
Abstract
Chiral scaffolds are essential to the advancement of asymmetric synthesis, yet the development of privileged motifs that more effectively communicate asymmetry constitutes a grand challenge for chemists. Here we describe a method using a confined chiral Brønsted acid catalyst to combine two inexpensive and widely available materials—indole and acetone—into a class of C₂-symmetric, spirocyclic compounds called SPINDOLE. SPINDOLEs extend the versatility of established frameworks by offering greater flexibility and ease of synthesis. The resulting chiral compounds can be readily modified to create diverse structures that excel in promoting highly selective reactions such as hydrogenation, allylic alkylation, hydroboration, and Michael addition. This work introduces a powerful strategy for advancing asymmetric catalysis, enabling the creation of versatile chiral frameworks with broad synthetic potential.
Similar content being viewed by others

Introduction
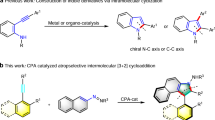
1,1’-Binaphthols (BINOLs)1,2,3 are widely regarded as “privileged” scaffolds4,5 in asymmetric catalysis due to their inherent axial chirality, modular design, and straightforward synthesis from inexpensive 2-naphthol6. 1,1’-Spirobisindane−7,7’-diols (SPINOLs) represent a conceptual synthetic evolution of BINOLs, incorporating a spirocyclic architecture that offers increased rigidity and stability7. These factors render SPINOL as an equally compelling platform for the development of chiral catalysts and ligands, providing solutions to complex problems in asymmetric chemistry8,9. Although their performance is notable, the study of SPINOLs has been impeded by their high cost and the paucity of efficient chemical synthesis methods10. Fundamental challenges arise in the key synthesis step that involves the two-fold intramolecular Friedel-Crafts alkylation-induced spirocyclization. The inherent site selectivity and low reactivity of this process necessitate additional steps to implement directing group strategies and the use of large amounts of hazardous acids (Fig. 1A). Moreover, the development of sufficiently active chiral catalysts capable of inducing asymmetry at the spiro quaternary carbon stereocenter while tolerating diverse electronic and steric functional groups are rare.

A The key synthesis step of forming spirocyclic architectures from phenol and naphthol is fundamentally challenging due to inherent site selectivity and low reactivity. These issues often require additional steps, such as employing directing group strategies and using large amounts of hazardous acids. B Strategies to achieve intramolecular spirocyclization of phenols include (i) Enhanced reactivity observed with ketals, (ii) A strategy involving 2-naphthol rather than phenol, (iii) Spirocyclization using Ir-catalyzed pre-formed chiral ketones, and (iv) Spirocyclization employing Rh-catalyzed pre-formed chiral ketones. C The construction of 1,1’-spirobisindane-6,6’-diols has also been explored by rearranging bisphenols formed via the condensation of phenols and acetone under acidic conditions. D Construction of Spiro-bisindoles with strategies as applied to SPINOLs. E In this work, a simpler approach is introduced using a confined chiral Brønsted acid-catalyzed process with readily available and inexpensive indole and acetone. This method provides a direct route to C2-symmetric spirocycles, addressing the thermodynamic challenges of bis-indole structures, as demonstrated by relative energy data.
Approaches using asymmetric catalysis are typically lengthy, involving specialized starting materials that necessitate multistep syntheses, complex catalysts, and excessive hazardous reagents (Fig. 1Bi, ii)11,12. These factors contribute to significant synthesis costs, thereby limiting their widespread application. Substrate-controlled enantioselectivity is well precedented in diastereoselective additions to pre-formed chiral ketones (Fig. 1Biii, iv)13,14. An alternative approach involves rearranging bisphenols formed via the condensation of phenols and acetone under acidic conditions (Fig. 1C)15,16. While the abundant and inexpensive supply of the starting materials render this strategy intuitively attractive, the laborious post-synthesis derivatization and harsh conditions limit its practicality for scalable catalytic integration.
By altering the primary aromatic constituent within the spirocyclic structure, we considered whether these synthetic approaches could be united in a fundamentally unique way. We envisaged that indoles should, in principle, constitute excellent substrates, as the inclusion of the ring nitrogen would be expected to offer simpler syntheses and derivative approaches, while retaining the desirable features for catalysis. To that extent, these heteroaromatic substrates not only react efficiently with acetone but also exhibit inherent site-selectivity that eliminate the need for post-synthesis modifications. During the course of our study, the Tan17 and Sun18 group reported strategies as applied to SPINOLs to prepare spiro‐bisindoles respectively (Fig. 1D), highly supporting our envisioned approach (Fig. 1E).
In this context, indole has been applied to the Brønsted acid-induced condensation of acetone on a few occasions; however, the major products of these reactions are bis(indolyl)propanes19 and cyclopenta[b]indoles20,21, with the spirocycle occurring in trace amounts20. As little work on this transformation had been previously described, we performed preliminary calculations to recognize any potential challenges that may be responsible for its difficulty. Quantum mechanical (QM) calculations predict that this reaction has a complex thermodynamic profile, with as little as 2.2 kcal/mol separating the bis-indole structures (see Supplementary pages 4–5 for details). Consequently, selective formation of the desired spirocyclic product requires a catalyst capable of suppressing competing low-energy pathways and destabilizing unwanted intermediates or side products. We reasoned that a chiral catalyst with carefully optimized steric and electronic properties could achieve this by selectively stabilizing the target product and enabling high enantiocontrol through noncovalent interactions (Fig. 1E).
Inspired by the profound structural diversity of the potential product structures ranging from achiral, planar forms to chiral structures with varied spatial configuration and key synthetic studies on chiral Brønsted acids (BAs) in selective catalysis, we initially hypothesized that that phosphoric acids (PAs) with phenyl-derived substituents in the 3,3’-positions, in particular, could offer the necessary shape complementarity to effectively stabilize the targeted structure. Moreover, the extensive availability of established or custom-designed PAs with well-defined chiral pockets provides a robust platform for further catalyst structure optimization22. We herein report that these fundamental acid-mediated transformations between indole and acetone can be expanded to produce spirocyclic bis-indoles (hereafter referred to as SPINDOLEs) in high levels of enantioselectivity, enabled by a confined BA catalyst featuring unique aromatic groups.
Results
Reaction optimization
Brønsted acids (BAs) are user-friendly catalysts, valued for their stability in the presence of oxygen and water, as well as their long-term storability. From a practical standpoint, BAs are environmentally benign and well-suited for large-scale synthesis, making them an appealing chemotype for exploration23,24. Since their pioneering application by Terada and Akiyama25,26, chiral phosphoric acids (CPAs) have proven to be particularly effective BA catalysts, especially in reactions involving basic electrophiles and protic nucleophiles27. CPAs have been widely applied in enantioselective transformations such as Friedel-Crafts alkylations28,29 and Pictet-Spengler reactions30, which are particularly relevant to our system under study. Their catalytic effectiveness derives from their ability to form multiple hydrogen bonds with substrates and their modular, tunable frameworks that enable precise enantioselective control in previously challenging reaction spaces. These features, combined with their established success across diverse reaction types, ultimately motivated their use as a starting point for catalyst optimization in this work.
As part of our initial screening efforts, we discovered that the reaction between indole and acetone at 80 °C in toluene with CPA A7 catalyzed the formation of the targeted spirocycle 4a, achieving modest yield but encouraging enantioselectivity 56% ee (Fig. 2). To explore the impact of catalyst properties, we evaluated an expanded inventory of CPAs with varied 3,3’-substituents, which influence both steric effects and pKa. A clear trend emerged: stronger CPAs (lower pKa values) improved transformation efficiency (see Supplementary Fig. S1). However, the formation of the by-product cyclopenta[b]indole 3a occurred to varying degrees and appeared unavoidable with this catalyst class. Notably, CPA A14, previously shown by Tan and Sun to deliver optimal performance for the asymmetric synthesis of SPINOL11 and SPHENOL12, did not exhibit efficient reactivity or selectivity in our system. Marked improvements in yield were obtained using N-triflyl phosphoric acids (NPAs), which possess lower pKa values31. Under these stronger acidic conditions, the undesired cyclopenta[b]indole 3a was virtually eliminated, although enantioselectivity did not improve. This outcome suggests that the higher proton availability of NPAs plays a critical role in driving spirocycle 4a formation. Ultimately, selective access to either cyclopenta[b]indole 3a or targeted spirocycle 4a can be effectively controlled by tuning the Brønsted acid structure based on pKa.

The reaction of 1a (0.1 mmol), acetone (0.5 mmol), and stated amount of catalysts was carried out in 0.5 ml of toluene in a 4 mL heavy wall vessel. The vessel was sealed and stirred at the stated temperature for 3 days (more details see Supplementary Figs. S1–S5). Isolated yields are shown. The ee values were determined by chiral SFC analysis.
Given the limitations in enantiofacial discrimination with CPAs and NPAs, we turned to more confined Brønsted acids—specifically imidodiphosphoric acids (IDP)32, iminoimidodiphosphoric acids (iIDP)33, and imidodiphosphorimidates (IDPi)34,35. These catalysts, characterized by their low pKa values and sterically encumbered active sites, have been shown to enhance both reactivity and enantioselectivity in asymmetric transformations, offering catalytic sites within chiral microenvironments that enhance both reactivity and enantioselectivity in asymmetric transformations36. Through extensive evaluation of these catalyst classes, we identified the iIDP catalyst D4 as optimal, achieving 90% ee and 94% yield at 60 °C in toluene. Further optimization led to the following conditions: treating 1a (0.2 mmol) with acetone (5.0 equiv.) in the presence of D4 (2.5 mol%) in THF (0.2 M) at 60 °C for 5 days yielded 4a with 86% yield and 96% ee. Excess acetone was essential to drive the reaction to completion, as it serves as the electrophile in the sequential Friedel–Crafts alkylation leading to the spirocyclic framework. The C10F7 group in the catalyst is crucial for the enantiocontrol, as the C10F7-substituted catalyst (D4, E4) significantly boosted the enantioselectivity from 0–7% ee to 80–90% ee when comparing to the analogous catalysts (D1-D3, E1-E3). Notably, D4 was synthesized via a palladium-free route, incorporating a 3,3’-C10F7 group on the BINOL backbone37. This design offers a sustainable and cost-effective catalytic process, with water as the sole by-product.
Substrate scope
With the optimized conditions in place, we evaluated a broad range of indoles with varying electronic properties and ring substitutions (Fig. 3). The transformation showed compatibility with C5 and C6 substitutions on the indole ring, yielding the desired spirocyclic products (4a–4y) in moderate to good yields (63–94%) with high enantioselectivity (up to 99% ee). Electron-rich, electron-deficient, and sterically demanding substituents—including alkyl, alkoxy, halides, and ester groups—at C5 or C6 were well-tolerated, consistently delivering products with 90–99% ee (e.g. 4a–4c, 4f-4o, 4q–4x). In addition, we observed that indoles bearing electron-deficient or bulky substituents faced challenges in reactivity and selectivity. Specifically, substrates with -Br, -I, or bulky groups (4d–4e, 4p–4q, 4s, 4u) achieved enantioselectivities of 73–85% ee. Meanwhile, electron-deficient indoles (-Cl, -Br, -I, -COOMe) exhibited reduced reactivity under our conditions, necessitating the use of toluene as a solvent and elevated temperatures for effective transformation. Substrates with -CF3 or -CN groups failed to yield the desired products, while acyl and -CH2OH substituents led to polymerization (Supplementary Fig. S10). We anticipated that substituents near the reaction site, particularly at the functional group binding to the catalyst, might alter the preferred substrate conformation, potentially affecting key interactions with the chiral catalyst. Halides (-Cl, -Br, -I) at C7 (Supplementary Fig. S10) did not yield desired products, even at elevated temperatures, while -Me and -COOMe at C4 (Supplementary Fig. S10) showed no conversion, with the starting material recovered. Electron-donating groups (-Me, -OBn) at C7 (4z–4bb) yielded low to moderate conversion with enantioselectivities of 24% and 13% ee, respectively. Encouragingly, indole bearing a -F group at C4 (4y) provided the desired product with a high enantioselectivity (82% yield, 93% ee). Interestingly, multisubstituted indoles at C5 and C6 consistently delivered exceptional results, achieving up to 99% ee (4t-4x), representing some of the highest enantioselectivities reported for confined catalysts in asymmetric transformations (99% ee, 4v).

Condition: 1a-1bb (0.2 mmol, 1.0 eq), acetone (1.0 mmol, 5.0 eq) iIDP D4 (9.6 mg, 0.005 mmol, 2.5 mol%), toluene or THF (1.0 mL), stirred at indicated temperature for the indicated days. Isolated yields given. Enantioselectivities (ee) were measured by SFC. Absolute configurations confirmed by the X-ray crystallographic analysis after recrystallization of 4d. The stereochemistry of the remainder of the entries is assigned by analogy. (a) Reactions were conducted in toluene. (b) Reactions were conducted in THF. (c) Reactions were conducted at 60 °C. (d) Reactions were conducted at 80 °C. (e) Reactions were conducted at 100 °C. (f) Reactions were conducted for 4 days. (g) Reactions were conducted for 5 days. (h) Reactions were conducted for 6 days. (i) Reactions were conducted for 7 days.
To demonstrate the practicality and scalability of this protocol, preparative synthesis of 4a was carried out on a 40 mmol (4.68 g) scale of indole. The desired product, 4a, was obtained in 84% yield (5.94 g) with 90% ee after 5 days of stirring in toluene. Enantiopure (R)-4a could be easily obtained by recrystallization in Et2O and hexanes, with 88% recovery and >99% ee. The opposite enantiomer, (S)-4a, was achieved with similar results by using (S,S)-D4 as the catalyst.
Chiral ligand and catalyst development
As noted above, the conformational rigidity, chemical stability, and modularity of axially spirocyclic scaffolds render them important backbones for chiral catalysts and ligands. Building on these advantages, the developed indole-modified spirocyclic scaffold 4a not only offers a straightforward synthesis approach using inexpensive indole and acetone as starting materials but also introduces milder and simpler derivatization methods for constructing diverse and potentially useful catalyst and ligand frameworks, comparable to BINOL and SPINOL analogs (Fig. 4). The weakly acidic NH functionality facilitates various substituent reactions through cost-effective, noble-metal-free method, which is particularly important in the cases of phosphine ligands38. This modularity is demonstrated by the synthesis of diverse compounds, including chiral urea (6a), thiourea (6b, 6c) bearing aromatic groups, and dihydrooxazoles (10a, 10b), which were obtained in high yields through efficient single-step reactions. Bisphosphines (7a, 7b) and monophosphine (8) were obtained by controlling the amount of nBuLi and the reaction temperature, and these were easily converted to corresponding phosphites (14, 16) by H2O2 oxidation. The monophosphine scaffold can be further diversified with various N1,1’-substituents (15, 17), underscoring the scaffold’s flexibility. Additionally, 11a was synthesized by reducing 4a with NaCNBH3, and can be evaluated as a secondary amine catalyst. The stereochemistry of 11a was assigned based on its analog 11b, confirmed by X-ray crystallography. Furthermore, the NH groups of the indole rings function as directing groups in Ir-catalyzed C7,7’-diborylation (12), demonstrating potential for diversifying the C7,7’-positions of SPINDOLEs and addressing limitations of C7-substituted indoles. Notably, none of these processes showed any evidence of racemization, highlighting the robustness of the approach.

A Straightforward transformation of 4a to catalysts and ligands. Conditions: (a) 4a (1.0 eq), NaH (3.0 eq), DMF, 0 °C, 1 h; PhNCO (5a)/PhNCS (5b)/3,5-(CF3)2C6H3-NCS (5c) (3.0 eq), rt, 16 h. (b) 4a (1.0 eq), nBuLi (2.2 eq), THF, 0 °C, 1 h; Ph2PCl or Xyl2PCl (2.2 eq), rt, 16 h. (c) 4a (1.0 eq), nBuLi (1.1 eq), THF, −78 °C, 1 h; Ph2PCl (1.1 eq), rt, 2 h. (d) 4a (1.0 eq), NaH (3.0 eq), DMF, 0 °C, 1 h; 2-(chloromethyl)-4,5-dihydrooxazole (9a) or (R)-2-(chloromethyl)-4-ethyl-4,5-dihydrooxazole (9b) (3.0 eq), rt, 16 h. (e) 4a or 4 d (77% ee) (1.0eq), NaCNBH3 (10. 0 eq), HOAc (10.0 eq), DCM, 0 °C, 4 h. (f) 4a (1.0eq), B2pin2 (2.0 eq), [Ir(cod)(OMe)]2 (1.5 mol%), dtbpy (3.0 mol%), THF, reflux, 24 h. B Transformation of 7a. Conditions: (g) 7a (1.0 eq), Pd(PhCN)2Cl2 (1.0 eq), Benzene, 16 h. (h) 7a (1.0 eq), H2O2 (10.0 eq), DCM, rt, 1 h. C Transformation of 8. Conditions: (i) 8 (1.0 eq), NaH (2.0 eq), DMF/THF (v/v = 1/4), 0 °C, 0.5 h; 3,5-(CF3)2C6H3-NCS (5c) (2.0 eq), rt, 3 h. (j) 8 (1.0 eq), H2O2 (5.0 eq), DCM, rt, 1 h. (k) 16 (1.0 eq), nBuLi (1.1 eq), THF, −78 °C, 1 h; Ph2PCl (1.1 eq), rt, 16 h.
Building on the success of using a single structural scaffold to generate a diverse range of functionalized components, we were compelled to investigate whether these tailored structures could extend effectively into catalytic applications. The versatility observed in structural modification underscores the potential of this scaffold as a platform for both ligand development and organocatalysis. With this in mind, we aimed to assess whether the achieved structural diversity could translate into meaningful catalytic activity, establishing these compounds as versatile tools for asymmetric synthesis.
Our initial focus was on bisphosphine derivatives due to their established importance in catalysis, particularly in stabilizing palladium (II) complexes. Given the distinct structural features of our scaffold (e.g. P-P distance, τ4, Bite angle, more details see Supplementary Table S3), we anticipated that it might exhibit unique properties relative to widely used BINAP39 and SDP40 ligands (Fig. 5A). As a preliminary assessment, we conducted a detailed comparison of key structural parameters—such as dihedral angles, 31P NMR shifts, and bite angles—to understand how our scaffold fits within established chemical space. If our structures demonstrate unique properties through these metrics, they could hold significant promise for producing reaction outcomes in organic synthesis.

A Comparing structural properties of Pd(II) complexes. B X-ray of Pd(II) complex 13. C Rhodium/7a catalyzed asymmetric hydroboration/cyclization. Condition: 18 (1.0 eq), HBpin (1.5 eq), Rh(cod)2BF4 (1.0 mol%), SIDP 7a (1.2 mol%), DCM, 0 °C, 24 h. (D) Rhodium/7b catalyzed asymmetric hydrogenation. Condition: 20 (1.0 eq), Rh(cod)2SbF6 (1.0 mol%), SIDP 7b (1.2 mol%), H2 balloon, DCM, rt, 6 h. E Palladium/7b catalyzed asymmetric allylic alkylation. Condition: rac-22 (1.0 eq), Dimethyl Malonate (2.0 eq), [Pd(allyl)Cl]2 (2.5 mol%), SIDP 7b (6 mol%), ZnEt2 (2.0 eq), THF, 0 oC, 24 h. (F) Secondary amine 11a catalyzed asymmetric Michael addition reaction. Condition: (1) 24 (1.0 eq), 25 (2.0 eq), 11a (5 mol%), TFA (10 mol%), DCM/IPA, 0 oC, 48 h; (2) NaBH4, MeOH, rt, 10 min.
To this end, we obtained the crystal structure of the palladium complex 13 and compared it to reported BINAP- and SDP-coordinated palladium complexes (Fig. 5B). Notably, the bite angle (P–Pd–P) in PdCl₂(SIDP) 13 complex is 100.4°, substantially larger than that in PdCl₂(BINAP) (92.7°) and PdCl₂(SDP) (96.0°), with a correspondingly longer P–P distance (3.49 Å) while maintaining similar Pd–P bond lengths. This increased bite angle likely reflects the inherent rigidity of our scaffold, potentially imparting unique steric and electronic properties in catalytic applications.
Further analysis showed that the dihedral angle between the phosphine phenyl rings in PdCl₂(SIDP) (68.1°) closely resembles that in PdCl₂(BINAP) (68.4°) but is significantly larger than in PdCl₂(SDP) (61.4°). Additionally, PdCl₂(SIDP) 13 has a lower geometry index parameter (τ₄ = 0.107) compared to BINAP and SDP analogs, indicating a less skewed square-planar geometry, which may influence reactivity and selectivity in asymmetric reactions. DFT analysis and 31P NMR studies further underscore these structural differences, likely due to the N1,1’ configuration of the indole backbone (see Supplementary Table S3 for details). Moreover, the NH bonds in SPINDOLE are positioned closer to the spirocenter compared to SPINOL (2.65 Å vs. 3.04 Å, as measured from X-ray crystallographic structures of 4d and SPINOL41), potentially resulting in a deeper active site for catalysis.
The distinct structural features of our scaffold raise intriguing questions about its performance in catalytic applications, especially in comparison to traditional ligands like BINAP and SDP. Motivated by these differences, we first evaluated compound 7a as a ligand in the Rh-catalyzed asymmetric hydroboration/cyclization of 1,6-enyne. Remarkably, the desired product was obtained in 90% ee with a 95% yield (Fig. 5C), while both BINAP and SDP showed limited reactivity and selectivity in similar reactions42. In the Rh-catalyzed asymmetric hydrogenation of 20 (Fig. 5D), we were delighted to observe 99% ee for product 21 when using 7b as the ligand. In contrast, the same reaction with BINAP resulted in low enantioselectivity and barely proceeded with SDP, as referenced in reported studies43. We further assessed our scaffold in a Pd-catalyzed asymmetric allylic alkylation reaction (Fig. 5E), where 7b again performed well, achieving 91% ee—comparable to SDP44 and BINAP40.
At this stage, we envisaged that a compelling demonstration of the potential of these structures would be to successfully apply it to a different class of compound entirely. Given the broad utility of secondary amine compounds in catalysis45, we considered the asymmetric Michael addition of cinnamaldehyde 24 and N-methylindole 25 to be of significant practical value (Fig. 5F). Using TFA as a co-catalyst of 11a, we achieved 91% ee, underscoring the scaffold’s versatility in catalysis. These promising results highlight the strong potential of SPINDOLE-based catalysts and ligands in asymmetric transformations. The unique structural features of this scaffold can indeed translate into significant catalytic advantages across diverse reaction types.
Mechanism studies
The optimization process yielded significant mechanistic insights through the isolation of intermediates 2a and 3a at different conversion levels. Subjecting 2a and racemic 3a to standard conditions in separate experiments produced the same ee and comparable yields (Fig. 6A). Combining 3f and 2a under standard conditions yielded a mixture of 4a, 4f, and 4cc (Fig. 6B), suggesting that the steps preceding cyclopenta[b]indole formation, specifically those involving 3a and 3f, are reversible. Although these intermediates we isolate are not necessarily on the most productive pathway, the reversibility of many steps in the process is evident. Stacked 1H NMR spectra of the reaction in toluene-d8 revealed that compound 2a was rapidly formed and consumed in the initial stages, followed by the generation of 3a and the gradual emergence of the desired spirocycle 4a. These observations suggest that intermediates 2a, 3a, and 4a are formed and possibly converted sequentially (Fig. 6C).

A Results of performing reactions with intermediates 2a and/or 3a under optimal conditions. B Positive cross over demonstrates reversibility. C Monitoring the concentration of 2a, 3a and 4a over time. D Experiment reads out primary kinetic isotope effect. E Evaluating SPINDOLE 4a catalytic reactivity. F Proposed stereochemical model for explaining the selectivity determined at the deprotonation step. G Proposed mechanism on the basis of evidence presented in A-F.
To elucidate the enantioselectivity-determining step, several careful experiments were conducted. A primary kinetic isotope effect (KIE) of 2.11 was measured using 1a and its deuterated analogue, 2D-1a, as probes. This suggests a potentially complex mechanism in which the spirocyclization step may be reversible. The relative energies of diastereomeric intermediates and the barriers to their deprotonation likely both contribute to the excellent observed enantioselectivities (Fig. 6D). Notably, the use of 2D-1a led to a significant decrease in enantioselectivity to 14% ee, supporting the notion that deprotonation is the most likely enantioselectivity-determining step.
Given the structural similarity between SPINDOLE and the catalyst, we explored whether the product could influence the stereochemistry-determining step. To test this, we replaced iIDP with SPINDOLE under otherwise identical conditions. The absence of catalytic activity confirmed that SPINDOLE does not participate in the reaction (Fig. 6E). Additionally, a linear relationship between the enantiopurity of the catalyst and the product was observed, suggesting that a single iIDP molecule is involved in the enantioselectivity-determining step (see Supplementary Fig. S9).
To rationalize the observed enantioselectivity, density functional theory (DFT) calculations were performed at the IEFPCM(THF)-ωB97XD/6-31G(d,p)//ωB97XD/6-31G(d) level of theory on diastereomeric SPINDOLE-iIDP complexes involving 4a and D4. The lowest-energy ground-state complex aligns with the experimentally determined enantiomer, suggesting that the stabilizing interactions in this structure are also likely relevant in the preceding transition state, consistent with the Hammond postulate. Notably, the calculations highlight key interactions between the aromatic groups on the catalyst and the substrate as likely determinants of enantioselectivity during the deprotonation step (see Supplementary pages 5–8 for details). Ultimately, these calculations provided the basis for constructing a preliminary mechanistic model that connects catalyst-intermediate interactions with the stereochemical outcome as shown in Fig. 6F.
Based on experimental results, we propose the following mechanism for the chiral Brønsted acid–catalyzed formation of the axially chiral spirocycle, SPINDOLE (Fig. 6G). The reaction begins with the protonation of acetone by catalyst D4, inducing the condensation of indole (1a) to form bis(indolyl)propane (2a). Under acidic conditions, the indole nitrogen facilitates C(sp³)–C(indolyl) bond cleavage, generating alkene-substituted indole species Ia and Ib. These intermediates react to form carbocation II, which cyclizes to form indolyl-cyclopenta[b]indole (3a). Upon heating, 3a rearranges into alkene species III, which reacts with Ib to form carbocation IV. This intermediate undergoes cyclization, followed by a Pictet- Spengler-type migration and deprotonation, producing the desired product (4a) and regenerating D4.
Discussion
In summary, we have developed SPINDOLE, a versatile chiral framework that creates opportunities in asymmetric synthesis. This approach leverages commercially available, inexpensive indoles and acetone to provide an efficient and cost-effective route to enantiopure indole-based axially spirocyclic scaffolds. The methodology offers rapid access to SPINDOLEs in good to excellent yields (up to 94%) and exceptional enantioselectivities (up to 99% ee), demonstrating broad functional group compatibility. Encouraged by these promising results, we have initiated the construction of a comprehensive catalyst and ligand library based on this innovative axially spirocyclic backbone. The SPINDOLE scaffold presents significant potential as a robust platform for the design of organocatalysts and ligands, serving as a complementary alternative to the widely used BINOL and SPINOL frameworks. Ongoing studies in our laboratory are focused on further exploring the utility of SPINDOLE as an axially chiral ligand and organocatalyst, with findings to be reported in future publications.
Methods
General procedure for enantioselective reactions
A 4 mL was charged with 1.0 mL of toluene or THF, 1a-1bb (0.2 mmol, 1.0 eq), acetone (58.0 mg, 1.0 mmol, 5.0 eq), and iIDP D4 (9.1 mg, 0.005 mmol, 2.5 mol%) were added. The vessel was sealed and stirred at 60 °C to 100 °C for 4–7 days. The reaction mixture was then subjected to silica gel column chromatography (Petroleum ether: AcOEt = 20:1 to 5:1) to afford compound 4a-4bb.
Data availability
All data are available in the main text or the supplementary information (including computational details, experimental details, NMR data, SFC and NMR spectrums). Source data are provided with this paper. All data are available from the corresponding author upon request. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC-2408120 for 4d; CCDC-2408122 for 11b; CCDC-2408121 for 13. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Source data are provided with this paper.
References
Chen, Y., Yekta, S. & Yudin, A. K. Modified BINOL ligands in asymmetric catalysis. Chem. Rev. 103, 3155–3212 (2003).
Brunel, J. M. Update 1 of: BINOL: a versatile chiral reagent. Chem. Rev. 107, PR1–PR45 (2007).
Shibasaki, M. & Matsunaga, S. Design and application of linked-BINOL chiral ligands in bifunctional asymmetric catalysis. Chem. Soc. Rev. 35, 269–279 (2006).
Yoon, T. P. & Jacobsen, E. N. Privileged chiral catalysts. Science 299, 1691 (2003).
Zhou, Q. L. Privileged Chiral Ligands and Catalysts (Wiley-VCH, Weinheim, Germany, 2011), pp. 1–462.
Akimoto, H., Shioiri, T., Iitaka, Y. & Yamada, S. I. Determination of the absolute configuration of 1, 1′-binaphthyl and its derivatives by x-ray diffraction. Tetrahedron Lett. 1, 97–102 (1968).
Birman, V. B., Rheingold, A. L. & Lam, K. C. 1, 1′-Spirobiindane-7, 7′-diol: a novel, C2-symmetric chiral ligand. Tetrahedron Asymmetry 10, 125–131 (1999).
Zhou, Q. L. & Xie, J. H. Chiral Spiro Catalysts. Top. Organomet. Chem 36, 1–28 (2011).
Zhu, S. F. & Zhou, Q. L. in Ligand Design in Metal Chemistry: Reactivity and Catalysis (Wiley‐VCH, Weinheim, Germany, 2016), pp. 66–103.
Zhang, Q., Tang, L. & Jin, P. Construction of Spiro Skeletons in 2, 2’, 3, 3’-Tetrahydro-1, 1’-spirobi [1H-indene]-7, 7’-diol (SPINOL) and Analogues. Chin. J. Org. Chem. 41, 3400–3413 (2021).
Li, S., Zhang, J. W., Li, X. L., Cheng, D. J. & Tan, B. Phosphoric acid-catalyzed asymmetric synthesis of SPINOL derivatives. J. Am. Chem. Soc 138, 16561–16566 (2016).
Zhang, R., Ge, S. & Sun, J. SPHENOL, a new chiral framework for asymmetric synthesis. J. Am. Chem. Soc 143, 12445–12449 (2021).
Yin, L. et al. Enantioselective Synthesis of 3, 3′‐Diaryl‐SPINOLs: Rhodium‐Catalyzed Asymmetric Arylation/BF3‐Promoted Spirocyclization Sequence. Angew. Chem. Int. Ed 58, 2474–2478 (2019).
Zheng, Z. et al. Chiral cyclohexyl-fused spirobiindanes: practical synthesis, ligand development, and asymmetric catalysis. J. Am. Chem. Soc 140, 10374–10381 (2018).
Molteni, V. et al. A New Class of HIV-1 Integrase Inhibitors: The 3, 3, 3 ‘, 3 ‘-Tetramethyl-1, 1 ‘-spirobi (indan)-5, 5 ‘, 6, 6 ‘-tetrol Family. J. Med. Chem 43, 2031–2039 (2000).
Zhou, Q., Pan, R., Shan, H. & Lin, X. Synthesis and Optical Resolution of 3, 3, 3′, 3′-Tetramethyl-1, 1′-spirobiindane-7, 7′-diol. Synthesis 51, 557–563 (2019).
Tan, B. et al. Organocatalytic Asymmetric Construction and Application of Axially Chiral Spiro‐bisindoles. Angew. Chem. Int. Ed 64, e202422951 (2025).
Wang, J. Y. & Sun, J. Design, Synthesis and Application of Chiral Spirocyclic Bisindoles. Angew. Chem. Int. Ed. 64, e202424773 (2025).
Khanna, L., Yadav, M. S., Misra, N. & Khanna, P. “In water” synthesis of bis (indolyl) methanes: a review. Synth. Commun. 51, 2892–2923 (2021).
Bergman, J., Norrby, P. O., Tilstam, U. & Venemalm, L. Structure elucidation of some products obtained by acid-catalyzed condensation of indole with acetone. Tetrahedron 45, 5549–5564 (1989).
Nadkarni, S. V. & Nagarkar, J. M. Synthesis of highly substituted indoles in presence of solid acid catalysts. Green Chem. Lett. Rev. 4, 121–126 (2011).
Betinol, I. O., Lai, J., Thakur, S. & Reid, J. P. A data-driven workflow for assigning and predicting generality in asymmetric catalysis. J. Am. Chem. Soc. 145, 12870–12883 (2023).
Akiyama, T. Stronger brønsted acids. Chem. Rev. 107, 5744–5758 (2007).
Akiyama, T. & Mori, K. Stronger Brønsted acids: recent progress. Chem. Rev. 115, 9277–9306 (2015).
Akiyama, T., Itoh, J., Yokota, K. & Fuchibe, K. Enantioselective Mannich‐type reaction catalyzed by a chiral Brønsted acid. Angew. Chem. Int. Ed 43, 1566–1568 (2004).
Uraguchi, D. & Terada, M. Chiral Brønsted acid-catalyzed direct Mannich reactions via electrophilic activation. J. Am. Chem. Soc 126, 5356–5357 (2004).
Reid, J. P., Simón, L. & Goodman, J. M. A practical guide for predicting the stereochemistry of bifunctional phosphoric acid catalyzed reactions of imines. Acc. Chem. Res. 49, 1029–1041 (2016).
S. Bhadury, P. & Pang, J. Chiral BrOnsted Acid-Catalyzed Friedel-Crafts Reaction of Indoles. Curr. Org. Chem. 18, 2108–2124 (2014).
Wu, H., He, Y. P. & Shi, F. Recent advances in chiral phosphoric acid catalyzed asymmetric reactions for the synthesis of enantiopure indole derivatives. Synthesis 47, 1990–2016 (2015).
Biswas, A. Organocatalyzed Asymmetric Pictet‐Spengler Reactions. ChemistrySelect 8, e202203368 (2023).
Caballero-García, G. & Goodman, J. M. N-Triflylphosphoramides: highly acidic catalysts for asymmetric transformations. Org. Biomol. Chem. 19, 9565–9618 (2021).
Čorić, I. & List, B. Asymmetric spiroacetalization catalysed by confined Brønsted acids. Nature 483, 315–319 (2012).
Liu, L., Kaib, P. S., Tap, A. & List, B. A general catalytic asymmetric Prins cyclization. J. Am. Chem. Soc 138, 10822–10825 (2016).
Kaib, P. S., Schreyer, L., Lee, S., Properzi, R. & List, B. Extremely active organocatalysts enable a highly enantioselective addition of allyltrimethylsilane to aldehydes. Angew. Chem. Int. Ed. 55, 13200–13203 (2016).
Lai, J. & Reid, J. P. A Bulky Imidodiphosphorimidate Brønsted Acid Enables Highly Enantioselective Prins-semipinacol Rearrangements. ACS Catal 14, 8518–8527 (2024).
Albrecht, L., Albrecht, A. & Dell’Amico, L. eds, Asymmetric Organocatalysis: New Strategies, Catalysts, and Opportunities (John Wiley & Sons, 2022), pp. 19–36.
Grimm, J. A. et al. Catalytic asymmetric synthesis of cannabinoids and menthol from neral. Nature 615, 634–639 (2023).
Xu, L. et al. RETRACTED ARTICLE: The amine-catalysed Suzuki–Miyaura-type coupling of aryl halides and arylboronic acids. Nat. Catal. 4, 71–78 (2021).
Ozawa, F. et al. Palladium-catalyzed asymmetric arylation of 2, 3-dihydrofuran with phenyl triflate. A novel asymmetric catalysis involving a kinetic resolution process. Organometallics 12, 4188–4196 (1993).
Xie, J. H. et al. Application of SDP Ligands for Pd‐Catalyzed Allylic Alkylation. Adv. Synth. Catal. 346, 625–632 (2004).
Huo, X. H., Xie, J. H., Wang, Q. S. & Zhou, Q. L. The Synthesis of Spirobitetraline Phosphoramidite Ligands and their Application in Rhodium‐Catalyzed Asymmetric Hydrogenation. Adv. Synth. Catal 349, 2477–2484 (2007).
Hou, F. et al. Rhodium-Catalyzed Asymmetric Hydroboration/Cyclization of 1,6-Enynes Enabled by Spirosiladiphosphine Ligands: Constructing Chiral Five-Membered Rings with a Boron Handle. Org. Lett. 25, 7810–7815 (2023).
Li, H. et al. SPSiPs, a class of diphosphine ligands based on SPSiOL with a large dihedral angle. Org. Lett. 25, 3859–3863 (2023).
Fuji, K., Kinoshita, N. & Tanaka, K. Et2Zn as a base: zinc enolate free from other metals significantly enhances the enantiomeric excess in palladium-catalyzed allylic alkylation. Chem. Commun. 18, 1895–1896 (1999).
Brazier, J. B., Tomkinson, N. C. Secondary and Primary Amine Catalysts for Iminium Catalysis, Asymmetric Organocatalysis (John Wiley & Sons, 2009), pp. 281–347.
Acknowledgements
Financial support to J.P.R. was provided by the University of British Columbia, the Natural Sciences and Engineering Research Council of Canada (NSERC) and the CFI John R. Evans Leaders Fund. Computational resources were provided from the Digital Research Alliance of Canada and the Advanced Research Computing (ARC) center at the University of British Columbia. We thank Brian Patrick (UBC) for solving the crystal structure. We thank Professor Jonathan Goodman (University of Cambridge) for insightful discussions regarding the proposed mechanism.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
J.P.R and J.L. performed the calculations. J.L. performed the experiments. B.L. supported the catalyst optimization process. J.P.R and J.L. wrote the manuscript. J.P.R. supervised the project and acquired project funding.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xiaowei Dou, Genping Huang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lai, J., List, B. & Reid, J.P. Bis-indole chiral architectures for asymmetric catalysis. Nat Commun 16, 3676 (2025). https://doi.org/10.1038/s41467-025-58313-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58313-4
