
Abstract
Ceftazidime-avibactam is a β-lactam/β-lactamase inhibitor combination restricted for the treatment of multidrug-resistant infections of Pseudomonas aeruginosa non-susceptible to ceftazidime and resistant to carbapenems. Crucially, it has not been studied if its use could allow the design or application of new or stablished evolution-based strategies that exploit the increased susceptibility that emerges when resistance is acquired (collateral sensitivity, CS). Works in the field have focused on the study of CS in model strains, but to be exploited it must robustly emerge in pre-existing resistant mutants that can coexist in a patient. This is the first analysis of CS robustness on this last-resort drug. We evolved 15 clinical isolates on ceftazidime-avibactam and in absence of inhibitor, and here we show that we found no robust -exploitable- pattern of CS. This, together with the selection of cross-resistance and the impossibility of using previously described CS-based strategies, supports that avibactam should be restricted for the treatment of particular genotypes.
Similar content being viewed by others


Introduction
Antibiotic-resistant bacteria are a growing threat to human health1. Among the bacterial pathogens that cause great concern, Pseudomonas aeruginosa stands out2,3. This opportunistic pathogen is a major cause of nosocomial and chronic infections in patients with cystic fibrosis or chronic obstructive pulmonary disease4,5,6. This ESKAPE bacterium presents low intrinsic susceptibility to antibiotics7,8 and can acquire antibiotic resistance by mutations9,10. Of special concern is the existence of high-risk epidemic clones of P. aeruginosa that present multidrug and extensively drug resistance (MDR and XDR respectively) phenotypes11,12, for which the activity of ceftazidime is increasingly compromised, particularly in mutants overproducing the chromosomal β-lactamase AmpC13. The combination ceftazidime-avibactam, a third-generation cephalosporin and a non-β-lactam β-lactamase inhibitor14, respectively, has high activity against Class A, Class C, and some Class D β-lactamases15. Therefore, the combination ceftazidime-avibactam is reserved for the treatment of multidrug-resistant strains non-susceptible to ceftazidime and resistant to carbapenems13,16,17,18. However, it is unknown the extent to which the use of avibactam may alter the development of resistance, the selection of specific ceftazidime resistance mutations and, importantly, the acquisition of collateral sensitivity (CS), by which increased resistance to a drug leads to susceptibility to another opening alternative therapeutic strategies19, something addressed here.
In an scenario in which we are increasingly pushed to use last resort therapies, new therapeutic strategies that improve the efficacy of available antibiotics20 and that -counterintuitively- are based on the cause of the problem, bacterial evolution, are urgently needed21. A possibility is finding robust evolutionary trade-offs, as CS, that emerge in the presence of a drug in different genomic backgrounds of P. aeruginosa (i.e., mutants previously resistant to antibiotics or varied clinical strains), something scarce in the field22,23,24. The reason for looking for conserved susceptible phenotypes is that patients with cystic fibrosis infected by P. aeruginosa can harbor different antibiotic resistant mutants25, due to their prolonged exposure to different antibiotic therapies10. Therefore, each of these mutants -presenting a different mutational resistome9,26- may evolve differently in the presence of the same drug when the patient is treated. This is due to epistatic and pleiotropic phenomena that shape the fitness costs of antibiotic resistance mutations, limiting those that can be fixed in each particular genomic background27,28,29,30. In fact, small genetic alterations can cause important changes in the evolutionary trajectories of antibiotic resistance and in their associated CS31. This explains why, in order to identify effective treatments against P. aeruginosa infections based on CS robustness, it is necessary to understand the different resistance mechanisms that may arise in different clinical strains exposed to an antibiotic, and the associated trade-offs.
In this work, we performed adaptive laboratory evolution (ALE) assays on ceftazidime and ceftazidime-avibactam in 15 clinical isolates of P. aeruginosa presenting 10 different sequence types (STs) and, having different antibiotic susceptibility profiles and mutational resistomes. We observed that the combination selects mechanisms of resistance generally different from those selected in the absence of inhibitor, that avibactam reduces the level of resistance to ceftazidime acquired in an important number of clinical strains but selects cross-resistance to other drugs (i.e., ciprofloxacin), and that, unfortunately, no conserved patterns of CS emerge that might allow us to propose new evolutionary-based strategies making a rational use of current antibiotics. Moreover, we observed that the use of ceftazidime-avibactam may prevent the application of a previously described combined treatment based on CS (ciprofloxacin-tobramycin)22,23 that is capable of eradicating various resistant mutants and clinical strains of P. aeruginosa. This indicates that the use of the last-resort drug ceftazidime-avibactam, in case of failure, may also close the door to other therapies. For all these reasons, we propose that the use of avibactam should be restricted to limited infection cases.
Results
Evolution of resistance after short-term evolution on ceftazidime or ceftazidime-avibactam
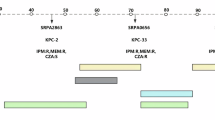
Fifteen ceftazidime susceptible clinical isolates from different Spanish hospitals and geographical areas (Table 1), belonging to ten different STs and presenting different antibiotic susceptibility profiles and mutational resistomes, were used in this work. In addition to studying ten different STs, we decided to include six different isolates of ST253, which differ in a range between 110 and 460 alleles, based in a whole genome multilocus sequence typing (MLST) analysis (Methods). The reason for including six ST253 strains is that the susceptible model strain of P. aeruginosa PA14, which is regularly used in predictive studies of antibiotic resistance evolution, belongs to ST253, an epidemic P. aeruginosa clone32. Consequently, this set of strains would allow us to evaluate, using ALE assays, the effect of intra-clonal and inter-clonal variations on the robustness of ceftazidime and ceftazidime-avibactam resistance evolution in P. aeruginosa, as well as to validate previous results obtained using PA14 as a model22,24,33,34,35. For that, four biological replicates of the 15 clinical isolates were submitted to ALE during 3 days in presence of ceftazidime, ceftazidime-avibactam or absence of antibiotic (control populations), with daily sequential dilutions in test tubes (180 populations). To analyze the evolution of ceftazidime resistance in absence and presence of inhibitor, ceftazidime minimal inhibitory concentrations (MICs) were measured in the final populations and they were compared with those of the parental strains. MIC variations above 2-fold (analyzed using E-test strips, which discriminate slight MIC variations) indicated biologically relevant changes of resistance level, as previously described22,36. An increase of ceftazidime MICs, from 2-fold up to 85-fold, was observed in the populations evolved in the presence of ceftazidime or ceftazidime-avibactam with respect to their parental strains (Fig. 1; Supplementary Data 1). In contrast, no statistically or biologically significant changes of ceftazidime MICs, with respect to their parental strains, were observed in the control populations submitted to ALE in the absence of antibiotic (Fig. 1; Supplementary Data 1). Importantly, 43 out of 60 ceftazidime evolved populations presented a ceftazidime MIC above EUCAST clinical breakpoint (8 µg/ml) after ALE, while 21 of 60 ceftazidime-avibactam evolved populations presented a ceftazidime MIC above the clinical breakpoint (Supplementary Data 1). These differences (p < 0.001) support that the use of avibactam may reduce, to a certain degree, the evolution of high levels of ceftazidime resistance. In this regard, it is important noticing that in ceftazidime evolved populations 47 out of 60 populations presented a biologically-significant reduction in the MIC to ceftazidime plus avibactam (Fig. 1, Supplementary Data 1), while only 21 out of 60 ceftazidime-avibactam evolved populations showed a significant reduction of the MIC to ceftazidime plus avibactam, being these differences statistically significant (p < 0.001). These results indicate that while the combination of ceftazidime-avibactam may be useful against ceftazidime-resistant populations arisen after ceftazidime use, the effectiveness may rapidly drop after its use as illustrated by the ceftazidime-avibactam selected populations, suggesting that avibactam should be used with great caution.

Resistance to ceftazidime and ceftazidime-avibactam was analyzed in 15 clinical isolates submitted to ALE in the presence of ceftazidime (CAZ; left panel) or ceftazidime-avibactam (CZA; right panel) for 3 days, 4 replicate populations each (120 populations). Only variations in MICs greater than 2-fold were considered biologically relevant to classify a population as “resistant” (purple), since control populations evolved in the absence of antibiotic (60 populations) may present subtle changes (below 2-fold) in their susceptibility to antibiotics with respect to the MIC of parental strains (22). MIC values (µg/ml) of all the evolved populations are included in Supplementary Data 1. Log2 (fold change MIC) values are represented in the diagram.
On the other hand, it is important noticing that the greatest increase of ceftazidime resistance occurred in the isolates ARA2-005, MAD04-002 and GAL02-002 after short term ALE on ceftazidime (p < 0.05) while the highest increase occurred in the isolates ARA02-005 and FQSE11-1010 after ceftazidime-avibactam ALE (p < 0.05). These results indicate that some genomic backgrounds, such as ARA02-005 (from ST267), are more prone to acquire a higher level of ceftazidime resistance than others in the presence of ceftazidime or ceftazidime-avibactam, something that does not appear to be determined by specific ST. This raises the question of whether these differences in resistance levels observed between the different isolates were acquired through the same mechanism of resistance, leading to different resistance levels depending on the original genomic background, or whether different resistance mechanisms were selected in the different clinical strains.
Lack of conservation of collateral sensitivity in the evolved populations
To analyze the possible emergence of cross-resistance and CS to other antibiotics in the set of clinical isolates of P. aeruginosa submitted to the short-term ALE on ceftazidime-avibactam and in the absence of inhibitor, MICs of different antibiotics were determined for each final population and its parental strain (Fig. 2; Supplementary Data 1). Setting a 2-fold change in MIC (analyzed by E-test strips) as biologically significant, as previously described22,37, we observed that most of the populations evolved in the presence of either ceftazidime or ceftazidime-avibactam presented cross-resistance to other β-lactams (aztreonam or meropenem), with no statistically-significant differences between treatments. In addition, cross-resistance to ciprofloxacin was observed in 9 populations evolved on ceftazidime and in 23 populations evolved on ceftazidime-avibactam. This higher frequency of cross-resistance to ciprofloxacin acquired after the use of the combination (p < 0.001), possibly explained by the mechanisms of ceftazidime resistance fixed in these populations, further highlights the risks associated with the use of the combination ceftazidime-avibactam instead of ceftazidime alone. CS to fosfomycin and, to a lesser degree, tobramycin was observed in some of the evolved populations (Fig. 2; Supplementary Data 1), but these patterns were far from robust, especially in the populations evolved in the presence of avibactam, something that contributes to discourage its use in the design of rational -alternated or combined- therapies based on evolution. In particular, CS to fosfomycin emerged in the genetic backgrounds MAD02-004, MAD04-002, CLE03-004, CAT09-003, FQSE11-1010, BAL02-001, and GAL02-002 after short term ALE on ceftazidime, as well as in the strains AST01-034, MAD02-004, CLE03-004, GAL02-002, and FQSE10-0503 after the use of ceftazidime-avibactam. This supports that, for some infections and specific clones, the alternation or combination of ceftazidime with fosfomycin might be promising, as we previously suggested33,34. However, the fact that ceftazidime-avibactam use leads to CS to tobramycin or fosfomycin only in 20% and 37%, respectively, of the populations, makes difficult to suggest the alternation or combination of ceftazidime-avibactam with tobramycin or fosfomycin. In this sense, a much more promising strategy for the treatment of diverse clinical strains, as those analyzed in this study, would be the use of the ciprofloxacin-tobramycin combination, since it has been previously shown that ciprofloxacin resistance evolution, despite occurring through different genetic mechanisms, robustly converges in an increase of susceptibility to tobramycin23. However, as we see here when using ceftazidime-avibactam, CS to fosfomycin is far from emerging robustly and, therefore, great efficacy is unlikely to be expected when using ceftazidime-avibactam-fosfomycin strategies to treat heterogeneous infections. The reason for differences in robustness of CS associated with the use of ceftazidime-avibactam and ciprofloxacin may be that the variety of resistance mechanisms selected on the first is high (see below) compared to those selected on ciprofloxacin22,23 and most of them do not present an associated CS pattern. In fact, from an evolutionary point of view, it is unlikely -but clinically relevant- that different resistance mechanisms, acquired “at a whim” of historical contingency in different isolates, may lead to the same pattern of CS (but see refs. 21,22,23).

MICs to five antibiotics were measured in populations from 15 clinical isolates (4 replicate populations each) submitted to ALE in the presence of ceftazidime (CAZ: left panel) or ceftazidime-avibactam (CZA: right panel) for 3 days (120 populations). Intensity of the color is proportional to the log-transformed fold change of MIC of the evolved populations with respect to the MIC of the respective parental strain. Variations of MICs above or below an increase or a decrease of 2-fold, respectively, were considered biologically relevant to classify a population as “resistant” (purple) or “susceptible” (orange) (22). MIC values (µg/ml) are included in Supplementary Data 1. Log2 (fold change MIC) values are represented in the diagram. TOB tobramycin, FOF fosfomycin, CIP ciprofloxacin, ATM aztreonam, MER meropenem.
Genetic variations responsible for resistance in the evolved clinical strains of P. aeruginosa
To understand the genetic causes of the acquisition of ceftazidime and ceftazidime-avibactam resistance, as well as the reasons for the lack of conservation of CS in these populations, they were subjected to whole-genome sequencing, together with a representative set of control populations (evolved in the absence of antibiotic) which did not acquire resistance mutations. The mutational resistome of each evolved population was compared to that of the respective parental strain, identifying the genetic variations acquired during evolution. Acquired mutations for each of the evolved populations in the presence of ceftazidime or ceftazidime-avibactam are shown in Supplementary Data 1. When genetic variations were detected in frequencies below an established breakpoint (10% of reads), individual clones were isolated, phenotyped (determination of MICs to ceftazidime and ceftazidime-avibactam) and sequenced to validate the population analysis. Importantly, we observed that the acquisition of genetic variations in these genes is dependent on the genomic background, even for the six isolates belonging to ST253, which do not present a common pattern (Supplementary Data 1; Fig. 3): dacB variants were acquired in 10 and 4 different clinical isolates evolved on ceftazidime and ceftazidime-avibactam, respectively, ampR variants were detected in 4 and 1 different clinical isolates evolved on ceftazidime and ceftazidime-avibactam, respectively, mexR variants were detected in 2 and 5 different clinical isolates evolved on ceftazidime and ceftazidime-avibactam, respectively and galU genetic variations were acquired in 4 and 3 different clinical isolates evolved on ceftazidime and ceftazidime-avibactam, respectively. In addition, variations in other genes (i.e., PBP3, mpl, cpxS, PA1438-1436) were acquired in specific clinical strains (Fig. 3).

Classical mutations regularly implicated in ceftazidime resistance in clinical settings were acquired (color boxes) during 3 days of ALE in the presence of ceftazidime (CAZ: green) or ceftazidime-avibactam (CZA: purple) in, at least, one population belonging to 15 different clinical isolates. The main mechanism of resistance selected in the absence of avibactam is the hyper-production of the β-lactamase AmpC, caused by mutations in dacB, ampR, ampD, or mpl. In contrast, the most frequent mechanism of ceftazidime resistance selected in the presence of avibactam is the hyper-production of the efflux pump MexAB-OprM, caused by mutations in its regulators mexR, nalC, or nalD. As observed, genomic background restricts early steps of resistance evolution (p = 0.024) (Supplementary Data 1).
A comparative analysis of the resistance mechanisms selected in the presence of ceftazidime, with or without avibactam, in the different populations was performed too (Fig. 4). As shown, the main mechanism of resistance selected in the absence of avibactam was the hyper-production of the β-lactamase AmpC, caused by mutations in dacB (most frequently), ampR, ampD, or mpl (Fig. 3; Supplementary Data 1). Selection of AmpC hyper-producer mutants was however strain-dependent, consistent with our previous findings, indicating that the β-lactam resistance response driven by DacB or AmpD inactivation varies among genetically-diverse strains13,38,39,40,41,42. It is also important noticing that, in some clinical strains, the presence of avibactam did not avoid the selection of mutations leading to the hyper-production of the β-lactamase AmpC (ARA02-005, AST01-034, CAT09-004, and FQSE11-1010). In contrast, the most frequent mechanism of ceftazidime resistance selected in the presence of avibactam was the hyper-production of the efflux pump MexAB-OprM, caused by mutations in its regulators mexR, nalC, or nalD43,44,45, which generally display a higher ciprofloxacin resistance (p < 0.001) as well. Moreover, in agreement with previous works34,35,41,46, some strains evolved in the presence of ceftazidime or ceftazidime-avibactam developed the loss of galU, encoding an enzyme whose inactivation reduces susceptibility to ceftazidime and other β -lactams47 and often linked to the loss of hmgA and mexXY, that cause pyomelanine production48 and CS to aminoglycosides24,49, respectively. Other mutations acquired during short-term ALE on ceftazidime-avibactam occurred in the PA1435-PA1436 (PA14_45910-45890) and PA1437-PA1438 (PA14_45880-45870) operons, encoding a probable RND efflux transporter and a two-component system, respectively, the last likely regulating the expression of the first, as previously described in ALE assays of the model strain of P. aeruginosa PA14 in the presence of ceftazidime-avibactam35. Moreover, 3 out of 4 populations from the clinical strain GAL02-002 exposed to ceftazidime-avibactam evolved towards an hyper-mutator phenotype due to the acquisition of a frameshift in the gene encoding the DNA mismatch repair protein MutS50, therefore causing an increase in the number of acquired mutations in the populations and presenting, among others, mutations in the mentioned PA1437-PA1438 operon. This is something we previously observed in the presence of ceftazidime-avibactam in ALE assays of the model strain of P. aeruginosa PA1434 but, the fact that hyper-mutators only emerged in 1 out of 15 isolates evolved on ceftazidime-avibactam (and just in 3 out of 4 populations from the mentioned isolate) and the fact that these mutants were not selected in isolates from ST253 (to which PA14 belongs), question the potential ability of avibactam to robustly promote this phenotype in P. aeruginosa. Finally, we detected in two different clinical strains a mutation in cpxS, encoding the sensor of the two-component system CpxSR involved in cellular hysteresis to β -lactam antibiotics and resistance to novel antipseudomonal β -lactams, as ceftolozane/tazobactam and cefiderocol (50-52)51,52,53. Overall, these results indicate that, while the mechanisms of resistance selected by ceftazidime and ceftazidime-avibactam are, in principle, limited (Fig. 4), the involved gene mutations are numerous (Fig. 3) and therefore, the emergence of a robust -exploitable- pattern of CS is less frequent than with the association to other drugs, such as ciprofloxacin22,23.

Pathways evolved in, at least, 2 of 4 populations for each of the 15 different clinical isolates are shown in the graph. The arrow indicates gene overexpression and Δ indicates gene deletion. Ceftazidime-avibactam (CZA) selected for mechanisms of resistance generally different from the ones selected in the absence of inhibitor (CAZ), which frequently lead to the over-expression of the ampC β-lactamase. However, mutations leading to this overexpression were acquired in the presence of ceftazidime-avibactam in some specific clinical strains. The overexpression of genes encoding efflux pumps was the main mechanism of resistance selected in ceftazidime-avibactam. Statistical significance (is indicated with an asterisk (ampC, p = 0.0011; mexAB, p < 2.2e-16).
Incapacity of a previously described collateral sensitivity-based evolutionary strategy to drive ceftazidime-avibactam resistant strains to extinction
In this work we did not identify a robust CS pattern that could emerge in distinct resistant isolates of P. aeruginosa (Table 1). Having in mind that clinicians may use ceftazidime-avibactam for the treatment of difficult infections and that resistance to this last resort drug rapidly emerges, we analyzed if it could be possible to apply the previously described antibiotic combination ciprofloxacin-tobramycin22,23 to ceftazidime-avibactam resistant populations. The reason for choosing this strategy is that we have recently described a great efficacy of the combination ciprofloxacin-tobramycin (associated with robust CS to tobramycin) against varied pre-existing antibiotic-resistant mutants and clinical strains of P. aeruginosa22,23. For that, we chose as representative population 1 from 5 different clinical isolates (MAD02-004, CLE03-004, BAL02-001, CLM02-004 and MAD04-002) previously evolved on ceftazidime-avibactam, presenting a robust increase of ceftazidime-avibactam and of ciprofloxacin resistance (being still susceptible based on clinical standards, MICs below 0.5 µg/ml). In addition, we included population 1 from the same 5 clinical isolates (MAD02-004, CLE03-004, BAL02-001, CLM02-004 and MAD04-002) previously evolved on ceftazidime. We evolved these populations in the presence of ciprofloxacin, tobramycin or the ciprofloxacin-tobramycin combination, being the last our previously described CS-based therapeutic option22,23. We submitted 4 replicate populations from each ceftazidime-avibactam or ceftazidime resistant population to the drug combination ciprofloxacin-tobramycin (40 populations), as well as to the 2 antibiotics separately, using the same concentrations present in each drug pair (80 control populations) (Methods). As shown in Fig. 5A, only 30% of the ceftazidime-avibactam resistant populations (6 out of 20) submitted to short-term ALE in the presence of ciprofloxacin-tobramycin became extinct (p < 0.001), while all the ceftazidime resistant populations (20 out of 20) became extinct (Fig. 5B) (p < 0.001). As expected, every control population thrived. In addition, we measured susceptibility levels to tobramycin and ciprofloxacin in final ceftazidime-avibactam resistant populations that grew on ciprofloxacin-tobramycin, confirming that they acquired resistance to both drugs during the ALE assays. In particular, the MIC of tobramycin and ciprofloxacin in BAL02-001, MAD02-004, MAD04-002 and CLM02-004 increased from 1.5 to 6 µg/ml, 0.38 to 0.75 µg/ml, 3 to 6 µg/ml or 1 to 6 µg/ml, and from 0.19 to 0.5 µg/ml, 0.25 to 0.5 µg/ml, 0.5 to 1 µg/ml or 0.38 to 0.75 µg/ml, respectively. These worrisome results indicate that the clinical use of the last resort combination ceftazidime-avibactam may not only have limited effectiveness, but may also limit the efficacy of other promising CS-based therapeutic strategies for the treatment of various resistant mutants and clinical strains of P. aeruginosa. Interestingly, loss-of function mutations in mexR are present in the three ceftazidime-avibactam resistant populations where the ciprofloxacin-tobramycin therapy failed, suggesting that these mutations may limit the efficacy of our previously described CS-based therapeutic strategy. To test this hypothesis, we included in this analysis the population 4 from the clinical isolate BAL02-001 previously evolved on ceftazidime, which acquired a genetic variation in mexR. None of the four BAL02-001 CAZ.4 replicate populations became extinct in the presence of the combination ciprofloxacin-tobramycin, while all the BAL02-001 CAZ.1 replicate populations, lacking mexR mutations, became extinct. These results support that MexAB overproducer mutants (very frequently selected by ceftazidime-avibactam) may escape from the ciprofloxacin-tobramycin strategy. Nevertheless, it will be necessary to delve deeper into this to determine the robustness of said evolutionary restriction and understand the molecular causes.

A Short-term evolution of population 1 from 5 different clinical isolates (MAD02-001, CLE03-004, CLM02-004, BAL02-001, and MAD04-002) previously evolved on ceftazidime-avibactam (CZA), represented as bacterial cells with different colors, 4 replicate populations of each parental population, was performed during 3 days in the presence of the ciprofloxacin-tobramycin (CIP-TOB) combination. Growth of the 40 control populations was confirmed in the 2 drugs independently used, at the same concentrations used for the drugs combinations. Extinct populations at the end of the experimental evolution are represented in black, while surviving populations are represented in gray. 6 out of 20 ceftazidime-avibactam resistant populations 1 submitted to short-term ALE in the presence of the antibiotic combination became extinct. These results are statistically significant (p = 0.002) and indicate that the ciprofloxacin-tobramycin CS-based therapeutic strategy previously proposed for the treatment of concern clinical strains may be ineffective on strains resistant to the last-resort combination ceftazidime-avibactam; especially those presenting mutations in mexR. B Short-term evolution of population 1 from 5 different clinical isolates (MAD02-001, CLE03-004, CLM02-004, BAL02-001 and MAD04-002) previously evolved on ceftazidime (CAZ), represented as bacterial cells with different colors, 4 replicate populations of each parental population, was performed during 3 days in the presence of the ciprofloxacin-tobramycin (CIP-TOB) combination. In addition, population 4 from the ceftazidime resistant clinical isolate BAL02-001 was added (BAL02-001 CAZ4), since this population acquired a genetic variation in mexR, something that frequently happened on ceftazidime-avibactam (CLM02-004 CZA, BAL02-001 CZA and MAD04-002 CZA). Growth of the 48 control populations was confirmed in the 2 drugs independently used, at the same concentrations used for the drugs combinations. Extinct populations at the end of the experimental evolution are represented in black, while surviving populations are represented in gray. All the populations 1 submitted to short-term ALE in the presence of the antibiotic combination became extinct while the treatment was ineffective for all the populations from BAL02-001 CAZ4, containing a genetic variation in mexR. These results are statistically significant (p = 8.415e-14) and indicate that the ciprofloxacin-tobramycin CS-based therapeutic strategy previously proposed for the treatment of concerned clinical strains may be ineffective on strains presenting mutations in mexR.
Discussion
The existing crisis of antibiotic resistance requires the design of new therapeutic strategies that improve the efficacy of available antibiotics20. For that, it is crucial to know the effect of the genomic background and epistasis on the evolution of antibiotic resistance, since these also constraint acquired evolutionary trade-offs, such as CS (22-24, 54). Although it has been proposed that CS is a exploitable trade-off for the rational design of therapeutic -alternated or combined- strategies against bacterial infections54,55,56,57,58,59,60,61,62,63,64,65, this and some previous works21,24,55,62 demonstrate that its exploitation largely depends on its robustness in different genomic backgrounds. In this sense, we show that CS to fosfomycin only emerges in populations from the isolates AST01-034, MAD02-004, CLE03-004, GAL02-002 and FQSE10-0503, after the use of ceftazidime-avibactam. This indicates that, for infections involving specific clones, the alternation or combination of ceftazidime-avibactam with fosfomycin might be an interesting therapeutic strategy to be used, as previously suggested33,34,66. However, the fact that different resistant isolates of P. aeruginosa may emerge in cystic fibrosis patients due to their exposure to prolonged antimicrobial therapies9,10,26, and the fact that CS to fosfomycin emerged in only 37% of the populations evolved on ceftazidime-avibactam, will possibly limit the translation of these results to clinical settings23. This highlights the importance of the genetic background in determining the evolution of antibiotic resistance and the choice of possible antibiotic therapies. Thus, it seems highly recommendable to establish personalized treatments that consider not only the characteristics of the patient/infection but also the genotype(s) of the infecting strain.
The reason for the selection of different mechanisms of resistance depending on the strain is that the same mutation may lead to different fitness and/or resistance levels depending on the genomic backgrounds where it is fixed27,28,29,30. In other words, the effect of each genetic variation is variable and depends on the physiological state of the bacteria in which it is acquired, which in turn has suffered a process of historical contingency. As shown here, this complicates therapeutic choices, since previous treatments with one antibiotic may result in the appearance of different resistance mutations, leading to divergent evolutionary lines following use of subsequent drugs in a patient. In particular, we observe that ceftazidime generally selects for mutations leading to the overproduction of the β-lactamase AmpC, associated with mutations in dacB, ampR, ampD, or mpl39,40,42,47,67. In contrast, the most frequent mechanism of ceftazidime-avibactam resistance is the hyper-production of efflux pumps (mostly MexAB-OprM), caused by mutations in their regulator encoding genes mexR, nalC, or nalD43,44,45 but also the hyper-production of a probable RND efflux transporter encoded by the operon PA1435-PA1436 likely associated to mutations in the operon PA1437-PA1438, encoding a two component system. Similar results, in which high-level of ampicillin resistance was acquired through mutations in genes involving efflux pumps have been previously described, in an Escherichia coli BW25113 strain from the Keio collection lacking the β-lactamase AmpC68. In addition, both treatments, ceftazidime and ceftazidime-avibactam, selected large chromosomal deletions containing galU, whose inactivation reduces the susceptibility to different β-lactams, including ceftazidime and meropenem47, hmgA, causing pyomelanine production48, and mexXY, causing aminoglycoside CS49, as previously described24,35,37. This indicates that there are a non-negligible number of genes (9 in the case of ceftazidime-avibactam; Fig. 3) whose mutations result in a limited type of resistance mechanisms (Fig. 4).
Our results show that ceftazidime-avibactam does not lead to a conserved pattern of CS in our set of 15 clinical strains (even in the case of isolates belonging to the same clone, ST253), as observed when ciprofloxacin was used for the treatment of 25 clinical strains22,23. A possible explanation is that there is a higher number of mutated genes on ceftazidime-avibactam (Fig. 3), compared to ciprofloxacin (mexS, nfxB and gyrA/B)22,23, making more improbable the acquisition of the same -exploitable- pattern of CS when using ceftazidime-avibactam. In addition, it is also possible that few of the genetic variations acquired on ceftazidime-avibactam lead to CS to fosfomycin (i.e., large chromosomal deletions33), while all the possible mechanisms selected by ciprofloxacin lead to CS to tobramycin22,23. A case in point: six of the isolates submitted to ceftazidime-avibactam ALE during this work (MAD04-002, CAT09-004, CAT09-003, ARA02-005, FQSE11010, and MAD05-008) were previously subjected to ALE on ciprofloxacin. While all of them showed a robust pattern of CS to tobramycin after short-term ALE on ciprofloxacin23, none of them showed a robust pattern of CS to a second drug (i.e., fosfomycin) after short-term of ALE on ceftazidime-avibactam (Fig. 2). This indicates that, at least for these isolates, the use of the ciprofloxacin-tobramycin combination is much more reasonable than the use of ceftazidime-avibactam-fosfomycin. The reason is that ciprofloxacin resistance evolution, despite occurring through different genetic mechanisms, converges in a robust increase of susceptibility to tobramycin23. When using ceftazidime-avibactam, however, susceptibility to fosfomycin is not as likely to be expected, since it extremely depends on the isolate(s) present in the infected patient. In fact, it is important to remark that the use of ceftazime-avibactam selects a relevant high number of cases of cross-resistance to non-β-lactam antibiotics, as ciprofloxacin, something that suggests that the use of avibactam may preclude the use of the ciprofloxacin-tobramycin CS-based combination. In fact, ALE assays on ciprofloxacin, tobramycin or the combination, showed that, for the treatment of these particular strains, it is not possible to apply this CS-based therapeutic strategy, since this causes extinction of just 30% of the ceftazidime-avibactam resistant populations analyzed (Fig. 5A). Although we previously described that the combination ciprofloxacin-tobramycin is extremely effective driving extinction of clinical strains resistant to different antibiotics, our results show that ceftazidime-avibactam resistant populations may escape this strategy. Therefore, we should be aware that the use of ceftazidime-avibactam may be riskier than expected since it would be difficult to find a therapeutic solution if this last-resort treatment failed. As it happens with most recent antibiotics, it is generally accepted that the use of ceftazidime-avibactam should be restricted to those isolates that do not respond to older beta-lactams13,16,17,18. This statement is based just in the preservation of the most novel antibiotics, that are more active than older ones, against resistant bacteria. However, our results indicate that, in addition, the consequences of acquiring resistance to these novel drugs must be explored as well. In this respect, our data support that ceftazidime is superior than ceftazidime-avibactam in some aspects, mainly because the mechanisms of resistance and trade-offs associated to the use of these antibiotics are different. As stated, ceftazidime alone renders less cross-resistance than ceftazidime-avibactam. In addition, previously described combination therapies22, based in information on CS, are more effective against ceftazidime resistant mutants than against ceftazidime-avibactam resistant ones. These findings further support that the use of ceftazidime-avibactam should be restricted to those situations in which no other beta-lactam is active13,16,17,18. Having said that, it is important to remark that, while our in vitro data highlights potential significant risks associated with the use of ceftazidime-avibactam, the translation of this information into clinical practice may be affected by other factors and should be assessed by clinicians before establishing strategic recommendations. Finally, it is worth to emphasize the potential that studies of purely evolutionary nature have to rationally guide the choice of future antibiotic strategies. This study suggests that this last-resort drug combination should be restricted for particular personalized treatments that consider not only the characteristics of the patient/infection but also the genotype(s) of the infecting strains. We strongly feel that the time has come to translate evolution, which is the origin of antibiotic resistance, into therapy.
Methods
Growth conditions and antibiotic susceptibility tests
Bacteria were grown in Lysogeny Broth (LB) (Lenox, Pronadisa) at 37 °C with shaking at 250 rpm in glass tubes. MICs of ceftazidime, ceftazidime-avibactam, tobramycin, fosfomycin, ciprofloxacin, aztreonam and meropenem were determined at 37 °C, in Mueller Hinton agar, using E-test strips (MIC Test Strip, Liofilchem®).
Short-term adaptive laboratory evolution experiments
ALE assays in the presence of ceftazidime, ceftazidime-avibactam or absence of antibiotic were performed as previously described22,23,24. 180 bacterial populations from stock cultures of 15 P. aeruginosa clinical isolates -4 replicates of each- from various Spanish hospitals (Table 1) were submitted to short-term ALE in presence of ceftazidime, ceftazidime-avibactam or absence (control populations) of drug during 3 days. Replicate populations were grown at 37 °C and 250 rpm in independent glass tubes to avoid cross-contamination. The cultures were diluted (1/100) every day, adding 10 µl of bacteria to 1 ml of fresh LB containing the concentration of ceftazidime (close to MIC) that hindered -but allowed- the growth of each P. aeruginosa isolate under these culture conditions (1 µg/ml for CAT09-004 and CLM02-004; 1.5 µg/ml for MAD04-002, CAT09-003, FQSE11-1010, BAL02-001, CAT03-005 and MAD05-008; 2 µg/ml for CLE03-004, GAL02-002, AST01-004 and MAD02-004; 3 µg/ml for FQSE10-0505; 8 µg/ml for GAL06-003), being this concentration half of the MIC of each parental strain or higher, or without antibiotic (control populations). The avibactam concentration was maintained constant, as used in clinical tests69, at 4 µg/ml. During 3 days, the concentration of antibiotic was maintained. At the end of the ALE assay, every replicate population was preserved at −80 °C. When needed, resistant clones were isolated by plating resistant populations on LB agar supplemented with ceftazidime and selecting individual colonies after overnight growth at 37 °C. The phenotype of the selected clones was confirmed determining their susceptibility to ceftazidime and ceftazidime-avibactam by E-Test.
Combination of ciprofloxacin with tobramycin in short-term adaptive laboratory evolution assays
ALE assays in the presence of ciprofloxacin, tobramycin or the antibiotic combination were performed as previously described22,23. 20 bacterial populations resistant to ceftazidime-avibactam from population 1 of 5 different isolates -4 replicates of each- and 20 bacterial populations resistant to ceftazidime from population 1 of the same clinical isolates were grown from glycerol stocks. Replicate populations were grown at 37 °C and 250 rpm in independent glass tubes to avoid cross-contamination. During 3 days, the cultures were diluted (1/100) in fresh LB medium containing a ciprofloxacin-tobramycin combination (40 populations) or each single drug (80 control populations). Each antibiotic was added at the concentration (close to MIC) that hinders -but allows- the growth of bacterial population of P. aeruginosa under these culture conditions. In the case of ceftazidime-avibactam resistant populations, the ciprofloxacin concentration used were: 0.05 µg/ml for BAL02-001, 0.075 µg/ml for MAD02-004, 0.1 µg/ml for CLM02-004 and MAD04-002, and 0.2 µg/ml for CLE03-004; the tobramycin concentration used were: 0.2 µg/ml for MAD02-004, 0.4 µg/ml for CLE03-004, 0.5 µg/ml for BAL02-001 and 1 µg/ml for CLM02-004 and MAD04-002. In the case of ceftazidime resistant populations, the ciprofloxacin concentration used were: 0.01 µg/ml for BAL02-001, 0.05 µg/ml for MAD04-002 and CLM02-004, 0.1 µg/ml for MAD02-004 and CLE03-004; the tobramycin concentration used were: 0.5 µg/ml for CLE03-004, 1 µg/ml for MAD02-004 and CLM02-004, 1.5 µg/ml for BAL02-001 and 2 µg/ml for MAD02-004. For the BAL02-001 CAZ.4 resistant population, the ciprofloxacin and tobramycin concentrations used in four replicate populations were 0.05 and 1 µg/ml, respectively. Extinction of the populations was determined by plating out 100 μl of final cultures on LB agar to look for viable cells. The absence of colonies was interpreted as extinction of the population.
Statistical analysis
Normality (Shapiro-Wilk) and homocedasticity (Bartlett) tests were run on the raw and log-transformed data to check parametricity. Following unsuccessful parametricity checks, we carried out a Kruskal-Wallis test to confirm the presence of significant differences in the dataset. We next carried a comparison of the parental and LB-evolved strains using Mann-Whitney-Wilcoxon U test with FDR correction for multiple comparison. After confirming that strains grown in LB showed no significant differences with the parental strain, we used these as control to compare each strain MICs after evolution in ceftazidime or ceftazidime-avibactam, using a many-one Mann-Whitney U test applying an FDR correction for the total (all strains and antibiotics) number of comparisons. To avoid considering statistically yet not biologically significant differences, we identified the later using an effect in fold changes two times above or below the reference; the counts were tabulated and compared using exact binomial tests with FDR correction to compare cross-resistance and CS among populations evolved in ceftazidime against ceftazidime-avibactam. Differences in extinction data of strains grown in ceftazidime or ceftazidime-avibactam, and evolved in the presence of ciprofloxacin plus tobramycin, was analyzed using the log-likelihood ratio test. Dependence of resistance mechanisms evolved after short term ALE in presence of ceftazidime or ceftazidime-avibactam was analyzed using an omnibus log-likelihood ratio test followed by binomial test comparisons corrected with FDR for multiple comparisons. Two sided (bilateral) tests were used in all cases. All statistical analyses were done using R Statistical Software v4.4.2 (R Core Team, 2021), with packages PMCMRplus and Deducer70.
Whole-genome sequencing and identification of genetic variations
To analyse the relatedness of the studied ST253 clinical isolates, their sequences, already deposited at PRJEB40140 were analyzed. Reads were de novo assembled using SPAdes v3.15 with default options. Output files in fasta format were compared through wgMLST using the cano-wgMLST_BacCompare software (http://baccompare.imst.nsysu.edu.tw)71.
To analyze genomic variations acquired during ALE assays, the extraction of the genomic DNA of 143 populations (60 ceftazidime, 60 ceftazidime-avibactam evolved populations and 23 control populations evolved in the absence of antibiotic) and the DNA quality analysis was performed by the Translational Genomics Unit (Instituto Ramón y Cajal de Investigación Sanitaria-Hospital Ramón y Cajal from Madrid). Genomic DNA of each population was extracted by ChemagicTM DNA Bacterial Kit H96 (CMG-799 Chemagic™) using the equipment Chemagic™ 360/MSMI (PerkinElmer). The assay of DNA quality was performed using an Agilent 2200 TapeStation System. Libraries construction and whole-genome sequencing were performed by the Oxford Genomics Centre. Pair-end libraries (2×150) were sequenced using an Illumina NovaSeq6000 system (Illumina Inc., USA). Coverage was higher than 150x for all samples. Genomic DNA for selected clones was obtained with a commercially available extraction kit (High Pure PCR Template Preparation Kit, Roche Diagnostics). Indexed paired-end libraries were generated by using the Illumina DNA Prep library preparation kit (Illumina Inc, USA) and then sequenced on an Illumina MiSeq® benchtop sequencer with MiSeq reagent kit v3 (Illumina Inc., USA), resulting in 300 bp paired-end reads. Coverage was higher than 30x for all clones.
All the Paired-end reads were mapped to the P. aeruginosa PAO1 reference genome (GenBank accession: NC_002516.2) with Bowtie 2 v2.2.6 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)72 and pileup, and raw files were obtained by using SAMtools v0.1.16 (https://sourceforge.net/projects/samtools/files/samtools/)73 and PicardTools v1.140 (https://github.com/broadinstitute/picard), using the Genome Analysis Toolkit (GATK) v3.4.46 (https://www.broadinstitute.org/gatk/)74 for realignment around InDels. Single-nucleotide polymorphisms (SNPs) were extracted from the raw files if at least 100 reads covered such positions and if the change was present in at least 5% of the reads. Micro-indels were extracted from the totalpileup files if also at least 100 reads covered such positions and but if the change was present in at least 10% of the reads. Filtered SNP files were converted to vcf and annotated with SnpEff v4.3 (http://snpeff.sourceforge.net/index.html)75 to study the genes involved in ceftazidime and ceftazidime-avibactam resistance, disregarding all mutations already present in the original isolates. To study large chromosomal deletions, number of reads per position in entire genome previously obtained in totalpileups, was introduced in RStudio v4.3.176 platform and plotted by using the ggplot package77 Those regions showing a decrease of the 50% the number of reads or that were not covered were considered as large chromosomal deletions.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Sequence files of derivatives obtained in this study under ceftazidime or ceftazidime-avibactam treatment and controls were deposited in the European Nucleotide Archive under study number PRJEB85283. The genomic sequences of the original strains can be found in projects: PRJEB24151 and PRJEB40140. Data needed to evaluate the conclusions of this work are present in the manuscript or the Supplementary Materials.
References
Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655, https://doi.org/10.1016/S0140-6736(21)02724-0 (2022).
Santajit, S. & Indrawattana, N. Mechanisms of antimicrobial resistance in ESKAPE pathogens. Biomed. Res. Int. 2016, 2475067 (2016).
Rello, J. et al. A global priority list of the TOp TEn resistant Microorganisms (TOTEM) study at intensive care: a prioritization exercise based on multi-criteria decision analysis. Eur. J. Clin. Microbiol. Infect. Dis. 38, 319–323 (2019).
Martinez-Solano, L., Macia, M. D., Fajardo, A., Oliver, A. & Martinez, J. L. Chronic Pseudomonas aeruginosa infection in chronic obstructive pulmonary disease. Clin. Infect. Dis. 47, 1526–1533 (2008).
Talwalkar, J. S. & Murray, T. S. The approach to Pseudomonas aeruginosa in cystic fibrosis. Clin. Chest Med. 37, 69–81 (2016).
Tummler, B., Wiehlmann, L., Klockgether, J. & Cramer, N. Advances in understanding Pseudomonas. F1000Prime Rep. 6, 9 (2014).
Olivares, J. et al. The intrinsic resistome of bacterial pathogens. Front. Microbiol. 4, 103 (2013).
Bellido, F., Martin, N. L., Siehnel, R. J. & Hancock, R. E. Reevaluation, using intact cells, of the exclusion limit and role of porin OprF in Pseudomonas aeruginosa outer membrane permeability. J. Bacteriol. 174, 5196–5203 (1992).
Lopez-Causape, C., Cabot, G., Del Barrio-Tofino, E. & Oliver, A. The versatile mutational resistome of Pseudomonas aeruginosa. Front. Microbiol. 9, 685 (2018).
Lopez-Causape, C. et al. Evolution of the Pseudomonas aeruginosa mutational resistome in an international cystic fibrosis clone. Sci. Rep. 7, 5555 (2017).
Oliver, A., Mulet, X., Lopez-Causape, C. & Juan, C. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist. Updates 21-22, 41–59 (2015).
Del Barrio-Tofino, E., Lopez-Causape, C. & Oliver, A. Pseudomonas aeruginosa epidemic high-risk clones and their association with horizontally-acquired beta-lactamases: 2020 update. Int. J. Antimicrob. Agents 56, 106196 (2020).
Horcajada, J. P. et al. Epidemiology and treatment of multidrug-resistant and extensively drug-resistant Pseudomonas aeruginosa infections. Clin. Microbiol. Rev. 32, https://doi.org/10.1128/CMR.00031-19 (2019).
Coleman, K. Diazabicyclooctanes (DBOs): a potent new class of non-beta-lactam beta-lactamase inhibitors. Curr. Opin. Microbiol. 14, 550–555 (2011).
Aktas, Z., Kayacan, C. & Oncul, O. In vitro activity of avibactam (NXL104) in combination with beta-lactams against Gram-negative bacteria, including OXA-48 beta-lactamase-producing Klebsiella pneumoniae. Int. J. Antimicrob. Agents 39, 86–89 (2012).
Sader, H. S., Flamm, R. K., Carvalhaes, C. G. & Castanheira, M. Antimicrobial Susceptibility of Pseudomonas aeruginosa to Ceftazidime-Avibactam, Ceftolozane-Tazobactam, Piperacillin-Tazobactam, and Meropenem Stratified by U.S. Census Divisions: Results from the 2017 INFORM Program. Antimicrob. Agents. Chemother. 62, https://doi.org/10.1128/AAC.01587-18 (2018).
Huband, M. D. et al. In vitro activity of ceftazidime-avibactam against contemporary Pseudomonas aeruginosa isolates from U.S. Medical Centers by Census Region, 2014. Antimicrob. Agents Chemother. 60, 2537–2541 (2016).
Lopez-Montesinos, I. et al. Suboptimal concentrations of Ceftazidime/Avibactam (CAZ-AVI) may select for CAZ-AVI resistance in extensively drug-resistant Pseudomonas aeruginosa: in vivo and in vitro evidence. Antibiotics 11, https://doi.org/10.3390/antibiotics11111456 (2022).
Szybalski, W. & Bryson, V. Genetic studies on microbial cross resistance to toxic agents. I. Cross resistance of Escherichia coli to fifteen antibiotics. J. Bacteriol. 64, 489–499 (1952).
Laxminarayan, R. Antibiotic effectiveness: balancing conservation against innovation. Science 345, 1299–1301 (2014).
Sanz-Garcia, F. et al. Translating eco-evolutionary biology into therapy to tackle antibiotic resistance. Nat. Rev. Microbiol. 21, 671–685 (2023).
Hernando-Amado, S., Laborda, P., Valverde, J. R. & Martinez, J. L. Mutational background influences P. aeruginosa ciprofloxacin resistance evolution but preserves collateral sensitivity robustness. Proc. Natl. Acad. Sci. USA 119, e2109370119 (2022).
Hernando-Amado, S. et al. Rapid phenotypic convergence towards collateral sensitivity in clinical isolates of Pseudomonas aeruginosa presenting different genomic backgrounds. Microbiol. Spectr. e0227622, https://doi.org/10.1128/spectrum.02276-22 (2022).
Hernando-Amado, S., Sanz-Garcia, F. & Martinez, J. L. Rapid and robust evolution of collateral sensitivity in Pseudomonas aeruginosa antibiotic-resistant mutants. Sci. Adv. 6, eaba5493 (2020).
Diaz Caballero, J. et al. Mixed strain pathogen populations accelerate the evolution of antibiotic resistance in patients. Nat. Commun. 14, 4083 (2023).
Armstrong, D. et al. Evidence for spread of a clonal strain of Pseudomonas aeruginosa among cystic fibrosis clinics. J. Clin. Microbiol. 41, 2266–2267 (2003).
Hernando-Amado, S., Sanz-Garcia, F. & Martinez, J. L. Antibiotic resistance evolution is contingent on the quorum-sensing response in Pseudomonas aeruginosa. Mol. Biol. Evol. 36, 2238–2251 (2019).
Vogwill, T., Kojadinovic, M. & MacLean, R. C. Epistasis between antibiotic resistance mutations and genetic background shape the fitness effect of resistance across species of Pseudomonas. Proc. Biol. Sci. 283, https://doi.org/10.1098/rspb.2016.0151 (2016).
Apjok, G. et al. Limited evolutionary conservation of the phenotypic effects of antibiotic resistance mutations. Mol. Biol. Evol. https://doi.org/10.1093/molbev/msz109 (2019).
Schenk, M. F. & de Visser, J. A. Predicting the evolution of antibiotic resistance. BMC Biol. 11, 14 (2013).
Gambello, M. J. & Iglewski, B. H. Cloning and characterization of the Pseudomonas aeruginosalasR gene, a transcriptional activator of elastase expression. J. Bacteriol. 173, 3000–3009 (1991).
Fischer, S., Dethlefsen, S., Klockgether, J. & Tummler, B. Phenotypic and genomic comparison of the two most common ExoU-positive Pseudomonas aeruginosa clones, PA14 and ST235. mSystems 5, https://doi.org/10.1128/mSystems.01007-20 (2020).
Laborda, P., Martinez, J. L. & Hernando-Amado, S. Convergent phenotypic evolution towards fosfomycin collateral sensitivity of Pseudomonas aeruginosa antibiotic-resistant mutants. Microb. Biotechnol. 15, 613–629 (2022).
Laborda, P., Martinez, J. L. & Hernando-Amado, S. Evolution of habitat-dependent antibiotic resistance in Pseudomonas aeruginosa. Microbiol. Spectr. e0024722, https://doi.org/10.1128/spectrum.00247-22 (2022).
Sanz-Garcia, F., Hernando-Amado, S. & Martinez, J. L. Mutation-driven evolution of Pseudomonas aeruginosa in the presence of either ceftazidime or ceftazidime/avibactam. Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.01379-18 (2018).
Hernando-Amado, S., Laborda, P., Valverde, J. R. & Martinez, J. L. Rapid decline of ceftazidime resistance in antibiotic-free and sublethal environments is contingent on genetic background. Mol. Biol. Evol. 39, https://doi.org/10.1093/molbev/msac049 (2022).
Hernando-Amado, S., Laborda, P., Valverde, J. R. & Martínez, J. L. Rapid decline of ceftazidime resistance in antibiotic-free and sub-lethal environments is contingent on genetic background. Mol. Biol. Evol. https://doi.org/10.1093/molbev/msac049 (2022).
Zamorano, L., Moyá, B., Juan, C. & Oliver, A. Differential beta-lactam resistance response driven by ampD or dacB (PBP4) inactivation in genetically diverse Pseudomonas aeruginosa strains. J. Antimicrob. Chemother. 65, 1540–1542 (2010).
Cabot, G. et al. Genetic markers of widespread extensively drug-resistant Pseudomonas aeruginosa high-risk clones. Antimicrob. Agents Chemother. 56, 6349–6357 (2012).
Juan, C. et al. Molecular mechanisms of beta-lactam resistance mediated by AmpC hyperproduction in Pseudomonas aeruginosa clinical strains. Antimicrob. agents Chemother. 49, 4733–4738 (2005).
Cabot, G. et al. Evolution of Pseudomonas aeruginosa antimicrobial resistance and fitness under low and high mutation rates. Antimicrob. Agents Chemother. 60, 1767–1778 (2016).
Moya, B. et al. Beta-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 5, e1000353 (2009).
Morita, Y., Cao, L., Gould, V. C., Avison, M. B. & Poole, K. nalD encodes a second repressor of the mexAB-oprM multidrug efflux operon of Pseudomonas aeruginosa. J. Bacteriol. 188, 8649–8654 (2006).
Sobel, M. L., Hocquet, D., Cao, L., Plesiat, P. & Poole, K. Mutations in PA3574 (nalD) lead to increased MexAB-OprM expression and multidrug resistance in laboratory and clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 49, 1782–1786 (2005).
Pan, Y. P., Xu, Y. H., Wang, Z. X., Fang, Y. P. & Shen, J. L. Overexpression of MexAB-OprM efflux pump in carbapenem-resistant Pseudomonas aeruginosa. Arch. Microbiol. 198, 565–571 (2016).
Cabot, G., Florit-Mendoza, L., Sanchez-Diener, I., Zamorano, L. & Oliver, A. Deciphering beta-lactamase-independent beta-lactam resistance evolution trajectories in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 73, 3322–3331 (2018).
Alvarez-Ortega, C., Wiegand, I., Olivares, J., Hancock, R. E. & Martinez, J. L. Genetic determinants involved in the susceptibility of Pseudomonas aeruginosa to beta-lactam antibiotics. Antimicrob. Agents Chemother. 54, 4159–4167 (2010).
Rodriguez-Rojas, A. et al. Inactivation of the hmgA gene of Pseudomonas aeruginosa leads to pyomelanin hyperproduction, stress resistance and increased persistence in chronic lung infection. Microbiology 155, 1050–1057 (2009).
Masuda, N. et al. Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-oprM efflux pumps in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44, 3322–3327 (2000).
Oliver, A., Baquero, F. & Blazquez, J. The mismatch repair system (mutS, mutL and uvrD genes) in Pseudomonas aeruginosa: molecular characterization of naturally occurring mutants. Mol. Microbiol. 43, 1641–1650 (2002).
Gomis-Font, M. A., Sastre-Femenia, M. A., Taltavull, B., Cabot, G. & Oliver, A. In vitro dynamics and mechanisms of cefiderocol resistance development in wild-type, mutator and XDR Pseudomonas aeruginosa. J. Antimicrob. Chemother. 78, 1785–1794 (2023).
Agyeman, A. A. et al. Ceftolozane/tazobactam plus tobramycin against free-floating and biofilm bacteria of hypermutable Pseudomonas aeruginosa epidemic strains: resistance mechanisms and synergistic activity. Int J. Antimicrob. Agents 62, 106887 (2023).
Roemhild, R. et al. Cellular hysteresis as a principle to maximize the efficacy of antibiotic therapy. Proc. Natl. Acad. Sci. USA 115, 9767–9772 (2018).
Imamovic, L. et al. Drug-driven phenotypic convergence supports rational treatment strategies of chronic infections. Cell 172, 121–134.e114 (2018).
Hernando-Amado, S., Laborda, P. & Martinez, J. L. Tackling antibiotic resistance by inducing transient and robust collateral sensitivity. Nat. Commun. 14, 1723 (2023).
Barbosa, C., Beardmore, R., Schulenburg, H. & Jansen, G. Antibiotic combination efficacy (ACE) networks for a Pseudomonas aeruginosa model. PLoS Biol. 16, e2004356 (2018).
Munck, C., Gumpert, H. K., Wallin, A. I., Wang, H. H. & Sommer, M. O. Prediction of resistance development against drug combinations by collateral responses to component drugs. Sci. Transl. Med. 6, 262ra156 (2014).
Jahn, L. J. et al. Compatibility of evolutionary responses to constituent antibiotics drive resistance evolution to drug pairs. Mol. Biol. Evol. https://doi.org/10.1093/molbev/msab006 (2021).
Imamovic, L. & Sommer, M. O. Use of collateral sensitivity networks to design drug cycling protocols that avoid resistance development. Sci. Transl. Med. 5, 204ra132 (2013).
Kim, S., Lieberman, T. D. & Kishony, R. Alternating antibiotic treatments constrain evolutionary paths to multidrug resistance. Proc. Natl. Acad. Sci. USA 111, 14494–14499 (2014).
Barbosa, C., Romhild, R., Rosenstiel, P. & Schulenburg, H. Evolutionary stability of collateral sensitivity to antibiotics in the model pathogen Pseudomonas aeruginosa. eLife 8, https://doi.org/10.7554/eLife.51481 (2019).
Podnecky, N. L. et al. Conserved collateral antibiotic susceptibility networks in diverse clinical strains of Escherichia coli. Nat. Commun. 9, 3673 (2018).
Barbosa, C. et al. Alternative evolutionary paths to bacterial antibiotic resistance cause distinct collateral effects. Mol. Biol. Evol. 34, 2229–2244 (2017).
Lazar, V. et al. Bacterial evolution of antibiotic hypersensitivity. Mol. Syst. Biol. 9, 700 (2013).
Lazar, V. et al. Genome-wide analysis captures the determinants of the antibiotic cross-resistance interaction network. Nat. Commun. 5, 4352 (2014).
Hamou-Segarra, M. et al. Synergistic activity of fosfomycin, beta-lactams and peptidoglycan recycling inhibition against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 72, 448–454 (2017).
Aguilera Rossi, C. G., Gomez-Puertas, P. & Ayala Serrano, J. A. In vivo functional and molecular characterization of the Penicillin-Binding Protein 4 (DacB) of Pseudomonas aeruginosa. BMC Microbiol. 16, 234 (2016).
Gross, R., Yelin, I., Lazar, V., Datta, M. S. & Kishony, R. Beta-lactamase dependent and independent evolutionary paths to high-level ampicillin resistance. Nat. Commun. 15, 5383 (2024).
Zhanel, G. G. et al. Ceftazidime-avibactam: a novel cephalosporin/beta-lactamase inhibitor combination. Drugs 73, 159–177 (2013).
Fellows, I. Deducer: a data analysis GUI for R. J. Stat. Softw. 49, 1–15 (2012).
Liu, Y. Y., Lin, J. W. & Chen, C. C. cano-wgMLST_BacCompare: a bacterial genome analysis platform for epidemiological investigation and comparative genomic analysis. Front. Microbiol. 10, 1687 (2019).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43, 11.10.11–11.10.33 (2013).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
R-Core-Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing (2023).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer Verlag, 2016).
Acknowledgements
We thank to our colleague and friend Teresa Coque (Instituto Ramón y Cajal de Investigación Sanitaria-Hospital Ramón y Cajal, Madrid) for advising and helping us contacting both, the Translational Genomics Unit (Instituto Ramón y Cajal de Investigación Sanitaria-Hospital Ramón y Cajal, Madrid) and the Oxford Genomics Centre. We also thank to Leticia Olavarrieta, from the Translational Genomics Unit, for the extraction of the genomic samples. We also thank to Biel Taltavull and to Carla López-Causapé from Hospital Son Espases-IdISBa for their support and advice with whole genome sequencing and bioinformatics analysis, respectively. This work was supported by MICIU/AEI/10.13039/501100011033 and FEDER (UE) -grant PID2023-149913OA-I00- to S.H.A. and by the Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación and Unión Europea—NextGenerationEU -grants PI21/00017 and Personalized and precision medicine grant MePRAM Project PMP22/00092- to A.O. The MICIU/AEI/ 10.13039/501100011033 is also acknowledged for supporting the National Center for Biotechnology through the “Severo Ochoa” grant CEX2023-001386-S.
Author information
Authors and Affiliations
Contributions
S.H.A. designed the study and performed the experimental work. M.G.F. performed the whole genome sequencing of the isolated clones and the bioinformatics analysis of the whole genome sequence data of the populations. J.R.V. performed the statistical analysis. A.O. and J.L.M. participated in the design of the study. S.H.A. wrote the first draft of the manuscript, which was afterwards reviewed by all authors. All authors approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hernando-Amado, S., Gomis-Font, M.A., Valverde, J.R. et al. Ceftazidime-avibactam use selects multidrug-resistance and prevents designing collateral sensitivity-based therapies against Pseudomonas aeruginosa. Nat Commun 16, 3323 (2025). https://doi.org/10.1038/s41467-025-58597-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58597-6
