
Abstract
Understanding compound-protein interactions is crucial for early drug discovery, offering insights into molecular mechanisms and potential therapeutic effects of compounds. Here, we introduce GraphBAN, a graph-based framework that inductively predicts these interactions using compound and protein feature information. GraphBAN effectively handles inductive link predictions for unseen nodes, providing a robust solution for predicting interactions between entirely unseen compounds and proteins. This capability enables GraphBAN to transcend the constraints of traditional methods that are typically limited to known contexts. GraphBAN employs a knowledge distillation architecture through a teacher-student learning model. The teacher block leverages network structure information, while the student block focuses on node attributes, enhancing learning and prediction accuracy. Additionally, GraphBAN incorporates a domain adaptation module, increasing its effectiveness across different dataset domains. Empirical tests on five benchmark datasets demonstrate that GraphBAN outperforms ten baseline models, while a case study analysis with the Pin1 protein further supports the model’s effectiveness in real world scenarios, making it as a promising tool for early drug discovery.
Similar content being viewed by others

Introduction
Drug discovery is the process of selecting, modifying, and advancing chemical molecules into therapeutic agents1. To discover potential drug candidates for target diseases, it is fundamental to identify chemical compounds that interact and bind a target protein involved in a disease in order to understand their therapeutic effect. As experimental identification of compound-protein interactions (CPIs) is generally costly and time-consuming, computational approaches have been developed to streamline the discovery process through in-silico CPI predictions, including molecular docking and molecular dynamics simulations. Molecular docking helps to investigate CPIs by estimating the binding affinity between a compound and its protein target2. By modeling the movement of molecules over time, molecular dynamics simulations allow for a better understanding of dynamic interactions3. However, these methods come with challenges. They can be resource-intensive, especially when identifying potential drug candidates from large compound libraries2,4. Also, to achieve reliable results, these approaches require high-quality molecular structures as inputs, which can be challenging as experimentally validated structures are not always accessible, especially for less-studied molecules3,5. These limitations can reduce the utility of these in-silico approaches for screening extensive molecular libraries to identify desired CPIs and pinpoint potential drug candidates.
Machine learning (ML) and deep learning (DL) developments offer complementary in-silico approaches for predicting CPIs, effectively reducing the burdens of time and cost by filtering out thousands of extraneous compounds in large databases. Current ML and DL methods for CPI predictions stand out in two critical aspects: the data model they accept for training and prediction, and the extent of similarity between the training and testing datasets.
The first aspect of CPI predictions involves choosing the appropriate input data types, which can be either network-based or tabular-based. Network-based inputs include bi-partite networks, which are two-layer network structures where compounds and proteins are distinct node types, linked by edges if there is an active interaction between them. In contrast, tabular inputs consist of discrete data points that lack an inherent relational structure.
Moreover, the effectiveness of a model’s performance evaluation depends on whether the approach is transductive or inductive. Transductive predictions target interactions among compounds and proteins observed during training, whereas inductive predictions seek to forecast interactions involving entirely unseen entities. While models such as CGINet6 and HGDTI7 use network-based data without fully addressing inductive link prediction, others such as AI-Bind8, DrugBAN9, MolTrans10, MFR-DTA11, and CPInformer12, utilize tabular data for inductive CPI predictions.
Given the practical importance of inductive predictions and the neighboring or connecting information provided by network-based data, there is a pressing need for models that can adeptly handle both inductive link predictions and the utilization of network-based input to enhance predictive accuracy.
The second aspect of advancements in CPI predictions focuses on assessing compound-protein similarity between the test set and the training set by categorizing analyses into in-domain and cross-domain. In-domain analysis applies when the training and test sets share similar descriptors (domains), enabling the model to utilize prior knowledge effectively. However, cross-domain analysis is required when descriptors differ significantly, challenging traditional predictive models that depend on similarity. Recent research introduced models using long short-term memory (LSTM) and convolutional neural networks (CNNs)13 ensemble deep learning approaches14 and using domain adaptation modules9 for CPI predictions in cross-domain settings. This distinction is essential for improving model generalization, especially when adapting to limited labeled training data for cross-domain test sets.
In this work, we introduce GraphBAN, an inductive model designed for compound-protein interaction (CPI) predictions that is capable of handling bi-partite network inputs for both transductive and inductive link predictions across in-domain and cross-domain test sets. At its core, GraphBAN employs a knowledge distillation (KD) process with teacher and student modules. The teacher module is trained on network properties using a graph autoencoder (GAE), which enables the model to manage unseen datasets where compound-protein links are unknown. The learned knowledge is transferred to the student module via a KD loss function, ensuring effective pattern learning from the initial features of compounds and proteins. By operating in a binary prediction mode, GraphBAN classifies interactions as “active” or “inactive”, which is crucial for biological and pharmaceutical applications. The architecture integrates large language model (LLM)-based and convolutional neural network (CNN)-based models for feature representation, along with a bilinear attention network (BAN) module and a cross-domain adaptation module, to enhance prediction accuracy and generalization across training and testing datasets.
Results
Overview of GraphBAN framework
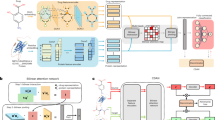
GraphBAN’s schematic is shown in Fig. 1a. Given compounds in the form of simplified molecular input line entry system (SMILES)15 and proteins as sequence of amino acids, the model generates a bi-partite network where compounds and proteins are represented as nodes and their active interactions are represented as edges. The bi-partite network also includes features of each compound and protein. We first use compound and protein feature extractor modules to embed compounds and proteins separately. The compound and protein features are generated through a fusion of four different methods (two methods for compounds and two methods for proteins) (Fig. 1b). The first method for extracting compounds’ features utilizes structural graph convolutional network (GCN) properties and the second employs a pre-trained LLM named ChemBERTa16. The protein features are extracted from a CNN layer plus a LLM named evolutionary scale modeling (ESM)17. Then, we use the KD architecture to extract network’s structural features (neighboring features) within the teacher block and distill this knowledge to the student block. The student block generates a joint representation of nodes’ features and learns local interactions between encoded compound and protein features with the BAN module. Lastly, we add a conditional domain adversarial network (CDAN) module (Fig. 1c) to increase the ability of our model to deal with cross-domain compound-protein pairs.

a The input compound molecules are encoded by graph convolutional network (GCN) layers and ChemBERTa separately, while protein sequences are encoded by 1D layer convolutional neural network (CNN) and evolutionary scale modeling (ESM). b Fusion of features with same architecture for both compounds and proteins. Fusion module shows how we bring the extracted features in the same dimensionality with the Linear Transform Layers. The feature fusion includes two “MatMul” layers that operate element-wise multiplication and one “Addition” layer that do the element-wise addition. c The CDAN module. It receives input from the bilinear attention network (BAN) layer and generates concatenation of the compound and protein features and SoftMax logits “g” for source and target domains into a joint conditional representation using the discriminator module. The discriminator has two fully connected layers with an adversarial loss to minimize the classification error between the source and target domains.
Evaluation strategies and metrics
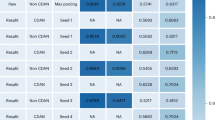
We study the performance of the model’s inductive link prediction on five previously assembled, experimental CPI collections, BindingDB18, BioSNAP19, KIBA20, PDBbind 201621, and C.elegans22. Our approach utilizes a fully inductive and cross-domain evaluation strategy, essential for assessing the robustness and real-world applicability of the model in drug discovery. By evaluating the model’s performance across unseen test sets and representing the CPIs from different domains, we gain a more comprehensive understanding of its generalization capabilities. For cross-domain evaluation, we use a clustering-based pair-splitting strategy that employs the single-linkage algorithm on extended connectivity fingerprints (ECFP) features for compounds and k-mer frequency features for proteins. We randomly select 60% of the compound and protein clusters derived from the clustering step for the source domain data. The remaining 40% of clusters of compound-protein pairs are used as target domain data. We adhere to the standard domain adaptation setting, using all labeled source domain data and 80% of the unlabeled target domain data for model training, with the remaining 20% of the labeled target domain data forming the fully inductive target test set. The details of the clustering strategy are provided in Supplementary Section 1. The transductive analysis is reported in the Supplementary Section 4. We employ three key evaluation metrics: the area under the receiver operating characteristic curve (AUROC), the area under the precision-recall curve (AUPRC), and the F1 score. To ensure reliability, we conduct five different runs with distinct random seeds and report the average scores along with t-statistic and p-value analyses that are presented in Supplementary Section 5.
Analysis of performance on public datasets
The disparities of the training and test data distributions encountered in real-world scenarios, underline the need for an inductive analysis. To address this challenge, we embed the CDAN module within the GraphBAN framework, which enables us to effectively manage the distinct data distributions. The performance assessment is presented across all distinct datasets (Table 1). In line with our findings, GraphBAN consistently outperforms other state-of-the-art models. Specifically, it exhibits superior performance compared to DrugBAN, boasting improvements of 9.32%, 5.46%, 3.32%, 2.76%, and 0.72% in AUROC across the BioSNAP, BindingDB, KIBA, C.elegans, and PDBbind 2016 datasets, respectively.
The overall higher performance observed with the C.elegans dataset compared to other datasets could be attributed to its relatively smaller size, which may inadvertently introduce a hidden bias. This bias could simplify the model learning process by revealing more distinct, easily recognizable patterns between active and inactive links, thereby enhancing their ability to train effectively. More details on how the CPIs’ diversity exists in different datasets are provided in the Supplementary Section 1. Additionally, the lower AUPRC scores for the KIBA dataset may be attributed to the high imbalance between active and inactive labels present in this dataset. The performance of the GraphDTA method indicates that a simple GNN-based model that uses molecular graph data as input can perform competitively, as GraphDTA gets the second-best AUROC score with the BioSNAP and PDBbind 2016 datasets for inductive predictions.
Ablation study
In this section, we evaluate the impact of the teacher module and KD workflow, feature fusion module, and the BAN + CDAN blocks in inductive link prediction, three of which are integrated into GraphBAN. To ascertain the effectiveness of each module in GraphBAN, we established a baseline model for comparison. The key distinctions between the baseline and the GraphBAN model are as follow. The baseline model employs a three-layer GCN network for extracting compound features and a three-layer CNN network for protein features. Also, the baseline model simply concatenates the compound and protein features without applying the BAN layer. It utilizes a combination of linear and batch normalization layers for the joint representation of features without the CDAN module. The remaining elements of the baseline model are consistent with GraphBAN.
The results, which compare our baseline model and variations with different configurations of our proposed components, are presented in Table 2. These configurations are tested using the two datasets: BioSNAP and BindingDB. Model-1 is composed of the baseline plus the feature fusion (FF) module. Model-2 consists of the baseline plus the BAN + CDAN (BC) modules. Model-3 is the GraphBAN without the teacher module.
The results demonstrate that the integration of all tested modules, namely teacher, FF and BC, enhances the baseline performance across two benchmark datasets. This substantiates our hypothesis that the fusion of GCN and CNN features with LLM-based features, coupled with the application of the BAN + CDAN block on network’s embedding in a KD framework, markedly augments CPI predictions efficacy. Specifically, the FF and BC modules contribute to an increase in the AUROC score on BindingDB dataset by 5.43%, 9.31%, respectively. Furthermore, Model-3 demonstrates the importance of the teacher module that can increase the overall performance of GraphBAN on BindingDB and BioSNAP by 9.46% and 6.07%, respectively. Most notably, GraphBAN, which integrates all the analyzed components, benefits synergistically from these modules, achieving the most optimal outcomes across all evaluated parameters.
Analysis of performance on a case study
To demonstrate the utility of GraphBAN in a real scenario of drug discovery, we focused on the peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin1)23, an essential enzyme involved in many cellular processes, including cell cycle regulation, development, and signaling pathways24,25,26. Because of its role in the cell cycle, Pin1 is a prime target for therapeutics of various types of cancer24,25. To predict compounds that interact with Pin1, we deployed around 250,000 compounds from the ZINC-250K dataset to GraphBAN to identify potential binding compounds. ZINC-250K is a comprehensive library of small molecules for virtual screening27. CPI values for the ZINC-Pin1 dataset are unavailable and our model predicts these interactions in an inductive manner.
Our model, GraphBAN, incorporates an unsupervised domain adaptation module to predict CPI interactions without CPI labels for the ZINC-Pin1 dataset. We use three high-quality datasets for training, BioSNAP, BindingDB, and KIBA; while excluding smaller datasets, C.elegans and PDBbind 2016, to mitigate underfitting risks. Each dataset brings distinct chemical and biological profiles, enabling us to compare predictions and utilize domain adaptation on CPI predictions for ZINC-Pin1 across models trained on three different baseline datasets. The training procedure is detailed in the Supplementary Section 8. As summarized in Fig. 2, after training, and deploying Pin1 and the ZINC compound pairs for predictions, GraphBAN identified 134 compounds with interaction probabilities with Pin1 above 0.5 that are in-common across models trained on three different datasets, respectively. The list of 134 compounds with their probabilities are provided in Supplementary Data 1.

a shows the steps are taken to reduce the number of top candidates predicted by GraphBAN. b, c depict the binding sites of selected compound 1 (with SMILES CC1(C)C[C@@H]([NH2 + ]CCc2cc3ccccc3[nH]2)C(C)(C)O1), (d, e) show the binding sites of selected compound 2 (with SMILES CCCn1nc(C)c(N)c1N1C[C@@H](C)[C@@H](C)C1), (f, g) show selected compound 1 and compound 2, both of which are predicted to have active interactions with Pin1. Binding sites in (c, e) are colored based on attention weights captured by GraphBAN, which are normalized to a range of 0 to 1. Colors transition from blue (lower attention weights and lesser importance to the final prediction) to white (medium) to red (higher attention weights and greater importance to the final prediction). ChimeraX60 was used for visualizing binding sites. For compounds in (f, g), the colouring also reflects normalized attention weights, with colours ranging from yellow (low) to orange (medium) to red (high), visualized using ChemDraw. Additionally, atoms that participate in hydrogen bonding interactions with the respective binding sites are enclosed in dashed boxes.
To narrow down the 134 top predicted compounds we excluded 10 candidates with poor solubility, permeability and chemical properties that favor false positives, applying the Lipinski’s rule-of-five28 and pan assay interference compounds (PAINS)29 filters (Fig. 2a). Then, the drug-likeness properties, including absorption, distribution, metabolism, excretion, and toxicity (ADMET) of the remaining candidates, were further evaluated using ADMET predictions from the ML platform ADMET-AI30. We filtered out 115 compounds predicted to have a high likelihood of clinical toxicity (probability > 0.25), a negative half-life (<0 h), low plasma protein binding rate (<50%), suboptimal lipophilicity (outside the range of 1–3, as suggested by ref. 31), and poor oral bioavailability (probability <0.5). The final nine candidates with suitable drug-likeness properties are listed in Supplementary Data 2.
To further highlight the relevance of the nine selected potential Pin1 inhibitors, we visualized two compounds that were chosen based on their top rankings according to the quantitative estimate of drug-likeness (QED)32. The QED quantifies a compound’s drug-likeness by evaluating its desirability across eight key physicochemical properties. As shown in Fig. 2b–e, these binding regions of the two chosen compounds were predicted through blind docking using AutoDock Vina33, with protein residues selected based on having at least one atom within 5 Å of any atom in the analyzed compound34. The majority of residues within these binding sites are assigned medium to high attention weights by our model, allowing these residues to have a greater contribution to the model’s predictions. Also, according to Fig. 2f, g, compound atoms that form hydrogen bonds with the corresponding binding site residues are assigned medium to high attention weights by the model, which supports our model’s capability to capture relevant bonding interactions for CPI predictions. In summary, these results support the conclusion that our model effectively captures the binding site regions.
BJP-06-005-3 has been identified as an active inhibitor of Pin124. To evaluate the potential of the two selected compounds, we calculated the Tanimoto similarity35 using molecular access system (MACCS) fingerprints36 between each compound and BJP-06-005-3. Both compounds exhibited moderate similarity in functional group features with BJP-06-005-3 (0.437 for compound 1 and 0.475 for compound 2), suggesting that they may interact with Pin1 in a similar manner, potentially yielding comparable inhibitory effects. While this similarity supports the idea that the selected compounds may share functional properties with BJP-06-005-3, it also highlights the distinctiveness of the identified compounds in their potential interactions. This further demonstrates the effectiveness of GraphBAN in identifying compound candidates that could offer favorable interactions with the target protein, thus advancing the development of more effective therapeutics.
Discussion
Effective structure-based drug discovery relies on the identification of compounds that bind to protein targets. Emerging advances in DL aiming at predicting unrecognized drug-target interactions can lower the high cost and time-consuming experimental approaches. Our model GraphBAN achieves higher scores when predicting inductive links between proteins and compounds compared to baseline models. The scores achieved reflect some ongoing challenges. The performance, while positive, could be influenced using cross-domain splits in dataset preparation. This, along with the natural biological variability of protein targets and the complex chemistry of the compounds, presents obstacles for the model in generalizing effectively across diverse biological contexts. Despite these limitations, GraphBAN’s performance indicates it can manage these challenges more effectively than traditional models by leveraging its integrated KD, BAN, and CDAN modules. Moreover, the details of the protein family analysis provided in Supplementary Section 6 indicate that the model is not sensitive to the specific type of CPIs.
The case study applied GraphBAN to predict compound-Pin1 interactions using an unsupervised approach on the ZINC-250K library. The top two compound candidates, which have not been tested in any biological screen, are selected after coupling with drug-likeness and ADMET properties predictions, similar filtering strategies have been successfully applied in previous studies37,38, further validating the robustness of our approach. These strategies ensure that the selected compounds meet key drug-likeness criteria and align with established filtering pipelines. Also, the attention weights visualization supports that the model effectively captures binding interactions relevant to Pin1 interactions, demonstrating GraphBAN’s potential to help identify promising molecular inhibitors.
Integrating incremental learning algorithms would enable GraphBAN to continuously adapt to unseen data without requiring extensive retraining, thereby improving both efficiency and adaptability. Furthermore, broadening the model’s capacity to process larger and more varied datasets through transfer learning could enhance its overall performance. Additionally, incorporating multi-modal data, such as genomic and transcriptomic information, could enrich the model’s insights into complex biological interactions’ networks, potentially leading to more precise predictions.
GraphBAN’s ability to outperform traditional models, despite facing the challenges of diverse and complex datasets, highlights its value as a tool in computational drug discovery. Future improvements, particularly in real-time learning capabilities and data integration, could significantly boost its applicability in personalized medicine and therapeutic development.
In conclusion, our contribution to the field of CPI predictions is encapsulated in the DL-based model, GraphBAN. This model adeptly processes CPI data in the form of a bi-partite network, enabling transductive and inductive link predictions across in-domain and cross-domain scenarios. GraphBAN incorporates KD, which includes a GAE that serves as a teacher module, imparting structural and neighboring information from the CPIs network to the student model. The student model effectively concatenates the separated features of compounds and proteins involved in interactions using the BAN layer. Furthermore, GraphBAN addresses the distribution gap between the training data and cross-domain test data through the CDAN module.
Our proposed GraphBAN exhibits considering performance by providing acceptable CPI prediction accuracy. When evaluated across five diverse datasets, it surpasses ten other state-of-the-art methods found in the literature. This research offers an advancement in CPI predictions and promises substantial practical implications in drug discovery.
Methods
Datasets
We evaluate GraphBAN on five public CPI datasets: BindingDB18, BioSNAP19, KIBA20, PDBbind 201621, and C.elegans22. BindingDB18 is a public dataset that contains over a million data entries of experimental CPIs with numerical affinities, primarily derived from scientific articles and US patents. As we need a low-biased version of BindingDB, we use a selection of the dataset previously created10 that includes around 50,000 CPIs. The BioSNAP dataset19 encompasses 13,741 drug-target pairs derived from the DrugBank39 database, showcasing a comprehensive network of proteins involved in various pharmacological processes essential for drug discovery10. The KIBA dataset developed by the various bioactivity types comes with around 118,000 CPIs, with different interaction types, including IC50, K(i), and K(d)20. The Caenorhabditis elegans (C.elegans) dataset is enriched with data that includes around 6,500 CPIs with positive interactions sourced from DrugBank 4.1 and Matador40 databases, providing a reliable basis for studying drug-target interactions. To enhance the utility for model training and testing, it also incorporates highly credible negative CPI samples derived through a systematic screening framework described by22. The PDBbind 201621 dataset comprises a curated collection of protein-ligand complex structures from the Protein Data Bank, annotated with experimentally measured binding affinities, and consists of 8,107 interactions. These interactions have been binarized into active and inactive classes using a negative sampling strategy to distinguish lab-confirmed bindings. The preprocessing steps taken to make the datasets ready for training and their statistics were added in Supplementary Section 2.
Knowledge distillation architecture
The KD architecture is effective in inductive learning for graph analysis, including tasks like node classification and link prediction41,42. The method utilizes a teacher-student model, structured as follows:
-
1.
The teacher model learning: The teacher model, \({{{\rm{T}}}}\), processes an input graph, \({{{\rm{G}}}}(V,E)\), where \(V\) represents nodes and \(E\) represents edges. The teacher learns the node features, \({{{\bf{F}}}}\), of the graph \({{{\bf{F}}}}={{{\rm{T}}}}({{{\rm{G}}}}\left(V,E\right))\) where \({{{\bf{F}}}}\) includes the learned features that capture the graph’s structure.
-
2.
Knowledge distillation to the student model: The student model, \({{{\rm{S}}}}\), learns from the teacher block’s knowledge, focusing on the joint representation of node features, \({{{\bf{J}}}}\), influenced by the graph’s connectivity, \({{{\bf{J}}}}={{{\rm{S}}}}({{{\bf{F}}}})\). The knowledge from the teacher to the student is distilled by optimizing a loss function such as mean squared error (MSE) and cosine similarity. This process ensures that the student model accurately learns from the teacher.
-
3.
Application to inductive tasks: The essence of KD in inductive graph analysis is its ability to generalize to unseen or test nodes, \({V}_{{test}}\). In scenarios where only node attributes are available, and not their connections, the student model employs its learned joint representation to perform tasks like node classification or link prediction. This can be represented as, \({{{\rm{Task}}}}\left({V}_{{test}}\right)={{{\rm{S}}}}({{{{\bf{J}}}}}_{{Vtest}})\) Here, task could represent either node classification or link prediction functions.
GAE
The GAE is a DL model that takes advantage of GCNs to learn representations of graph-structured data. The GAE can be illustrated by three main sections: the encoder that made by Sage layers, the latent space that contains the generated embedding, and the decoder with linear layers43 to reconstruct the graph. The encoder section of the GAE transforms the input graph data with node features into a lower-dimensional latent space representation. This latent space represents the key features and relationships presented in the graph data. The decoder section, on the other hand, is responsible for reconstructing the original graph data from the learned latent space representation. We optimize the embedding generation with binary cross-entropy loss (BCE loss). The details of loss functions used in GraphBAN provided in Supplementary Section 7.
CNN for protein sequence
As shown in Fig. 1a, the three-layer CNN block is tailored for protein feature extraction and employs an approach that initializes a learnable embedding matrix with representations for all 23 amino acids. This proactive initialization equips the network with essential biochemical information to discern amino acid sequences in the interactions. Subsequently, the CNN block specializes in extracting local residue patterns from the matrix of protein features, a pivotal step in capturing nuanced dependencies within protein sequences. Notably, this CNN architecture conceptualizes a protein sequence as a sequence of overlapping 3-mer amino acids, enabling it to capture both short-range and long-range interactions within the protein structure. Additionally, the network adheres to a maximum allowed length for protein sequences. The sequences exceeding this length are truncated, while shorter sequences are padded with zeros. This approach reflects a fusion of domain-specific knowledge and DL, showcasing its potential for decoding protein structures and features while handling varying sequence lengths. The protein encoder layer is defined as
where \({{{{\bf{W}}}}}_{{{{\bf{c}}}}}^{({{{\bf{l}}}})}\) and \({{{{\bf{b}}}}}_{{{{\bf{c}}}}}^{({{{\bf{l}}}})}\) are the learnable weight matrices (filters) and bias vector in the \({l}^{{th}}\) CNN layer. \({H}_{p}^{l}\) denotes as \({H}_{p}^{(0)}={{{{\bf{X}}}}}_{{{{\bf{p}}}}}.{{{\rm{\sigma }}}}(\cdot )\) where \({{{{\bf{X}}}}}_{{{{\bf{p}}}}}\) is the corresponding feature matrix for each protein sequence \(P\) and \({{{\rm{\sigma }}}}(\cdot )\) represents the ReLU activation function.
GCN
In the initialization phase of the atom nodes within GCN model, the GCN21 is employed to systematically assign a 128-dimensional integer vector to each atom in the molecules. This vector encapsulates a comprehensive array of chemical properties essential for defining the atom’s characteristics and its interactions within the molecule. These properties encompass several characteristics: the atom type, which specifies the chemical element of the atom; atom degree, reflecting the number of direct covalent bonds with neighboring atoms; and the number of implicit hydrogen atoms, accounting for hydrogen atoms assumed to be bonded but not explicitly represented. Additional properties include atom hybridization, indicating orbital mixing that impacts bonding configurations; radical electrons, denoting unpaired electrons contributing to reactivity; and formal charge, representing the atom’s electric charge from electron surplus or deficit. Lastly, total hydrogen atoms encompass both explicit and implicit atoms, and aromaticity identifies atoms in an aromatic ring with shared electrons in a stable configuration. This detailed vector representation equips the model with a nuanced understanding of each atom’s potential behavior and interactions, thereby enhancing the predictive capability of the GCN model regarding molecular properties and activities.
ChemBERTa and ESM models
The ChemBERTa model16 is an advanced adaptation of the transformer architecture, meticulously crafted for predicting molecular properties. It leverages a substantial dataset of SMILES sequences for self-supervised pretraining, allowing it to internalize rich representations that encapsulate diverse chemical properties. This pretraining phase involves training the model to predict masked tokens within SMILES sequences, thereby enabling it to understand and encode complex molecular structures and their inherent characteristics.
Following the self-supervised pretraining, ChemBERTa undergoes a fine-tuning phase where it is specifically adapted to targeted molecular property prediction tasks. This dual-phase training approach ensures that the model not only learns general chemical patterns but also tailors its learning to specific applications, significantly enhancing its predictive accuracy.
The ESM17 is a type of language model, similar to those used in natural language processing, but adapted for the sequences of amino acids that make up proteins. By training on a massive dataset of 250 million protein sequences, the ESM model learns to predict the contextual relationships between amino acids in a sequence, capturing biologically relevant features such as protein structure and function directly from the sequence data. This model encodes deep biological insights, allowing it to perform tasks like predicting the effects of mutations or the structure of proteins, solely based on their amino acid sequence.
Feature fusion module
In this study, we introduce an innovative feature fusion module designed to enhance CPI predictions by integrating features from multiple sources12 for both compounds and proteins. This module is central to our approach, as depicted in Fig. 1b, and involves the strategic combination of molecular features derived from ChemBERTa and a GCN block, alongside protein features extracted from an ensemble of ESM (an LLM for proteins) and CNN modules. This fusion of feature sets enhances the comprehensiveness of our molecule and protein representations, offering a holistic view of their structures. The fusion block (with details based on compounds‘ features) is defined as
where \({{{{\bf{F}}}}}_{{{{\bf{c}}}}}\in {R}^{n\times 128}\) is the fused compound feature that n is number of atoms, \({{{{\bf{F}}}}}_{{{{\bf{g}}}}}\in {R}^{n\times 128}\) is the GCN-based molecular feature, \({{{{\bf{F}}}}}_{{{{\bf{p}}}}}\in {R}^{1\times 128}\) is the ChemBERTa feature, and \({{{\rm{t}}}}(\cdot )\) is the transition function. The transition function added as the initial LLM-based group fingerprint is in the shape of \({{{{\bf{F}}}}}_{{{{\bf{p}}}}}\in {R}^{1\times 384}\) and we use three fully connected layers (with the parameter sizes of 384 × 512, 512 × 256, and 256 × 128) to reduce the dimensionality of the fingerprint to \({{{{\bf{F}}}}}_{{{{\bf{p}}}}}\in {R}^{1\times 128}\). The \({{{{\rm{d}}}}}_{{{{\rm{out}}}}}\) is dropout function applies to prevent overfitting during the training process.
In the feature fusion process for proteins, the concept of using different embedding sizes is utilized similarly. Here, \(n\) represents the number of amino acids. The embedding from the ESM model has a size of 1280. These embeddings are fused with CNN-based features, which have a size of 128. This fusion results in a final embedding size of 128 for the protein features.
Bilinear attention neural network
The BAN which was previously introduced9 is an integral component of our model. The BAN was also previously used in visual question answering problems44, which proved to be helpful in the CPI tasks9. It is designed to capture pairwise local interactions between compounds and proteins. The BAN comprises two key elements: the bilinear interaction map, formed by combining hidden compounds and proteins representations to create an attention-weighted matrix, and the bilinear pooling layer, which extracts a unified compound-protein representation. Pairwise interaction learning is achieved through the bilinear attention mechanism, enhancing the model’s predictive capabilities.
Bilinear interaction map
The first component of BAN, the bilinear interaction map, plays a crucial role in modeling the pairwise interactions. It is constructed using the hidden representations of both drugs and targets. This process results in a pairwise interaction matrix, which encapsulates the attention weights assigned to each compound-protein pair. The values in this matrix denote the strength of interaction or relevance between specific compounds and proteins, thereby facilitating the identification of critical compound-protein associations. The single head pairwise interaction matrix \({{{\bf{I}}}}\in {R}^{M{{{\rm{x}}}}N}\) comes from the protein and compound fusion modules that generating the hidden protein and drug representations where M and N show the number of encoded substructures in an amino acid sequence of a protein and atoms/bonds in a compound. The \({{{\bf{I}}}}\) in the \({i}^{{th}}\) and \({j}^{{th}}\) column represents as follows:
Where \({h}_{d}^{i}\) is the \({i}^{{th}}\) substructure of the last compound fusion block’s layer and \({h}_{p}^{j}\) is the \({j}^{{th}}\) substructure of the last layer of the protein CNN block. The \({{{\bf{U}}}}\in {R}_{d}^{D}\times K\) and \({{{\bf{V}}}}\in {R}_{P}^{D}\times K\) are learnable weight matrices for compound and protein representations, where D is the dimension of features, \({{{\bf{q}}}}\in {R}^{K}\) is a learnable weight vector, and (∘) denotes element-wise product.
Bilinear pooling layer
The second component of BAN is the pooling layer to obtain the joint representation \({{{\bf{f}}}}\in {R}^{k}\), applying over the interaction map \({{{\bf{I}}}}\). The \({K}^{{th}}\) element of \({{{{\bf{f}}}}}^{{\prime} }\) is computed as
where \({U}_{k}\) and \({V}_{k}\) denote the \({k}^{{th}}\) column of weight matrices \({{{\bf{U}}}}\) and \({{{\bf{V}}}}\). Moreover, to obtain more compact feature map, we have a sum pooling on the joint representation vector
where the SumPool(⋅) function is a sum pooling operation with stride \(s\) and it converts the dimensionality of \({{{{\bf{f}}}}}^{{{{\prime} }}}\in {R}^{k}\) to \({{{\bf{f}}}}\in {R}^{k/s}\). In the last step we feed the joint representation \({{{\bf{f}}}}\) into a fully connected layer to classify the inputs and the objective of training is to minimize the BCE with logit loss function.
Cross-domain adaptation to enhance generalization
In cross-domain analysis, we tend to train our model with CPIs and to ensure that the trained model can perform well in real-world cases where the compounds and proteins are different from the nodes in the training set based on their features distributions. As a result of this scenario, it will become hard for simple ML/DL models to perform well on cross-domain data in the test sets. To overcome this distribution shift issue between the training and test data, a model is proposed45 that uses the CDAN to combine adversarial networks with multilinear feature mapping. As it was demonstrated that using CDAN can improve CPI prediction accuracy9, we also embed the CDAN into GraphBAN to enhance the performance of inductive cross-domain CPI prediction.
As shown in Fig. 1c, the BAN layer generates the source domain joint representation of compound-protein pairs called \({{{{\bf{X}}}}}_{{{{\bf{s}}}}}\) with true labels \({{{{\bf{Y}}}}}_{{{{\bf{s}}}}}\) plus the target domain joint as \({{{{\bf{X}}}}}_{{{{\bf{t}}}}}\) without any label. The CDAN’s workflow starts with the component \({{{\rm{f}}}}(\cdot )\) as the feature extractor that generates concatenation of separate initial features of the nodes and the BAN’s output for source and target domains separately as follows, \({{{{\bf{f}}}}}_{{{{\bf{s}}}}}={{{\rm{F}}}}\left({{{{\bf{X}}}}}_{{{{\bf{s}}}}}\right),{{{{\bf{f}}}}}_{{{{\bf{t}}}}}={{{\rm{F}}}}({{{{\bf{X}}}}}_{{{{\bf{t}}}}})\). The next module is a classifier layer called \({{{\rm{G}}}}(\cdot )\) which works as the generator part in the adversarial loss, that generates \({{{{\bf{g}}}}}_{{{{\bf{s}}}}}={{{\rm{G}}}}\left({{{{\bf{X}}}}}_{{{{\bf{s}}}}}\right)\) for the source and \({{{{\bf{g}}}}}_{{{{\bf{t}}}}}={{{\rm{G}}}}\left({{{{\boldsymbol{X}}}}}_{{{{\boldsymbol{t}}}}}\right)\) for the target data.
To be able to apply the domain discriminator, we need to have the joint conditional representation h of the g and f defined as
where \((\bigoplus )\) is the outer product.
Based on the CDAN’s workflow, we align the joint conditional representation h for both the source and target domains by the domain discriminator module \({{{\rm{D}}}}(\cdot )\). The task of \({{{\rm{D}}}}\left(\cdot \right)\) function is to learn how to distinguish between the joint conditional representation h generated from the source and target data domains. As the final goal of the conditional adversarial network, the \({{{\rm{F}}}}(\cdot )\) and \({{{\rm{G}}}}(\cdot )\) functions are trained to minimize the source domain cross entropy loss \({{{\rm{L}}}}\left(\cdot \right)\) by having the true label information and simultaneously generate h in an indistinguishable way for \({{{\rm{D}}}}\left(\cdot \right)\) function. The following loss functions represent the cross-entropy loss (\({{{{\rm{L}}}}}_{{{{\rm{s}}}}}\)) and adversarial loss (\({{{{\rm{L}}}}}_{{{{\rm{adv}}}}}\)) considering \({{{\rm{E}}}}\left(\cdot \right)\) represents the expectation over the empirical source domain data distribution.
Following the procedure of optimization for the adversarial loss we need to yield the maxmin() model and define the final representation of CDAN’s loss function (\({{{{\rm{L}}}}}_{{{{\rm{CDAN}}}}}\)) as
where \(\beta > 0\) is a hyperparameter to weight \({{{{\rm{L}}}}}_{{{{\rm{adv}}}}}\).
Experimental setting
Implementation
The proposed method is developed using Python 3.8 and PyTorch 1.7.146, DGL 0.7.147, Scikit-learn 1.0.2, Numpy 1.20.248, Pandas 1.2.449, and RDKit 2021.03.2 libraries. The full list of detailed hyperparameter settings is included in Supplementary Section 3. The hyperparameters are set using a combination of optimization techniques (random search) and experimental analysis. For instance, GraphBAN runs with a batch size set to 32 and the optimizer set to Adam in both the student and teacher submodules. The learning rate for optimizing the GAE in the teacher module is 1e-3, and in the student module, it is 1e-4. To achieve the best possible results with the validation set, GraphBAN runs for a maximum of 250 epochs in the teacher module and 50 epochs in the student module.
Baselines
Our baseline framework encompasses the random forest RF50 model, which has input augmented with ECFP features, alongside the utilization of k-mer frequency embeddings for the effective representation of compound and protein data. DeepConv-DTI51 presents a DL model that employs CNNs to identify local residue patterns in raw protein sequences for predicting drug target interactions51. GraphDTA52 leverages GNNs to encode compound molecular graphs and employs CNNs for encoding protein sequences. MolTrans10 is a DL model that innovatively adapts the transformer network to encode both compound and protein information, enhancing its predictive capabilities through a CNN-based interactive module designed to capture sub-structural interactions. The GraphsformerCPI53 introduces a DL model that leverages graph transformers to predict CPIs more accurately. This model incorporates dual attention mechanisms and structure-enhanced self-attention techniques to effectively integrate semantic and spatial structural features of molecules. Moreover, we introduce DrugBAN9, a model rooted in our student framework. The model has no feature fusion module, but it concatenates compound and protein features derived from the GCN and CNN modules, and further incorporates BAN and CDAN for cross-domain analysis without the inclusion of network analysis within its architecture. The SPVec-SGCN-CPI54 model utilizes a sequence-based approach based on skip-gram model55 to embed the SMILES and amino acid sequences and generates a graph with the compound and protein features to do compound-protein link prediction. The DSANIB model56 captures local interactions between CPI pairs of compounds and proteins each captured with GCN and CNN modules respectively. Furthermore, focusing on providing effective embeddings of compounds and proteins and concatenate the embeddings using inter-view attention network. The PocketDTA model57 is a multimodal architecture designed for drug-target affinity (DTA) prediction by integrating diverse data modalities. It leverages Morgan fingerprints and ESM-2 pre-trained embeddings for drug and protein sequence representations. The model fuses these features through a BAN layer. Finally, we have FusionDTI58 that is a bi-encoder model for drug-target interaction prediction that integrates self-referencing embedded strings (SELFIES) for drugs and structure-aware vocabulary for proteins, incorporating 3D features from AlphaFoldDB via Foldseek. It employs BAN and Cross Attention Network (CAN) to capture fine-grained token-level interactions. BAN models pairwise interactions, while CAN uses multi-head and cross-attention mechanisms for deeper dependencies. The fused outputs are processed through an multi layer perceptron for binary interaction prediction.
These models collectively represent renowned algorithms in the realm of CPI prediction. However, they do not function in the same way as our proposed methods, as they do not focus on analyzing the input training data as a network and performing inductive cross-domain analyses all together.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw data used as benchmark are BindingDB available at http://www.bindingdb.org, BioSNAP available at https://snap.stanford.edu/biodata/datasets/10015/10015-ChG-TargetDecagon.html, KIBA available at https://researchportal.helsinki.fi/en/datasets/kiba-a-benchmark-dataset-for-drug-target-prediction, PDBbind 2016 avaliable at http://www.pdbbind.org.cn, and C.elegans avaliable at https://snap.stanford.edu/data/C-elegans-frontal.html. The data splits used to train GraphBAN are included in the GitHub repository at https://github.com/HamidHadipour/GraphBAN/tree/main/Data. The predicted probabilities generated by GraphBAN for the 134 candidates identified in the case study are provided in Supplementary Data 1. All calculated properties of the selected compounds in the case study are available in Supplementary Data 2. The trained models used to make predictions in the case study analyses are hosted on Zenodo at https://doi.org/10.5281/zenodo.14813233. The input and output docking files, as well as attention weight files used for compound colouring are stored in the GitHub repository at https://github.com/HamidHadipour/GraphBAN/tree/main/case_study. Source data are provided with this paper as a Source Data file.
Code availability
The code used to develop the model, perform the analyses, and generate results in this study plus the information on the computational costs in terms of computation time is publicly available and has been deposited in GitHub at https://github.com/HamidHadipour/GraphBAN under MIT license. The specific version of the code associated with this publication is archived in Zenodo and is accessible via https://zenodo.org/records/1498470759.
References
Singh, N. et al. Drug discovery and development: introduction to the general public and patient groups. Fronti. Drug Discov. https://doi.org/10.3389/fddsv.2023.1201419 (2023).
Sadybekov, A. V. & Katritch, V. Computational approaches streamlining drug discovery. Nature 616, 673–685 (2023).
Hollingsworth, S. A. & Dror, R. O. Molecular Dynamics Simulation for All. Neuron 99, 1129–1143 (2018).
Durrant, J. D. & McCammon, J. A. Molecular dynamics simulations and drug discovery. BMC Biol. 9, 71 (2011).
Hughes, J., Rees, S., Kalindjian, S. & Philpott, K. Principles of early drug discovery. Br. J. Pharmacol. 162, 1239–1249 (2011).
Wang, W., Yang, X., Wu, C. & Yang, C. CGINet: graph convolutional network-based model for identifying chemical-gene interaction in an integrated multi-relational graph. BMC Bioinformatics. 21, 544 (2020).
Yu, L. et al. HGDTI: predicting drug–target interaction by using information aggregation based on heterogeneous graph neural network. BMC Bioinformatics. 23, 126 (2022).
Chatterjee, A. et al. Improving the generalizability of protein-ligand binding predictions with AI-Bind. Nat. Commun. 14, 1989 (2023).
Bai, P., Miljković, F., John, B. & Lu, H. Interpretable bilinear attention network with domain adaptation improves drug–target prediction. Nat. Mach. Intell. 5, 126–136 (2023).
Huang, K., Xiao, C., Glass, L. M. & Sun, J. MolTrans: molecular interaction transformer for drug–target interaction prediction. Bioinformatics 37, 830–836 (2021).
Hua, Y., Song, X., Feng, Z. & Wu, X. MFR-DTA: a multi-functional and robust model for predicting drug–target binding affinity and region. Bioinformatics 39, btad056 (2023).
Hua, Y. et al. CPInformer for efficient and robust compound-protein interaction prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 20, 285–296 (2023).
Abbasi, K. et al. DeepCDA: Deep cross-domain compound-protein affinity prediction through LSTM and convolutional neural networks. Bioinformatics 36, 4633–4642 (2020).
Kao, P. Y., Kao, S. M., Huang, N. L. & Lin, Y. C. Toward drug-target interaction prediction via ensemble modeling and transfer learning. Proc. IEEE Int. Conf. Bioinformatics Biomed. 2021, 2384–2391 (2021).
Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988).
Chithrananda, S., Grand, G. & Ramsundar, B. ChemBERTa: Large-scale self-supervised pretraining for molecular property prediction. ArXiv https://doi.org/10.48550/arXiv.2010.09885 (2020).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA 118, e2016239118 (2021).
Gilson, M. K. et al. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 44, D1045–D1053 (2016).
Zitnik, M., Sosič, R., Maheshwari, S. & Leskovec, J. BioSNAP Datasets: Stanford Biomedical Network Dataset Collection. https://snap.stanford.edu/biodata (2018).
Tang, J. et al. Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis. J. Chem. Inf. Model 54, 735–743 (2014).
Cang, Z., Mu, L. & Wei, G.-W. Representability of algebraic topology for biomolecules in machine learning based scoring and virtual screening. PLoS Comput. Biol. 14, e1005929 (2018).
Liu, H., Sun, J., Guan, J., Zheng, J. & Zhou, S. Improving compound-protein interaction prediction by building up highly credible negative samples. Bioinformatics 31, i221–i229 (2015).
Ranganathan, R., Lu, K. P., Hunter, T. & Noel, J. P. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 89, 875–886 (1997).
Pinch, B. J. et al. Identification of a potent and selective covalent Pin1 inhibitor. Nat. Chem. Biol. 16, 979–987 (2020).
Lu, K. P. & Zhou, X. Z. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 8, 904–916 (2007).
Chen, Y. et al. Prolyl isomerase Pin1: a promoter of cancer and a target for therapy. Cell Death Dis. 9, 883 (2018).
Gómez-Bombarelli, R. et al. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 4, 268–276 (2018).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings 1PII of original article: S0169-409X(96)00423-1. The article was originally published in advanced drug delivery reviews 23 (1997) 3–25. 1. Adv. Drug Deliv. Rev. 46, 3–26 (2001).
Baell, J. B. & Holloway, G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53, 2719–2740 (2010).
Swanson, K. et al. ADMET-AI: a machine learning ADMET platform for evaluation of large-scale chemical libraries. Bioinformatics 40, btae416 (2024).
Waring, M. J. Lipophilicity in drug discovery. Expert Opin. Drug Discov. 5, 235–248 (2010).
Bickerton, G. R., Paolini, G. V., Besnard, J., Muresan, S. & Hopkins, A. L. Quantifying the chemical beauty of drugs. Nat. Chem. 4, 90–98 (2012).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010).
Rossi, A., Marti‐Renom, M. A. & Sali, A. Localization of binding sites in protein structures by optimization of a composite scoring function. Protein Sci. 15, 2366–2380 (2006).
Bajusz, D., Rácz, A. & Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 7, 20 (2015).
Durant, J. L., Leland, B. A., Henry, D. R. & Nourse, J. G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 42, 1273–1280 (2002).
Kralj, S., Jukič, M. & Bren, U. Molecular filters in medicinal chemistry. Encyclopedia 3, 501–511 (2023).
Liu, H., Shen, C., Li, H., Hou, T. & Yang, Y. Discovery of potent covalent CRM1 inhibitors via a customized structure-based virtual screening pipeline and bioassays. J. Chem. Inf. Model 64, 7422–7431 (2024).
Wishart, D. S. et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 36, D901–D906 (2008).
Günther, A. L. B., Remer, T., Kroke, A. & Buyken, A. E. Early protein intake and later obesity risk: which protein sources at which time points throughout infancy and childhood are important for body mass index and body fat percentage at 7 y of age? Am. J. Clin. Nutr. 86, 1765–1772 (2007).
Samy, A. E., Kefato, Z. T. & Girdzijauskas, S. Graph2Feat: inductive link prediction via knowledge distillation. Proc. ACM Web Conf. 2023, 805–812 (2023).
Zhang S., Liu Y., Sun Y., Shah N. Graph-less neural networks: teaching old mlps new tricks via distillation. arXiv https://doi.org/10.48550/arXiv.2110.08727 (2021).
Kipf, T. N. & Welling, M. Variational graph auto-encoders. arXiv https://doi.org/10.48550/arXiv.1611.07308 (2016).
Zhan, L.-M., Liu, B., Fan, L., Chen, J. & Wu, X.-M. Medical visual question answering via conditional reasoning. In Proc. 28th ACM International Conference on Multimedia 2345–2354 (2020).
Long, M., Cao, Z., Wang, J. & Jordan, M. I. Conditional adversarial domain adaptation. Adv. Neural Inf. Process. Syst. 31, 1647–1657(2018).
Paszke, A. et al. PyTorch: an imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Syst. 32, 8026–8037 (2019).
Wang, M. et al. Deep graph library: a graph-centric, highly-performant package for graph neural networks. Preprint at arXiv https://arxiv.org/abs/1909.01315 (2019).
Harris et al. Array programming with numpy. Nature 585, 357–362 (2020).
The pandas development team. pandas-dev/pandas: pandas 1.2.4. Zenodo https://doi.org/10.5281/zenodo.4681666 (2021).
Ho, T. K. Random decision forests. Proc. Int. Conf. Document Anal. Recognit. 1, 278–282 (1995).
Lee, I., Keum, J. & Nam, H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 15, e1007129 (2019).
Nguyen, T. et al. GraphDTA: predicting drug–target binding affinity with graph neural networks. Bioinformatics 37, 1140–1147 (2021).
Ma, J. et al. GraphsformerCPI: Graph transformer for compound–protein interaction prediction. Interdiscip. Sci. 16, 361–377 (2024).
Zhang, Y. et al. An end-to-end method for predicting compound-protein interactions based on simplified homogeneous graph convolutional network and pre-trained language model. J. Cheminform. 16, 67 (2024).
Gao, M. et al. GraphormerDTI: A graph transformer-based approach for drug-target interaction prediction. Comput. Biol. Med. 173, 108339 (2024).
Tian, Z. et al. DSANIB: drug-target interaction predictions with dual-view synergistic attention network and information bottleneck strategy. IEEE J. Biomed. Health Inform. 29, 1484–149 (2024)
Zhao, L., Wang, H. & Shi, S. PocketDTA: an advanced multimodal architecture for enhanced prediction of drug−target affinity from 3D structural data of target binding pockets. Bioinformatics 40, btae594 (2024).
Meng, Z., Meng, Z., Yuan, K. & Ounis, I. FusionDTI: fine-grained binding discovery with token-level fusion for drug-target interaction. arXiv https://doi.org/10.48550/arXiv.2406.01651 (2024).
Hadipour, H. et al. GraphBAN. Zenodo https://zenodo.org/records/14984707 (2025).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Acknowledgements
This research was supported in part by the Canadian Institutes of Health Research (grant #169121 to S.T.C., R.D., and P.H.; PLL185683 to P.H.; PJT 190272 to P.H.), the Canada Research Chairs Tier II Program (CRC-2021-00482 to P.H.), the Natural Sciences, Engineering Research Council of Canada (RGPIN-2021-04072 to P.H.) and The Canada Foundation for Innovation (CFI) John R. Evans Leaders Fund (JELF) program (grant #43481 to P.H.).
Author information
Authors and Affiliations
Contributions
H.H. and P.H. conceptualized the idea and designed the algorithms. P.H. and S.T.C. oversaw the project. H.H. conducted the experiments, implemented the algorithms, and drafted the manuscript. H.H. and Y.Y.L. created the figures and tables. Y.Y.L. performed the case study analysis and wrote the case study section of the manuscript. H.H., Y.S., and C.D. conducted the baseline experiment analysis. H.H. Y.S. and Y.Y.L. prepared the data. S.T.C., R.D. and L.L. provided guidance on data analysis and interpretation. S.T.C., P.H., and R.D. secured funding for the project. All authors contributed to the manuscript revisions and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Karim Abbasi, Dong-qing Wei and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hadipour, H., Li, Y.Y., Sun, Y. et al. GraphBAN: An inductive graph-based approach for enhanced prediction of compound-protein interactions. Nat Commun 16, 2541 (2025). https://doi.org/10.1038/s41467-025-57536-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57536-9
This article is cited by
-
GraphBAN-based identification of active ingredients and key gene targets in compound Chinese herbal medicines for polycystic ovary syndrome
Naunyn-Schmiedeberg's Archives of Pharmacology (2026)
-
Generalizable compound protein interaction prediction with a model incorporating protein structure aware and compound property aware language model representations
Communications Chemistry (2025)
-
Evidential deep learning-based drug-target interaction prediction
Nature Communications (2025)
