
Abstract
Interphases are critical in electrochemical systems, influencing performance by controlling ion transport and stability. This study explores a metal-organic interphase in the electrocatalytic reduction of CO2 (CO2RR) on Cu, extending the concept of interphases to CO2 conversion. Investigating organic modifications on CuOx, we discover metal-organic interphases over 10 nm thick in highly ethanol-selective systems, contrary to the expected monolayer adsorption. Using an automated platform, 1080 CO2RR experiments with 180 molecular modifiers identify functional groups affecting selectivity for ethanol and multi-carbon (C2+) products. We find that these modifiers consistently produce metal-organic interphases on the Cu or CuOx surface. These interphases modulate Cu coordination, CO2RR intermediates, and interfacial water configuration, significantly improving electrocatalytic performance. Testing across 11 CuOx-based catalysts validates this approach, culminating in the development of two electrocatalysts that achieve ~80% faradaic efficiency for C2+ products with ethanol partial current densities up to 328 and 507 mA cm−2. This study highlights the pivotal role of interphases in CO2RR, advancing CO2 conversion technologies.
Similar content being viewed by others
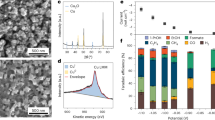
Introduction
The formation of interphases is integral to various electrochemical systems, such as lithium-ion batteries, where they can significantly influence performance by controlling ion transport and electrochemical stability and is crucial to the performance of electrochemical systems1,2,3,4. The interphase typically refers to the interfacial layer with certain thickness between the electrode and the electrolyte. Although there is massive research of interphase on lithium-ion batteries, there is little research in electrochemcial CO2 reduction (CO2RR).
CO2RR driven by renewable electricity offers significant opportunities to produce energy feedstocks and fuels5,6, with copper-based catalysts in CO2RR standing out for its ability to catalyze the formation of C2+ products7,8. Molecular tuning, which involves controlling the steric and electronic properties near active sites, shows great potential for directing CO2 electrolysis pathways9. By immobilizing organic molecules to interact with surface-bound intermediates, this method can help finetune the CO2RR activity and selectivity10 and establish structure-selectivity relationships11. Molecules with X(N/S/O) terminals, such as amines, thiols, and N-heterocyclic carbenes, are known to bind to the Cu surface and alter CO2RR product selectivity9,12,13,14,15,16,17,18,19. The hydrophobicity of the modified surface, especially in thiol-modified Cu catalysts, has been frequently emphasized15,20,21,22,23,24. However, a systematic investigation of various modifiers is needed to determine if other factors also play a significant role, and such investigation is still challenging.
In this study, we used our previously developed automatic electrocatalysis platform25 and systematically investigated 180 molecular modifiers, which either have N/S/O coordination sites or are water-insoluble, enabling them to remain on the Cu surface during electrolysis. This design is quite different from our previous work that used organic additives to tune catalyst preparation process26. We found that the S-terminal modifier shows a more significant C2+ selectivity dependence on molecular hydrophobicity/hydrophilicity than the others. More strikingly, for several of the catalysts highly selective for ethanol production, we observed the formation of metal-organic interphase over 10 nm in thickness instead of a monolayer of modification. This interphase may influence the species distribution in the electrochemical double layer, affecting CO2 and intermediate transport, interfacial water structure, and the exposed electrochemical facets, all of which are critical to the electrocatalytic process.
Results
Automated electrocatalysis experiments for screening organic modifiers on CuOx
The automatic electrocatalysis platform used in this work has undergone iterative optimization and updates since our previous publication25,27. The major updates include the installation of a catalyst replacement and positioning module (Supplementary Fig. 1) and a catholyte collection unit (Supplementary Fig. 2) for collecting CO2RR liquid products during the measurements. The system now features an automatic electrolysis unit with an automatically assembled flow cell, a catalyst replacement module, a compliant peristaltic pump for transporting 1.0 M KOH, a catholyte collection unit, and an online micro-fast chromatography (GC) for gas products analysis (Fig. 1a). The platform reduces assembly and testing time by nearly 10 times compared to commercial flow cells while simultaneously enhancing the precision and repeatability of evaluation results.

a Schematic illustration of automatic electrocatalysis platform for CO2RR. b Linear sweep voltammetry (LSV) curves without IR compensation of CuOx, 1,8-octanedithiol-CuOx, and 2-pyridinemethanethiol-CuOx catalysts. The cell resistances (R) were measured by electrochemical impedance spectroscopy (EIS) under open circuit potentials to be 1.2 ± 0.2 Ω after CO2RR. Figure 1 mainly focuses on data from the automated platform. Source data for Fig. 1b are provided as a Source Data file.
We selected CuOx catalysts synthesized by a facile solvothermal method as the substrate for molecular modification due to the superior CO2-to-C2+ performance of oxide-derived Cu (OD-Cu) compared to pure Cu in alkaline environments28,29,30,31. To modify the catalysts, we chose molecules containing N/S/O terminals that can coordinate to the CuOx surface through lone pair electrons or water-insoluble organics that bind via non-bonding interactions.
Most modifications led to a decrease of the total current density during electrolysis compared to bare CuOx, possibly due to a decreased area of exposed Cu and slightly increased charge-transfer resistance (Rct) (Supplementary Fig. 3), but the decreases in current density ranging from 11% to 18% over 0 to −1.2 V vs reversible hydrogen electrode (RHE) were mostly less than 20%, keeping the activity acceptable (Fig. 1b).
We calculated relative Faradaic efficiencies (FEs) by comparing the optimized FE of a molecule-modified CuOx with that of the unmodified CuOx catalyst as the following Eq. (1):
where i represents different products like C2H4, EtOH, and C2+ products.
The accuracy of the data collected by the automated platform (Fig. 1) was manually validated on a commercial flow electrolysis cell platform by selecting 1,8-octanedithiol CuOx for ethanol selectivity, 2-pyridinemethanethiol-CuOx for formate selectivity, and CuOx catalyst (Fig. 2).

FEs for all products over a CuOx catalyst, b 1,8-octanedithiol-CuOx catalyst, and c 2-pyridinemethanethiol-CuOx catalyst. d The ratio of faradaic efficiencies of ethanol and ethylene (FEEtOH/FEC2H4) and partial ethanol current density of CuOx and 1,8-octanedithiol-CuOx. e The C2+ FE plots of CuOx, 1,8-octanedithiol-CuOx, and 2-pyridinemethanethiol-CuOx catalysts. The experiments at all the applied current density were performed at least three times and the results were displayed as mean ± standard deviation. The data in Fig. 2 were collected manually on the commercial flow cell platform. Source data are provided as a Source Data file.
Modifiers promoting EtOH production
From the data collected by the automation platform (Fig. 1), it is worth noting that 15 S-terminal modifiers exhibited significantly enhanced selectivity for ethanol with relative FE above +70%. Among these, 1,8-octanedithiol achieved a maximum Faradaic efficiency for ethanol that was 2.4 times that of the CuOx catalyst at −0.62 V vs RHE (Supplementary Fig. 4). These 15 S-molecules maintained FEC2H4 while reducing FECO, suggesting a common catalytic pathway directing more *CO coupling toward ethanol. Notably, the CO2RR results manually verified on commercial flow cell platform (Fig. 1 and Supplementary Figs. 5 and 6) confirmed the enhancement of FEEtOH from 17% on CuOx to 29% on 1,8-octanedithiol-CuOx at applied current density of 400 mA cm−2, with a FEEtOH/FEC2H4 ratio of ~0.9, which is 2.3-fold higher than that over the pristine CuOx (~0.4). FECO was suppressed (31%–13% of CuOx vs 9%–2% of 1,8-octanedithiol-CuOx), and the FEC2+ was also enhanced from 36%–61% (CuOx) to 53%–65% (1,8-octanedithiol-CuOx) at applied current density ranging from 100 to 400 mA cm−2 (Fig. 2a, b, d, e). In contrast, the automatic electrocatalysis platform showed that another thiol-containing compound which inhibited FEC2+, 2-pyridinemethanethiol, increased FEformate, which was also confirmed on the commercial flow cell (from 8% on CuOx to 43% on 2-pyridinemethanethiol-CuOx) (Fig. 2c, e).
Hydrophobicity/hydrophilicity of the modifiers
Previous reports suggested hydrophobicity of the modifier can affect the hydrophobicity of the electrode for preserving water/gas interface, which is beneficial for generating C2+ products15,22,23,24. In search such relationship, we measured the contact angle of a water droplet on various modified electrodes using alkylthiols with various alkyl chain lengths, pyridinethiols, and aromatic thiols. However, we did not observe a correlation between the FEs with the contact angles either before or after the reaction (Supplementary Figs. 7 and 8). For example, bare CuOx has a contact angle of 145.9° before reaction and 98.7° after reaction, while the 1,8-octanedithiol modified electrode has a contact angle of 146.1° before reaction and 103.2° after reaction. Similarly, 2-pyridinemethanethiol, which leads to high FE for formate and low FE for C2+ products, has a contact angle of ~142.4° before reaction and 100.1° after reaction.
We categorized all modifiers into eight groups based on the type of N/O/S coordination terminals and the hydrophilicity/hydrophobicity of the connected functional groups (FGs) (Fig. 3a and Supplementary Table 1). The violin plot reflects data distribution, central tendency, and variability. Plotting the relative/absolute FE of CO2RR products for each molecular category highlights their regulatory effects on product selectivity. Statistical analysis of data of all the modifiers showed that hydrophobic FGs were more likely to promote ethylene/ethanol/C2+ production for molecules with similar coordination terminals (Fig. 3b–d). The ‘S terminal + hydrophobic FGs’ displayed the best promotion effect, but there were significant variations within each category. Some molecules with hydrophilic functional groups still promoted ethanol production, such as 4-nitrobenzaldehyde with relative FEEtOH of +86% and l-Cystine with relative FEEtOH of +72%. Only a few modifiers could increase FEEtOH while maintaining FEC2H4 at levels similar to bare CuOx. This indicates that factors other than hydrophobicity/hydrophilicity may influence the performance.

a Schematic illustration for the division rules of molecules tested according to types of coordination terminals and the hydrophilicity and hydrophobicity of functional groups. b Violin plot of relative FEC2H4 for each molecular category with a row of example molecular structures for each category (N/O/S terminals are plotted in blue, red, and yellow, respectively, hydrophobic groups in black, hydrophilic groups in red, and halogen atoms in green). c Violin plot of relative FEEtOH for each molecular category. d Violin plot of relative FEC2+ for each molecular category. e Violin plot of highest FEEtOH on tested S-molecules in different categories with a row of example molecular structures for each category. The gray dashed line represents the highest FEEtOH of the pristine CuOx catalyst. The embedded dashed black lines within each violin plot indicates the 25th and 75th percentiles with the median represented by the center solid black line. f SHAP analysis diagram based on the optimal combination of selected features in EtOH Faradaic efficiency using MLP model. The annotations connected by arrows in the figure are factor categories related to physicochemical property descriptors, and the remaining annotations connected by solid lines are related physicochemical property categories. Source data for Fig. 3b–e are provided as a Source Data file.
These data showed that the hydrophobicity of the functional groups in the modifier promotes ethanol but does not directly correlate with the hydrophobicity on the electrode level.
Machine learning analysis revealing factors influencing EtOH production
First focusing on sulfur-containing modifiers including 82 tested samples, we found that most S-modifiers enhanced FECH4, FEEtOH, and FEacetate but decreased FEC2H4 and FEn-propanol (Fig. 3e and Supplementary Figs. 9–12, Supplementary Table 2). We then used machine learning to analyze the dataset. The featurization was achieved using a combination of molecular fragment fingerprints (MFFs) and descriptors of molecular physicochemical properties (RDkit and LoFFi/LoFFi-MOE32) (see ‘Supplementary Methods’ section in the Supplementary Information for more details). Using various machine learning models, a pipeline of stepwise forward feature selection, cross validation, and SHAP analysis was established for analysis. To predict the FEs of each CO2RR product to be low or high, the best performance was achieved using a multi-layer perceptron (MLP) model with test set accuracy ranging from 60% to 79% (Supplementary Figs. 13–15, Supplementary Tables 3–7) and area under the receiver operating characteristic curve (AUC–ROC) ranging from 62% to 81%. The FEEtOH prediction by this model was 64% in accuracy and 70% in AUC–ROC for the test set.
The important features for each CO2RR product were identified by a SHAP analysis (Fig. 3f and Supplementary Figs. 16–23), and the interpretation of many of the physicochemical descriptors were achieved by correlating them to structural features of the molecules in the dataset using factor analysis (Supplementary Figs. 24–26 and Supplementary Tables 8–17), a new interpretation method tailor-designed for this work (see ‘Supplementary Discussion’ section in the Supplementary Information for more details). Using this method, we found several of the descriptors surprisingly represent something in the small dataset totally different from their intended general definition, an issue that was overlooked in previous works. For example, the size of the conjugation part of the molecules accidentally correlates with the presence of aniline in this small dataset, which can otherwise give misleading interpretation. A bunch of structural features beyond hydrophobicity (represented by LogP)15,22 was identified to promote EtOH production (Fig. 3f, Supplementary Figs. 24–26 and Supplementary Table 15):
-
(1)
Linear alkyl chains (represented by ‘Kappa2’) promoted ethanol production, while branched alkylthiols (‘VSA_EState8’) disfavored EtOH production, indicating critical steric effect around the thiol group.
-
(2)
Larger and round-shaped modifiers favored EtOH production (represented by ‘Long/Sum size ratio’, ‘Molecular Radius’, ‘Farthest Distance’, ‘Molecular Size Long’, ‘Num_Rotatable_Bonds’).
-
(3)
Disulfide, ‘Aryl bromide’ and ‘Aryl fluoride’ (represented by ‘Mid-Fun_Partial Equalization of Orbital Electronegativities (PEOE)-Charge_Sum’) disfavored EtOH production as compared to thiol-groups, possibly due to poor coordination capability of them.
-
(4)
The presence of N/O atoms (‘NO count’, ‘NHOH count’, ‘VSA_EState7’), C = S/thiazole (negatively correlated to ‘Atom1_ESP Maximal value’ and ‘Atom1_ESP Pi’33,34,35), or aniline (related to ‘Num of Conju-All-Atoms’ and ‘Conju-Part Wt (Max Conju)’), all related to hydrophilicity, reduces ethanol production.
-
(5)
The presence of S = O (‘PEOE_VSA12’) that is also hydrophilic positively impact the FEEtOH, violating the hydrophobicity requirement.
The hydrophobicity of the functional groups at the microscopic level of the S-modifier is an important but not the only factor affecting the selectivity for ethanol. This complexity challenges the simplistic understanding of hydrophobicity-promoted ethanol production. The role of the hydrophobicity of functional groups in S-modifiers in enhancing ethanol selectivity is discussed in the following sections.
Observation of metal–organic interphase
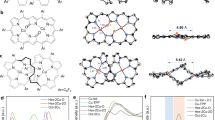
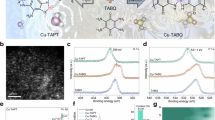
To further investigate the structure of the CuOx-electrode modified by different molecules, we performed transmission electron microscopy (TEM) imaging and elemental energy-dispersive X-ray spectroscopy (EDX) mapping. Surprisingly, we observed a continuous interphase over 10 nm in thickness on CuOx modified by 1,8-octanedithiol-CuOx, instead of a single layer of coordinated 1,8-octanedithiol (Fig. 4a–f). EDX mapping showed that the interphase contains both Cu and S. Scanning electron microscopy (SEM) images before or after CO2RR (Supplementary Figs. 27 and 28) showed minimal changes in macroscopic morphology under the modification of 1,8-octanedithiol, except for the surface becoming smoother. Scanning transmission electron microscopy (STEM) by spherical aberration corrected electron microscopy (Fig. 4g–l and Supplementary Fig. 29) further revealed Cu-rich areas of a few nanometers intercalated with regions deficient in Cu. The distribution of S was more uniform. This is consistent with a picture of very small Cu-based nanoparticles or nanoclusters, coordinated by the 1,8-octanedithiol-CuOx catalyst, aggregated into an interphase of >10 nm thickness. This 1,8-octanedithiol-Cu interphase remained after the CO2RR reaction (Supplementary Fig. 30). To probe whether this interphase is porous and electrochemically active, we added HAuCl₄ to the KOH electrolyte during the electrolysis using 1,8-octanedithiol-CuOx catalyst. The Au reduced from AuCl₄− at the negative potential on the electrode can serve as a probe for EDX mapping to explore the accessibility of the interphase during CO2RR. We observed significant Au electrodeposition on Cu in the interphase and on the Cu surface buried under the interphase after CO2RR (Supplementary Figs. 31 and 32), together with K distribution in the interphase. These observations suggested that the interphase is porous and the Cu within the interphase and buried below the interphase are both electrochemically active.

a, b TEM image of as prepared 1,8-octanedithiol-CuOx catalyst, c HAADF image of 1,8-octanedithiol-CuOx catalyst and corresponding EDX mapping results of d Cu, e S elements, and f overlapping plot of Cu and S elements. g DF-STEM image. h The enlarged BF-STEM image of 1,8-octanedithiol-CuOx of the red frame in (g). i Magnified DF-STEM image of a localized region within the interphase for 1,8-octanedithiol-CuOx and its corresponding EDX mapping of j Cu, k S, and l Cu and S overlapping. m, n TEM image of as prepared 2-pyridinemethylthiol-CuOx catalyst, o HAADF image of 2-pyridinemethylthiol-CuOx catalyst and corresponding EDX mapping results of p Cu, q S elements, and r overlapping plot of Cu and S elements. s XRD patterns of CuOx, 1,8-octanedithiol-CuOx, and 2-pyridinemethanethiol-CuOx catalysts at open circuit potential (OCP) and after CO2RR. t XPS spectra of Cu 2p of as prepared CuOx, 1,8-octanedithiol-CuOx, and 2-pyridinemethanethiol-CuOx catalysts. XPS spectra of S 2p (The solid line represents S 2p3/2, the dashed line represents S 2p1/2) of as prepared u 1,8-octanedithiol-CuOx catalyst, and v 2-pyridinemethanethiol-CuOx catalyst. Source data for Fig. 4s–v are provided as a Source Data file.
The thickness and composition of interphase on 1,8-octanedithiol-CuOx catalyst would be affected by electrolyte and temperature. We used TEM and EDX mapping to examine 1,8-octanedithiol-CuOx catalysts after CO2RR in 1.0 M KOH, 0.1 M KHCO3 and 1.0 M KCl at room temperature and 80 °C, respectively (Supplementary Figs. 30, 33–37). Decreasing the pH or increasing temperature in electrolysis destabilized the interphase on 1,8-octanedithiol-CuOx catalysts. Related to this, the relative ethanol FEs of 1,8-octanedithiol-CuOx catalysts in 1.0 M KOH at room temperature (~10 nm interphase consisting of Cu and S), 1.0 M KOH at 80 °C (~10 nm interphase consisting of Cu and O), 0.1 M KHCO3 at room temperature (~10 nm interphase consisting of Cu and S), 0.1 M KHCO3 at 80 °C (no obvious interphase), 1.0 M KCl at room temperature (no obvious interphase), and 1.0 M KCl at 80 °C (no obvious interphase) were +144%, +79%, +91%, +52%, +55% and +30%, respectively, at the applied current density of 100 mA cm−2 (Supplementary Figs. 38 and 39).These results are consistent with the notion that the presence of ~10 nm interphase consisting of Cu and S elements on 1,8-octanedithiol-CuOx catalyst correlated with the enhancement of FEEtOH.
The interphase is not unique to 1,8-octanedithiol. For example, 1-octanethiol, a FEEtOH promoter, also formed an interphase of similar thickness (Supplementary Fig. 40). In contrast, 2-pyridinemethanethiol, a FEformate promoter, has a layer thickness of ~1.4 nm (Fig. 4m–r). Although this value is also thicker than a single molecular layer, considering the complexity of metal/metal oxide surface and the measurement inaccuracies, it may still correspond to a monolayer modification.
Interestingly, for 1-octadecanethiol-CuOx, we observed mostly an organic phase over 100 nm in thickness with undetectable Cu signals (Supplementary Fig. 41), which may explain why 1-octadecanethiol lacks promoting effect seen with other alkylthiols. The 1-octadecanethiol also decreased the water droplet contact angle on the electrode to 122.4°, possibly due to oxidized thiol group on the surface of the organic layer (Supplementary Fig. 7j). We further changed the amount of 1-octadecanethiol to tune the interphase thickness (Supplementary Figs. 42 and 43). When adding only 50% of the initial amount of 1-octadecanethiol, we observed the interphase thickness decreases to 60 nm, and the relative FEEtOH increased to +94% (Supplementary Figs. 42 and 44). When further decreasing the 1-octadecanethiol to 15% of its initial value, an incomplete interphase coverage of the 15%-1-octadecanethiol-CuOx was observed, and the relative FEEtOH decreased to 57% (Supplementary Figs. 43 and 44). This shows that the thickness of the interphase is also critical to the performance.
Further examination of 1,4-benzenedithiol, tritylmethanethiol, 2,2′:5′,2′′-terthiophene, l-cystine, 4-chlorobenzyl alcohol and d(+)-tryptophan modified CuOx did not reveal a clear metal-organic interphase (Supplementary Figs. 45–50). None of these were effective FEEtOH promoters. However, 4-nitrobenzaldehyde, which lacks thiol groups presented a ~ 50 nm thick interphase (Supplementary Fig. 51) and achieved a relative FEEtOH of +86%, making it a decent FEEtOH promoter.
For a more controlled comparison to 1,8-octanedithiol, we prepared CuOx modified by 1,8-octanediol and 1,8-diaminooctane. TEM and EDX mapping showed that none of them formed a complete interphase around the CuOx particles (Supplementary Figs. 52–54). The modification of 1,8-octanediol on CuOx mainly increased FECO and decreased FEC2H4 and FEEtOH at the applied current density of 100 mA cm−2 (Supplementary Fig. 55). On the other hand, the 1,8-diaminooctane-treated CuOx can only achieve a relative FEEtOH of 44%, as compared to 144% for 1,8-octanedithiol-treated CuOx at the applied current density of −100 mA cm−2 (Supplementary Fig. 55). This shows the importance of the coordination group in forming the metal-organic interphase and tuning the CO2RR activity and selectivity.
Based on these results, we hypothesize that the formation of an effective interphase for FEEtOH enhancement requires:
-
(1)
The presence of Cu-coordination terminals.
-
(2)
Reductive functional groups that can react with surface Cu2+ to release Cu+ for interphase formation.
-
(3)
Multiple coordination sites for forming coordination networks, or the presence of both a coordination site and hydrophobic alkyl chains of proper length for forming a bi-continuous phase.
Characterization of interphase vs. monolayer modification
We characterized the interphase vs. monolayer modification using 1,8-octanedithiol-CuOx and 2-pyridinemethanethiol-CuOx as examples (Supplementary Notes). The S content determined by inductively coupled plasma optical emission spectrometry (ICP-OES) was about 1.12 wt% and 0.49 wt% for the two samples, respectively (Supplementary Table 19). Fourier transform infrared spectroscopy (FTIR) of both the samples confirmed the presence of the thiol molecules and the disappearance of the S–H vibration signal at 2563 cm−1 for 1,8-octanedithiol and 2552 cm−1 for 2-pyridinemethanethiol (Supplementary Fig. 56), suggesting the formation of S–Cu bonds. X-ray diffraction (XRD) did not reveal any crystalline phase other than Cu2O for both samples, indicating that the interphase may be amorphous. After the catalysis, XRD showed that Cu2O partially converted to Cu0 (Fig. 4s).
X-ray photoelectron spectroscopy (XPS) measurements showed that modification with 1,8-octanedithiol and 2-pyridinemethanethiol converted CuII (Cu 2p3/2 at 934.8 eV) in CuOx to CuI. The CuI peak position shifted from Cu 2p3/2 at 932.6 eV and Cu LMM at 570.0 eV in bare CuOx to 933 eV and 571 eV in the modified samples (Fig. 4t and Supplementary Fig. 57), likely due to the formation of CuI–S bonds. Additionally, S 2p spectra (Fig. 4u, v and Supplementary Table 20) confirmed the presence of metal-bound thiol (S 2p3/2 at 162.8 eV), unbound thiol (S 2p3/2 at ~164 eV) and oxidized sulfur (S 2p3/2 at ~168–169 eV)11,36. The unbound thiol in Fig. 4u, v refers to disulfide36. After CO2RR, the metal-bound thiol accounted for ~60% on 1,8-octanedithiol-CuOx surfaces, while the oxidized sulfur accounted for ~62% on 2-pyridinemethanethiol-CuOx surface, reflecting the higher Cu–S binding strength of 1,8-octanedithiol (Supplementary Fig. 58 and Supplementary Table 21). Cu LMM spectra of catalysts after electrolysis demonstrated partial transformation from oxide Cu to metallic Cu0 (Supplementary Fig. 59), noting that Cu0 can be re-oxidzed to CuI during sample transfer for testing. Based on previous literature reports37,38,39, the CuO on the surface of CuOx first undergoes a redox reaction with the reducing –SH terminal of alkylthiols (RSH), resulting in the formation of CuI and disulfide (RS–SR), which became the disordered stacking of the organic part in the interphase. Subsequently, the CuI species, originating both from Cu2O and reduced CuO, further coordinate with excess alkylthiol molecules on the CuOx surface, leading to the formation of multilayer metal-thiol (CuI–S) compounds, which is the metal coordination layers in the interphase. The reducing and coordinating abilities of the S terminals can be modulated by the attached functional groups. For instance, pyridinethiol is less reducible due to the electron-withdrawing effect of the pyridine ring. Other molecular modifiers with terminal groups that possess reducing and coordinating capabilities can also form metal–organic interphases, as discussed in the ‘Observation of metal–organic interphase’ section.
The interphases may contain metal coordination layers and disordered stacking of the organic part. This explains many of the experimental observations. First, the hydrophobicity may encourage the stacking of the modifier-Cu complex or nanostructure in water, helping in the formation of the metal-organic interphase40,41. Second, the reason why aromatic thiols are less effective than alkyl thiols in promoting ethanol production may be due to the rigidity of aromatic rings, which presents a barrier to form disordered continuous interphase. Third, the beneficial effect of large molecular size and round shape of the molecule to ethanol production may be related to the tendency of large and round molecules to stack into flexible and porous interphase via van der Waals interaction. Fourth, the steric effect around the thiol-group may interfere with its ability to form multiple S‒Cu bonds for forming a continuous interphase.
In situ Raman spectroscopy
We conducted in situ Raman spectroscopy in a flow cell with continuously pumped CO2 and 1.0 M KOH, scanning from open circuit potential (OCP) to −0.8 V vs. RHE (Supplementary Fig. 60). The Raman spectra confirmed that 1,8-octanedithiol remained on the catalyst during catalysis, as evidenced by persistent and enhanced (surface plasmonic effect of Cu) vibrational bands of the molecule at 630, 697, 1015, 1078, 2855 and 2924 cm−1. These bands correspond to vibrational modes of v(C–S)G, v(C–S)T, v(C − C)G, v(C–C)T, vs(−CH2) and vas(−CH2), where subscript ‘G’ or ‘T’ denote trans or gauche conformations) (Supplementary Figs. 61, 62). Notably, the S − H stretching mode was absent throughout the process, indicating that the Cu–S linkage remained intact. The vibrational peak positions and relative intensities suggested that the molecules primarily adopted a trans conformation, differing from the mixed conformation in the molecular solid42,43.
Both CuOx and 1,8-octanedithiol-CuOx exhibited 2–3 distinct C≡O stretching bands of adsorbed *COatop between 2040 and 2010 cm−1. Two of these bands can be assigned to *COHFB (~2090 cm−1) and *COLFB (~2050 cm−1) following literature convention (Fig. 5a, b)44,45. The Raman peak area ratio (A*COHFB/A*COLFB) of *COHFB and *COLFB was significantly higher for 1,8-octanedithiol-CuOx compared to CuOx, and this ratio increased at more negative potentials for 1,8-octanedithiol-CuOx, whereas it decreased for CuOx below −0.4 V vs RHE (Fig. 5c). The *COHFB band is typically associated with low coordination sites, which favor C1 product generation, while *COLFB is associated with terrace sites, which favor further *CO reduction and C‒C coupling45,46,47. The higher *COHFB/*COLFB ratio for 1,8-octanedithiol-CuOx may reflect that *COLFB is consumed more rapidly in downstream conversion processes than *COHFB. The *COHFB and *COLFB peak positions shifted as a function of voltage due to the stark effect from the local electric field within the electrochemical double layer (EDL). The Stark slopes of 1,8-octanedithiol-CuOx (14.3 cm−1 V−1 for *COHFB and 29.6 cm−1 V−1 *COLFB) were close to that of CuOx (17.0 cm−1 V−1 for *COHFB and 20.3 cm−1 V−1 *COLFB) (Supplementary Fig. 63).

In situ Raman spectroscopy in the region from 1900 to 2200 cm−1 for a CuOx and b 1,8-octanedithiol-CuOx catalyst in flow cell with continuously pumped CO2 and 1.0 M KOH from open circuit potential (OCP) to −0.8 V vs RHE. Although there is some debate on whether the nature of the *CO band at ~2000 cm‒1 is attributed to *CObridge, this does not affect our analysis. c Area ratio of *COHFB to *COLFB over applied potential window calculated from in situ Raman spectroscopy. In situ ATR-SEIRAS spectra in the region of 2200 to 1700 cm−1 for d CuOx, e 1,8-octanedithiol-CuOx catalyst, and f 2-pyridinemethanethiol-CuOx catalyst from 0 to −1.4 V vs RHE in CO2-saturated 0.5 M KHCO3. In situ ATR-SEIRAS spectra in the region of 3800 to 2800 cm−1 for g CuOx, h 1,8-octanedithiol-CuOx catalyst, and i 2-pyridinemethanethiol-CuOx catalyst from 0 to −1.4 V vs RHE in CO2-saturated 0.5 M KHCO3. The potentials converted to RHE were presented as non-iR corrected values. Source data are provided as a Source Data file.
We observed an interfacial water band at 3364 cm−1 only on CuOx, possibly thanks to surface-enhanced Raman effect by metallic Cu. However, we did not observe interfacial water signal on 1,8-octanedithiol-CuOx, suggesting that the water is further away from the Cu surface and fails to gain enough enhancement in the presence of the interphase. Due to intense hydrogen production on the 2-pyridylmethanethiol-CuOx surface, the Raman signals were significantly affected (Supplementary Fig. 64), preventing detailed analysis.
In situ IR spectroscopy
We also conducted in situ attenuated total reflectance surface enhanced infrared absorption spectroscopy (ATR-SEIRAS) from 0 to −1.4 V vs RHE in CO2-saturated 0.5 M KHCO3 in a single cell (Supplementary Fig. 65). Although performed at a different pH from the catalytic condition, ATR-SEIRAS results were consistent with in situ Raman. The bands of *COatop on pristine CuOx (Fig. 5d) comprised of *COHFB (~2075 cm−1) and COLFB (~2024 cm−1)44. The different wavenumbers compared to the Raman data are likely due to pH differences. A *CObridge signal at ~1800 cm−1 was also observed on CuOx under this condition48. Modification with 1,8-octanedithiol and 2-pyridinemethylthiol both inhibited the formation of *CObridge (Fig. 5e, f) whose role in C–C coupling was still controversial49,50. The modification likely reduced the number of available bridge sites. Similar to in situ Raman results, significantly more *COHFB than *COLFB was observed on 1,8-octanedithiol-CuOx (Fig. 5e), while *COLFB was prevalent on 2-pyridinemethylthiol-CuOx (Fig. 5f).
The bidentate CO(*)O(*)− species (~1400 cm−1) with oxygen coordinated on Cu, an intermediate for HCOOH generation, and C-bound intermediate *COOH (~1380 cm−1), an intermediate for *CO production, were both observed on CuOx and 2-pyridinemethylthiol-CuOx (Supplementary Figs. 66 and 67)51,52. These two peaks grow at a similar rate on CuOx as the potential scanned negatively, while CO(*)O(*)− intensity increased much faster than *COOH on 2-pyridinemethylthiol-CuOx. In contrast, only *COOH was observed on 1,8-octanedithiol-CuOx (Supplementary Fig. 68), suggesting it suppresses the HCOOH formation pathway53. The dissociation of *COOH to generate *CO is also considered to be one of the important rate-determining steps affecting the *CO coverage and C–C coupling54.
The band between 3000 and 3800 cm−1 represents the stretching vibration mode of the O–H bond, associated with the hydrogen bonding environments of the interfacial water55,56. Peaks at ~3250 cm−1, ~3450 cm−1, and ∼3550 cm−1 correspond to strong hydrogen bond (HB) networks comprising 4-coordinated water (4-HB⋅H2O), weak HBs of 2-coordinated water (2-HB⋅H2O), and isolated water, respectively57. The surface modification by both 1,8-octanedithiol and 2-pyridinemethanethiol increased the percentage of isolated water (Supplementary Figs. 69a, 70a, 71a)58.
The modification of 1,8-octanedithiol reduced the Stark slopes from 53.9 cm−1 V−1 (isolated water), 97.9 cm−1 V−1 (2-HB⋅H2O) and 74.2 cm−1 V−1 (4-HB⋅H2O) on CuOx (Fig. 5g and Supplementary Fig. 69b) to 16.9 cm−1 V−1, 29.5 cm−1 V−1 and 23.1 cm−1 V−1 (Fig. 5h and Supplementary Fig. 70b), respectively59,60. This reduction of the slopes by ~3.1-3.3 times is very similar to the measured decrease in electrical double-layer (EDL) capacitance (1.24 mF cm−2 for 1,8-octanedithiol-CuOx vs 4.11 mF cm−2 for CuOx, showing a ~3.3-fold reduction, Supplementary Fig. 72). Both the reduction in Stark slopes of surface water and EDL capacitance can be attributed to the increase in EDL thickness after modification. The change in dielectric constant of the EDL may also contribute to the difference.
The presence of 2-pyridinemethanethiol also decreased the Stark slopes of isolated water (33.3 cm−1 V−1), 2-HB⋅H2O (49.9 cm−1 V−1) and 4-HB⋅H2O (21.3 cm−1 V−1), smaller than the CuOx values by 1.6–3.3 times (Fig. 5i and Supplementary Fig. 71b), while the EDL capacitance ratio of CuOx to 2-pyridinemethanethiol-CuOx was 2.4 times (Supplementary Fig. 72).
The different trends of the Stark slope changes between interfacial water and adsorbed *CO intermediates may be due to the fact that *CO is directly adsorbed on Cu, while interfacial water is at a distance from the Cu site in the EDL. It is reported that a lower stark slope represents a longer distance from the metal surface and/or a more restricted water molecule with a slower reorientation58,59,60.
Exploring exposed Cu facets via OH electro-adsorption
OH electro-adsorption measurements, which are more surface-sensitive, showed adsorption peaks at ~0.37 V, ~0.42 V and ~0.48 V vs RHE on bare CuOx-derived catalyst, corresponding to adsorption on Cu(100), Cu(110), and trace amount of Cu(111) facets, respectively61. Modification of 2-pyridinemethylthiol left an adsorption peak at ~0.48 V for Cu(111) during electrolysis, while the 1,8-octanedithiol-CuOx showed a dominant adsorption peak at ~0.42 V for Cu(110) (Fig. 6a), and maintaining a 2-fold higher Cu(110):Cu(100) ratio than CuOx after CO2RR (Supplementary Fig. 73). Previous reports indicate that Cu(111) favors C1 product formation, Cu(100) favors C2H4, and Cu(110) favors C2+ oxygenates (acetate and ethanol)62. Therefore, favorable Cu(110) activity of 1,8-octanedithiol-CuOx is consistent with its promotion of ethanol. Density functional theory (DFT) calculation showed that 1,8-octanedithiol anchored the Cu surface through Cu–S bonds and can interact with Cu atom at the sublayer on Cu(110) (Fig. 6b), while 1,8-octanedithiol only coordinated to the outermost layer on Cu(100) (Fig. 6c), which might leave more exposed Cu sites on Cu(110) than Cu(100). Moreover, the average adsorption energies of 1,8-octanedithiol on Cu(100) and Cu(110) are −3.66 eV and −3.36 eV, respectively. Such a small difference in adsorption energy is within the DFT calculation error. We also considered solvent effect on catalyst state during electrolysis (Supplementary Fig. 74). Water molecules outside the alkyl chain did not influence the coordination of 1,8-octanedithiol on Cu(100) and Cu(110) but change the average adsorption energies to −3.08 eV and −3.50 eV for Cu (100) and Cu (110), respectively, with the difference being minimal and within the margin of DFT error. The modifiers occupying and blocking certain facets is consistent with CO stripping experiments (Fig. 6d), which showed reduced CO stripping peak areas and thus reduced amount of adsorbed *CO after modification. However, this facet analysis using OH-adsorption peaks should be considered cautiously, as surface modification of the same facet can very likely shift the peak position. Moreover, the Cu surface under reaction condition is quite dynamic. Surface reorganization can quickly transform one facet to another upon adsorption of intermediates like *CO and *H during the reaction.

a OH− electro-adsorption measurements of CuOx, 1,8-octanedithiol-CuOx (current density was magnified 1.5 times) and 2-pyridinemethanethiol-CuOx catalysts in Ar-saturated 1.0 M KOH after 200 s reduction at −0.69 V vs RHE. b The side view of the 1,8-octanedithiol coordinated on Cu(110). c The side view of the 1,8-octanedithiol coordinated on Cu(100). Atomic color codes: Cu (brown), S (yellow), C (gray), O (red), and H (white). d CO stripping of CuOx, 1,8-octanedithiol-CuOx and 2-pyridinemethanethiol-CuOx catalysts. e Correlation coefficients and hierarchical clustering results between FE of each CO2RR product. Comparison of CO2RR and CORR product distributions on f CuOx, g 1,8-octanedithiol-CuOx, and h 2-pyridinemethanethiol-CuOx catalysts at applied current density of 100 mA cm−2 and 200 mA cm−2, respectively. The experiments of CO2RR at all the applied current density were performed at least three times and the results were displayed as mean ± standard deviation. The CORR data was tested once. The potentials converted to RHE were presented as non-iR corrected values. Source data for Fig. 6a, d, f–h data are provided as a Source Data file.
Possible consequences of forming a metal–organic interphase
On the one hand, the metal–organic interphase would change interfacial water structure and H transfer process. The presence of the interphase likely disrupts the interfacial water structure. As revealed by the water O‒H stretching band in the in situ Raman and IR measurements, water molecules may be further away from the Cu surface in the interphase. Moreover, upon thiol group modification, the percentage of isolated water increased, promoting H transfer from H2O to Cu or species on Cu surface. This explains why the majority of S-containing modifiers, especially pyridinethiols, promoted H2 evolution (up to FEH2 ~ 100%) (Supplementary Fig. 75). Consistent with this, the pyridinethiols enhanced formate formation, which also requires H addition (Supplementary Figs. 76 and 77). We noticed a correlation between FEformate and FEH2 among the different modifiers (Pearson correlation coefficient of 0.35) (Fig. 6e), indicating H-transfer is part of the rate-determining step of formate formation. There is also a correlation between FECH4 and FEs of C2H4 and EtOH (Pearson correlation coefficients of 0.44–0.49). The formation of CH4, C2H4, and EtOH all require the addition of H onto *CO as a common step. Their correlation suggests that the H-to-*CO transfer is a critical step in these systems.
These arguments converge into a coherent picture regarding interfacial water: the modifiers on Cu surface in general disrupts the regular hydrogen-bonded interfacial water structure and increases the percentage of isolated water. These water molecules are prone to provide H to the Cu surface to increase surface *H occupation as well as H addition to surface species, leading to increased H2 generation. On the other hand, the formation of a metal-organic interphase presents some restrictions on the interfacial water and pushing them further away from the Cu surface. This hinders the H-to-Cu transfer (to generate *H) and also decreases H-to-*CO transfer rate but to a less degree, balancing *CO hydrogenation and C‒C coupling rates to enhance EtOH formation.
On the other hand, the metal-organic interphase would affect CO adsorption and C‒C coupling. To explore the effect of *CO coverage, we switched the reactant from CO2 to CO to increase CO adsorption (Fig. 6f–h). For both bare CuOx and 2-pyridylmethanethiol-CuOx, FEs of all C2 compounds (ethylene, ethanol and acetate) increased with no specific selectivity for a single C2 product. However, abundant *CO on 1,8-octanedithiol-CuOx primarily enhanced FEacetate with only slight promotion or even inhibition for other C2 products at 100–200 mA cm−2, which is consistent with several literature results of promoted FEacetate in CORR63,64,65,66.
This indicates that high *CO occupation does not lead to high FEEtOH or FEC2H4 for 1,8-octanedithiol-CuOx. This would be consistent with the limited H-to-*CO transfer rate in the interphase of 1,8-octanedithiol-CuOx. As the *CO occupation is high, the H-to-*CO transfer is not fast enough to generate highly hydrogenated products like CH3CH2OH and C2H4 and thus promotes the less hydrogenated CH3CO2− as the product.
However, under CO2RR condition, 1,8-octanedithiol modification induces faster *COLFB conversion than *COHFB conversion upon scanning to negative potential as shown in the Raman spectra. The Raman peak position of the CO stretching modes did not change as compared to bare CuOx, suggesting unaltered adsorption strengths and back-bonding to the *CO. The *COLFB/*COHFB conversion rate difference is likely due to different rate of H-transfer to *CO. Notably, OH electro-adsorption measurement showed that the interphase in 1,8-octanedithiol-CuOx left the Cu (110) facet accessible and blocked the other facets, which can be responsible for this *COLFB vs *COHFB difference as well as the EtOH selectivity.
FEEtOH and FEC2H4 among different modifiers are strongly correlated (Pearson correlation coefficient of 0.80 for S-modifiers) (Fig. 6e), suggesting that ethylene and ethanol likely share a common formation pathway and only diverge at the end of the electrochemical process67,68,69. C2H4 formation can occur on multiple pathways, the most referenced being through H2C = CHO* + H+ + e− → H2C = CH2 + O*, which is associated with the formation of surface-bound O*67,70. In contrast, the Cu surfaces of weaker oxygen affinity promote C2H5OH formation by preserving the C–O bond71. Based on the different ratios of C-absorbed *COOH and O-absorbed CO(*)O(*)− on CuOx and that modified by 1,8-octanedithiol and 2-pyridinemethylthiol (Supplementary Figs. 66 and 68), the order of oxygen affinity on their surfaces can be inferred: 1,8-octanedithiol-CuOx < CuOx < 2-pyridinemethylthiol-CuOx. The surface modification, change of interfacial water structure, preservation of a certain facet may interfere with the O* formation, responsible for the general modification effect on CH3CH2OH/C2H4 ratio as compared to bare CuOx.
In addition, the similar position of electrochemical CO stripping peaks of CuOx with or without modification indicate that the Cu–CO adsorption strengths on the three catalyst surfaces are similar.
Applying 1,8-octanedithiol to other CuOx-based catalysts
We further used the discovered 1,8-octanedithiol modifier to treat 11 different CuOx-based CO2RR catalysts to boost ethanol production on these catalysts. Some of these CuOx-based catalysts were synthesized according to literature reports25,72,73,74,75, while others were synthesized using our own methods (detailed synthesis methods are listed in the Supplementary Information). Interestingly, the 1,8-octanedithiol modifier enhanced FEEtOH and FEC2+, as well as the FEC2H4/FEEtOH ratio, in all of them, indicating a general promoting effect (Fig. 7a and Supplementary Table 22). Notably, the promotion of Cd-modified CuOx (Cd@CuOx) and Eu-modified CuOx (Eu@CuOx) achieved 80% FEC2+ (jC2+ = 640 mA cm−2) with jEtOH of 328 mA cm−2 at −0.77 V vs RHE and 76% FEC2+ (jC2+ = 988 mA cm−2) with jEtOH of 507 mA cm−2 at −0.85 V vs RHE, respectively, which are comparable or better than recently reported state-of-the-art performance of CO2-to-EtOH/C2+ conversion using Cu-based electrocatalysts (Fig. 7b and Supplementary Table 23).

a The 1,8-octanedithiol functionalization approach verified on diverse oxide derived Cu substrates synthesized by 11 methods and the optimized CO2-to-EtOH/C2+ performance. b Comparison of CO2RR-to-EtOH/C2+ with state-of-the-art Cu-based catalysts reported recently in 1.0 M KOH flow cell. The experiments with error bars were performed at least three times and the results were displayed as mean ± standard deviation. Source data are provided as a Source Data file.
We also conducted characterizations on Cd@CuOx and Eu@CuOx catalysts with or without 1,8-octanedithiol modification. The modification of 1,8-octanedithiol did not change the phase structure (Supplementary Fig. 78) or electrode surface hydrophilicity/hydrophobicity (Supplementary Fig. 79) of Cd@CuOx and Eu@CuOx catalysts. Although the electrochemical facets compositions of Eu@CuOx and Cd@CuOx catalysts are slightly different, the selectively exposed Cu(110) electrochemical facet on 1,8-octanedithiol-Eu@CuOx and 1,8-octanedithiol-Cd@CuOx can be clearly observed in the OH electro-adsorption measurement (Supplementary Fig. 80). Importantly, the modification also formed >10 nm metal–organic interphase (Supplementary Figs. 81–84). The XPS results of these two optimal CO2-to-EtOH catalysts demonstrated the conversion of CuII to CuI after 1,8-octanedithiol modification and the presence of CuI-S bonds (Supplementary Figs. 85–88).
Discussion
In summary, we compiled a comprehensive dataset comprising 1080 experimental data points on CuOx modified by 180 organic compounds featuring coordinated N/S/O terminals or exhibiting water-insolubility. Our analysis highlights the structural prerequisites of modifiers that enhance ethanol production via the carbon dioxide reduction reaction (CO2RR). Notably, 1,8-octanedithiol emerged as one of the most effective promoters, boosting ethanol production across 11 distinct CuOx-based catalysts.
A pivotal discovery in the study is that modifiers conducive to ethanol production universally induce the formation of metal-organic interphases thicker than 10 nm. This contrasts starkly with non-promoting modifiers, which typically generate monolayer adsorption approximately 1 nm thick on Cu surfaces. Such thick metal–organic interphases, exemplified by the highly effective 1,8-octanedithiol, selectively expose the Cu(110) facet while blocking others. This facet-specific exposure is crucial as the Cu(110) is particularly favorable for ethanol production. In situ Raman and IR spectroscopy further demonstrated that low-frequency atop CO (*COLFB) was consumed more rapidly than the high-frequency one (*COHFB) in the presence of the interphases. The interphase which also disrupted the interfacial water structure, likely altering the hydrogen transfer rate to the Cu surface, a critical factor in controlling product selectivity.
By establishing a standardized database and employing machine learning techniques, we have begun to extract key features that impact structure-activity relationships, paving the way for targeted development of high-performance catalysts. Our findings on the role of metal–organic interphases in promoting ethanol production provide insights that could influence further research in catalysis and surface science. This systematic data science approach uncovers underlying dynamics of complex catalytic systems and contributes to the field of advanced catalyst development.
Methods
Chemicals and materials
Glyphosate (98%) was purchased from Jinan Henghua Sci. & Tec. Co. Ltd. 2,2’-Bipyridine-4,4’-dicarboxylic acid (98%) was purchased from Accela ChemBio Co. Ltd. Dimethylglyoxime (98%), l(+)-ascorbic acid (99.7%), l-glutamic acid (98.5%), l-cysteine (98.5%), thiourea (99%), thioacetamide (99%), Cu(NO3)2·3H2O (99%), CuCl (97%), CuCl2·2H2O (99%), potassium bicarbonate (KHCO3, 99.95%), potassium hydroxide (KOH, 99.99%) and N,N-dimethylformamide (DMF, 99.5%) were purchased from Sinopharm Chemical Reagent. 2,2’-Bipyridine (99%), 5-aminovaleric acid (98%), 4-aminobutyric acid (98%), 1-octene (99%), 2-fluorobenzoic acid (98%), 4-formylbenzoic acid (97%), 2-phenylpyridine (98%), 1,4-benzoquinone (99%), 4-chlorobenzonitrile (99%), 4-mercaptopyridine (98%), 2,2’:5’,2”-terthiophene (99%), 7-diethylamino-3-thenoylcoumarin (97%), glutathione (98%), l-threonine (99%), and 4-cyanopyridine (98%) were purchased from J&K Chemicals Reagent company. Acetohydroxamic acid (98%), 3-chloro-1-propanol (98%), 3-(carboxymethyl)benzothiazolium bromide (97%), 4,4’-dipyridine disulfide (98%), 2-pyridinemethanethiol (93%), 2,3-butanedithiol (98%), 1,8-octanedithiol (98%), 1,4-butanedithiol (98%), 4-chlorobenzyl alcohol (98%), 5,10,15,20-tetra(4-pyridyl)-21H,23H-porphine (97%), l(+)-2-aminobutyric acid (97%), 2-cyano-5-methylpyridine (98%), 4-nitrobenzaldehyde (97%), 6-quinolinecarboxylic acid (98%), cis-4-hydroxy-D-proline (95%), 1-mercaptooctane (98%), thiophene (99%), tetrahydrothiazolyl thione (98%), 2,2’-diaminodiphenyl disulfide (98%), bis(2-nitrophenyl) disulfide (97%), phenyl disulfide (95%), p-toluenesulfonamide (98%), dl-dithiothreitol (98%), 1,4-benzenedithiol (95%), 2-bromo-5-methylthiophene (98%), tert-dodecylthiol (98%), 3,3’-dimercapto-[1,1’-biphenyl]-4,4’-dicarboxylic acid (98%), 2-amino-4-chloro thiophenol (95%), 4,4’-diaminodiphenylsulfone (98%), 2-bromothiophene (95%), diallyl sulfide (98%), [4-(4-phosphonophenyl)phenyl]phosphonic acid (97%), cyclohexyl mercaptan (98%) and 1H,1H,2H,2H-perfluorooctyl mercaptan (98%) were purchased from Acmec Biochemical company. Tetrahydroxyquinone (98%), 3-iodopyridine (98%), 2,5-dihydroxybenzaldehyde (98%), 2-chlorobenzyl alcohol (98%), 3-pyridinemethanethiol (95%), 10-methylphenothiazine (95%), 2-chlorophenothiazine (98%), 1,10-decanedithiol (98%), tetrahydrothiopyran-4-one (98%), 4-(methylsulfanyl)benzaldehyde (98%), 4-(methylthio)phenol (98%), 2-amino-6-bromobenzothiazole (98%), 2-amino-6-fluorobenzothiazole (96%), glycol sulfite (98%), di(thiophen-2-yl)methanone (97%), 2,3,5,6-tetrafluorothiophenol (98%), triphenylmethanethiol (98%), di-tert-butyl disulfide (95%), 1-adamantanethiol (95%), [4,2’:6’,4”-terpyridine]-4’-carboxylic acid (98%), 4-pyridinylphosphonic acid (97%), 1-methyl-2-imidazolidinethione (95%), 2,2’-dithiobis(ethylamine) (95%), 2-phenyl-1,3-dithiane (95%) and dimethylthioacetamid (98%) were purchased from Bide Pharmatech Co., Ltd. 4,5-Imidazoledicarboxylic acid (97%), methyl 4-bromobenzoate (98%), 2-(1H-pyrazol-3-yl)pyridine (98%), 5-methyl-3-(pyridin-2-yl)-1H-1,2,4-triazole (95%), 2-(1H-1,2,3,4-tetraazol-5-yl)pyridine (97%), methyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate (97%), 2-(trifluoromethyl)benzoic acid (98%), 2-bromo-5-(trifluoromethyl)pyridine (98%), 2-bromo-5-methylpyridine (98%), 1,4-phenylenediacetonitrile (99%), 2,2’-dithiodipyridine (98%), 4-thiazolecarboxylic acid (97%), 5-acetyl-2-thiopheneboronic acid (98%), 2-amino-1-ethanethiol (98%), p-tolyl disulfide (98%), 2-mercaptophenol (95%), 4-chlorothiophenol (98%), 1,3-diphenyl-2-thiourea (97%), diphenyl sulfone (99%) and 2-amino-5-methyl-1,3,4-thiadiazole (97%) were purchased from Energy Chemical Reagent Company. Risedronic acid (97%) was purchased from Hanhong company. 4-(Diphenylamino)phenylboronic acid (98%), 4-(phenylazo)benzoic acid (98%), 4-bromobenzaldehyde (98%), phosphonic acid (99%), 2,5-dibromopyridine (99%), 4-hydroxyphenylboronic acid (97%), 5-bromo-2-pyridinecarbonitrile (97%), pyridine-4-boronic acid (97%), methyl 5-formyl-2-hydroxybenzoate (98%), d-cystine (98%), d-2-aminobutyric acid (99%), 4-methoxybenzyl alcohol (98%), d-phenylalanine (98%), l-aspartic acid (99%), l-cystine (99.5%), dl-cystine (98%), 4-nitrobenzyl alcohol (98%), and 2-ethylhexyl bromide (99%) were purchased from Aladdin Chemical Reagent Company. Mono-methyl isophthalate (98%), l-alanine (99%), d-alanine (98%), chloranil (98%), malonic acid (99.5%), anthracene-9-carboxylic acid (98%), methyl 4-aminobenzoate (98%), glyoxal (40 wt% in water), 4-pyridinecarboxaldehyde (97%), dl-phenylalanine (98%) and N-fluorobenzenesulfonimide (97%) were purchased from Innochem Science & Technology company. 2-Mercaptopyridine (98%), ethanethiol (97%), thiazole (98%), methanethiole (10% in propanediol), 1,5-pentanedithiol (96%), 1,3-benzenedithiol (95%), cyclohexyl mercaptan (98%), benzyl mercaptan (98%), 5-(2-hydroxyethyl)-4-methylthiazole (98%) and 1,2,4-triazole (99%) were purchased from Macklin Biochemical Company. 4-Pyridinylphosphonic acid (97%), 1,4-benzenedimethanol (98%) and 1-[(Tert-butoxy)carbonyl]pyrrolidine-2-carboxylic acid (98%) were purchased from Shanghai Adamas Reagent Co., Ltd. Pyromellitic acid (99%) was purchased from Zhengzhou Ruke Biological Technology Co., Ltd. Poly(4-vinylpyridine) (Mw ~60,000) and 4-carboxy-p-terphenyl (97%) were purchased from Sigma-Aldrich Company. d(+)-tryptophan (99%), 2-thiopheneacetic acid (98%), 3-hydroxyphenylboronic acid (97%) and adamantine (90%) were purchased from Acros reagent. D-tyrosine (99%), 4-ethylbenzoic acid (97%), N-tert-butylacetamide (98%), triphenylamine (98%), 3,3’-dithiodipropionic acid (98%), agarose (Mr = 0.12) and N-acetyl-d-glucosamine (98%) were purchased from Alfa Aesar Reagent Company. 2,6-Dichloro-1,4-benzoquinone (98%), l-phenylalanine (98%), l-serine (98%), umbelliferone (99%), 4-(2-aminoethyl)pyridine (97%), bromanilic acid (98%), cis-cyclooctene (95%), 4-bromotoluene (99%), mercaptoacetic acid (98%), benzothiazole (96%), 4-mercaptobenzoic acid (95%), 6,6’-dithiodinicotinic acid (98%), 2-chlorobenzyl mercaptan (98%) and octadecanethiol (97%) were purchased from TCI (Shanghai) Development Co., Ltd. Erythorbic acid (99%) and 1,2-ethanedithiol (98%) was purchased from Tianjin HEOWNS Biochemical Technology Development Co., Ltd. 2-Anilinopyridine (97%) was purchased from Fluorochem Ltd. Trimethylhydroquinone (98%) was purchased from Rhawn Chemical Reagent Company. 1,2-benzenedithiol (98%) was purchased from Kawei Chemical Company. Polyvinyl pyrrolidone (PVP, Mw ~220,000) and pyrrole (99%) was purchased from Beijing OKA Biotechnology Co., Ltd. 2’,5’-Dihydroxyacetophenone (98%) was purchased from Jiuding Chemical (Shanghai) Technology Co., Ltd. Europium (III) nitrate hexahydrate (Eu(NO3)3·6H2O) (99.99%) was purchased from Beijing Hwrkchemical Company. Anion exchange membrane of ~130 μm thickness (fumasep FAA-PK-130), gas diffusion layer (GDL, YLS-30T), Nafion binder (5 wt%, DuPont 520), and nickel foam (thickness 1.0 mm) were purchased from Suzhou Sinero Technology company. The CO2 (99.999%) and CO (99.999%) gases were purchased from Messer Gas Products Co., Ltd. The pH of the 1.0 M KOH solutions were 13.80 ± 0.1, as measured with a pH meter (Mettler–Toledo International Inc.). Basically, 1.0 M KOH stored in volumetric bottle is consumed in one week and then re-prepared for use.
Preparation of CuOx substrate used for screening on automatic electrocatalysis platform
In total, 500 mg Cu(NO3)2·3H2O was added to 50 mL DMF. After ultrasonic for 10 min, the copper nitrate was completely dissolved, and then the copper nitrate solution was moved into the Teflon lined autoclave. The autoclave was sealed and placed in an oven for 4 h at 200 °C. After natural cooling to room temperature in ambient environment, the solid product was collected after centrifuging, washing with ethanol and ultrapure water, and drying in vacuum at 60 °C.
Preparation of molecules modified CuOx ink used for screening on automatic electrocatalysis platform
The CuOx substrates were first prepared into a 10 g L−1 dispersion in isopropanol–water–Nafion binder solution in a 30:10:1 volume ratio by sonicating for 1 h. Use 10 mL of isopropanol for each molecular modifier to prepare a 0.01 mol L−1 molecular dispersion. Finally, 200 μL of CuOx substrates dispersion and 315 μL of molecular modifier isopropanol-dispersion were mixed and ultrasonicated to form a homogeneous ink of molecules modified CuOx.
Preparation of ‘CuOx-160’ substrate and ‘1,8-octanedithiol-CuOx-160’ ink
For ‘CuOx-160’ substrate: based on previous report25, 500 mg Cu(NO3)2·3H2O was added to 50 mL DMF. After ultrasonic for 10 min, the copper nitrate was completely dissolved, and then the copper nitrate solution was moved into the Teflon lined autoclave. The autoclave was sealed and placed in an oven for 8 h at 160 °C. After natural cooling to room temperature in ambient environment, the solid product was collected after centrifuging, washing with ethanol and ultrapure water, and drying in vacuum at 60 °C.
For ‘1,8-octanedithiol-CuOx-160’ ink: the ‘CuOx-160’ substrates were first prepared into a 10 g L−1 dispersion in isopropanol-water-Nafion binder solution in a 30:10:1 volume ratio by sonicating for 1 h. Use 10 mL of isopropanol for 1,8-octanedithiol to prepare a 0.01 mol L−1 molecular dispersion. Finally, 200 μL of ‘CuOx-160’ dispersion and 50 μL of 1,8-octanedithiol isopropanol-dispersion were mixed and ultrasonicated to form a homogeneous ink.
Preparation of ‘Ag-Cu2O’ substrate and ‘1,8-octanedithiol-Ag-Cu2O’ ink
For ‘Ag-Cu2O’ substrate: based on previous report72, 0.5 mL NaOH solution (1.0 M) and 0.5 mL Cu(NO3)2·3H2O solution (0.1 M) were added to a 100 mL beaker under vigorous stirring for 5 min at RT to give a blue-color Cu(OH)2 suspension. 10 mL ascorbic acid aqueous solution (27 mg ascorbic acid) was added to the beaker under vigorous stirring. Following stirring for 30 min, 0.1 mL AgNO3 solution (0.01 M) was added to the beaker, and stirring continued for 30 min. Solid products were obtained and then collected by centrifugation and washed with EtOH and water, and dried in vacuum at 60 °C.
For ‘1,8-octanedithiol-Ag-Cu2O’ ink: the ‘Ag-Cu2O’ substrates were first prepared into a 10 g L−1 dispersion in isopropanol-water-Nafion binder solution in a 30:10:1 volume ratio by sonicating for 1 h. Use 10 mL of isopropanol for 1,8-octanedithiol to prepare a 0.01 mol L−1 molecular dispersion. Finally, 200 μL of ‘Ag-Cu2O’ dispersion and 150 μL of 1,8-octanedithiol isopropanol-dispersion were mixed and ultrasonicated to form a homogeneous ink.
Preparation of ‘CuCl–H2O–CuOx’ substrate and ‘1,8-octanedithiol-CuCl–H2O–CuOx’ ink
For ‘CuCl–H2O–CuOx’ substrate: based on previous report73, 0.03 g CuCl was added to 15 mL DMF under magnetic stirring, and then 0.1 mL ultra-pure water was added into the CuCl DMF suspension under vigorous stirring for 10 min. The mixture above was transferred into a Teflon-lined autoclave and maintained at 160 °C for 4 h. After natural cooling to room temperature in ambient environment, the solid product was collected after centrifuging, washing with ethanol and ultrapure water, and drying in vacuum at 50 °C.
For ‘1,8-octanedithiol-CuCl–H2O–CuOx’ ink: the ‘CuCl–H2O–CuOx’ substrates were first prepared into a 10 g L−1 dispersion in isopropanol–water–Nafion binder solution in a 30:10:1 volume ratio by sonicating for 1 h. Use 10 mL of isopropanol for 1,8-octanedithiol to prepare a 0.01 mol L−1 molecular dispersion. Finally, 200 μL of ‘CuCl–H2O–CuOx’ dispersion and 50 μL of 1,8-octanedithiol isopropanol-dispersion were mixed and ultrasonicated to form a homogeneous ink.
Preparation of ‘CuCl–CuOx’ substrate and ‘1,8-octanedithiol-CuCl–CuOx’ ink
For ‘CuCl–CuOx’ substrate: based on previous report73, 120 mg CuCl was added to 60 mL DMF under magnetic stirring, and then the CuCl DMF suspension was directly transferred into a Teflon-lined autoclave and maintained at 160 °C for 4 h. After natural cooling to room temperature in ambient environment, the solid product was collected after centrifuging, washing with ethanol and ultrapure water, and drying in vacuum at 50 °C.
For ‘1,8-octanedithiol-CuCl–CuOx’ ink: the ‘CuCl–CuOx’ substrates were first prepared into a 10 g L−1 dispersion in isopropanol–water–Nafion binder solution in a 30:10:1 volume ratio by sonicating for 1 h. Use 10 mL of isopropanol for 1,8-octanedithiol to prepare a 0.01 mol L−1 molecular dispersion. Finally, 200 μL of ‘CuCl–CuOx’ dispersion and 50 μL of 1,8-octanedithiol isopropanol-dispersion were mixed and ultrasonicated to form a homogeneous ink.
Preparation of ‘CuOx–H2O-sheet’ substrate and ‘1,8-octanedithiol-CuOx–H2O-sheet’ ink
For ‘CuOx–H2O-sheet’ substrate: based on previous report74, 850 mg of CuCl2·2H2O was dispersed in 50 mL of NaOH solution (1.0 M) and PVP (150 mg) with stirring for 20 min. The above solution was then transferred into a 100 mL Teflon-lined stainless-steel autoclave, sealed and maintained at 180 °C for 20 h, and cooled to room temperature naturally. Finally, the black precipitate was separated by centrifugation, washed several times with deionized water and dried in vacuum for 12 h.
For ‘1,8-octanedithiol-CuOx–H2O-sheet’ ink: the ‘CuOx–H2O-sheet’ substrates were first prepared into a 10 g L−1 dispersion in isopropanol–water–Nafion binder solution in a 30:10:1 volume ratio by sonicating for 1 h. Use 10 mL of isopropanol for 1,8-octanedithiol to prepare a 0.01 mol L−1 molecular dispersion. Finally, 200 μL of ‘CuOx–H2O-sheet’ dispersion and 50 μL of 1,8-octanedithiol isopropanol-dispersion were mixed and ultrasonicated to form a homogeneous ink.
Preparation of ‘CuOx-sheet’ substrate and ‘1,8-octanedithiol-CuOx-sheet’ ink
For ‘CuOx-sheet’ substrate: based on previous report74, 672.3 mg of CuCl2 was dispersed in 50 mL of NaOH solution (1.0 M) and PVP (150 mg) with stirring for 20 min. The above solution was then transferred into a 100 mL Teflon-lined stainless-steel autoclave, sealed and maintained at 180 °C for 20 h, and cooled to room temperature naturally. Finally, the black precipitate was separated by centrifugation, washed several times with deionized water and dried in vacuum for 12 h.
For ‘1,8-octanedithiol-CuOx-sheet’ ink: the ‘CuOx-sheet’ substrates were first prepared into a 10 g L−1 dispersion in isopropanol–water–Nafion binder solution in a 30:10:1 volume ratio by sonicating for 1 h. Use 10 mL of isopropanol for 1,8-octanedithiol to prepare a 0.01 mol L−1 molecular dispersion. Finally, 100 μL of ‘CuOx-sheet’ dispersion and 50 μL of 1,8-octanedithiol isopropanol-dispersion were mixed and ultrasonicated to form a homogeneous ink.
Preparation of ‘CuOx–GLU’ substrate and ‘1,8-octanedithiol-CuOx–GLU’ ink
For ‘CuOx–GLU’ substrate: based on previous report75, 50 mg Cu(acac)2 was added into a solution of 10 mL DMF. After vigorous stirring for 5 min, 2 mL H2O containing 5 mg glucose was added by dropwise with vigorous stirring. After half an hour, the mixture was transferred into a 100 mL Teflon-lined autoclave, sealed and heated at 160 °C for 4 h. Then the system was allowed to cool down to room temperature naturally, and the mixture was collected by centrifuging, washing with ethanol and water, and finally dried in vacuum.
For ‘1,8-octanedithiol-CuOx–GLU’ ink: the ‘CuOx–GLU’ substrates were first prepared into a 10 g L−1 dispersion in isopropanol–water–Nafion binder solution in a 30:10:1 volume ratio by sonicating for 1 h. Use 10 mL of isopropanol for 1,8-octanedithiol to prepare a 0.01 mol L−1 molecular dispersion. Finally, 200 μL of ‘CuOx–GLU’ dispersion and 100 μL of 1,8-octanedithiol isopropanol-dispersion were mixed and ultrasonicated to form a homogeneous ink.
Preparation of ‘CuBr–Cu’ substrate and ‘1,8-octanedithiol-CuBr–Cu’ ink
For ‘CuBr–Cu’ substrate: 174 mg CuBr was added to 60 mL DMF. After ultrasonic for 10 min, the CuBr was evenly dispersed, and then the CuBr dispersion was moved into the Teflon lined autoclave. The autoclave was sealed and placed in an oven for 4 h at 160 °C. After natural cooling to room temperature in ambient environment, the solid product was collected after centrifuging, washing with ethanol and ultrapure water, and drying in vacuum at 60 °C.
For ‘1,8-octanedithiol-CuBr–Cu’ ink: the ‘CuBr–Cu’ substrates were first prepared into a 10 g L−1 dispersion in isopropanol–water–Nafion binder solution in a 30:10:1 volume ratio by sonicating for 1 h. Use 10 mL of isopropanol for 1,8-octanedithiol to prepare a 0.01 mol L−1 molecular dispersion. Finally, 200 μL of ‘CuBr–Cu’ dispersion and 50 μL of 1,8-octanedithiol isopropanol-dispersion were mixed and ultrasonicated to form a homogeneous ink.
Preparation of ‘Cd-Cu’ substrate and ‘1,8-octanedithiol-Cd–Cu’ ink
For ‘Cd–Cu’ substrate: 120 mg Cd(NO3)2·4H2O and 500 mg Cu(NO3)2·3H2O were added to 50 mL DMF. After ultrasonic for 10 min, the cadmium nitrate and copper nitrate were completely dissolved, and then the mixed solution was moved into the Teflon lined autoclave. The autoclave was sealed and placed in an oven for 4 h at 160 °C. After natural cooling to room temperature in ambient environment, the solid product was collected after centrifuging, washing with ethanol and ultrapure water, and drying in vacuum at 60 °C.
For ‘1,8-octanedithiol-Cd–Cu’ ink: the ‘Cd–Cu’ substrates were first prepared into a 10 g L−1 dispersion in isopropanol–water–Nafion binder solution in a 30:10:1 volume ratio by sonicating for 1 h. Use 10 mL of isopropanol for 1,8-octanedithiol to prepare a 0.01 mol L−1 molecular dispersion. Finally, 200 μL of ‘Cd–Cu’ dispersion and 50 μL of 1,8-octanedithiol isopropanol-dispersion were mixed and ultrasonicated to form a homogeneous ink.
Preparation of ‘Cd@CuOx’ substrate and ‘1,8-octanedithiol-Cd@CuOx’ ink
For ‘Cd@CuOx’ substrate: 120 mg CuCl was added to 60 mL DMF. After ultrasonic for 10 min, the CuCl was evenly dispersed, and then the CuCl suspension was moved into the Teflon lined autoclave. The autoclave was sealed and placed in an oven for 4 hours at 160 °C. After natural cooling to room temperature in ambient environment, the solid product was collected after centrifuging, washing with ethanol and ultrapure water, and drying in vacuum at 60 °C. 100 mg Cd(NO3)2·4H2O was dissolved in 10 mL DMF, then 1 mL of this solution was added to 10 mg above solid product in a Teflon lined autoclave. The autoclave was sealed and placed in an oven for 1 h at 150 °C. After natural cooling to room temperature in ambient environment, the solid product was collected after centrifuging, washing with ethanol and ultrapure water, and drying in vacuum at 60 °C.
For ‘1,8-octanedithiol-Cd@CuOx’ ink: the ‘Cd@CuOx’ substrates were first prepared into a 10 g L−1 dispersion in isopropanol-water-Nafion binder solution in a 30:10:1 volume ratio by sonicating for 1 h. Use 10 mL of isopropanol for 1,8-octanedithiol to prepare a 0.01 mol L−1 molecular dispersion. Finally, 200 μL of ‘Cd@CuOx’ dispersion and 50 μL of 1,8-octanedithiol isopropanol-dispersion were mixed and ultrasonicated to form a homogeneous ink.
Preparation of ‘Eu@CuOx’ substrate and ‘1,8-octanedithiol-Eu@CuOx’ ink
For ‘Eu@CuOx’ substrate: 500 mg of Cu(NO3)2·3H2O and 60 mg Eu(NO3)3·6H2O were added to 50 mL of DMF. After ultrasonic for 10 min, the mixed solution was transferred into the Teflon lined autoclave. The autoclave was sealed and placed in an oven for 4 h at 160 °C. After natural cooling to room temperature in ambient environment, the solid product was collected after centrifuging, washing with ethanol and ultrapure water, and drying in vacuum at 60 °C.
For ‘1,8-octanedithiol-Eu@CuOx’: the ‘Eu@CuOx’ substrates were first prepared into a 10 g L−1 dispersion in isopropanol-water-Nafion binder solution in a 30:10:1 volume ratio by sonicating for 1 h. Use 10 mL of isopropanol for 1,8-octanedithiol to prepare a 0.01 mol L−1 molecular dispersion. Finally, 200 μL of ‘Eu@CuOx’ dispersion and 50 μL of 1,8-octanedithiol isopropanol-dispersion were mixed and ultrasonicated to form a homogeneous ink.
Fabrication of GDEs for automatic electrocatalysts testing platform
For screening test: 80 μL of the molecules modified CuOx ink was dropped onto a 1.2 cm × 1.2 cm gas diffusion layer (the actual test area is 1.0 cm2) to create a gas diffusion electrode (GDE) and then dried on a heating plate at 60 °C. The mass loading of catalyst was controlled at about 0.20 mg cm−2.
For verification of universality test: 36 μL of the 1,8-octanedithiol modified OD Cu ink was dropped onto a 1.2 cm × 1.2 cm gas diffusion layer (the actual test area is 1.0 cm2) to create a gas diffusion electrode (GDE) and then dried on a heating plate at 60 °C. The mass loading of catalyst was controlled at about 0.20 mg cm−2.
Fabrication of GDEs for commercial flow cell
For validation test of CuOx, 1,8-octanedithiol-CuOx and 2-pyridinemethanethiol-CuOx: 150 μL of the molecules modified CuOx ink was dropped onto a 2.5 cm × 1.0 cm gas diffusion layer (the actual test area is 0.8 cm2) to create a gas diffusion electrode (GDE) and then dried at 60 °C. The mass loading of catalyst was controlled at about 0.20 mg cm−2.
For verification of universality test: 100 μL of the 1,8-octanedithiol modified OD Cu ink was dropped onto a 2.5 cm × 1.0 cm gas diffusion layer (the actual test area is 0.8 cm2) to create a gas diffusion electrode (GDE) and then dried at 60 °C. The mass loading of catalyst was controlled at about 0.30 mg cm−2.
Electrochemical rapid screening measurements on the automatic electrocatalysts testing platform
The automated electrocatalysis platform is composed of several components, including automatic electrolysis unit, automatic catholyte collection unit, compliable peristaltic pumps, control program and an online micro-fast gas chromatograph equipped with TCD detector (Agilent 990). For automatic electrolysis unit, it consists of a 3D-printed flow cell reactor fixed on a screw slide platform A and an automatic catalyst replacement module fixed on another screw slide platform B. The detailed design 3D-printed flow cell has been presented in our previous work, comprising a gas chamber, a cathodic chamber, and an anodic chamber. The working electrode was positioned between the gas chamber and the cathodic chamber, while the counter electrode was placed in the anodic chamber. The 3D-printed flow cell is equipped with a GDE cathode, a Ag/AgCl reference electrode (3.5 M KCl), and a foamed nickel anode25. An anion exchange membrane of 1.3 × 1.3 cm2 in area effectively separated the cathodic and anodic chambers. The anion exchange membrane was soaked in 1.0 M KOH for 24 h and then rinsed with deionized water before use. The automatic catalyst replacement module (Supplementary Fig. 1) mainly consist of a catalyst array plate loaded with multiple silicone gaskets and a screw slide platform, which is used to move the catalyst array plate. After the GDEs are pasted on each silicone gasket through copper conductive glue, the array plate is then placed on the screw slide. For automatic catholyte collection unit, it mainly consists of a rotation disk loaded with electrolyte bottles and a slide rail connected with electrolyte pipelines (Supplementary Fig. 2). The move of screw slide platform is started by the initiation of the stepper motor and driving the rotation of the screw rod to induce displacement in the fixed objects. The online micro-fast GC has three channels, but we mainly use GC1A channel (Molecular Sieve 5A column) and GC1C channel (Plot Q column) to analyze CO2RR products FEs. The gas products were analyzed in 3 min, a high-sensitivity thermal conductivity detector (TCD) and an automatic sampling system. During the entire platform operation, ultrapure CO2 continues to flow into the gas chamber. The 1.0 M KOH aqueous solution was continuously pumped into the cathode and anode compartments at a flow rate of 6.0 mL min−1 as the electrolyte through two compilable peristaltic pumps, and high-purity carbon dioxide with a flow rate of about 32 mL min−1 was supplied to the cathode through a mass flow controller. The electrolysis process was controlled by a CHI 660E electrochemical workstation. To calculate the Faradaic efficiency of the gaseous products, a soap film flowmeter was connected to the cathode outlet of the flow cell to measure the real-time flow rate during the reaction.
The platform operation process is as follows:
First, run the automatic catholyte collection unit, that is, automatically lower the slide rail connected with 1.0 M KOH pipelines. Second, after the series of GDEs to be tested are loaded manually on the catalyst array plate through copper conductive glue and the array plate is fixed on the corresponding screw slide platform B manually, an automatically quick move of CO2 flow plate on the screw slide platform A to close the flow cell for the next screening experiment. Then 1.0 M KOH is continuously pumped into the electrolyte chambers of the 3D-printed automated flow cell via automatically operating compilable peristaltic pumps. Two reaction potentials are then tested for each catalyst ( − 0.56 and −0.62 V vs RHE) and after the catalyst was pre-reduced at each reaction potential for about 160 s, the gaseous products were automatically sampled three times in succession by online micro-fast GC, with an interval of 3 min for each sampling. The average value of basically three sampling data was taken as the result. It only takes about 20 min to complete the above test of an electrocatalyst. During the alternation of two test potentials, restart the automatic catholyte collection unit to automatically lift up the slide rail until all the circulating electrolyte to be recovered for additional measurements by 1H NMR and then automatically rotate the rotation disk to move to the next bottle of fresh electrolyte, then lower the electrolyte pipelines again for the next potential test. When the first catalyst evaluation process is completed, start the automatic catholyte collection unit again to recover the liquid product. Subsequently, a quick move of screw slide platform A is controlled to accomplish the automatic separation of CO2 flow plate and other fixed plates of automated flow cell. Finally, the array plate fixed on crew slide platform B automatically driven by a stepper motor moves a fixed distance to the next grid position of a new GDE for the next round of catalyst evaluation. All the automatic operations on this electrolysis platform are primarily controlled by the LabVIEW program.
Electrochemical measurements in commercial flow cell
The CO2RR test was performed in a custom-designed flow cell (Gaossunion) using chronopotentiometry method. This commercial three-compartment flow cell includes a gas chamber, a cathodic chamber, and an anodic chamber. The working electrode was positioned between the gas chamber and the cathodic chamber, while the counter electrode was placed in the anodic chamber. The cell is equipped with a gas diffusion electrode (GDE) as the working electrode, an anion exchange membrane measuring 2.5 cm × 1.0 cm to separate the cathodic and anodic chambers, a foamed nickel anode of the same dimensions (2.5 cm × 1.0 cm), and a reference electrode consisting of Ag/AgCl (3.5 M KCl). The anion exchange membrane was soaked in 1.0 M KOH for 24 h and then rinsed with deionized water before use. Pure CO2 gas flow was fed to the cathode gas chamber backside the GDE. The inlet CO2 flow rate was ~52 mL min−1 and the actual outlet CO2 rate measured by a soap film flowmeter connected to the cathode outlet of the flow cell under different applied current density was recorded to calculate the individual gaseous product Faradaic efficiency (FE). 10 mL min−1 of 1.0 M KOH aqueous electrolyte was pumped into cathode and anode chamber. The electrolysis was controlled by a CHI 660E electrochemical workstation equipped with a high current amplifier CHI 680 C. During CO2RR, the gas products was sampled at about 200 s and 1100 s at 15 min interval, respectively, and the average FE for individual CO2RR product of these two sampling results was recorded as the final data. The accumulated liquid product was collected after 1200 s of reaction. The FEs of gas products were analyzed by FULI online gas chromatography with a flame ionization detector (FID) and a thermal conductivity detector (TCD). The FEs of liquid products were detected by 1H-NMR spectroscopy (Bruker AVANCE AV III 500 MHz), in which 0.50 mL of electrolyte was mixed with 0.10 mL of DMSO (internal standard, diluted to 200 ppm (v/v) by deuterated water).
The electrochemical workstation parameters were not set to automatically compensate for IR during the CO2RR test, but the flow cell resistances (R) were measured by electrochemical impedance spectroscopy (EIS) under open circuit potentials (1.2 ± 0.2 Ω) after CO2RR. The electrode potential converted to RHE can be manually compensated for 85% of the IR to obtain as the following Eq. (2).
Electrochemical impedance spectroscopy (EIS) measurement
EIS was conducted in the frequency from 10 Hz to 100 kHz with a signal amplitude of 5 mV in commercial flow cell with 1.0 M KOH as electrolyte and continuously pumped CO2.
Faradaic efficiency (FE) of products calculation
The Faradaic efficiency (FE) of products was calculated as the following Eq. (3):
where z represents the number of electrons transferred during product formation, nnn denotes the moles of product determined by GC or 1H NMR, F is Faraday constant (96,500 C mol−1). I is the applied current (A) and t is the reaction time (s) measured by the electrochemical workstation.
Reference electrode calibration
A mixed aqueous solution of 0.4 mM potassium ferrocyanide and 0.5 M potassium chloride was used as the electrolyte, a glassy carbon electrode was used as the working electrode, a Pt wire was used as the counter electrode, and the reference electrode to be calibrated was used as the reference electrode. Cyclic voltammetry was performed at a scan rate of 0.05 V s−1 from −0.2 V to 0.7 V vs Ag/AgCl (3.5 M KCl), and the reference electrode potential was calibrated as the following Eq. (4):
Where Ep1 and Ep1 represent the potentials of the oxidation and reduction peaks, respectively.
Contact angle measurements
Contact angles of water droplets on the surfaces of gas diffusion layers loaded with CuOx or S-molecules modified CuOx catalysts were measured on an OCA20 contact angle measurement instrument (Dataphysics Instrument Co., Ltd., Germany) at room temperature.
Characterizations
X-ray diffraction (XRD) measurements were performed on a Rigaku Ultima IV diffractometer using Cu Ka radiation (40 kV, 30 mA). Scanning electron microscope (SEM) measurements were performed on a ZEISS Sigma operated at 15 kV. Transmission electron microscopy (TEM), high-resolution transmission electron microscope (HRTEM) and the energy-dispersive X-ray spectroscopy (EDX) elemental mapping measurements were carried out on a JEOL JEM-F200 electron microscope operated at an acceleration voltage of 200 kV. Inductively coupled plasma optical emission spectroscopy (ICP-OES) measurements were conducted on a ThermoFisher iCap 7000. X-ray Photoelectron Spectroscopy (XPS) and Auger spectroscopy measurements were performed in an ultrahigh vacuum system chamber equipped with an Thermo Scientific ESCALAB Xi+ XPS. In order to avoid the influence of S element in Nafion, Nafion binder is not added to the catalyst ink during SEM, TEM, EDX mapping, ICP, and XPS testing.
XPS measurements of catalysts after CO2RR
In order to exclude oxygen as much as possible in the sample transfer process after CO2RR, the electrochemical flow cell was immediately transferred to a glove box for the disassembly of the flow cell after the CO2RR test. GDEs were washed with ethanol and then sealed with thermoplastic sealing machine in the glove box. The sealed GDEs were finally transferred to the XPS chamber (Thermo Scientific ESCALAB Xi+ XPS).
Electrical double-layer (EDL) capacitance measurement
The catalyst was first reduced at −0.7 V vs RHE for 200 s and scanned in the potential range of −0.125 to −0.225 V vs RHE in Ar-saturated 1.0 M KOH at sweep rates of 20, 40, 60, 80, and 100 mV s−1, respectively. Ar was purged during the measurement. The anodic and cathodic current densities at −0.175 V vs RHE in the last scan cycle were recorded. The differences in these two current densities (Δj) at different sweep rates were then calculated and Δj/2 plotted against the sweep rates for each catalyst. The slopes of Δj/2 vs sweep rate curves were calculated via linear fitting, corresponding the double-layer capacitances for different catalysts.
Electrochemical OH− adsorption measurement
Electrochemical OH− adsorption was performed in a Ar-saturated 1 M KOH electrolyte with a linear sweep voltammetry method at a sweep rate of 100 mV s−1. The potential ranged from −0.2 to 0.6 V vs RHE. In addition to testing the catalysts after CO2RR reaction, all catalysts were reduced at −0.69 V versus RHE for 200 s before performing the OH− adsorption measurement.
CO stripping measurement
CO stripping measurement was performed in a H-cell. The GDEs were first reduced in 1.0 M KOH at −1.7 V vs Ag/AgCl (3.5 M KCl) for 200 s. Then the electrodes were washed by deionized water. Subsequently, the electrode was held at 0.1 V vs RHE in 0.1 M HClO4 solution for 15 min to allow CO to be adsorbed on the electrode surface under CO atmosphere, and then excess unadsorbed CO was removed by purging Ar gas for 20 min. Finally, CO was stripped by conducting LSV from 0 to 1.0 V vs RHE at 50 mV s−1.
In situ ATR-FTIRS measurement
The ink of CuOx, 1,8-octanedithiol-CuOx, and 2-pyridinemethanethiol-CuOx was directly dropped onto the reflecting plane of a Si prism. Then the Si prism was fixed upon a home-made ATR spectro-electrochemical cell. A platinum wire electrode and a Ag/AgCl electrode were used as the counter and reference electrodes, respectively. The electrolyte was CO2-saturated 0.5 M KHCO3 solution. The in situ ATR-FTIRS measurements were carried on a Nicolet 6700 FTIR spectrometer equipped with a liquid-nitrogen-cooled MCT-A detector and an EverGlo IR source. The electrode potential was held at 0 V vs RHE and a background spectrum was thus recorded. The electrode potential was scanned from 0 V to −1.4 V vs RHE stepwise, and in the meantime infrared spectra were recorded with a time resolution of 42 s per spectrum at a spectral resolution of 8 cm−1. The resulting spectra were reported as relative change in reflectivity as the following Eq. (5):
where R(ES) and R(ER) are single-beam spectra collected at the sample potential and the reference potential, respectively.
In situ Raman measurement
In situ Raman measurement was performed in a commercial Raman flow cell equipped with a quartz window to detect the working electrode GDE. The ink of CuOx, 1,8-octanedithiol-CuOx and 2-pyridinemethanethiol-CuOx was directly dropped onto the gas diffusion layers to fabricate GDEs. In situ Raman spectra were collected on a HORIBA LabRAM HR Evolution Raman system using a 633 nm laser as the excitation source. During the in-situ Raman testing, a 1.0 M KOH solution serving as both the catholyte and anolyte was pumped into the electrolyte compartment at a flow rate of 2.0 mL min−1, whereas CO2 (99.999%) was continuously supplied to the gas compartment at a flow rate of 5 mL/min. Potentials were applied (in potential holds) with respect to an Ag/AgCl reference electrode and reported with respect to RHE. In situ Raman spectra were obtained at 4 min after the potential was initially applied. Potential-dependent spectra were obtained from OCP to −0.8 V vs RHE at −0.1 V vs RHE intervals. Nickel foam was used as the counter electrode.
DFT calculation methods
To reveal the speculation of S-terminal coordination structure and CO intermediates adsorption behavior on 1,8-octanedithiol-CuOx and CuOx, the mechanism of CO2RR on the Cu(110) and Cu(110) with or without 1,8-octanedithiol were calculated by density functional theory (DFT) through Vienna Ab initio Simulation Package (VASP)76,77,78,79. The Projector Augmented Wave (PAW)80,81 in conjunct with the Perdew–Burke–Ernzerh (PBE) flavor82 were carried out in optimization calculations. The k-point sampling of (3,5,1) for Cu(110) and Cu(100) were employed. The convergence criteria were set to 1 × 10−4 eV, and the cutoff energy of plane wave basis was set to 400 eV. The threshold for force was set to −0.02 eV Å−1, and the Van der Waals (vdW) correction was adopted by Grimme (DFT + D3)83.
The Cu(110) and Cu(100) surfaces with or without 1,8-octanedithiol modification were modeled using five-layer slab within (5 × 4) surface unit cell, respectively. The top four layers and the adsorbates were fully relaxed, and the remaining two layers were fixed. To avoid the periodic interactions of the system, a vacuum region of 20 Å between two repeated slabs was used in the direction perpendicular to the surface.
The free energies of all intermediates of the electrochemical reactions were calculated by the computational hydrogen electrode model84. The ΔG0 was calculated at 298.15 K by the VASPKIT package85, which according to ΔG0 = ΔEDFT + ΔEZPE − TΔS. The EDFT, EZPE, and S indicates the electronic energy, zero-point energy and entropy, respectively.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files. The high-throughput screening data are listed in the Supplementary Information (Supplementary Table 24). The reproducibility of the collected CO2RR product dataset on autonomous electrocatalytic platform is supported by repeated experimental data for several selected molecular modifiers-CuOx catalysts (Supplementary Table 25). Source data are provided as a Source data file. Source data are provided with this paper. The raw data for the paper has been deposited in Figshare under accession code DOI link: https://doi.org/10.6084/m9.figshare.2650236486. Source data are provided with this paper.
References
Heiskanen, S. K., Kim, J. & Lucht, B. L. Generation and evolution of the solid electrolyte interphase of lithium-ion batteries. Joule 3, 2322–2333 (2019).
Esmaeilpour, M., Jana, S., Li, H., Soleymanibrojeni, M. & Wenzel, W. A solution-mediated pathway for the growth of the solid electrolyte interphase in lithium-ion batteries. Adv. Energy Mater. 13, 2203966 (2023).
Kim, M. et al. Thermal decomposition mechanism of lithium methyl carbonate in solid electrolyte interphase layer of lithium-ion battery. Energy Storage Mater. 70, 103517 (2024).
Cheng, K. et al. Material–electrolyte interfacial interaction enabling the formation of an inorganic-rich solid electrolyte interphase for fast-charging Si-based lithium-ion batteries. Energy Environ. Sci. 17, 2631–2641 (2024).
Shin, H., Hansen, K. U. & Jiao, F. Techno-economic assessment of low-temperature carbon dioxide electrolysis. Nat. Sustain. 4, 911–919 (2021).
Nitopi, S. et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610–7672 (2019).
Yu, J. et al. Recent progresses in electrochemical carbon dioxide reduction on copper-based catalysts toward multicarbon products. Adv. Funct. Mater. 31, 2102151 (2021).
Xue, Y., Guo, Y., Cui, H. & Zhou, Z. Catalyst design for electrochemical reduction of CO2 to multicarbon products. Small Methods 5, 2100736 (2021).
Nam, D.-H. et al. Molecular enhancement of heterogeneous CO2 reduction. Nat. Mater. 19, 266–276 (2020).
Wagner, A., Sahm, C. D. & Reisner, E. Towards molecular understanding of local chemical environment effects in electro- and photocatalytic CO2 reduction. Nat. Catal. 3, 775–786 (2020).
Creissen, C. E. et al. Molecular inhibition for selective CO2 conversion. Angew. Chem. Int. Ed. 61, e202206279 (2022).
Palui, G., Aldeek, F., Wang, W. & Mattoussi, H. Strategies for interfacing inorganic nanocrystals with biological systems based on polymer-coating. Chem. Soc. Rev. 44, 193–227 (2015).
Wan, M. et al. Enhanced CO2 reactive capture and conversion using aminothiolate ligand–metal interface. J. Am. Chem. Soc. 145, 26038–26051 (2023).
Cao, Z. et al. Chelating N-heterocyclic carbene ligands enable tuning of electrocatalytic CO2 reduction to formate and carbon monoxide: surface organometallic chemistry. Angew. Chem. Int. Ed. 57, 4981–4985 (2018).
Wakerley, D. et al. Bio-inspired hydrophobicity promotes CO2 reduction on a Cu surface. Nat. Mater. 18, 1222–1227 (2019).
Chen, X. et al. Electrochemical CO2-to-ethylene conversion on polyamine-incorporated Cu electrodes. Nat. Catal. 4, 20–27 (2021).
Iijima, G. et al. Methanethiol SAMs induce reconstruction and formation of Cu+ on a Cu catalyst under electrochemical CO2 reduction. ACS Catal. 10, 15238–15249 (2020).
Pankhurst, J. R., Iyengar, P., Loiudice, A., Mensi, M. & Buonsanti, R. Metal–ligand bond strength determines the fate of organic ligands on the catalyst surface during the electrochemical CO2 reduction reaction. Chem. Sci. 11, 9296–9302 (2020).
Zhang, J. et al. Molecular tuning for electrochemical CO2 reduction. Joule 7, 1700–1744 (2023).
Laibinis, P. E. & Whitesides, G. M. Self-assembled monolayers of n-alkanethiolates on copper are barrier films that protect the metal against oxidation by air. J. Am. Chem. Soc. 114, 9022–9028 (1992).
Ron, H., Cohen, H., Matlis, S., Rappaport, M. & Rubinstein, I. Self-assembled monolayers on oxidized metals. 4. Superior n-alkanethiol monolayers on copper. J. Phys. Chem. B 102, 9861–9869 (1998).
Lin, Y. et al. Tunable CO2 electroreduction to ethanol and ethylene with controllable interfacial wettability. Nat. Commun. 14, 3575 (2023).
Shirzadi, E. et al. Ligand-modified nanoparticle surfaces influence CO electroreduction selectivity. Nat. Commun. 15, 2995 (2024).
Perry, S. C. et al. Hydrophobic thiol coatings to facilitate a triphasic interface for carbon dioxide reduction to ethylene at gas diffusion electrodes. Faraday Discuss. 230, 375–387 (2021).
Xie, M. et al. Fast screening for copper-based bimetallic electrocatalysts: efficient electrocatalytic reduction of CO2 to C2+ products on magnesium-modified copper. Angew. Chem. Int. Ed. 61, e202213423 (2022).
Guo, Y. et al. Machine-learning-guided discovery and optimization of additives in preparing Cu catalysts for CO2 reduction. J. Am. Chem. Soc. 143, 5755–5762 (2021).
Shen, Y., Wang, Z., Wang, Y. & Wang, C. Rapid screening of copper-based bimetallic catalysts via automatic electrocatalysis platform: electrocatalytic reduction of CO2 to C2+ products on europium-modified copper. Artif. Intell. Chem. 2, 100056 (2024).
Peng, J. et al. Surface coordination layer passivates oxidation of copper. Nature 586, 390–394 (2020).
Kas, R. et al. Electrochemical CO2 reduction on Cu2O-derived copper nanoparticles: controlling the catalytic selectivity of hydrocarbons. PCCP 16, 12194–12201 (2014).
Yang, P. P. et al. Protecting copper oxidation state via intermediate confinement for selective CO2 electroreduction to C2+ fuels. J. Am. Chem. Soc. 142, 6400–6408 (2020).
Lei, Q. et al. Investigating the origin of enhanced C2+ selectivity in oxide-/hydroxide-derived copper electrodes during CO2 electroreduction. J. Am. Chem. Soc. 142, 4213–4222 (2020).
He, X. et al. Uncovering the influence of the modifier redox potential on CO2 reduction through combined data-driven machine learning and hypothesis-driven experimentation. J. Mater. Chem. A 11, 18106–18114 (2023).
Zhang, J. & Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. PCCP 23, 20323–20328 (2021).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Liu, Z., Lu, T. & Chen, Q. Intermolecular interaction characteristics of the all-carboatomic ring, cyclo[18]carbon: focusing on molecular adsorption and stacking. Carbon 171, 514–523 (2021).
Castner, D. G., Hinds, K. & Grainger, D. W. X-ray photoelectron spectroscopy sulfur 2p study of organic thiol and disulfide binding interactions with gold surfaces. Langmuir 12, 5083–5086 (1996).
Wang, Y., Im, J., Soares, J. W., Steeves, D. M. & Whitten, J. E. Thiol adsorption on and reduction of copper oxide particles and surfaces. Langmuir 32, 3848–3857 (2016).
Keller, H., Simak, P., Schrepp, W. & Dembowski, J. Surface chemistry of thiols on copper: an efficient way of producing multilayers. Thin Solid Films 244, 799–805 (1994).
Chang, Y., Teo, J. J. & Zeng, H. C. Formation of colloidal CuO nanocrystallites and their spherical aggregation and reductive transformation to hollow Cu2O nanospheres. Langmuir 21, 1074–1079 (2005).
Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 437, 640–647 (2005).
Whitesides, G. M. & Grzybowski, B. Self-assembly at all scales. Science 295, 2418–2421 (2002).
Bandyopadhyay, S. & Dey, A. Convenient detection of the thiol functional group using H/D isotope sensitive Raman spectroscopy. Analyst 139, 2118–2121 (2014).
Aleknavičienė, I., Talaikis, M., Budvytyte, R. & Valincius, G. The impact of an anchoring layer on the formation of tethered bilayer lipid membranes on silver substrates. Molecules 26, 6878 (2021).
Gunathunge, C. M., Li, J., Li, X., Hong, J. J. & Waegele, M. M. Revealing the predominant surface facets of rough Cu electrodes under electrochemical conditions. ACS Catal. 10, 6908–6923 (2020).
An, H. et al. Sub-second time-resolved surface-enhanced raman spectroscopy reveals dynamic CO intermediates during electrochemical CO2 reduction on copper. Angew. Chem. Int. Ed. 60, 16576–16584 (2021).
Fan, M. et al. Cationic-group-functionalized electrocatalysts enable stable acidic CO2 electrolysis. Nat. Catal. 6, 763–772 (2023).
Yin, J. et al. Customizable CO2 electroreduction to C1 or C2+ products through Cuy/CeO2 interface engineering. ACS Catal. 12, 1004–1011 (2022).
Katayama, Y. et al. An in situ surface-enhanced infrared absorption spectroscopy study of electrochemical CO2 reduction: selectivity dependence on surface C-bound and O-bound reaction intermediates. J. Phys. Chem. C 123, 5951–5963 (2019).
Li, F. et al. Molecular tuning of CO2-to-ethylene conversion. Nature 577, 509–513 (2020).
Gunathunge, C. M., Ovalle, V. J., Li, Y., Janik, M. J. & Waegele, M. M. Existence of an electrochemically inert CO population on Cu electrodes in alkaline pH. ACS Catal. 8, 7507–7516 (2018).
Zhu, S., Jiang, B., Cai, W.-B. & Shao, M. Direct observation on reaction intermediates and the role of bicarbonate anions in CO2 electrochemical reduction reaction on Cu surfaces. J. Am. Chem. Soc. 139, 15664–15667 (2017).
Feaster, J. T. et al. Understanding selectivity for the electrochemical reduction of carbon dioxide to formic acid and carbon monoxide on metal electrodes. ACS Catal. 7, 4822–4827 (2017).
Mi, Z., Wang, T., Xiao, L., Wang, G. & Zhuang, L. Catalytic peculiarity of alkali metal cation-free electrode/polyelectrolyte interfaces toward CO2 reduction. J. Am. Chem. Soc. 146, 17377–17383 (2024).
Zhengxiang, G. et al. Efficient electrocatalytic CO2 reduction to C2+ alcohols at defect-site-rich Cu surface. Joule 5, 429–440 (2021).
Schaefer, J., Backus, E. H. G., Nagata, Y. & Bonn, M. Both inter- and intramolecular coupling of O–H groups determine the vibrational response of the water/air interface. J. Phys. Chem. Lett. 7, 4591–4595 (2016).
Wen, Q. et al. Engineering a local free water enriched microenvironment for surpassing platinum hydrogen evolution activity. Angew. Chem. Int. Ed. 61, e202206077 (2022).
Gomes, R. J. et al. Modulating water hydrogen bonding within a non-aqueous environment controls its reactivity in electrochemical transformations. Nat. Catal. 7, 689–701 (2024).
Wang, Y.-H. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81–85 (2021).
Li, C.-Y. et al. In situ probing electrified interfacial water structures at atomically flat surfaces. Nat. Mater. 18, 697–701 (2019).
Wang, Y. et al. Strong hydrogen-bonded interfacial water inhibiting hydrogen evolution kinetics to promote electrochemical CO2 reduction to C2+. ACS Catal. 14, 3457–3465 (2024).
Wang, Y. et al. Catalyst synthesis under CO2 electroreduction favours faceting and promotes renewable fuels electrosynthesis. Nat. Catal. 3, 98–−106 (2019).
Hori, Y., Takahashi, I., Koga, O. & Hoshi, N. Electrochemical reduction of carbon dioxide at various series of copper single crystal electrodes. J. Mol. Catal. A 199, 39–47 (2003).
Ji, Y. et al. Selective CO-to-acetate electroreduction via intermediate adsorption tuning on ordered Cu–Pd sites. Nat. Catal. 5, 251–258 (2022).
Yan, X. et al. Synergy of Cu/C3N4 interface and Cu nanoparticles dual catalytic regions in electrolysis of CO to acetic acid. Angew. Chem. Int. Ed. 62, e202301507 (2023).
Ding, J. et al. Molecular tuning boosts asymmetric C-C coupling for CO conversion to acetate. Nat. Commun. 15, 3641 (2024).
Zhu, P. et al. Direct and continuous generation of pure acetic acid solutions via electrocatalytic carbon monoxide reduction. Proc. Natl Acad. Sci. USA 118, e2010868118 (2021).
Kortlever, R., Shen, J., Schouten, K. J. P., Calle-Vallejo, F. & Koper, M. T. M. Catalysts and reaction pathways for the electrochemical reduction of carbon dioxide. J. Phys. Chem. Lett. 6, 4073–4082 (2015).
Xiao, H., Cheng, T. & Goddard, W. A. 3rd Atomistic mechanisms underlying selectivities in C1 and C2 products from electrochemical reduction of CO on Cu(111). J. Am. Chem. Soc. 139, 130–136 (2017).
Li, Y. C. et al. Binding site diversity promotes CO2 electroreduction to ethanol. J. Am. Chem. Soc. 141, 8584–8591 (2019).
Calle-Vallejo, F. & Koper, M. T. Theoretical considerations on the electroreduction of CO to C2 species on Cu(100) electrodes. Angew. Chem. Int. Ed. 52, 7282–7285 (2013).
Li, Z. et al. Directing CO2 electroreduction pathways for selective C2 product formation using single-site doped copper catalysts. Nat. Chem. Eng. 1, 159–169 (2024).
Wang, P. et al. Boosting electrocatalytic CO2-to-ethanol production via asymmetric C–C coupling. Nat. Commun. 13, 3754 (2022).
Xu, S., Sun, S., Chen, G., You, T. & Song, X. Kinetically controlled synthesis of hollow Cu2O nanocubes under solvothermal condition. Cryst. Res. Technol. 44, 721–724 (2009).
Wang, H. et al. Efficient electrocatalytic reduction of CO2 to ethanol enhanced by spacing effect of Cu−Cu in Cu2-xSe nanosheets. Adv. Funct. Mater. 33, 2214946 (2023).
Ma, G. et al. A hydrophobic Cu/Cu2O sheet catalyst for selective electroreduction of CO to ethanol. Nat. Commun. 14, 501 (2023).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S. Density functional theory with London dispersion corrections. WIREs Comput. Mol. Sci. 1, 211–228 (2011).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Wang, V., Xu, N., Liu, J.-C., Tang, G. & Geng, W.-T. VASPKIT: a user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 267, 108033 (2021).
YanShen. Observation of metal-organic interphase in Cu-based electrochemical CO2-to-ethanol conversion. Figshare https://doi.org/10.6084/m9.figshare.26502364 (2025).
Wang-Group. Observation of metal-organic interphase in Cu-based electrochemical CO2-to-ethanol conversion. Zenodo https://doi.org/10.5281/zenodo.14711666 (2025).
Acknowledgements
The authors acknowledge funding support from the National Natural Science Foundation of China (22125502 C.W., 22071207 C.W., 22121001 Y.W.) and the Fundamental Research Funds for the Central Universities (20720240151 D.Z., 20720220011 C.W., 20720230023 D.Z.). Large language models were used for language polishing.
Author information
Authors and Affiliations
Contributions
Y. Shen conducted most of the experiments and analyzed the data. N.F. conducted in situ ATR-SEIRAS and analyzed the data. X.L. performed machine learning. Y.L. and B.Z. built and optimized the automation platform. Y. Su and H.L. extracted some descriptors. T.T. performed TEM test. F.C. conducted in situ Raman and analyzed the data. J.W. assisted in experimental testing. D.S. performed DFT calculations and data analysis. S.X. and Y.W. guided the writing of the article. D.Z. guided the machine learning. T.Z. and R.C. guided the writing of the article. C.W. supervised the project and co-wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Wan-Jian Yin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shen, Y., Fang, N., Liu, X. et al. Observation of metal-organic interphase in Cu-based electrochemical CO2-to-ethanol conversion. Nat Commun 16, 2073 (2025). https://doi.org/10.1038/s41467-025-57221-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57221-x
This article is cited by
-
Cavity-confined Au@Cu2O yolk-shell nanoreactors enable switchable CH4/C2H4 selectivity
Nature Communications (2025)
-
Electrocatalytic CO2 reduction: surface dynamic effects on conversion efficiency
Ionics (2025)
-
Advances in lithium carbon dioxide batteries: bridging the gap between fundamental research and practical applications
Ionics (2025)
