
Abstract
Rapid genotype-based drug susceptibility testing for the Mycobacterium tuberculosis complex (MTBC) relies on a comprehensive knowledgebase of the genetic determinants of resistance. Here we present a catalogue of resistance-associated mutations using a regression-based approach and benchmark it against the 2nd edition of the World Health Organisation (WHO) mutation catalogue. We train multivariate logistic regression models on over 52,000 MTBC isolates to associate binary resistance phenotypes for 15 antitubercular drugs with variants extracted from candidate resistance genes. Regression detects 450/457 (98%) resistance-associated variants identified using the existing method (a.k.a, SOLO method) and grades 221 (29%) more total variants than SOLO. The regression-based catalogue achieves higher sensitivity on average (+3.2 percentage points, pp) than SOLO with smaller average decreases in specificity (−1.0 pp) and positive predictive value (−1.6 pp). Sensitivity gains are highest for ethambutol, clofazimine, streptomycin, and ethionamide as regression graded considerably more resistance-associated variants than SOLO for these drugs. There is no difference between SOLO and regression with regards to meeting the target product profiles set by the WHO for genetic drug susceptibility testing, except for rifampicin, for which regression specificity is below the threshold of 98% at 97%. The regression pipeline also detects isoniazid resistance compensatory mutations in ahpC and variants linked to bedaquiline and aminoglycoside hypersusceptibility. These results inform the continued development of targeted next generation sequencing, whole genome sequencing, and other commercial molecular assays for diagnosing resistance in the MTBC.
Similar content being viewed by others

Introduction
Genotypic drug-resistance prediction for MTBC is rapidly becoming more favoured than phenotypic drug-susceptibility testing due to its comparative speed and reliability1 and has been endorsed by the World Health Organization for some drugs2. Freely available tools, such as Mykrobe3 and TBProfiler4, rely on rigorously validated catalogues of resistance-associated to make resistance predictions for user-supplied genome sequencing. Therefore, it is imperative that catalogues have both high sensitivity and specificity and are continually updated using newly collected data.
The current method used by the World Health Organization to build catalogues of mutations associated with antibiotic resistance focuses on univariate association between solitary mutations and binary resistance phenotype (resistant vs. susceptible), excluding isolates with multiple possibly causative resistance mutations. Additional confidence grading rules are applied to the output of this association analysis and integrated with independent data from the literature (e.g. allelic exchange data) to generate the final grading of mutations into 5 categories: Group (1) Associated with resistance, Group (2) Associated with Resistance - Interim, Group (3) Uncertain significance, Group (4) Not associated with Resistance - Interim, and Group (5) Not associated with resistance.
This univariate association method5,6 is based upon the assumption that most resistant strains have just one non-synonymous, non-lineage defining mutation in candidate genes whereas most susceptible strains have none5. Evidence implicating a mutation with resistance is derived solely from isolates in which it is the only mutation occurring in a drug-resistant isolate (after exclusion of a pre-specified list of neutral variants). This approach considers the mutation independently of potential additive effects with other mutations. Mutations that do not fit those criteria are graded using additional grading rules (i.e. predicted effect on protein function, proximity to the drug’s active site) or evidence from the literature. If none of the above is possible, then it is graded as uncertain7,8.
Different from univariate association, multivariable regression can perform phenotypic association of solitary or multiple co-occurring mutations, estimating the effect of each mutation on phenotype, conditional on the presence or absence of other mutations. Regression is used to generate polygenic risk scores for predicting human disease9. Similarly, given the known high heritability of antibiotic resistance in bacteria10,11, this approach can also be used to predict antibiotic resistance from genotypic data. Both binary and semi-continuous outcomes (e.g. minimum inhibitory concentrations, or MICs, are typically measured at serial doubling dilutions) can be predicted. However, genetic association using regression requires an assumption that the relationship between mutation presence and the probability of phenotypic resistance follows a logistic function and that the effects of co-occurring mutations on resistance are additive. Strong co-linearity between mutations can bias the fit of a regression model and affect its attribution of effects between linked mutations and the phenotype. Further, accurate association using regression requires genotypic and phenotypic data from a large and diverse sample set.
In this work, we compare the accuracy and predictive performance of a multivariable penalised regression model with the SOLO method currently used in the World Health Organization (WHO) catalogue of mutations in the Mycobacterium tuberculosis complex and their association with drug resistance (“WHO mutation catalogue”)8. The regression pipeline improves sensitivity over the existing catalogue and identifies variants associated with drug hypersusceptibility and compensatory variants in ahpC associated with isoniazid resistance. This approach may also lower dependence on literature sources external to the data to grade mutations and improve scalability and future automation of catalogue production.
Results
Overview of regression gradings
Abbreviations used throughout the work are in Table 1. We trained single-drug regression models for 15 drugs using the same genotypic and phenotypic data as the 2nd edition of the published WHO mutation catalogue8. The complete dataset across all 15 drugs is composed of 52,567 high-quality MTBC genomes with associated phenotypic data. For each model, we excluded all isolates with variant calls with an allele frequency (AF) > 0.25 and ≤0.75 or low-confidence (i.e., “missing”) variant calls in the candidate genes (Methods). After these exclusions, the remaining isolates and variants were used for training single-drug models (Supplementary Data 1). Variants were encoded as binary variables — present if AF > 0.75 and absent if AF ≤ 0.25.
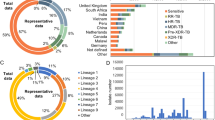
Binary phenotypic drug susceptibility testing (pDST) data were subset into two groups: the “WHO dataset” (N = 41,141) consists of higher confidence phenotypes tested using WHO-approved phenotypic testing methods, and the “ALL dataset” (N = 52,567) includes the entire WHO dataset as well as results measured using phenotypic approaches not endorsed by the WHO, such as UKMYC 96-well plates12. Phenotypic resistance frequency varied from 2% to 43% across the 15 drugs and differed between the WHO and ALL datasets especially for bedaquiline (BDQ) (32 percentage points, pp) (Fig. 1a). A large fraction of the bedaquiline pDST data in the WHO dataset came from a single national reference laboratory that received isolates for testing after suspicion of bedaquiline resistance in source laboratories with limited to no concurrent sampling of Bedaquiline-susceptible isolates8. About 80% of all isolates belonged to the Euro-American (L4) and East Asian (L2) genetic lineages (Fig. 1b).

a Percentages of phenotypically resistant (dark shades) and susceptible (light shades) isolates in the base models for the WHO (blue) and ALL (orange) datasets, across 15 drugs. b Lineage distribution for isolates in the base model for the ALL dataset only. Other category = M. bovis and L5-L7. The percentages for the “Other” category are not shown for readability. Only isolates with a single primary lineage according to the Coll 2014 scheme are shown in (b). In both panels, isolate counts for each bar are shown in parentheses. Bar colours: pink = L1, blue = L2, purple = L3, red = L4, white = all other lineages.
Regression was applied to 21,589 unique (drug, variant) pairs (Fig. 2). The resultant catalogue (Table 2, Fig. 3a, Supplementary Data 3) was benchmarked against the SOLO data-derived associations without application of additional grading rules (GR). We observe high agreement between the two methods for resistance (R)-associated variants, with 450/457 (98.5%) of SOLO R-associated variants also detected as such by regression (Fig. 3b). When GR are applied to regression, 1378/1383 (99.6%) of SOLO + GR R-associated variants are detected by the regression pipeline (Fig. 3c). Specifically, 738 individual LoF variants are upgraded to Group 2 because the pooled LoF variants for those genes are R-associated, and 147 variants are upgraded by other grading rules.

All p values were computed from the estimated coefficient and the distribution of coefficients from the 1000 permuted models (Supplementary Fig 1a, b), two-sided for the neutral test and one-sided for all others. False discovery rate (FDR) correction was performed using the Benjamini-Hochberg method. A variant is considered significant if it has an FDR-corrected p ≤ 0.05 for non-silent variants and FDR ≤ 0.01 for silent variants. LB lower bound in a binomial exact confidence interval. *: Relaxed thresholds for pncA are the same as in the SOLO algorithm -- Present in ≥2 PZA-resistant or susceptible isolates (depending on the sign of the OR) and PPV ≥ 0.5. +: Significance testing exception: raw p values and a cutoff of 0.05 for silent variants in the neutral permutation test.

Regression variant gradings for 15 drugs, coloured by number of variants in each cell. Group 3) Uncertain significance variants are not shown in (a). Grading comparison tables for regression vs. SOLO (b) and regression with GR vs. SOLO with GR (c). Variant colouring: Dark blue = variants graded “Uncertain” by SOLO, not “Uncertain” by regression; light blue = variants graded “Uncertain” by regression, not “Uncertain” by SOLO; red = major up-/down-grade discrepancies by regression; grey = group agreement; black = Group 1 or 2 by both regression and SOLO but not perfect agreement.
Sampling and variant co-occurrence affects grading
To understand discrepancies between the gradings, we first examined variants that may have been graded differently due to data or sampling differences between the WHO and ALL datasets. 12 variants were “Uncertain” in the ALL dataset and “Assoc w R” in the WHO dataset, resulting in a final grading of “Uncertain”. In SOLO, 6 of these are graded “Uncertain”, one Group 1, three Group 2, and two Group 5. The Group 1 variant is Rv0678_p.Met146Thr (BDQ), which is noted to have a potentially inflated PPV due to sampling bias8.
Downgrading variants that are “Assoc w R” in the WHO dataset but “Uncertain” in the ALL dataset to a final “Uncertain” grading is exemplified by the variant mmpS5_c.-74G > T (alias Rv0678_c.-11C > A), which has been experimentally associated with BDQ hypersusceptibility in previous work13,14. In the binary models, mmpS5_c.-74G > T was measured to have a significant positive association with BDQ resistance in both the WHO and ALL datasets. In the WHO dataset, the PPV LB was also high at 0.76. In the ALL dataset, the PPV LB was less than the “Assoc w R” threshold (0.25) at 0.18, and this resulted in an “Uncertain” grading. Given the larger size of the ALL dataset, the observed discrepancy raised the possibility of biased sampling in the WHO data, inflating the association with resistance. The WHO dataset is significantly enriched for BDQ resistance with limited to no sampling of BDQ-susceptible isolates from the same communities8. This bias is less pronounced in the ALL dataset as evidenced by the lower PPV, but as the WHO dataset is a subset of the ALL dataset, the bias was likely still present (Fig. 1a, Supplementary Fig. 3a). The MIC model, however, reproduced the variant’s known association with susceptibility (coef. = −0.046, p = 0.009) (Supplementary Data 5, Supplementary Fig. 3b). Notably, in the BDQ MIC dataset, only 4.4% of isolates are also in the BDQ WHO dataset (Supplementary Data 1). This assessment confirms that biased sampling can skew associations especially for the novel drugs to which resistance remains of limited prevalence globally and is potentially amplified by transmission15.
A related methodologically informative observation was that variants causal of resistance can be more diverse and individually rarer than linked polymorphism or mutations associated with hypersusceptibility such as mmpS5_c.-74G > T in the available data. A subset of resistant isolates with this polymorphism lacks potential resistance-causing variants in the regions studied. This results in the regression models assigning a positive effect on drug resistance to the polymorphism or hypersusceptibility variant. Specifically, there are 37 bedaquiline-resistant isolates with mmpS5_c.-74G > T. Of these, 14 carry one of eight Group 1 or 2 BDQ resistance variants according to the WHO mutation catalogue (Supplementary Data 10)8. In addition to mmpS5_c.-74G > T, all 37 isolates share the same three neutral polymorphisms in mmpL516: Asp767Asn, which is an L2.2.1 marker, Thr794Ile, and Ile948Val. Of the remaining 23 BDQ-R isolates without a known BDQ resistance marker, only one isolate has any variant in the Tier 1 genes for bedaquiline – atpE, mmpS5, mmpL5, Rv0678, and pepQ – and both variants in this isolate are graded “Uncertain” by both SOLO and regression. This leaves 22 BDQ-R isolates with wild-type sequences at the AF = 0.75 level except for mmpS5_c.-74G > T. The high proportion of BDQ-R without a potential causal variant suggests the need to look for low-frequency variants below an AF threshold of 0.25 or for variants outside of the evaluated genes for BDQ.
Discrepancies between SOLO and regression grading
We next compare gradings across the two methods, identifying no major downward discrepancies, one major upward discrepancy, seven minor downward discrepancies, and 228 minor upward discrepancies of regression compared with SOLO (Fig. 3b). There are two additional major discrepancies between regression and SOLO after grading rules are applied (Fig. 3c).
Major downward discrepancies (i.e., Group 4–5 by regression and Group 1–2 by SOLO): None
Major upward discrepancies (i.e., Group 1–2 by regression and Group 4–5 by SOLO): One variant, rrs_n.514 A > C (capreomycin, CAP). rrs_n.514 A > C is a known marker of streptomycin (STM) resistance17; After adding a control for STM resistance in the CAP regression model (by including an additional binary phenotypic STM resistance covariate), rrs_n.514 A > C was not significantly associated with CAP resistance, indicating confounding due to a high correlation between CAP and STM resistance as the cause of this discrepancy.
Minor downward discrepancies (i.e. Group 3 by regression and Group 1–2 by SOLO): Seven variants: Rv0678_p.Met146Thr (BDQ), pncA_p.Ala161fs, pncA_p.Arg154fs, pncA_p.Thr22fs, pncA_p.Val155Ala, pncA_p.Val93Ala, and pncA_p.Asp63_Ser67delinsGlu (pyrazinamide, PZA). Rv0678_p.Met146Thr was downgraded due to having a low PPV in the ALL dataset, similar to mmpS5_c.-74G > T. Many of the pncA variants fall slightly below the significance and PPV thresholds, and all are rare, occurring in up to 6 isolates. Although we relaxed the PPV and frequency grading criteria for pncA as in the WHO mutation catalogue to account for rarity of individual mutations (Fig. 2), additional relaxed criteria for pncA variants may be needed.
Minor upward discrepancies (i.e., Group 3 by regression and Group 4–5 by SOLO): There are 228 such variants. Of these, 54 variants are in rrs (16S) and were excluded from the regression testing because of higher rates of missing variant calls. This was due to heterozygosity in the sequencing read data and is an issue recognised to arise for ribosomal genes due to homology with other species. Due to rarity in the ALL dataset (observed in <5 isolates), 26 variants are graded “Uncertain” by regression, and of these, 16 have significant FDR ≤ 0.05 in the neutral permutation test. All 16 variants are in rrs for amikacin (AMK), capreomycin (CAP), and kanamycin (KAN). Seven silent variants have OR < 1 with an FDR p-value greater than 0.01 but less than or equal to 0.05, but they are graded “Uncertain” because we apply a stricter cut-off of 0.01 for silent variants. The remaining 141 variants meet the SOLO Neutral criterion of PPVR upper bound <10%4 but were not significant in either the regression permutation test or the neutral permutation test.
Major discrepancies between regression + GR and SOLO + GR
katG_c.12 A > G is graded Group 4 by grading rules because it is a silent variant. It is found in 13 isoniazid (INH)-R isolates and 0 INH-S isolates, none of which is a SOLO isolate. Eleven isolates have known resistance variants inhA_c.-777C > T (N = 10) or katG_p.Ser315Thr (N = 1), which are both correctly graded Group 1 by regression. Despite being highly correlated with other INH R-associated variants, katG_c.12 A > G is found to be significantly associated with R by regression, possibly due to the two INH-R isolates without other variants in the regions tested. This result is similar to that of mmpS5_c.-74G > T and suggests that additional statistical corrections may be required for very high correlations between rare variants. We note that the application of GR corrects this discrepancy for regression.
A single variant is classified as Group 4–5 by regression and Group 2 by SOLO + GR: ethA_p.Pro209fs. This variant only occurs in 26 ethionamide (ETO)-S isolates from one lineage (L4.3.4.2), but it is upgraded to Group 2 on grading rules based on the prediction of a frameshift in ethA (Supplementary Table 1). Given that ethA_p.Pro209fs is not observed in ETO-R isolates, it either does not have the expected disruption of ethA, or there is an alternative mechanism of activating ETO than the ethA-encoded enzyme in these isolates.
Regression grades 294 (+64%) more R-associated variants than SOLO, superseding the need to use additional grading rules for 79 variants
Regression grades 751 R-associated variants and 224 (161 non-silent) Not Assoc with R variants, compared to SOLO’s 457 and 297 (230 non-silent), respectively and leaves 221 fewer uncertain mutations than SOLO. The use of grading rules is superseded by regression for 79 R-associated variants and 46 Not Assoc with R variants (Supplementary Data 4). The 79 variants graded R-associated by regression or by grading rules but not by SOLO are most commonly missed due to a frequency of <5 in isolates without other potential candidate mutations (N = 27, 34%) or due to a non-significant association (FDR > 0.05 in Fisher’s exact test, N = 50, 63%).
The MIC models (Supplementary Data 5) were built on largely non-overlapping data with the WHO binary model (up to 15% overlap) (Supplementary Data 1) and so allow for partial validation of the binary regression results. Further, as MIC measures resistance more quantitatively, the MIC models can more finely measure the direction and size of effects on resistance, which may more directly link to biological effect11. Of the 372 variants graded as neither “Neutral” nor “Uncertain” by regression and “Uncertain” by SOLO (Fig. 4), 301 (81%) were tested in the MIC models; the remainder could not be tested due to rarity or the presence of missing or low frequency calls in available isolates with MIC data (MIC dataset set size ranged N = 1007–12,613 across the 15 drugs). 232 of these variants have a significant regression OR in the WHO dataset, and of these, 83 (36%) are also significantly associated with MIC (76 with resistance, 7 susceptibility), with all significant associations reproducing the direction of effect measured in the binary WHO models. Of the 232–83 = 149 remaining variants associated in the binary WHO dataset regression but not associated in the MIC models, 78 (52%) occurred in fewer than 5 isolates in the MIC datasets, which could explain the inability to detect these associations. An additional 9 variants are significantly associated with MIC at a less conservative FDR threshold of 0.1, and their directions of association are also congruent with those measured in the binary regression.
The seven drug-variant pairs with significant associations with drug susceptibility in the WHO and MIC datasets (Fig. 4a) are eis_c.-9T > C (AMK), eis_p.Met1? (loss of start codon) (AMK, KAN), eis_p.His150fs (AMK, KAN), mmpL5_LoF (BDQ), and mmpL5_p.Arg202fs (BDQ). The literature supports a causal relationship between drug hypersusceptibility and loss-of-function of these two genes. Eis is an enzyme that inactivates AMK and KAN, and mmpL5 is a component of a bedaquiline efflux pump18.

Neutral variants were excluded from this analysis. WHO dataset ORs vs. MIC coefficients for 232 variants graded Group 3 by SOLO and Groups 1–2 (b, N = 204) or 4–5 (a, N = 28) by regression, were tested in MIC models, and have a significant OR in the WHO dataset. Point colour reflects the direction of association in the MIC model and significance at FDR ≤ 0.05 for all variants (red = significant/positive, blue = significant/negative, grey = not significant). Coef: coefficient in the MIC model.
To further probe the newly graded R-associated variants by regression, we inspected rifampicin (RIF) and isoniazid (INH) variants graded R-associated by regression and “Uncertain” by SOLO + GR. There are 5 such variants for RIF in rpoB – F424V, I491L, I491M, I491T, and S493L – none of which has a significant association with MIC. These variants occur outside of the rifampicin resistance determining region (RRDR) but are predominantly found in RIF-R isolates. Due to the high rates of co-occurrence with known R-associated variants, most frequently H445N, L430P, and D435Y in rpoB, they have small estimated ORs in the ALL dataset, ranging from 1.07 to 1.11. They are close to RRDR residues S428 (4.5 Å from F424) and R447-L449 (within 4.6 Å of S493) (PDB ID: 5ZX2). For the three variants observed in codon 491, given the diversity of alleles at this site and the known association between I491F and RIF resistance19, these associations may be causal. For isoniazid, there are 38 variants graded R-associated by regression and “Uncertain” by SOLO + GR. Of these, 8 have significant positive associations with MICs as well, adding additional evidence to their associations. The remaining are 22 missense variants (20 katG, 1 ahpC, 1 inhA) and 8 upstream gene variants (5 ahpC, 2 inhA, and 1 katG). The five variants upstream of ahpC are known INH resistance compensatory mutations (Table 3).
Variants acquired in members of two or more independent lineages are more likely to be causal or compensatory for resistance20,21. The majority (178 out of 220) of the variants graded “Assoc w R” by regression and “Uncertain” by SOLO + GR are observed in at least 2 lineages (Supplementary Data 6, Supplementary Fig. 5). The remaining 42 variants are observed to be lineage-restricted, but of these, 17 are rare and found in fewer than 10 isolates, making it less likely to observe acquisition in multiple lineages. (Supplementary Data 6). Changing the grading of a mutation based on its lineage distribution has not been benchmarked because most resistance-associated mutations are homoplastic, occurring independently in multiple lineages. The use of lineage distribution to inform gradings is currently being investigated for the 3rd edition of the WHO resistance mutation catalogue.
Sensitivity, specificity, and PPV of the regression-based catalogue
We computed sensitivity, specificity and PPV of resistance diagnosis using the catalogues built using SOLO, SOLO + GR, regression, and regression + GR (Fig. 5). We expect these performance metrics to be optimistic because they are estimated on the same data from which the catalogues were derived but this derivation is less likely to invalidate the comparison between catalogues. The sensitivity of the regression-derived list is higher for 13/15 drugs than the SOLO-derived list (difference 0.081 to 10.2 percentage points (pp)). SOLO specificity and PPV are higher than regression for 12/15 drugs (specificity difference range: 0.011 to 3.70 pp, PPV difference range: 0.10 to 12.7 pp). For delamanid, the statistics are the same because both SOLO and regression graded the same two variants as R-associated. The average difference in sensitivity (+3.2 pp) between regression and SOLO is larger than the average differences in specificity (−1.0 pp) and PPV (−1.6 pp), and the F1 scores (harmonic mean of sensitivity and PPV) averaged across the 15 drugs are 69.2% for SOLO, 69.9% for SOLO + GR, and 70.2% for regression.

Sensitivity (a), specificity (b), and PPV (c) comparison between SOLO (green), SOLO + GR (purple), regression (orange), and regression + GR (grey) mutation lists. Bars are the computed sensitivity, specificity, and PPV, as percent, for each drug and model type. Error bars are 95% exact binomial confidence intervals computed using the Clopper-Pearson method. Source data are in Supplementary Data 7, including the dataset sizes.
The largest gains in sensitivity for regression are for ethambutol (\(\Delta\) = +10.2 pp), clofazimine (+9.6), streptomycin (+9.5), ethionamide (+9.4), rifampicin (+2.6), and isoniazid (+2.1). For four drugs, regression specificity is more than 1 percentage point lower than SOLO specificity: ethionamide (\(\Delta\) = -3.7 pp), streptomycin (-3.3), ethambutol (-2.7), and capreomycin (-2.2). The drop in capreomycin specificity is due to the rrs_n.514 A > C false positive. If this variant is removed from the regression list, the capreomycin model metrics are the same for both SOLO and regression, and the average F1 score for regression is 70.6% (Supplementary Data 8).
When grading rules are applied to the regression results, sensitivity increases for eight drugs, three of which have increases greater than 1 pp. The largest increase is 3.9 pp for amikacin due to eis_c.-14C > T and rrs_n.1402 C > T, which are upgraded due to WHO approval as resistance markers. Sensitivity increases for delamanid (+2.8 pp) and clofazimine (+1.9 pp) due to the inclusion of additional LoF mutations presumed to cause resistance.
The WHO has set minimum target product profiles (TPPs) for genetic drug susceptibility testing – ≥98% specificity for all drugs, and for sensitivity, >95% (RIF), >90% (INH, LFX, MXF), and >80% (BDQ, LZD, CFZ, DLM, AMK, PZA)22. For RIF, SOLO specificity is ≥98%, but specificities for SOLO + GR, regression, and regression + GR are 97%. For all other drugs, the four methods do not differ in whether they meet the sensitivity and specificity TPPs.
Lowering AF threshold to 25% improves sensitivities for bedaquiline, clofazimine, and fluoroquinolones
For eight drugs, lowering the AF cutoff to 25% (i.e., variant is considered present if AF > 25%) increased the sensitivity of these mutations for predicting resistant phenotypes by at least 2 pp (Supplementary Fig. 6, Supplementary Data 9). The largest increases are 10.3 pp for bedaquiline to 58.9% (95% CI, 55.9–61.9%), 4.7 pp for moxifloxacin to 90.3% (95% CI, 89.4–91.2%), 4.5 pp for levofloxacin to 90.0% (95% CI, 89.2–90.7%), and 3.8 pp for clofazimine to 27.8% (95% CI, 24.4–31.3%). The corresponding decreases in specificity are 0.29, 0.36, 0.27, and 0.30 pp, respectively. This confirms the importance of heteroresistance for fluoroquinolones and demonstrates that it is also relevant for bedaquiline and clofazimine. These results are consistent with SOLO, for which the largest increase in sensitivity is 10.2 pp for bedaquiline8.
Discussion
In this work, we demonstrate that logistic regression models can reproduce the results of SOLO, the current WHO catalogue resistance-association method in identifying R-associated variants from WGS and resistance phenotype data. Of the 457 R-associated variants graded by SOLO, 450 (98.5%) are also identified by regression. Regression graded 221 more variants and achieved higher sensitivity on average across 15 drugs than SOLO. Most importantly, regression does not require defining neutral variants before starting grading of mutations. Further, because regression is trained on all high-quality variant data and considers variant co-occurrence, it can reduce the need to use grading rules for variants that do not pass the SOLO algorithm’s inclusion criteria – 125 variants graded by regression required grading rules to be upgraded – or provide evidence against applying grading rules broadly, e.g. ethA_p.Pro209fs. Although the primary goal of this work was to benchmark the regression catalogue against the published catalogue, which only identified associations with variants in genes known or highly suspected to be relevant8, regression can be used to associate variants in additional loci to discover novel low-level contributors to resistance.
Using the regression pipeline, we identify LoF variants in eis and mmpL5 as associated with aminoglycoside and bedaquiline hypersusceptibility, respectively, which agrees with recently published work associating genotype with MICs11. We reproduce these associations in MIC models. Overall, we find the MIC models useful as adjunct evidence supporting the gradings derived from the more abundant binary resistance data (Results Section D). The use of MIC and lineage/homoplasy could in the future further reduce the need for grading rules.
Regression also associated six compensatory ahpC mutations with isoniazid resistance (Table 3). These co-occur with resistance-causing mutations to mitigate their fitness cost relative to wild-type. katG detoxifies reactive oxygen species (ROS) and activates isoniazid, which is a pro-drug23. To compensate for the loss of katG function through INH-resistance causing mutations, promoter mutations that increase the expression of ahpC, an antioxidant enzyme similar to katG, are frequently acquired, restoring the bacterium’s resistance to ROS24. All the ahpC promoter variants graded as R-associated by regression are in the region 47–92 base pairs upstream of ahpC that is targeted by the Xpert MTB/XDR test25, and four have experimental evidence of ahpC overexpression26,27,28,29, adding validity to the regression. Although ahpC mutations are not causative of resistance, they have proven useful diagnostically for resistance classification in several reports25,30. This is likely due to their rarity in susceptible isolates. Our results support the use of the six associated ahpC mutations (Table 3) to classify resistance because their inclusion changes INH resistance prediction sensitivity by +0.5 percentage points (pp) and specificity by −0.1 pp (Supplementary Data 8). Further research is needed to assess if multivariate models can offer more accuracy when combining resistance causing and compensatory mutations in diagnostics use.
The regression grading approach is not without its limitations. Fitting a regression model requires the missing data to be imputed or dropped. Because imputation can introduce bias, we removed isolates containing unfixed variants or variants that could not be called. For the same reasons, variants that were absent from the remaining isolates were also excluded. 2388 variants (11%) were not fit by the regression models and were automatically graded “Uncertain.” Most (2334; 98%) are also graded “Uncertain” by SOLO, and 54 (all rrs: aminoglycoside pairs) are graded Group 5. Future work in which regression is rerun on subsets of the rrs variants and isolates with complete data can address this limitation. Despite the improved ability to grade mutations without the need of additional grading rules, many variants (>400) classified by regression still require some adjunct evidence to be interpreted.
Associations determined by regression do not imply causality. Regression can make false positive associations in the setting of biased sampling, linkage disequilibrium, and high correlation between resistances to different related drugs, underlying its catalogue’s lower specificity and PPV compared with SOLO. Uncovering these limitations informs the application of regression for phenotype association and prediction across a range of systems including in human genetics. However, we note that the drops in specificity are small and occur for drugs where phenotypic DST is known to have limited accuracy or reproducibility and where mutations can have intermediate effects on resistance (e.g. ethambutol and ethionamide). Like the approach taken to correct the CAP resistance model for STM resistance (Results Section B), future models can correct for resistance to other drugs to reduce these false positives.
To take a completely data-driven approach to classify variants, it is critical to have datasets that are representative of circulating MTBC strains across the spectrum of the resistance phenotype. We observe a considerable range in the percentage of isolates with resistance across the drugs the WHO and ALL datasets with heavy oversampling of resistance compared with its known population prevalence (Fig. 1a, Supplementary Fig. 3a). This was especially the case for bedaquiline. Oversampling is intended to sufficiently sample resistant isolates, but when oversampling is biased, this can result in confounding. In the case of BDQ, we observed linkage between neutral polymorphisms indicative of genetic lineage and a diverse set of individually rare resistance variants. Furthermore, several isolates with BDQ resistance are still unexplained by candidate resistance markers (Results Section B). We used principal components (PC) to account for lineage effects limiting to the first 50 PCs as there is an expected trade-off between adding additional PCs and statistical power of grading. This PC correction will need to be re-evaluated as more data is available and is expected to be less impactful on power as more representative data becomes available. In the meantime, further analysis of variant lineage distributions and/or phylogenetic inference can help identify the potential false positives arising due to biased sampling and linkage disequilibrium.
In summary, we developed a regression-based pipeline to grade more than 21,000 genetic variants in the MTBC based on their association with resistance to 15 antitubercular drugs. The pipeline is flexible and can be adapted quickly as more data is collected. Although the results we present here are specific to MTBC, this approach provides a model for grading associations between genotype and phenotype for other organisms to use in diagnostic applications.
Methods
Definitions, abbreviations, and curation of MTBC data
Regression models were trained using the same genotypic and phenotypic data as the 2nd edition of the published WHO mutation catalogue8. These phenotypic and genotypic data were curated from published datasets, consortium initiatives, and direct submissions in response to public calls for contributions by the WHO Global Tuberculosis Programme31. Quality control was previously performed for the 2nd edition of the mutation catalogue (Supplementary Data 10), with 52,567 isolates passing both phenotypic and genotypic data quality control8. Data contributors and affiliations are listed in Supplementary Data 11.
The pDST data were subset into two groups: the “WHO dataset” (N = 41,141) consists of higher confidence phenotypes tested using WHO-approved phenotypic testing methods, and the “ALL dataset” (N = 52,567) includes the entire WHO dataset as well as pDST results measured using phenotypic approaches not endorsed by the WHO (e.g. UKMYC 96-well plates)12.
Candidate resistance genes were assigned to two tiers based on published literature, including the first edition of the catalogue7, and discussions among an international panel of expert advisors: Tier 1 is composed of genes and associated promoter regions deemed by the expert panel to contain resistance mutations with high probability and Tier 2 is composed of genes that may contain resistance mutations based on new or less established evidence8. We focused the regression on associating variants in Tier 1 genes (Supplementary Table 1) because evidence from the 2nd edition of the mutation catalogue did not support associations between drug resistance and variants in Tier 2 genes8. However, regression models can be fit on variants in both Tier 1 and 2 genes by adding an additional flag to the model scripts.
Variants not covered by at least 10 reads in the read alignment were labelled as missing calls8. For variants with at least 10 reads of support, a variant was considered present if it had a within-isolate allele frequency (AF) > 0.75 and absent if AF ≤ 0.25. We did not attempt to associate variants with intermediate allele frequencies with resistance in the regression models. Regression models cannot be trained on missing data, so for a given single-drug model, we excluded all isolates with variant calls with an AF in the range (0.25, 0.75] and isolates with missing variant calls (Supplementary Data 1). After association, resistance prediction to assess sensitivity, specificity, and PPV did use isolates with intermediate allele frequencies as described in the results section. The genes rrs and rrl are known to have a high degree of homology across bacteria, and false low frequency variant calls are common due to contamination in the sequencing process. For this reason, the ribosome-targeting drugs have the largest numbers of excluded isolates and associated variants (Supplementary Data 1).
Regression model design
To maintain high statistical power, we built a series of nested models (Supplementary Table 2). The “base” model for a given phenotypic group was fit on all non-silent variants. Definitions of silent variants and LoF mutations were the same as for the SOLO method (Table 1). Two additional models were fit: one in which LoF variants – frameshift, start_lost, stop_gained, and feature_ablation variants – were pooled on a per-gene basis into a single “LoF” feature for each gene, and another with silent variants in addition to the non-silent variants in the “base” model. For the pooled models, the LoF component mutations were removed from the variant matrix so that a variant was not multiply considered during model fitting. The expected effect size of silent variants is significantly smaller than those of non-silent mutations, so no model was trained only on silent variants.
Non-silent variants were tested in all three models in Supplementary Table 2, but we used only the results from the “unpooled” model to grade them. The only results derived from the models with LoF pooling and silent variants were the pooled LoF and silent variants themselves, respectively. We separately fit models on the WHO and ALL datasets. Models were trained to regress single-drug binary pDST on genotype. The MIC models were not used for grading but were instead used to add supportive evidence for associations between genotype and pDST.
We trained single-drug regression models with L2 penalties due to expected multi-collinearity in the data, which is caused by clonality and population structure in bacteria. We performed a series of statistical tests to grade mutations into five categories. Because genes were selected for inclusion into the models based on the literature and known or putative mechanisms of action, all mutations in the genes in Supplementary Table 1 were included. To select the strength of L2 regularisation for each model, we performed five-fold cross-validation using all powers of ten from 10−6 to 106 and selected the regularisation parameter with the lowest binary cross-entropy, with class balancing. Coefficient significance was determined using a permutation test with 1000 re-shufflings of phenotype across the full dataset and the same regularisation parameter determined on the original model (Supplementary Fig. 1a, b).
Principal component analysis
MTBC is a clonally evolving bacterium with strong population structure, which can lead to systematic false positives when estimating the effects of mutations on resistance that may not be sufficiently corrected for through L2 regularisation32,33. Therefore, we additionally adjusted for population structure using principal components analysis (PCA) on a kinship matrix. 6938 single nucleotide variants (SNVs) more than 50 base pairs away from any PE or PPE gene across the MTBC genome and with a variant allele frequency of at least 1% were included. We excluded previously identified homoplastic sites34, canonical drug resistance loci35, and sites at which more than 1% of isolates have a variant occurring at a within-isolate read frequency <75%, leaving 6190 SNV sites for the kinship matrix.
We performed PCA on the covariance matrix of these 6190 sites. The resulting principal components are latent variables the first of which are expected to describe major axes of ancestral variation (i.e., lineage) in the MTBC. We included the first 50 principal components, which explain 94.5% of the total variance in the genotype (Supplementary Fig. 2a), as covariates in every model. We ran fast-lineage-caller36 on the variant call format (VCF) files to obtain lineage designations (Supplementary Data 2). We considered an isolate’s lineage to be the most specific lineage that fast-lineage-caller identified. Eight isolates did not have a lineage by the Coll scheme37 but were classified as Mycobacterium canettii by the Lipworth scheme38. Several principal components are highly correlated with lineages and sublineages (Supplementary Fig. 2). Many of the principal components (Supplementary Fig. 2e–h) separate sublineages of L4, which is the most diverse of the MTBC lineages. 395/52,567 (0.75%) of isolates have more than one lineage assigned according to the Coll scheme37. These are excluded from Supplementary Fig. 2 but are in all models and analyses.
Likelihood ratio test
To further discriminate between the strengths of association of different mutations, we performed a likelihood ratio test (LRT). This test compares two nested models and determines if adding additional variants significantly improves a model’s goodness-of-fit. The LRT is more conservative than the permutation test as it more directly controls for collinearity in the mutations, i.e. for a mutation to demonstrate significance on the LRT it must improve the fit of the model when added to all other mutations in the model. This LRT statistic follows the chi-squared distribution (Supplementary Fig. 1c).
Test for mutation neutrality
We implemented a second permutation test that reverses the hypothesis test to determine if the evidence supports a lack of an association between a variant and drug resistance (i.e., test if a mutation is neutral with an OR ~ 1 on resistance). In the permutation test for mutation effect, if the proportion of permuted ORs that are more extreme than the estimated OR on the training dataset is less than α, then the variant has a significant OR on resistance (Supplementary Fig. 1a). Conversely, for the test for mutation neutrality, if the proportion of permuted ORs that are less extreme (i.e., closer to 1) than the estimated OR is less than α, then the variant is not associated with the drug resistance phenotype (Supplementary Fig. 1b).
Mutation classifications
We used five grading groups as in the WHO catalogue8, noting that these gradings were necessarily defined by different statistical criteria for regression. The flowchart in Fig. 2 details the grading for each variant in a single model (i.e., single drug, single phenotypic group). The gradings assigned in Fig. 2 are interim, after which the gradings were integrated between the two phenotypic datasets to a final grading (Supplementary Table 3).
To reduce grading of false associations among rare variants, regression-graded variants must be present in at least five isolates. For grading associations with R, we set the FDR q-value cutoff at 0.05 for non-silent variants (including pooled LoF variants), as this constituted the primary analysis, and 0.01 for silent variants to be more conservative in the latter secondary analysis. We additionally required candidate R-associated variants to pass the LRT to mitigate false associations with R, but did not require this for candidate variants not associated with R. For consistency with the SOLO algorithm, we required the lower bound of the PPV to be at least 0.25 for candidate variants associated (OR > 1, PPVR) and not associated with R (OR < 1, PPVS). PPVR = P(resistant | variant present), PPVS = P(susceptible | variant present), NPV = P(susceptible | variant absent).
The SOLO method set an FDR q-value cutoff of 0.05 for all significance testing, except the Neutral masking algorithm. For consistency with the Neutral masking algorithm of SOLO, we used raw p values and a cutoff of 0.05 for silent variants in the neutral permutation test.
Like the SOLO algorithm, we integrated associations identified using the WHO dataset (a more select data subset with higher confidence phenotypes) with those from the ALL dataset, which includes the WHO dataset plus additional isolates tested for resistance using phenotypic approaches not endorsed by the WHO. We note that due to differences in the underlying statistical methods, some of the rules for resolving category differences between the two phenotypic groups differ between regression and SOLO8.
Importantly, no variants had discrepant associations between the WHO and ALL datasets (i.e. graded “Assoc w R” in one dataset and “Not assoc w R” or “Neutral” in the other). 2388 variants (11%) were not tested in either model due to absence or co-occurrence with low-frequency or missing variant calls, so they were graded “Uncertain.” Of the 19,201 variants tested in the regression models, 6710 (35%) were found only in the ALL dataset. Because the WHO dataset is a strict subset of the ALL dataset, no variant was found only in the WHO dataset.
Among the 12,491 variants found in both the WHO and ALL datasets, 12,177 (98%) have the same grading in both datasets. The combined grading across the WHO and ALL datasets (Supplementary Table 3) reflected the prioritisation of resistance calls from the WHO phenotypic testing and associations, but the ability of the ALL dataset to add uncertainty to or upgrade variants that would benefit from higher power or wider sampling. We upgraded variants that were “Uncertain” in the WHO dataset to Group 2 if the ALL evidence suggested an association with R (Supplementary Table 3, row 7). However, in the reverse scenario, the variant was downgraded to “Uncertain” (Supplementary Table 3, row 5) due to the potential for overcalling R-associated variants in the WHO dataset. The SOLO algorithm graded Neutral variants only from the WHO dataset. We therefore prioritised calling Neutrals from the WHO dataset (Supplementary Table 3, row 3) by grading them into Group 5, but we additionally upgraded variants that were Neutral in the ALL dataset to Group 4 if they were also present in the WHO dataset (Supplementary Table 3, row 4).
Finally, we applied grading rules to the regression output to measure agreement between regression and SOLO + GR, which is the grading of the final published catalogue8. To apply grading rules to the regression catalogue, we upgraded variants that were graded “Uncertain” in both regression and SOLO but not “Uncertain” in SOLO + GR to the SOLO + GR grading.
Resistance prediction and assessing sensitivity and specificity
Both in this work and the published catalogues7,8, resistance predictions were made for the same dataset from which the gradings were derived. Gradings were used to predict isolates in the ALL dataset as resistant or susceptible with a presence cutoff of AF > 75%. All isolates were included; no isolates were excluded due to the presence of low-quality or intermediate frequency variants. An isolate containing any variant in Groups 1–2 is predicted resistant, correcting for epistasis for bedaquiline, amikacin, and kanamycin18, and an isolate lacking all such variants is predicted susceptible. If a pooled LoF variant was graded in Groups 1–2, then all isolates containing any component frameshift, start_lost, stop_gained, or feature_ablation variant in that gene were predicted resistant. We compared metrics between regression, regression + GR, SOLO, and SOLO + GR (Table 2) for each drug.
MIC models
We built genotypic models on MIC data to investigate and validate associations identified in the binary models. These models did not influence the regression-based grading. For some isolates, multiple MICs measurements were available in different media. We prioritised MICs measured in solid media over liquid media over plate-based assays as detailed in the hierarchy in Supplementary Table 4 when deduplicating isolates with multiple measured MICs.
All MICs were then normalised to the most common medium for a given drug, which was typically UKMYC due to the large proportion of isolates from the CRyPTIC study12. Normalisation was done by multiplying the measured MIC by the ratio of the critical concentrations in the most common medium and the medium of the measurement as was previously done10. An L2-penalised linear regression model was built regressing the log2-transformed MICs on the same genotypic inputs as for the logistic regression models. The same nested models were fit as for the binary phenotypes, and the regularisation parameters were selected by minimising the root mean squared error. MIC coefficient significance was determined using a permutation test with 1000 reshufflings and FDR thresholds of 0.05 for non-silent variants and 0.01 for silent variants.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All code, data, and original figures are available at https://github.com/farhat-lab/who-analysis. Variant matrices in long format are available in the “Releases” section of this repository. The full set of isolates used in this work, with internal IDs and public BioSample accessions if available, are in Supplementary Data 2. For information on the bioinformatics pipeline used to call variants and transform them to the variant matrices referenced above, please consult this repository: https://github.com/GTB-tbsequencing/mutation-catalogue-2023. Sequencing and phenotypic drug susceptibility testing data collected by the World Health Organization are available to download here: https://tbsequencing.who.int/download.
Code availability
All code, data, and original figures are available at https://github.com/farhat-lab/who-analysis. All computation was done in Python 3 using the numpy (v1.21.5), pandas (v1.4.2), scikit-learn (v1.0.2), and statsmodels (v0.13.5) packages. Package versions are available in the environment.yaml file in the repository.
References
Hall, M. B., Lima, L., Coin, L. J. M. & Iqbal, Z. Drug resistance prediction for Mycobacterium tuberculosis with reference graphs. Microb. Genom. 9, mgen001081 (2023).
WHO. The use of next-generation sequencing for the surveillance of drug-resistant tuberculosis: An implementation manual (World Health Organization, 2023).
Hunt, M. et al. Antibiotic resistance prediction for Mycobacterium tuberculosis from genome sequence data with Mykrobe. Wellcome open Res. 4, 191 (2019).
Phelan, J. et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 11, 41 (2019).
Miotto, P. et al. A standardised method for interpreting the association between mutations and phenotypic drug resistance in Mycobacterium tuberculosis. Eur. Respir. J. 50, 1701354 (2017).
Walker, T. M. et al. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study. Lancet Infect. Dis. 15, 1193–1202 (2015).
Walker, T. M. et al. The 2021 WHO catalogue of Mycobacterium tuberculosis complex mutations associated with drug resistance: a genotypic analysis. Lancet Microbe 3, e265–e273 (2022).
WHO. Catalogue of mutations in Mycobacterium tuberculosis complex and their association with drug resistance, second edition (World Health Organization, 2023).
Lewis, C.M. & Vassos, E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 12, 44 (2020).
Farhat, M. R. et al. GWAS for quantitative resistance phenotypes in Mycobacterium tuberculosis reveals resistance genes and regulatory regions. Nat. Commun. 10, 2128 (2019).
CRyPTIC Consortium. Quantitative measurement of antibiotic resistance in Mycobacterium tuberculosis reveals genetic determinants of resistance and susceptibility in a target gene approach. Nat. Commun. 15, 488 (2024).
CRyPTIC Consortium. Epidemiological cut-off values for a 96-well broth microdilution plate for high-throughput research antibiotic susceptibility testing of M. tuberculosis. Eur. Respir. J. 60, 2200239 (2022).
Chesov, E. et al. Emergence of bedaquiline resistance in a high tuberculosis burden country. Eur. Respir. J. 59, 2100621 (2022).
Villellas, C. et al. Unexpected high prevalence of resistance-associated Rv0678 variants in MDR-TB patients without documented prior use of clofazimine or bedaquiline. J. Antimicrob. Chemother. 72, 684–690 (2017).
Barilar, I. et al. Emergence of bedaquiline-resistant tuberculosis and of multidrug-resistant and extensively drug-resistant Mycobacterium tuberculosis strains with rpoB Ile491Phe mutation not detected by Xpert MTB/RIF in Mozambique: a retrospective observational study. Lancet Infect Dis. 24, 297–307 (2024).
Rivière, E. et al. Variants in Bedaquiline-Candidate-resistance genes: prevalence in bedaquiline-naive patients, effect on MIC, and association with mycobacterium tuberculosis lineage. Antimicrob. Agents Chemother. 66, e00322–e00322 (2022).
Jagielski, T. et al. Screening for streptomycin resistance-conferring mutations in Mycobacterium tuberculosis clinical isolates from Poland. PloS ONE 9, e100078 (2014).
Vargas, R. Jr et al. Role of Epistasis in Amikacin, Kanamycin, Bedaquiline, and Clofazimine resistance in Mycobacterium tuberculosis complex. Antimicrob. Agents Chemother. 65, e0116421 (2021).
André, E. et al. Novel rapid PCR for the detection of Ile491Phe rpoB mutation of Mycobacterium tuberculosis, a rifampicin-resistance-conferring mutation undetected by commercial assays. Clin. Microbiol. Infect. 23, 267.e5–267.e7 (2017).
Farhat, M. R. et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat. Genet. 45, 1183–1189 (2013).
Merker, M. et al. Phylogenetically informative mutations in genes implicated in antibiotic resistance in Mycobacterium tuberculosis complex. Genom. Med. 12, 27 (2020).
WHO. Target product profile for next-generation drug-susceptibility testing at peripheral centres (World Health Organization, 2021)
Ng, V. H., Cox, J. S., Sousa, A. O., MacMicking, J. D. & McKinney, J. D. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol. Microbiol. 52, 1291–1302 (2004).
Wong, C. F. et al. AhpC of the mycobacterial antioxidant defense system and its interaction with its reducing partner Thioredoxin-C. Sci. Rep. 7, 5159 (2017).
Information sheet: Practical considerations for implementation of the Cepheid Xpert MTB/XDR test (Stop TB Partnership, 2024).
Kelley, C. L., Rouse, D. A. & Morris, S. L. Analysis of ahpC gene mutations in isoniazid-resistant clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 41, 2057–2058 (1997).
Sherman, D. R. et al. Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science 272, 1641–1643 (1996).
Wilson, T. M. & Collins, D. M. ahpC, a gene involved in isoniazid resistance of the Mycobacterium tuberculosis complex. Mol. Microbio. 19, 1025–1034 (1996).
Zhang, Y., Dhandayuthapani, S. & Deretic, V. Molecular basis for the exquisite sensitivity of Mycobacterium tuberculosis to isoniazid. PNAS 93, 13212–13216 (1996).
Farhat, M. R. et al. Genetic determinants of drug resistance in Mycobacterium tuberculosis and their diagnostic value. Am. J. Respir. Critic. Care Med. 194, 621–630 (2016).
WHO. Public call for whole genome sequencing and phenotypic data from clinical isolates of M. tuberculosis complex (World Health Organization, 2022).
Gagneux, S. Ecology and evolution of Mycobacterium tuberculosis. Nat. Rev. Microbiol. 16, 202–213 (2018).
Earle, S. et al. Identifying lineage effects when controlling for population structure improves power in bacterial association studies. Nat. Microbiol. 1, 16041 (2016).
Vargas, R. Jr et al. Phase variation as a major mechanism of adaptation in Mycobacterium tuberculosis complex. PNAS 120, e2301394120 (2023).
Green, A. G. et al. A convolutional neural network highlights mutations relevant to antimicrobial resistance in Mycobacterium tuberculosis. Nat. Commun. 13, 3817 (2022).
Freschi, L. et al. Population structure, biogeography and transmissibility of Mycobacterium tuberculosis. Nat. Commun. 12, 6099 (2021).
Coll, F. et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 5, 4812 (2014).
Lipworth, S. et al. SNP-IT Tool for identifying subspecies and associated lineages of Mycobacterium tuberculosis Complex. Emerg. Infect. Dis. 25, 482–488 (2019).
Eilbeck, K. et al. The sequence ontology: a tool for the unification of genome annotations. Genome Biol. 6, R44 (2005).
Acknowledgements
We acknowledge data contributed by the World Health Organization sequencing network (Supplementary Data 11). We acknowledge Claudio Köser for detailed and helpful feedback on the analysis and writing of the results. This work was partially funded by the Wellcome Trust: T.M.W. was supported by a Clinical Research Career Development Fellowship (214560/Z/18/Z). S.G.K. was supported by a National Science Foundation Graduate Research Fellowship DGE2140743. L.C. was supported by the MRC Centre for Global Infectious Disease Analysis (reference MR/X020258/1), funded by the UK Medical Research Council. P.M. and T.C.R received funding through The Foundation for Innovative New Diagnostics (FIND) under Unitaid grant 2019-32-FIND MDR. M.R.F. was supported by a National Institute of Allergy and Infectious Diseases / National Institutes of Health grant R01AI155765. The authors have applied a CC BY public copyright license to this publication. The views expressed by the authors do not necessarily reflect the views of the funding agencies.
Author information
Authors and Affiliations
Contributions
M.R.F. and T.C.R. conceived the study and supervised the research. S.G.K. implemented the code and performed the analyses. S.L. curated and shared data from contributors and provided feedback on methods. P.M., L.C. and T.M.W. provided guidance on methods and results. C-M.N. and N.I. provided analysis consultation from the World Health Organization. S.G.K. and M.R.F. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
T.C.R. received funding support from FIND through a service contract with UC San Diego. T.C.R. received grant funding from NIH to develop and evaluate a tNGS solution for drug resistant TB (R01AI176401). T.C.R. is a co-founder, board member and unpaid shareholder of Verus Diagnostics Inc. T.C.R. is a co-inventor on patents pertaining to tNGS. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Benjamin Sobkowiak, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kulkarni, S.G., Laurent, S., Miotto, P. et al. Multivariable regression models improve accuracy and sensitive grading of antibiotic resistance mutations in Mycobacterium tuberculosis. Nat Commun 16, 2149 (2025). https://doi.org/10.1038/s41467-025-57174-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57174-1
