
Abstract
The direct photocatalytic oxidation of methane to value-added chemicals has garnered considerable interest in recent years. However, achieving high productivity while maintaining high selectivity at an appreciable methane conversion rate remains a formidable challenge. Here, we present photochemically-triggered and photothermally-enhanced oxidative coupling of methane to multi-carbon C2+ alkanes over an Au and CeO2 nanoparticle-decorated ZnO photocatalyst, which exhibits a record-breaking C2+ production rate of 17,260 μmol g−1 h−1 with ~90% C2+ selectivity under wide-spectrum light irradiation without a secondary source of heating. Comprehensive characterizations and computational studies reveal that CH4 activation is a photochemical reaction initiated by ultraviolet light-excited ZnO, and the introduction of CeO2 substantially enhances the activation of CH4 and O2 due to the cooperative interaction between Au and CeO2. Concurrently, Au nanoparticles capture visible and near-infrared light to generate localized heating, which greatly promotes the subsequent desorption of produced methyl radical for C–C coupling prior to undergoing further undesired overoxidation.
Similar content being viewed by others
Introduction
Methane is increasingly used as a feedstock for the production of chemicals, plastics, and fertilizers in industry1,2. Currently, the large-scale transformation of methane into liquid fuels and high-value chemicals is indirect and usually involves energy-intensive steam reforming3. Direct conversion of methane to chemicals has the potential for higher carbon and energy efficiency and is considered a sustainable process4. In recent years, significant progress has been made in thermocatalytic direct methane conversion processes5, particularly in the oxidative coupling of methane (OCM) to C2+ hydrocarbons (C2H6, C2H4, C3H8, etc.)6,7, which is not constrained by low equilibrium conversion thanks to its negative reaction free energy. However, the required high reaction temperatures in OCM inevitably lead to immense energy consumption, severe catalyst deactivation, and the overoxidation of desired products to thermodynamically favored CO and CO2, thereby limiting their applicability8. Photocatalytic direct methane conversion using photonic energy to trigger reactions is expected to take place at low or even room temperature and can thus potentially solve such issues9,10,11,12,13. Recent studies on photocatalytic OCM (POCM) represent one of the most promising methods to convert methane to ethane with selectivity up to 90% under mild reaction conditions14,15,16,17. Despite much impressive progress, the photocatalytic conversion of CH4 is a great challenge owing to the limited utilization of solar light and the lack of effective photocatalysts for C–H bond activation, a key step in CH4 conversion.
As is well known, the solar spectrum consists of approximately 5% ultraviolet (UV) light, 45% visible light, and 50% near-infrared (NIR) light. At present, most of the photocatalysts reported to date for methane conversion are UV-light-responsive oxide semiconductors, such as Ga2O3, TiO2, and ZnO18,19,20. In view of the theoretically efficient solar light utilization, developing long-wavelength-responsive photocatalysts with narrow band gaps for methane conversion is a common way. However, these photocatalysts typically provide low driving forces for photoredox reactions, which deliver low reaction rates or may not drive methane oxidation, primarily attributed to the unfavorable thermodynamics and sluggish kinetics of C2+ hydrocarbons production stemming from the strong C–H bond, multi-electron transfer, and C–C coupling21. In addition to generating charge carriers to directly drive catalytic reactions (photochemical effect), light illumination ubiquitously induces intense lattice vibration and thereby results in the heating of the photocatalyst to high temperatures (photothermal effect)22. However, previous photocatalytic methane oxidation studies have mainly focused on photochemical effects, while overlooking or disregarding photothermal effects9,10. By utilizing and optimizing both photochemical and photothermal effects from light irradiation, it is possible to fully harness the solar spectrum, achieve maximum solar energy efficiency, and enable tunable product selectivity. Hence, investigating the influence of light-induced heating on the formation of C2+ products is rather important for POCM, yet it remains rarely explored.
Zinc oxide (ZnO), characterized by its high redox potential, is photocatalytically active towards methane oxidation16,20. Using noble metals such as Au as cocatalysts have been found to improve the activity and selectivity of POCM16. However, these catalysts still suffer from relatively low inherent photocatalytic performance, primarily due to inadequate capability to dissociate the C–H bonds in CH4 and O–O bonds in O2. Therefore, it is highly desirable to develop effective ZnO-based photocatalysts under wide-spectrum light irradiation. CeO2 has garnered significant attention as a promising catalyst component in heterogeneous catalysis, particularly in oxidation reactions, thanks to its distinctive redox properties, exceptional oxygen storage capacity, and strong interaction with metals23. The appropriate combination of ZnO and CeO2, together with the incorporation of Au, presents a potential strategy to enhance C–H bond cleavage and O2 capture/dissociation, and finally facilitate photocatalytic methane conversion.
Motivated by the aforementioned analysis, we herein report a hybrid photochemical–photothermal catalytic system based on a ZnO-supported Au and CeO2 photocatalyst that exclusively utilizes solar energy to efficiently drive the OCM to C2+ hydrocarbons. Photogenerated charge carriers from the excitation of ZnO by UV light enable the photochemical activation of methane to methyl radical. The synergy of Au and CeO2 nanoparticles on ZnO greatly facilitates the activation of CH4 and O2. Meanwhile, Au nanoparticles can efficiently convert the absorbed visible and near-infrared light into thermal energy to drive the desorption of methyl radicals and accelerate C–C coupling. The integration of photochemical catalysis with photothermally generated heat significantly enhances C2+ hydrocarbon production, achieving a C2+ production rate of 17,260 μmol g−1 h−1 with approximately 90% C2+ selectivity.
Results
Performance dependent on CeO2 amount and light intensity
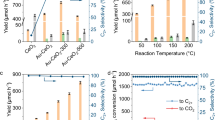
CeO2/ZnO mixed oxide catalysts with different Ce/(Ce + Zn) molar ratios were synthesized using a coprecipitation method. Pure ZnO and CeO2 were also prepared using a similar method as references. We previously reported that Au was an excellent cocatalyst for POCM16. Therefore, Au nanoparticles (NPs) were loaded on the as-prepared oxides with a 1.5 weight percent (wt%). The resultant samples are denoted as Au/x%CeO2/ZnO (x% stands for the molar percentage of Ce, x = 0.5, 1, 3, 10), Au/ZnO or Au/CeO2. The actual amounts of Au and Ce in the catalysts determined by inductively coupled plasma-optical emission spectrometry (ICP-OES) are very close to the designed values (Supplementary Table 1). Photocatalytic performance was evaluated in a continuous-flow reactor by flowing methane and air under UV and visible light (300–800 nm) irradiation. As shown in Fig. 1a, the CH4 photooxidation products observed were C2-3 (C2H6, C2H4, C3H8, C3H6) and CO2, with only trace amounts of CO (concentration below 10 ppm). The performance of the photocatalyst varied greatly with the mole ratio of Ce/(Ce + Zn). The photocatalytic activity of the catalyst for the production rate of C2-3 (r(C2-3)) was considerably improved and then gradually decreased with an increase in the Ce/(Ce + Zn) mole ratio (Fig. 1a). Au/1%CeO2/ZnO showed the highest r(C2-3) of 9446 μmol g−1 h−1 with a C2-3 selectivity of up to 95%, representing a 2.3-fold improvement compared with Au/ZnO (4043 μmol g−1 h−1, 98% selectivity). Comparatively, only a small amount of C2H6 was produced with 40% selectivity on Au/CeO2, probably due to the narrower band gap of CeO2 compared to ZnO24. In addition, the r(C2-3) on Au/1%CeO2/ZnO was around 2.6 times that over mechanically mixed Au/CeO2 and Au/ZnO with the same amount of CeO2, suggesting a synergetic effect between these three composites in the photocatalytic CH4 oxidation. The performance optimization test confirmed that the ideal Au amount was around 1.5 wt% (Supplementary Fig. 1).

a Photocatalytic performance of Au/CeO2/ZnO catalyst with different Ce/(Ce + Zn) molar ratio under 380 mW cm−2 light irradiation. b, c Dependence of photocatalytic performance on light intensity over Au/1%CeO2/ZnO (b) and Au/ZnO (c). d The long-term stability test of Au/1%CeO2/ZnO under 670 mW cm−2 light irradiation. Reaction condition in (a–d): 197 mL min−1 of CH4 and 3 mL min−1 of air, 20 mg catalyst, 300 W Xe light (300–800 nm). e Dependence of catalyst temperature on light intensity over Au/1%CeO2/ZnO and Au/ZnO. f Photocatalytic performance of Au/1%CeO2/ZnO under the irradiation of full solar spectrum, UV light, and visible-IR light from the concentrated simulated sunlight. Reaction condition in (f): 197 mL min−1 of CH4 and 3 mL min−1 of air, 20 mg catalyst. Error bars represent standard deviations obtained from three independent measurements.
Strikingly, r(C2-3) on Au/1%CeO2/ZnO increased substantially with the increase of light intensity and reached a maximum at the threshold of 670 mW cm−2, followed by a decrease with further increasing light intensity (Fig. 1b). Whereas, the production rate of CO2 r(CO2) increased continuously with light intensity, thus leading to a continuous decrease in C2-3 selectivity. A similar trend in r(C2-3) and r(CO2) was also observed on Au/ZnO (Fig. 1c), but with much lower activity than Au/1%CeO2/ZnO. Consequently, the CH4 conversion rate of Au/1%CeO2/ZnO and Au/ZnO exhibited a gradual increase with increasing light intensity, eventually reaching a plateau above 670 mW cm−2 (Supplementary Fig. 2). The methane conversion on Au/1%CeO2/ZnO was 0.47% under 670 mW cm−2 light irradiation. Au/1%CeO2/ZnO delivered a record-breaking r(C2-3) of 17,260 μmol g−1 h−1 with 90% selectivity under 670 mW cm−2 light irradiation, which was 2.8 times that on Au/ZnO under the same conditions and prominently exceeded that on the best-performing photocatalysts for OCM (Supplementary Table 2)14,15,16,17,25,26,27,28,29,30,31,32,33,34,35,36,37. The TON (turnover number) was calculated to be 523 in 1 h with respect to the amount of Au cocatalyst under the optimized reaction conditions, which is, to our knowledge, the largest value among reported photocatalysts under similar reaction conditions (Supplementary Table 2). The Au/1%CeO2/ZnO retained more than 94% of its initial performance with >90% selectivity in a long-term operational stability test under continuous 670 mW cm−2 illumination for 50 h (Fig. 1d), revealing the good stability of Au/1%CeO2/ZnO for POCM. A similar stability performance is also observed on Au/ZnO (Supplementary Fig. 3). Carbon isotope-tracing experiments confirmed that the product C2H6 originated from CH4 (a peak at m/z = 32 attributable to 13C2H6 was observed by gas chromatography-mass spectrometry) (Supplementary Fig. 4).
It is worth noting that light illumination commonly increases the temperature of the catalyst due to the photothermal effect22, especially for the gas-solid phase reaction systems without temperature control. In view of this, the equilibrium temperature at the center of the illuminated surface of the catalyst was measured by a built-in contact thermocouple affixed to the photocatalyst surface under the reaction conditions. Figure 1e shows that the surface equilibrium temperature of Au/1%CeO2/ZnO and Au/ZnO consistently increased with increasing light intensity, and the temperature observed on Au/1%CeO2/ZnO was approximately 6–10 °C higher compared to that on Au/ZnO, probably due to the stronger light absorption capability of Au/1%CeO2/ZnO than Au/ZnO (Supplementary Fig. 5). The equilibrium temperature of Au/1%CeO2/ZnO was about 116 °C at 380 mW cm−2, increased to 162 °C at 670 mW cm−2, and reached 193 °C when increasing the intensity to 870 mW cm−2. This means that increasing light intensity not only excites more electrons and holes to facilitate the POCM reaction but also elevates the temperature at the catalyst surface, likely further promoting the reaction. Inspired by this, we used simulated sunlight with wavelength from UV to IR light to perform POCM on Au/1%CeO2/ZnO, considering that Au NPs show strong localized surface plasmon resonance (LSPR) in visible light region22,38, facilitating efficient light-to-heat conversion (Supplementary Table 3). Under the irradiation of the full solar spectrum from the concentrated simulated sunlight (10 Suns), the r(C2-3) was 2926 μmol g−1 h−1 with a catalyst temperature of 168 °C (Fig. 1f), while the r(C2-3) decreased to 1372 μmol g−1 h−1 with a temperature of 39 °C under UV light (λ < 400 nm) irradiation and no C2-3 was produced under visible and IR light (λ > 440 nm) irradiation with a temperature of 128 °C. These results demonstrate that POCM is solely triggered by UV light excitation of CeO2/ZnO semiconductors, and illumination-induced heat is advantageous for C2-3 production. The impact of elevated temperature on the performance of POCM will be discussed in the following sections.
Positive effect of heat on POCM
To study the effect of heat, the CH4 oxidation reaction was carried out at different elevated temperatures with and without light illumination. Experiments were conducted using a vertically oriented reactor, wherein the catalyst can be electrically heated to the desired temperature and exposed to a 360 nm monochromatic light with a low intensity of 20 mW cm−2, aiming to minimize light-induced heating effect37 (the surface temperatures of Au/1%CeO2/ZnO and Au/ZnO were measured to be only approximately 28 °C under the irradiation of this light source, demonstrating that the photothermal effect is negligible under such low-intensity light irradiation). Figure 2 shows that the catalytic performance exhibited a notable correlation with the reaction temperature for both Au/1%CeO2/ZnO and Au/ZnO catalysts. For Au/1%CeO2/ZnO, with increasing reaction temperature, r(CO2) increased consistently, while the production rate of C2H6 r(C2H6) first underwent a linear increase (from 80 to 160 °C), followed by a gradual increase (160 to 200 °C), reaching a maximum at 200 °C, and ultimately decreasing with further increases in temperature (Fig. 2a). Likewise, Au/ZnO also exhibited an initial gradual increase in r(C2H6) followed by a decline with increasing reaction temperature (Fig. 2b), except that it obtained the maximum C2H6 production rate at a temperature at least 20 °C lower than that of Au/1%CeO2/ZnO. In sharp contrast, without light irradiation, no C2H6 production was detected on both Au/1%CeO2/ZnO and Au/ZnO, with the sole product being CO2 at temperatures exceeding 200 °C (Fig. 2c). This suggests that light completely alters the reaction pathways and is an indispensable factor for C2H6 production in POCM, consistent with our previous observations16. In addition, a similar trend in the dependence of performance on reaction temperature was observed under different light intensity (Fig. 2d), showing that an increase in light intensity led to an increase in r(C2H6) at a constant reaction temperature.

a, b Photocatalytic activity of Au/1%CeO2/ZnO (a) and Au/ZnO (b) at various temperatures and under 360 nm light irradiation with an intensity of 20 mW cm−2. c Catalytic activity of Au/1%CeO2/ZnO and Au/ZnO at various temperatures without light irradiation. d C2H6 production rate of Au/1%CeO2/ZnO at various temperatures and under 360 nm light irradiation with various intensity. e AQE for C2H6 production over Au/1%CeO2/ZnO and Au/ZnO at various temperatures. f Dependence of AQE for C2H6 production over Au/1%CeO2/ZnO on the incident light wavelength at 200 °C, together with the diffuse reflectance spectrum of Au/1%CeO2/ZnO for comparison. Reaction conditions: 197 mL min−1 of CH4 and 3 mL min−1 of air, 10 mg catalyst, a flow reactor that can provide external heating and allow illumination. Error bars represent standard deviations obtained from three independent measurements.
The favorable influence of heat on the production of C2H6 has a significant impact on the apparent quantum efficiency (AQE) of the reaction. The AQE with respect to the C2H6 production as a function of reaction temperature was measured (Fig. 2e and Supplementary Tables 4 and 5). The AQE of Au/1%CeO2/ZnO increased from 18.4% at 80 °C to a maximum of 38.7% at 200 °C, and the AQE of Au/ZnO increased from 10.9% at 80 °C to a maximum of 19.8% at 180 °C. At the same reaction temperature, the AQE of Au/1%CeO2/ZnO was higher than that of Au/ZnO, in line with the results of the pure photocatalytic process. For Au/1%CeO2/ZnO, the AQE measured without a secondary source of heating was 11.2%, which, to the best of our knowledge, is the highest value reported for POCM (Supplementary Table 2). Elevating the reaction temperature from room temperature to 200 °C provided a 3.5-fold increase in the AQE (38.7%), highlighting the critical role of heat in this process. The wavelength-dependent C2H6 production rate of Au/1%CeO2/ZnO was studied by measuring the AQE under different monochromatic light illumination at the optimized temperature of 200 °C (Fig. 2f and Supplementary Table 5). The calculated AQE aligned with the absorption curve of Au/1%CeO2/ZnO. In addition, there was a clear linear dependence of C2H6 production rate on 360 nm light intensity at 200 °C (Supplementary Fig. 6). These observations further suggest that the POCM reaction is driven by light-induced charge carriers through bandgap transitions in the semiconductor.
These results in Fig. 3 evidently show that within an appropriate temperature range, a higher reaction temperature leads to an increased C2H6 production rate, largely owing to less energy required at higher temperatures to overcome the activation barrier of the methane coupling reaction. Au/1%CeO2/ZnO performs more efficiently than Au/ZnO at elevated reaction temperatures and meanwhile achieves its optimal performance at a temperature 20 °C higher than that of Au/ZnO. The two factors act to yield a higher C2-3 production in POCM.

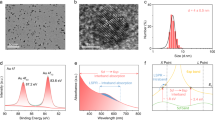
a–c, TEM (a), STEM (b) and HRTEM (c) images of 1%CeO2/ZnO. d–f, HRTEM (d), STEM (e), and the corresponding elemental mapping (f) images of Au/1%CeO2/ZnO. g XRD patterns of Au/1%CeO2/ZnO, 1%CeO2/ZnO, CeO2 and ZnO. h, i, The Ce LIII-edge XANES (h) and the Fourier transform EXAFS (i) spectra of Au/1%CeO2/ZnO and reference samples.
Structural characterizations
To explore the excellent performance of Au/1%CeO2/ZnO in POCM, we conducted a series of structural characterizations. Transmission electron microscopy (TEM) image (Fig. 3a) shows that the size of ZnO NPs was around 10–30 nm. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and high-resolution TEM (HRTEM) images (Fig. 3b, c and Supplementary Fig. 7) show that CeO2 NPs with an average diameter of 2.6 nm were formed on the ZnO surface. The obvious lattice spacings with distances of 0.281 and 0.325 nm were attributed to the ZnO (100) and CeO2 (111) facets, respectively. After loading Au NPs on 1%CeO2/ZnO, in addition to the Au/ZnO and CeO2/ZnO interface, Au/CeO2/ZnO triple interfaces were also observed (Fig. 3d, more images can be found in Supplementary Fig. 8). STEM images (Fig. 3e and Supplementary Fig. 9) show that the mean particle size of Au was 3.8 nm. The corresponding energy-dispersive X-ray elemental (EDX) mapping images (Fig. 3f and Supplementary Fig. 9) show that Au and Ce were well dispersed over Au/1%CeO2/ZnO; more importantly, most of the Au signals were overlapped with the Ce signals, which indicates that Au NPs were mainly in the vicinity of CeO2 NPs, further confirming the formation of Au/CeO2/ZnO triple interfaces. The synergistic interactions at the triple interface likely play an important role in enhancing the photocatalytic performance (Supplementary Fig. 10). Characterizations of the spent Au/1%CeO2/ZnO catalyst after 50 h of reaction show no noticeable carbon deposition on the surface of the catalyst with minor Au NPs sintering (Supplementary Figs. 11–13). The slight decrease in photocatalytic performance (Fig. 1d) is likely attributed to the minor sintering of Au NPs.
X-ray diffraction (XRD) patterns (Fig. 3g) of Au/1%CeO2/ZnO and 1%CeO2/ZnO show characteristic peaks of ZnO; no diffraction peaks assigned to CeO2 and/or Au were detected, probably owing to the small NP sizes and low loading amounts of CeO2 and Au. The Ce LIII−edge X-ray absorption near-edge structure (XANES) spectrum (Fig. 3h) of Au/1%CeO2/ZnO shows a white line profile with two main peaks (located at ca. 5731.4 and 5738.1 eV) that were similar to that of pure CeO2 reference39, suggesting that the structure of CeO2 remained in Au/1%CeO2/ZnO. In comparison with pure CeO2, the lower white line intensity and redshift edge position indicate a decrease in the Ce valence states. This suggests that the Ce species in Au/1%CeO2/ZnO were CeO2 phase with a slight reduction, namely, the co-existence of Ce4+ and Ce3+ with + 4 as the predominant oxidation state. The X-ray photoelectron spectroscopy (XPS) analysis further confirmed the presence of Ce4+ and Ce3+ in Au/1%CeO2/ZnO (Supplementary Fig. 14), and the ratio of Ce3+ to Ce4+ was estimated to be 21% based on the curve fitting of the spectrum40. As demonstrated by the Fourier transformed extended X-ray absorption fine structure (FT-EXAFS) spectra (Fig. 3i) of these samples, the two major peaks at ca. 1.83 and 3.58 Å were assigned to the first Ce–O shell scattering as well as the second Ce–Ce shell and third Ce–O shell scatterings in CeO239, respectively. The intensity of peak at 3.58 Å in the EXAFS spectrum of Au/1%CeO2/ZnO decreased obviously. In addition, on the basis of the fitting structural parameters (Supplementary Fig. 15 and Supplementary Table 6), the coordination number in the first Ce–O shell was 8 over pure CeO2, while the Ce–O coordination number decreased to 5.4 over Au/1%CeO2/ZnO. From these results, we can conclude that CeO2 is highly dispersed in Au/1%CeO2/ZnO with a small particle size, consistent with the STEM and HRTEM observations.
Mechanistic investigations for charge separation and reaction intermediates
To investigate the role of CeO2 in promoting photocatalytic performance, the dynamics of photo-generated electrons in the samples were evaluated. Au/1%CeO2/ZnO displayed drastically weaker photoluminescence (PL) intensity compared with pristine ZnO and Au/ZnO (Fig. 4a), confirming reduced recombination of separated photogenerated carriers. In line with this, femtosecond transient absorption (fs-TA) spectroscopy investigations show the photoexcited electrons transfer from ZnO to Au and CeO2 (Supplementary Fig. 16). The density of states (DOSs) shows that CeO2 created midgap states for capturing photo-excited electrons (Supplementary Fig. 17). These results validate that loading Au and CeO2 on ZnO could accelerate the transfer and separation of photoexcited electrons from ZnO. The photo-generated electron transfer was further investigated by in situ irradiated XPS. As shown in Fig. 4b and Supplementary Fig. 18, in the presence of O2 and CH4, the Au 4 f and Ce 3 d peaks evidently shifted to the low binding energy under light irradiation, which revealed that the photogenerated electrons on ZnO migrated to Au and CeO2 in photocatalytic CH4 oxidation, in good agreement with fs-TA analysis.

a PL spectra of ZnO, Au/ZnO and Au/1%CeO2/ZnO. b In situ ESR spectra of Au/1%CeO2/ZnO in the presence of O2 and the mixture of O2 and CH4 in the dark and under illumination. c Quasi in situ XPS spectra of Au 4 f for Au/1%CeO2/ZnO exposing to a gas mixture composed of 0.3 mbar CH4 and 0.015 mbar O2 in dark and under light irradiation.
In situ electron spin resonance (ESR) tests were further carried out to explore the charge separation and resulting generation of reaction intermediates during photocatalytic CH4 oxidation. Figure 4c shows that a symmetric ESR peak at g = 1.959 assigned to Zn+ species41 was observed on Au/1%CeO2/ZnO under illumination and exposure to O2, which was generated by the capture of photogenerated electrons by Zn2+. The intensity of this signal was weak compared with Au/ZnO (Supplementary Fig. 19), indicative of the transfer of photogenerated electrons from ZnO to CeO2 NPs. The ESR peaks at g = 2.001 and g = 2.020 were probably attributed to reaction oxygen species (O2– and/or O– species)41,42. O2– and O– species were produced through the consumption of photogenerated electrons by O2 and the trapping of photogenerated holes by the ZnO lattice oxygen, respectively. After the introduction of CH4, the intensity of the ESR signals corresponding to reactive oxygen species decreased clearly (Fig. 4c), revealing that the reaction of CH4 with reactive oxygen species occurred, demonstrating the central role of reactive oxygen species in activating methane. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements was employed to study the reaction process and intermediates of POCM. After exposure to the mixed gas of CH4 and O2, the peaks at 3015 and 1305 cm−1 were ascribed to the characteristic frequencies of gas-phase CH4, and the peak at 1540 cm−1 corresponded to the vibrations of adsorbed CH4 (Supplementary Fig. 20)43. Upon light irradiation, one set of bonds at 1474 and 1448 cm−1 assigned to the C–H deformation vibration of CH3* and two peaks at 1436 and 1418 cm−1 assigned to the CH2/CH3 deformation vibrations emerged (Supplementary Fig. 21)28,30. In addition, the peaks at 1686 and 1272 cm−1 attributed to the vibrational modes of CH2O* species, as well as the peaks at 1558 and 1376 cm−1 attributed to COOH* species were observed25,44,45,46. That is, three intermediate species, CH3*, CH2OH* and COOH* were detected, suggesting the potential reaction process of ethane formation and overoxidation in POCM (CH4 → CH3* → C2H6; CH4 → CH3* → CH2O → COOH → CO2).
Surface catalytic properties and kinetic studies
The O2 adsorption abilities and acidic properties of catalytic sites play important roles in governing catalytic activity47,48. To study the O2 adsorption abilities of catalysts, O2-temperature programmed desorption (O2-TPD) mass spectroscopy experiments were carried out. As shown in Fig. 5a, two distinct oxygen desorption peaks were observed. The first peak shown below 200 °C was assigned to chemisorbed oxygen species (Oads) weakly bonded on the surface of oxides47, potentially O2– as supported by ESR results. The second peak detected around 250-350 °C belonged to superficial active lattice oxygen species (Olatt)47,48. The Au/ZnO catalyst displayed a nearly negligible Oads desorption peak at temperatures below 200 °C, whereas the Au/1%CeO2/ZnO catalyst exhibited a broad Oads desorption peak in the temperature range of 100–200 °C, suggesting a higher concentration of Oads. Furthermore, the area of Olatt desorption peak on Au/1%CeO2/ZnO was larger than that on Au/ZnO, indicating the presence of more Olatt. These results reveal that the loading of CeO2 enhanced the oxygen adsorption ability of the catalyst, and thus Au/1%CeO2/ZnO contained a higher proportion of weakly bound oxygen species correlated with oxygen vacancies, which is conducive to the photocatalytic CH4 oxidation reaction.

a, b O2-TPD-MS profiles (a) and NH3-TPD-MS profiles (b) of Au/ZnO and Au/1%CeO2/ZnO. c, d Effects of CH4 (c) and O2 (d) partial pressure on photocatalytic CH4 conversion rate. Reaction condition: 20 mg photocatalyst, atmospheric pressure, 100 mL min−1 total flow rate balanced by N2. e, f Photocatalytic CH4 conversion rates on Au/ZnO and Au/1%CeO2/ZnO under (e) CH4/CD4 + O2 and f CH4 + 16O2/18O2. Reaction condition: 20 mg photocatalyst, 20 mL min−1 CH4/CD4, 1 mL min−1 16O2/18O2, 29 mL min−1 N2. Error bars represent standard deviations obtained from three independent measurements.
The acidic properties of the catalysts were characterized by NH3−TPD. Figure 5b shows that Au/ZnO and Au/1%CeO2/ZnO exhibited similar desorption features in a wide temperature range (100–500 °C) with two peaks centered at 150 and 400 °C, corresponding to NH3 desorption from weak and medium acid sites48,49, respectively. Clearly, the intensities of these two peaks on Au/1%CeO2/ZnO were higher than those on Au/ZnO, suggesting that the surface of Au/1%CeO2/ZnO was rich in acid sites. These acid sites on the surface of Au/1%CeO2/ZnO catalyst could improve the adsorption of CH4 and facilitate C − H bond cleavage, thereby contributing to enhanced activity in the photocatalytic CH4 oxidation reaction.
Kinetic measurements were conducted to obtain molecular-level understandings of the mechanism of CH4 photo-activation. Figure 5c and d show that the CH4 conversion rate increased with increasing O2 partial pressure (0–15 kPa), yet the dependence exhibited the beginning of a saturation trend at pressure >10 kPa, suggesting high surface coverage of O2 on Au/1%CeO2/ZnO. By contrast, the methane conversion rate shows a sharp increase with the CH4 partial pressure (0–70 kPa), which indicates that the CH4 partial pressure has a stronger effect on the CH4 conversion than the O2 partial pressure, likely owing to the weak adsorption of CH4 on catalyst surfaces caused by its intrinsic nonpolar and symmetrical geometry nature. Moreover, more CO2 was produced as the O2 partial pressure was increased, while more C2-3 was produced as the CH4 partial pressure was increased (Supplementary Fig. 22). These results reveal that the adsorption of CH4 and O2 are important steps in photocatalytic CH4 oxidation, and CH4 adsorption plays the predominant role.
In order to study the kinetics of photo-activation of CH4 and O2 on Au/1%CeO2/ZnO, we performed isotope labeling experiments using CD4 and 18O2. The kinetic isotope effect (KIE) value of Au/1%CeO2/ZnO was calculated as 2.12 for CH4 + O2 versus CD4 + O2, which was lower than that of Au/ZnO (2.33). While the KIE values of Au/1%CeO2/ZnO (1.03) and Au/ZnO (1.04) were quite similar for CH4 + 16O2 versus CH4 + 18O2. These observations manifest that C–H bond cleavage is involved in the rate-limiting step. Furthermore, it validates that the introduction of CeO2 species can enhance the photocatalytic CH4 conversion performance of ZnO and improve the C2-3 production rate because of the promotion of C–H bond activation in the reaction process.
Computational investigations on reaction mechanism
To understand the excellent photocatalytic CH4 oxidation performance of Au/1%CeO2/ZnO, we performed density functional theory (DFT) calculations for analyzing the key steps of CH4 oxidation to C2H6 on Au/ZnO and Au/1%CeO2/ZnO. Two structural models of cluster Au10/ZnO(101) and clusters Au10/Ce4O8/ZnO(101) were built to simulate Au/ZnO and Au/CeO2/ZnO catalysts (Supplementary Fig. 23). Gibbs free energy diagrams for the O2 dissociation to atomic O*, CH4 activation to CH3*, and the subsequent CH3* desorption to were simulated at 25 °C (see Supplementary Figs. 24–27 for detailed structures). The adsorption energies of O2 on Au10/ZnO(101) and Au10/Ce4O8/ZnO(101) were 32.6 and 12.0 kJ mol−1 (Fig. 6a), respectively. The O2 dissociation reaction on Au10/ZnO(101) and Au10/Ce4O8/ZnO(101) was exothermic by − 20.9 and − 88.6 kJ mol−1 with reaction barriers of 49.9 and 23.2 kJ mol−1, respectively. These results means that O2 is thermodynamically and kinetically favorable to dissociate on Au10/Ce4O8/ZnO(101) relative to Au10/ZnO(101), revealing the important role of CeO2 in facilitating O2 dissociation. This is probably because, compared with Au10/ZnO(101), more electrons transferred to O2 weaken and elongate the O−O bond on Au10/Ce4O8/ZnO(101) (Supplementary Fig. 28). Furthermore, the O2 dissociation occurs preferentially on Au rather than on CeO2, owing to the lower reaction energy and reaction barrier (Supplementary Fig. 29).

a, b Energy profiles for O2 activation (a) and CH4 activation and CH3 desorption (b) Au10/ZnO(101) and Au10/Ce4O8/ZnO(101) at 25 °C and 150 °C. The optimized structures of the transition state (TS) of CH4 activation and CH3 desorption on Au10/Ce4O8/ZnO(101) are shown inset in (b). * represents the catalytic site. (g) label represents gas phase. Gray, Zn; yellow, Ce; orange, Au; O in ZnO, red; H, white; brown, C. c Comparisons of the reaction barrier of O2 activation, CH4 activation, and CH3 desorption on Au10/ZnO(101) and Au10/Ce4O8/ZnO(101) at 25 °C and 150 °C. d The projected crystal orbital Hamilton population (pCOHP) plots of the H3C–Au bond on Au10/ZnO(101) and Au10/Ce4O8/ZnO(101). e Schematic illustration of the reaction mechanism of POCM on Au/CeO2/ZnO The projected crystal orbital Hamilton population (pCOHP) plots of H3C–Au bond on Au10/ZnO(101) and Au10/Ce4O8/ZnO(101).
Figure 6b illustrates the energy diagram of CH4 activation, in which CH4 was first chemisorbed on ZnO, followed by C–H bond cleavage to form CH3* and H*, and lastly, CH3* desorption. The activation barrier for the first C–H bond cleavage, a rate-limiting step in CH4 conversion, on Au10/ZnO(101) was 72.0 kJ mol−1. Interestingly, on Au10/Ce4O8/ZnO(101), the activation barrier can be greatly reduced to as low as 4.5 kJ mol−1, indicating that the first C–H bond of CH4 could be easily broken by Au and CeO2-decorated ZnO catalyst, which is consistent with KIE results. We note that such a low reaction barrier obtained is attributed to the migration of an Au atom adjacent to the Au10/Ce4O8 interface during the C–H bond activation (the migrated Au atom is marked in the red circle in the inset of Fig. 6b). This Au atom moved away from the Au10 cluster to promote C–H bond cleavage by accepting CH3 and subsequently reintegrated into the Au10 cluster after CH4 dissociation. This dynamic behavior under reaction conditions is in line with the previously observed dynamic formation of single-atom Au catalytic active sites on Au/CeO2 interface50. The desorption of CH3 species, a key step for C2H6 formation, undergoes two steps; CH3 species are initially transferred from the Au atom adjacent to ZnO to the top Au atom, and then the transferred CH3 species desorb in the gas phase to form CH3• radicals, which can couple to generate C2H6. The maximum reaction energy for these two steps on Au10/Ce4O8/ZnO(101) (70.0 kJ mol−1) was lower than that on Au10/ZnO(101) (105.6 kJ mol−1), suggesting that CH3 desorption is thermodynamically easier to take place on Au10/Ce4O8/ZnO(101). The enhanced CH3 desorption is attributed to the increased occupancy of the antibonding orbitals associated with C–Au bonds (Fig. 6c), showing a weakened interaction strength of CH3 absorbed on Au. This likely arises from the greater electron transfer from Au to CeO2 (Supplementary Fig. 30).
To clarify the role of heat in the enhanced catalysis, the corresponding Gibbs free energies were also calculated at a high temperature of 150 °C, at which highest C2-3 production rates were observed on Au/ZnO and Au/1%CeO2/ZnO (Fig. 1e). It is found that elevating the temperature led to higher adsorption energies of O2*/CH4* and O* + O*/CH3* + H* because of stronger molecular vibrations at high temperature. Although this temperature variation did not significantly affect the reaction energies of O2 and CH4 dissociation, the reaction energies for CH3 desorption on Au10/Ce4O8/ZnO(101) and Au10/ZnO(101) were reduced by ~27.7% (from 70.0 to 50.6 kJ mol−1) and 19.9% (from 105.6 to 84.6 kJ mol−1), respectively. As summarized in Fig. 6d, the CeO2 modification plays a key role in reducing the reaction barriers for CH4 and O2 activation at both 25 °C and 150 °C, while higher reaction temperature primarily lowers the reaction barrier for CH3 desorption. The main contribution of photo-induced heating is to promote CH3 desorption, thereby enhancing C2H6 production. In addition, the reaction barriers for *CH3 species reacting with *O or *O2 are substantially higher than the desorption energy of *CH3, revealing that the desorption is more favorable than reaction with either *O or O2 on the Au/CeO2/ZnO system (Supplementary Fig. 31). Therefore, compared with Au/ZnO, reduced reaction barriers for O2 and CH4 dissociation on Au/CeO/ZnO, along with lower reaction energy for CH3 desorption at an elevated temperature, are expected to be responsible for the enhancement of POCM activity.
In light of these results, a reaction mechanism for the photochemcially-triggered and photothermally-enhanced OCM on Au/CeO2/ZnO is proposed (Fig. 6e). ZnO absorbs UV light to excite electrons and holes, while Au NPs serve as a photothermal material, absorbing visible and NIR light through LSPR to transform light energy into heat, thereby elevating the photocatalyst temperature. Electrons are transferred to Au and CeO2 to participate in O2 reduction, leaving long-lived holes on ZnO to activate CH4. Due to the unique properties of Au, CH4 is readily and selectively dissociated into CH3 species, which subsequently absorb onto the Au NPs surface. These CH3 species then desorb to produce •CH3 radicals that undergo homogeneous coupling in the gas phase to form C2H6. In situ ESR experiments demonstrate the formation of •CH3 radicals (Supplementary Fig. 32). The formation of other hydrocarbons follows a similar coupling mechanism. For instance, the production of C3H8 occurs through the coupling of CH4 and C2H6. Both CH4 and C2H6 were activated by reaction oxygen species (O2– and O–), wherein the O2– and O– species were generated through the reduction of O2 by photogenerated electrons and the trapping of photogenerated holes by the lattice oxygen of ZnO, respectively51. The Au NPs bound to CeO2 clusters can substantially promote the activation and conversion of CH4 and O2 by decreasing the intrinsic barriers for dissociation of the first C–H bond in CH4 and O–O bond in O2, mostly arising from the cooperative interactions between Au and CeO2, which is responsible for the boosted POCM performance on Au/CeO2/ZnO. Moreover, the local heating at the Au surface generated from the LSPR effect of Au greatly facilitates the desorption of •CH3 radicals for subsequent C–C coupling before further undesired overoxidation takes place. Therefore, the photo-induced charge carriers from ZnO guarantee the occurrence of POCM, while the photo-induced heating accelerates the POCM reaction. The photochemical effect works synergistically with the photothermal effect to enhance the sunlight-driven photocatalytic production of C2-3 under full solar spectrum irradiation.
Discussion
In summary, we have demonstrated that optimizing the interplay of photochemical catalysis and photothermal catalysis can remarkably enhance C2+ hydrocarbon production in photo-driven OCM reactions without a secondary source of heating. ZnO-supported Au and CeO2 photocatalysts were found to be capable of utilizing the full solar-light spectrum to transform CH4 into C2+ hydrocarbons with O2. A series of in situ techniques reveal that Au and CeO2 can capture photogenerated electrons from the excitation of ZnO by UV light for accelerating O2 activation. Meanwhile, the synergistic effect of Au and CeO2 considerably promotes CH4 activation by reducing the first C–H bond dissociation reaction barrier, thus facilitating the rate-determining step, as evidenced by KIE studies and DFT calculations. The heat generated by Au NPs with visible and NIR light irradiation could contribute to the facile desorption of the produced •CH3 radicals to form C2H6 before undesired overoxidation occurs. Consequently, the synergistic interplay of photochemical and photothermal effects resulted in a substantial improvement in C2+ yield and selectivity. The results presented in this work not only offer a highly selective and efficient photocatalytic system for direct oxidation of CH4 to C2+ hydrocarbons with O2 but also pave the way for the high-efficiency utilization of the full solar spectrum to drive catalytic reactions with enhanced activity and selectivity.
Methods
Catalyst synthesis
Zn(SO4)2·6H2O (99%), Ce(NO3)3·6H2O (99%), Na2CO3 (99.5%), NaBH4 (98%), HAuCl4·3H2O (99.9%) were purchased from Shanghai Aladdin Bio-Chem Technology Co., LTD. CeO2/ZnO mixed oxide catalysts were synthesized by a facile coprecipitation method. Typically, 6.5 g of Zn(SO4)2·7H2O and a calculated amount of Ce(NO3)3·6H2O were dissolved in 80 ml of deionized water. Then, 60 ml of Na2CO3 aqueous solution (1 M) was added dropwise to the aforementioned solution drop by drop. The suspension was continuously stirred for 3 h, followed by centrifugating and washing with deionized water for 10 times. Then the product was dried at 80 °C under vacuum for 10 h and calcined at 350 oC in the air for 4 h. Finally, the obtained CeO2/ZnO mixed oxide catalyst was denoted as x%CeO2/ZnO, x% representing the molar percentage of Ce. The CeO2/ZnO catalysts with different molar ratios of Ce/(Ce+Zn) were obtained by tuning the amount of Ce(NO3)3·6H2O. Specifically, 0.5%CeO2/ZnO, 1%CeO2/ZnO, 3%CeO2/ZnO, and 10%CeO2/ZnO were prepared by adding 0.05 g, 0.1 g, 0.31 g and 1.09 g of Ce(NO3)3·6H2O, respectively. Pure ZnO and CeO2 were obtained using a similar method except without adding Ce(NO3)3·6H2O and Zn(SO4)2·7H2O, respectively. Au NPs were loaded on x% CeO2/ZnO via a facile NaBH4 reduction method, and the obtained samples were denoted as Au/x%CeO2/ZnO. In this manuscript, unless otherwise specified, the amount of Au in Au/x% CeO2/ZnO is 1.5 wt%. For comparison, an Au/CeO2/ZnO sample with weaker interface interactions was also prepared. Au NPs were first deposited on ZnO by the same NaBH4 reduction method, followed by impregnation with an aqueous Ce(NO3)3 solution, drying at 60 °C for 12 h, and calcining in air at 300 °C for 2 h. The obtained sample was denoted as Au/CeO2/ZnO-IMP. The loading amounts of Au and Ce was 1.6 wt% and 1.5 wt%, respectively, as determined by ICP-OES.
Catalytic testing
The photocatalytic activity tests of the samples for the OCM reaction were performed in a continuous-flow reactor equipped with a quartz window for illumination. 20 mg of photocatalyst was uniformly placed on the sample holder. The reactant gas mixture with a CH4/synthetic air (20 vol.% O2/N2) ratio of 197/3 was flowed into the reactor. The gas flow rate was kept constant at 200 mL min−1. Prior to the reaction, the reactant gas was introduced into the reactor for at least 1 h. The light from a 300 W Xenon lamp or an AM 1.5 light source was focused into the reactor, and the light output intensity was tuned by adjusting the lamp current. A K-type thermocouple was inserted at the center of the catalyst for measuring the temperature of the irradiated catalyst during the reaction. The effluents were analyzed by a Shimadzu GC-2014 gas chromatograph (GC) equipped with a flame ionization detector (FID) and methanizer.
The CH4 conversion and C2-3 selectivity were calculated using Eqs. 1 and 2, respectively.
The catalytic performance for OCM at elevated temperatures and with or without illumination was conducted in a homemade flow reactor. 10 mg of samples were uniformly dispersed in the reaction chamber. Light irradiation provided by an LA-251 Xe lamp equipped with a 360 nm monochromatic filter was introduced into the reactor through a quartz window with an irradiation diameter of 8.5 mm. The light intensity was tuned by adjusting the lamp current. The temperature of the sample was maintained at the desired temperature by a resistive heater equipped with a temperature controller, and the photo-induced heating was alleviated. The feed gas of CH4 and air (CH4/air, 197/3 v/v) was introduced into the reaction chamber with a space velocity of 200 mL min−1. Products were quantified by a Shimadzu GC-2014 GC equipped with FID and methanizer.
Catalyst characterizations
Powder XRD data was collected using a Rigaku Ru-200b X-ray diffractometer operated at 40 kV and 100 mA equipped with Cu Kα radiation (λ = 1.5418 Å). TEM and HRTEM images were obtained on a Hitachi Model H-800 microscope. HAADF-STEM images and elemental mapping were performed on a JEOL ARM-200F microscope. The contents of Au and Ce in the samples were determined using inductively coupled plasma-optical emission spectrometry (ICP-OES, Thermo Scientific iCAP PRO). UV-visible diffuse reflectance spectra were acquired by a Shimadzu 3600 spectrophotometer. The surface area was measured by the BET method using an analyzer (QUADRASORB SI). XAS measurement for the Ce LIII-edge was collected in fluorescence mode at beamline BL14W1 of the Shanghai Synchrotron Radiation Facility. The XANES and EXAFS data analysis were conducted using the Athena software. XPS measurements were conducted using a Thermo Scientific Escalab 250Xi spectrometer, with binding energies referenced by setting the carbon 1 s peak to 284.8 eV. O2-TPD and NH3−TPD measurements was performed on an Auto Chem II 2920 system (Micromeritics Instrument Corp.). fs-TA spectra were acquired using a Spectra-Physics Spitfire Ace femtosecond amplifier system. This system generated 35 fs laser pulses centered at 800 nm with a 1 kHz repetition rate. A pellet was prepared for measurement by thoroughly mixing the sample with KBr powder. NAP XPS analysis was carried out with a SPECS NAP-XPS system. The SPECS μ-FOCUS 600 NAP X-ray source provided monochromatic Al Kα radiation (1486.6 eV). The chamber was filled with a gas mixture consisting of 0.3 mbar CH₄ and 0.015 mbar O₂. Following the initial XPS data collection, the sample was subjected to Xe lamp exposure, and subsequent XPS spectra were obtained to monitor changes in the sample’s electronic structure. In situ ESR spectra were obtained using a JEOL JES-FA200 spectrometer. Before measurement, the sample was subjected to evacuation at 120 °C for 2 h. The ESR spectra were collected at a temperature of 120 K in O2 or a mixture of CH4 and O2 gases under dark and under light irradiation. In situ, DRIFTS measurements were performed using a Nicolet 6700 spectrometer equipped with a conventional IR cell coupled to a gas handling system. Prior to measurement, the sample was subjected to a heat treatment at 150 °C to eliminate adsorbed water and surface contaminants. All spectra were recorded under dark and light irradiation within a reactor fed with a CH4:O₂ mixture (197:3) at a flow rate of 200 mL min−1.
Computational details
All spin-polarized theoretical simulations were performed using the Kohn–Sham formalism with the generalized gradient approximation (GGA) and Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional, as implemented in the VASP code.52,53 The plane-wave basis sets of 400 eV cutoff kinetic energy to approximate the valence electron densities and the projector augmented wave (PAW) method to account for the core−valence interaction.54 The Γ-point approximation was employed for the Brillouin zone integration because of the significant size of the supercell. All the structures were relaxed until the forces on each ion were less than 0.05 eV/ Å, and the convergence criterion for energy was set as 10−5 eV. Gaussian smearing was used for the electronic states of our models, and the width was set to 0.05 eV. The DFT + U method was used by applying the Hubbard terms on the Zn 3 d (U = 5.0 eV) and Ce 4 f (U = 4.5 eV) states. The DFT-D3 method of Grimme was also used to describe van der Waals interactions,55 to obtain a better description of the interactions between molecules. The transition states during the reaction pathway were evaluated by the climbing-image nudged elastic band (CI-NEB) combined with minimum-mode following dimer method.56,57,58 All the transition state structures were identified by vibrational frequency analysis. A wurtzite ZnO(101)-p(3 × 6) surface slab with three Zn–O layers was used to model the ZnO substrate, only the bottom one layer is was kept frozen. This surface model contains 108 Zn and 108 O atoms with a 20 Å vacuum layer, and the lattice parameters were a = 18.3661 Å, b = 19.3714 Å, c = 25.8664 Å, α = β = 90°, γ = 105.29°. Such a large model is enough to accommodate two clusters, Ce4O8 and Au10, and reduce the interactions between neighboring mirrors of clusters. The two optimized computational models, Au10/ZnO(101) and Au10/Ce4O8/ZnO(101), are provided in Supplementary Fig. 23.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files. Source data are provided as a source data file with this paper and are available from the corresponding authors upon request. Source data are provided in this paper.
References
Tang, Y., Li, Y. & Tao, F. Activation and catalytic transformation of methane under mild conditions. Chem. Soc. Rev. 51, 376–423 (2022).
Freakley, S. J. et al. Methane oxidation to methanol in water. Acc. Chem. Res. 54, 2614–2623 (2021).
Ravi, M., Ranocchiari, M. & van Bokhoven, J. A. The direct catalytic oxidation of methane to methanol—A critical assessment. Angew. Chem. Int. Ed. 56, 16464–16483 (2017).
Schwach, P., Pan, X. & Bao, X. Direct conversion of methane to value-added chemicals over heterogeneous catalysts: Challenges and prospects. Chem. Rev. 117, 8497–8520 (2017).
Blankenship, A., Artsiusheuski, M., Sushkevich, V. & van Bokhoven, J. A. Recent trends, current challenges and future prospects for syngas-free methane partial oxidation. Nat. Catal. 6, 748–762 (2023).
Arinaga, A. M., Ziegelski, M. C. & Marks, T. J. Alternative oxidants for the catalytic oxidative coupling of methane. Angew. Chem. Int. Ed. 60, 10502–10515 (2021).
Wortman, J., Igenegbai, V. O., Almallahi, R., Motagamwala, A. H. & Linic, S. Optimizing hierarchical membrane/catalyst systems for oxidative coupling of methane using additive manufacturing. Nat. Mater. 22, 1523–1530 (2023).
Farrell, B. L., Igenegbai, V. O. & Linic, S. A viewpoint on direct methane conversion to ethane and ethylene using oxidative coupling on solid catalysts. ACS Catal. 6, 4340–4346 (2016).
Song, H. & Ye, J. Direct photocatalytic conversion of methane to value-added chemicals. Trends Chem. 4, 1094–1105 (2022).
Li, X., Wang, C. & Tang, J. Methane transformation by photocatalysis. Nat. Rev. Mater. 7, 617–632 (2022).
Song, H., Meng, X., Wang, Z.-j, Liu, H. & Ye, J. Solar-energy-mediated methane conversion. Joule 3, 1606–1636 (2019).
An, B. et al. Direct photo-oxidation of methane to methanol over a mono-iron hydroxyl site. Nat. Mater. 21, 932–938 (2022).
Fan, Y. et al. Selective photocatalytic oxidation of methane by quantum-sized bismuth vanadate. Nat. Sustain. 4, 509–515 (2021).
Zhai, G. et al. Highly efficient, selective, and stable photocatalytic methane coupling to ethane enabled by lattice oxygen looping. Sci. Adv. 10, eado4390 (2024).
Li, X. et al. Efficient hole abstraction for highly selective oxidative coupling of methane by Au-sputtered TiO2 photocatalysts. Nat. Energy 8, 1013–1022 (2023).
Song, S. et al. A selective Au-ZnO/TiO2 hybrid photocatalyst for oxidative coupling of methane to ethane with dioxygen. Nat. Catal. 4, 1032–1042 (2021).
Yu, X. et al. Stoichiometric methane conversion to ethane using photochemical looping at ambient temperature. Nat. Energy 5, 511–519 (2020).
Han, C. et al. Selective ceavage of chemical bonds in targeted intermediates for highly selective photooxidation of methane to methanol. J. Am. Chem. Soc. 145, 8609–8620 (2023).
Jiang, Y. et al. Elevating photooxidation of methane to formaldehyde via TiO2 crystal phase engineering. J. Am. Chem. Soc. 144, 15977–15987 (2022).
Song, H. et al. Direct and selective photocatalytic oxidation of CH4 to oxygenates with O2 on cocatalysts/ZnO at room temperature in water. J. Am. Chem. Soc. 141, 20507–20515 (2019).
Olivos-Suarez, A. I. et al. Strategies for the direct catalytic valorization of methane using heterogeneous catalysis: Challenges and opportunities. ACS Catal. 6, 2965–2981 (2016).
Cui, X. et al. Photothermal nanomaterials: A powerful light-to-heat converter. Chem. Rev. 123, 6891–6952 (2023).
Montini, T., Melchionna, M., Monai, M. & Fornasiero, P. Fundamentals and catalytic applications of CeO2-based materials. Chem. Rev. 116, 5987–6041 (2016).
Yu, D. et al. Solar photocatalytic oxidation of methane to methanol with water over RuOx/ZnO/CeO2 nanorods. ACS Sustain. Chem. Eng. 10, 16–22 (2022).
Zhang, J. et al. Regulation of oxygen activation pathways to optimize photocatalytic methane oxidative coupling selectivity. ACS Catal. 14, 3855–3866 (2024).
Chen, Y. et al. Continuous flow system for highly efficient and durable photocatalytic oxidative coupling of methane. J. Am. Chem. Soc. 146, 2465–2473 (2024).
Wang, C. et al. Synergy of Ag and AgBr in a pressurized flow reactor for selective photocatalytic oxidative coupling of methane. ACS Catal. 13, 3768–3774 (2023).
Jiang, W. et al. Pd-modified ZnO–Au enabling alkoxy intermediates formation and dehydrogenation for photocatalytic conversion of methane to ethylene. J. Am. Chem. Soc. 143, 269–278 (2021).
Li, X., Xie, J., Rao, H., Wang, C. & Tang, J. Platinum- and CuOx-decorated TiO2 photocatalyst for oxidative coupling of methane to C2 hydrocarbons in a flow reactor. Angew. Chem. Int. Ed. 59, 19702–19707 (2020).
Zhang, W. et al. High-performance photocatalytic nonoxidative conversion of methane to ethane and hydrogen by heteroatoms-engineered TiO2. Nat. Commun. 13, 2806 (2022).
Wang, G. et al. Light-induced nonoxidative coupling of methane using stable solid solutions. Angew. Chem. Int. Ed. 60, 20760–20764 (2021).
Wu, S. et al. Ga-doped and Pt-loaded porous TiO2–SiO2 for photocatalytic nonoxidative coupling of methane. J. Am. Chem. Soc. 141, 6592–6600 (2019).
Meng, L. et al. Gold plasmon-induced photocatalytic dehydrogenative coupling of methane to ethane on polar oxide surfaces. Energy Environ. Sci. 11, 294–298 (2018).
Li, L. et al. Synergistic effect on the photoactivation of the methane C-H bond over Ga3+‐modified ETS‐10. Angew. Chem. Int. Ed. 51, 4702–4706 (2012).
Li, L. et al. Efficient sunlight‐driven dehydrogenative coupling of methane to ethane over a Zn+‐modified zeolite. Angew. Chem. Int. Ed. 50, 8299–8303 (2011).
Yuliati, L., Hamajima, T., Hattori, T. & Yoshida, H. Highly dispersed Ce (III) species on silica and alumina as new photocatalysts for non-oxidative direct methane coupling. Chem. Commun. 38, 4824–4826 (2005).
Singh, S. P. et al. A Pd-Bi dual-cocatalyst-loaded gallium oxide photocatalyst for selective and stable nonoxidative coupling of methane. ACS Catal. 11, 13768–13781 (2021).
Song, H. et al. Light-enhanced carbon dioxide activation and conversion by effective plasmonic coupling effect of Pt and Au nanoparticles. ACS Appl. Mater. Interfaces 10, 408–416 (2018).
Fang, J. et al. Interfacial and surface structures of CeO2−TiO2 mixed oxides. J. Phys. Chem. C. 111, 19078–19085 (2007).
Sakpal, T. & Lefferts, L. Structure-dependent activity of CeO2 supported Ru catalysts for CO2 methanation. J. Catal. 367, 171–180 (2018).
Qi, G. et al. Low-temperature reactivity of Zn+ ions confined in ZSM-5 zeolite toward carbon monoxide oxidation: Insight from in situ DRIFT and ESR spectroscopy. J. Am. Chem. Soc. 135, 6762–6765 (2013).
Kumar, C. P., Gopal, N. O., Wang, T. C., Wong, M.-S. & Ke, S. C. EPR Investigation of TiO2 nanoparticles with temperature-dependent properties. J. Phys. Chem. B 110, 5223–5229 (2006).
Wu, Y., Hu, Q., Chen, Q., Jiao, X. & Xie, Y. Fundamentals and challenges of engineering charge Polarized active sites for CO2 photoreduction toward C2 products. Acc. Chem. Res. 56, 2500–2513 (2023).
Wu, J. et al. Regulated photocatalytic CO2-to-CH3OH pathway by synergetic dual active sites of interlayer. J. Am. Chem. Soc. 146, 26478–26484 (2024).
Bera, P. & Vohs, J. M. Reaction of CH3OH on Pd/ZnO(0001) and PdZn/ZnO(0001) model catalysts. J. Phys. Chem. C. 111, 7049–7057 (2007).
He, Y. et al. In situ identification of reaction intermediates and mechanistic understandings of methane oxidation over hematite: A combined experimental and theoretical study. J. Am. Chem. Soc. 142, 17119–17130 (2020).
Meng, Q. et al. Active oxygen species in Lan+1NinO3n+1 layered perovskites for catalytic oxidation of toluene and methane. J. Phys. Chem. C. 120, 3259–3266 (2016).
Huang, Z. et al. Highly efficient oxidation of propane at low temperature over a Pt-based catalyst by optimization support. Environ. Sci. Technol. 56, 17278–17287 (2022).
Yang, J. et al. Oxygen vacancies and Lewis acid sites synergistically promoted catalytic methane combustion over perovskite oxides. Environ. Sci. Technol. 55, 9243–9254 (2021).
Sun, J.-J. & Cheng, J. Solid-to-liquid phase transitions of sub-nanometer clusters enhance chemical transformation. Nat. Commun. 10, 5400 (2019).
Wang, P. et al. Photocatalytic ethylene production by oxidative dehydrogenation of ethane with dioxygen on ZnO-supported PdZn intermetallic nanoparticles. Nat. Commun. 15, 789 (2024).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, https://doi.org/10.1063/1.3382344 (2010).
Henkelman, G. & Jónsson, H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 111, 7010–7022 (1999).
Henkelman, G. & Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 113, 9978–9985 (2000).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 22372118, 22002060, U23A20139 and 22378017), the State Key Laboratory of Clean Energy Utilization (Open Fund Project No. ZJUCEU2023018), the State Key Laboratory of Silicon and Advanced Semiconductor Materials (Open Fund Project No. SKL2023-09), the State Key Laboratory of Chemical Resource Engineering (Open Fund Project No. CRE-2023-03-01), and the Young Scientists Fund of the Natural Science Foundation of Tianjin (No. 23JCQNJC01790), the technical support for Nano-X from Suzhou Institute of Nano-Tech and Nano-Bionics, Chinese Academy of Sciences (SINANO).
Author information
Authors and Affiliations
Contributions
H.S. conceived the ideas, performed the catalysts synthesis, characterization, and photocatalytic tests, and wrote the manuscript. K.S. conducted ESR measurement. H.H. conducted DFT calculations. S.N. carried out the in situ DRIFTS experiment. S.W. performed the XAFS measurement. Z.W. and Y.W. performed fs-TA measurements. Y.C. and Y.L. performed quasi in situ XPS measurements. X.W. conducted TEM measurement. D.W. and L.L. helped with the data analysis. Z.-j.W. and J.Y. supervised the research and co-wrote the paper. All authors contribute to the discussion and revision of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, H., Sun, K., Huang, H. et al. Integrating photochemical and photothermal effects for selective oxidative coupling of methane into C2+ hydrocarbons with multiple active sites. Nat Commun 16, 2831 (2025). https://doi.org/10.1038/s41467-025-58101-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58101-0
This article is cited by
-
Formation of charge-polarized regions at dual single-atom sites for C-H bond activation in methane
Nature Communications (2026)
-
Orchestrated multi-physics field-engineering toward valorized C2+ chemicals from CO2/CH4
Science China Materials (2026)
