNA was isolated from these cells, and their gene expression profiles were analyzed using next-generation sequencing (NGS) (Fig. 6a).

a Schematic of screening of the genes that responded after the uptake of FITC-sIL1β-mRNA_OMe_AE. FITC-positive CD56+CD3- NK and CD3+ T cells were sorted for RNA sequencing. b Summary of the expression profile related to inflammatory cytokines. Data shows RPKM (reads per kilobase million) values of inflammation-related genes. For comparison, ACTB data is added in the top row. c GZMB were upregulated in CD56+CD3- NK and CD8+ T cells 0.5 hr after the uptake of FITC-sIL1β-mRNA_OMe_AE. N = 2 biological replicates. d, e IHC analysis of GZMB expression in CD56dimCD3- NK cells 6 hr after stimulation with sIL1β-mRNA_OMe_AE. d Representative images of GZMB staining and (e) quantification of GZMB expression. Scale bar: 10 μm. N = 50 cells per group. Student’s two-sided t test. Mean ± SEM. f Schematic of electroporation and GZMB staining. g–j Quantitative analysis of tumor cells (Caco-2, DLD-1, and MDA-MB231) killed by sIL1β-mRNA_OMe_AE-activated CD56dimCD3- NK cells or CD8+ T cells after electroporation of siRNA -control (g, i), -GZMB (h, j). N = 3 independent experimental replicates. One-way ANOVA with Bonferroni correction was used for multiple comparisons. Mean ± SEM. Source data are provided as a Source Data file.
Gene Ontology (GO) analysis was performed based on the gene expression changes resulting from s-mRNA stimulation in the NK and T-cell populations (Supplementary Fig. 18b). The enrichment analyses of transcriptional regulatory relationships, as revealed by sentence-based text mining (TRRUST) and transcription factor targets, appeared to differ between the two groups (Supplementary Fig. 18c, d). GO analyses of upregulated genes ( > 2-fold) revealed that both NK and T cells exhibited activation of ‘natural killer cell-mediated cytotoxicity’ and ‘positive regulation of defense response’ (Supplementary Fig. 19a). In addition, GO analyses of downregulated genes (< 0.5-fold) indicated that ‘regulation of cell activation’ and ‘inflammatory regulation’ were suppressed (Supplementary Fig. 19b), which is consistent with the fact that a massive inflammatory response was prevented in the s-mRNA treatment group. When visualizing the cytokine response as pie charts, the percentages of sIL1β-mRNA_OMe_AE-induced individual responses were found to be within the background levels (Supplementary Fig. 19c). The inflammatory cytokine-related heatmap showed that stimulation of NK and T cells with sIL1β-mRNA_OMe_AE did not affect the levels of many inflammatory cytokines, including IL-2, IL-6 and TNFα (Fig. 6b and Supplementary Fig. 19d). Furthermore, we examined signal transduction by western blot analyses and found that p38 phosphorylation occurred after sIL1β-mRNA stimulation (Supplementary Fig. 19e), which is related to cell migration24.
Supplementary Table 1 shows the genes whose expression was upregulated compared to that in the unstimulated group as a control. We focused on two genes, granzyme B (GZMB) and Fc epsilon receptor Ig (FCER1G), as they are abundant in immune cells37,38. To confirm the NGS results, we quantified the expression levels of GZMB and FCER1G in CD56+CD3- NK cells and CD8+ T cells using quantitative polymerase chain reaction (qPCR) (Fig. 6c and Supplementary Fig. 20a). According to reports, GZMB is a central protease derived from cytotoxic lymphocytes, such as NK cells and CTLs, that is responsible for killing target cells39. The production of GZMB from cytotoxic lymphocytes was increased by sIL1β-mRNA_OMe_AE (Fig. 6d, e). In addition, sIL1β-mRNA_OMe_AE stimulation moderately upregulated TNFSF10 (TRAIL)40 in NK cells (Fig. 6b). To clarify the function of the upregulated genes in tumoricidal activity, we knocked down GZMB and TRAIL in NK cells and CTLs using small interfering (si) RNAs41 (Fig. 6f and Supplementary Fig. 20b–g). The killing ability associated with s-mRNA was prevented in CD56+CD3- NK cells and CD8+ T cells transfected with GZMB siRNA (Fig. 6g–j), and the knockdown with TRAIL siRNA did not change this ability (Supplementary Fig. 21a, b). For verification, we performed this experiment using PBMCs from different donors and obtained the same results (Supplementary Fig. 22a–c and 23a–c). These results suggest that GZMB is a central molecule involved in s-mRNA-mediated tumoricidal activity.
sIL1β-mRNA promoted tumoricidal activity in NK cells and CTLs from cancer patients
The composition and status of PBMCs differ between cancer patients and healthy donors. Specifically, GZMB is reportedly decreased in the CTLs of cancer patients compared to those of healthy donors42,43. We investigated whether sIL1β-mRNA_OMe_AE could activate CTLs derived from colon cancer patients. CD56+CD3- NK and CD8+ T cells were prepared from five patients who were diagnosed and treated based on their cancer stage (Fig. 7a, b). The results indicated that compared with the control sPoly A-mRNA_OMe_AE, sIL1β-mRNA_OMe_AE induced cytotoxicity in CD56+CD3- NK and CD8+ T cells against the colon cancer cell lines DLD-1 and Caco-2 (Fig. 7c–f). Notably, s-mRNA maintained its killing ability, whereas IL-12 failed to do so, especially during anticancer drug treatment or in the case of multiple cancers (Fig. 7g). In addition, ~ 70% of cancer cells were eliminated by sIL1β-mRNA_OMe_AE-primed NK cells and CTLs (Fig. 7c, d). Furthermore, the groups of DLD-1 cells treated with s-mRNA, in addition to the anti-PD1 antibody exhibited enhanced killing ability compared to the group treated with the anti-PD1 antibody alone (Supplementary Fig. 24a–c).

a Schematic of blood collection from colorectal cancer patients. b Summary of the characteristics of patients with colorectal cancer. c–g Quantitative analysis of colon cancer cells (Caco-2 and DLD-1) killed by sIL1β-mRNA_OMe_AE-activated CD56dimCD3- NK cells and/or CD8+ T cells from patient-1 (c), patient−2 (d), patient-3 (e), patient-4 (f), and patient-5 (g). N = 3 independent experimental replicates. One-way ANOVA with Bonferroni correction was used for multiple comparisons. Mean ± SEM. Source data are provided as a Source Data file.
In summary, the modified short mRNA is imported into the nucleus of NK cells and CTLs via the RNA binding protein receptor ZC3H12D, which induces a common tumor-killing molecule, GZMB. Its booster-like effects allow for multiple doses without fatigue. The treatment can be administered systemically and exhibits antitumor effects without severe side effects, such as the substantial production of inflammatory cytokines (Fig. 8).

Since the chemically synthesized short mRNA (IL1β-mRNA_OMe_AE) is RNase resistant, it can be used to stimulate NK and CTL via the ZC3H12D receptor in vivo. These immune cells enhance its Granzyme B-dependent tumoricidal activity without inducing inflammatory responses that cause severe side effects.
Discussion
sIL1β-mRNA drives tumor killing
Recent progress has been made in the development of immunotherapies, including ACT and immune checkpoint blockade (ICB)44. The combination of the innate and adaptive immune systems accounts for both the nonspecific and specific immune responses in the attack against tumor cells45. Traditionally, cytotoxic lymphocytes such as NK cells and CTLs play central roles in innate and adaptive anticancer immunity by recognizing the loss or gain of tumor-derived peptide antigens on major histocompatibility complex class I (MHC-I) molecules on tumor cells, respectively13. In addition, CD4+ T cells, monocytes, and macrophages collaborate to participate in tumoricidal activity46,47,48. Various inducers that can mobilize both systems have been discovered. The IL-12 protein is known for its ability to strongly activate NK cells and CTLs. Clinical trials of IL-12 administration with improved drug delivery to mitigate severe toxic side effects are underway29,33. Clinical trials of CpG oligos, which indirectly promote the killing of tumor cells by NK cells and CTLs, are also ongoing49,50.
Our previous study suggested that mRNA is a potential activator for immune cells in vitro17. The difficulty in delivering mRNA into the body is that it is degraded by RNase in its naked state. Therefore, we introduced chemical modifications into a short-length mRNA that can be synthesized in an oligonucleotide synthesizer to increase RNase resistance while maintaining its biological activity to suppress tumor metastasis in vivo. This study indicates that sIL1β-mRNA is a potent inducer of NK cells and CTLs. The expression of GZMB was upregulated in cells that received s-mRNA, and this upregulation enhances their cytotoxic activity. Notably, s-mRNA stimulation does not cause serious side effects, such as a cytokine storm. Unlike many immunostimulants, including IL-12, s-mRNA does not upregulate IFNα/β and only moderately induces IFNγ, a key activator of cytotoxicity. The levels of the IRF-1 transcription factor51, which is responsible for IFN gene expression, did not increase upon sIL1β-mRNA stimulation (GEO or Supplementary Table 1). Importantly, chronic inflammatory stimulation can change the characteristics of immune cells from antitumor to protumor in the tumor microenvironment. Therefore, the modulation of immune cells to prevent excessive inflammation is necessary for effective treatment. The advantage of s-mRNA treatment is that it can be administered in multiple doses without causing unwanted inflammatory side effects. Antimetastasis therapy requires long-term treatment, making periodic injections of therapeutic reagents inevitable. Thus, we conclude that s-mRNA is suitable for sustainable treatment to suppress metastasis.
Again, the mechanism clarified in this paper is that extracellular chemically modified short mRNA regulates signal transduction through p38 phosphorylation and induction of GZMB-dependent tumoricidal activity, without significant induction of inflammatory cytokines. Correlations between the fact that the s-mRNA complex is transported into the nucleus and that s-mRNA induces antimetastatic effects without serious side effects need to be addressed in the future.
Sequence-dependent tumoricidal activity via ZC3H12D
Our previous studies unveiled three important facts related to the sequence-dependent IL1β-mRNA antimetastatic activity. First, we found that the biological function of IL1β-mRNA as a signaling molecule is translation-independent because the introduction of stop codon mutations in the coding sequence of IL1β-mRNA did not spoil its activity17. This is the first observation in the field of mammalian biology. Second, the IL1β-mRNA signal is received by the ZC3H12D protein, which recognizes AU-rich elements in the 3’UTR region of IL1β-mRNA. This is why β-actin and Poly A-mRNA were unable to stimulate antimetastatic immune cells. Third, antimetastatic immune cells expressing ZC3H12D were first found as a population that migrated from the liver to the lung10, and the population showed strong tumor-killing activity. Although the whole mechanism of this biology is still unknown, in this paper, we revealed part of the mechanism responsible for the tumoricidal activity.
sIL1β-mRNA is an antimetastatic agent
Many targets associated with metastasis could be candidates for antimetastatic drugs2. Host tissues have been shown to dictate the extent to which a tumor can metastasize. The tumor microenvironment is complex, varies extensively among different organs, and is influenced by tumor-host cell interactions, physical and metabolic changes, and secreted cytokines, chemokines, and growth factors4,5,6. Many preclinical tumor models are more suitable for testing agents with a direct antitumor effect on primary tumors rather than clinically relevant effects on metastasis1. In addition, making it is difficult to determine its effect in previous clinical studies52. Therefore, early-stage molecules related to premetastatic and early metastatic stages may be stronger candidates. As demonstrated in this study, s-mRNA was found to be a stimulator of NK cells and CTLs in metastasis. Metastatic cancers are a complex and heterogeneous group of cancers that evade the immune response. The MHC molecules on the surface of cancer cells are also heterogeneous53. Furthermore, CTLs derived from colon cancer patients who received chemotherapy or had multiple types of cancer showed exhaustion without exerting a tumoricidal effect (Fig. 7e–g). These data suggest that protecting against escaping cancer cells is challenging for the immune system. Moreover, s-mRNA, but not IL-12, elicited cytotoxic effects in these cells (Fig. 7e–g). The s-mRNA-primed cytotoxic lymphocytes may prevent metastasis.
In this study, we evaluated lung metastasis, but other metastasis including liver metastasis, could not be evaluated due to the limitations of the metastasis model. As for macroscopic metastasis to other organs, such as the liver, we need further elaboration of stable assessment for this issue. In the future, we expect to discover a tumor-associated liver factor that attracts ZC3H12D-positive immune cells in the liver or a liver-specific RNA ligand. These factors are likely to complement the s-mRNA based antimetastatic therapy.
Thus, we conclude that s-mRNA is suitable for sustainable treatment to suppress metastasis. In terms of clinical application, we need to continue to carefully investigate factors related to side effects such as cytokine storm in vivo.
Methods
Ethical approval
This research complies with all relevant ethical regulations. Blood samples were purchased from Lonza (CC-2702) or collected from healthy donors as part of an ethically approved study (approved by the Ethics Committee of the Shinshu University School of Medicine, Approval No.4816). A random sample from multiple donors was used for this research. In addition, blood samples were collected from five colorectal cancer patients during a standard therapeutic procedure (approved by the Ethics Committee of the Shinshu University School of Medicine, Approval No.5167). The mean age of the patients was 68 years (ranging from 56 to 79 years). Written informed consent was obtained from all patients for the analysis of this study. The investigation was conducted in accordance with the principles of the Declaration of Helsinki. All animal procedures were performed according to the guidelines of the Animal Research Committee of Shinshu University, after the approval of a protocol by the Committee (permission numbers: 021029-1, 024023, 024032). In this study, the tumor size did not exceed 13 mm, which is the maximum tumor size permitted by the Committee.
Materials
Human cell culture: Anti-human IL-2 (BioLegend, 589102, 200 U/mL); Anti-human IL-12 (Proteintech, HZ-1256, 20 ng/mL); Anti-human GM-CSF (BioLegend, 572902, 1000 U/mL); Anti-human IL-4 (BioLegend, 574002, 1000 U/mL); Anti-human CD279 (PD-1) (BioLegend, 329902, 2.5 μg/mL).
Mouse in vivo: Anti-asialo GM1 (Wako, 014-09801, 50 μL/animal); Anti-mouse CD4 (BioLegend, 100457, 250 μg/animal); Anti-mouse CD8a (BioLegend, 100763, 250 μg/animal).
Human cell sorting: PE anti-human CD56 (NCAM) (BioLegend, 362524, dilution 1:50–100), and isotype control, mouse IgG1, κ (BioLegend, 400112, dilution 1:50–100); Alexa Fluor 488 anti-human CD3 (BioLegend, 300454, dilution 1:100), and isotype control, mouse IgG1, κ (BioLegend, 400132, dilution 1:100); Alexa Fluor 647 anti-human CD4 (BioLegend, 300523, dilution 1:100), and isotype control, mouse IgG1, κ (BioLegend, 400130, dilution 1:100); PE anti-human CD8 (BioLegend, 344705, dilution 1:100), and isotype control, mouse IgG1, κ (BioLegend, 400112, dilution 1:100); Anti-ZC3H12D (Thermo, PA5-110590, dilution 1:200), and isotype control, rabit (DA1E) mAb IgG XP (Cell Signaling Technology, #3900, dilution 1:200). Human Fc receptor blocking solution (BioLegend, 422302, dilution 1:40) was used to block nonspecific antibody bindings.
Mouse cell sorting: Brilliant Violet 421 anti-mouse NK-1.1 (BioLegend, 108741, dilution 1:100), and isotype control, Brilliant Violet 421 mouse IgG2a, κ (BioLegend, 400260, dilution 1:100); PE anti-mouse CD4 (BioLegend, 100511, dilution 1:100), and isotype control, PE rat IgG2a, κ (BioLegend, 400507, dilution 1:100); FITC anti-mouse CD8a (BioLegend, 100706, dilution 1:100), and isotype control, FITC rat IgG2a, κ (BioLegend, 400505, dilution 1:100); APC-Cy7 anti-mouse TCRβ chain (BD Bioscience, 560656, dilution 1:100), and isotype control, APC-Cy7 hamster IgG2a, λ1 (BD Bioscience, 560604, dilution 1:100). Anti-mouse CD16/CD32 monoclonal antibody (BioLegend, 156604, dilution 1:20) was used to block nonspecific antibody bindings.
Cell staining: Anti-ZC3H12D (Abcam, ab1000862, dilution 1:100–200), and isotype control, rabit (DA1E) mAb IgG XP (Cell Signaling Technology, #3900, dilution 1:200); Anti-IFNγ (BioLegend, 506501, dilution 1:200), and isotype control, mouse IgG1, κ (MBL, M075-3, dilution 1:200); Anti-GZMB (CST, 17215, dilution 1:400); Zombie Green Fixable Viability Kit (BioLegend, 423111, dilution 1:600); PKH26 red fluorescent cell linker mini kit (Sigma-Aldrich, MINI26-1KT, dilution 1:200); DAPI (BioLegend, 422801, dilution 1:1200) or ProLong™ Diamond Antifade Mountant with DAPI (Thermo, P36962); Anti-Ki67 (Sigma-Aldrich, SAB5700770, dilution 1:50), and isotype control, normal rabbit IgG (Wako, 148-09551, dilution 1:200); Anti-mouse CD45 (BioLegend, 103101, dilution 1:200), and isotype control, rat IgG2b, κ (BioLegend, 400601, dilution 1:200).
Western blot: Anti-TLR7 (Cell Signaling Technology, #82658, dilution 1:1000); Anti-GAPDH (MBL, #M171-3, dilution 1:1000); Anti-Akt (Cell Signaling Technology, #9272, dilution 1:1000); Anti-phospho-Akt (Cell Signaling Technology, #4060, dilution 1:1000); Anti-p38 MAPK (Cell Signaling Technology, #2371, dilution 1:1000); Anti-phospho-p38 MAPK (Cell Signaling Technology, #4631, dilution 1:1000); Anti-IκBα (Cell Signaling Technology, #9242, dilution 1:1000); Anti-phospho-IκBα (Cell Signaling Technology, #9241, dilution 1:1000); Anti-ERK1/2 (Proteintech, #51068-1-AP, dilution 1:1000); Anti-phospho-ERK1/2 (Proteintech, #28733-1-AP, dilution 1:1000).
Preparation of mRNA
Enzymatically synthetic RNAs were generated using RiboMAX large-scale RNA production systems (T7 RNA polymerase). The templates used were genes cloned in pCR-Blunt II and linearized by SpeI or BamHI restriction endonuclease. The resulting synthetic RNAs were treated with DNase I and purified using the RNA Basic kit spin column kit (Nippon Genetics, Tokyo, Japan). We used the Agilent 2100 Bioanalyzer (Agilent Technologies Japan, Ltd., Tokyo, Japan) to analyze synthesized RNAs.
Solid-phase oligonucleotide synthesis
The DNA/RNA synthesis was performed using a phosphoramidite method on a synthesizer. After synthesis, the controlled-pore glass (CPG) beads were treated with 10% dimethylamine in CH3CN for 5 min, followed by a rinse with CH3CN to selectively remove cyanoethyl groups. The oligomers were then cleaved from the CPG beads and deprotected by treatment with a concentrated solution of NH3/40% methylamine (1:1, v/v) for 10 min at 65 °C. The 2’-O-t-butyldimethylsilyl groups were eliminated using Et3N·3HF (125 µL) + DMSO (100 µL) at 65 °C for 1.5 hr. The reaction was quenched with 0.1 M Triethylammonium acetate buffer (pH 7.0), and the mixture was desalted using a Sep-Pak C18 cartridge. The oligonucleotides were purified using 15% polyacrylamide gel electrophoresis (500 V, 20 mA).
RNA-seq
Human PBMCs were purchased from Roche. Thawed PBMCs were maintained in an HPLM (Thermo) medium supplemented with 10% FCS. The next day, FITC-labeled IL1β-mRNA (10 ng/mL) was added to the media, and 0.5 h or 6 h later, cells were FACS-sorted to isolate NK cells (CD56+CD3-) and CD3+ T cells. The isolated cells were immediately added to Sepasol®-RNA I Super G (Nacalai Tesque, Inc., Kyoto, Japan, 09379-97) reagent to obtain total RNA. Library preparation for RNA-Seq was performed using the TAKARA SMART-Seq® HT PLUS Kit (Takara Bio Inc., Shiga, Japan, R400748). A portion (0.4–1.4 ng) of total RNA was reverse transcribed and amplified by 12 cycles of PCR. Then, 3 ng of each complementary DNA (cDNA) was subjected to fragmentation, Illumina index attachment at the ends, and PCR amplification (15 cycles). Finally, the combined library was sequenced using HiSeq X Ten and Illumina Hiseq 2 × 150 bp sequencing configuration, per lane (~ 350 M reads) (Azenta Japan Corp., Tokyo, Japan). Reads were trimmed and clipped in Trimmomatic v0.39 and imported into STAR v2.7.6a for alignment to BAM files using the human genome sequence (hg38, GRCh38). BAM files were imported into FeatureCounts (in Subread v2.0.6), and PRKM counts were calculated. Based on the comparisons between (NK control = 0 hr) and (NK 0.5 hr or 6 hr) or between (T control = 0 hr) and (T 0.5 hr or 6 hr), genes upregulated (> 2-fold) in the RNA-stimulated cells were selected and the gene list was imported into Metascape, a gene annotation and analysis resource54 (https://metascape.org/gp/index.html#/main/step1). Heatmap was generated using Next-Generation Clustered Heat Map (NG-CHM) Viewer (https://bioinformatics.mdanderson.org/public-software/ngchm/)55.
Electrophoretic mobility shift assay (EMSA)
Human ZC3H12D protein, purified from HEK293T cells transfected with hZC36 expression vector17, was mixed with FITC-labeled mouse IL1β-mRNA 50-mer or mouse IL1β-mRNA_OMe_AE 50-mer in a reaction buffer (10% Glycerol, 0.1 mg/mL tRNA, 50 mM phosphate, 10 mM Hepes, 30 mM NaCl, 40 mM arginine, 20 mM KCl, 1 mM MgCl2, pH 7.4) and run in a 5–20% gradient polyacrylamide gel (WAKO). Fluorescent signals from FITC-labeled mRNA were detected by a ChemiDoc Touch Gel Imaging System (Bio-Rad Laboratories, Hercules, California, USA), and data are analyzed by Image Lab 6.1 software (Bio-Rad).
To demonstrate sequence-specific interaction between ZC protein and IL1β-mRNA, non-labeled IL1β-mRNA 50-mer or unrelated RNA 50-mer was added to the samples.
Nuclease resistance of mRNA
Fluorescein-labeled RNAs (300 pmol) were dissolved in 37.5 μL of Opti-MEM. Then, 60 μL of bovine serum was added, and the solution was incubated at 37 °C for the specific time intervals (0 and 5 min and 1, 3, 6, 12, 24 hr, and 48 hr). Aliquots of 2 μL were diluted with a stop solution (10 mM EDTA, 90% formamide, 10 μL). The samples underwent electrophoresis in 20% denaturing PAGE*1 (500 V, 20 mA) and were analyzed using a Luminescent Image analyzer LAS-4000 (Fujifilm) and Image Lab 6.1 software (Bio-Rad).
FITC-labeled RNA purification from mouse lung
Mouse lungs were homogenized in 4 mL extraction buffer (50 mM sodium phosphate buffer, pH 7.6, 300 mM NaCl, 1% Triton X-100, 100 µg/mL Proteinase K (Wako)). The homogenate was centrifuged, and the supernatant was passed through a microspin column (Cytiva). Ethanol was then added to the solution to precipitate the RNA. The precipitate was dissolved in an aqueous buffer containing MgCl2 and treated with DNase I (Sigma) for 1 hr at room temperature (RT), followed by treatment with Proteinase K (1 hr at RT). After these reactions, the RNA was retrieved by ethanol precipitation. The precipitate was further purified using NucleoSpin miRNA (Takara), and the purified small RNA fraction was analyzed on a 16% urea-polyacrylamide gel.
Animals
In the animal study, 6- to 10-week-old female or male C57BL/6 mice and NOD.Cg-PrkdcscidIl2rgtm1Sug/ShiJic (NOG) mice were used. Mice were purchased from SLC (Shizuoka, Japan) and Clea Japan (Tokyo, Japan), respectively. The Zc3h12d KO mice26 were also used in the experiments. The mice were maintained in a specific pathogen-free condition in a controlled environment with a constant temperature (22 °C) and humidity (50%) under a 12 hr light/dark cycle.
Tumor cell lines and TCM
Mouse E0771 breast cancer cells (American Type Culture Collection (ATCC), CRL-3461) were originally established by Dr Sirotnak (Memorial Sloan-Kettering Cancer Center, New York, NY, USA) and provided by Dr Mihich (Roswell Park Memorial Institute, Buffalo, NY, USA). MC-38 murine colon carcinoma cells (T8291) were purchased from Applied Biological Materials Inc. (Richmond, BC, Canada). RAW264.7 (RCB0535) was purchased from RIKEN Cell Bank (Tokyo, Japan). E0771 cells and RAW264.7 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin G sodium, and 100 μg/mL streptomycin sulfate. MC-38 cells were maintained in DMEM supplemented with 10%FBS, 2 mM glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 10 mM Hepes, and 50 μg/mL gentamicin sulfate.
Human breast cancer cells MDA-MB-231 (HTB-26) and human kidney carcinoma cells 786-O (CRL-1932) were purchased from the ATCC. Human colorectal cancer cells Caco-2 (CVCL_0025), DLD-1 (CVCL_0248), and human monocytic cells THP-1 (RCB1189) were purchased from RIKEN Cell Bank (Tokyo, Japan) and Cell Resource Center for Biomedical Research Institute of Development, Aging and Cancer, Tohoku University, respectively. 786-O cells were cultured in DMEM/Ham’s F-12 medium supplemented with 10%FBS, 100 units/mL penicillin G sodium, and 100 μg/mL streptomycin sulfate. MDA-MB-231 cells, DLD-1 cells, and THP-1 cells were maintained in RPMI1640 supplemented with 10%FBS, 100 units/mL penicillin G sodium, and 100 μg/mL streptomycin sulfate. Caco-2 cells were cultured in DMEM supplemented with 10%FBS, 100 units/mL penicillin G sodium, and 100 μg/mL streptomycin sulfate. The cell lines were tested negative for mycoplasma contamination by using MycoAlert Mycoplasma Detection Kit (Lonza, LT-07-218). Cells were not authenticated. TCM was obtained by incubating cells overnight in a serum-free medium without cell death. The collected TCMs were centrifuged at 1000 × g for 5 min, and the supernatant was further centrifuged at 20,000 × g for 20 min. The TCMs were then stored at − 80 °C before experiments.
Tumor metastasis models
E0771 cells or MC-38 cells were implanted in 6- to 10-week-old female or male C57BL/6 mice via mammary fat pads implantation or subcutaneous implantation, respectively. A total of 1 × 106 tumor cells were implanted to generate tumor-bearing mice. In some cases, TCM obtained from E0771 cells was injected intraperitoneally to mimic tumor-bearing mice. For the tumor cell homing assays, 1 × 106 PKH26-labeled cells were suspended in 100 μL of PBS and intravenously injected. Lungs were excised 48 hr after the injection. In the case of long-term observation assay, 1 × 105 or 5 × 104 E0771 cells were injected. Random lung tissue fragments (2 mm in diameter) were embedded in an Optimal Cutting Temperature compound (Sakura Finetek Japan, Tokyo, Japan) and frozen at − 80 °C. Sections (10 μm thickness) were obtained from the frozen samples using a CM3050S or CM1950 cryostat (Leica Biosystems, Nussloch, Germany). The sections were then examined under confocal microscopy (Leica TCS SP8, Heidelberg, Germany). The labeled tumor cell counts were normalized based on the total tissue surface area.
Xenograft model
We used NOG mice as a xenograft model. To simulate a tumor-bearing environment, we administered TCM derived from MDA-MB-231 cells intraperitoneally. We injected 1 × 105 human CD56+CD3- NK cells or CD8+ T cells from a healthy donor intravenously. Then, 1 × 105 PKH26-labeled MDA-MB-231 cells were injected intravenously. After 48 h, the lungs were excised for analysis.
Orthotopic and survival models
E0771 cells were implanted into female C57BL/6 mice via the mammary fat pad. A total of 1 × 106 tumor cells were used to generate tumor-bearing mice. Fourteen days after injection, the tumor was excised to create lung metastasis models. For the orthotopic model, the lungs were excised 21 days after tumor removal, and the number of macroscopic lung metastases was counted. In the survival model, mice were monitored until death or weight loss of up to 20%.
Flow cytometry and data analysis
To collect NK cells and T cells from human peripheral blood mononuclear cells (PBMCs) and splenocytes from mice, cells were purified using a cell sorter (FACSAria™ IIu or FACSAria™ III (BD Bioscience)). FACSCelesta (BD Bioscience) was also used for flow cytometry, and data analyses were conducted with FACSDiva v8.0.2 (BD Bioscience) and FlowJo v10.6.2 (FlowJo LLC).
RNA uptake assay for flow cytometry
Prior to mRNA application, primary cells were pre-incubated in serum-free culture medium for 3 hr and supplemented with 1% TCM from MDA-MB-231 cells or E0771 cells for 30 min. Subsequently, 10 ng/mL of mRNAs was added to the medium for 1 h. To halt mRNA uptake, 4% paraformaldehyde was added for 5 min (2% final concentration) and washed three times. After the staining of CD56+, CD3+, CD4+, CD8+, NK1.1+, or TCRβ+ cells were isolated using BD FACSCelesta (BD Bioscience). Data analyses were performed using FACSDiva v8.0.2 (BD Bioscience) and FlowJo v10.6.2 (FlowJo LLC).
RNA uptake assay for immunohistochemistry
The cells fixed with 4% paraformaldehyde in the RNA uptake assay were treated with 0.1% Triton X-100 and stained with anti-ZC3H12D and a fluorescent-labeled secondary antibody. Confocal z-stack images were obtained using Leica TCS SP8 confocal microscopy. The top and bottom of the nucleus were oriented by the 4′,6-diamidino-2-phenylindole (DAPI) signal, and a series of 10 images from the top to the bottom of the nucleus were captured and constructed into 3D images for a cell. For precise comparisons among samples, image data were acquired within 2 days. The image data were analyzed using Leica Application Suite X (LAS X v3, Leica). All cells were checked for RNA uptake in the nucleus using a single image of the central portion of the nucleus.
Migration assay
Cell migration was assessed with a chemotaxis Boyden chamber (AP48, Neuro Probe) using a 5 μm pore-size polyvinylpyrrolidone-free polycarbonate filter (Neuro Probe). Various mRNAs were added to the bottom wells. A 50 μL aliquot of the 786-O cell suspension (1 × 106 cells/mL) was seeded into each of the top wells and incubated for 3 hr. Following incubation, the filter membrane was stained with Diff Quick (Sysmex, 16920). Cells on the upper surface of the filter were removed by wiping, and the migrated cells on the lower surface were counted.
IFNγ induction and tumoricidal assay for NK cells
Human CD56+CD3- NK cells were primed with 10 ng/mL of mRNAs or 20 ng/mL of human IL-12 as a positive control in a Human Plasma-Like Medium (HPLM) (Gibco) supplemented with 200 IU/mL of IL-2. After priming, NK cells were stained with an anti-human IFNγ antibody 24 hr later. For the tumoricidal assay, mRNA was removed by washing to eliminate any impact on the tumor cells. The dead PKH26-stained tumor cells were detected 18–24 hr after incubation with human CD56dimCD3- NK cells that were primed with 10 ng/mL mRNA for 24 hr, using Zombie Green Fixable Viability Kit (BioLegend).
IFNγ induction and tumoricidal assay for T cells
Human CD4+ T cells and CD8+ T cells were primed with 10 ng/mL of mRNAs or 20 ng/mL of human IL-12. After 24 hr, the mRNA was removed by washing. The T cells were then co-cultured with dendritic cells (DCs) primed with cancer cell lysate, as described below, for 24 hr. After this, the DCs were removed, and cells were stained with an anti-human IFNγ antibody. For the tumoricidal assay, the DC-stimulated T cells were co-cultured with tumor cells for 18–24 hr. Dead tumor cells were detected using the Zombie Green Fixable Viability Kit.
Generation of monocyte-derived dendritic cells
Monocytes were isolated from PBMCs by adhering them to culture dishes for 2 hr of incubation at 37 °C and 5%CO2. Non-adherent cells were removed, and adherent PBMCs were cultured in RPMI1640, containing 10%FBS, 1000 U/mL of GM-CSF, and 1000 U/mL of IL-4. Half of the medium containing GM-CSF and IL-4 was exchanged every 3 days. After 6 days of culture, the cells were incubated with freeze-thawed tumor cell lysate (10%) for 24 hr. The non-adherent and loosely adherent cells of tumor cell lysate-primed DCs were harvested and washed twice. Then the DC cells were co-incubated with the CD8+ T cells at a ratio of 1:20 in HPLM, containing 10% FBS and 200 U/mL IL-2 for assay of cytotoxic T lymphocyte activity.
Preparation of tumor cell lysate
To prepare the lysate, tumor cells (1 × 106 cells) were rapidly frozen at − 80 °C for 5 min and then thawed at 37 °C six times. The lysate was then centrifuged (10,000 × g, 5 min) to remove crude debris. The resulting supernatants was filtered through on a 0.2 μm filter (Minisart, Sartorius) and stored in aliquots at − 80 °C until use.
Side effect examination
Mice were anesthetized, and blood was collected through cardiac puncture. Subsequently, the lung, liver, spleen, and kidney were extracted. The serum was separated by centrifugation (1500 × g for 10 min) twice immediately after sample collection. The following values were measured using FUJI DRI-CHEM NX700 according to the manufacturer’s instructions: aspartate aminotransferase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN), and creatinine (CRE). Inflammatory cytokines were measured by using a bead-based immunoassay with the LEGENDplex™ Mouse Inflammation Panel (13-plex) (BioLegend) following the manufacturer’s instructions and analyzed on a flow cytometer (FACSCelesta). The data were analyzed with the LEGENDplex Software (Version 8.0). To ensure accurate comparisons, mean fluorescence intensity was evaluated as the limits of detection for cytokines were higher than the actual values. The extracted organs were fixed in 10% formaldehyde and embedded in paraffin. Subsequently, 4 μm thick serial sections were cut from these blocks and stained with haematoxylin and eosin.
ELISA for IFNα
Mouse splenocytes were incubated with or without 10 ng/mL of mRNA or 10 μg/mL of Poly (I:C). After 24 hr, the supernatants were collected, and IFNα secreted into the medium was measured using the IFN-α ELISA kit (ELISA MAX™ Deluxe Set Mouse IFN-α1, 447904, BioLegend). A 96-well ELISA plate was coated with diluted capture antibody (Mouse IFN-α1 ELISA MAX™ Capture Antibody, dilution 1:200) incubated overnight at 4 °C. After washing four times with PBS containing 0.05% Tween 20 (PBS-T), Assay Diluent A was added to the wells to block nonspecific binding and reduce background. The plate was incubated for 1 hr at RT and washed four times. Then, standards or samples were added to each well and further incubation was carried out for 2 hr at RT. The plate was washed four times. A diluted detection antibody (Mouse IFN-α1 ELISA MAX™ Detection Antibody, dilution 1:200) was added to each well and incubated for 1 hr at RT. The plate was washed four times, and diluted Avidin-HRP solution (1:1000) was added to each well and incubated for 30 min at RT. After washing five times, TMB substrate solution was added to each well and incubated for 10 min at RT in the dark, and the reaction was stopped by using hydrochloric acid (3 M). Absorbance was recorded at 450 and 570 nm on a plate reader (SpectraMax iD5, Molecular Devices) with SoftMaxPro 7.1 (Molecular Devices).
Quantitative PCR
To generate cDNA from total RNA was performed with SMART-Seq® HT PLUS Kit (Takara Bio Inc) as RNA Seq. Quantitative polymerase chain reaction (PCR) analysis was performed using PowerTrack™ SYBR Green Master Mix for qPCR (Applied Biosystems, Foster City, CA, A46109), THUNDERBIRD® SYBR™ qPCR Mix (Toyobo Co., Ltd., Osaka, Japan, QPS-201) or TaqMan™ Gene Expression Master Mix (Applied Biosystems, 4369016) in a detection system (StepOnePlus, Applied Biosystems). The gene expression levels were calculated from Ct values, and normalized to that of 18S of ribosomal RNA in each sample. The analysis of FcεR1G, GZMB, IL-2, IL-6, TNFα, and 18S, were done with the primer sets and TaqMan Assay described below.
FcεR1G 5’-ACCAGGAACCAGGAGACTTA-3’
5’- GGAAGAACCAGAAGCCAAAGA-3’
GZMB 5’-CGACAGTACCATTGAGTTGTGC-3’
5’-GCCACCTTGTTACACACAAGAG-3’
TNFα 5’-CCAGGGACCTCTCTCTAATCA-3’
5’-TCAGCTTGAGGGTTTGCTAC-3’
IL-2 5’-CTCACCAGGATGCTCACATTTA-3’
5’-CCTCCAGAGGTTTGAGTTCTTC-3’
IL-6 5’-GGAGACTTGCCTGGTGAAA-3’
5’-CTGGCTTGTTCCTCACTACTC-3’
18S Hs03928990_g1 (Applied Biosystems, 4331182)
Transfection for SiRNA
To knockdown TLR7 for the migration assay, RAW264.7 cells were treated with siRNA [ON-TARGET plus Mouse Tlr7 (170743) siRNA (SMARTpool)] using Lipofectamine™ RNAiMAX transfection reagent (Thermo Fisher Scientific, USA). ON-TARGET plus Nontargeting pool (Dharmacon Horizon Discovery) was used as control siRNA.
To knockdown GZMB and TRAIL, human CD56+CD3- NK cells or CD8+ T cells were treated with 0.5 µM siRNA [ON-TARGET plus GZMB (3002) siRNA and TRAIL (8743) siRNA (SMARTpool)] using electroporation with an Amaxa Nucleofector system (Human T Cell Nucleofector Kit, Lonza, Basel, Switzerland, VPA-1002) by using program U-014. The control siRNA used was ON-TARGET plus Nontargeting pool. The efficiency of knockdown at the protein level was examined with IHC staining. An RNA uptake assay was carried out 48 hr after the application of 0.5 µM siRNA.
Checkpoint blockade experiments
To analyze the impact of the PD-1 signaling pathway on T cell anti-tumor response and its synergistic effect with s-mRNA, we used blocking monoclonal antibody (anti-human CD279 (PD-1), BioLegend, 329902). The anti-PD1 antibody was added to the target wells at a concentration of 2.5 μg/mL during co-culture with tumor cells. The efficacy of the blockade was assessed using the zombie assay as well as described above.
Statistics & Reproducibility
The data are presented as mean ± standard error of the mean. Statistical analysis was conducted as specified. P < 0.05 was considered statistically significant. All Student’s t tests were unpaired and two-sided. Analysis of variance was always performed with Bonferroni’s correction. P-values were calculated using Prism v8 software (GraphPad Software, San Diego, CA, USA). The experiments were repeated at least twice to obtain representative images. No statistical method was used to predetermine the sample size. No data were excluded from the analyses. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The next-generation sequencing data [Fig. 6b and Supplementary Fig. 18b–d and 19a–c, and Supplementary Table 1] have been deposited in the NCBI Gene Expression Omnibus as GSE262314. All other data supporting the findings in this work are included in the main article, supplementary information, or source data file. Source data are provided in this paper.
References
Jianhui, Z. et al. Global trends in incidence, death, burden and risk factors of early-onset cancer from 1990 to 2019. BMJ Oncol. 2, e000049 (2023).
Anderson, R. L. et al. A framework for the development of effective anti-metastatic agents. Nat. Rev. Clin. Oncol. 16, 185–204 (2019).
Gonzalez, H., Hagerling, C. & Werb, Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 32, 1267–1284 (2018).
Fares, J., Fares, M. Y., Khachfe, H. H., Salhab, H. A. & Fares, Y. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct. Target Ther. 5, 28 (2020).
Lambert, A. W., Pattabiraman, D. R. & Weinberg, R. A. Emerging biological principles of metastasis. Cell 168, 670–691 (2017).
McAllister, S. S. & Weinberg, R. A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 16, 717–727 (2014).
Peinado, H. et al. Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer 17, 302–317 (2017).
Hiratsuka, S. et al. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell 2, 289–300 (2002).
Han, Y. et al. Citrullinated fibrinogen-SAAs complex causes vascular metastagenesis. Nat. Commun. 14, 4960 (2023).
Hiratsuka, S. et al. Hepato-entrained B220(+)CD11c(+)NK1.1(+) cells regulate pre-metastatic niche formation in the lung. EMBO Mol. Med. 10, https://doi.org/10.15252/emmm.201708643 (2018).
Granot, Z. et al. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 20, 300–314 (2011).
Correia, A. L. Locally sourced: site-specific immune barriers to metastasis. Nat. Rev. Immunol. 23, 522–538 (2023).
Algarra, I., Garrido, F. & Garcia-Lora, A. M. MHC heterogeneity and response of metastases to immunotherapy. Cancer Metastasis Rev. 40, 501–517 (2021).
Wicks, E. E. & Semenza, G. L. Hypoxia-inducible factors: cancer progression and clinical translation. J. Clin. Invest. 132, https://doi.org/10.1172/JCI159839 (2022).
Shaver, K. A., Croom-Perez, T. J. & Copik, A. J. Natural killer cells: The Linchpin for successful cancer immunotherapy. Front. Immunol. 12, 679117 (2021).
Sun, L. et al. Activating a collaborative innate-adaptive immune response to control metastasis. Cancer Cell 39, 1361–1374 (2021).
Tomita, T. et al. Extracellular mRNA transported to the nucleus exerts translation-independent function. Nat. Commun. 12, 3655 (2021).
Kaplan, R. N. et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438, 820–827 (2005).
Maeda, K. & Akira, S. Regulation of mRNA stability by CCCH-type zinc-finger proteins in immune cells. Int. Immunol. 29, 149–155 (2017).
Kano, T., Katsuragi, Y., Maeda, Y. & Ueno, Y. Synthesis and properties of 4’-C-aminoalkyl-2’-fluoro-modified RNA oligomers. Bioorg. Med. Chem. 26, 4574–4582 (2018).
Inoue, H. et al. Synthesis and hybridization studies on two complementary nona(2’-O-methyl)ribonucleotides. Nucleic Acids Res. 15, 6131–6148 (1987).
Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 24, 374–387 (2014).
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
Hiratsuka, S., Watanabe, A., Aburatani, H. & Maru, Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 8, 1369–1375 (2006).
Minagawa, K. et al. Inhibition of G(1) to S phase progression by a novel zinc finger protein P58(TFL) at P-bodies. Mol. Cancer Res. 7, 880–889 (2009).
Minagawa, K. et al. Posttranscriptional modulation of cytokine production in T cells for the regulation of excessive inflammation by TFL. J. Immunol. 192, 1512–1524 (2014).
Poli, A. et al. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology 126, 458–465 (2009).
Tugues, S. et al. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 22, 237–246 (2015).
Nguyen, K. G. et al. Localized interleukin-12 for cancer immunotherapy. Front. Immunol. 11, 575597 (2020).
Koh, S. K. et al. Natural killer cell expansion and cytotoxicity differ depending on the culture medium used. Ann. Lab Med. 42, 638–649 (2022).
Tello-Lafoz, M. et al. Cytotoxic lymphocytes target characteristic biophysical vulnerabilities in cancer. Immunity 54, 1037–1054 (2021).
Yu, Y. et al. Optogenetic-controlled immunotherapeutic designer cells for post-surgical cancer immunotherapy. Nat. Commun. 13, 6357 (2022).
Chen, P. et al. An IL-12-Based nanocytokine safely potentiates anticancer immunity through spatiotemporal control of inflammation to eradicate advanced cold tumors. Adv. Sci. 10, e2205139 (2023).
Brunda, M. J. et al. Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J. Exp. Med. 178, 1223–1230 (1993).
Karki, R. et al. Synergism of TNF-alpha and IFN-gamma triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 184, 149–168 (2021).
Hiratsuka, S. et al. C-X-C receptor type 4 promotes metastasis by activating p38 mitogen-activated protein kinase in myeloid differentiation antigen (Gr-1)-positive cells. Proc. Natl. Acad. Sci. USA 108, 302–307 (2011).
Prakash, M. D. et al. Granzyme B promotes cytotoxic lymphocyte transmigration via basement membrane remodeling. Immunity 41, 960–972 (2014).
Morrish, E. & Ruland, J. Cytotoxic FCER1G(+) innate-like T cells: new potential for tumour immunotherapy. Signal Transduct. Target Ther. 7, 204 (2022).
Voskoboinik, I., Whisstock, J. C. & Trapani, J. A. Perforin and granzymes: function, dysfunction and human pathology. Nat. Rev. Immunol. 15, 388–400 (2015).
Cardoso Alves, L., Corazza, N., Micheau, O. & Krebs, P. The multifaceted role of TRAIL signaling in cancer and immunity. FEBS J. 288, 5530–5554 (2021).
Pan, R., Ryan, J., Pan, D., Wucherpfennig, K. W. & Letai, A. Augmenting NK cell-based immunotherapy by targeting mitochondrial apoptosis. Cell 185, 1521–1538 (2022).
Assy, L., Khalil, S. M., Attia, M. & Salem, M. L. IL-12 conditioning of peripheral blood mononuclear cells from breast cancer patients promotes the zoledronate-induced expansion of gammadelta T cells in vitro and enhances their cytotoxic activity and cytokine production. Int. Immunopharmacol. 114, 109402 (2023).
Zhang, Y. et al. Pan-cancer scRNA-seq analysis reveals immunological and diagnostic significance of the peripheral blood mononuclear cells. Hum. Mol. Genet. 33, 342–354 (2024).
Demaria, O. et al. Harnessing innate immunity in cancer therapy. Nature 574, 45–56 (2019).
Gajewski, T. F., Schreiber, H. & Fu, Y. X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 (2013).
Lee, H. G., Cho, M. J. & Choi, J. M. Bystander CD4(+) T cells: crossroads between innate and adaptive immunity. Exp. Mol. Med. 52, 1255–1263 (2020).
Hartl, C. A. et al. Combination therapy targeting both innate and adaptive immunity improves survival in a pre-clinical model of ovarian cancer. J. Immunother. Cancer 7, 199 (2019).
Kruse, B. et al. CD4(+) T cell-induced inflammatory cell death controls immune-evasive tumours. Nature 618, 1033–1040 (2023).
Chen, W. et al. CpG-Based nanovaccines for cancer immunotherapy. Int. J. Nanomed. 16, 5281–5299 (2021).
Kawarada, Y. et al. NK- and CD8(+) T cell-mediated eradication of established tumors by peritumoral injection of CpG-containing oligodeoxynucleotides. J. Immunol. 167, 5247–5253 (2001).
Galon, J., Sudarshan, C., Ito, S., Finbloom, D. & O’Shea, J. J. IL-12 induces IFN regulating factor-1 (IRF-1) gene expression in human NK and T cells. J. Immunol. 162, 7256–7262 (1999).
Steeg, P. S. Targeting metastasis. Nat. Rev. Cancer 16, 201–218 (2016).
Garrido, F. & Aptsiauri, N. Cancer immune escape: MHC expression in primary tumours versus metastases. Immunology 158, 255–266 (2019).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Ryan, M. C. et al. Interactive Clustered Heat Map Builder: An easy web-based tool for creating sophisticated clustered heat maps. F1000Res. 8, https://doi.org/10.12688/f1000research.20590.2 (2019).
Acknowledgements
This study was supported by the following grants: Japan Society for the Promotion of Science (JSPS) KAKENHI Grant-in-Aid for Scientific Research 22H02900 (S.H.), and 23K05693 (T.T.): Shiga Prefecture Governor, grant number #PT330033 (T.C.): The Aiba Works Medical Research Grant (H.H. and N.A.). This work was the result of using research equipment shared in the MEXT Project for promoting public utilization of advanced research infrastructure (Program for supporting construction of core facilities) Grant Number JPMXS0441000023. Illustrations in the figures were taken from TogoTV (©2016 DBCLS TogoTV/CC-BY-4.0).
Author information
Authors and Affiliations
Contributions
H.H., T.T., and S.H. designed the study and analysis. Experiments were performed by H.H., S.S., T.T., M.Kato, N.A., M.Kawade, C.Y., H.Sakai, H.T., M.T., M.I., Y.U., and S.H. Data interpretation analysis were performed by T.C., H.Sanjo, S.T., Y.N., Y.S., and S.H. This manuscript was prepared by H.H., T.T., and S.H. with input from all authors.
Corresponding author
Ethics declarations
Competing interests
T. Chano is the president and CEO of GF Mille Co. Ltd. M. Kawade served as a member of GF Mille Co. Ltd. Y. Ueno is a senior advisor of GF Mille Co. Ltd. S. Hiratsuka has received research funding from GF Mille Co. Ltd. and Bourbon Corp., and a manuscript fee from Nippon Boehringer Ingelheim Co. Ltd. Y. Soejima reports grant to his institution from Kaken Pharmaceutical Co. Ltd. and has received fees from Eli Lilly Japan K.K., AstraZeneca K.K., Eisai Co. Ltd., Miyarisan Pharmaceutical Co. Ltd., Merck & Co. Inc., TAIHO Pharmaceutical Co. Ltd., Novartis Pharma K.K., Takeda Pharmaceutical Co. Ltd., EA Pharma Co. Ltd., Chugai Pharmaceutical Co. Ltd., TEIJIN Pharma Ltd., TSUMURA Co. Ltd., Otsuka Pharmaceutical Factory Inc., Guardant Health Japan Corp., and ONO Pharmaceutical Co. Ltd. Y. Nakazawa and M. Tanaka are shareholders of A-SEEDS Co. Ltd. Y Nakazawa reports grants to his institution from A-SEEDS Co. Ltd., BrightPath Biotherapeutics Co. Ltd., AGC Inc., Toshiba Corp., Daiichi Sankyo Co. Ltd., Bourbon Corp., Cell Science & Technology Institute, Inc., and Sekisui Chemical Co. Ltd, and has received fees from Bourbon Corp., Novartis Pharma K.K., Ajinomoto Co. Inc., Daiichi Sankyo Co. Ltd., and AstraZeneca K.K. All other authors report no competing interests.
Peer review
Peer review information
Nature Communications thanks Wei Tao, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hayashi, H., Seki, S., Tomita, T. et al. Synthetic short mRNA prevents metastasis via innate-adaptive immunity. Nat Commun 16, 1925 (2025). https://doi.org/10.1038/s41467-025-57123-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57123-y
