
Abstract
Integration of multi-omics data can provide information on biomolecules from different layers to illustrate the complex biology systematically. Here, we build a multi-omics atlas containing 132,570 transcripts, 44,473 proteins, 19,970 phosphoproteins, and 12,427 acetylproteins across wheat vegetative and reproductive phases. Using this atlas, we elucidate transcriptional regulation network, contributions of post-translational modification (PTM) and transcript level to protein abundance, and biased homoeolog expression and PTM in wheat. The genes/proteins related to wheat development and disease resistance are systematically analyzed, thus identifying phosphorylation and/or acetylation modifications for the seed proteins controlling wheat grain quality and the disease resistance-related genes. Lastly, a unique protein module TaHDA9-TaP5CS1, specifying de-acetylation of TaP5CS1 by TaHDA9, is discovered, which regulates wheat resistance to Fusarium crown rot via increasing proline content. Our atlas holds great promise for fast-tracking molecular biology and breeding studies in wheat and related crops.
Similar content being viewed by others


Introduction
Dissection and improvement of crop traits, such as yield, quality and stress tolerance, are among the most urgent challenges for humans to tackle in order to ensure efficient crop production and food security. However, these tasks are hard to accomplish because plant traits are usually controlled by polygenes and affected by changing environmental factors, with the outcomes shaped by complex gene transcriptional regulations and functional interactions of diverse proteins that act in multiple biochemical and physiological processes1,2,3,4. Furthermore, post-translational modifications (PTMs), such as phosphorylation, acetylation, ubiquitination, and glycosylation, often occur to cellular proteins in response to developmental and environmental cues5,6,7,8, adding further complexities to understanding trait formation and variation9. Fortunately, the advent of genomics, especially the rapid development of functional multi-omics resources, e.g., transcriptome, proteome, phosphoproteome, and acetylproteome, have greatly facilitated trait studies, with substantial progress made in the model plants (e.g., Arabidopsis) and a few crop species with relatively simple genomes10,11,12.
For example, comprehensive transcriptome, proteome, and phosphoproteome datasets have been produced and integratively analyzed in Arabidopsis, which are highly valuable for discovering important proteins, tissue-specific protein complexes, and signalling processes involving phosphorylation events10. By integrating transcriptome, proteome, and phosphoproteome research, robust gene regulatory networks (GRNs) useful for systemic gene functional analysis were built for developing maize11. In-depth analysis of a multi-omics atlas constructed with transcriptome, proteome, phosphoproteome, and acetylproteome datasets provided insights into the mechanisms regulating symbiosis in Medicago truncatula12. The upsurge of systemic multi-omics research demonstrates the high value of this type of resources for accelerating trait analysis and crop improvement in the genomics era13. However, high quality multi-omics resources are still very limited for the plants with complex genome. These include hexaploid common wheat (Triticum aestivum, 2n = 6x = 42, AABBDD), a major staple food crop whose efficient improvement is of immense importance to humans in a world with fast population growth, dwindling agricultural resources, and intensifying climate changes. The genome of common wheat is comprised of 21 chromosome pairs organized in three sub-genomes (A, B, and D)14. Its large genome size ( ~ 17 Gb) and allohexaploidy, characterized by high degree duplications of gene and repetitive elements15, hampered functional studies of complex traits. For example, repetitive elements account for approximately 85% of the genome; more than 90% of the genes has two or three sub-genomic homoeologs16, with more than 22,000 gene triads annotated. However, with the development of high-throughput sequencing and assembly technologies, whole genome sequence has been determined for over 30 cultivars from different parts of the world, with relatively more genome resources generated for the landrace cultivar Chinese Spring (CS)16,17,18,19,20. However, the available transcriptome and proteome data were usually generated using limited plant samples and the coverage of proteins are often low21. More importantly, there is still scarce information on wheat protein PTM omics (PTMomics hereafter). This will become a bottleneck for deeply dissecting and improving wheat traits, considering PTMs have been found to play a pivotal role in fine tuning protein activities in response to developmental and environmental changes and have profound impacts on phenotypic diversities and trait variations5,6,7,8.
In this work, in recognizing the fundamental need for systematic and integrated multi-omics resources in common wheat, we generate transcriptome, proteome, phosphoproteome, and acetylproteome data sets using 20 samples across wheat vegetative and reproductive phases, by combining high-throughput RNA sequencing (RNA-seq) and liquid chromatography tandem mass spectrometry (LC-MS/MS). We explore relative contributions of PTMs and transcript level to protein abundance control at genome-wide level, predict developmentally regulated gene regulatory networks (GRNs), and examine biased homoeolog expression and PTMs. The usefulness of our multi-omics atlas is demonstrated by unraveling transcripts, proteins, and rich phosphorylation and acetylation PTMs for the genes related to wheat development and disease resistance. In addition, an important protein module entailing TaP5CS1 de-acetylation by TaHDA9 is discovered, which regulate wheat resistance to Fusarium crown rot (FCR) via increasing proline content. The insights gained demonstrate that our multi-omics atlas can be used to accelerate molecular biology and breeding studies in common wheat.
Results
Overview of omics data sets
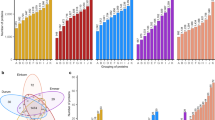
To produce the data required for constructing a high-quality multi-omics atlas for common wheat, we profiled the transcripts, proteins, as well as the proteins with phosphorylation or acetylation sites, in each of the 20 sets of samples collected at the seedling, jointing, booting, heading, or grain filling stages (hereafter referred as SS, JS, BS, HS, and FS, respectively) of a strong gluten wheat Yunong 268 (Fig. 1a). Root (RT) tissues were profiled for all stages. Leaf (LF) tissues were examined for SS, JS, BS, and HS plants, with flag leaves used at HS. Moreover, basal stem (BPST) tissues were analyzed for JS, BS, HS, and FS plants, immature and flowering spike (SP) tissues for BS and HS plants, stalk and leaf sheath (SL and SH) tissues for HS plants, and developing seeds at 14, 28, and 35 days post anthesis (DPA) for FS plants (Fig. 1a).

a A schematic overview of 20 wheat tissues for the atlas across wheat vegetative and reproductive phases. b Circular proteome maps depict the similarities and differences in the tissue-specific proteomes based on proteome analysis of 20 tissues. The protein/transcript numbers were displayed in the bars of each tissue. The number of the identified proteins associated with each tissue was displayed in the bars that span each distinct tissue region. The color gradient within these bars reflected the number of proteins identified in each tissue. The relative protein abundance within each tissue is represented by the histograms (black) that span the length of each bar. Specific PTMs proteins, tissue-specific proteins, and acetylation proteins, phosphorylation proteins, and transcript were indicated by the gray, red, purple, yellow, and blue bars, respectively. c The numbers of the genes/transcripts/proteins and their PTM (post-translational modification) sites and triads. SS_RT: root of seedling stage; SS_SG: seedling of seedling stage; JS_LF: leaf of jointing stage; JS_RT: root of jointing stage; JS_BPST: basal part of stem of jointing stage; BS_LF: leaf of booting stage; BS_RT: root of booting stage; BS_BPST: basal part of stem of booting stage; BS_SP: spike of booting stage; HS_LF: flag leaf of heading stage; HS _RT: root of heading stage; HS _BPST: basal part of stem of heading stage; HS_ SL: stalk of heading stage; HS_ SH: sheath of heading stage; HS _SP: spike of heading stage; FS _RT: root of filling stage; FS _BPST: basal part of stem of filling stage; FS_14d_seed: seed of 14 day post anthesis (DPA) of filling stage; FS_28d_seed: 28 DPA of filling stage; FS_35d_seed: 35 DPA of filling stage. The same below.
After integrative analysis of all data sets using the genome information of Chinese Spring (CS) as a reference, we identified 132,570 full-length transcripts from 106,914 genes (Fig. 1b, c; Supplementary Fig. 1a; Supplementary Data 1), including the transcripts for 22,097 gene triads (Supplementary Data 1). The transcribed genes/triads we identified approached the range of high confidence genes (106,925) and triads (22,097) annotated for common wheat (http://wheat.cau.edu.cn/TGT/download.html), demonstrating the high genome-wide coverage of our transcriptome data. In the proteomics data set, we identified 44,473 proteins, including 19,970 phosphoproteins with 69,364 phosphorylation sites and 12,427 acetylproteins with 34,974 acetylation sites (Fig. 1b, c; Supplementary Fig. 1a–h; Supplementary Data 1). Of the 44,473 proteins, 32,256 were identified in the conventional proteome analysis, which had intensity-based absolute quantification (iBAQ, see Methods) values, 8220 and 3015 were specifically found in phosphoproteome and acetylproteome experiments, respectively, but without protein iBAQ quantification (Supplementary Fig. 1h). Therefore, the 32,256 proteins were used as the main proteome data set in subsequent analyses requiring protein abundance information. Overall, the numbers of proteins, phosphoproteins, and acetylproteins we revealed are over 4-17 times higher than those reported for common wheat by previous studies21,22. These numbers were also substantially higher than those described for similar data sets in A. thaliana, Z. mays, and M. truncatula10,11,12 consistent with the hexaploid nature of common wheat.
Of the 132,570 transcripts, 33,452 showed relatively high abundance with their average TPM (transcripts per million) values greater than 0.5, which specified 81% of the 32,256 proteins (identified in conventional proteome analysis), 78% of the 19,970 phosphoproteins, and 77% of the 12,427 acetylproteins (Supplementary Fig. 1i). Among the 69,364 phosphorylation sites, phosphorylated serine, threonine, and tyrosine (pSer, pThr, and pTyr) accounted for 85.3%, 14.0%, and 0.7%, respectively (Supplementary Fig. 1j), which were comparable to similar data obtained in A. thaliana, Z. mays, and M. truncatula10,11,12. The numbers of transcripts, proteins, phosphoproteins, and acetylproteins varied considerably among the 20 samples, with at least 81,719 transcripts, 16,847 proteins, 7236 phosphoproteins, and 3374 acetyl-proteins found across this series of samples (Supplementary Fig. 1a; Supplementary Data 1).
Shared transcriptome and proteome (presented in all samples) were composed of 27,149 (20.5% of 132,570) transcripts and 4002 (12.4% of 32,256) proteins, respectively (Supplementary Data 2). We calculated entropy index for the 32,256 proteins (see Methods) and applied a one-tenth percentile cutoff (Supplementary Fig. 1k), which showed that 1633 proteins (5.1% of 32,256) were highly sample (tissue) specific (Fig. 2a; Supplementary Data 2). An overall protein expression pattern in the 20 samples was shown in Fig. 1b. Greater numbers of specific transcripts and proteins were detected in the three stages of developing grains (Fig. 1b), corresponding to abundant accumulation of SSPs known to specifically expressed in wheat grains23,24. According to iBAQ values, the shared proteome exhibited substantially high abundance (average iBAQ: 626,205 ± 27,446) over tissue specific proteins (98,727 ± 13,176). Gene ontology (GO) enrichment analysis indicated that the shared proteome was predominantly involved in tricarboxylic acid (TCA) cycle, protein transport and translation (Fig. 2b). In contrast, the 1633 tissue-specific proteins were mainly enriched for response to water and reproductive growth (Fig. 2c). Additionally, we found that the shared proteins were more likely modified by phosphorylation and acetylation and carried relatively (Fig. 2d, e).

a The number of tissue-specific proteins, P (phosphorylation)-sites, K (acetylation)-sites and transcripts in 20 tissues. b Heatmaps illustrate the significantly enriched biological processes by gene ontology (GO) enrichment analysis with the shared proteins from each tissue. c Biological processes of tissue-specific proteins by GO enrichment. (d) The proportion and (e) distribution of phosphorylation and acetylation of shared proteins and tissue-specific proteins. f Scatter plot of protein-versus-transcript abundance (median across 20 tissues). r denotes Pearson’s coefficient of correlation. TPM, transcripts per million. g Genes with high and low PTRs (protein-to-mRNA ratio) are defined as two standard deviations outside the median PTR distribution. FDR, false discovery rate. The same abbreviations were used in subsequent figure legends. Source data are provided as a Source Data file.
We developed a database and web-interface named WheatPro (https://www.csuligroup.com/WheatPro/), providing access to transcripts, proteins (including phospho-proteins and acetyl-proteins), PTM sites, contribution ratios of PTMs and transcripts (PTC) values, co-expression modules, and gene regulatory networks revealed through analyzing 20 distinct wheat samples across five developmental stages. In addition, all of our data will also be made available via ProteomicsDB (https://www.ProteomicsDB.org), which offers a large number of interactive views, including visualization of individual mass spectrometry (MS) data and superimposition of identified PTMs on experimental or predicted protein structures.
Landscapes of gene expression
Using the four omics data sets obtained, we explored the overall characteristics, relationships, and patterns of genes expression at the transcript, protein, phosphorylation, and acetylation levels (Supplementary Fig. 1l–n and Supplementary Fig. 2a–g). Protein abundance spanned six orders of magnitude, whereas transcript abundance only spanned four (Supplementary Fig. 1l–m). Protein phosphorylation and acetylation were identified across the entire protein abundance (Supplementary Fig. 1m–n), indicating that phospho-peptides or acetyl-peptides were highly enriched. By analyzing eight representative samples, gene expression at the protein and transcript levels differed substantially (r = 0.25 ~ 0.53; Supplementary Fig. 2e), with almost no overlapping in protein and transcript levels as well as in the most abundant transcripts and proteins (Supplementary Fig. 2g). In the developing grains, γ-gliadins were predominant in 35 d seeds and reached 2.1% of the total proteins quantified by iBAQ; the top five types of proteins, i.e., low-molecular-weight glutenin subunits (LMW-GSs), γ-gliadins, histone H4, trypsin inhibitor CMc, and the large subunit of ribulose-1,5-bisphosphate carboxylase/oxygenase (RbcL), accounted for 5.3% (Supplementary Fig. 2f, right). As expected, RbcL, accounting for 2.3%–3.9% of the total leaf proteins quantified, was also the most abundant protein in all leaf samples (Supplementary Fig. 2f, right).
Some genes with similar transcript levels exhibited over 100-fold difference in the abundance of their proteins (Fig. 2f). Variation of protein-to-mRNA ratio (PTR) in wheat samples exhibited similar distributions (Fig. 2g), which is consistent to what was found in A. thaliana10. The genes with high PTRs were involved mainly in the tricarboxylic acid (TCA) cycle and carbon fixation in photosynthetic organisms (Supplementary Fig. 2h) that were possibly associated with housekeeping functions25,26. In contrast, genes with low PTRs were enriched mainly in autophagy and plant hormone signal transduction processes that generally involved protein degradation27,28 (Supplementary Fig. 2h). Whereas over 90% genes had stable PTRs, implying these genes probably under similar regulation in different tissues (Supplementary Fig. 2i).
Correlation analysis across the 20 samples indicated that protein abundance had the greatest variation among the four omics data sets (Supplementary Fig. 2j–o). Although phosphorylation/acetylation levels fluctuated among the samples, stable ratios between protein abundance and phosphorylation/acetylation sites, i.e., the higher the abundance, the higher the PTM levels, were consistently observed (Supplementary Fig. 2l–o). This consistency was possibly resulted from similar trends of variations in the abundance of cognate proteins.
Transcription factors (TFs) are key regulators of plant growth, development, and cell fate29. Based on Plant Transcription Factor Database30, we identified 11,516 transcripts, 3470 proteins, 2664 phosphoproteins, and 1488 acetylproteins in 57 TF families (Supplementary Data 3); the majority of them exhibited temporal and spatial expression patterns (Supplementary Data 3; Supplementary Fig. 3a–b). To further explore the regulatory patterns of gene expression across wheat development, we generated gene regulatory networks (GRNs), which are directed networks of TFs and their target genes11. Previous study indicated that the incorporation of TF phosphorylation increased GRN quality11, therefore, we constructed an over 0.9 million GRN (total GRN) combining mRNA, protein, and phosphorylation GRNs using GRNboost2 (Supplementary Data 4). Moreover, 20 individual GRNs were constructed in the same tissue type-based manner (TTBM), and they were involved in at least 1459 TFs and over 0.7 million regulation relationships (Supplementary Data 4; Supplementary Fig. 4). The total GRN was evaluated using the published data for the well-known wheat TF TaNAC019 (NAC_290, TraesCS3B03G0216600.1) that regulated the expression of various genes31. In the GRN of TaNAC019, we observed 179 target genes that were involved in diverse biological processes (Supplementary Fig. 5a; Supplementary Data 5), and 82 of them were previously reported31. As the largest TF family in plants, MYB played important roles in regulating plant growth and development32. In the top 500 TFs using GRNboost2 predictions, 48 MYB were covered and they regulated 12,767 target genes (Supplementary Fig. 5b; Supplementary Data 6).
To examine the patterns of all gene types throughout wheat development, we used gene co-expression network analysis (WGCNA)33 to assign common functionality to genes detected as mRNAs, proteins, phospho-sites, or acetyl-sites in at least three tissues. Of these genes, 58,263, 8231, 15,930, and 11,033 were assigned to 57, 29, 60, and 26 modules, respectively (Supplementary Data 7-8). Nine modules had similar expression patterns in different omics (Supplementary Data 7). Besides, 39,558 hub genes (Supplementary Data 9) were identified. These resources could provide valuable cues for further studies of wheat gene expression.
Contribution of PTMs and transcript level to protein abundance
To study the importance of PTMs and transcriptional regulation on protein abundance, we calculated contribution ratios of PTMs and transcripts (PTC) to protein abundance with 22,941 proteins whose transcripts and protein products were detected in at least eight samples (Fig. 3a, b; see Methods). Based on PTC values, we defined three categories of proteins: transcript dominant (TD) with PTC of 0–40%, balanced (BL) with PTC of 40%–60%, and PTM dominant (PD) with PTC of 60%–100% (Fig. 3c, d). For phosphorylation (n = 6661) and acetylation (n = 5383), proteins in the PD category accounted for a higher proportion (47.5% and 54.5%) than the BL and PT categories (Fig. 3d; Supplementary Data 10). GO enrichment revealed that phosphorylation or acetylation proteins in TD categories were mainly involved in RNA modification and small GTPase-mediated signal transduction, while proteins in PD category was mainly involved in sucrose metabolic processes and microtubule-based movement (Supplementary Data 11). Furthermore, we analyzed the PTC of TFs and identified different PTC types in the same TF family (Fig. 3e; Supplementary Data 10), indicating the complexity of gene regulation in hexaploid wheat. Some biological processes were simultaneously involved in TD and PD (Fig. 3f; Supplementary Data 11). These PTC data could be an important reference for dissecting PTM or transcription regulation on wheat protein abundance.

a The formula definition of contribution ratio of PTM and transcript (PTC). PCC, Pearson correlation coefficient. Trans, Transcript. b The diagram illustrates the process involved in the modulation of PTC in the phosphoproteome and acetylproteome datasets. c The PTC distributions of phosphoproteome and acetylproteome. d Violin plots show the three defined categories of balanced (BL), transcript dominant (TD), and PTM dominant (PD) categories. n is the protein number. In the violin plots, the upper and lower whiskers represent maxima and minima, respectively, the middle line represents the median, and the upper and lower lines represent the third quartile and first quartile, respectively. The bounds of the plot represent data density. e The proportions of proteins from the TD, BL and PD categories at phosphorylation and acetylation levels, respectively, and all transcription factors (TFs) and their top 10 in our atlas were examplized to detect the proportion of three categories. f, TD and PD proteins were functionally annotated using GO categories (Hypergeometric Test, Benjamini-Hochberg adjusted P value < 0.05). Colored bars represent the proportion of TD and PD proteins in each enriched category. Source data are provided as a Source Data file.
Regulatory enzymes of protein phosphorylation and acetylation
It is very important to elucidate the regulatory enzymes of PTMs for understanding the controlling mechanisms and functions of PTMs34. Here, we detected 2199 regulatory enzymes likely involved in modulating protein phosphorylation, including 1713 kinases and 486 phosphatases, from the 33,256 proteins with quantitative information (Supplementary Fig. 6a, b; Supplementary Data 12). We then constructed a regulatory network of protein kinases and their putative substrates we detected in the same tissue. This network predicted 5415 regulatory relationships on 1592 phosphorylated sites of 362 proteins (Supplementary Fig. 6c; Supplementary Data 13). These proteins included shared histones and a wide range of other nuclear, chloroplast, cytosolic, and mitochondrial proteins (Supplementary Data 13) involved in microtubule-based movement autophagy, protein catabolic process, ribosome biogenesis, and translational initiation (Supplementary Data 14). This network contained the kinases predicted to phosphorylate a large number of well-known wheat proteins. For example, a TKL kinase is predicted to phosphorylate sucrose transporter 2 A (SUT2, TraesCS5D03G0002100.1); a CMGC kinase might phosphorylate heat shock protein 90 (TraesCS2A03G1324400.1). Furthermore, the number of phosphorylation sites varied widely among the 19,970 phosphoproteins identified by us (Supplementary Fig. 6d). Aided by the motif-X software, we computed 321 phosphorylation motifs that were grouped into ‘proline-directed’, ‘acidic’, ‘basic’ or ‘other’ motif classes (Supplementary Fig. 6e; Supplementary Data 15). Evaluation of solvent accessibility indicated that phosphorylation residues (pSer, pThr, and pTyr) were less surface accessible compared to the unmodified Ser, Thr, and Tyr residues. Analysis of the distribution patterns of phosphorylated residues indicated that there was structural preference compared with the unmodified residues (Supplementary Fig. 6f).
A similar investigation was performed on the enzymes regulating protein acetylation. We detected 51 histone acetyltransferases (HATs) and 30 histone deacetylases (HDACs), which are key enzymes in catalyzing protein acetylation or de-acetylation34, in our proteome data set (Supplementary Fig. 6g, h; Supplementary Data 12). Relatively high numbers of HATs and HDACs were found expressed in the roots and the developing grains at 14 DPA (Supplementary Fig. 6g, h), implying that these tissues may have intensive biological processes involving protein acetylation or de-acetylation events. We detected 178 acetyl-motifs by Motif-X, which were similar to the phosphorylation motifs in site variation, solvent accessibility, structural preference (Supplementary Fig. 6i–k; Supplementary Data 15). More acetylated proteins were assigned to the chloroplast, followed by the cytoplasm, nucleus, and mitochondria (Supplementary Fig. 6l). The shared histones, including histone 1 (H1), H2A, H2B, H3, and H4, were highly modified by acetylation (Supplementary Data 15). For instance, H2B (TraesCS1A03G0948000.1) was modified in 18 acetylation sites. Interestingly, the proteins associated with sucrose metabolism, tricarboxylic acid cycle, fatty acid metabolic process, carbon fixation, and chitin catabolic process were widely modified by acetylation (Supplementary Data 14).
The protein phosphorylation and acetylation events and the relevant regulatory enzymes, especially those showing tissue and/or developmental phase specificities, can aid deeper dissection of gene functions.
Diversity and bias of homoeologous protein expression
In polyploid wheat, differences in homoeologous gene expression have important influences on transcriptional landscapes and gene function divergence35, but this phenomenon is complex and requires further analysis especially at multi-omics level and with respect to developmental phase regulation. Here, we explored the diversity and bias in homoeolog expression at four omics layers. Principal component analysis revealed separations of protein abundance according to both sub-genomes and developmental stages, with subgenomes A, B, and D playing a more prominent role in driving the separation (Supplementary Fig. 7a, b).
We identified 3565 triads that exhibited three homoeologous genes in three (A, B, and D) sub-genomes and were detected to be expressed in at least one of 20 samples in our atlas, which were used to analyze homoeolog protein expression bias across wheat development in more detail. A total of seven protein expression patterns were observed (Supplementary Fig. 7c; Supplementary Data 16). The balanced triads had their A, B, and D homoeologs expressed with similar abundance. The three types of dominant triads had their A, B, or D homoeolog showing strongly elevated expression relative to the other two homoeologs, whereas the reverse was found for the three kinds of suppressed triads. Obviously, more than 93% of the 3,565 examined triads showed expression bias in homoeologous protein expression in the 20 samples analyzed (Supplementary Fig. 7d; Supplementary Data 17). The range of protein abundance was highest for D homoeologs (Supplementary Fig. 7e; Supplementary Data 18). Within the 3565 triads, 1,786 and 425 sets had their three homoeologs all phosphorylated or acetylated (Supplementary Data 18). More than half of the 1786 triads had biased accumulation of phosphorylated homoeologs (Supplementary Fig. 7f–h; Supplementary Data 17-18). The percentage of balanced triads with their acetylated A, B and D homoeologs accumulated to similar abundance varied considerably (32.8%-83.3%) across the 20 samples; the remaining triads (16.7%-67.2%) exhibited either suppressed or increased accumulation of A, B or D acetylproteins (Supplementary Fig. 7i–k; Supplementary Data 16–18). In 18,919 triads identified in our atlas with relatively high transcript levels, D genome showed biased expression (Supplementary Data 16–18).
Collectively, the above data suggest that biased homoeolog transcript accumulation, protein expression, phosphorylation, and acetylation occur extensively in hexaploid wheat. Biased homoeolog protein expression was prevalent across wheat development, with the D genome homoeologs tending to be more strongly expressed. This is consistent with the previous finding of comparatively low DNA methylation level in the D subgenomes35.
Regulation of wheat development by PTMs
Protein phosphorylation and acetylation are crucial PTMs for regulating metabolism and biological processes6,8. In this study, we identified 35,545 transcripts, 16,323 proteins, 69,364 phosphorylation sites, and 34,974 acetylation sites that exhibited developmental stage (SS, JS, BS, HS, and FS, in Fig. 1a)-specific expression, respectively (Supplementary Fig. 5c; Supplementary Data 19). The abundant variations of PTMs during wheat development stages are likely to be associated with specific biochemical processes (Supplementary Data 19). The grain-filling stage from anthesis to maturity is crucial for seed development and yield formation36. Altogether, 6761 phosphoproteins and 3730 acetylproteins had relatively high abundance in the grain filling stage, which were mainly involved in glucose metabolism, amino acid biosynthesis, and response to water, implying that these pathways were highly modulated by PTMs (Supplementary Fig. 5d, e; Supplementary Data 19). In general, the leaf, root, stem, and grain samples were regulated by phosphorylation and acetylation with different expression patterns (Supplementary Fig. 5f, g), which suggests that PTMs play important regulatory roles in tissue-specific expression proteins.
The quantity and composition of seed storage proteins (SSPs) have strong influences on the processing and nutritional qualities of wheat grains23,24. Wheat SSPs mainly consist of low- and high-molecular-weight glutenin subunits (LMW-GSs and HMW-GSs, respectively) and gliadins23. In this atlas, we identified 18 transcripts, 16 proteins, nine phosphoproteins, and five acetylproteins for glutenin subunits (Fig. 4a; Supplementary Data 20). The finding of phosphorylation and acetylation in both HMW-GSs and LMW-GSs is highly interesting, because such PTMs have not been identified for these proteins despite of over 60 years of intensive research on them23. For the five HMW-GSs expressed in Yunong 268 grains, phosphorylation and acetylation were both detected in 1Bx7, 1By9, 1Dx5, and 1Dy10, with only acetylation found for 1Ax1; for the 11 expressed LMW-GSs, five were phosphorylated, with none of them showing acetylation modification (Fig. 4a; Supplementary Data 20). Thus, at least 62.5% (10/16) of the glutenin subunits were affected by phosphorylation and/or acetylation PTMs in Yunong 268 grains. For the nine subunits affected by phosphorylation, the percentages of Ser, Thr, and Tyr residues phosphorylated (2.1%-31.2%) differed substantially, with those in 1Dx5, 1Bx7, and 1Dy10 being relatively higher; for the five HMW-GSs affected by acetylation, the percentages of Lys residues acetylated (4.5%–50%) also varied widely (Fig. 4b, c). Collectively, these findings suggest that many glutenin subunits are modified by phosphorylation and/or acetylation in wheat grains, although the extent of such PTMs may differ widely among different subunits (Fig. 4d). Like glutenin subunits, many α-, β-, γ-, and ω-gliadins also exhibited phosphorylation and/or acetylation modifications in our PTMomics experiments (Supplementary Fig. 5h; Supplementary Data 20).

a Expression patterns and PTMs of low- and high-molecular-weight glutenin subunits (LMW-GSs and HMW-GSs) at the protein level. b Scatter plot of the number of phosphorylation Ser-Thr-Tyr (STY) residues in a GS sequence versus the proportion of STY residues phosphorylated. c Scatter plot of the number of lysine (K) residues in a protein lysine sequence versus the proportion of K residues acetylated. d The primary structure and the PTM sites of 10 LMW-GSs and HMW-GSs in our atlas. e Venn diagram of numbers of differentially expressed genes (DEGs), proteins (DEPs), and acetylation proteins (DEKPs) in response to Fusarium crown rot. f Expression analysis of TaP5CS1 in 20 tissues at transcript and protein levels, respectively. iBAQ, intensity-based absolute quantification. g TaP5CS1 showed the multiple acetylation sites. Source data are provided as a Source Data file.
During grain development in cereals, SSP synthesis is primarily controlled at transcriptional level37. Hence, we constructed a GRN of wheat SSP genes using our multi-omics data. Altogether 10 genes from different TF families (e.g., MYB, WRKY, bHLH, M-type-MADS, NAC_290 and ERF) were predicted to regulate SSP gene expression (Supplementary Fig. 5i, j; Supplementary Data 4). According to the GRN constructed here, the previously reported NAC_290 may regulated 22 genes, which was considerably more numerous than those identified in the original study31. Many TF genes implicated in the GRN have not been demonstrated to regulate SSP biosynthesis, thus representing diverse targets for understanding and improving the functionality of SSPs in wheat quality control.
Analysis of PTMs for the genes functioning in disease resistance
Diseases constitute a major limitation to worldwide wheat production, which is worsening as climate change intensifies38. Understanding the functions disease resistance-related genes (DRRGs) is a perquisite for efficient breeding of resistant cultivars. An increasing number of prominent studies have shown that phosphorylation plays a crucial role in DRRG function39,40,41. Evidence for modulation of DRRG function by acetylation is also emerging42,43. In this context, it is timely to develop a multi-omics atlas of DRRG transcripts, proteins, phosphoproteins, and acetylproteins to facilitate deeper studies of DRRGs.
Using a disease resistance gene analog (RGA) prediction pipeline44, we identified 7225 transcript, 1113 proteins, 850 phosphoproteins, and 239 acetylproteins for DRRGs in our atlas (Supplementary Data 21). These DRRGs were predicted to encode nucleotide-binding-site (NBS) proteins, NBS-leucine-rich-repeat (LRR) proteins (NBS-LRRs), coiled-coil NBS-LRR proteins (CNLs), coiled-coil NBS (CC-NBS) proteins, receptor-like kinases (RLKs), receptor-like proteins (RLPs), or transmembrane coiled-coil (TM-CC) proteins. The DRRGs were distributed on 21 chromosomes, with a higher proportion of them located on group 2 chromosomes (Supplementary Fig. 5k). Moreover, we found that 6570 transcripts, 552 proteins, 850 phosphoproteins, and 239 acetylproteins derived from DRRGs exhibiting tissue-specific expression (Supplementary Fig. 5l; Supplementary Data 21).
Further inspection revealed complex tissue-specific variations in DRRGs with respect to transcript and protein numbers and phosphorylation and acetylation levels of the proteins (Supplementary Fig. 8a). Notably, relatively more RLKs were phosphorylated and a higher proportion of the TM-CC proteins were acetylated (Supplementary Fig. 8b). Although it is anticipated that RLKs are more likely affected by phosphorylation due to their kinase activity, the finding of abundant acetylation of TM-CC proteins is remarkable and provides a crucial clue for further functional analysis of these poorly studied proteins. In addition, more R genes were found expressed in roots based on proteomic data (Supplementary Fig. 8b, c), possibly due to greater numbers of genes and proteins identified in roots in our atlas (Supplementary Fig. 1a).
GRN network analysis showed that DRRGs were likely regulated by the genes belonging to different TF families, e.g., bHLH, bZIP, and HD-ZIP (Supplementary Fig. 8d; Supplementary Data 4). Interestingly, biased expression appeared to affect DRRGs, with the D homoeolog tending to be more highly expressed (Supplementary Fig. 8e). These observations, plus the analysis above, will serve to accelerate functional and mechanistic studies of DRRGs to aid disease resistance breeding in wheat.
Identification of the TaHDA9-TaP5CS1 module mediating wheat Fusarium crown rot resistance
Wheat Fusarium crown rot (FCR) disease, caused mainly by F. pseudograminearum (F. pg) and damaging primarily wheat coleoptile, leaf sheath and stem base, is a persistent and serious soil-borne disease causing huge yield losses in many regions of the world45,46. Wheat resistance to FCR was controlled by quantitative trait loci (QTLs)47,48,49. Although cloning more QTL genes for pyramiding breeding is widely recognized as an effective strategy for developing resistant varieties urgently needed for FCR control, very few such genes have been characterized47,50. Therefore, we set out to mine diverse FCR resistance genes with the aid of our multiomics atlas.
As the first step of this effort, we performed transcriptome and proteomics analyses using the Yunong 268 tissues collected at 0, 24, 48, 72, and 96 hours post inoculation (hpi) with F. pg. This resulted in the identification of 8998 transcripts, 1162 proteins, 2076 phosphoproteins, and 1310 acetylproteins as differentially expressed genes (DEGs) or proteins (phosphoproteins, or acetylproteins) (DEPs, DEPPs, or DEAPs) (Supplementary Data 22–25), which were mainly involved in defense response, reactive oxygen species, pyruvate metabolism, and ribonucleoside triphosphate biosynthetic process (Supplementary Data 26). The 8998 DEGs were all present in the multi-omics atlas constructed beforehand; the DEPs, DEPPs, and DEAPs were also found in the multi-omics atlas in high percentages (being 81%, 79%, and 70%, respectively) (Supplementary Fig. 9a–d). This not only supports the high quality of our multi-omics atlas but also indicates that the findings obtained from analyzing the atlas may be utilized to accelerate the mining of FCR resistance genes.
To explore the potential effect of PTMs on FCR resistance, we integrated DEGs, DEPs, DEPPs, and DEAPs, and identified 309 proteins in response to phosphorylation and/or acetylation in least two omics levels. As no gene/protein was overlapped in four omics data, we next focused on six overlapped genes in three omics data (transcript, protein, acetylation) (Fig. 4e). Of these six genes, three DEAPs, i.e., TaPOD1 (a putative peroxidase), TaSUS1 (a predicted sucrose synthase), and TaP5CS1 (a potential Δ1-pyrroline-5-carboxylate synthetase), were highly expressed in the basal stems at multiple stages (Fig. 4e, f). They were thus selected as candidate proteins for regulating wheat FCR resistance. The cognate genes of TaPOD1, TaSUS1, and TaP5CS1 were silenced, respectively, using barley stripe mosaic virus vector-mediated gene silencing (BSMV-VIGS) (Supplementary Fig. 9e, f), followed by F. pg inoculation. Compared to the wheat plants treated with the empty vector (BSMV0), those with TaPOD1, TaSUS1, or TaP5CS1 silenced exhibited significantly increased disease index (DI), with the highest DI value recorded for TaP5CS1-silenced plants (Supplementary Fig. 9g). Consequently, TaP5CS1 (Supplementary Fig. 9h, i) likely plays an import role in promoting wheat FCR resistance. In line with this proposition, TaP5CS1 was significantly induced at both transcriptome and proteome levels after F. pg inoculation (Supplementary Fig. 9j, k). A PPI network analysis with F. pg responsive DEPs, DEPPs, and DEAPs showed that TaP5CS1 is a vital protein involved in three top clusters (metabolic pathways, biosynthesis of secondary metabolites, and biosynthesis of amino acids) (Supplementary Fig. 9m; Supplementary Data 27).
As a rate-limiting enzyme in the biosynthesis of the amino acid proline, P5CS1 has been commonly found in stress-induced proline biosynthesis in plants51, but its regulation of wheat FCR resistance has not been reported before. Therefore, we isolated one mutant of P5CS1 with abnormal translation from the EMS-mutagenized Kronos library. To exclude the interference of other mutation sites (Supplementary Fig. 10a), we developed BC2 populations for this mutant using wild-type (WT) Kronos as recurrent parent. After F. pg inoculation, TaP5CS1 mutant showed significantly increased DI in both greenhouse and field tests compared with WT (Supplementary Fig. 10b, c). We further prepared transgenic plants overexpressing TaP5CS1 using the FCR susceptible wheat cultivar Fielder as the recipient. Relative to WT control, the TaP5CS1- overexpressed (OE) lines (Supplementary Fig. 9n) had sharply decreased DI in both greenhouse and field tests (Fig. 5a, b; Supplementary Fig. 9o). Accordingly, the OE plants had much less pathogen hyphal growth as examined by scanning electron microscopy (SEM), fungal biomass, and cell death during F. pg infection (Fig. 5c–e). These data confirm that TaP5CS1 positively contributes to wheat FCR resistance. Moreover, TaP5CS1-OE lines did not show penalties in agronomic traits relative to WT control (Supplementary Data 28).

a-c The phenotype comparison of TaP5CS-overexpressed (OE) and wild-type (WT) plants infected by F. pseudograminearum (F. pg) in (a) greenhouse (12 plants per replicate) and (b) field and (c) scanning electron microscope (SEM) image. Scale bar = 50 μm (the length of ten lattices). The hyphal (HY) growth was marked with red arrow. d, Fungal biomass quantification by qPCR in SEM. e Trypan blue staining of necrotic cell death in TaP5CS1-OE plants infected by the F. pg. TaP5CS1-OE showed a decreased cell death in compared with control. f-i, Interaction analysis of TaP5CS1 and TaHDA9 proteins by (f) yeast two-hybrid, (g) a firefly luciferase complementation imaging in tobacco, (h) co-immunoprecipitation and (i) pull-down. j The recombinant TaP5CS1-GST protein purified from E. coli was acetylated with or without Trichostatin A (TSA) and β-nicotinamide (NAM). k, TaP5CS1 was deacetylated by TaHDA9 in vitro. l–m TaHDA9 reduced the (l) acetylation (Kac) level and (m) accumulation of TaP5CS1 protein in transient expression tobacco. n, Immunoblots detected decreased Kac level of TaP5CS1K634R but not TaP5CS1K221R and TaP5CS1K231R in transient expression tobacco. o, Immunoblots detected decreased protein abundance of TaP5CS1K634R in transient expression tobacco. p-r, The phenotype comparison of TaHDA9-OE and WT plants infected by F. pg in (p) greenhouse (12 plants per replicate), (q) field and (r) SEM image. Scale bar = 200 μm or 50 μm. HY growth was marked with red arrow. s Fungal biomass quantification by qPCR in the SEM. t, Trypan blue staining of necrotic cell death in TaHDA9-OE plants infected by the F. pg. TaHDA9-OE showed a rapid induction of cell death. In (a) and (p), the experiments were conducted with five biological replicates; in (c), (e), (f–o), (r), and (t), the experiments were conducted with at least three biological replicates showing similar results (Each sample was loaded twice). In (d) and (s), data are presented as the mean values ± SD (*P < 0.05, **P < 0.01; P = 0.0049, 0.0090, 0.0089 in (d); P = 0.0151, 0.0163, 0.0232 in (s); unpaired two-sided t-test; n = 3 biologically independent experiments). Source data are provided as a Source Data file.
Because TaP5CS1 was acetylated (Fig. 4g), and belonged to the PD type based on its calculated PTC value in the atlas (Supplementary Data 10), we hypothesized that its acetylation may be modulated by HAT and/or HDAC enzymes. Through screening a yeast-two-hybrid (Y2H) library with TaP5CS1 as a bait, we identified a histone deacetylase, i.e., TaHDA951, that interacted with TaP5CS1. We further verified the interaction between TaP5CS1 and TaHDA9 by Y2H, luciferase complementation imaging assay (LCI), co-immunoprecipitation (Co-IP), and pull-down assays (Fig. 5f–i). TaP5CS1 could be acetylated when ectopically expressed in E. coli cells (Fig. 5j), but the level of TaP5CS1 acetylation was substantially decreased when a His-TaHDA9 fusion protein was co-expressed (Fig. 5k). Additionally when a Flag-tagged TaP5CS1-GFP protein was co-expressed with TaHDA9-GFP-His in tobacco cells, TaP5CS1 acetylation was strongly reduced, accompanied by lowered protein abundance (Fig. 5l, m). Of the 11 acetylation sites in TaP5CS1 (Fig. 4g), three (K221, K231, and K634) responded positively to FCR disease (Supplementary Fig. 9l). However, mutational analysis in tobacco cells indicated that only the mutant TaP5CS1K634R had drastically decreased acetylation level and reduced protein abundance compared with WT TaP5CS1 (Fig. 5n, o), indicating that K634 is a key site of TaP5CS1 de-acetylation by TaHDA9. Although TaHDA9 was expressed in all 20 samples with its protein distributed throughout the cell, its expression level was substantially decreased after F. pg inoculation (Supplementary Fig. 10d–f). Altogether, these data support the deacetylation of TaP5CS1 by TaHDA9 both in vitro and in vivo, which down-regulates TaP5CS1 accumulation and may thus negatively regulate wheat FCR resistance. TaHDA9 expression declines in F. pg infected plants, probably resulting in stabilization of TaP5CS1 acetylation level and thus FCR resistance.
To verify the above proposition, we isolated four mutants of HDA9 with abnormal translation from the EMS-mutagenized Kronos library. To exclude the interference of other mutation sites, we developed BC2 populations for each of the four mutants using WT Kronos as recurrent parent (Supplementary Fig. 10g). After F. pg inoculation, all four mutants showed significantly decreased DI than WT control in both greenhouse and field tests (Supplementary Fig. 10h, i). Concomitantly, we prepared three maize ubiquitin gene promoter-directed TaHDA9-OE lines, with Fielder (FCR susceptible, see above) as recipient. As anticipated, the TaHDA9-OE plants (Supplementary Fig. 9q) displayed significantly higher DI in both greenhouse and field tests (Fig. 5p–q; Supplementary Fig. 9r), and supported more fungal hyphal growth than WT controls (Fig. 5r–t). These results confirm that TaHDA9 negatively regulates wheat FCR resistance.
Further to the above experiments, we determined that the transgenic lines overexpressing TaP5CS or TaHDA9 had significantly increased or decreased proline contents relative to WT control (Supplementary Fig. 9p, s), pointing to the involvement of proline content in the regulation of FCR resistance by TaP5CS or TaHDA9. Consequently, we observed that increasing proline concentrations in the culture medium gradually inhibited the growth of F. pg and caused abnormal hyphae, but this was not found when valine, which resembles proline in being nonpolar and hydrophobic, was added to the media (Supplementary Fig. 11a–c). Lastly, exogenous application of proline enhanced the resistance of wheat plants to FCR disease (Supplementary Fig. 11d, e). Judging from these findings, it is highly likely that the acetylated TaP5CS1 promoting wheat FCR resistance through maintaining proline content. Down-regulation of TaHDA9 expression upon F. pg infection is a natural mechanism for stabilizing TaP5CS1 acetylation level and protein abundance, which is conducive for plant defense to FCR.
Discussion
As plant science research enters the genomics era, development and utilization of high-quality multi-omics resources10,11,52,53,54 has become a prerequisite for efficient dissection and improvement of crop traits. It is now highly the time to create complementary and comprehensive multi-omics resources for accelerating trait enhancement. Here we succeeded in building a unique multi-omics atlas for a strong gluten wheat (cv Yunong 268) using transcriptome, proteome, phosphoproteome, and acetylproteome data sets generated with 20 samples encompassing five developmental stages. The quality and application of our atlas are attested by its high coverage of common wheat genes and proteins annotated by genome sequence projects and by the provision of both phosphoproteome and acetylproteome data sets that are not available for common wheat before.
Importantly, we conducted multiple types of integrative analysis with the four omics data sets, and shed light on the expression, regulation, and protein dynamics and PTMs for hundreds and thousands of common wheat genes in a broad range of tissues across both vegetative and reproductive phases. We proposed the concept of PTC for assessing the relative contributions of PTMs and transcript level in determining protein abundance, whose value was validated in guiding us to focus on testing the functional importance of acetylated TaP5CS1 in promoting wheat FCR resistance. Fascinatingly, we discovered the occurrence of phosphorylation and/or acetylation PTMs for the three main types of wheat SSPs (HMW-GSs, LMW-GSs, and gliadins), which will inspire diverse approaches for enhancing the functionalities of these proteins in refining wheat processing and nutritional qualities. Our analysis represents a prelude in the mining of the four data sets. Further exploration of the integrated multi-omics atlas by interested researchers will likely yield even more innovative principles governing wheat growth and development.
As sessile organisms, plants have to constantly and rapidly adjust their gene expression programs and growth behavior in response to environmental changes55,56. Ideally, a comprehensive multi-omics atlas should include transcriptomic, proteomic, and PTMomics changes resulting from diverse stresses. This points to an important direction for further strengthening our multi-omics atlas in the future. Notably, in the separate F. pg infection experiment conducted in this work, the great majority of the DEGs, DEPs, DEPPs, and DEAPs identified were found included in our multi-omics atlas. The TaHDA9-TaP5CS1 module subsequently defined provides important clues for more deeply studying and improving the genetic basis of wheat resistance against FCR disease that is currently devastating worldwide wheat production. Thus, the present atlas may be consulted for identifying and analyzing gene functions regulating plant responses to various stresses.
Finally, to facilitate the access and utilization of this resource by international research communities, the original data and the valuable findings obtained by us are all deposited in the WheatPro database (https://www.csuligroup.com/WheatPro/) and the public Protein Data Bank. With its rich information and high utility, our multi-omics atlas will become a treasured asset for accelerating both basic and applied trait studies (e.g., growth and development, abiotic and biotic stresses, processing quality, yield-related traits) in wheat and related crops.
Methods
Plant materials and growth conditions
We selected a common wheat, Yunong 268, released in 2023, as a middle-strong gluten wheat cultivar owing to its excellent comprehensive agronomic traits and great resistance to disease and stress. Yunong 268 was grown in pot culture at 25/15 °C day/night before the heading stage and at 32/26 °C day/night until the filling stage with a 16/8 h light/dark photoperiod in a greenhouse. The positions of plants in the growth chamber were randomly shuffled during plant growth to minimize the microenvironmental effect. The plants with normal and consistent growth were sampled for each experiment. We collected 20 different developmental stages across wheat vegetative and reproductive phases, including roots and seedlings at the seedling stage (SS_RT, SS_SG), leaves, roots, and the basal part of stems at the jointing stage (JS_LF, JS_RT, JS_BPST), leaves, roots, basal part of stems, and spikes at the booting stage (BS_LF, BS_RT, BS_BPST, BS_SP), flag leaves, roots, basal part of stems, stalks, sheaths, and spikes at the heading stage (HS_LF, HS _RT, HS _BPST, HS_ SL, HS_ SH, HS _SP), and roots, basal part of stems, and seeds at 14 days post anthesis (DPA), 28 DPA, and 35 DPA at the filling stage (FS _RT, FS _BPST, FS_14d_seed, FS_28d_seed, FS_35d _seed). After the samples were collected, immediately flash frozen in liquid nitrogen, and stored at −80 °C for protein and RNA extraction.
Fusarium species isolation and inoculation on wheat
The wheat cultivar Yunong 268 was planted in a greenhouse and inoculated with a prevalent Chinese F. pseudograminearum (F. pg) isolate, namely FP-Ta5. FP-Ta5 is a highly aggressive strain isolated from infected wheat crowns in northwest of the Yellow and Huai wheat regions of China. The strain was routinely cultured on potato dextrose agar (PDA) at 25°C. When seedlings grew to 3 cm long, 8 ~ 10 seedlings per cultivar that grew uniformly were inoculated, then 0.4 g colonized millet grains were scattered to each plastic container48. All seedlings were grown in a greenhouse under 16/8 h day/night conditions at 25/20°C day/night with 60–80% relative humidity. After F. pg inoculation for 0, 24, 48, 72, and 96 hours at the one-leaf stage, basal stems with three biology replicates were collected, and frozen in liquid nitrogen, and stored at −80 °C for further use.
Protein extraction of 20 samples from wheat developmental stages
To extract tissue proteins, lysis buffer (10 mM dithiothreitol, 1% TritonX-100, 50 μM PR-619, 50 mM NAM, 3 μM TSA, 2 mM EDTA, and 1% Protease Inhibitor Cocktail) was used57. The proteins were re-dissolved in 8 M urea, and a BCA Protein Assay Kit (Catalog No. P0012, Beyotime, Beijing, China) was used to determine the protein concentration according to the manufacturer’s instructions.
Trypsin digestion
For initial digestion, trypsin was added at a ratio of 1:50 trypsin to protein mass overnight. Next, the sample was reduced with 5 mM dithiothreitol for 60 min at 37 °C and alkylated with 11 mM iodoacetamide for 45 min at room temperature in the dark. Finally, Strata X SPE column was used to desalt the peptides.
High-performance liquid chromatography (HPLC) fractionation
The tryptic peptides were fractionated using high-pH reverse-phase HPLC with Agilent 300 Extend C18 column (5 μm particles, 4.6 mm ID, 250 mm length). The process involved separating peptides into 60 fractions using a gradient of 2% to 60% acetonitrile in 10 mM ammonium bicarbonate (pH 10) over 60 min. The peptides were then combined into 24, 10, and four fractions for proteome, phosphoproteome, and acetylproteome, respectively, and dried by vacuum centrifuging.
Phospho-peptide enrichment
Peptide mixtures were subjected to incubation with a suspension of immobilized metal affinity chromatography (IMAC) microsphere, accompanied by vibration in the loading buffer (50% acetonitrile/0.5% acetic acid). In order to eliminate non-specifically adsorbed peptides, a sequential washing procedure was employed suing 50% acetonitrile/0.5% acetic acid and 30% acetonitrile/0.1% trifluoroacetic acid solutions on the IMAC microspheres. After adding an elution buffer containing 10% NH4OH and shaking the mixture, the phospho-peptides supernatant was collected and lyophilized for LC-MS/MS analysis.
Acetyl-lysine enrichment
The tryptic peptides were dissolved in NETN buffer (100 mM NaCl, 1 mM EDTA, 50 mM Tris-HCl, 0.5% NP-40, pH 8.0) and incubated with anti-acetyl-lysine antibody agarose conjugated antibody beads (PTM Biolabs, Lot number PTM-104) at 4 °C overnight with gentle shaking to enrich acetylation-modified peptides. The bound peptides were eluted from the beads using 0.1% trifluoroacetic acid, and then the beads were washed four times in NETN buffer and twice in deionized water. The eluted fractions were combined and vacuum-dried. The resulting peptides were cleaned with C18 ZipTips (Millipore) as per the manufacturer’s instructions.
Tandem mass spectroscopy (LC-MS/MS) analysis
The tryptic peptides were dissolved in solvent A (0.1% formic acid, 2% acetonitrile in water) and then directly loaded onto a reversed-phase analytical column (25 cm length, 100 μm i.d.). For the proteome, peptides were separated using a nanoElute UHPLC system (Bruker Daltonics) with a 6% to 24% gradient of solvent B (0.1% formic acid in acetonitrile) over 42 min, 24% to 32% over 12 min, climbing to 80% over 3 min, and held at 80% for the last 3 min, with a constant flow rate of 450 nL/min the whole time. For the phosphoproteome, peptides were separated with a 2% to 22% gradient of solvent B over 44 min, 22% to 35% over 10 min, climbing to 90% over 3 min, and held at 90% for the last 3 min, with a constant flow rate of 450 nL/min on a nanoElute UHPLC system (Bruker Daltonics). For the acetylproteome, peptides were separated with a 6% to 24% gradient of solvent B over 40 min, 24% to 32% over 12 min, climbing to 80% over 4 min, and held at 80% for the last 4 min, all at a constant flow rate of 450 nL/min on a nanoElute UHPLC system (Bruker Daltonics).
The peptides were subjected to a capillary source followed by timsTOF Pro (Bruker Daltonics) mass spectrometry (4D Mass Spectrometer). For the proteome, the electrospray voltage applied was 1.70 kV. Precursors and fragments were analyzed with a TOF detector, with an MS/MS scan range of 100–1700 m/z. The timsTOF Pro was operated in parallel accumulation serial fragmentation (PASEF) mode. Precursors with charge states 0 to 5 were selected for fragmentation, with 10 PASEF-MS/MS scans acquired per cycle. The dynamic exclusion was set to 30 s. For the phosphoproteome, the electrospray voltage applied was 1.75 kV, with the dynamic exclusion set to 24 s and the other parameters in line with the proteome. For the acetylproteome, the dynamic exclusion was set to 24 s, with the other parameters in line with the proteome.
Database search
The resultant data from MS/MS were processed using the MaxQuant search engine (v.1.6.15.0). Tandem mass spectra were searched against the latest version (IWGSC RefSeq v2.1)19 of the International Wheat Genome Sequencing Consortium (IWGSC) concatenated with reverse decoy database. Trypsin/P was specified as the cleavage enzyme, allowing up to two missing cleavages. The mass tolerance for precursor ions was set as 20 ppm for both the initial and subsequent searches, with the mass tolerance for fragment ions set as 0.02 Da. Peptide length was restricted to no less than 7, and no more than 5 peptide modifications were allowed. A fixed modification for the proteome included carbamidomethyl on cysteine (Cys), whereas variable modifications included acetylation on the protein N-terminal and oxidation on methionine (Met). For the phosphoproteome, carbamidomethyl on Cys was specified as a fixed modification, with acetylation on the protein N-terminal, oxidation on Met, and phosphorylation on serine (Ser), threonine (Thr), and tyrosine (Tyr) specified as variable modifications. For the acetylproteome, a fixed modification included a carbamidomethyl on Cys and three variable modifications included acetylation on the protein N-terminal, oxidation on Met, and acetylation on lysine (Lys). The false discovery rate (FDR) was adjusted to <1%. When calculating total protein, if unique peptides = 0, the total number of peptides identified should be at least 2, and a protein should be determined by two peptides to ensure the reliability of the identification; If unique peptides ≥ 1, we did not screen total peptides.
Fusarium crown rot responsive multi-omics
To find Fusarium crown rot (FCR) responsive proteins (proteins with ≥ 2 unique peptides), the wheat differential multi-omics were conducted with three biological replicates to examine the differentially expressed proteins (DEPs), phosphorylation proteins (DEPPs), and acetylation proteins (DEAPs) after F. pg inoculation for 0, 24, 48, 72, and 96 hours using the above method. Fold changes were determined using the abundance ratio of these proteins identified at 24, 48, 72, and 96 hours relative to those at 0 hour. Moreover, significant differences were determined between means of control (0 hour) and each of four inoculation times (24, 48, 72, 96 hours), respectively, using a two-sample t-test. DEPs, DEPPs, and DEAPs were defined based on thresholds of >1.5- or <0.67-fold change ratios and P < 0.05 at 24, 48, 72, and 96 hours compared to those at 0 hour.
RNA sequencing
Trizol® Reagent Plant RNA Reagent (Invitrogen) was used to extract total RNA from 20 tissue and FCR assay samples with three biological replicates. The sequencing depth is 10 G of a single sample. After the DNase I (TaKara) treatment, RNA-seq libraries were constructed using a TruSeqTM RNA sample preparation Kit (Illumina, USA), with 150 bp paired-end sequences generated using an Illumina DNBSEQ-T7 sequencer. SeqPrep and Sickle with default parameters were used to trim and quality control the raw paired-end reads. The clean reads were aligned to the reference genome (IWGSC RefSeq v2.1)19 with orientation mode using HISAT2 software58. StringTie was used to assemble the mapped reads of each sample in a reference-based approach59. According to the transcripts per million reads (TPM) method, the expression level of each transcript was calculated by RSEM60 to quantify gene abundance. DESeq261 provided statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution. The resulting P-values were adjusted using the Benjamini and Hochberg’s approach to control the FDR. Genes with |log2FC | >1 and FDR < 0.05 were considered differentially expressed genes (DEGs).
Transcript abundance distributions
For all 132,570 transcripts, a kernel density estimation of log2 (TPM) distribution was calculated. Similarly, other kernel density estimates were calculated for the subsets of transcripts with 30,772 proteins, 18,912 phosphoproteins, and 11,751 acetylproteins. The distributions were plotted using R.
Tissue-wise mRNA-to-protein correlations
Pearson’s correlation coefficient (r) was calculated for the gene expression of all pairwise tissue combinations to assess tissue similarity. Correlations were displayed as separate heat maps at the protein level and transcript level. For the shared data, including 27,149 shared_transcript, 4002 shared_protein, and 2751 shared_intersection data, the Pearson’s correlation values between protein and transcript were calculated using genes from the 2,751 shared_intersection dataset.
Gene expression analysis
Gene expression analysis was conducted at transcript and protein levels10. We used proteins (intensity-based absolute quantification, iBAQ) and transcripts (TPM) from highest to lowest for the cumulative abundance calculation of eight representative tissues. The six most abundant transcripts or proteins are listed. The contribution of each gene representing the most abundant protein in at least one tissue was determined by taking the associated protein abundance value (iBAQ) and dividing it by the total number of protein abundance values (∑iBAQ) in that tissue.
Bioinformatics
We constructed the circular proteome map (CPM) using Circos62 normalizing the organ LFQ values (Lt) for each protein, as follows:
where \({gen}{e}_{i,j}\) represents the organ LFQ value of the protein i in organ j or time j, and n represents the number of organs or times.
Shannon entropy was used to assign organ specificity to the proteome, phosphoproteome, and acetylproteome63,64. We calculated a relative expression value for each protein, as follows:
where m represents the number of proteins.
For each protein, the specificity score was calculated based on its relative expression, as follows:
where i represents the protein i.
Each protein has a specificity score, with the density curve made to determine the specificity threshold and screen out the specific protein. Phosphoproteome and acetylproteome applied a five-tenth percentile cutoff. All statistical analyses were performed using R. Gene ontology (GO) enrichment was carried out, with only GO terms with P < 0.05 (p.adjust/p-value < 0.05) considered significant. We analyzed the model of amino acid sequences in specific positions of phospho-12-mers or acetyl-20-mers in all protein sequences using the Motif-X software65. The wheat proteome was used as the background database, with the parameters set as occurrences = 20, Bonferroni corrected P = 0.000001, and other parameters set to default values. Subcellular localization was predicted using WoLFPSORT (http://www.genscript.com/wolf-psort.html). Protein pathways were annotated using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database66; P < 0.05 (p.adjust/FDR < 0.05) was the threshold for pathway significance. The protein domain functional description was annotated using InterPro67 (http://www.ebi.ac.uk/interpro/). Protein-protein interactions of FCR responsive proteins were determined from the protein (STRING) database v10.5, with the network visualized in Cytoscape68.
Prediction of kinase-substrate regulations
The prediction of kinase-substrate regulations using proteome data was conducted by Group-based Prediction System (GPS) 5.069 (http://gps.biocuckoo.cn), which indemnified the primary specificity based on the theory of short linear motifs around phosphorylation sites (p-sites). The corresponding kinase proteins in the kinase family were acquired through a comparison with the kinase sequence in the IEKPD2.0 database. A “medium” threshold was chosen in GPS 5.0. One kinase can regulate multiple substrates, and a phosphorylation site can be regulated by more than one kinase. Therefore, a regulatory network diagram of protein kinase-substrate in the same tissue was constructed.
Protein/mRNA relation
We determined the relationship between protein and mRNA according to the previously reported method10. In brief, for more than eight tissues (n = 18,194), we calculated the r between median protein (median iBAQ of 20 tissues) and median transcript (median TPM of 20 tissues abundance. Protein-to-mRNA ratio (PTR) values are calculated for each tissue individually by constructing the ratio between the corresponding transcript abundance (TPM) and protein abundance (iBAQ) for each gene. The Tissue PTR values are then utilized to determine the median and median absolute deviation (MAD) of PTR across all 20 developmental stages. Further, quantiles (Q1–Q5) were created using the PTR MAD values for the median PTR distribution. Both phosphoproteome and acetylproteome were developed in a similar way.
Co-expression analysis
Co-expression networks were constructed using the weighted gene co-expression network analysis (WGCNA) R package33 as described in the prior study11. We constructed a set of four networks to capture all available information and use all available genes detected in at least three tissues. These networks comprised 58,263 mRNA, 8,231 proteins, 15,930 phospho-sites, and 11,033 acetyl-sites. Due to excessive transcriptome data flux, one parameter used for transcript (minModuleSize = 150) differed from other omics (minModuleSize = 30), with all other parameters used for each network identical. To get rid of differences in sequencing depth between studies, the count expression level of each gene was normalized with the variance stabilizing transformation from DESeq261. The soft power threshold was calculated to exceed a scale-free topology fit index of 0.9 for each network separately. The soft power threshold SEDs were: transcript = 16, proteome =18, phosphoproteome = 24, and acetylproteome = 1. The topographical overlap matrices (TOM) were calculated block-wise. The module function using TOM Type = “signed”, with the minimum module size set to 30. Co-expression modules were constructed through hierarchical clustering of the TOM distance (1-TOM) using the hclust function with method = “average”. A parameter merge Cut Height = 0.15 was used to merge similar modules. Using these four omics data sets, we identified 172 modules (including 39,558 hub genes) by WGCNA (Supplementary Data 7–9).
Identifying highly connected hub genes
The signedKME function of the WGCNA R package was employed to compute hub genes for each module. Genes that exhibited a stronger absolute correlation with the eigengene (absolute correlation coefficient > 0.8) were classified as hub genes.
Gene regulatory network
For the gene regulatory networks (GRNs) inference, we generated three individual networks from transcriptome, proteome and phospho-proteome in our atlas, respectively. Transcription factors (TFs) were predicted using PlantTFDB (Plant Transcription Factor Database)30. A set of 24,955 transcripts, 24,442 proteins, and 7187 phospho-proteins (Supplementary Data 4) were used as potential targets regulated by the predicted TFs. These transcripts, proteins, and phospho-proteins were observed in at least 2 omics (TPM > 0, iBAQ > 0, intensity > 0) in at least six samples. TFs, which were composed of 2414 transcripts, 2729 proteins, and 1049 phospho-proteins detected in at least 2 omics in at least six samples, were selected potential regulators (Supplementary Data 4). Then, three full GRNs were constructed by GRNboost270 (version 0.1.6) with default parameters. For each inferred GRN, we retained only edges with an importance score ≥1. Based on previous study11, regulatory edges presented in more than two GRNs were incorporated to the total GRN, where its importance score was determined by the average. GO functional enrichment (p. value < 0.05) was performed for target genes of some TFs by using an R Bioconductor package cluster profiler (version 4.4.4). To investigate the tissue-specific regulatory mechanism, we employed a sample-specific network (SSN) method71 to construct tissue-specific GRN based on transcriptome data (TFs) and the total GRN. The regulatory edges with P value ≤ 0.01 were regarded as tissue-specific. These huge data of GRNs were deposited in our WheatPro (https://www.csuligroup.com/WheatPro/#/).
Quantitative contribution of post-translational modifications and transcript
The data subset with genes detected in at least eight tissues on a protein level (n = 2,2941) was used for all further calculations involving the quantitative contribution of post-translational modifications (PTM) and transcripts (PTC). For the phosphoproteome and acetylproteome, Pearson’s correlation coefficients (PCC) for phosphoprotein and protein, transcript and protein, and acetyl-protein and protein were calculated; proteins with at least one kind of modification were retained (Fig. 3c). To standardize the relative ratio between PCCs, we normalized the PCC for each protein with absolute values, defining PTC as follows:
For proteins modified by phosphorylation and acetylation simultaneously, the type with the higher PTC was selected as the dominant modification type. Three linearization categories were defined based on PTC values: balanced (BL), with PTC from 40–60%, transcript dominant (TD), with PTC from 0–40%, and PTM dominant (PD), with PTC from 60–100%. GO enrichment was undertaken, with only GO terms with P < 0.05 considered significant.
Principal component analysis and correlation coefficient analysis
We used categorized protein abundance values to visualize sub-genome and tissue expression-relatedness. These expression values (iBAQ) were averaged across tissues and log2-transformed. The principal component analysis employed singular value decomposition via the prcomp function in R. Pearson’s correlation coefficients were determined with the Euclidean distance and complete linkage methods used to implement hierarchical clustering.
Relative expression levels of sub-genome homologs across triads
The analysis focused exclusively on the gene triads with a 1: 1: 1 correspondence across the three homologous sub-genomes, including 22,097 syntenic triads (66,291 genes) (http://wheat.cau.edu.cn/TGT/download.html). For relative expression levels of sub-genome homologs across triad, the transcripts/proteins with gene ID.1 (for instance, TraesCS1A02G002100.1) were selected. Protein abundance was normalized (riBAQ) by the ratios of the corresponding protein abundance value (iBAQ) in all protein abundance values (∑iBAQ). A protein triad was defined when the sum of the A, B, and D sub-genome homologs was > 0.01 riBAQ. Using this criterion, we defined 10,695 proteins (3565 triads) that were considered expressed (Supplementary Data 16). To standardize the relative expression of each homolog across the triad, we normalized the absolute riBAQ for each protein within the triad as follows:
where A, B, and D represent the gene corresponding to the A, B, and D homologs in the triad. The normalized expression was calculated for each tissue, and the average across all tissues. In addition, we defined 56,757 genes (18,919 triads) considered expressed at the mRNA level (Supplementary Data 16)35.
For the phosphoproteome and acetylproteome, modification abundance was normalized (rintensity) by the ratios of their corresponding modification abundance value (intensity) in all modification abundance values (∑intensity), with 5358 phosphoproteins (1786 triads) and 1275 acetylproteins (425 triads) considered expressed. Protein abundance was normalized (rintensity) by the ratios of their corresponding modification abundance value (intensity) in all modification abundance values.
Definition of homolog expression bias categories
The ideal normalized expression bias for the seven categories was defined in Supplementary Data 16. The homolog expression bias category for each triad by selecting the shortest distance35 for each of the intermediate tissue as well as for the average across all expressed tissues (combined analysis).
RNA isolation and quantitative real-time PCR (qRT-PCR)
After isolating total RNA with TRIZOL isolation reagent (Yeasen), we utilized the one-step ReScript II RT All-in-One Mix (with dsDNase) (Nobelab) to conduct reverse transcription procedures in accordance with the manufacturer’s instructions. The endogenous control was the β-actin gene (GenBank No. AB181991) (Supplementary Data 29).
Subcellular localization in wheat protoplasts
pJIT163-Ubi: delta-1-pyrroline-5-carboxylate synthase (TaP5CS1, TraesCS1B03G0812300)-hGFP, pJIT163-Ubi: histone deacetylase 9 (TaHDA9, TraesCS2B02G309700.1) -hGFP (Supplementary Data 29) and pJIT163-Ubi:hGFP were transfected into wheat mesophyll protoplast cells using a PEG-mediated method72. After incubation for 20 h in the dark at 22 °C, confocal microscopy (Carl Zeiss, LSM710) was used to examine the protoplasts.
Yeast two-hybrid (Y2H) assays
The CDS of the TaP5CS1 gene was cloned into the bait vector pGBKT7. A cDNA library was constructed from seedlings of wheat variety Aikang 5873 to screen interaction proteins of TaP5CS1 using a yeast two-hybrid (Y2H) assay according to the manufacturer’s protocol (Clontech, USA). Yeast colonies were amplified by PCR and sequenced by T7-specific primers (Supplementary Data 29). After sequencing, the client genes were identified. The CDS of a histone deacetylase (TaHDA9) was amplified and fused to the prey vector pGADT7 to confirm protein interaction with the bait in the Y2H screening (Supplementary Data 29). The bait and prey vectors were co-transformed into yeast strain AH109 and screened on SD/Leu-Trp and SD/Trp-Leu-His-Ade + X-a-Gal medium.
Luciferase complementation imaging assay (LCI)
The CDSs of TaHDA9 and TaP5CS1 were subcloned into pCAMBIA1300-cLUC and pCAMBIA1300-nLUC to form TaHDA9-cLUC and TaP5CS1-nLUC, respectively. The recombinant vectors were transformed into Agrobacterium strain GV3101. Different vector combinations were co-transfected into the lower epidermis of N. benthamiana leaves. TaP5CS1-nLUC and cLUC, nLUC and TaHDA9-cLUC, cLUC and nLUC were used as negative controls. Luminescence images were captured using a plant-living imaging system (Berthold, Night Shade LB 985).
Prokaryotic expression of TaP5CS1
The TaP5CS1 was cloned into the prokaryotic expression vector pGEX-6P-1 using BamH1 and Not1 as restriction enzyme cutting sites (Supplementary Data 29). E. coli BL21 (DE3) was used to express recombinant TaP5CS1. TaP5CS1 protein was purified using a GST-tag Protein Purification Kit (Beyotime, China, Catalog No. P2262). The collected TaP5CS1 recombinant protein was analyzed by immunoblotting using anti-GST (PTMbio, PTM 5046) and pan anti-acetyl-lysine (anti-Kac antibody, Cell Signaling Technology, Catalog No. 9681) antibodies.
Transient expression assays
35S: TaP5CS1-GFP-Flag and 35S: TaHDA9-GFP-His or 35S: GFP were injected into four-week-old tobacco (N. benthamiana) leaf cells. Then, the total proteins extracted in lysis buffer [20 mM Tris-HCl (pH 8.0), 2 mM DTT, 1% Triton X-100, 800 μM PMSF, 250 mM sucrose] containing protease inhibitor cocktail. After washing73, we performed immunoprecipitation using anti-Flag antibody (Abcam, M20008) and Protein A/G PLUS-Agarose (Santa Cruz, Lot number sc-2003) to purify TaP5CS1-GFP-Flag proteins from the above extracts. Eluted proteins were detected by immunoblotting with anti-Flag (Abcam, M20008) and anti-Kac (Cell Signaling Technology, Catalog No. 9681) antibodies using the ECL SuperSignal system.
Protein extracts of tobacco leaves with transient expression of 35S: TaP5CS1-GFP-Flag and 35S: TaHDA9-GFP-His or 35S: GFP were analyzed by immunoblotting with anti-Flag antibody (Abcam, M20008) to examine whether TaHDA9 could regulate the expression of TaP5CS1 protein. Anti-actin antibody (Abbkine, Lot number A01050) was used as a loading control.
Co-immunoprecipitation (Co-IP) assays
We transfected 35S:TaP5CS1-GFP-Flag and 35S: TaHDA9-GFP-His or 35S: GFP into tobacco leaf cells to extract the total proteins. TaP5CS1-GFP-Flag proteins from the above extracts were IP with anti-Flag antibody (Abcam, M20008) and Protein A/G PLUS-Agarose (Santa Cruz, Lot number sc-2003). The eluted proteins were detected by immunoblotting with anti-Flag antibody (Abcam, M20008) and anti-His antibody (Abmart, M20001), respectively.
Pull-down protein interaction assays
TaHDA9-His and TaP5CS1-GSTor GST protein were expressed in E. coli. Purification of TaP5CS1-GST or GST proteins was accomplished with a GST-tag Protein Purification Kit (Beyotime, China, Catalog No. P2262) and beyoGoldTM GST-tag Purification Resin in buffer (50 mM Tris, 150 mM NaCl, 10 mM GSH, pH 8.0). The pulled-down proteins were mixed with the SDS sample buffer and detected by immunoblotting with anti-GST (PTMbio, PTM5046) and anti-His (Abmart, M20001) antibodies.
In vitro lysine deacetylase enzyme activity assay
To determine whether TaHDA9 can deacetylate TaP5CS1 in vitro, E. coli was transformed with TaP5CS1-GST and TaHDA9-His or a His vector. The TaP5CS1-GST recombinant protein was isolated from E. coli cultures that were grown both with and without the deacetylation inhibitors 5 nM trichostatin A (TSA, MedChemExpress, Catalog No. HY-15144) and 5 mM β-nicotinamide (NAM, MedChemExpress, Catalog No. HY-B0150). Anti-GST (PTMbio, PTM5046) and pan anti-Kac (Cell Signaling Technology, Catalog No. 9681) antibodies were used for immunoblotting detection of the isolated proteins.
Site-directed mutagenesis
The 35S: TaP5CS1-GFP-Flag plasmid was subjected to site-directed mutagenesis of K221, K231, and K634 (K221R, 231R, K634R, AAA-AGA, AAG-AGG, AAA-AGA) using the Fast Site-Directed Mutagenesis Kit (Tiangen, China, Catalog No. KM101). PCR was conducted using site-specific primers (Supplementary Data 29). Mutations were verified by DNA sequencing. Four-week-old tobacco (N. benthamiana) leaf cells were used to express recombinant wild-type and three mutants of TaP5CS1, respectively. Proteins were purified by IP and analyzed by immunoblotting with anti-Flag (Abcam, M20008) and anti-pan-Kac (Cell Signaling Technology, Catalog No. 9681) antibodies. In addition, TaP5CS1 and TaP5CS1K634R were transiently expressed in tobacco, respectively, and analyzed by immunoblots with an anti-Flag antibody (Abcam, M20008) to examine the expression of wild-type and three mutants of TaP5CS1. An anti-actin antibody (Abbkine, Lot number A01050) was used as the loading control.
Barley strip mosaic virus (BSMV) induced gene silencing
Virus-induced gene silencing (VIGS) experiments were conducted on the middle-sensitivity to BSMV wheat cultivar Yunong 268. The primers for peroxidase (TaPOD, TraesCS2B03G1538300.1), sucrose synthase (TaSUS, TraesCS7D03G0082900.2), and TaP5CS1 were generated from 312, 259, and 232 base pair fragments, respectively, using Primer 3.0 (Supplementary Data 29). The α, β, and γ RNAs of the BSMV genome were synthesized from linearized plasmids, using RiboMAXTM Large Scale RNA Production System-T7 (Promega, P1300) and Ribo m7G Cap Analog (Promega, P1712). The positive control used plants inoculated with BSMV: PDS (phytoene desaturase). Transcripts of each vector (α, β, γ, or recombinant γ-gene) were mixed in a 1:1:1 ratio for inoculating the wheat cultivar. Ten days after the wheat plants were infected with F. pg, the mixture RNA virus was added to the FES buffer for inoculation of the second leaves of wheat seedlings, and after darkness for 24 h, all plants were grown at 23 °C in 60–80% relative humidity. After two weeks, the FCR resistance of wheat plants was investigated, with the third and fourth leaf tissues were collected to determine the efficiency of silencing of TaPOD, TaSUS, and TaP5CS1.
Ethyl methanesulfonate mutants of TaHDA9
Seeds of the tetraploid wheat Kronos were mutagenized by Jorge Dubcovsky’s team using the chemical mutagen ethyl methanesulfonate (EMS)74. The mutant lines P5CS1-1B (K3196: C/T, mutation effect = stop gained), HDA9-2A (K4505: C/T, mutation effect = splice_acceptor_variant), HDA9-2A (K4236: C/T, mutation effect = stop gained), HDA9-2B (Kronos733:A497G, mutation effect = splice_acceptor_variant), and HDA9-2B (Kronos2217, C/T, mutation effect = stop gained) were sequenced using specific primers (Supplementary Data 29). The positive mutant individual plants selected for backcrossing with the wild-type to obtain BC2 mutants for functional verification.
Overexpressed transgenic plants of TaP5CS1 and TaHDA9
The CDSs of TaP5CS1 and TaHDA9 were cloned into the LGY-OE3 vector with the Ubi promoter to produce TaP5CS1-OE and TaHDA9-OE plants, respectively. The vectors containing targeted genes were transformed by Agrobacterium-mediated infection into immature embryos of Fielder (FCR-susceptible) to obtain TaP5CS1-OE and TaHDA9-OE lines. T0 transgenic plants detected as positive by PCR using specific primers (Supplementary Data 29) were further self-pollinated. The T2 non-segregating plants of three lines with high expression levels were selected for further analysis.
Disease index (DI) investigation
F. pg was routinely cultured on potato dextrose agar (PDA) at 25°C. Millet grains were used as the pathogen medium to inoculate the wheat seedlings. All seedlings were grown in a greenhouse under 16 h/8 h day/night conditions at 25/20°C day/night temperatures with 60-80% relative humidity. The severity of FCR was evaluated at the fourth week after being inoculated with F. pg and the resistance of the plants was estimated with the DI, which was calculated on a scale from 0 to 9 for every plant. The detailed method is based on our previous study48. Briefly, a total of 12 infection stems were counted for each experiment. Five independent biological repeats were performed. The field test of the FCR resistance was using inoculated millet grains as a pathogen medium concurrent with the planting material, and the severity of the disease was investigated at wheat grain filling stage, which was calculated by the DI with a scale from 0 to 475.
Microscopic analysis of pathogen infection
To observe F. pg development on wheat plants, we sampled the stems of the overexpressed transgenic wheat and wild type after F. pg inoculation for four weeks, then stained them with 2.5% glutaraldehyde. The F. pg fungals were observed with a scanning electron microscope (TEM, Hitachi, HT7700). A total of 12 infection stems were counted from for each experiment. Three independent biological repeats were performed.
We measured the amount of F. pg biomass on wheat plants by taking samples from the stems of overexpressed transgenic wheat and wild type wheat that had been infected with F. pg for four weeks. We then extracted the total DNA. The semi-quantitative PCR was conducted using tubulin gene from wheat (GenBank No. MG852130.1) and F. pg (GenBank No. CP102997.1), respectively. A total of 12 infection stems were counted for each experiment. Three independent biological repeats were performed.
To assess the cellular response of the overexpressed transgenic wheat and wild type to F. pg infection, cell necrotic death at the infection site was stained by trypan blue39. The inoculated stems were detached from the plants and stained in a 0.5% solution of trypan blue in lactophenol for 2 days at room temperature and mounted in a solution of 2.5 mg/mL chloral hydrate. The stained segments were observed and photographed by a stereomicroscope (Olympus, Tokyo, Japan). A total of 12 infection stems were counted for each experiment. Three independent biological repeats were performed.
Proline content measurement
Proline content was quantified in the overexpressed transgenic wheat and wild type using the acid ninhydrin assay. Briefly, proline was extracted by boiling 0.5 g of plant material in 2 mL of distilled water before adding 500 μL of 0.2 mM sodium citrate (pH 4.6) and 2 mL of 1% ninhydrin to 0.5 μL of plant extract. The mixture was boiled for 1 h before adding 2 mL toluene for extraction and centrifuging. A standard curve was prepared by measuring the absorbance of the standard proline (Himedia, India). Proline content was measured at 520 nm using a spectrophotometer (Shimadzu, Japan). All experiments were conducted in triplicate.
Assay of proline inhibition against F. pg growth
To evaluate the effect of exogenous proline on F. pg growth, 0, 1, 3, 5, 7, 9, and 10 mM proline were separately added to potato dextrose agar (PDA) medium. Since valine has a similar structure to proline, it was selected as a control. Subsequently, F. pg mycelia were inoculated into each pore of PDA and incubated at 25°C for 6 days. From the first to sixth day post-inoculation (DPI), we recorded the diameter of the mycelial colony and hyphal morphology. The mycelium growth inhibition rate was determined by measuring the colony diameter76:
A total of five treated PDA were counted for each experiment. Three independent biological repeats were performed.
Effect of exogenous proline on wheat Fusarium crown rot resistance
After wheat germination, 10, 20, and 30 mM proline were separately added to wheat plot culture and an equal amount water was used as a control. After F. pg inoculation for four weeks, disease phenotype of wheat seedlings was evaluated, with DI calculated as above. Twelve infection stems were counted for each experiment. Five independent biological repeats were performed.
Quantification and statistical analysis
Two-tailed Student’s t-test (*P < 0.05, **P < 0.01) was used for statistical analysis of experimental and control groups. As for multiple samples, one-way analysis of variance (ANOVA) with Tukey’ multiple comparisons test was conducted.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the findings of this work are available within the article and Supplementary Information files. The RNA-seq data have been deposited in the NCBI database under accession PRJNA1037187. The mass spectrometry proteomics data were deposited in the ProteomeXchange Consortium via the iProX partner repository under accessions PXD041703 and PXD042492. The raw data of identified proteins, phosphorylation sites, and lysine acetylation sites in 20 wheat tissues (Fig. 1b) were deposited at repository Zenodo [https://zenodo.org/records/14822020]. The raw GRN data were deposited at repository Zenodo [https://zenodo.org/records/14625876]. The Chinese Spring wheat reference genome (IWGSC RefSeq v2.1) is publicly available at IWGSC data repository hosted by URGI-INRAE [http://wheat-urgi.versailles.inra.fr/Seq-Repository/Assemblies] and in the NCBI database under accession PRJNA669381. Source data are provided with this paper.
References
Demirjian, C., Vailleau, F., Berthomé, R. & Roux, F. Genome-wide association studies in plant pathosystems: success or failure? Trends Plant Sci. 28, 471–485 (2023).
Zhu, J. K. Abiotic Stress Signaling and Responses in Plants. Cell 167, 313–324 (2016).
McWhite, C. D. et al. A pan-plant protein complex map reveals deep conservation and novel assemblies. Cell 181, 460–474 (2020).
Huang, D., Kosentka, P. Z. & Liu, W. Synthetic biology approaches in regulation of targeted gene expression. Curr. Opin. Plant Biol. 63, 102036 (2021).
Zhao, S., Zhang, X. & Li, H. Beyond histone acetylation-writing and erasing histone acylations. Curr. Opin. Struct. Biol. 53, 169–177 (2018).
Choudhary, C., Weinert, B. T., Nishida, Y., Verdin, E. & Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550 (2014).
Figlia, G., Willnow, P. & Teleman, A. A. Metabolites Regulate Cell Signaling and Growth via Covalent Modification of Proteins. Dev. Cell 54, 156–170 (2020).
Millar, A. H. et al. The scope, functions, and dynamics of posttranslational protein modifications. Annu Rev. Plant Biol. 70, 119–151 (2019).
Bradley, D. The evolution of post-translational modifications. Curr. Opin. Genet Dev. 76, 101956 (2022).
Mergner, J. et al. Mass-spectrometry-based draft of the Arabidopsis proteome. Nature 579, 409–414 (2020).
Walley, J. W. et al. Integration of omic networks in a developmental atlas of maize. Science 353, 814–818 (2016).
Marx, H. et al. A proteomic atlas of the legume Medicago truncatula and its nitrogen-fixing endosymbiont Sinorhizobium meliloti. Nat. Biotechnol. 34, 1198–1205 (2016).
Della Coletta, R., Qiu, Y., Ou, S., Hufford, M. B. & Hirsch, C. N. How the pan-genome is changing crop genomics and improvement. Genome Biol. 22, 3 (2021).
Chenu, K. et al. Contribution of crop models to adaptation in wheat. Trends Plant Sci. 22, 472–490 (2017).
A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345, 1251788 (2014).
Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361, 7191 (2018).
Walkowiak, S. et al. Multiple wheat genomes reveal global variation in modern breeding. Nature 588, 277–283 (2020).
Shi, X. et al. Comparative genomic and transcriptomic analyses uncover the molecular basis of high nitrogen-use efficiency in the wheat cultivar Kenong 9204. Mol. Plant 15, 1440–1456 (2022).
Zhu, T. et al. Optical maps refine the bread wheat Triticum aestivum cv. Chinese Spring genome assembly. Plant J. 107, 303–314 (2021).
Jiao, C. et al. Pan-genome bridges wheat structural variations with habitat and breeding. Nature (2024).
Guo, W. et al. Proteome and lysine acetylome analysis reveals insights into the molecular mechanism of seed germination in wheat. Sci. Rep. 10, 13454 (2020).
Ma, L. et al. Proteomic and phosphoproteomic analyses reveal a complex network regulating pollen abortion and potential candidate proteins in tcms wheat. Int J. Mol. Sci. 23, 6428 (2022).
Lafiandra, D. & Shewry, P. R. Wheat Glutenin polymers 2. The role of wheat glutenin subunits in polymer formation and dough quality. J Cereal Sci. 106 (2022).
Wang, D. et al. Molecular genetic and genomic analysis of wheat milling and end-use traits in China: progress and perspectives. Crop J. 6, 68–81 (2018).
Schuller, J. M. et al. Redox-coupled proton pumping drives carbon concentration in the photosynthetic complex I. Nat. Commun. 11, 494 (2020).
Scagliola, A., Mainini, F. & Cardaci, S. The tricarboxylic acid cycle at the crossroad between cancer and immunity. Antioxid. Redox Signal 32, 834–852 (2020).
Yang, M., Ismayil, A. & Liu, Y. Autophagy in plant-virus interactions. Annu Rev. Virol. 7, 403–419 (2020).
Santner, A. & Estelle, M. The ubiquitin-proteasome system regulates plant hormone signaling. Plant J. 61, 1029–1040 (2010).
Kaufmann, K., Pajoro, A. & Angenent, G. C. Regulation of transcription in plants: mechanisms controlling developmental switches. Nat. Rev. Genet 11, 830–842 (2010).
Jin, J. et al. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45, 1040–1045 (2017).
Gao, Y. et al. The endosperm-specific transcription factor TaNAC019 regulates glutenin and starch accumulation and its elite allele improves wheat grain quality. Plant Cell 33, 603–622 (2021).
Millard, P. S., Kragelund, B. B. & Burow, M. R2R3 MYB transcription factors - functions outside the DNA-Binding domain. Trends Plant Sci. 24, 934–946 (2019).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 559 (2008).
Shvedunova, M. & Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 23, 329–349 (2022).
Ramírez-González, R. H. et al. The tra The transcriptional landscape of nscriptional landscape of polyploid wheat. Science 361, 6089 (2018).
Ma, C. et al. Contribution of the flag leaf to lead absorption in wheat grain at the grain-filling stage. Ecotoxicol. Environ. Saf. 225, 112722 (2021).
Boudet, J. et al. The bZIP transcription factor SPA heterodimerizing protein represses glutenin synthesis in Triticum aestivum. Plant J. 97, 858–871 (2019).
Savary, S. et al. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 3, 430–439 (2019).
Wang, N. et al. Inactivation of a wheat protein kinase gene confers broad-spectrum resistance to rust fungi. Cell 185, 2961–2974 (2022).
Wang, L. et al. The OsBDR1-MPK3 module negatively regulates blast resistance by suppressing the jasmonate signaling and terpenoid biosynthesis pathway. Proc. Natl Acad. Sci. USA 120, e2211102120 (2023).
Wang, S. et al. Phosphorylation and ubiquitination of OsWRKY31 are integral to OsMKK10-2-mediated defense responses in rice. Plant Cell 35, 2391–2412 (2023).
Ding, B., Bellizzi Mdel, R., Ning, Y., Meyers, B. C. & Wang, G. L. HDT701, a histone H4 deacetylase, negatively regulates plant innate immunity by modulating histone H4 acetylation of defense-related genes in rice. Plant Cell 24, 3783–3794 (2012).
Walley, J. W., Shen, Z., McReynolds, M. R., Schmelz, E. A. & Briggs, S. P. Fungal-induced protein hyperacetylation in maize identified by acetylome profiling. Proc. Natl Acad. Sci. USA 115, 210–215 (2018).
Li, P. et al. RGAugury: a pipeline for genome-wide prediction of resistance gene analogs (RGAs) in plants. BMC Genom. 17, 852 (2016).
Smiley, R. W., Gourlie, J. A., Easley, S. A. & Patterson, L. M. Pathogenicity of Fungi Associated with the Wheat Crown Rot Complex in Oregon and Washington. Plant Dis. 89, 949–957 (2005).
Hogg, A. C. et al. Monitoring fusarium crown rot populations in spring wheat residues using quantitative real-time polymerase chain reaction. Phytopathology 100, 49–57 (2010).
Yang, X. et al. A loss-of-function of the dirigent gene TaDIR-B1 improves resistance to Fusarium crown rot in wheat. Plant Biotechnol. J. 19, 866–868 (2021).
Yang, X. et al. Investigation and genome-wide association study for Fusarium crown rot resistance in Chinese common wheat. BMC Plant Biol. 19, 153 (2019).
Su, J. et al. Genetics of Resistance to Common Root Rot (Spot Blotch), Fusarium Crown Rot, and Sharp Eyespot in Wheat. Front Genet 12, 699342 (2021).
Lv, G. et al. A cell wall invertase modulates resistance to fusarium crown rot and sharp eyespot in common wheat. J. Integr. Plant Biol. 65, 1814–1825 (2023).
Niu, Y. et al. HISTONE DEACETYLASE 9 transduces heat signal in plant cells. Proc. Natl Acad. Sci. USA 119, e2206846119 (2022).
Han, L. et al. A multi-omics integrative network map of maize. Nat. Genet 55, 144–153 (2023).
Depuydt, T., De Rybel, B. & Vandepoele, K. Charting plant gene functions in the multi-omics and single-cell era. Trends Plant Sci. 28, 283–296 (2023).
Bressan, D., Battistoni, G. & Hannon, G. J. The dawn of spatial omics. Science 381, 4964 (2023).
Zhang, H., Zhu, J., Gong, Z. & Zhu, J. K. Abiotic stress responses in plants. Nat. Rev. Genet 23, 104–119 (2022).
Salse, J., Barnard, R. L., Veneault-Fourrey, C. & Rouached, H. Strategies for breeding crops for future environments. Trends Plant Sci. 29, 303–318 (2023).
Zhang, N. et al. Global profiling of 2-hydroxyisobutyrylome in common wheat. Genom. Proteom. Bioinf 20, 688–701 (2021).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 12, 323 (2011).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. circlize Implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812 (2014).
Xie, W. et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153, 1134–1148 (2013).
Schwartz, D. & Gygi, S. P. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 23, 1391–1398 (2005).
Chou, M. F. & Schwartz, D. Biological sequence motif discovery using motif-x. Curr. Protoc. Bioinforma. Chapter 13, 15–24 (2011).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, 353–361 (2017).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 49, 344–354 (2021).
Cline, M. S. et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2, 2366–2382 (2007).
Wang, C. et al. GPS 5.0: An Update on the prediction of kinase-specific phosphorylation sites in proteins. Genom. Proteom. Bioinf 18, 72–80 (2020).
Moerman, T. et al. GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics 35, 2159–2161 (2019).
Liu, X., Wang, Y., Ji, H., Aihara, K. & Chen, L. Personalized characterization of diseases using sample-specific networks. Nucleic Acids Res. 44, 164 (2016).
Yoo, S. D., Cho, Y. H. & Sheen, J. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat. Protoc. 2, 1565–1572 (2007).
Zhang, N. et al. Global crotonylatome and GWAS revealed a TaSRT1-TaPGK model regulating wheat cold tolerance through mediating pyruvate. Sci. Adv. 9, eadg1012 (2023).
Henry, I. M., Nagalakshmi, U. & Lieberman, M. C. Efficient genome-wide detection and cataloging of EMS-induced mutations using exome capture and next-generation sequencing. Plant Cell 26, 1382–1397 (2014).
Zhou, H. et al. Diversity of the Fusarium pathogens associated with crown rot in the Huanghuai wheat-growing region of China. Environ. Microbiol 21, 2740–2754 (2019).
Tian, L. et al. First report of antifungal activity conferred by non-conventional peptides. Plant Biotechnol. J. 19, 2147–2149 (2021).
Acknowledgements
This project was funded by the National Key Research and Development Program (2023YFD1200401 to N.Z.), the National Natural Science Foundation (U23A20191 to F.C. and 62225209 to M. L.), and the Henan University Science and Technology Innovation Talent Project (24HASTIT055 to N.Z.) of China.
Author information
Authors and Affiliations
Contributions
F.C., M. L., D.W.W., and N.Z. designed the research. S.S.W., D.Y.C., L.R.Z. and N.Z performed sample preparations. L.T., Y.C.Z., R.Q.Z., W.K.W., S.S.W., D.Y.C. C.W.S. and N.Z. led the omics data analysis. Y.C.Z. and A. S. constructed the database. S.G.L., L.L., M.J.G., S.S.W., L.R.Z., D.Y.C., X.Y., Y. R. and N.Z. performed experiments. N.Z., D.W.W., M.W. and F.C. wrote the manuscript. All the authors edited and proofed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xiaojie Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, N., Tang, L., Li, S. et al. Integration of multi-omics data accelerates molecular analysis of common wheat traits. Nat Commun 16, 2200 (2025). https://doi.org/10.1038/s41467-025-57550-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57550-x
This article is cited by
-
Multi-Network Co-expression Analysis Enhances Biological Insights from Single-Cell Gene Expression
Interdisciplinary Sciences: Computational Life Sciences (2026)
-
Advances in genetic and molecular mechanisms of crop resistance to stalk rot
Stress Biology (2026)
-
The pathogenicity and multi-organ proteomic profiles of Mpox virus infection in SIVmac239-infected rhesus macaques
Nature Communications (2025)
-
A transcriptomic profiling across tissues, developmental stages, and types of Nicotiana tabacum
Scientific Data (2025)
-
Wheat genomics frontiers for gene discovery and breeding applications
WheatOmics (2025)
