
Abstract
Direct electro-epoxidation of propylene (D-EOPO) with a membrane electrode assembly (MEA) system represents a sustainable approach for producing propylene oxide, which can reduce ohmic losses and simplify product separation. To address the challenges of selectivity and activity, we develop an Ag/V catalyst and integrate it into the “liquid-free” MEA reactor for continues D-EOPO. The V in the catalyst facilitates the formation of Ag-O active centers, thereby reducing the generation energy of *O radicals. Meanwhile, V doping also results in a downshift of the d-band center of the Ag sites. Consequently, the formation of the crucial intermediate (*OC3H6) is significantly accelerated through the coupling *O with adsorbed propylene, thereby markedly improving propylene oxide (PO) production. The MEA reactor, integrated with the developed Ag/V catalyst, can maintain a stable production rate of PO at 227 μmol/h over a period of 78 hours. Thus, the “liquid-free” electro-epoxidation protocol developed here exhibits greater industrial applicability.
Similar content being viewed by others

Introduction
Propylene oxide (PO) is a vital industrial chemical, widely used in the manufacture of polyurethanes1, propylene glycol2, and non-ionic surfactants3. Traditional PO production methods, which rely on strong oxidants such as hydrogen peroxide and chlorine, pose safety risks and generate substantial amounts of corrosive and environmentally harmful byproducts4,5. Recently, the electrochemical oxidation of propylene (EOPO) has emerged as a promising alternative6,7. This method uses water as the oxidant source and enables PO synthesis under mild conditions. As depicted in Fig. 1a, a chloride-mediated EOPO process has been previously explored8,9,10. Initially, chloride ions in the electrolyte are oxidized to molecular chlorine (Cl2) at the anode, which is then rapidly converted into hypochlorous acid (HClO) with water. Subsequently, propylene is oxidized by the HClO-containing electrolyte to form chloropropanol. Finally, the synthesized chloropropanol is mixed with an alkali solution and converted to PO. The technology enables a renewable, electricity-driven EOPO process that leverages clean energy and achieves a high conversion rate. Nevertheless, several challenges still hinder the continuous production of PO, including the corrosive ClO− byproducts and the dissolution of PO into the electrolyte.

a Schematic of chloride-mediated electrochemical propylene epoxidation setup. b Schematic of membrane electrode device for water-mediated electrochemical propylene epoxidation. c Possible reaction pathway for electrochemical propylene epoxidation on Ag-V-O/GDL and Ag-O/GDL (direct water mediation).
In comparison (Fig. 1b), D-EOPO in the chloro-free electrolyte is an alternative approach to produce PO. In this method, water in the electrolyte is oxidized to active oxygen species on the anode, which can oxidize propylene without generating environmentally harmful byproducts11,12,13. However, D-EOPO currently faces limitations due to side reactions, such as the over-oxidation to acrylic acid and eventually to CO214. In addition, the poor solubility of propylene in water hinders the effective mass transport during D-EOPO in traditional H-cell, leading to drastic oxygen evolution as a competitive reaction12. To overcome these issues, the flow cell with gas-diffusion electrode has been employed to balance the mass transport on the gas-liquid-solid interface of electrode. Under the prolonged application of oxidation potential, flooding inevitably occurs on the gas diffusion electrode in flow cells, resulting in poor diffusion efficiency, PO production rate and stability11. In comparison, liquid-free membrane electrode assembly (MEA) reactors have gained widespread application in fuel cells15,16,17 and CO2 electroreduction18,19,20 due to their low resistance, high mass transport efficiency and excellent anti-flooding capability. More importantly, the PO generated via electro-catalytic oxidation of propylene in liquid-free MEA reactors can be directly separated from gas products by condensation, which greatly reduces the cost of separation process. Thus, the liquid-free MEA system for direct electrochemical epoxidation represents a promising approach to synthesize PO in quantities of practical relevance.
Compared to traditional reactors, the residence time of reactants on the catalyst surface is reduced in MEA reactors, which requires high active catalyst with promoted selectivity for catalyzing EOPO. Despite extensive research on catalyst design and the electrochemical production mechanisms of PO, the selectivity and production of D-EOPO are still unsatisfactory. Previously, *O (* denotes the adsorbed species on the surface) has been widely recognized as a crucial intermediate in olefin epoxidation reactions21,22,23. During EOPO, H2O on electrode surface is initially oxidized to generate active oxygen species *O21, which exhibits strong electrophilic properties, effectively activating the C = C bonds. Consequently, *O rapidly reacts with adsorbed propylene molecules on the active centers of catalyst, and epoxidizes them into propylene oxide. However, the catalytic activation of H2O to form *O involves a high energy barrier, which becomes the main obstacle to increase EOPO efficiency. Thus, developing electrocatalysts with negative binding energies with *O is crucial for enhancing the efficiency of D-EOPO.
Precious metal catalysts (Pt, Ag etc.) have been proven highly activity in thermal catalytic epoxidation, which have also been adopted for catalyzing EOPO. For instance, a high surface area Pd electrode has been developed by electrodeposition strategy14, which preferentially activates the C-H bond on the allyl carbon, leading to the formation of acrolein. Manthiram et al13. reported an oxidized palladium-platinum alloy (PdPtOx/C) catalyst, achieving a PO Faradaic efficiency (FE) of 66 ± 5% for EOPO. Although the selectivity for PO is improved, the long-term stability and lifespan still limited. In contrast, Ag-based materials are more inclined to break the C = C double bond in propylene, promoting the formation of C-O bonds, which is beneficial for the efficient production of PO24,25. Geng et al11. reported an efficient Ag3PO4 cubic electrocatalyst, achieving a yield of 5.3 gPO m-2 h-1 in a flow cell. However, during long-time operation, the transformation of the gas diffusion electrode from hydrophobic to hydrophilic hinder propylene diffusion, leading to a decrease in PO production.
Aforementioned, the employment of liquid-free MEA reactors for continuous D-EOPO to produce PO presents considerable efficiency benefits. Nonetheless, this technology relies on the advancement of D-EOPO catalysts to improve activity and selectivity. Our prior research has illustrated that V has a distinct valence electron configuration, capable of introducing new energy levels within the oxide lattice, which in turn diminishes the energy barrier for the migration of charge carriers11. Therefore, we have developed an Ag-V-O/GDL bimetallic oxide catalyst with negative O binding energy, which has been successfully integrated into a liquid-free MEA system for stable D-EOPO to produce PO (Fig. 1b, c). The incorporation of V also can modulate the electronic distribution at Ag sites, forming new trapping centers for electrons or holes, equilibrating the adsorption and desorption of oxygen intermediates, and consequently reducing the reaction energy barrier11. Moreover, V doping can also enhance the effective separation of electrons and holes, increasing conductivity and reducing energy consumption during D-EOPO14. Ultimately, the Ag/V bimetallic catalyst can steadily catalyze D-EOPO in MEA reactor with a stable PO yield of 227 μmol/h over 78 hours (almost the highest among current reports).
Results
Characterization of Ag-V-O/GDL catalyst
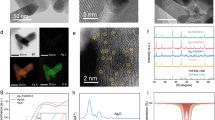
The Ag/V electrocatalyst was prepared via a co-precipitation method (detailed in the “Methods” section). The catalyst was synthesized at the optimized nV/nAg of 1, which exhibits the highest PO FE and production and is denoted as Ag-V-O/GDL (Supplementary Fig. 1). X-ray diffraction (XRD) analysis indicates that Ag-V-O/GDL is consisted of Ag3VO4 and Ag4V2O7 as main components (Fig. 2a, the XRD pattern is in the inset of SEM image; Supplementary Fig. 2)26. In addition, the Ag oxide electrocatalyst Ag-O/GDL was synthesized as a control sample by using ammonia instead of ammonium metavanadate and adjusting the pH with nitric acid. XRD pattern of Ag-O/GDL indicates the catalyst tends to expose Ag2O(111) facet (Supplementary Fig. 3). Scanning electron microscopy (SEM) reveals that both Ag-V-O/GDL and Ag-O/GDL are composed of irregular microspheres, with a diameter of approximately 1 μm (Fig. 2a and Supplementary Fig. 4). SEM-mapping image of Ag-V-O/GDL indicates the uniform distribution of Ag, V, and O elements, further confirming the successful doping of V (Supplementary Fig. 5). Compared to Ag-O/GDL, a new V 2p XPS peak is observed in Ag-V-O/GDL, indicating the successful doping of V (Supplementary Fig. 6). To determine the chemical structural variation of V doping, the XPS characteristic peaks of Ag 3 d in Ag-V-O/GDL and Ag-O/GDL were compared (Fig. 2b). The binding energy of Ag 3 d in Ag-V-O/GDL increases by 0.15 eV after incorporating V, indicating the decreasing of electron cloud of Ag through oxidation with doped V27,28. Furthermore, Density functional theory (DFT) calculations were used to analyze the partial density of states (PDOS) of Ag (Fig. 2c), which demonstrates a downshift of 0.17 eV in the d-band center of Ag after V doping. Consequently, the adsorption and desorption of intermediates onto the Ag sites can be optimized, thereby affecting the EOPO process.

a SEM image and XRD pattern of Ag-V-O/GDL. b Ag 3 d XPS spectra of Ag-V-O/GDL and Ag-O/GDL. c PDOS diagrams of Ag element in Ag-V-O/GDL and Ag-O/GDL. d EIS spectra of Ag-V-O/GDL and Ag-O/GDL (H-cell, Ag-O/GDL: Rs = 13.88 Ω cm2, Rct = 35.53 Ω cm2; Ag-V-O/GDL: Rs = 13.87 Ω cm2, Rct = 20.91 Ω cm2). e Mössbauer effect curves of Ag-V-O/GDL and Ag-O/GDL. f TDOS diagrams of Ag-V-O/GDL and Ag-O/GDL. All potentials were not iR corrected. The a.u. stands for arbitrary units. Source data are provided as a Source Data file.
Moreover, the impact of V doping on the conductivity of the Ag-O catalyst was further evaluated using electrochemical impedance spectroscopy (EIS). The charge transfer resistance (Rct) of the Ag-V-O/GDL catalyst (Fig. 2d and Supplementary Table 1) is significantly lower than that of Ag-O/GDL (20.91 Ω cm2 vs. 35.53 Ω cm2). The enhanced conductivity of the Ag-V-O/GDL catalyst can reduce polarization and allow the epoxidation reaction to occur at lower overpotentials, consequently minimizing side reactions and enhancing the catalyst’s activity29,30. In addition, the carrier mobility of Ag-V-O/GDL and Ag-O/GDL was compared using Mott-Schottky curves (Fig. 2e). The results indicate that the Ag-V-O/GDL catalyst possesses higher carrier mobility, suggesting that the addition of V improves the catalyst’s conductivity, consistent with the EIS test results. In Fig. 2f, DFT calculation analyzes the changes of the density of states (DOS) after V doping. Due to the bandgap effect of the Fermi level, Ag-O/GDL exhibits semiconductor properties. Upon V doping, the Ag-V-O/GDL catalyst introduces new orbitals at the Fermi level (0 eV), exhibiting metallic characteristics and thus increasing electrode conductivity.
Electrochemical performance and mechanism
The EOPO performance of as-prepared catalysts was investigated through linear sweep voltammetry (LSV) in an H-cell (Fig. 3 and Supplementary Fig. 7). Compared to Ag-O/GDL, Ag-V-O/GDL exhibits a lower overpotential by 0.07 V at the current density of 3 mA/cm². In addition, the normalized current density by the electrochemically active surface area (ECSA) of Ag-V-O/GDL is higher than that of Ag-O/GDL (Supplementary Fig. 8 and Supplementary Table 2). Thus, the Ag-V-O/GDL demonstrates higher activity during catalyzing D-EOPO. Furthermore, after constantly catalyzing EOPO at 3 mA/cm², Ag-V-O/GDL also shows higher FE (18.3% vs. 10.7%) and production rate (10.2 μmol·h⁻¹ vs. 6.0 μmol·h⁻¹) of PO than those of Ag-O/GDL (Fig. 3b). In addition to oxygen, other by-products such as carbon dioxide, carbon monoxide, propylene glycol, methanol, methanal and acetaldehyde were identified (Supplementary Table 3). The XRD patterns reveal that the dominant exposed facets of the Ag-V-O/GDL catalyst remain unchanged after the reaction (Supplementary Fig. 9). Auger electron spectroscopy (AES) of Ag MNN also indicates that the Ag-V-O/GDL catalyst is mainly consisted of Ag+ species (356.6 eV) both before and after reaction (Supplementary Fig. 10)31.

a LSV curves of propylene epoxidation on Ag-V-O/GDL and Ag-O/GDL (H-cell, 0.1 M PBS, 25 oC, scan rate: 20 mV s−1, without iR correction, Ag-O/GDL: Rs = 13.88 Ω cm2; Ag-V-O/GDL: Rs = 13.87 Ω cm2). b PO production and Faradaic efficiencies (FE) of Ag-V-O/GDL and Ag-O/GDL at 3 mA/cm² (H-cell, 0.1 M PBS, 25 oC). c In situ Raman spectra of propylene epoxidation on Ag-V-O/GDL catalyst (700–900 cm⁻¹). d In situ Raman spectra of propylene epoxidation on Ag-V-O/GDL catalyst (200–500 cm⁻¹). e In situ Raman spectra of propylene epoxidation on Ag-O/GDL catalyst (300–900 cm⁻¹). All potentials were not iR corrected. The a.u. stands for arbitrary units. Error bars represent the standard deviation from at least three independent measurements. Source data are provided as a Source Data file.
To further elucidate the catalytic mechanism of EOPO, in situ, Raman spectroscopy was employed to confirm the roles of different oxide species during the epoxidation reaction (Fig. 3c, d and Supplementary Fig. 11)32,33,34,35,36. The Raman peak, around 782 cm⁻¹, is attributed to the O-O vibration yielded by adsorbed O2 (*O2) on Ag catalyst (i.e., Ag4-O2)36. The Raman spectra reveal an increased peak intensity around 782 cm⁻¹ for the Ag-V-O/GDL catalyst, attributing to the increased oxygen concentration during the formation of Ag4-O2 on the catalyst surface under oxidation potentials. In addition, new characteristic peaks are observed at 322, 375, and 420 cm⁻¹, which indicate the formation of Ag-O species in Ag-V-O/GDL32,33. Correspondingly, a new Raman peak also appears around 782 cm⁻¹ in the Ag-O/GDL profile (Fig. 3e), with much weaker characteristic peaks of Ag-O at 322, 375, and 420 cm⁻¹. Thus, more Ag4-O2 species are generated on the surface of activated Ag-O/GDL under the oxidation potential, and the Ag-O species are obviously reduced36. Meanwhile, Raman peaks of Ag-V-O/GDL at 782 cm⁻¹ are narrower than those of Ag-O/GDL, which is attributed to increased crystallinity after V doping26,37,38. The Raman peak around 600 cm⁻¹ is ascribed to the O-Ag-O structure formed on the surface of Ag oxides39. In comparison, the Ag4-O2 species will decompose and diffuse out as O2 at elevated voltage, leading to over-oxidation of some adsorbed propylene to CO2. Thus, the CO2 and O2 production and FE of Ag-O/GDL are significantly higher than those of Ag-V-O/GDL (Supplementary Fig. 12). Moreover, in situ time-resolved Raman spectroscopy of Ag-V-O/GDL at different potentials was conducted (Supplementary Fig. 13). The intensity of Ag-O Raman peak increases over time, which is attributed to the continuous generation of *O on the catalyst surface, resulting in an increase in Ag-O active sites and thus an improvement in catalytic activity. Moreover, as the potential increases, the Raman peak intensity of Ag-O gradually increases, indicating that more Ag-O active sites are generated under higher overpotential. Hence, the presence of V can promote the formation of Ag-O sites during the activation, thereby facilitating the EOPO process.
To pinpoint the active components within the Ag-V-O/GDL catalyst, we synthesized the pure Ag4V2O7 and Ag3VO4 catalysts for comparative analysis (detailed in the “Methods” section). In situ Raman spectra indicate that Ag3VO4, Ag4V2O7, and Ag-V-O/GDL ultimately form Ag-O reactive species (Fig. 3d and Supplementary Figs. 14, 15). Besides, Ag-O peaks are preferentially observed on Ag-V-O/GDL and Ag4V2O7 surfaces under more negative potentials, indicating the generation of more Ag-O species. (Fig. 3d and Supplementary Figs. 14, 15). Furthermore, the PO production and FE of Ag4V2O7 are higher than those of Ag3VO4, but lower than those of Ag-V-O/GDL (Supplementary Fig. 16). EIS spectra reveal that the inferior performance of Ag4V2O7 compared to Ag-V-O/GDL can be attributed to its higher electrolyte resistance (Rs) and Rct (Supplementary Fig. 17 and Supplementary Table 4). Moreover, the normalized current density by ECSA of Ag4V2O7 is higher than that of Ag3VO4, indicating that Ag4V2O7 presents a higher catalytic activity compared to Ag3VO4. (Supplementary Fig. 18 and Supplementary Table 5). Therefore, Ag4V2O7 is the main active species of EOPO in the Ag-V-O/GDL catalyst.
Reaction process of EOPO
To verify the supposed EOPO reaction process, in situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) was conducted. In situ ATR-SEIRAS spectra of Ag-O/GDL (Fig. 4a) show a higher peak around 1608 cm⁻¹, corresponding to the *C3H5 intermediate on the catalyst surface through dehydrogenating adsorbed propylene (C3H6) at elevated voltage40. Based on in situ Raman results, the surface of Ag-O/GDL intends to form Ag4-O2 sites with H2O activation at increased potential. The peroxy group within the Ag4-O2 complex decomposes and transforms into reactive oxygen species, which are capable of oxidizing the *C3H5 intermediates. However, this reaction can lead to excessive oxidation products, thereby reducing the yield of PO (Fig. 4b left). Correspondingly, the in situ ATR-SEIRAS spectra of Ag-V-O/GDL (Fig. 4c) reveal new peaks at 1506 cm⁻¹, which corresponds to the formation of C-C bonds on the Ag-V-O/GDL catalyst surface after the breakage of C = C in propylene41. This step is essential for the formation of the *OMC intermediate (oxametallacycle, *CH3-CH(CH2)-O) on the surface Ag-O sites, which subsequently results in a higher conversion rate to PO with excellent selectivity (Ag-V-O/GDL: 74.7%; Ag-O/GDL: 45.8%) (Fig. 4b right and Supplementary Table 6).

a In situ ATR-SEIRAS spectra of propylene epoxidation on Ag-O/GDL catalyst. b Schematic diagram of the reaction process of EOPO on Ag-O/GDL and Ag-V-O/GDL. c In situ ATR-SEIRAS spectra of propylene epoxidation on Ag-V-O/GDL catalyst. d Scheme of the reaction pathways of EOPO on Ag-V-O/GDL (*OMC: *CH3-CH(CH2)-O). e Free energy diagrams for propylene epoxidation on Ag-V-O/GDL and Ag-O/GDL (insert: energy barriers for the epoxidation between*O and *C3H6 on Ag-V-O/GDL and Ag-O/GDL. *OMC: *CH3-CH(CH2)-O). All potentials were not iR corrected. The a.u. stands for arbitrary units. Source data are provided as a Source Data file.
To confirm the reaction pathway of EOPO, the Gibbs free energy for the coupling of *OH and *O with *C3H6 on Ag-V-O/GDL catalyst was compared (Supplementary Fig. 19). The energy barrier of the key coupling steps of the *OH pathway (0.993 eV) is higher than that of the *O pathway (0.702 eV), indicating that *C3H6 is more likely to couple with *O. The activated *C3H6 can undergo a coupling reaction with *O to form *OMC, which is subsequently converted to PO. Besides, when the -CH3 in propylene is more active, *OMC will not preferentially generate. Alternatively, propylene can undergo -CH3 dehydrogenation through *O to form *C3H5, thereby generating CO2 and H2O42. Comparing energy barriers for *C3H6 dehydrogenation and coupling of Ag-V-O/GDL and Ag-O/GDL catalysts (Supplementary Fig. 20), Ag-V-O/GDL is more prone to undergo coupling reaction to form *OMC, while Ag-O/GDL is more prone to undergo -CH3 dehydrogenation (Supplementary Fig. 21).
Thus, the reaction pathway of Ag-V-O/GDL has been supposed in Fig. 4d and Supplementary Fig. 22, which includes the adsorption and dehydrogenation of H2O on Ag sites to form the *OH radical, followed by further dehydrogenation of *OH on the catalyst surface to *O radical. Subsequently, propylene adsorbs on the surface of the catalyst will couple with *O radical to *OC3H6, leading to the formation of the five-membered rings intermediate *OMC (*CH3-CH(CH2)-O), similar to the thermal epoxidation process. Finally, the *OMC intermediate is transformed to adsorbed PO (*PO) before desorption and then starts the next reaction cycle. During the EOPO process, the H⁺ ions generated on the anode will migrate to the cathode through the membrane for hydrogen evolution reaction (HER). Figure 4b shows the relevance of intermediate structural on the catalyst surface with the reaction pathway of EOPO. When propylene is adsorbed on the catalyst surface and converted into *C3H5 intermediate, which intends to be over-oxidized and produces CO2 after several steps. In comparison, the formation of the *OMC intermediate is crucial for the production of PO, which necessitates the modulation of the catalyst’s structure to optimize the interfacial mass and charge transfer.
In addition, to clarify the role of the Ag-V-O/GDL catalyst, which contains Ag-O species, in facilitating the reaction, the ΔG on the proposed reaction pathway was calculated by density functional theory (DFT). For this, the main crystal facet is selected to model the EOPO process, as depicted in Fig. 4e. The formation of *O requires the highest ΔG as the rate-determining step of EOPO. The ΔG of the rate-determining step (*O formation) in the EOPO process is lower with Ag-V-O/GDL compared to Ag-O/GDL (0.805 eV vs. 0.954 eV). Furthermore, the energy barrier for the generation of *O in Ag-V-O/GDL is 1.569 eV, which is lower than the 2.770 eV in Ag-V-O/GDL, indicating that *O is more easily generated on Ag-V-O/GDL (Supplementary Fig. 23). In addition, the energy barrier for epoxidation between *C3H6 and *O in Ag-V-O/GDL is lower than that in Ag-O/GDL (inset in Fig. 4e). Thus, the high catalytic activity of Ag-O to reduce the energy barrier during *O formation, further promoting the epoxidation step between *O and *C3H6, ultimately enhancing the activity of EOPO.
Epoxidation reaction in the membrane electrode assembly reactor
Since the reaction potential of EOPO is close to that of oxygen evolution reaction (OER), OER inevitably occurs as a side reaction during EOPO. In flow-cell and H-cell, the generated O2 bubbles will dislodge the catalyst from the electrode, deteriorating the performance during EOPO. To address these issues, we developed a MEA reactor for EOPO (Fig. 5a). In the flow channel on the anode side of the MEA reactor (Fig. 5b, c), propylene gas containing saturated water was introduced, where H2O molecules decomposed on the catalyst surface to produce reactive oxygen species, followed by in situ epoxidation of propylene to PO. On the cathode side, the aqueous catholyte was pumped into the flow channel of the cathode at a certain flow rate using a peristaltic pump, where HER occurred on a foam Ni electrode. The EOPO reaction (Fig. 5d) was carried out at 35 oC, and the produced PO was carried out from the MEA reactor by the gas phase and then collected by condensation in a condenser (Supplementary Figs. 24, 25).

a, b Device diagram of MEA reactor. c Physical image of MEA reactor. d Overall process chart of EOPO in MEA reactor. e PO production and FEs of EOPO on Ag-V-O/GDL catalyst in MEA reactor under different catholyte solutions. f, g Effect of ion exchange membranes on electrocatalytic propylene epoxidation on Ag-V-O/GDL catalyst in MEA reactor. h Effect of propylene flow rates on electrocatalytic propylene epoxidation on Ag-V-O/GDL catalyst in MEA reactor. i Effect of current densities on electrocatalytic propylene epoxidation on Ag-V-O/GDL catalyst in MEA reactor. j Stability test of Ag-V-O/GDL for electrocatalytic propylene epoxidation at 10 mA/cm² in MEA reactor. k Comparison of electrochemical performance of EOPO in three types of electrocatalytic reactors. The electrochemical tests in MEA reactors were conducted at 35 oC. All potentials were not iR corrected. Error bars represent the standard deviation from at least three independent measurements. Source data are provided as a Source Data file.
Therefore, a MEA reactor with a 2*2 cm2 electrode area was selected for continuous EOPO. To optimize experimental conditions, the following catholytes were compared initially: 0.1 M hydrochloric acid solution, 0.1 M phosphate buffer solution, 0.1 M KOH solution, and 0.1 M NaCl solution (Fig. 5e). When adopting KOH solution as the catholyte, the high pH of the catholyte hinders the HER on the cathode, thereby affecting the efficiency of EOPO on the anode. In comparison, although hydrochloric acid as the catholyte can accelerate HER at the cathode, the pH on the anode side is also decreased due to the suppressed penetration of OH−, leading to a reduction in FE during EOPO. A neutral catholyte can effectively balance the cathodic and anodic electrochemical reactions, improving the EOPO efficiency in MEA reactors. Therefore, a 0.1 M neutral NaCl aqueous solution was chosen as the low-cost catholyte for the MEA reactors. As the core component of MEA reactors, three types of membranes are compared in further: Nafion 117 proton exchange membrane, FKB-PK-130 cation exchange membrane, and FAA-3-PK-130 anion exchange membrane. Based on the experiment results (Fig. 5f, g), the acidic environments of the Nafion 117 membrane leads to further hydrolysis of PO to produce propylene glycol, thereby reducing the production and FE of PO (Supplementary Fig. 26). Moreover, the alkaline environments (FAA-3-PK-130 membrane) favors EOPO but also promotes the OER side reaction, leading to the fall of the FE towards PO (Supplementary Fig. 27). Therefore, the catalyst is more conducive to generate PO in a neutral environment, and FKB-PK-130 membrane exhibits the best performance. Subsequently, the effect of catholyte on the EOPO reaction was studied in MEA reactors with FKB-PK-130 membrane.
The constant current density and flow rate of propylene were optimized for effectively produce PO. The results, illustrated in Fig. 5h, indicate that the highest FE (30.4%) and production (227 μmol/h) of PO are achieved at a propylene flow rate of 10 standard cubic centimeters per minute (sccm) with the current density of 10 mA/cm2, both of which are the highest in MEA reactors among current reports (Supplementary Table 7). The FE and production of O2 are 27.9% and 209 μmol/h, respectively, indicating that the OER is the main side reaction during EOPO (Supplementary Fig. 28). As the current density increases, the production of PO gradually increases, while FE initially increases and then decreases. This decrease of FE is attributed to the enhanced OER at higher current densities. Compared to the Ag-O/GDL catalyst, the OER of the Ag-V-O/GDL catalyst is suppressed (Fig. 5i). In addition, the reaction temperature (35 oC) and propylene humidity (100%) were also optimized (Supplementary Figs. 29, 30). Long-term stability tests were conducted under the optimized reaction conditions (Fig. 5j). The Ag-V-O/GDL catalyst in the MEA reactor maintains an FE of over 27% for more than 78 hours. To demonstrate the unique advantages of MEA reactors, we compared EOPO in the MEA reactor to that in an H-cell and a flow cell (Supplementary Fig. 31) with the same catalyst. The EOPO reaction in H-cells exhibits a short duration of only 2-3 h without sustained production of PO (Supplementary Fig. 32). Although the efficiency of EOPO is enhanced in flow cells, its durability remains limited to ~ 4-5 h (Supplementary Fig. 33). The PO production in the MEA reactor is several times higher (22.2-fold vs. H-cell and 7.8-fold vs. flow cell) than that in the two other electrolytic cells (Fig. 5k and Supplementary Fig. 34).
Comparing with H-cells or flow cells, the MEA system integrates electrodes is separated by a solid electrolyte membrane, eliminating the need for ion transfer through the liquid electrolyte, which fundamentally avoids the issue of interfacial flooding. In addition, the solid electrolyte membrane can shorten the ion diffusion distance, providing lower resistance and efficient ion conduction, thus achieving higher energy utilization efficiency43,44,45. In MEA reactors, propylene can directly adsorb onto the solid/solid surface from the gas diffusion layer, reducing the impact of gas on the reaction rate, which is conducive to enhancing the stability and efficiency of the reaction. Besides, the developed “liquid-free” MEA system with the new design Ag-V bimetallic oxide electrocatalyst exhibits exceptional stability to prepare PO through continuous electro-epoxidation (activity does not decline after 78 h of electrocatalysis). Compared with previous works, our work avoids the use of organic or inorganic electrolytes, thereby reducing ohmic resistance and environmental pollution. More importantly, pure PO can be directly separated by cooling the gas products, a significant advantage not achieved in earlier works. Thus, the MEA system offers advantages in reducing energy losses and enabling continuous production, which are essential in practical industrial operations.
Discussion
This work successfully integrates a V-activated Ag-V-O/GDL bimetallic oxide catalyst into the MEA system, enabling the continuous production of PO through D-EOPO without involving any chloride species. In situ spectroscopy and theoretical calculations reveal the critical role of introduced V, which can accelerate the formation of Ag-O to decrease the generation energy barriers of O* from adsorbed water on the catalyst surface. Moreover, V doping induces a positive shift in the d-band center of Ag, facilitating the adsorption of propylene on the active center. Consequently, the formation of the key intermediate (OC3H6*) by coupling O* with adsorbed propylene is synergistically enhanced, thereby improving the efficiency of D-EOPO. In addition, the Ag-V-O/GDL catalyst exhibits higher conductivity compared to the Ag-O/GDL catalyst, which can reduce energy consumption during D-EOPO. As a result, stable PO production exceeding 227 μmol/h with an FE of 30.4% is achieved for over 78 h in the MEA reactor. Under higher current (40 mA) operation, the PO production in the MEA reactor is several times higher (22.2-fold vs. H-cell and 7.8-fold vs. flow cell) than that in the two other electrolytic cells, which is attributed to the smaller interelectrode gap and lower internal resistance. Our work offers new insights into catalyst and reactor design for electrocatalytic propylene epoxidation, suggesting that future research may focus on the design of electrocatalysts and device structures in MEA reactors to enhance conversion rate and selectivity.
Methods
Preparation of Ag-O catalyst
The pH of 50 mL of 0.02 mol/L NH3·H2O (25%, Sinopharm Chemical Reagent Co., Ltd) was adjust to 9 by using nitric acid (65%, Sinopharm Chemical Reagent Co., Ltd). Subsequently, 10 mL of 0.1 mol/L AgNO3 aqueous solution was slowly dripped into the above mixture, followed by stirring for 30 min. The resulting solution was sonicated in an ultrasonic cleaner (SK5200H, Shanghai Kudos Ultrasonic Instrument Co., Ltd.) for 6 h. The precipitate was washed with distilled water and anhydrous ethanol (99.5%, Sinopharm Chemical Reagent Co., Ltd) (reduce catalyst aggregation caused by water tension), then separated by centrifugation. The obtained sample was dried at 60 °C in an oven for 12 h to yield the Ag-O catalyst.
Preparation of Ag-V-O catalyst
10 mL of 0.1 mol/L AgNO3 (99%, Aladdin Co. Ltd.) aqueous solution was dripped into 50 mL of 0.02 mol/L of NH4VO3 (99%, Aladdin Co. Ltd.), whose pH had been adjusted to 9 using NH3·H2O (25%, Sinopharm Chemical Reagent Co., Ltd.). The mixture was stirred for 30 min. The resulting solution was sonicated in an ultrasonic cleaner (SK5200H, Shanghai Kudos Ultrasonic Instrument Co., Ltd.) for 6 h. The precipitate was washed with distilled water and anhydrous ethanol (99.5%, Sinopharm Chemical Reagent Co., Ltd.) and then separated by centrifugation. The obtained sample was dried at 60 °C in an oven for 12 h to yield the Ag-V-O catalyst.
Preparation of Ag4V2O7 catalyst
10 mL of 0.1 mol/L AgNO3 (99%, Aladdin Co. Ltd.) aqueous solution was dropped into 50 mL of 0.02 mol/L NH4VO3 (99%, Aladdin Co. Ltd.) aqueous solution. The pH value of the above solution was adjusted to 10 with NH3·H2O (25%, Sinopharm Chemical Reagent Co., Ltd.) and stirred for 30 min. After ultrasonic treatment (SK5200H, Shanghai Kudos Ultrasonic Instrument Co., Ltd.) for 6 h, the mixed solution was centrifuged to obtain a precipitate, which was then washed with distilled water and anhydrous ethanol (99.5%, Sinopharm Chemical Reagent Co., Ltd.). The obtained sample was dried in a drying oven at 60 oC for 12 h to obtain an Ag4V2O7 catalyst.
Preparation of Ag3VO4 catalyst
1 mmol of Na3VO4 (99%, Aladdin Co. Ltd.) was dissolved in 50 mL of water and stirred for 10 min to obtain the Na3VO4 solution. 10 mL of 0.1 mol/L AgNO3 (99%, Aladdin Co. Ltd.) aqueous solution was dropped into the aforementioned Na3VO4 solution and stirred for 30 minutes. After ultrasonic treatment for 6 hours, the mixed solution was centrifuged to obtain a precipitate, which was then washed with distilled water and anhydrous ethanol (99.5%, Sinopharm Chemical Reagent Co., Ltd.). The obtained sample was dried in a drying oven at 60 oC for 12 h to obtain Ag3VO4 catalyst.
Preparation of electrodes
2 mg of the catalyst, 10 µL of Nafion solution (5 wt%, 3AChem Co., Ltd.), and 1 mL of anhydrous ethanol (99.5%, Sinopharm Chemical Reagent Co., Ltd.) were mixed to form an ink, which was homogenized by the ultrasonic cleaner (SK5200H, Shanghai Kudos Ultrasonic Instrument Co., Ltd.). The ink was uniformly coated onto the carbon paper (YLS-30T, Suzhou Sinero Technology Co., Ltd.) with a loading of 0.5 mg/cm2. Ag-O/GDL, Ag-V-O/GDL, Ag4V2O7, and Ag3VO4 electrodes were obtained after drying.
Characterization
Scanning electron microscopy (SEM) images were acquired on a ZEISS Gemini 300 scanning electron microscope operating at an accelerating voltage of 1.5 kV. XRD pattern was collected on a Bruker AXS D8 X-ray diffractometer with Cu K ( = 1.54056 Å, 100 mA, and 40 kV) under a scan rate of 2 degrees/min. XPS spectra were first deoxygenated under Ar for 1 h and then investigated using a Thermo Scientific ESCALAB 250XI spectrometer equipped with an Al KX-ray source, and the spectra were calibrated with a binding energy of 284.6 eV for C 1 s.
Preparation of electrolytes
(1) The chemical reagents were weighed/measured.
0.1 M PBS (pH = 7): 1.8591 g of NaH₂PO₄ and 1.3852 g of Na₂HPO₄ (AR, Aladdin Co. Ltd.);
0.1 M NaCl (pH = 7): 1.4625 g NaCl (AR, Aladdin Co. Ltd.);
0.1 M HCl (pH = 1): 2.1 mL of HCl (37%, Aladdin Co. Ltd.);
0.1 M NaOH (pH = 13): 1 g NaOH (99%, Aladdin Co. Ltd.).
(2) The deionized water was added to dissolve the aforementioned chemical reagents completely. After cooling, the solution was transferred into a 250 mL volumetric flask using a glass rod. The beaker was then washed 2-3 times with deionized water, and the washings were transferred into the volumetric flask. Deionized water was slowly added to the volumetric flask until the liquid level was 1–2 cm below the calibration mark. Subsequently, deionized water was added dropwise using a dropper until the bottom of the meniscus was exactly tangent to the calibration line. The prepared solution should be stored in a fume hood and was used within 3 days.
Treatment of membranes
(1) The cation exchange membrane FKB-PK-130 (Fumatech, 2.5 × 2.5 cm2, 110–140 µm) was stored in a 1 wt% NaCl (AR, Aladdin Co. Ltd.) solution at 25 °C and rinsed with 0.5 M NaCl (AR, Aladdin Co. Ltd.) solution before use.
(2) The anion exchange membrane FAA-3-PK-130 (Fumatech, 2.5 × 2.5 cm2, 110–130 µm) was stored in a 1 M KOH (95%, Aladdin Co. Ltd.) solution at 25 °C and rinsed with deionized water before use.
(3) The proton exchange membrane Nafion 117 (Chemours, 2.5 × 2.5 cm2, 183 µm) was first treated with 5 wt% H₂O₂ (30 wt%, Aladdin Co. Ltd.) at 80 °C for 1 h, then rinsed repeatedly with deionized water and soaked for 30 min. Subsequently, it was treated with 5 wt% H₂SO₄ (95.0–98.0%, Sinopharm Chemical Reagent Co., Ltd.) solution at 80 °C for 1 h, rinsed thoroughly with deionized water, and finally soaked in deionized water.
Electrochemical measurement
All electrochemical experiments were conducted using the electrochemical workstation (CS310MA, Wuhan Corrtest Instrument Co., Ltd.) for all electrochemical measurements. The potential measured against the Ag/AgCl electrode can be converted to the reversible hydrogen electrode (RHE) scale using ERHE = EAg/AgCl + 0.197 + 0.0591 × pH. The reference electrode was calibrated in a 0.5 M H₂SO₄ solution. Prior to testing, the solution was saturated with hydrogen gas for 30 minutes. Platinum foil was used as both the working and counter electrodes, while an Ag/AgCl electrode served as the reference electrode. The cyclic voltammetry (CV) test was conducted at a scan rate of 1 mV/s. The onset potential of the hydrogen evolution reaction (HER) was used as the zero potential reference relative to the RHE. All potentials were not iR corrected. Error bars represent the standard deviation from at least three independent measurements.
H-cell
The homemade catalyst was employed as the working electrode (1 × 1 cm²). A platinum rod (Φ1 mm, Tianjin Ida Technology Development Co., Ltd.) and an Ag/AgCl electrode (Tianjin Ida Technology Development Co., Ltd.) were used as the counter and reference electrodes, respectively. The cathode and anode compartments were separated by a cation exchange membrane (FKB-PK-130, Fumatech). Each compartment contained 25 mL of 0.1 M PBS as the electrolyte. Before testing, the anolyte was saturated with propylene for 30 min. Propylene was introduced into the anode at the flow rate of 10 sccm during the reaction. The electrochemical tests were conducted at 25 oC.
Flow cell (microfluidic)
The homemade catalyst was used as the working electrode (1 × 1 cm2). A platinum rod (Φ1 mm, Tianjin Ida Technology Development Co., Ltd.) and an Ag/AgCl electrode (Tianjin Ida Technology Development Co., Ltd.) were used as the counter electrode and reference electrode, respectively. The cathode and anode compartments were separated by a cation exchange membrane (FKB-PK-130, Fumatech). Both compartments were continuously fed with 50 mL of 0.1 M PBS at a flow rate of 20 mL/min using a peristaltic pump. Before testing, the anolyte was saturated with propylene for 30 min. Propylene was introduced into the anode gas chamber at the flow rate of 10 sccm during the reaction. The electrochemical tests were conducted at 25 oC.
Membrane electrode assembly reactor
The homemade catalyst and nickel foam (1.5 × 210 × 250 mm, Suzhou Sinero Technology Co., Ltd.) were used as the work electrode (2 × 2 cm2) and counter electrode, respectively. The cathode and anode compartments were separated by a cation exchange membrane (FKB-PK-130, Fumatech). The cathode was continuously fed with 50 mL of 0.1 M NaCl at a flow rate of 20 mL/min using a peristaltic pump. Propylene, humidified by a humidifier (deionized water injected into the humidifier at a flow rate of 2 µL/min), was introduced into the anode gas chamber at a certain flow rate (5, 10, 20 sccm). The generated propylene oxide exited the membrane electrode reactor as a gas and was collected using the cold trap device. The electrochemical tests were conducted at 25, 35, 45, and 55 oC. The cell voltage of the MEA reactor in this paper was full-cell voltage.
Product analysis
The obtained propylene oxide (PO) and propylene glycol (PG) were quantified through gas chromatography (GC9790PLUS). The internal standard method was employed to detect the PO and PG production. Ethylene glycol dimethyl ether (C4H10O2, AR, Shanghai Mucklin Biochemical Technology Co., Ltd.) was used as the internal standard due to its chemical inertness and water solubility. The amount of the PO and PG can be calculated based on the relative correction factor. The calculation formula is given as following Eq. (1) and Eq. (2):
Relative correction factor:
Product mass:
Where f is the relative correction factor. As and ms are the peak area and mass of the added internal standard, respectively. Ar and mr is the peak area and mass of the added product, Ai and mi are the peak area and mass of the product in the test solution, respectively. To determine the relative correction factor f, standard samples were prepared as follows: 10 μL of PO/PG and 10 μL of the internal standard were added to 20 mL of electrolyte. 1 μL of this standard sample was analyzed by gas chromatography. The relative correction factor was calculated based on the average value obtained from multiple replicate analyses.
The quantification of O2 is carried out by adding Na2SO3 and BaCl2 solution to the cold trap device. The product enters the cold trap device to form a BaSO4 precipitate. The mass of the dried precipitate is measured to calculate the amount of O2. The quantification of CO2 is performed by adding Na₂SiO₃ solution into the cold trap device. The product enters the cold trap device to form H₂SiO₃ precipitate. The mass of the dried precipitate is measured to calculate the amount of CO2. Other by-products (carbon monoxide, methanol, methanol, and acetaldehyde) were quantified using a gas chromatography-mass spectrometry (GCMS-QP2020NX).
The calculation formulas for production and Faradaic efficiency are given as following Eq. (3), Eq. (4), and Eq. (5):
The molar amount of the product:
Production:
Faradaic efficiency:
Where ni represents the molar amount of the product in the test solution, Mi is the relative molecular mass of the product, FE is the Faradaic efficiency, z is the number of electrons involved in the reaction, F is the Faraday constant, I is the reaction current, and t is the reaction time.
The selectivity for the target product is determined by dividing the molar amount of the target product by the total molar amount of all products derived from propylene (PO, carbon dioxide, carbon monoxide, propylene glycol, methanol, methanol, and acetaldehyde). The calculation formula for selectivity is given as following Eq. (6):
Note: All data are averaged through at least three independent measurements.
Electrochemical impedance spectroscopy (EIS)
EIS measurements were performed in an H-cell or MEA reactor at 25 oC using the electrochemical workstation (CS310MA, Wuhan Corrtest Instrument Co., Ltd.). A platinum rod (Φ1 mm, Tianjin Ida Technology Development Co., Ltd.) and an Ag/AgCl electrode (Tianjin Ida Technology Development Co., Ltd.) were used as the counter electrode and the reference electrode, respectively. The EIS measurements were conducted in a 0.1 M PBS in the H-cell. The impedance spectra were recorded with an amplitude from 10 mV of 0.01–100 kHz. The data obtained from the EIS measurements were fitted by the Zview software (Version 3.1, Scribner Associates, USA).
ECSA measurements
The ECSA was measured by the double-layer capacitance (Cdl) of electrodes in 0.1 M PBS electrolyte in an H-cell using the electrochemical workstation (CS310MA, Wuhan Corrtest Instrument Co., Ltd.) (ECSA=Cdl/Cs, Cs is the specific capacitance). Firstly, electro-epoxidation was conducted at 10 mA/cm2 for 1 h. After electrolysis, the propylene feeding was immediately switched to Ar feeding, and then cyclic voltammetry was performed after the electrolyte stopped flowing. The scan rate ranged from 20 to 140 mV s−1. The function of current and scan rate was established to determine the Cdl.
In situ Raman spectroscopy
In situ Raman spectroscopy was performed using an inVia Reflex Raman spectrometer with laser excitation at 532 nm. The electrode was encased in a PEEK fitting, exposing a circular geometric surface area of ~ 1 cm2. A carbon paper coated with the catalyst was used as the anode. A platinum rod (Φ1 mm, Tianjin Ida Technology Development Co., Ltd.) and an Ag/AgCl electrode (Tianjin Ida Technology Development Co., Ltd.) were used as the counter electrode and the reference electrode, respectively46. The cathode and anode compartments were separated by a cation exchange membrane (FKB-PK-130, Fumatech). 0.1 M PBS was used as the electrolyte. The anolyte was saturated with propylene for 30 minutes before the test. The measurement range was recorded from 0.25 to 1.75 V vs. Ag/AgCl at intervals of 0.25 V (Supplementary Fig. 38).
In situ ATR-SEIRAS measurements
In situ ATR-SEIRAS spectroscopy was conducted using the MCT-A detector on the Thermo Scientific 8700 instrument. The catalyst loaded on Si prism, a platinum rod (Φ1 mm, Tianjin Ida Technology Development Co., Ltd.) and an Ag/AgCl electrode (Tianjin Ida Technology Development Co., Ltd.) were served as the working electrode, counter electrode, and reference electrode, respectively. The cathode and anode compartments were separated by a cation exchange membrane (FKB-PK-130, Fumatech). 0.1 M PBS was used as the electrolyte. The anolyte was saturated with propylene for 30 min before the test. The spectra were recorded within the range of 0.25 to 1.75 V (vs. Ag/AgCl) at intervals of 0.25 V (Supplementary Fig. 38).
Density functional theory (DFT) methods
All calculations are based on density functional theory (DFT) using the Vienna Ab initio Simulation Package (VASP)47. The Perdew-Burke-Ernzerhof functional and the projector augmented wave method are employed to describe exchange-correlation potentials and electron-nucleus interactions48,49,50. Ag4V2O7 is the main active component in the Ag-V-O/GDL catalyst for EOPO. XRD pattern indicates that the Ag-V-O/GDL tends to expose the Ag4V2O7(040) facet. Thus, the Ag4V2O7(040) facet was selected as the slab. The (040) facet of Ag4V2O7 contained 2 layers with a supercell formula of Ag32V16O56。 The energy cutoff is set at 400 electron volts, and convergence thresholds of 0.03 electron volts per angstrom for forces and 10^-5 electron volts for energies. A vacuum space of 15 Å was added in the Z-direction to avoid the interactions between each slab, part of the atoms in the Ag4V2O7 layer were fixed, and all other atoms including adsorbates, were allowed to relax. Weak van der Waals interactions in layered structures were corrected using the BJ damping DFT-D3 method48. The self-interaction error (SIE) affecting DFT when applied to tightly localized electrons is treated using the DFT + U approach within Dudarev’s rotationally invariant approximation implemented in VASP. An on-site correction (U-J) of 2.7 eV is applied to the V 3 d manifold51,52. The Gibbs free energy changes (ΔG) for the elementary steps in the proposed mechanism are calculated as follows: ΔG = ΔEDFT + ΔZPE -TΔS + eURHE. where ΔE represents the total energy difference, ΔZPE is the zero-point energy correction, ΔS is the entropy change, T was the ambient temperature, U(RHE) was the potential correction relative to the reversible hydrogen electrode (including pH correction). In the calculations, the Gibbs free energy was determined using the equation with T = 298.15 K and U(RHE) = 0 V. According to the model of computational hydrogen electrode, at standard conditions, the free energy change of the surface deprotonation process is equivalent to the hydrogen production, namely H+ + e− = 1/2 H253. The atomic coordinates of the optimized computational models in electronic structure calculations and the initial and final configurations in transition state calculations are provided in Supplementary Data 1.
Data availability
The data generated in this study are provided in the Supplementary Information and Source Data file. Source data are provided in this paper.
References
Childers, M. I. et al. Isospecific, chain shuttling polymerization of propylene oxide using a bimetallic chromium catalyst: A new route to semicrystalline polyols. J. Am. Chem. Soc. 139, 11048–11054 (2017).
Martín, C. et al. Recent advances in the catalytic preparation of cyclic organic carbonates. ACS Catal. 5, 1353 (2015).
Exerowa, D. & Platikanov, D. Thin liquid films from aqueous solutions of non-ionic polymeric surfactants. Adv. Colloid Interface Sci. 147, 74–87 (2009).
Nijhuis, T. A. et al. The production of propene oxide: Catalytic processes and recent developments. Ind. Eng. Chem. Res. 45, 3447–3459 (2006).
Gordon, C. P. et al. Efficient epoxidation over dinuclear sites in titanium silicalite-1. Nature 586, 708–713 (2020).
Zhang, P., Wang, T. & Gong, J. Advances in electrochemical oxidation of Olefins to epoxides. CCS Chem. 5, 1028 (2023).
Jin, W. et al. Selective electrochemical oxidation of alkene: Recent progress and perspectives. Chinese J. Catal. 53, 34–51 (2023).
Leow, W. R. et al. Chloride-mediated selective electrosynthesis of ethylene and propylene oxides at high current density. Science 368, 1228–1233 (2020).
Chung, M. J. et al. Mechanism of chlorine-mediated electrochemical ethylene oxidation in saline water. ACS Catal. 10, 14015–14023 (2020).
Li, Y. et al. Redox-mediated electrosynthesis of ethylene oxide from CO2 and water. Nat. Catal. 5, 185–192 (2022).
Ke, J. et al. Facet-dependent electrooxidation of propylene into propylene oxide over Ag3PO4 crystals. Nat. Commun. 13, 932 (2022).
Iguchi, S. et al. Direct epoxidation of propylene with water at a PtOx anode using a solid-polymer-electrolyte electrolysis cell. Catal. Sci. Tech. 12, 469 (2022).
Chung, M. J. et al. Direct propylene epoxidation via water activation over Pd-Pt electrocatalysts. Science 383, 49–55 (2024).
Winiwarter, A. et al. Towards an atomistic understanding of electrocatalytic partial hydrocarbon oxidation: propene on palladium. Energy Environ. Sci. 12, 1055–1067 (2019).
Dustin, B. et al. Critical advancements in achieving high power and stable nonprecious metal catalyst–based MEAs for real-world proton exchange membrane fuel cell applications. Sci. Adv. 4, eaar 7180 (2018).
Breitwieser, M. et al. Tailoring the membrane-electrode interface in PEM fuel cells: A review and perspective on novel engineering approaches. Adv. Energy Mater. 8, 1701257 (2017).
Fan, J. et al. Bridging the gap between highly active oxygen reduction reaction catalysts and effective catalyst layers for proton exchange membrane fuel cells. Nat. Energy 6, 475–486 (2021).
Lees, E. W. et al. Gas diffusion electrodes and membranes for CO2 reduction electrolysers. Nat. Rev. Mater. 7, 55–64 (2022).
Zhang, Z. et al. Membrane electrode assembly for electrocatalytic CO2 reduction: Principle and application. Angew. Chem. Int. Ed. 135, 202302789 (2023).
Kwak, H. et al. Emerging halide superionic conductors for all-solid-state batteries: Design, synthesis, and practical applications. ACS Energy Lett. 7, 1776–1805 (2022).
Lin, X. et al. Direct oxygen transfer from H. Angew. Chem. Int. Ed. 61, e202207108 (2022).
Jin, K. et al. Epoxidation of cyclooctene using water as the oxygen atom source at manganese oxide electrocatalysts. J. Am. Chem. Soc. 141, 6413–6418 (2019).
Chung, M. et al. Tuning single-atom dopants on manganese oxide for selective electrocatalytic cyclooctene epoxidation. J. Am. Chem. Soc. 144, 17416–17422 (2022).
Lei, Y. et al. Increased silver activity for direct propylene epoxidation via subnanometer size effects. Science 328, 224–228 (2010).
Ghosh, S. et al. Selective oxidation of propylene to propylene oxide over silver-supported tungsten oxide nanostructure with molecular oxygen. ACS Catal. 4, 2169–2174 (2014).
Guo, J. et al. Efficient visible-light driven photocatalyst, silver (meta) vanadate: synthesis, morphology and modification. Chem. Eng. J. 352, 782–802 (2018).
Wang, S. et al. Fabrication of a novel bifunctional material of BiOI/Ag3VO4 with high adsorption–photocatalysis for efficient treatment of dye wastewater. Appl. Catal. B Environ. 168, 448–457 (2015).
Zhu, T. et al. Synthesis of g-C3N4/Ag3VO4 composites with enhanced photocatalytic activity under visible light irradiation. Chem. Eng. J. 271, 96–105 (2015).
Li, Z. et al. V “Bridged” Co-O to eliminate charge transfer barriers and drive lattice oxygen oxidation during water-splitting. Adv. Funct. Mater. 31, 2008822 (2021).
Li, Z. et al. Anion-modulated HER and OER activities of 3D Ni–V-based interstitial compound heterojunctions for high-efficiency and stable overall water splitting. Adv. Mater. 31, 1901174 (2019).
Ferraria, A. M., Carapeto, A. P. & do Rego, A. M. B. X-ray photoelectron spectroscopy: Silver salts revisited. Vacuum 86, 1988–1991 (2012).
Chen, D., Kang, P. L. & Liu, Z. P. Active site of catalytic ethene epoxidation: machine-learning global pathway sampling rules out the metal sites. ACS Catal. 11, 8317–8326 (2021).
Teran, J. et al. Propylene epoxidation using molecular oxygen over copper- and silver-based catalysts: A review. ACS Catal. 10, 13415–13436 (2020).
Cao, X. et al. Strong p‐d orbital hybridization on bismuth nanosheets for high performing CO2 electroreduction. Adv. Mater. 36, 2309648 (2024).
Yang, J. et al. Vanadium hexacyanoferrate prussian blue analogs for aqueous proton storage: Excellent electrochemical properties and mechanism insights. Small 20, 2305386 (2023).
Tiancheng, P. et al. Revealing the nature of active oxygen species and reaction mechanism of ethylene epoxidation by supported Ag/α-Al2O3 catalysts. ACS Catal. 14, 406–417 (2024).
Thyr, J. & Edvinsson, T. Evading the illusions: Identification of false peaks in micro‐raman spectroscopy and guidelines for scientific best practice. Angew. Chem. Int. Ed. 62, e202219047 (2023).
Marinov, A. D. et al. Ex situ characterization of 1T/2H MoS2 and their carbon composites for energy applications, a review. ACS Nano 17, 5163–5186 (2023).
Liu, C. et al. Computational and experimental insights into reactive forms of oxygen species on dynamic Ag surfaces under ethylene epoxidation conditions. J. Catal. 405, 445–461 (2022).
Xiong, W. et al. Author Correction: Fine cubic Cu2O nanocrystals as highly selective catalyst for propylene epoxidation with molecular oxygen. Nat. Commun. 12, 6429 (2021).
Liu, X. C. et al. Reaction mechanism and selectivity tuning of propene oxidation at the electrochemical interface. J. Am. Chem. Soc. 144, 20895–20902 (2022).
Huš, M. et al. Going beyond silver in ethylene epoxidation with first‐principles catalyst screening. Angew. Chem. Int. Ed. 62, e202305804 (2023).
García de Arquer, F. Pelayo et al. CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2. Science 367, 661–666 (2020).
Xing, Z. et al. Enhancing carbon dioxide gas-diffusion electrolysis by creating a hydrophobic catalyst microenvironment. Nat. Commun. 12, 136 (2021).
Zhong, Y. et al. An artificial electrode/electrolyte interface for CO2 electroreduction by cation surfactant self‐assembly. Angew. Chem. Int. Ed. 59, 19095–19101 (2020).
Wang, J. et al. Selective electrochemical oxidation of alkene: Recent progress and perspectives. Chinese J. Catal. 53, 34–51 (2023).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set - ScienceDirect. Comp. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Kieron, B. & Matthias, E. Generalized gradient approximation made simple. Phys. Rev. Lett. 78, 1396 (1997).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 59, 1758 (1999).
Huang, S. et al. Strain-tunable van der Waals interactions in few-layer black phosphorus. Nat. Commun. 10, 2447 (2019).
Grimme, S. et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Zhao, C. X. et al. Multiscale construction of bifunctional electrocatalysts for long-lifespan rechargeable Zinc–air batteries. Adv. Funct. Mater. 11, 1789 (2022).
NΦrskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. C. 108, 17886–17892 (2004).
Acknowledgements
We acknowledge the National Key R&D Program of China (SQ2022YFE020274, Z.L.), the National Natural Science Foundation of China (21572269, Z.L. and 52403355 Y.L.), the China Postdoctoral Science Foundation (BX20240188, Y.L. and 2024M751616, Y.L.), and the Fundamental Research Funds for Chinese Central Universities (27RA2304013, Z.L).
Author information
Authors and Affiliations
Contributions
Z.L. supervised the project. Z.L., Y.L., H.L., and X.M. conceptualized the project. Y.L., H.L., X.M., X.Y., W.C., and M.W. conducted the experiments and the related data processing. H.R. and Y.S. carried out the theoretical calculations. All authors participated in the discussion of the research.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, Y., Li, H., Miao, X. et al. V activated electro-epoxidation catalyst in membrane electrode assembly system for the production of propylene oxide. Nat Commun 16, 3113 (2025). https://doi.org/10.1038/s41467-025-58486-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58486-y
