
Abstract
Although mutations in human patatin-like phospholipase PNPLA6 are associated with hereditary retinal degenerative diseases, its mechanistic action in the retina is poorly understood. Here, we uncover the molecular mechanism by which PNPLA6 dysfunction disturbs retinal homeostasis and visual function. PNPLA6, by acting as a phospholipase B, regulates choline mobilization from phosphatidylcholine and subsequent choline turnover for phosphatidylcholine regeneration in retinal pigment epithelial cells. PNPLA6-driven choline is supplied from retinal pigment epithelial cells to adjacent photoreceptor cells to support their survival. Inhibition of this pathway results in abnormal morphology, proliferation, metabolism, and functions of retinal pigment epithelial and photoreceptor cells, and mice with retina-specific PNPLA6 deletion exhibit retinitis pigmentosa-like retinal degeneration. Notably, these abnormalities are entirely rescued by choline supplementation. Thus, PNPLA6 plays an essential role in retinal homeostasis by controlling choline availability for phospholipid recycling and provide a framework for the development of an ophthalmic drug target for retinal degeneration.
Similar content being viewed by others

Introduction
Retinitis pigmentosa, a genetic ophthalmological disease with an incidence of approximately 1 in 4000–8000 people, causes visual loss, visual field constriction, and night blindness due to the degeneration of retinal pigment epithelial (RPE) and photoreceptor cells. Retinitis pigmentosa is regarded as a hereditary retinal degenerative disease caused by mutations in more than 100 genes, many of which are suggested to be involved in the visual cycle or the structure and function of RPE or photoreceptor cells1; however, the precise mechanisms underlying its pathogenesis are still incompletely understood. Clinically, there have currently been few established treatments for retinitis pigmentosa, and supportive care with drugs, carotenoids, vitamins, and other medications is used to improve retinal circulation and delay retinal degeneration. Depending on the mutations, genetic treatment approaches, such as gene therapy targeting RP GTPase regulator (RPGR), have recently been developed2. Nevertheless, the discovery of drugs and therapeutic methods that will contribute to innovative and preventive treatment strategies for retinitis pigmentosa are needed.
Current evidence suggests that glycerophospholipid (hereafter phospholipid) metabolism is crucial for the maintenance of normal ocular function. Phospholipids are synthesized de novo via the Kennedy pathway and undergo remodeling of their fatty acyl chains via the Lands cycle. Multiple mutations in choline phosphate cytidyltransferase 1 A (PCYT1A/CCT-α), an enzyme involved in phosphatidylcholine (PC) biosynthesis via the Kennedy pathway, have been identified in retinitis pigmentosa3,4. Dietary intake of fish oil, which is rich in ω3 polyunsaturated fatty acids (PUFAs), is considered beneficial for photoreceptor function, and docosahexaenoic acid (DHA; C22:6), an ω3 PUFA, is abundantly esterified at the sn-2 position of retinal phospholipids5,6. Mice deficient in lysophosphatidic acid (LPA) acyltransferase 3 (LPAAT3), which incorporates DHA into phospholipids mainly via the Kennedy pathway, display retinal abnormalities, such as the degeneration of disc structure and mitochondria in photoreceptor cells7. Mice lacking lysophosphatidylcholine (LPC) acyltransferase 1 (LPCAT1/LPLAT8), which transfers palmitic acid (C16:0) to LPC to produce dipalmitoyl-PC via the Lands cycle8, and rd11 mice, which have a frameshift mutation in the Lpcat1 gene, also display retinal degeneration9. LPCAT1 controls mitochondrial reactive oxygen species (ROS) generation and retinal photoreceptor cell survival10. These observations provide compelling evidence that phospholipid synthesis and remodeling are critical for proper retinal function in vivo, although our understanding of phospholipid metabolism in the retina is limited.
Phospholipids undergo hydrolytic degradation by various phospholipases in addition to their de novo biosynthesis and remodeling. The phospholipase A2 (PLA2) superfamily, which contains a group of enzymes that generally hydrolyze the sn-2 ester bond of phospholipids to produce fatty acids and lysophospholipids, is structurally and functionally subdivided into several families, including cytosolic PLA2s (cPLA2s), Ca2+-independent or patatin-like PLA2s (iPLA2s/PNPLAs), secreted PLA2s (sPLA2s), and several others11,12,13. Currently, the main focus on PLA2 has been its role as a key regulator of arachidonic acid metabolism for lipid mediator generation14,15. However, recent studies using gene-manipulated mice for various PLA2 enzymes in combination with comprehensive lipid metabolome analysis (lipidomics) have demonstrated that, beyond arachidonic acid metabolism, they participate in diverse lipid-metabolic pathways based on the expression profiles and enzymatic properties of individual PLA2s, thereby regulating various biological responses ranging from homeostasis to disease12. Moreover, several enzymes in the PLA2 superfamily display phospholipase A1 (PLA1), lysophospholipase, neutral lipid lipase, or transacylase activity, rendering our understanding of PLA2-mediated lipid metabolism more complex than previously thought.
Genetic mutations in human PNPLA6 (also called neuropathy target esterase (NTE) or iPLA2δ) are associated with hereditary retinal degenerative diseases summarized as “PNPLA6-related disorders”. These include autosomal recessive inherited diseases, such as Oliver–McFarlane, Laurence–Moon, Boucher–Neuhäuser, and Gordon–Holmes syndromes, which harbor retinitis pigmentosa associated with central or peripheral neuropathy and hormonal abnormalities16,17,18. In these diseases, most of the PNPLA6 mutations are found in the catalytic patatin domain or the three regulatory nucleotide-binding domains of this protein. PNPLA6 exhibits lysophospholipase or phospholipase B (PLB; PLA1/2 + lysophospholipase) activity in yeast, flies, and vertebrates19. Although the biological functions of PNPLA6 have been analyzed mainly in relation to the nervous system20, its precise role and underlying mechanism in the retina remain largely unexplored.
Here, we aimed to clarify the role of PNPLA6-driven lipid metabolism in the retina and elucidate the molecular mechanism by which PNPLA6 dysfunction causes retinal degeneration. We provide evidence that PNPLA6, by acting as a PLB, regulates PC catabolism to mobilize endogenous choline, whose recycling into PC regeneration plays an essential role in RPE and photoreceptor homeostasis.
Results
PNPLA6 is abundantly expressed in RPE cells in the retina
Quantitative PCR (qPCR) revealed that the phospholipid-hydrolyzing PNPLA isoforms PNPLA6–9 were expressed in the human retina, with the expression of PNPLA6 being the highest (Supplementary Fig. 1a). Immunostaining of PNPLA6 protein in human retina showed a positive signal for PNPLA6 in RPE cells, whereas its staining in the sensory retina, which includes photoreceptor cells, was rather faint (Fig. 1a). Among the individual ocular structures that were anatomically isolated from C57BL/6 mice, Pnpla6 expression was the highest in RPE cells (Fig. 1b), although it was also detected at lower levels in the cornea, lens, sensory retina, and optic nerve (Supplementary Fig. 1b), consistent with public single-cell RNA sequencing datasets (e.g., https://proteinpaint.stjude.org/F/2019.retina.scRNA.html and https://www.proteinatlas.org/ENSG00000032444-PNPLA6/single+cell+type/eye). Expression of the RPE marker Rpe65 was detected exclusively in RPE cells (Fig. 1b), confirming proper tissue isolation. In human and mouse primary RPE cells, expression of Pnpla6 and Pnpla8 was higher than that of Pnpla7 and Pnpla9 (Supplementary Fig. 1c). Thus, PNPLA6 is highly expressed in RPE cells in human and mouse retinas.

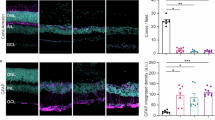
a HE staining and immunostaining for PNPLA6 (red, PNPLA6; blue, DAPI) in the RPE and sensory retina cells of healthy human retinas. Scale bar, 100 µm. Boxed areas in the left panels are magnified in the right panels. b qPCR analysis of Pnpla6 and the RPE marker Rpe65 in fractionated mouse eye tissue (n = 4 independent animals). c Tamoxifen administration protocol in Pnpla6fl/flCreER(−) or Pnpla6fl/flCreER(+) mice. d qPCR analysis of Pnpla6 in RPE cells isolated from Pnpla6fl/flCreER(−) and Pnpla6fl/flCreER(+) mice treated with tamoxifen, with Gapdh as a normalization control (n = 5 independent animals). e HE staining of retina sections from Pnpla6fl/flCreER(−) and Pnpla6fl/flCreER(+) mice. Brackets, ONL. Scale bar, 50 μm. f Quantification of retinal thickness (n = 6 from e). g Vertical number of nuclei in the ONL (n = 6 from e). h TEM images of tamoxifen-inducible Pnpla6fl/flCreER(−) and Pnpla6fl/flCreER(+) retinas. RPE, RPE and photoreceptors, outer segment of photoreceptors, and cell body of photoreceptors are shown, from left to right, respectively. White arrows, mitochondria of RPE; arrowheads, outer segment; red arrows, inner segment; yellow arrow, disc structure of photoreceptors; blue arrow, mitochondria of photoreceptors. Scale bar, 1 μm. i Thickness of the RPE layer (n = 5 from h). j Number of mitochondria in RPE (n = 5 from h). k Thickness of inner and outer photoreceptor segments (n = 5 from h). l Number of abnormal mitochondria in photoreceptors (n = 4 from h). m TEM images of RPE cells demonstrating phagocytosis of photoreceptors; arrowheads, microvilli. Scale bar, 1 μm. n Quantitative values of the number (n = 5 from m) and length ratio (n = 10 from m) of microvilli. Data are presented as mean ± SEM and representative of at least two independent experiments. P-values were derived from one-way ANOVA with Tukey’s multiple comparison test (b) and two-tailed unpaired-t test (d, f, g, i–l, and n). Source data are provided as a Source Data file.
Conditional PNPLA6 deficiency leads to retinal degeneration in mice
Because systemic PNPLA6 deficiency is lethal in mice in utero and cannot be analyzed in adults21,22, we crossed Pnpla6-floxed mice (Pnpla6fl/fl) with heterozygous CAG-CreER transgenic mice to generate Pnpla6fl/flCAG-CreER(+) and control Pnpla6fl/flCAG-CreER(−) mice (Supplementary Fig. 1d and e). In Pnpla6fl/flCAG-CreER(+) mice, Pnpla6 was systemically deleted following intraperitoneal (i.p.) administration of tamoxifen (Fig. 1c). qPCR using isolated RPE cells after 1 month of tamoxifen treatment confirmed that Pnpla6 expression was markedly reduced in knockout Pnpla6fl/flCAG-CreER(+) mice compared to that in control Pnpla6fl/flCAG-CreER(−) mice (Fig. 1d). Hematoxylin and eosin (HE) staining revealed that the retinas of tamoxifen-treated, Pnpla6-deficient Pnpla6fl/flCAG-CreER(+) mice were significantly thinner than those of control Pnpla6fl/flCAG-CreER(−) mice (Fig. 1e and f). Particularly, the thinning of the outer nuclear layer (ONL) was more pronounced in Pnpla6-deficient Pnpla6fl/flCAG-CreER(+) mice, in which there were fewer photoreceptor cell nuclei, than in control Pnpla6fl/flCAG-CreER(−) mice (Fig. 1g). Since both male and female Pnpla6fl/flCAG-CreER(+) mice displayed similar retinal thinning, we used male mice throughout this study. A schematic representation of the photoreceptor cell layer adjacent to RPE cells is provided in Supplementary Fig. 1f. Transmission electron microscopy (TEM) revealed that the RPE cell layer in the outermost retina was thinner and the number of mitochondria in RPE cells was fewer in Pnpla6-deficient mice than in the controls (Fig. 1h–j). Compared to those in the controls, the outer and inner segments of the photoreceptor cell layer were thinned, and the photoreceptor disc structures in the outer segment exhibited abnormal morphology, such as dissociation or atrophy, in Pnpla6-deficient mice (Fig. 1h, k and l). Mitochondrial abnormalities were observed in photoreceptor cells of Pnpla6-deficient mice, similar to those in RPE cells (Fig. 1h). Microvilli, which are crucial for phagocytic removal of the outer segments of old photoreceptor cells, were reduced in RPE cells of Pnpla6-deficient mice (Fig. 1m and n), suggesting lower phagocytotic activity. Therefore, inducible Pnpla6 deletion in adult mice following i.p. treatment with tamoxifen leads to retinal degeneration.
PNPLA6 is widely expressed in various tissues across animal species, and patients with hereditary retinal degenerative diseases with PNPLA6 mutations, such as Laurence–Moon and Boucher–Neuhäuser syndromes, also have abnormalities in several organs other than the retina17,18. Tamoxifen-inducible systemic deletion of Pnpla6 in adult mice resulted in a marked reduction of Pnpla6 expression in several organs, including the liver, kidney, and skeletal muscle (Supplementary Fig. 1g), and was accompanied by reduced survival rate, weight loss, decreased blood glucose level, and loss of visceral and subcutaneous fats within a month after tamoxifen treatment (Supplementary Fig. 1h–l). Thus, beyond the embryonic lethality in global Pnpla6−/− mice21, global Pnpla6 deletion in adult mice markedly perturbs systemic metabolism, leading to early death.
To clarify whether retinal degeneration induced by systemic PNPLA6 deficiency was attributable to the retina-intrinsic loss of PNPLA6 or whether it was a secondary consequence of abnormalities in other organs, we altered the tamoxifen treatment route by administering tamoxifen eyedrops to Pnpla6fl/flCAG-CreER(+) and control Pnpla6fl/flCAG-CreER(−) mice three times daily for 5 days (Fig. 2a). One week after treatment, the protein and mRNA expression of PNPLA6 in RPE cells was almost completely abolished in Pnpla6fl/flCAG-CreER(+) mice (Fig. 2b and c), whereas there was no difference in Pnpla6 expression in the brain (Fig. 2c) and other organs (Supplementary Fig. 2a) between the two groups, indicating that PNPLA6 was selectively lost in the eye. Histological analysis revealed retinal thinning in ocular-selective Pnpla6-deficient mice (Fig. 2d), as in systemic Pnpla6-deficient mice (Fig. 1e). Noninvasive optical coherence tomography (OCT) analysis showed that the ONL of the retina was thinner in ocular-selective Pnpla6-deficient mice than in controls (Fig. 2e and f). Therefore, the retinal degeneration observed in systemic Pnpla6-deficient mice was most likely due to PNPLA6 deficiency in the retina. In contrast to the observations in ocular-selective Pnpla6-deficient mice, thinning of the ONL was not evident in mice with tamoxifen-inducible systemic deletion of PNPLA7 (Pnpla7fl/flCre-ER(+)) or those with global deletion of PNPLA8/iPLA2γ (Pnpla8−/−) or PNPLA9/iPLA2β (Pnpla9−/−) (Supplementary Fig. 2b).

a Protocol for ocular-specific Pnpla6 deletion. b Immunostaining for PNPLA6 in retinas from tamoxifen-treated Pnpla6fl/flCreER(−) and Pnpla6fl/flCreER(+) mice. Red, PNPLA6; blue, DAPI; arrow, RPE. Scale bar, 50 μm. c qPCR of Pnpla6 in isolated RPE or brain from Pnpla6fl/flCreER(−) and Pnpla6fl/flCreER(+) mice, with Gapdh or Hprt1 as a control (n = 3 independent animals). d HE staining of Pnpla6fl/flCreER(−) and Pnpla6fl/flCreER(+) retinas; brackets, ONL. Scale bar, 50 μm. e OCT analysis of Pnpla6fl/flCreER(−) and Pnpla6fl/flCreER(+) retinas. Brackets, ONL. f Quantified values in OCT analysis of Pnpla6fl/flCreER(−) and Pnpla6fl/flCreER(+) retinas (n = 4 from e). g TEM images of Pnpla6fl/flCreER(−) and Pnpla6fl/flCreER(+) retinas. Arrowheads, disc structure of photoreceptors. Scale bar, 1 μm. h ERG analysis of Pnpla6fl/flCreER(−) and Pnpla6fl/flCreER(+) mice under a dark condition (150.3 cd.s/m2). i Amplitudes of a-wave and b-wave (n = 4 from h). j Amplitudes of a-wave and b-wave of the ERG under dark conditions (n = 7 independent animals). k qPCR of Pnpla6 in isolated RPE or brain from 3-month-old Pnpla6fl/fl and Best1-CrePnpla6fl/fl mice (n = 3 independent animals). l HE staining of Pnpla6fl/fl and Best1-CrePnpla6fl/fl retinas. Scale bar, 50 μm. m TEM images of Pnpla6fl/fl and Best1-CrePnpla6fl/fl retinas. Arrows, microvilli of RPE; arrowheads, disc structure of photoreceptors. Scale bar, 1 μm. n OCT analysis of ONL thickness in Pnpla6fl/fl and Best1-CrePnpla6fl/fl mice (n = 3 independent animals). o TUNEL staining of Pnpla6fl/fl and Best1-CrePnpla6fl/fl retinas. Blue, DAPI; Red arrow, TUNEL-positive cells. Scale bar, 50 μm. p Amplitudes of a-wave and b-wave of the ERG under a dark condition (150.3 cd.s/m2) (n = 4 independent animals). Data are presented as mean ± SEM and representative of at least two independent experiments. P-values were derived from two-way ANOVA with Tukey’s multiple comparison test (f, j, and n) and two-tailed unpaired-t test (c, i, k, and p). *P < 0.05, **P < 0.01, and ***P < 0.001, versus control mice (f, j, and n). Source data are provided as a Source Data file.
In addition, TEM analysis showed a trend toward abnormalities in the photoreceptor disc structure of the retinal outer segment in ocular-selective Pnpla6-deficient mice (Fig. 2g), similar to that in systemic Pnpla6-deficient mice (Fig. 1h). We then used electroretinogram (ERG) to analyze whether the retinal degeneration observed thus far would also affect visual function. Photoreceptor cells are composed of two types of cells: rod cells, which are involved in sensing brightness, and cone cells, which are responsible for color vision. In a dark-conditioned ERG, in which the function of rod cells is mainly evaluated, a typical waveform consisting of a-waves generated by retinal photoreceptor cells and b-waves generated by bipolar and Müller cells was observed in the control group. In contrast, both a-wave and b-wave amplitudes were attenuated in ocular-selective Pnpla6-deficient mice compared to those in controls, with marked attenuation of the a-wave (Fig. 2h and i). We further analyzed the amplitudes of the a- and b-waves under light and dark conditions and found that the amplitudes of both waves at various light intensities in the dark were markedly reduced in ocular-selective Pnpla6-deficient mice (Fig. 2j). Even though it was less obvious in the light, a trend toward reduced a-wave amplitudes was observed at multiple light intensities in the null mice (Supplementary Fig. 2c). Thus, ocular-selective Pnpla6-deficient mice display morphological and functional retinal degeneration with impaired visual function.
To further confirm whether PNPLA6 expressed in RPE cells is essential for retinal homeostasis, we generated RPE-specific Pnpla6 knockout mice by crossing Pnpla6fl/fl mice with Best1-Cre mice (Pnpla6fl/flBest1-Cre), in which Pnpla6 was congenitally reduced specifically in RPE cells even without tamoxifen treatment (Fig. 2k and Supplementary Fig. 2d). Histological analysis of the retina demonstrated that 3-month-old Pnpla6fl/flBest1-Cre mice, compared with the control Pnpla6fl/fl mice, displayed retinal thinning with decreased nuclei in the ONL and inner nuclear layer (INL) (Fig. 2l). TEM revealed decreased mitochondria and microvilli in RPE cells and disturbed outer segment disc structure in photoreceptor cells in Pnpla6fl/flBest1-Cre mice (Fig. 2m and Supplementary Fig. 2e), similar to those seen in tamoxifen-inducible global or ocular-selective Pnpla6-deficient mice (Figs. 1h and 2g). Furthermore, the ONL was thinned (Fig. 2n) and TUNEL-positive dead cells were increased (Fig. 2o) in the retina of Pnpla6fl/flBest1-Cre mice. ERG analysis showed significant decreases of a- and b-waves in Pnpla6fl/flBest1-Cre mice under scotopic and photopic conditions (Fig. 2p and Supplementary Fig. 2f and g). Similar retinal thinning was also observed in 6-months-old Pnpla6fl/flBest1-Cre mice (Supplementary Fig. 2h). Based on histological analysis, permanent RPE-specific deletion of PNPLA6 in Pnpla6fl/flBest1-Cre mice over 2–6 months of age (Supplementary Fig. 2i and j) and inducible, ocular-selective deletion of PNPLA6 in adult Pnpla6fl/flCAG-CreER(+) mice after tamoxifen eyedrops for 1-2 weeks (Supplementary Fig. 2k and l) displayed comparable retinal thinning. Collectively, multiple models of ocular PNPLA6 deficiency revealed that PNPLA6 plays a key role in maintaining RPE and adjacent photoreceptor cell homeostasis, and its absence leads to retinal degeneration with loss of visual function.
PNPLA6 displays PLB activity to mobilize endogenous choline in RPE cells
To elucidate the mechanism underlying PNPLA6 deficiency-induced retinal degeneration, we evaluated the enzymatic activity of PNPLA6 using the human RPE cell line ARPE-19. Our initial attempt to establish PNPLA6-knockout ARPE-19 cells using the CRISPR/Cas9 system was unsuccessful as the knockout cells hardly proliferated and were difficult to culture. Therefore, we introduced PNPLA6 siRNA into ARPE-19 cells to acutely reduce PNPLA6 expression by > 90% (Fig. 3a). Lipidomics analysis using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) revealed that PNPLA6-knockdown cells contained significantly more total PC and LPC than did control cells (Fig. 3b). In general, phospholipids have saturated or monounsaturated fatty acids at the sn-1 position and mono- or polyunsaturated fatty acids (PUFAs) at the sn-2 position, and the PLA1 and PLA2 reactions release these fatty acids from the sn-1 and sn-2 positions, respectively. Various PC and LPC molecular species with saturated and unsaturated fatty acids were increased in PNPLA6-knockdown cells relative to those in control cells (Fig. 3c). Hydrolysis of LPC by lysophospholipase gives rise to glycerophosphocholine (GPC), which is then metabolized to choline by glycerophosphodiester phosphodiesterase (GDE). The levels of GPC and choline were decreased by nearly half in PNPLA6-knockdown ARPE-19 cells compared to those in control cells (Fig. 3d), suggesting that PNPLA6 is critically involved in the conversion of LPC to GPC. In addition, most molecular species of phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylinositol (PI), and phosphatidylglycerol (PG), as well as those of lysophosphatidylethanolamine (LPE), were also increased in PNPLA6-knockdown cells relative to those in control cells (Fig. 3e and f).

a qPCR of PNPLA6 in PNPLA6-knockdown (si PNPLA6) and control (scramble) ARPE-19 cells, with GAPDH as an internal control (n = 6 biological replicates). b PC and LPC levels in PNPLA6-knockdown ARPE-19 cells relative to control cells, with the levels in control cells as 1 (n = 3 biological replicates). c Lipidomics of PC and LPC molecular species in PNPLA6-knockdown ARPE-19 cells and control cells (n = 4 biological replicates). d GPC and choline levels in PNPLA6-knockdown ARPE-19 cells relative to control cells, with the levels in control cells as 1 (n = 4 biological replicates). e Lipidomics of PE and LPE molecular species in PNPLA6-knockdown ARPE-19 cells and control cells (n = 4 biological replicates). f Heatmap representation of the ratio of individual phospholipids and lysophospholipids in PNPLA6-knockdown ARPE-19 cells relative to control cells (n = 4 biological replicates). g PC and LPC levels in RPE cells isolated from tamoxifen-inducible, ocular-selective Pnpla6-deficient mice relative to those from control mice, with the levels in control mice as 1 (n = 3 biological replicates). h Lipidomics of PC and LPC molecular species in Pnpla6-deficient RPE cells and control cells (n = 3 biological replicates). i Choline levels in Pnpla6-deficient RPE cells relative to control cells, with the levels in control cells as 1 (n = 4 biological replicates). j Heatmap representation of the ratio of individual phospholipids and lysophospholipids in Pnpla6-deficient RPE cells relative to control cells (n = 4 biological replicates). Data are presented as mean ± SEM and representative of at least two independent experiments. P-values were derived from two-way ANOVA with Tukey’s multiple comparison test (c–e, h and i) and two-tailed unpaired-t test (a, b, and g). Source data are provided as a Source Data file.
Conversely, overexpression of PNPLA6 in ARPE-19 cells, in which PNPLA6 expression was increased approximately 100-fold over that in control cells (Supplementary Fig. 3a), resulted in marked reductions in total PC and LPC species (Supplementary Fig. 3b). Virtually all PC and LPC species, as well as most other phospholipids and lysophospholipids, were reduced in PNPLA6-overexpressing ARPE-19 cells relative to those in control cells (Supplementary Fig. 3c–g). Exceptionally, rather than decreasing, several PE species remained unchanged or increased slightly in PNPLA6-overexpressing cells (Supplementary Fig. 3d and e), probably because PE hydrolysis might be decreased or PE biosynthesis might be complementarily increased owing to the decrease in other phospholipids through an unknown mechanism. Thus, PNPLA6 exhibits PLB activity in ARPE-19 cells, hydrolyzing both the sn-1 and sn-2 positions of various phospholipids and lysophospholipids, depending on its expression level. At an endogenous expression level, PNPLA6 hydrolyzes PC/LPC and possibly PE/LPE, PS, and PI, whereas at an excess expression level, PNPLA6 broadly hydrolyzes various phospholipids and lysophospholipids.
To obtain the in vivo relevance of these observations, we next analyzed phospholipids in the ocular tissue. However, overall changes in phospholipids in the whole eyes of ocular-selective Pnpla6-deficient mice compared to control mice were marginal (Supplementary Fig. 3h and i), likely because various cell types other than RPE cells were also present in the ocular tissue. We, therefore, performed lipidomics of the RPE layer isolated from the ocular tissue. In agreement with the in vitro results as above, there were significant increases in PC and LPC and a decrease in choline by ocular-selective PNPLA6 deficiency (Fig. 3g–j). Most other phospholipid and lysophospholipid species (except PE and LPE) were also elevated in RPE cells from ocular-selective Pnpla6-deficient mice relative to those from control mice (Fig. 3j). Taken together, these in vitro and in vivo results led us to conclude that PNPLA6 acts as a PLB, catalyzing sequential PLA1/2 and lysophospholipase reactions to hydrolyze various phospholipids and lysophospholipids, particularly PC and LPC to generate GPC, which is then metabolized to choline by GDEs (see below), in RPE cells (Supplementary Fig. 3j).
PNPLA6 regulates growth, adhesion, phagocytosis, and mitochondrial function in RPE cells
Scanning electron microscopy (SEM) revealed that PNPLA6-knockdown ARPE-19 cells were smaller than control cells (Fig. 4a and b). Microarray gene expression profiling followed by Gene Ontology (GO) analysis showed that PNPLA6 knockdown altered the expression of a group of genes mainly related to cell proliferation and adhesion (Fig. 4c). Although PNPLA6 knockdown did not cause cell death, cell proliferation and intracellular choline concentration were decreased in knockdown cells compared with those in control cells (Fig. 4d and e). Culturing PNPLA6-knockdown cells in a medium containing a 10-fold concentration of choline fully restored cell proliferation and intracellular choline concentration to levels similar to those in control cells (Fig. 4d and e). Conversely, PNPLA6 overexpression in ARPE-19 cells facilitated cell proliferation and increased intracellular choline levels (Supplementary Fig. 4a and b). Supplementing parental ARPE-19 cells with exogenous choline increased intracellular choline levels and promoted cell proliferation (Supplementary Fig. 4c and d), confirming the dependence of cell growth on choline. Cell cycle analysis through flow cytometry showed that the cells in the G1 phase were increased, while those in the G2/M phase were decreased, in PNPLA6-knockdown cells compared to control cells (Fig. 4f and Supplementary Fig. 4e). When the PNPLA6-knockdown cells were cultured in a high-choline medium, the cell cycle progressed from the G1 phase to G2/M phase similar to the case in control cells (Fig. 4f and Supplementary Fig. 4e). Thus, PNPLA6-driven mobilization of endogenous choline may be crucial for cell cycle progression and ultimately cell proliferation.

a SEM images of PNPLA6-knockdown (si PNPLA6) and control (scramble) ARPE-19 cells. Scale bar, 1 μm. b Cell sizes of PNPLA6-knockdown and control cells, with control cells as 1 (n = 7 from a, biological replicates). c Microarray of PNPLA6-knockdown cells relative to control cells. GO analysis of genes altered in knockdown relative to control cells (left) and a heatmap of proliferation-related genes that were decreased in knockdown relative to control cells (right). d Proliferation in normal medium (containing 64.3 μM choline) or in medium containing 10-fold excess choline (643 μM), with the normal medium as 1 (n = 3 biological replicates). e Cellular choline levels (n = 3 biological replicates). f Cell cycle analysis of control cells (scramble), PNPLA6-knockdown cells (si PNPLA6), and PNPLA6-knockdown cells with excess choline (si PNPLA6 + choline) (n = 3 biological replicates). g TEER analysis (n = 8 biological replicates). h Immunostaining of ZO-1. Green, ZO-1; blue, DAPI. Scale bar, 10 μm. Arrows indicate areas where ZO-1 was disturbed in the cell periphery. i Phagocytosis of FITC-labeled (red) porcine POS. Scale bar, 10 μm. j Quantification of phagocytosis, with control cells as 1 (n = 3 biological replicates). k JC-1 staining. Scale bar, 10 μm. Bottom right, magnified image. l The red/green ratio of JC-1 staining (n = 5 biological replicates). m Oxygen consumption rate determined by an extracellular flux analyzer (n = 4 biological replicates). n Cellular ATP levels (n = 4 biological replicates). o Immunoblotting of mitochondrial proteins (n = 3 biological replicates). p Densitometry of mitochondrial proteins relative to β-actin, with control cells as 1 (n = 3 from o, biological replicates). q Heatmap of water-soluble metabolites (n = 4 biological replicates). Data are presented as mean ± SEM and compiled from or representative of at least two independent experiments. P-values were derived from two-way ANOVA (m) and one-way ANOVA with Tukey’s multiple comparison test (d–g and n) and two-tailed unpaired-t test (b, j, l, and p). *P < 0.05 versus control cells (n). Source data are provided as a Source Data file.
To assess the impact of PNPLA6 silencing on cell adhesion, the transepithelial electrical resistance (TEER) of confluent PNPLA6-knockdown ARPE-19 cells was compared to that of control cells. PNPLA6-knockdown cells showed a lower TEER than did control cells (Fig. 4g), suggesting that cell-cell adhesion is sparse in the former. When PNPLA6-knockdown cells were cultured in a high-choline medium, the TEER was recovered to the normal level seen in control cells (Fig. 4g). In PNPLA6-knockdown cells, immunofluorescent staining of zonula occludens/tight junction protein 1 (ZO-1), a tight junction marker, was disturbed at the cell periphery and dispersed in the cytoplasm (Fig. 4h), suggesting insufficient formation of tight junctions owing to the mislocalization of ZO-1. Furthermore, microarray analysis revealed that desmocollin-3 (DSC3), a transmembrane protein of the cadherin family that together with desmoglein is involved in the formation of the adhesion core of desmosomes23, was decreased in PNPLA6-knockdown cells and conversely increased in PNPLA6-overexpressing cells (Supplementary Fig. 4f). Consistently, qPCR confirmed that DSC3 expression was markedly lower in PNPLA6-knockdown cells than in control cells (Supplementary Fig. 4g). DSC3-knockdown cells had a lower TEER than did control cells (Supplementary Fig. 4h and 4i), suggesting that the impairment of cell adhesion in PNPLA6-knockdown cells could, at least in part, be due to the reduced expression of DSC3.
RPE cells have long microvilli and maintain photoreceptor homeostasis by phagocytosing aged or damaged photoreceptor outer segments (POS)24. Since the number of microvilli was decreased in RPE cells of Pnpla6-deficient mice in vivo (Fig. 1m and n) and in PNPLA6-silenced ARPE-19 cells in vitro (Supplementary Fig. 4j), we examined the effect of PNPLA6 knockdown on the phagocytotic function. Phagocytosis of fluorescence-labeled porcine POS was markedly impaired in PNPLA6-knockdown ARPE-19 cells relative to that in control cells (Fig. 4i and j). This defective phagocytosis was restored by supplementation with a high concentration of choline in PNPLA6-knockdown cells (Supplementary Fig. 4k).
Evaluation of the mitochondrial membrane potential of ARPE-19 cells by JC-1 staining showed higher green fluorescence (Supplementary Fig. 4l) and lower red/green fluorescence intensity ratio (Fig. 4k and l) in PNPLA6-knockdown cells than in control cells, suggesting that PNPLA6 knockdown decreased the mitochondrial membrane potential. Analysis of the cellular metabolic state using an extracellular flux analyzer revealed that the maximal respiration (oxygen consumption rate; OCR) upon treatment with the mitochondrial uncoupler FCCP (carbonyl cyanide-p-trifluoromethoxyphenylhydrazone) was lower in PNPLA6-knockdown cells than in control cells (Fig. 4m). Accordingly, ATP production was reduced by nearly half in knockdown cells relative to that in control cells (Fig. 4n). These metabolic changes were rescued by culturing PNPLA6-knockdown cells in high-choline medium (Fig. 4 m and n). In contrast, the extracellular acidification rate (ECAR), which reflects lactate production via glycolysis, did not differ between PNPLA6-knockdown and control cells (Supplementary Fig. 4m). Immunoblotting of mitochondrial proteins revealed significant reduction of mitofusin 1 (MFN1) and dynamin-related protein 1 (DRP1), but not cytochrome c oxidase subunit 4 (COX IV), in PNPLA6-knockdown cells compared to control cells (Fig. 4o and p), suggesting disturbed mitochondrial fusion and fission by PNPLA6 deficiency.
Metabolome analysis showed that several glycolytic metabolites, such as glucose-6-phosphate, 3-phosphoglycerate, and phosphoenol pyruvate, and metabolites involved in the TCA cycle, such as acetyl-CoA, citrate, cis-aconitate, and isocitrate, were substantially decreased in PNPLA6-knockdown ARPE-19 cells relative to control cells (Fig. 4q). However, in agreement with the unaltered ECAR (Supplementary Fig. 4m), lactate (the end byproduct of glycolysis) was not affected by PNPLA6 deficiency (Fig. 4q), suggesting that its level might be counterbalanced at the step of lactate dehydrogenase or through other mechanisms. The accumulation of ribose-related metabolites in the pentose phosphate pathway and the decrease in phosphoribosyl-2-phosphate (PRPP) in PNPLA6-knockdown cells might reflect the reduced utilization of ribose metabolites for the synthesis of the nucleotide precursor PRPP, which may be related to reduced nucleotide synthesis and, ultimately, decreased cell proliferation. In addition, nucleotide triphosphates, including ATP, CTP, GTP, and UTP, were decreased, whereas nucleotide mono- and diphosphates were increased, in knockdown cells (Fig. 4q), which is consistent with the observed decreases in mitochondrial potential and energy production. Decreases in GPC and ethanolamine phosphate in PNPLA6-knockdown cells likely mirrored the reduced breakdown of PC and PE via the loss of PNPLA6 (Fig. 4q), validating the metabolome analysis. Conversely, most of these metabolites were increased in PNPLA6-overexpressing ARPE-19 cells (Supplementary Fig. 4n). These metabolic changes were largely, if not solely, recapitulated in the eyes of tamoxifen-inducible, ocular-selective Pnpla6-deficient mice (Supplementary Fig. 4o). These results suggest that PNPLA6-driven phospholipid catabolism affects various metabolic pathways, including the TCA cycle, glycolysis, pentose phosphate pathway, mitochondrial oxidative phosphorylation for ATP production, and nucleotide biosynthesis, in ARPE-19 cells. The decreases in various glycolytic and TCA cycle metabolites in PNPLA6-knockdown cells may be due to the reduced metabolic flux from GPC, a metabolite immediately downstream of PNPLA6 (Supplementary Fig. 4p).
PNPLA6 knockdown increases oxidative stress in RPE cells in vitro and in vivo
PNPLA6-knockdown ARPE-19 cells had increased ROS (Supplementary Fig. 5a and b) and accumulated malondialdehyde, a marker of lipid peroxidation (Supplementary Fig. 5c). Microarray analysis of RPE cells isolated from tamoxifen-treated, ocular-selective Pnpla6-deficient mice compared with control mice revealed notable alterations in oxidative stress-related pathways (Supplementary Fig. 5d). These results suggest that perturbed phospholipid metabolism and mitochondrial dysfunction caused by PNPLA6 deficiency increase the susceptibility of membrane phospholipids in RPE cells to oxidative stress.
We, therefore, investigated the effects of PNPLA6 deficiency in a NaIO3-induced retinal degeneration model, in which NaIO3-induced ROS damage in RPE cells rapidly induces retinal degeneration25,26. Pnpla6fl/flCAG-CreER(+) and control Pnpla6fl/flCAG-CreER(−) mice were treated with a suboptimal concentration of tamoxifen via eyedrops (Supplementary Fig. 5e), a condition under which Pnpla6 expression in RPE cells was incompletely ablated by 64% (Supplementary Fig. 5f) and therefore retinal degeneration did not occur spontaneously in Pnpla6fl/flCAG-CreER(+) mice (Supplementary Fig. 5g and h). In control Pnpla6fl/flCAG-CreER(−) mice, modest thinning of the ONL was observed only at the highest concentration (50 mg/kg) of NaIO3, whereas the ONL was markedly thinned even at a lower concentration (25 mg/kg) of NaIO3 in Pnpla6fl/flCAG-CreER(+) mice (Supplementary Fig. 5g and h), indicating that the ocular-selective decrease in PNPLA6 expression exacerbated ROS-dependent retinal degeneration.
PNPLA6-derived choline recycling into PC re-biosynthesis through the Kennedy pathway is crucial for RPE cell homeostasis
Since the mobilization of choline from PC is crucial for RPE cell homeostasis, we attempted to identify GDE isoforms downstream of PNPLA6 that catalyze the conversion of GPC to choline. Among the seven GDE isoforms, GDE1 was the most abundantly expressed, followed by GDE2/GDPD5 and GDE5/GPCPD1, whereas the expression levels of GDE3/GDPD2, GDE4/GDPD1, GDE6/GDPD4, and GDE7/GDPD3 were minimal, in ARPE-19 cells (Supplementary Fig. 6a). Knockdown of GDE1, GDE2, and GDE5 by respective siRNAs decreased their expression levels by 80–90% (Supplementary Fig. 6b), which resulted in significant reductions in intracellular choline levels and cell proliferation, with GDE2 knockdown having the greatest effect (Supplementary Fig. 6 c and d). When these knockdown cells were cultured in a high-choline medium, intracellular choline levels and cell proliferation were recovered to levels equivalent to those in control cells (Supplementary Fig. 6c and d). In contrast, both intracellular choline levels and cell proliferation in GDE3-, GDE4-, GDE6-, and GDE7-knockdown cells were comparable to those in control cells (Supplementary Fig. 6e and f). Thus, GPC produced from PCs by PNPLA6 is metabolized to choline by GDE2 and, to a lesser extent, by GDE1 and GDE5 in ARPE-19 cells.
In the Kennedy pathway for de novo PC biosynthesis, choline is phosphorylated by the choline kinase CHKα or CHKβ to be converted to phosphocholine, which is then metabolized to CDP-choline by the choline phosphate cytidylyltransferase PCYT1A/CCT-α and then to PC by the choline phosphotransferase CHPT1. Therefore, we speculated that the choline pool endogenously mobilized from PC by the sequential action of PNPLA6 and GDE1/2/5 would be utilized for the regeneration of PC through this metabolic pathway. Metabolomics of whole ocular tissues demonstrated substantial decreases of choline, phosphorylcholine, and CDP-choline in tamoxifen-inducible, ocular-selective Pnpla6-deficient mice relative to those in control mice (Supplementary Fig. 4o), substantiating this hypothesis. Since ARPE-19 cells expressed CHKA, PCYT1A, and CHPT1 (Supplementary Fig. 6g), we knocked down these enzymes by > 90% using their respective siRNAs (Supplementary Fig. 6h). In the knockdown cells, intracellular choline levels were unchanged or even elevated, whereas cell proliferation was significantly suppressed, relative to those in control cells (Supplementary Fig. 6i and j). Intracellular choline levels were increased markedly when these knockdown cells were cultured in high-choline medium, possibly because the metabolic flow from choline to PC through the Kennedy pathway was impeded (Supplementary Fig. 6i); nonetheless, cell proliferation of knockdown cells was not restored and remained lower than that of control cells (Supplementary Fig. 6j). Thus, PC biosynthesis mediated by CHKα, PCYT1A, and CHPT1 through the Kennedy pathway, but not choline per se, is critical for cell proliferation. In corroboration, as in the case of PNPLA6 knockdown (Fig. 3k and l), CHKA knockdown also decreased the mitochondrial membrane potential in ARPE-19 cells (Supplementary Fig. 6k and l).
In the PC-catabolic pathway, GPC is split into choline and glycerol-3-phosphate (G3P), which is sequentially converted to LPA, PA, diacylglycerol (DAG), and triacylglycerol (TAG) by G3P acyltransferases (GPATs), LPA acyltransferases (LPAATs), PA phosphatases (lipins), and DAG acyltransferases (DGATs), respectively. GPAT4, AGPAT1, LPIN1, and DGAT1 are the main isoforms expressed in APRE-19 cells (Supplementary Fig. 6m). However, PNPLA6 knockdown did not affect cellular TAG levels (Supplementary Fig. 6n). Choline can be linked to the methionine cycle in the liver and acetylcholine synthesis in neurons27; however, the contribution of these choline-metabolic routes was unlikely in APRE-19 cells since the key enzymes responsible for these processes (BHMT and CHAT, respectively) were minimally expressed (Supplementary Fig. 6o). Collectively, these results suggest that a pool of choline mobilized from PC by the sequential action of PNPLA6 and GDE1/2/5 is utilized to regenerate PC through the Kennedy pathway and that impairment of this PC recycling hampers RPE cell homeostasis (Supplementary Fig. 6p).
PNPLA6-mediated choline turnover is required for primary RPE homeostasis
Next, we evaluated whether the results observed in the transformed RPE cell line APRE-19 could also be applicable to primary human RPE cells. As in ARPE-19 cells, knockdown of PNPLA6 by > 90% in primary RPE cells (Fig. 5a) resulted in increases in PC (Fig. 5b) and LPC (Fig. 5c), accompanied by a decrease in choline (Fig. 5d). Furthermore, PNPLA6 knockdown in primary RPE cells reduced cell proliferation (Fig. 5e), transepithelial barrier (Fig. 5f), phagocytosis (Fig. 5g and h), and mitochondrial membrane potential (Fig. 5i and j). TEM revealed reduced mitochondria and microvilli in the knockdown cells (Fig. 5k and l). Furthermore, although the growth inhibition of PNPLA6-silenced primary RPE cells was rescued by exogenous choline, this effect was abrogated by MN58b, a CHKα-specific inhibitor (Fig. 5m), suggesting that the flux of PNPLA6-driven choline into the Kennedy pathway for PC synthesis is critical for cell proliferation. Thus, various defects caused by PNPLA6 knockdown in transformed ARPE-19 cells are recapitulated in primary RPE cells.

a qPCR analysis of PNPLA6 in PNPLA6-knockdown (si PNPLA6) and control (scramble) primary human RPE cells, with GAPDH as an internal control (n = 3 biological replicates). b, c Lipidomics of PC (n = 4 biological replicates) (b) and LPC (n = 3 biological replicates) (c) in PNPLA6-knockdown and control cells. d Changes in choline level in PNPLA6-knockdown cells relative to control cells, with the levels in control cells as 1 (n = 5 biological replicates). e Proliferation of PNPLA6-knockdown cells relative to control cells, with the levels in control cells as 1 (n = 3 biological replicates). f TERR analysis of PNPLA6-knockdown and control cells (n = 3 biological replicates). g Phagocytosis of FITC-labeled (red) porcine POS in PNPLA6-knockdown and control cells. Scale bar, 10 μm. h Quantitative values of phagocytosis, with control cells set as 1 (n = 4 biological replicates). i JC-1 staining of PNPLA6-knockdown and control cells. Scale bar, 10 μm. j Red/green ratio of JC-1 staining (n = 4 biological replicates). k, l TEM images of PNPLA6-knockdown and control cells. Representative images (left) with quantification of mitochondria number (n = 5 biological replicates) (right) (k) and microvilli (n = 5 biological replicates) (l) are shown. Arrows, mitochondria. Scale bar, 1 μm. (m) Effects of choline and CHKα inhibitor (MN58b) on the proliferation of PNPLA6-knockdown cells (n = 8 biological replicates). Data are presented as mean ± SEM and representative of at least two independent experiments. P-values were derived from one-way ANOVA with Tukey’s multiple comparison test (m) and two-tailed unpaired-t test (a–f, h, j, k, and l). Source data are provided as a Source Data file.
Paracrine supply of PNPLA6-driven choline from RPE cells is critical for photoreceptor cell survival
Although PNPLA6 is primarily expressed in RPE cells within the retina, mice with systemic, ocular-selective, and RPE-specific PNPLA6 deletion developed photoreceptor degeneration, which led to impaired visual function (Figs. 1 and 2). We hypothesized that RPE cells might support adjacent photoreceptor cell homeostasis by supplying PNPLA6-mobilized choline as an essential nutrient to photoreceptor cells in a paracrine manner. When 661 W cells, a mouse retinal cone photoreceptor cell line, were cultured in choline-depleted medium, cell proliferation was reduced compared to that in choline-sufficient medium (Fig. 6a). Intracellular choline levels were decreased (Fig. 6b) and TUNEL-positive dead cells were increased (Fig. 6c and d) in choline-depleted 661 W cells relative to those in choline-sufficient 661 W cells. Microarray gene profiling revealed that out of 26,487 genes, 1970 (7.4%), including a panel of apoptosis-related genes, showed more than 2-fold changes in their expression in choline-depleted 661 W cells relative to choline-sufficient 661 W cells (Fig. 6e and f). TEM analysis showed that the number of mitochondria was decreased in choline-depleted 661 W cells relative to that in choline-sufficient 661 W cells (Fig. 6g and h), consistent with the in vivo results in which mitochondrial abnormalities and TUNEL-positive signals were observed in photoreceptor cells of ocular-selective Pnpla6-deficient mice (Fig. 2o). Pnpla6 expression was low in photoreceptor cells, and its knockdown did not affect cell proliferation or morphology (Supplementary Fig. 7a–c), suggesting that the survival and proliferation of photoreceptor cells depend largely on exogenous, not endogenous, choline.

a, b Proliferation (a; n = 6 biological replicates) and intracellular choline levels (b; n = 3 biological replicates) in 661 W cells cultured in choline-sufficient (+) or choline-deficient (−) medium. c TUNEL staining of 661 W cells. d Quantification of dead cells per field of view (n = 4 biological replicates). Scale bar, 50 μm. e, f Microarray analysis of 661 W cells. Scattered plot (e) and heatmap of apoptosis-related genes (f) in choline-deficient relative to choline-sufficient cells. g TEM images of 661 W cells. Arrowheads, mitochondria. Scale bar, 1 μm. h The number of mitochondria in choline-deficient (n = 6 biological replicates) and -sufficient (n = 4 biological replicates) cells per image in (g). i Choline concentrations in medium of PNPLA6-knockdown (si PNPLA6; n = 3 biological replicates) and control (scramble; n = 4 biological replicates) ARPE-19 cells cultured in choline-deficient medium. j Photos of 661 W cells cultured in a conditioned medium from PNPLA6-knockdown or control ARPE-19 cells. Scale bar, 50 μm. k Proliferation of 661 W cells (n = 3 biological replicates). l Proliferation of 661 W cells cultured in a conditioned medium from PNPLA6-knockdown or control ARPE-19 cells with or without 10-fold excess choline (n = 3 biological replicates). m qPCR of Slc44a1 in Slc44a1-knockdown or control 661 W cells (n = 4 biological replicates). n Proliferation of Slc44a1-knockdown or control 661 W cells, with control cells as 1 (n = 5 biological replicates). o Proliferation of Slc44a1-knockdown or control 661 W cells cultured with conditioned medium from PNPLA6-knockdown or control ARPE-19 cells (n = 3 biological replicates). p Proliferation of 661 W cells cultured in conditioned medium from PNPLA6-knockdown or control ARPE-19 cells with or without 1 μM MN58b or 10-fold excess choline (n = 3 biological replicates). Data are presented as mean ± SEM and representative of at least two independent experiments. P-values were derived from one-way ANOVA with Tukey’s multiple comparison test (l, o, and p) and two-tailed unpaired-t test (a, b, d, h, i, k, m, and n). Source data are provided as a Source Data file.
Control ARPE-19 cells cultured in choline-depleted medium for 72 h released choline into the culture medium, whereas this release was markedly decreased in PNPLA6-knockdown ARPE-19 cells (Fig. 6i). Proliferation of 661 W photoreceptor cells cultured in conditioned medium obtained from PNPLA6-knockdown ARPE-19 cells was significantly lower than that of cells cultured with conditioned medium obtained from control ARPE-19 cells (Fig. 6j and k). This decrease in proliferation was entirely rescued by choline supplementation to the medium (Fig. 6l), suggesting that the choline released from RPE cells in a PNPLA6-dependent manner supports photoreceptor cell proliferation and survival. Cellular uptake of choline is mediated by choline transporters, including CHT1 with high affinity, SLC44A1-5 with intermediate affinity, and SLC22A1-3 with low affinity and poor specificity28. In addition, FLVCR1 and FLVCR2 have recently been identified as high-affinity choline transporters29. Microarray analysis revealed that 661 W cells expressed Slc44a1, Slc44a2, Flvcr1, and Flvcr2, among which only Slc44a1 expression was increased (4.1-fold) upon choline deficiency (Supplementary Fig. 7d). Knockdown of Slc44a1 reduced 661 W cell proliferation by ~ 30% (Fig. 6m and n), accompanied by reduced SLC44A1 immunostaining (Supplementary Fig. 7e), implying that SLC44A1 is responsible for choline uptake in these cells. Although the reduced proliferation of 661 W cells after culture with the conditioned medium from PNPLA6-knockdown ARPE-19 cells was reversed by supplementation with exogenous choline, this rescue effect was abrogated upon Slc44a1 silencing in 661 W cells (Fig. 6o). In 661 W cells cultured in a choline-depleted medium, the levels of several PC species were reduced, whereas those of PE species remained unchanged or even complementarily increased, compared with those in cells cultured in choline-sufficient medium (Supplementary Fig. 7f), suggesting that exogenous choline incorporated into photoreceptor cells is used to generate PC. We confirmed that exogenous d9-choline was taken up by 661 W cells and promptly converted to d9-choline-containing PC within 2 h (Supplementary Fig. 7g). Furthermore, inhibition of PC biosynthesis using the CHKα inhibitor MN58b blocked the proliferation of 661 W cells and canceled the rescue effect of exogenous choline on cells cultured with the conditioned medium from PNPLA6-knockdown RPE cells (Fig. 6p). Collectively, RPE cells release PNPLA6-mobilized choline extracellularly, which is acquired by photoreceptor cells via SLC44A1 and utilized for PC biosynthesis to maintain photoreceptor homeostasis. Impairment of this paracrine process leads to decreased proliferation and increased death of photoreceptor cells, with alterations in the expression of numerous genes.
Choline supplementation prevents progressive retinal degeneration in vivo
Having known that choline functions as a key metabolite downstream of PNPLA6, we speculated that retinal degeneration caused by PNPLA6 deficiency in vivo could be ameliorated by choline supplementation. Therefore, we administered 2% (w/v) choline solution and tamoxifen to Pnpla6fl/flCreER(+) mice via eyedrops and analyzed the retinal pathology after 2 weeks (Fig. 7a). Strikingly, choline supplementation largely restored retinal thinning (Fig. 7b and c), dissociation and atrophy of the optic discs in photoreceptor cells (Fig. 7d), and visual function in the dark and light (Fig. 7e) in Pnpla6fl/flCreER(+) mice to levels similar to those in control Pnpla6fl/flCreER(−) mice. Thus, retinal degeneration in ocular-selective Pnpla6-deficient mice is ameliorated by topical choline supplementation. Together, PNPLA6-produced choline in RPE cells is transported into photoreceptor cells to maintain photoreceptor homeostasis, whereas PNPLA6 deficiency in RPE cells hampers both RPE and photoreceptor functions, leading to retinal degeneration (Fig. 7f).

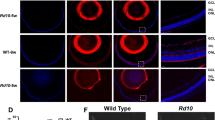
a Protocol for topical choline application to the eyes of ocular-selective Pnpla6-deficient mice. b HE staining of retinas from tamoxifen-treated Pnpla6fl/flCreER(−) and Pnpla6fl/flCreER(+) mice with or without choline eyedrops. Scale bar, 50 µm. c OCT analysis of ONL thickness of retinas in Pnpla6fl/flCreER(−) and Pnpla6fl/flCreER(+) mice with or without choline eyedrops (n = 6 independent animals). d TEM analysis of retinas in Pnpla6fl/flCreER(−) and Pnpla6fl/flCreER(+) mice. Arrowheads, disc structure of photoreceptors. Scale bar, 1 µm. e ERG analysis of Pnpla6fl/flCreER(−) and Pnpla6fl/flCreER(+) mice with or without choline eyedrops. Amplitudes of a-wave (left) and b-wave (right) under scotopic conditions are shown (n = 3 independent animals). f Schematic presentation of the function of PNPLA6 in the retina. In RPE cells, mobilization of choline from PC by PNPLA6 is crucial for RPE homeostasis. PNPLA6-produced choline in RPE cells is transported into photoreceptor cells to maintain photoreceptor homeostasis. PNPLA6 deficiency in RPE cells hampers both RPE and photoreceptor functions, leading to retinal degeneration. Data are presented as mean ± SEM and representative of at least two independent experiments. P-values were derived from two-way ANOVA with Tukey’s multiple comparison test (c and e). *P < 0.05, **P < 0.01, and **** P < 0.0001 versus control mice (c, e). Source data are provided as a Source Data file.
Mutations in PNPLA6-related disorders impairs enzymatic function of PNPLA6
Point mutations in human PNPLA6 in PNPLA6-related disorders are often found in the nucleotide-binding domains or patatin domain, where the active center residues are found16,17. Herein, we analyzed the point mutations T1058I and G726R in Boucher–Neuhäuser and Laurence–Moon syndromes, respectively. We constructed and overexpressed these two PNPLA6 point mutants, the active center mutant S1014A, and wild-type human PNPLA6 in ARPE-19 cells (Fig. 8a and b). Although wild-type PNPLA6 increased intracellular choline levels and proliferation compared to that in control cells, S1014A, T1058I, and G726R failed to do so (Fig. 8c and d). Therefore, the two disease-associated PNPLA6 point mutants (T1058I and G726R) lost their enzymatic activity.

a PNPLA6 point mutations reported in human retinal degeneration. We generated three-point mutants, G726R (found in Laurence-Moon syndrome), T1058I (found in Boucher-Neuhäuser syndrome), and S1014A (the catalytic center), of human PNPLA6. b–d qPCR of PNPLA6 relative to GAPDH (b), intracellular choline levels (c), and cell proliferation (with the value of mock cells as 1) (d) in ARPE-19 cells transfected with WT or mutant PNPLA6 (n = 3 biological replicates). e 3D structural model of PlpD of Pseudomonas aeruginosa used a template. Amino acid sequences are shown in ribbons, and serine (S) and aspartate (D) in the active site are indicated by space-filling models (red, oxygen; yellow, carbon). f 3D structural model of residues 977−1260 including the patatin domain of PNPLA6, constructed by homology remodeling using SWISS-MODEL. Green, lid structure; light blue, α-helix; blue, β-strand. g The surface of the patatin domain of PNPLA6. Left, lid is pictured as ribbon; Right, all components are pictured as superficial structure. Pink, S1014; yellow, hydrophobic residues around the channel; green, lid. h Structure of the vicinity of the active center in the patatin domain of WT PNPLA6. Left, full image; Right, magnified image of the lid. i Structure of the vicinity of the active center in the patatin domain of T1058I mutant. Left, full image; Right, magnified image of the lid; dotted lines, hydrophobic interaction. Data are presented as mean ± SEM and representative of at least two independent experiments. P-values were derived from one-way ANOVA with Tukey multiple comparison test (b, c, and d). Source data are provided as a Source Data file.
To understand how PNPLA6 activity is impaired by T1058I mutation, homology modeling of the putative three-dimensional (3D) structure of PNPLA6 (residues 977–1260 encompassing the patatin domain) was performed in silico using the SWISS-MODEL30,31, with Pseudomonas aeruginosa Patatin-like protein (PlpD, UniProt: Q9HYQ6), whose tertial structure of residues 20–333 that includes the patatin domain had been solved31,32,33, as a suitable template (Fig. 8e and f). In a steady state, the catalytic site of PlpD is covered by a lid structure; however, in an active state, it undergoes a conformational change, moving away from the catalytic channel and thereby relieving it from inhibition of activity by the lid31 (Fig. 8g). In wild-type PNPLA6, T1058, which resides at the C-terminal side of residues 1041–1057 in the lid region, is located at the tip of the channel where the lid covers the catalytic site and the channel entrance (Fig. 8h). The T1058I mutation at the base of the lid is likely to reduce the flexibility of the lid by strengthening the hydrophobic interaction between I1058 and V1050 or L1047 within the α-helix of the lid (Fig. 8i). Thus, our in silico model suggests that the catalytic domain of PNPLA6 is regulated by the lid: when the lid is open, the active center becomes accessible to phospholipid substrates, whereas a T1058I mutation could enhance the hydrophobic interaction with the lid, thereby closing the catalytic center and leading to loss of enzymatic activity. Although the mechanism by which a G726R mutation affects the structure of PNPLA6 is unknown, our results indicate that the third nucleotide-binding domain is crucial for choline mobilization by this enzyme.
Discussion
Using genetic, histological, cell biological, transcriptome, metabolome, and in silico structural analyses, this study has provided unequivocal evidence that PNPLA6 plays an essential role in retinal homeostasis by controlling choline availability for phospholipid recycling. In the retina, PNPLA6 is predominantly expressed in RPE cells and contributes to the mobilization of choline from membrane PC through its PLB activity. Retina-specific conditional depletion of PNPLA6 in mice through multiple strategies led to retinal degeneration reminiscent of retinitis pigmentosa. PNPLA6 knockdown markedly perturbed the proliferation, adhesion, mitochondrial metabolism, and phagocytic function of cultured RPE cells. Furthermore, PNPLA6-driven choline is secreted from RPE cells and incorporated into adjacent photoreceptor cells to support their proliferation, survival, optic disc structure, and function. Choline supplementation can efficiently rescue these abnormalities in vivo and in vitro. Moreover, PNPLA6 mutations relevant to Boucher–Neuhäuser and Laurence–Moon syndromes impair the ability of PNPLA6 to generate choline. These results collectively suggest that the primary role of PNPLA6 in the retina is to maintain retinal homeostasis in RPE and adjacent photoreceptor cells by hydrolyzing membrane PC to mobilize choline that is recycled to the membrane as a component of newly synthesized PC.
Since PNPLA6 is ubiquitously expressed in various organs, with high expression in the central nervous system, lymphoid tissue, kidney, lung, and testis34, its mutation or deficiency in both mice and humans could be associated with diverse phenotypes, including neurodegeneration17,18,34,35. Inducible deletion of PNPLA6 in the entire body of conditional Pnpla6-deficient mice after i.p. tamoxifen treatment compromised systemic metabolism and led to early death, indicating the critical role of PNPLA6 in multiple tissues. Although Pnpla6-deleted Drosophila are normal immediately after eclosion, photoreceptor cells in the retina displayed abnormal features at 3 weeks, suggesting that retinal degeneration is not merely a secondary outcome caused by neurodegeneration that occurs at a later stage18. Here, we found that PNPLA6 is expressed abundantly in RPE cells but only at lower levels in the neural retina, including photoreceptor cells. Our approaches using inducible Pnpla6fl/flCAG-CreER mice treated with tamoxifen eyedrops (ocular-selective Pnpla6-deficient mice) and Pnpla6fl/flBest1-Cre mice (RPE-specific Pnpla6-deficient mice), as well as gene silencing of PNPLA6 in cultured RPE cells, have revealed that retinal degeneration due to PNPLA6 deficiency is intrinsic to RPE cells. The impairment of RPE homeostasis by PNPLA6 deficiency eventually perturbs the function of adjacent photoreceptor cells, leading to optic disc degeneration and visual loss. It should be noted that we deleted Pnpla6 in the C57BL/6 N strain carrying rd8 mutation in the Crb1 gene, which causes mild retinal degeneration with reduced response to light over 10 months of ERG analysis36. Nevertheless, our results showed that tamoxifen-inducible Pnpla6 deletion in this strain gave more profound defects in retinal structure and functions only within 1 week and that its transient knockdown in cultured human RPE cells markedly perturbed cellular proliferation, epithelial barrier, phagocytosis, and mitochondrial functions, implying the essential role of PNPLA6 in RPE homeostasis.
In RPE cells, PNPLA6 catalyzes the PLB reaction, removing two fatty acyl chains from the sn-1 and sn-2 positions of PC and LPC to generate GPC, which is then metabolized by GDE1/2/5 to choline. In cells, choline constitutes the hydrophilic base of PC and sphingomyelin and is utilized for neurotransmitter synthesis, signal transduction, lipid transport, and methyl group metabolism37. In vivo, choline can be supplied via an endogenous pathway through PC degradation or an exogenous pathway derived from dietary sources. Although the endogenous pathway alone has long been considered insufficient to supply choline and the dietary intake of choline as an essential nutrient is desirable38, our studies using mice deficient in PNPLA6 in the retina, or those deficient in PNPLA7 (the closest paralog of PNPLA6) in the liver39, have provided insights into the critical role of endogenous choline mobilized from membrane PC by these two closely-related PLB/lysophospholipases in tissue and cellular homeostasis. In RPE cells, the metabolic flux of PNPLA6-driven endogenous choline into PC regeneration via the Kennedy pathway is crucial for cell proliferation, adhesion, phagocytosis, and mitochondrial homeostasis, whereas in hepatocytes, PNPLA7 participates in supplying endogenous choline to replenish the methionine cycle with methyl groups39. The former view is compatible with the findings that a PNPLA6/7 ortholog in the yeast Saccharomyces cerevisiae acts as an ER-resident PLB to produce GPC for PC re-biosynthesis via CDP-choline19 and that silencing of PNPLA6–9 in HeLa cells disturbs the turnover of PC and other phospholipids, resulting in the suppression of cell cycle progression40.
Thus, beyond the well-recognized roles of the PLA2 family as regulators of lipid mediator generation11,15,41 and phospholipid remodeling through the Lands cycle41,42,43, PNPLA6 is a critical regulator of phospholipid recycling by bridging phospholipid catabolism and regeneration to maintain cellular membrane homeostasis. Considering that PNPLA6 also hydrolyzes phospholipids/lysophospholipids other than PC/LPC, the perturbed turnover of other phospholipids might also underlie, at least in part, the severe degenerative phenotypes associated with PNPLA6 deficiency. Reportedly, alterations in the amount or composition of phospholipids caused by mutations or deletion of genes related to their degradation, biosynthesis, remodeling, and transport lead to disturbance of mitochondrial biogenesis and function not only in the retina7,8,9,10, but also in the liver, muscle, and macrophages29,44,45,46,47,48,49. ER and mitochondria are connected via the ER-mitochondria contact site, through which de novo synthesized phospholipids in the ER are delivered to mitochondria50,51. Therefore, it is not surprising that PNPLA6 deletion, which shuts off PC recycling, also results in mitochondrial degeneration. Disturbance of this fundamental metabolic pathway may also result in the retention of old or deteriorated phospholipids, such as oxidized phospholipids, and thereby increase oxidative stress, which can lead to cellular dysfunction and death52. Indeed, we observed an accumulation of oxidized phospholipids in PNPLA6-depleted RPE cells in vitro and an increase of dead cells in Pnpla6-deficient retinas in vivo. Since PNPLA9/iPLA2β, whose mutations in humans cause neurodegenerative diseases such as infantile neuroaxonal dystrophy (INAD), neurodegeneration with brain iron accumulation (NBIA), and parkinsonism (PARK14)53,54,55, plays a crucial role in preventing neuronal ferroptosis through the hydrolytic removal of oxidized phospholipids56, we speculate that PNPLA6 may also contribute to the protection against neuronal or retinal cell death by counteracting ferroptosis.
Since photoreceptor cells have no direct contact with blood vessels, many of their required ions and metabolites are supplied from adjacent RPE cells. Therefore, abnormalities in RPE cells can hamper an adequate nutrient supply to photoreceptor cells, resulting in photoreceptor dysfunction. We found that PNPLA6-driven choline is secreted from RPE cells, taken up by photoreceptor cells via the choline transporter SLC44A1, and utilized for PC synthesis in these cells and that this process is essential for photoreceptor homeostasis. Current evidence suggests that de novo synthesis of PC via the Kennedy pathway or fatty acid remodeling of PC via the Lands cycle is involved in the regulation of photoreceptor cells. For instance, mutations in the PCYT1A gene have been identified in retinitis pigmentosa4,57, and knockout or mutations of the lysophospholipid acyltransferases LPCAT1 and LPAAT3 profoundly perturb retinal homeostasis, leading to optic disc degeneration and visual function impairment7,9,58. Alternatively, choline can be metabolized to acetylcholine, which functions as a neurotransmitter or as a signaling molecule for cell growth, differentiation, and adhesion37,59. Although the α7 nicotinic acetylcholine receptor nAChR expressed in RPE cells is involved in Müller cell differentiation60, there has been no evidence that acetylcholine synthesized in the retina is involved in photoreceptor homeostasis. Since choline acetyltransferase is expressed in photoreceptor cells but not in RPE cells, the possibility that exogenous choline supplied from RPE cells to photoreceptor cells is converted to acetylcholine and contributes to retinal homeostasis should also be taken into consideration.
Choline metabolism and ocular diseases are closely related from a clinical standpoint. For example, choline or PC synthesized by choline metabolism constitutes the tear fluid oil layer and promotes tear fluid secretion via acetylcholine synthesis; as such, choline is involved in the protection against dry eye and maintenance of tear fluid on the ocular surface. Moreover, oral administration of choline alphoscerate can improve dry eye59,61. Choline is also essential for the normal development and function of retinal ganglion, amacrine, horizontal, and cone cells62, and CDP-choline treatment prevents retinal ganglion cell injury63. Since choline insufficiency can have various impacts on the anterior and posterior eyes, choline administration might have prophylactic and/or therapeutic effects on various ocular diseases. Circulating choline levels are approximately 10 μM when the recommended amounts of choline (550 and 425 mg/day in males and females, respectively) are ingested64. The concentration of choline in the culture medium used in this study is 64.3 µM, which is sufficient to rescue the abnormalities in PNPLA6-knockdown RPE cells. The concentration of choline used in our eyedrops to treat Pnpla6-deficient mice was set at 2% (= 191 mM) based on previous findings in vivo administration experiments65; however, the validity of this treatment method for human ocular diseases should be verified in the future. Although there have been no reports on in vivo choline concentrations in patients with PNPLA6-related disorders or those with reduced PNPLA6 expression or activity, systemic or ophthalmic supplementation with choline might be effective at preventing these diseases.
Neuronal deletion of PNPLA6 in mice, zebrafish, and flies leads to degeneration of motor neurons with abnormal axons and demyelination in the central nervous system17,18,34,35. Mutations in the human PNPLA6 gene lead to PNPLA6-related disorders with spastic paraplegia, ataxia, hypogonadism, and chorioretinal dystrophy16,17,18,20. Thus, although we focused on the function of PNPLA6 in RPE cells in this study, the possibility that loss of PNPLA6 in non-RPE cells, such as neural cells and Müller cells, also contributes to retinal pathology cannot be fully ruled out and should be addressed in the future study. While this manuscript is under revision, two additional high-affinity choline transporters, FLVCR1 and FLVCR2, have been identified29. FLVCR1-driven uptake of extracellular choline and ethanolamine is coupled with de novo PC and PE synthesis via the Kennedy pathway in various cells66, while FLVCR2 is expressed specifically in endothelial cells at the blood-brain barrier and is responsible for choline uptake into the brain67. Interestingly, FLVCR1 mutations in humans lead to posterior column ataxia and retinitis pigmentosa with loss of vision, abnormal proprioception, and muscle atrophy68, similar to the symptoms of PNPLA6-related disorders16,17,18,20. Thus, although beyond the scope of this study, FLVCR1, in addition to SLC44A1 as shown here, may also take part in choline uptake for PC synthesis in photoreceptor cells. Moreover, the similarity between the FVLCR1 and PNPLA6 mutations further supports our conclusion that PNPLA6 is associated with PC turnover for choline mobilization.
PNPLA6 was initially identified as an esterase that degrades organophosphorus compounds (OPs) and, therefore, named NTE69. Chemical modification of PNPLA6/NTE by OPs inhibited its carboxylesterase activity (measured by hydrolysis of phenyl valerate) and caused long axon degeneration in adults69,70 and dendrite growth alteration in vitro71. Glynn and coworkers isolated, cloned, and genetically characterized this enzyme, and described that PNPLA6/NTE has a phospholipase activity and is involved in phospholipid turnover72. Although inhibition of PNPLA6/NTE by OPs did not seem by itself to have a profound impact on early cell differentiation related to embryonic neurodevelopment73,74, subsequent genetic approaches clearly demonstrated that it plays an essential role in an early stage of embryonic development73,74 and axonal maintenance in adults70. Whole-body deletion of PNPLA6 in vivo caused embryonic lethality21 and its silencing in vitro perturbed the differentiation of mouse and human embryonic stem cells74. Neuronal phospholipid deacylation by this enzyme is essential for axonal and synaptic integrity75. Thus, the mechanism we have disclosed herein, i.e., PNPLA6-driven mobilization of choline through phospholipid turnover and the increase in oxidative stress by its loss, could be associated not only with retinal homeostasis but also with axonal maintenance and embryonic development. Further, given that chromaffin cells also have high PNPLA6 expression76, PNPLA6-mediated choline turnover might also affect the state of chromaffin cells in the adrenal medulla. These points, besides the retina, may merit future studies.
In conclusion, we have demonstrated that PNPLA6 acts as a PLB in RPE cells by producing choline through PC catabolism, promoting regeneration of membrane PC, and regulating the function of RPE and neighboring photoreceptor cells. PNPLA6 dysfunction disrupts the homeostasis of RPE cells and, secondarily, that of photoreceptor cells, resulting in progressive retinal degeneration. Given our present findings that choline supplementation prevents the progression of retinal degeneration related to PNPLA6 deficiency, the activation of choline metabolism or delivery may serve as an ophthalmic drug strategy for retinal degeneration.
Methods
Mouse breeding
All animal experiments were conducted in accordance with the University of Tokyo’s Regulations for the Conduct of Animal Experiments, the Statement for the Use of Animals in Ophthalmic and Vision Research of the Association for Research in Vision and Ophthalmology, and the Japanese Guide for the Care and Use of Laboratory Animals. All animals were kept in a specific pathogen-free environment in a 12 h light/dark cycle (light: 8:00–20:00, dark: 20:00–8:00), at a room temperature of 23 ± 1 °C and a humidity of 50 ± 10%. The animals were fed a CE-2 diet (Clea Japan) and were provided ultra-filtrated water, ad libitum.
Conditional Pnpla6-deficient mice
The targeting vector for the Pnpla6 gene was obtained from the Knockout Mouse Project (KOMP) Repository (PG00244_Z_7_C03; European Mouse Mutant Cell Repository (EuMMCR). The conditional-ready mutant allele was composed of two FRT sites flanking an internal ribosome entry site (IRES):lacZ-trapping cassette and a floxed human ACTB (β-actin) promoter-driven Neo cassette inserted into the sixth intron of the Pnpla6 gene and an additional third loxP site inserted at downstream of exon 8. The targeting vector was transferred into C57BL/6 N mouse-derived embryonic stem (ES) cells (RENKA; provided by Dr. Kenji Sakimura, Niigata University) via electroporation, and the cells that contained the correctly targeted Pnpla6 locus were identified through PCR and Southern blot analysis. Chimeric embryos were generated by aggregation of the recombinant ES cells and 8-cell stage ICR mouse embryos. Two F0 mice with 100% ES cell-derived coat color were mated with C57BL/6 N mice (Japan SLC) to achieve germline transmission. A conditional knockout mouse strain was generated by crossing heterozygous mice with CAG-Flp mice (Jackson Laboratory) expressing the flippase recombinase under the control of the chicken β-actin promoter (CAG). This resulted in the excision of the IRES:lacZ and the neo cassettes and the generation of a floxed allele. These floxed mice were mated with CAG-CreER transgenic mice in which the Cre-loxP system can be systemically activated through tamoxifen administration under the control of the CAG promoter77 to obtain inducible Pnpla6-deficient mice (Pnpla6fl/flCAG-CreER(+)) and their controls (Pnpla6fl/flCAG-CreER(−)). Mouse genotypes were determined through PCR using the tail biopsy (see the “Mouse genotyping” section). These mice, as well as mice deficient in PNPLA7, PNPLA8 or PNPLA939,78,79, on the C57BL/6 N background (male, 8–12 weeks) were used for experiments.
To conditionally delete PNPLA6, Pnpla6fl/flCreER(+) and control Pnpla6fl/flCreER(−) mice were treated with tamoxifen (Toronto Research Chemicals) dissolved in corn oil. For systemic PNPLA6 deficiency, 2 mg/day (2 mg/100 µl) of tamoxifen was administered intraperitoneally for 5 consecutive days. For ocular-selective PNPLA6 deficiency, these mice were treated with 0.6 mg/day (0.2 mg/10 µl three times a day) of tamoxifen by eyedrops continuously for 5 days. For RPE-specific PNPLA6 deficiency, Pnpla6fl/fl mice were crossed with Best1-Cre mice (C57BL/6-Tg1Jdun/J, strain number 017557, RRID: IMSR_JAX:017557; Jackson Laboratory).
Mouse genotyping
Individual mice were identified through ear punching. The mouse tails were cut into 3–4 mm pieces and lysed in 30 µl of Allele-In-One–Mouse Tail Direct Lysis Buffer (Allele Biotech) containing proteolytic enzymes by shaking at 55 °C overnight. The genotyping PCR reaction was run on an Applied Biosystems 9800 Fast Thermal Cycler (Applied Biosystems) using GeneAmp Fast PCR Master Mix (Thermo Fisher Scientific) to amplify the target gene from the template DNA. Primers used to detect Pnpla6 were as follows: forward, 5’-ACTTGCCTGTTATGTGTAGG-3’; reverse, 5’-TGATGACATCAAGGATGCTC-3’. Primers used to detect CreER were as follows: forward, 5’-ACTGGTCGAGCGATGGATTTCCGT-3’; reverse, 5’-CTTGTCCAGGACACGGTGGATGTG-3’. The PCR products were analyzed through electrophoresis on 1% (w/v) agarose gels containing ethidium bromide (Nippon gene).
Isolation and fractionation of mouse retina
Pnpla6fl/flCAG-CreER(+), Pnpla6fl/flCAG-CreER(−), Pnpla6fl/flBest1-Cre, and Pnpla6fl/fl mice were euthanized through cervical dislocation, after which their eyeballs were isolated, the periocular connective tissue was removed, and the optic nerve was isolated. The cornea was isolated at the corneal ring using a Vannas shear (World Precision Instruments), after which the iris and lens were removed. Using a microsurgical crescent knife (MANI) under a dissecting microscope (SZX7, Olympus), intact RPE sheets were peeled off from the underlying Bruch’s membrane. After washing with cultured medium twice, RPE cells were carefully collected in a 1.5 ml centrifuge tube, avoiding collection of other tissues such as retina debris or choroid, and carefully resuspended using a P200 micropipette80.
OCT analysis
Mydriasis of the pupils of Pnpla6fl/flCAG-CreER(+), Pnpla6fl/flCAG-CreER(−), Pnpla6fl/flBest1-Cre, and Pnpla6fl/fl mice was performed using 1 drop of 0.5% (w/v) tropicamide and 0.5% (w/v) phenylephrine ophthalmic solution (Midorin P, Santen Pharmaceutical Co.). Their heads were anesthetized, kept warm, and fixed using a finger to take cross-sectional retinal tomograms through the optic nerve papilla with an optical coherence tomograph (OCT Bi-µ, Kowa)81. Image J analysis software (National Institutes of Health) was used to quantify retinal and outer granular layer thickness.
NaIO3-induced retinal degeneration
Twelve-week-old Pnpla6fl/flCreER(+) or Pnpla6fl/flCreER(−) mice were treated with suboptimal tamoxifen (0.15 mg/day; 0.05 mg/10 μl in one eye, three times per day) for 5 consecutive days. One week after the last treatment, NaIO3 (Sigma-Aldrich) dissolved in phosphate-buffered saline (PBS) was administered intraperitoneally at concentrations of 10, 25, and 50 mg/kg82,83. After 1 week, OCT and histology analysis of the retina were performed.
ERG analysis
Mydriasis of the left eye of mice was performed using one drop of 0.5% (w/v) tropicamide and 0.5% (w/v) phenylephrine, and the ERG was measured under general anesthesia using a stimulation device (LS-W; Mayo) in dark and light conditions84. Each stimulus in the light condition was a flash with an intensity, and the response waveforms were recorded. The difference from the baseline to the minimum of the a-wave was measured as the amplitude of the a-wave, and the difference from the minimum of the a-wave to the maximum of the b-wave was measured as the amplitude of the b-wave; both of which were analyzed through LabChart 7 software (ADInstruments). The average of three stimuli under dark conditions and 32 stimuli under light conditions was calculated.
Histology and immunohistochemistry
Mouse retinas were fixed in 4% (w/v) paraformaldehyde (Fujifilm-Wako) at 4 °C overnight. Paraffin sections of human normal retinas were purchased from Paraffin Tissue Section – Human Adult Normal: Eye: Retina (T2234110, Biochain). The fixed tissues were dehydrated in 70% (v/v) ethanol and placed in Tissue-Tek VIP-5-Jr. (Sakura) for paraffin replacement. The paraffin-embedded sections (4 µm thick) were deparaffinized and stained with HE.
For immunostaining, deparaffinized sections were antigen-activated with 20 μg/ml proteinase K (Thermo Fisher Scientific-Invitrogen). Endogenous peroxidase was inactivated by treatment with 3% (v/v) H2O2 solution (Fujifilm-Wako) for 5 min and blocked with Block Ace (DS Pharma Biomedical) for 1 h. Then the sections were incubated with rabbit anti-human PNPLA6 antibody at 1:500 dilution (ab228683, Abcam)18 or mouse SLC44A1 antibody at 1:500 dilution (14687-1-AP, Proteintech) overnight at 4 °C, and then with Alexa Flour 647-labeled goat anti-rabbit IgG at 1:1000 dilution (ab15008, Thermo Fisher Scientific) for 1 h at room temperature. The tissue was sealed in VECTASHIELD Mounting Medium with DAPI (Vector Laboratories) and observed with an all-in-one fluorescence microscope (BZ-X710, Keyence, Excitation/Emission = 490/530 nm).
Electron microscopy
Pnpla6fl/flCAG-CreER(+), Pnpla6fl/flCAG-CreER(−), Pnpla6fl/flBest1-Cre, and Pnpla6fl/fl mice were euthanized and perfused with 4% paraformaldehyde through the heart. Perfusion-fixed eyeballs were removed and washed with PBS twice. Then, the ocular tissue was immersed in 2.5% (w/v) glutaraldehyde fixative solution at 4 °C overnight, after which the tissue was washed three times every 30 min with 0.1 M cacodylate buffer. ARPE-19, primary human RPE, and 661 W cells cultured in 6-well plates were fixed with 2.5% glutaraldehyde fixative solution (2 ml/well) for 30 min, after which they were washed three times with 0.1 M cacodylate buffer. The subsequent processes of block preparation, ultrathin section preparation, and photography for TEM and SEM were outsourced to Applied Medical Research for further analysis.
Cell culture
The human RPE cell line ARPE-19 (CRL-2302, ATCC) and the mouse retinal cone photoreceptor cell line 661 W (an SV40-inactivated clone was provided by Dr. Ryo Terao, Washington University) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 (Sigma-Aldrich) and DMEM high glucose (Sigma-Aldrich), respectively, with penicillin streptomycin (100 units/ml, Thermo Fisher Scientific) and 10% (v/v) fetal bovine serum (FBS, Gibco) in a CO2 incubator under 5% (v/v) CO2 at 37 °C. Although 661 W cells were reported to have a property of retinal ganglion precursor-like cells expressing both cone- and ganglion-specific markers85, probably because of mistaken cross-contamination of the two cell lines, our microarray analysis (Fig. 6e) showed that 661 W cells we used in this study did express the cone cell markers Rom1 and Otx2, but not the retinal ganglion cell marker Brn3a, confirming that our 661 W cells, which had also been used in our current studies81,86, were indeed cone photoreceptor cells. Human primary RPE cells (#6540, Sciencell Research Laboratories) was cultured with epithelial cell medium (#4101, Sciencell Research Laboratories) in a CO2 incubator under 5% (v/v) CO2 at 37 °C. The medium was changed every 2 days, and upon reaching confluence, the cells were washed with PBS, treated with 0.05% (w/v) trypsin and 0.53 mM EDTA (Fujifilm-Wako), and passaged at appropriate cell densities. As required for subsequent experiments, the cells were cultured in the high-choline or choline-deficient medium for 3 days (Research Institute for the Functional Peptides).
PNPLA6 overexpression
Human PNPLA6 cDNA (accession no. NM_001166111.1) were subcloned into the mammalian cell expression vector pcDNA3.1/Hygro(+) (GenScript) and transfected into ARPE-19 cells by lipofection using Lipofectamine 3000 (Thermo Fischer Scientific). The cells were used for the subsequent experiment 24 hours after the lipofection.
Gene silencing
Target-specific Silencer® Select siRNAs (Thermo Fischer Scientific) (Supplementary Table 1) or a Silencer® Select Negative Control No. 1 siRNA (4390843, Thermo Fischer Scientific) were transfected into ARPE-19, primary human RPE, or 661 W cells with Lipofectamine 3000.
Phagocytosis assay
POS were isolated from normal porcine eyes (Tokyo Shibaura Organ Corporation) under red dim light87. Collected POS were washed and treated with PBS containing 2.5% (w/v) sucrose and added to fluorescein isothiocyanate (FITC) (Dojindo) to obtain a final concentration of 1 mg/ml. After incubation in the dark at 4 °C for 1 h, FITC-labeled POS were washed with PBS and centrifuged at 800 × g for 10 min, dissolved in PBS containing 2.5% sucrose, and added to ARPE-19 cells or primary human RPE cells at a concentration of 10 POS/cell88. After incubation at 37 °C for 12 h, the cells were washed four times with PBS and treated with 0.4% (w/v) trypan blue (Sigma-Aldrich) in PBS for 10 min to stop the binding effect of FITC-labeled POS and to observe only internalized FITC-labeled POS89. After washing with PBS, the cells were fixed with cold 4% paraformaldehyde and observed using the all-in-one fluorescent microscope. Upon quantification, the background values obtained from cells without FITC-labelled POS was subtracted to eliminate the effect of autofluorescence.
Site-directed PNPLA6 mutagenesis
We generated point mutants of PNPLA6, G726R (c.2176 G > C), T1058I (c.3173 C > T)), and S1014A (c.3040 T > G)16,17. Using the human PNPLA6/pcDNA3.1/Hygro(+) as a template, inverse PCR was performed using the primers with the above-mentioned mutations and the KOD-Plus-Mutagenesis Kit (Toyobo), according to the manufacturer’s instructions. The template plasmid was cut by DpnI, and the DNA strand amplified through inverse PCR was self-ligated and transformed into the JetGiga Competent Cell (K12 strain DH5a, BioDynamics Laboratory). Escherichia coli clones with the gene product of interest were selected through colony PCR using Ex Taq Kit (Takara), and the plasmids were purified using PureYield Plasmid Miniprep System (Promega). The correct insertion of the mutation of interest was confirmed through DNA sequencing.
qPCR analysis
Total RNAs from the eye tissues or cultured cells were prepared using TRIzol Reagent (Thermo Fisher Scientific–Invitrogen) and dissolved in RNase-free water. First-strand cDNA synthesis by reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific–Applied Biosystems), according to the manufacturer’s instructions. Human normal retinal cDNA was obtained from Human Adult Normal Tissue First Strand cDNA - Eye: Retina (C1234110-10, Biochain). Quantitative PCR was performed using a pre-designed primer-probe set (TaqMan Gene Expression Assay) and TaqMan Gene Expression Master Mix (Thermo Fisher Scientific–Applied Biosystems), or PrimeTime pre-designed primer-probe set and PrimeTime Gene Expression Master Mix (Integrated DNA Technologies), on a StepOnePlus Real-Time PCR System (Supplementary Table 2). The mRNA expression level was calculated as the ΔCt value (threshold cycle, Ct value of target gene − Ct value of endogenous control gene), and 2-ΔCt was defined as the relative gene expression level. Gapdh/GAPDH (glyceraldehyde-3-phosphate dehydrogenase) or Hprt1/HPRT1 (hypoxanthine phosphoribosyltransferase 1) was used as an internal control.
Western blotting
Cell lysates were prepared using 1% (v/v) NP-40 RIPA buffer containing protease inhibitor cocktail (Nacalai Tesque). Proteins (1.5 × 105 cells/lane) were separated by SDS-PAGE and transferred to Immobilon PVDF membranes (Merck Millipore). The membranes were blocked with Block Ace and probed with rabbit antibodies for human MFN1 (13798-1-AP, Proteintech), DRP1 (12957-1-AP, Proteintech), and COX IV (11242-1-AP, Proteintech), followed by goat-anti-rabbit IgG (H + L) (Merck Sigma-Aldrich). Signals were visualized with the ECL Prime Western Blotting Detection Reagents (Cytiva) on FUSION Solo S Western blot and chemiluminescence imaging system (Vilber-Smart Imaging).
Microarray analysis
All instruments and reagents used in the following analyses were purchased from Agilent Technologies. Total RNA was extracted from tissues or cells, and RNA quality was evaluated using a 2100 Bioanalyzer. Reverse transcription and Cy3 labeling through in vitro transcription were performed using the Low Input QuickAmp Labeling Kit. The prepared cRNA was hybridized overnight with an oligo DNA array (Whole Mouse or Human Genome DNA Microarray Kit, 4 × 44 K) carrying approximately 44,000 genes. After washing, the array was scanned using a SureScan Microarray Scanner, and the fluorescence intensity of each spot was converted to data using Feature Extraction software (Agilent Technologies). GeneSpring GX software (Agilent Technologies). was used for the standardization of raw data, inter-group comparison of gene expression, and GO analysis.
Cell proliferation assay
PNPLA6-overexpressing, -silenced, or mock-transfected ARPE-19 or human primary RPE cells were re-seeded at 4 × 104 cells/well in 6-well plates 24 h after transfection. After 72 h, the cells were washed with PBS, trypsinized, and collected. Viable and dead cells were counted through trypan blue staining. Conditioned medium was collected from plates of PNPLA6-silenced or mock-transfected ARPE-19 cells 6 h after culture in a choline-deficient medium (Research Institute for the Functional Peptides). The 661 W cells were seeded at 4 × 104 cells/well in 6-well plates with the conditioned medium, incubated for 72 h, trypsinized, and collected for cell counting. For CHKα inhibition, 661 W cells were treated with 1 μM MN58b (MedChem Express).
Quantification of choline
Choline concentrations were determined using the Choline/Acetylcholine Quantification Kit (BioVision) according to the manufacturer’s instructions. Cells were incubated with 50 μl of Choline Assay Buffer (BioVision) for 10 min on ice and centrifuged 1000 × g for 5 min to sediment the debris. The supernatants were incubated with a choline probe, acetylcholinesterase, and an enzyme mix at room temperature for 30 min under a light-shielded condition. Fluorescence at 570 nm was measured using an absorbance microplate reader (Multiskan FC Thermo Fisher Scientific).
Quantification of TAG
The TAG concentration in ARPE-19 cells was determined using the Triglyceride E-Test Wako (Fujifilm-Wako). Briefly, 3 ml of chromogenic reagent was mixed with 20 µl of cell lysate and incubated at 37 °C for 5 min. Fluorescence at 600 nm was measured using an absorbance microplate reader (Multiskan FC).
Quantification of ROS and peroxidized lipids
ROS was detected using a ROS Assay Kit (Dojindo), and fluorescence was measured using a microplate reader (SynergyH1, Agilent Technologies; Excitation/Emission = 490/530 nm). Lipid peroxidation was evaluated using the Thiobarbituric Acid Reactive Substances (TBRAS) assay kit (Cayman Chemical Company) according to the manufacturer’s instructions.
Flow cytometry (FACS analysis)
PNPLA6-silenced or control cells were seeded in 6-well plates at 4 × 104 cells/well. After 24 h, the medium was changed to 1% (v/v) FBS-DMEM/f12 to synchronize the cell cycle. The following day, the medium was changed to 10% (v/v) FBS-DMEM/f12, and the cells were collected after 36 h of culture, fixed with 4% paraformaldehyde, and stained with FxCycle PI/RNase Staining Solution (Thermo Fisher Scientific-Invitrogen) for 30 min. After adding 5 mM EDTA-3Na (Dojindo), the cells were passed through a 40 µm cell strainer and resuspended in 500 µl of FACS buffer for cell cycle analysis using the BD FACSMelody Cell Sorter (BD Biosciences).
TEER analysis
ARPE-19 cells or human primary RPE cells were grown in 6-well hanging inserts (4.2 cm2, Corning) and incubated at 37 °C for 72 h. Then, the degree of cell-cell adhesion was quantified by measuring the TEER inside and outside the hanging insert using a Millicell ERS-2 resistance measurement system90.
Metabolic flux analysis
All instruments and reagents used in the following analyses were purchased from Agilent Technologies. PNPLA6-silenced or control cells were cultured in 96-well microplates at 0.8 × 105 cells/well. After 16 h, the plates were washed with analysis medium, and sensor cartridges, which were hydrated with Seahorse XF Calibrant Solution the day before and kept at 37 °C on the day of incubation, were placed on the plates. The analysis was conducted using a basic medium containing D-glucose (3151 mg/l), pyruvate (55 mg/l), and L-glutamine (365 mg/l). Oligomycin, FCCP, rotenone, and antimycin were placed in each port of the sensor cartridge, and intracellular energy metabolism was measured using the XF mitostress kit on the XFe96 extracellular flux analyzer. Data analysis was performed using the ATP Rate Assay Report Generator (Wave ver. 2.6.1.53, Agilent technologies)91.
Cell staining
ARPE-19 cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min. After washing with PBS, the cells were permeabilized with 0.2% (v/v) Triton X-100 for 10 min and blocked with 3% (w/v) bovine serum albumin for 15 min. The cells were incubated with rabbit anti-human ZO-1 polyclonal antibody at 1:500 dilution (61-7300; Thermo Fisher Scientific) for 1 h and then with Alexa Fluor 488-labeled goat anti-rabbit IgG at 1:1000 dilution (A-11070; Thermo Fisher Scientific) for 1 h at room temperature. After washing with PBS, the fluorescent signals were observed using an all-in-one fluorescence microscope.
For TUNEL staining, cells were fixed with 4% paraformaldehyde for 10 min, incubated with 100 μl of Permeabilization Buffer from the in situ Apoptosis Detection Kit (Takara), placed on ice for 5 min, added to labeling reaction solution in a wet box at 37 °C for 60 min, and then observed under a fluorescence microscope.
For JC-1 staining, PNPLA6-silenced or control cells were seeded in 96-well plates at 5 × 103 cells/well. After culturing for 1 h, the cells were incubated with a JC-1 working solution for 30 min using the JC-1 MitoMP Detection Kit (Dojindo) according to the manufacturer’s instructions. The cells were washed twice with DMEM/f12 medium, after which an imaging buffer solution (Dojindo) was added. Fluorescence intensities (green fluorescence, excitation: 485 nm/535 nm; red fluorescence, excitation: 540 nm/595 nm) were quantified using a microplate reader (2030-0030 J ARVO X3; PerkinElmer).
Metabolome analysis
Metabolome analysis of low-molecular-weight hydrophilic compounds was performed using capillary electrophoresis-mass spectrometry (CE-MS)92,93. For the cell-based analysis, ARPE-19 cells were seeded at 5 × 104 cells/ml in culture dishes, and after 72 h, the cells were washed twice with 5% (v/v) mannitol solution for 10 min at room temperature. Then, 1 ml of methanol containing internal standards (25 μM L-methionine sulfone (Fujifilm-Wako), 25 μM 2-morpholinoethanesulfonic acid (Dojindo), and 25 μM D-shono-10-sulfonic acid (Fujifilm-Wako)) was added. The cells were collected using a cell scraper and vortexed for 30 s. Then, 400 μl of chloroform and 200 μl of deionized water were added to 400 μl of the collected supernatant, which was then agitated thoroughly and centrifuged at 10,000 × g for 3 min at 4 °C. Then, 400 μl of the aqueous phase was collected, transferred to a Human Metabolome Technologies (HMT) 5-kDa ultrafiltration tube (UFC3LCCNB-HMT, Ultrafree-MC-PLHCC 250/pk; Human Metabolome Technologies), and centrifuged at 9100 × g for 2 h at 20 °C. For tissue-based analysis, mouse eyes were homogenized in methanol and mixed with cholesterol and water, and the aqueous phase was filtered as detailed above. The filtered solution was dried in a centrifugal evaporator at 1500 rpm at 1000 Pa and resuspended in 50 μl of ultrapure water before metabolome analysis using CE-MS on an Agilent 7100 capillary electrophoresis system coupled to a 6224 time-of-flight liquid chromatography-mass spectrometer (CE-TOFMS) for cationic compounds and a capillary electrophoresis tandem mass spectrometer (CE-MS/MS) for anionic compounds (Agilent Technologies). Peaks detected in CE-TOFMS analysis were extracted using automatic integration software (MasterHands ver. 2.16.0.15; Keio University), and those in CE-MS/MS analysis were extracted using automatic integration software (MassHunter Quantitative Analysis B.06.00; Agilent Technologies). Putative metabolites were then assigned from the HMT metabolite database based on m/z and migration time. Metabolite concentrations were calculated by normalizing the peak area of each metabolite with respect to the area of the internal standard and using a standard curve obtained through three-point calibrations.
Measurement of GPC and choline
The levels of GPC and choline in cells or tissues were determined using a Nexera ultra-high performance LC system and an LC/MS 8080 triple quadrupole mass spectrometer (Shimadzu). Samples were dissolved in 1 ml of methanol, 50 µl of which were diluted 2-fold with methanol containing internal standards and used for analysis. Choline-d9 chloride (C432652; Toronto Research Chemicals) and GPC-d9 (G598702; Toronto Research Chemicals) were used as internal standards at final concentrations of 0.1 and 1 µM, respectively. The ratio of each area value to the area value of the internal standard was calculated.
Quantification of phospholipids
Total lipids were extracted using the Bligh and Dyer method94. After the samples were dried, 175 µl of digestion reagent (60% HClO: H2SO4 = 1:1 (v/v)) was added to the samples or a phosphorus standard (NaH2PO4), stirred, and scorched using a gas burner. After cooling at room temperature, 125 µl of water, 1 ml of 1% (w/v) ammonium molybdate, and 50 µl of reducing reagent were added and the samples were boiled for 10 min. The reaction was stopped on ice, and the absorbance was measured at 750 nm95.
LC-MS/MS analysis
Lipidomics analysis was performed using an LC-MS/MS system96. For LC separation conditions, we used an LC-30AD system (autosampler: SIL-30AC (Shimadzu), column: SeQuant ZIC-HILIC column (2.1 × 250 mm; 3.5 µm, Merck). To obtain the mobile layer and flow rate, the sample retained on the column was eluted by a mixture of solvent A (acetonitrile/water = 95/5 (v/v), 10 mM ammonium acetate) and solvent B (acetonitrile/water = 50/50, 20 mM ammonium acetate). Then, a triple quadrupole lipid molecular device (4500 QTRAP LC-MS/MS/MS system; AB Sciex) was used to measure each lipid molecule. Lipid samples (5 nmol of phospholipids) or standard lipids (Avanti Polar Lipids), which were prepared as described above, were transferred to a vial, and the internal standards for each lipid were added. Next, the solvent was evaporated with nitrogen gas and finally solved in 50 µl of isopropanol: methanol = 1:1 (v/v) and set in an autosampler. Lipid profiling was performed using a simultaneous quantification system for phospholipids and lysophospholipids through multiple reaction monitoring (MRM). The data were analyzed using MultiQuant software (AB Sciex) to obtain peak area under the curve (AUC) values for each lipid molecule. The individual lipid molecules were quantified based on the calibration curves generated by AUCs found at the same LC elution times as for the standard lipids.
Choline tracing in 661 W cells
The 661 W cells were cultured at 2 × 104 cells/ml in 6-well plates. After 3 days, d9-choline (DLM-549-1; Cambridge Isotope Laboratories) was added to the medium at 10 µM of the final concentration. After 2 h, the cells were collected, placed in methanol, and analyzed through LC-MS/MS, as described above.
In silico structural analysis of PNPLA6
The amino acid sequence of human PNPLA6 was obtained from UniProt Q8IY17 (PLPL6_HUMAN). As there are four variants of human PNPLA6 with selective splicing, isoform-4 (identifier: Q8IY17-4), in which point mutations in PNPLA6 (T1058I and G726R) in PNPLA6-related disorders were reported16,17, was used. Protein conformational data were obtained from the Protein Data Bank. The 3D conformational modeling of PNPLA6 was performed using homology remodeling with the SWISS-MODEL (Altif Laboratories, Inc.). The comparison between wild-type and mutant models, measurement of interatomic distances, and display of 3D structures were analyzed using the Waals software (Altif Laboratories, Inc.).
Statistical analysis
Sample sizes were chosen based on previous experiences in our laboratory. Data are expressed as mean ± standard error of the mean (SEM). Differences in statistical analyses between the two groups were determined through the unpaired t-test or Mann–Whitney U-test, depending on the variance. Differences in statistical analyses between multiple groups were determined through one-way analysis of variance (ANOVA) with post-hoc Tukey’s test or two-way ANOVA with post-hoc Sidak analysis. Differences in distribution between the two groups were analyzed using the χ2 test. Statistical significance was set at P < 0.05 (*), P < 0.01 (**), P < 0.001 (***), and P < 0.0001 (****). GraphPad Prism software (GraphPad, CA, USA) was used for statistical analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data and materials are available in the paper and the supplementary information. Microarray datasets are publicly available under the GEO accession numbers GSE253526 and GSE253529. Metabolome datasets are publicly available at Metabolomics Workbench under the accession numbers ST003648 and ST003658. Additional requests for data may be obtained from the corresponding authors upon request. Source data are provided in this paper.
Code availability
The present study did not generate any unique code.
References
Hartong, D. T., Berson, E. L. & Dryja, T. P. Retinitis pigmentosa. Lancet 368, 1795–1809 (2006).
Cehajic-Kapetanovic, J. et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat. Med. 26, 354–359 (2020).
Hoover-Fong, J. et al. Mutations in PCYT1A, encoding a key regulator of phosphatidylcholine metabolism, cause spondylometaphyseal dysplasia with cone-rod dystrophy. Am. J. Hum. Genet. 94, 105–112 (2014).
Yamamoto, G. L. et al. Mutations in PCYT1A cause spondylometaphyseal dysplasia with cone-rod dystrophy. Am. J. Hum. Genet. 94, 113–119 (2014).
Neuringer, M., Connor, W. E., Lin, D. S., Barstad, L. & Luck, S. Biochemical and functional effects of prenatal and postnatal omega 3 fatty acid deficiency on retina and brain in rhesus monkeys. Proc. Natl. Acad. Sci. USA 83, 4021–4025 (1986).
Neuringer, M., Connor, W. E., Van Petten, C. & Barstad, L. Dietary omega-3 fatty acid deficiency and visual loss in infant rhesus monkeys. J. Clin. Invest. 73, 272–276 (1984).
Shindou, H. et al. Docosahexaenoic acid preserves visual function by maintaining correct disc morphology in retinal photoreceptor cells. J. Biol. Chem. 292, 12054–12064 (2017).
Nakanishi, H. et al. Cloning and characterization of mouse lung-type acyl-CoA:lysophosphatidylcholine acyltransferase 1 (LPCAT1). Expression in alveolar type II cells and possible involvement in surfactant production. J. Biol. Chem. 281, 20140–20147 (2006).
Friedman, J. S. et al. Loss of lysophosphatidylcholine acyltransferase 1 leads to photoreceptor degeneration in rd11 mice. Proc. Natl Acad. Sci. USA 107, 15523–15528 (2010).
Nagata, K. et al. Lysophosphatidylcholine acyltransferase 1 controls mitochondrial reactive oxygen species generation and survival of retinal photoreceptor cells. J. Biol. Chem. 298, 101958 (2022).
Dennis, E. A., Cao, J., Hsu, Y. H., Magrioti, V. & Kokotos, G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 111, 6130–6185 (2011).
Murakami, M. et al. Recent progress in phospholipase A2 research: from cells to animals to humans. Prog. Lipid Res. 50, 152–192 (2011).
Lambeau, G. & Gelb, M. H. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu. Rev. Biochem. 77, 495–520 (2008).
Kita, Y., Ohto, T., Uozumi, N. & Shimizu, T. Biochemical properties and pathophysiological roles of cytosolic phospholipase A2s. Biochim. Biophys. Acta 1761, 1317–1322 (2006).
Murakami, M., Sato, H. & Taketomi, Y. Updating phospholipase A2 biology. Biomolecules 10, 1457 (2020).
Hufnagel, R. B. et al. Neuropathy target esterase impairments cause Oliver-McFarlane and Laurence-Moon syndromes. J. Med. Genet. 52, 85–94 (2015).
Synofzik, M. et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 137, 69–77 (2014).
Kmoch, S. et al. Mutations in PNPLA6 are linked to photoreceptor degeneration and various forms of childhood blindness. Nat. Commun. 6, 5614 (2015).
Zaccheo, O., Dinsdale, D., Meacock, P. A. & Glynn, P. Neuropathy target esterase and its yeast homologue degrade phosphatidylcholine to glycerophosphocholine in living cells. J. Biol. Chem. 279, 24024–24033 (2004).
Rainier, S. et al. Neuropathy target esterase gene mutations cause motor neuron disease. Am. J. Hum. Genet. 82, 780–785 (2008).
Moser, M. et al. Placental failure and impaired vasculogenesis result in embryonic lethality for neuropathy target esterase-deficient mice. Mol. Cell Biol. 24, 1667–1679 (2004).
Quistad, G. B., Barlow, C., Winrow, C. J., Sparks, S. E. & Casida, J. E. Evidence that mouse brain neuropathy target esterase is a lysophospholipase. Proc. Natl. Acad. Sci. USA 100, 7983–7987 (2003).
Spindler, V. et al. Desmocollin 3-mediated binding is crucial for keratinocyte cohesion and is impaired in pemphigus. J. Biol. Chem. 284, 30556–30564 (2009).
Bonilha, V. L. et al. The retinal pigment epithelium apical microvilli and retinal function. Adv. Exp. Med. Biol. 572, 519–524 (2006).
Nilsson, S. E., Knave, B. & Persson, H. E. Changes in ultrastructure and function of the sheep pigment epithelium and retina induced by sodium iodate. I. The ultrastructure of the normal pigment epithelium of the sheep. Acta Ophthalmol. 55, 994–1006 (1977).
Sorsby, A. Experimental pigmentary degeneration of the retina by sodium iodate. Br. J. Ophthalmol. 25, 58–62 (1941).
Li, Z. & Vance, D. E. Phosphatidylcholine and choline homeostasis. J. Lipid Res. 49, 1187–1194 (2008).
Inazu, M. Choline transporter-like proteins CTLs/SLC44 family as a novel molecular target for cancer therapy. Biopharm. Drug Dispos. 35, 431–449 (2014).
Kenny, T. C. et al. Integrative genetic analysis identifies FLVCR1 as a plasma-membrane choline transporter in mammals. Cell Metab. 35, 1057–1071 (2023).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Wijeyesakere, S. J., Richardson, R. J. & Stuckey, J. A. Crystal structure of patatin-17 in complex with aged and non-aged organophosphorus compounds. PLoS ONE 9, e108245 (2014).
Rydel, T. J. et al. The crystal structure, mutagenesis, and activity studies reveal that patatin is a lipid acyl hydrolase with a Ser-Asp catalytic dyad. Biochemistry 42, 6696–6708 (2003).
da Mata Madeira, P. V. et al. Structural basis of lipid targeting and destruction by the type V secretion system of Pseudomonas aeruginosa. J. Mol. Biol. 428, 1790–1803 (2016).
Richardson, R. J. et al. Neuropathy target esterase (NTE/PNPLA6) and organophosphorus compound-induced delayed neurotoxicity (OPIDN). Adv. Neurotoxicol. 4, 1–78 (2020).
Salgado, P. et al. Gordon Holmes syndrome due to compound heterozygosity of two new PNPLA6 variants - A diagnostic challenge. eNeurologicalSci 14, 9–12 (2019).
Aleman, T. S. et al. Human CRB1-associated retinal degeneration: comparison with the rd8 Crb1-mutant mouse model. Invest. Ophthalmol. Vis. Sci. 52, 6898–6910 (2011).
Zeisel, S. H. & da Costa, K. A. Choline: an essential nutrient for public health. Nutr. Rev. 67, 615–623 (2009).
Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate OBV, and Choline. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. In: Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline) (1998).
Hirabayashi, T. et al. Hepatic phosphatidylcholine catabolism driven by PNPLA7 and PNPLA8 supplies endogenous choline to replenish the methionine cycle with methyl groups. Cell Rep. 42, 111940 (2023).
Hermansson, M., Hanninen, S., Hokynar, K. & Somerharju, P. The PNPLA-family phospholipases involved in glycerophospholipid homeostasis of HeLa cells. Biochim. Biophys. Acta 1861, 1058–1065 (2016).
Shimizu, T. Lipid mediators in health and disease: enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu. Rev. Pharm. Toxicol. 49, 123–150 (2009).
Gibellini, F. & Smith, T. K. The Kennedy pathway-De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 62, 414–428 (2010).
McMaster, C. R. From yeast to humans - roles of the Kennedy pathway for phosphatidylcholine synthesis. FEBS Lett. 592, 1256–1272 (2018).
Moon, S. H. et al. Genetic deletion of skeletal muscle iPLA2γ results in mitochondrial dysfunction, muscle atrophy and alterations in whole-body energy metabolism. iScience 26, 106895 (2023).
Selathurai, A. et al. The CDP-ethanolamine pathway regulates skeletal muscle diacylglycerol content and mitochondrial biogenesis without altering insulin sensitivity. Cell Metab. 21, 718–730 (2015).
Capelo-Diz, A. et al. Hepatic levels of S-adenosylmethionine regulate the adaptive response to fasting. Cell Metab. 35, 1373–1389 (2023).
Mitsuhashi, S. et al. A congenital muscular dystrophy with mitochondrial structural abnormalities caused by defective de novo phosphatidylcholine biosynthesis. Am. J. Hum. Genet. 88, 845–851 (2011).
Tavasoli, M. et al. Mechanism of action and therapeutic route for a muscular dystrophy caused by a genetic defect in lipid metabolism. Nat. Commun. 13, 1559 (2022).
Sanchez-Lopez, E. et al. Choline uptake and metabolism modulate macrophage IL-1β and IL-18 Production. Cell Metab. 29, 1350–1362 (2019).
Hernández-Alvarez, M. I. et al. Deficient endoplasmic reticulum-mitochondrial phosphatidylserine transfer causes liver disease. Cell 177, 881–895 (2019).
Naón, D. et al. Splice variants of mitofusin 2 shape the endoplasmic reticulum and tether it to mitochondria. Science 380, eadh9351 (2023).
Forcina, G. C. & Dixon, S. J. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics 19, e1800311 (2019).
Morgan, N. V. et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat. Genet. 38, 752–754 (2006).
Kurian, M. A. et al. Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). Neurology 70, 1623–1629 (2008).
Tomiyama, H. et al. PLA2G6 variant in Parkinson’s disease. J. Hum. Genet. 56, 401–403 (2011).
Sun, W. Y. et al. Phospholipase iPLA2β averts ferroptosis by eliminating a redox lipid death signal. Nat. Chem. Biol. 17, 465–476 (2021).
Haider, A. et al. PCYT1A regulates phosphatidylcholine homeostasis from the inner nuclear membrane in response to membrane stored curvature elastic stress. Dev. Cell 45, 481–495 (2018).
Wu, J. et al. Molecular screening of the LPCAT1 gene in patients with retinitis pigmentosa without defined mutations in known retinitis pigmentosa genes. Mol. Med. Rep. 12, 5983–5988 (2015).
Hwang, J. S. & Shin, Y. J. Role of choline in ocular diseases. Int. J. Mol. Sci. 22, 4733 (2021).
Webster, M. K. et al. Evidence of BrdU-positive retinal neurons after application of an α7 nicotinic acetylcholine receptor agonist. Neuroscience 346, 437–446 (2017).
Choi, J. J., Hwang, J. S. & Shin, Y. J. Effect of oral choline alfoscerate on patients with Keratoconjunctivitis sicca. Nutrients 12, 1526 (2020).
Trujillo-Gonzalez, I. et al. Low availability of choline in utero disrupts development and function of the retina. FASEB J. 33, 9194–9209 (2019).
Oshitari, T., Fujimoto, N. & Adachi-Usami, E. Citicoline has a protective effect on damaged retinal ganglion cells in mouse culture retina. Neuroreport 13, 2109–2111 (2002).
Savendahl, L., Mar, M. H., Underwood, L. E. & Zeisel, S. H. Prolonged fasting in humans results in diminished plasma choline concentrations but does not cause liver dysfunction. Am. J. Clin. Nutr. 66, 622–625 (1997).
Roberti, G. et al. A preliminary study of the neuroprotective role of citicoline eye drops in glaucomatous optic neuropathy. Indian J. Ophthalmol. 62, 549–553 (2014).
Son, Y., Kenny, T. C., Khan, A., Birsoy, K. & Hite, R. K. Structural basis of lipid head group entry to the Kennedy pathway by FLVCR1. Nature 629, 710–716 (2024).
Cater, R. J. et al. Structural and molecular basis of choline uptake into the brain by FLVCR2. Nature 629, 704–709 (2024).
Rajadhyaksha, A. M. et al. Mutations in FLVCR1 cause posterior column ataxia and retinitis pigmentosa. Am. J. Hum. Genet. 87, 643–654 (2010).
Johnson, M. K. The delayed neuropathy caused by some organophosphorus esters: mechanism and challenge. Crit. Rev. Toxicol. 3, 289–316 (1975).
Read, D. J., Li, Y., Chao, M. V., Cavanagh, J. B. & Glynn, P. Neuropathy target esterase is required for adult vertebrate axon maintenance. J. Neurosci. 29, 11594–11600 (2009).
Henschler, D., Schmuck, G., van Aerssen, M. & Schiffmann, D. The inhibitory effect of neuropathic organophosphate esters on neurite outgrowth in cell cultures: A basis for screening for delayed neurotoxicity. Toxicol. Vitr. 6, 327–335 (1992).
Glynn, P. A mechanism for organophosphate-induced delayed neuropathy. Toxicol. Lett. 162, 94–97 (2006).
Sogorb, M. A., Pamies, D., Estevan, C., Estévez, J. & Vilanova, E. Roles of NTE protein and encoding gene in development and neurodevelopmental toxicity. Chem. Biol. Interact. 259, 352–357 (2016).
Pamies, D., Vilanova, E. & Sogorb, M. A. Functional pathways altered after silencing Pnpla6 (the codifying gene of neuropathy target esterase) in mouse embryonic stem cells under differentiation. Vitr. Cell Dev. Biol. Anim. 50, 261–273 (2014).
Glynn, P. Neuronal phospholipid deacylation is essential for axonal and synaptic integrity. Biochim. Biophys. Acta 1831, 633–641 (2013).
Quesada, E., Sogorb, M. A., Vilanova, E. & Carrera, V. Bovine chromaffin cell cultures as model to study organophosporus neurotoxicity. Toxicol. Lett. 151, 163–170 (2004).
Hayashi, S. & McMahon, A. P. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 244, 305–318 (2002).
Yoda, E. et al. Mitochondrial dysfunction and reduced prostaglandin synthesis in skeletal muscle of Group VIB Ca2+-independent phospholipase A2γ-deficient mice. J. Lipid Res. 51, 3003–3015 (2010).
Shinzawa, K. et al. Neuroaxonal dystrophy caused by group VIA phospholipase A2 deficiency in mice: a model of human neurodegenerative disease. J. Neurosci. 28, 2212–2220 (2008).
Shang, P., Stepicheva, N. A., Hose, S., Zigler, J. S. Jr. & Sinha, D. Primary cell cultures from the mouse retinal pigment epithelium. J. Vis. Exp. 133, 56997 (2018).
Terao, R. et al. Light stress-induced increase of sphingosine 1-phosphate in photoreceptors and its relevance to retinal degeneration. Int. J. Mol. Sci. 20, 3670 (2019).
Kiuchi, K., Yoshizawa, K., Shikata, N., Moriguchi, K. & Tsubura, A. Morphologic characteristics of retinal degeneration induced by sodium iodate in mice. Curr. Eye Res. 25, 373–379 (2002).
Machalinska, A. et al. Sodium iodate selectively injuries the posterior pole of the retina in a dose-dependent manner: morphological and electrophysiological study. Neurochem. Res. 35, 1819–1827 (2010).
Koso, H. et al. Conditional rod photoreceptor ablation reveals Sall1 as a microglial marker and regulator of microglial morphology in the retina. Glia 64, 2005–2024 (2016).
Sayyad, Z., Sirohi, K., Radha, V. & Swarup, G. 661W is a retinal ganglion precursor-like cell line in which glaucoma-associated optineurin mutants induce cell death selectively. Sci. Rep. 7, 16855 (2017).
Shirahama, S. et al. Long noncoding RNA U90926 is crucial for herpes simplex virus type 1 proliferation in murine retinal photoreceptor cells. Sci. Rep. 10, 19406 (2020).
Ueta, T. et al. Intense physiological light upregulates vascular endothelial growth factor and enhances choroidal neovascularization via peroxisome proliferator-activated receptor γ coactivator-1α in mice. Arterioscler. Thromb. Vasc. Biol. 32, 1366–1371 (2012).
Roggia, M. F. & Ueta, T. αvβ5 integrin/FAK/PGC-1α pathway confers protective effects on retinal pigment epithelium. PLoS ONE 10, e0134870 (2015).
Mazzoni, F., Safa, H. & Finnemann, S. C. Understanding photoreceptor outer segment phagocytosis: use and utility of RPE cells in culture. Exp. Eye Res. 126, 51–60 (2014).
Dunn, K. C., Aotaki-Keen, A. E., Putkey, F. R. & Hjelmeland, L. M. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp. Eye Res. 62, 155–169 (1996).
Shu, D. Y., Butcher, E. R. & Saint-Geniez, M. Suppression of PGC-1α drives metabolic dysfunction in TGFβ2-induced EMT of retinal pigment epithelial cells. Int. J. Mol. Sci. 22, 4701 (2021).
Satoh, K. et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc. Natl. Acad. Sci. USA 114, E7697–E7706 (2017).
Soga, T. et al. Metabolomic profiling of anionic metabolites by capillary electrophoresis mass spectrometry. Anal. Chem. 81, 6165–6174 (2009).
Bligh, E. G. & Dyer, W. J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 (1959).
Lee, S. et al. Impaired retrograde membrane traffic through endosomes in a mutant CHO cell defective in phosphatidylserine synthesis. Genes Cells 17, 728–736 (2012).
Akagi, S. et al. Lysophosphatidylcholine acyltransferase 1 protects against cytotoxicity induced by polyunsaturated fatty acids. FASEB J. 30, 2027–2039 (2016).
Acknowledgements
The authors sincerely thank N. Kono and H. Arai (the University of Tokyo) for their valuable advice, T. Iwagawa (the University of Tokyo) for ERG examination, R. Terao (Washington University) for providing 661 W cells, which had been originally established by M. Al-Ubaidi (University of Houston), and K. Sakimura (Niigata University) for providing the RENKA ES cells. This work was supported by JP24gm1210013 (to M.M.) from the Japan Agency for Medical Research and Development (AMED) and JSPS KAKENHI JP20H05691 (to M.M.), JP24K19806, JP22K20954 and JP20J12307 (to Takashi Ono) from Japan Society for the Promotion of Science (JSPS), and Takeda Science Foundation 2024044197 (to Takashi Ono). For open access purposes, the authors have applied a CC BY public copyright license to any author-accepted manuscript version arising from this submission.
Author information
Authors and Affiliations
Contributions
T.O. (Takashi Ono), Y.T., T.H., and H.S. conceptualized and designed the research project. T.H., Y.N., S.T., Y.O., and T.S. conducted metabolomics analysis. T.U., T.M., and S.W. performed retinal analysis. Y.N. conducted microarray analysis. T.O. (Tomio Ono), C.T., S.A., and T.H. produced and analyzed laboratory animals. C.M. assisted cell culture and immunoblotting. T.O. (Takashi Ono) and Y.T. conducted the statistical analysis of the data. T.O. (Takashi Ono), Y.T., T.H., H.S., and S.T. obtained the data and wrote the manuscript. M.A. and M.M. supervised the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Julia Busik, Eugenio Vilanova, and Heping Xu for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ono, T., Taketomi, Y., Higashi, T. et al. PNPLA6 regulates retinal homeostasis by choline through phospholipid turnover. Nat Commun 16, 2221 (2025). https://doi.org/10.1038/s41467-025-57402-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57402-8
