
Abstract
The protozoan parasite Trypanosoma brucei undergoes a complex life cycle, moving between its mammalian host and the blood-feeding tsetse fly vector. The two major surface proteins expressed by procyclic forms in the insect midgut, EP and GPEET procyclin, are transcribed from a polycistronic transcription unit by RNA polymerase I. Here we identify a long non-coding RNA, TblncRNA-23, that is encoded between the two procyclin genes. TblncRNA-23 localizes to the nucleolus and also associates with polysomes. Overexpression of TblncRNA-23 and its down regulation by RNAi or knockout (KO) identify EP and GPEET mRNAs as targets, among other mRNAs that changed abundance in the transition from early to late procyclic forms and from procylic to the metacylic forms, suggesting its role in regulating gene expression which accomapines or dictates of the parasite transitions within in its insect host. TblncRNA-23 interacts with its substrates via base-pairing using different domains. Purification of TblncRNA-23-associated proteins by RaPID identifies hundreds of proteins, including proteins translated from its target mRNAs, suggesting its association with translating ribosomes. Early and late procyclic forms differ in their social motility (SoMo) capabilities, which is essential for migration away from the insect midgut to enable parasite transmission. Overexpression of TblncRNA-23 results in hypermotility, whereas KO compromises this capacity, suggesting a regulatory role in SoMo. Moreover, silencing of the RNA abrogates the ability of the parasite to transform from procylic to the metacyclic forms affecting the parasite’s potential to cycle between its hosts.
Similar content being viewed by others
Introduction
The parasite Trypanosoma brucei causes sleeping sickness in humans and Nagana in domestic animals. The parasite cycles between a mammalian host and the tsetse fly. In the mammalian host, parasites propagate as slender bloodstream forms (BSF). Upon ingestion by the fly, the parasite differentiate into early procyclic forms (PCF) in the lumen of the insect midgut1. In addition to major changes in morphology and metabolism, parasites replace the variant surface glycoprotein (VSG) with two insect-specific coat proteins known as GPEET and EP procyclins2. At this point in the infection cycle, there are two possible outcomes. Either the early PCF is eliminated by the fly, or the parasites reach the other side of the peritrophic matrix and colonize the ectoperitrophic space. Once the infection is established, parasites in the midgut are considered to be late PCF2, which express EP, but not GPEET2. Transcriptomic analysis of early and late PCF in culture identified 73 transcripts that are differentially regulated3 between the early and late stages. In addition, a phenotypic difference between early and late PCF is their ability to engage in social motility (SoMo) when the parasites are plated on semi-solid plates4. This motility was recently shown to comprise a chemotactic response to self-generated pH gradients5. To complete the life cycle in the fly, parasites undergo further rounds of differentiation and move to the proventriculus, where they transform into epimastigotes and then colonize the salivary glands, culminating in the production of metacyclic forms, which can be transmitted to a new mammalian host1. Overexpression of the RNA binding protein RBP6 in PCF induces their transformation to metacyclic forms6 and this transition involves changes in the expression of hundreds of transcripts7,8. An RNAi screen identified factors that are essential for the transition to either epimastigotes or to metacyclic forms9.
GPEET and two isoforms of EP (EP1, EP3) are marker proteins for the transition from BSF to early and late PCF. The 3’ UTR of both mRNAs has a conserved structure that is composed of three stem-loops (LI-III) that play a role in stage-specific expression10,11,12,13. Two domains, a 26-mer present in LII and a 16-mer present in LIII, are conserved in all EP and GPEET 3’ UTRs10,13. The 26-mer is a destabilizing element in all life-cycle stages. The 16-mer is implicated in RNA stability and translational control10. TbZFP3 is the only RNA binding protein that was shown to date to bind LII in EP1, and regulates translation by competing out one or more unidentified repressor proteins14.
EP and GPEET mRNAs are derived from a polycistronic RNA precursor transcribed by RNA polymerase I (pol I)15,16. The level of these transcripts is reduced to almost undetectable levels in the BSF10. This downregulation is controlled by three mechanisms: transcriptional down-regulation, rapid degradation of the mRNA, and translational regulation10.
Apart from EP and GPEET, which are transcribed by pol I17, all other mRNA coding genes are constitutively transcribed by pol II forming polycistronic units which are processed by the concerted action of trans-splicing and polyadenylation18. The differential gene expression of transcripts from the same polycistronic transcription unit, as well as stage-specific gene expression, is mediated by hundreds of RNA-binding proteins19.
Little is known about non-coding RNAs that regulate gene expression in trypanosomes20. These parasites appear to lack microRNAs21. However, our group recently identified abundant ncRNAs that are developmentally regulated and/or respond to stress22. RNA sequencing led to the identification of 1428 previously uncharacterized lncRNA transcripts. One of these is the grumpy lncRNA23. A small nucleolar RNA is processed from this lncRNA to form the snoGRUMPY, which binds to mRNAs and regulates their expression. Grumpy overexpression reduces parasitemia in infected mice23.
In this study, we reveal the function of a pol I-transcribed lncRNA that regulates the expression of genes associated with the transition from early to late procyclic forms and from procyclic to metacyclic forms, identify its targets, and demonstrate how it controls social motility of these parasites.
Results
TblncRNA-23 is a long-noncoding RNA transcribed by RNA pol I
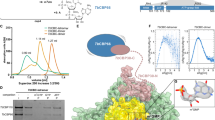
A screen conducted to identify novel ncRNAs in T. brucei identified TbsRNA-23, which is preferentially expressed in PCF22. Analysis of RNA-seq data of polysomal (translating) and sub-polysomal (non-translating) mRNAs24 revealed that TbsRNA-23 was encoded within the procyclin loci located on chromosomes 6 and 10 (Fig. 1A) and that it is a long non-coding RNA. We therefore changed its name to TblncRNA-23. The procyclin loci are transcribed by RNA pol I16,25 into a polycistronic RNA precursor, giving rise to EP and GPEET mRNAs, and TblncRNA-23 which is trans-spliced and polyadenylated24. It is currently unknown if pol II is also involved in transcription within this locus. Inspection of its expression in transcriptomes obtained from parasites during infection in the fly, and in culture-derived slender and stumpy BSF showed that TblncRNA-23 expression is highest during midgut infection (days 7-28), but it is not detectable in BSF (Fig. 1B)3,26. Its expression was verified by Northern blot analysis (Fig. 1C). The exact size of 404 bases was determined from its sequence in polyA primed libraries27 (Supplementary Fig. 1). The localization of TblncRNA-23 was determined by in situ hybridization combined with immunofluorescence using antibodies against the nucleolar protein, NHP2 (Fig. 1D)28, and suggested that TblncRNA-23 RNA is localized in the nucleolus, where EP and GPEET were shown to be transcribed29. TblncRNA-23 distribution was examined on RNPs separated by sucrose-gradient centrifugation that revealed one major peak that is larger in size than snoRNPs ( ~10S), as well as two less abundant peaks. Based on the fractionation of TB11Cs2C2, a snoRNA involved in rRNA processing30, a minor fraction of TblncRNA-23 appeared to co-migrate with rRNA-processing complexes, as well with polysomal fractions where translating GPEET mRNA fractionated (Fig. 1E). Despite the localization of the TblncRNA-23 in the cytoplasm on translating ribosomes, a signal was not observed in the cytoplasm by in situ hybridization, most likely because it is not as concentrated as in the nucleolus.

A Genomic organization and TblncRNA-23 levels in polysomal and post-polysomal RNA in PCF and BSF parasites. The location of genes is indicated. Distribution of the transcript reads across the polycistronic units is given in reads per kilobase per million (RPKM). The different transcripts are color coded and their identity is presented. The position of the SL addition site is indicated by a red line. The read data are taken from Tinti et al.24. B Differential expression of TblncRNA-23 during the various developmental stages. Distribution of the transcript reads across the coding region for the RNA is given as RPKM, and was derived from data published in PCF3,26; the different samples are color coded. MG-Midgut, PV-proventriculus, SG- salivary gland C TblncRNA-23 expression in PCF and BSF. Total T. brucei from PCF and BSF (20 μg) was separated on denaturing gels and probed with32 P-labeled anti-sense RNA probes. 32P-labeled pBR322 DNA MspI digest was used as a size marker. The experiment presents one replicate. The expression of TblncRNA-23 in PCF and BSF was determined in three biological replicates by real-time PCR, with statistical analysis provided in Supplementary Fig. 2. Source data are provided as a Source Data file. D High-resolution fluorescence in situ hybridization. The in situ hybridization coupled with immunofluorescence was performed for the indicated RNAs (green), and NHP2 (red). The scale bar is indicated. Additional images from independent experiments are presented in Supplementary Fig. 3. E Fractionation of TblncRNA-23. Whole cell extract from procyclic cells (2 × \({10}^{9}\)) was fractionated on (10–30 %) sucrose gradient at 70,000 rpm for 3 h using a Beckman SW41 rotor. The fractions were deproteinized, and the RNA was separated on a 6% polyacrylamide-denaturing gel and subjected to Northern analysis using anti-sense RNA probes. The optical density of the fractions is presented. The experiment presents one replicate, an additional replicate is presented in Supplementary Fig. 4. Source data are provided as a Source Data file.
Interactions of TblncRNA-23 with GPEET and other mRNAs
We next determined whether TblncRNA-23 interacts with mRNAs. To this end, we used interactomes that were prepared from cells exposed to psoralen (AMT) and subjected to UV irradiation to crosslink interacting RNA molecules22. The chimeric molecules containing TblncRNA-23 are presented in Fig. 2A. We observed cross-linking between TblncRNA-23 and the EP-coding region, and with the GPEET 3’ UTR. Additional mRNAs were identified as potential substrates (Fig. 2A). The levels of these mRNAs changed upon overexpression of TblncRNA-23 or its silencing, suggesting that they are regulated by TblncRNA-23 (see below). To examine if EP and GPEET are regulated by TblncRNA-23, the lncRNA was targeted by dsRNA, which resulted in ~80% reduction in the level of the RNA (Fig. 2B i). To achieve a better depletion of the RNA, the gene was knocked out using CRISPR-Cas9. Knockout (KO) resulted in the complete elimination of cellular TblncRNA-23, suggesting that the gene was removed from both copies of chromosome 6 and chromosome 10 (Fig. 2B ii). TblncRNA-23 silencing led to a concomitant reduction in the level of GPEET protein (Fig. 2C i) and an increase in the expression of EP protein (Fig. 2C ii). Likewise, KO also led to a reduction in the level of GPEET (Fig. 2E i). In contrast, the EP protein level was not increased as expected, but was reduced to 40% of its level in the control and its migration was retarded compared to the parental strain (PS) (Fig. 2F i). However, the level of EP mRNA was increased as expected (Fig. 2f ii). The complete elimination of TblncRNA-23 in the KO may have affected the protein’s post-translational modification, altering its stability.

A Ciros plot showing TblncRNA-23 mRNA chimeric molecules. The data are from Rajan et al.22. B (i) TblncRNA-23 silencing. Cells were silenced for indicated days and the RNA was subjected to Northern analysis. Data from n = 3 experiments are presented as mean ± S.E.M., and p value was determined by Student’s t-test one tailed distribution. (ii) TblncRNA-23 in KO cells. RNA from two independent KO clones was subjected to Northern analysis. C (i) Expression of GPEET following TblncRNA-23 silencing. Lysates from induced ( + TET) and uninduced (-TET) cells were subjected to western analysis with the indicated antibodies (left). The expression level is presented as mean ± S.E.M. The p values are indicated (n = 3) and determined by Student’s t-test one-tailed distribution. ZC3H41 was used as a loading control. (ii) Expression of EP following TblncRNA-23 silencing. Whole cell lysates from induced ( + TET) and uninduced (-TET) cells were subjected to western analysis with the indicated antibodies. The expression level is presented as mean ± S.E.M (n = 3). The p values were determined by Student’s t-test one tailed distribution. PTB2 was used as a loading control. D (i) Cells were silenced for the indicated number of days, and subjected to Northern analysis. The experiments (n = 3) are presented as mean ± S.E.M., and p value was determined by Student’s t-test one tailed distribution. (ii) Northern analysis of EP mRNA in cells silenced for TblncRNA-23. Cells were silenced for the indicated number of days, and RNA was subjected to Northern analysis. The data (n = 3) are presented as mean ± S.E.M. and p value was determined by Student’s t-test one tailed distribution. E, F Expression of GPEET and EP following TblncRNA-23 KO. Extracts from PS and KO cells were subjected to Western analysis and RNA was subjected to Northern analysis. The expression level is presented as mean ± S.E.M (n = 3). The p values were determined by Student’s t-test one tailed distribution. ZCH341 and 7SL RNA were used as loading controls. Source data for all panels are provided in the Source Data file.
To further examine if the changes in expression were due to changes in the levels of mRNA and/or regulation of translation, the levels of GPEET and EP mRNA were determined by Northern analyses (Fig. 2D i and ii). While the reduction in GPEET protein could be explained almost entirely by the change in mRNA level, the EP protein level was increased by 6-fold and that of mRNA by 4-fold, suggesting that enhanced translation may also contribute to its increase. The changes in GPEET and EP levels, which are inversely correlated, resemble the changes that take place during the transition from early to late PCF, when GPEET mRNA and proteins decrease, and EP mRNA and proteins increase2,31.
TblncRNA-23 interacts with GPEET mRNA by base-pairing with its 3’ UTR, and the interaction domain is essential for its regulation
Bioinformatic analysis suggested a potential for base pairing between TblncRNA-23 and L II of the GPEET 3’ UTR (Fig. 3A i). To further study the validity of the interaction domain, TblncRNA-23 was tagged to monitor its expression by inserting a boxB (15 nt) at position 133 (Fig. 3A ii). Three types of mutations were introduced into the tagged TblncRNA-23 gene (Fig. 3A ii): a mutation removing the entire interaction domain, and two substitution mutations. Cells overexpressing wild-type and mutated TblncRNA-23 were generated, and a ~ 3-fold and 2.5-fold increase in the level of wild-type and mutant transcripts, respectively, were observed (Fig. 3B). Overexpression (OE) of the wild-type TblncRNA-23 induced a > 10-fold increase in the level of GPEET protein, and the EP protein was reduced to undetectable levels (Fig. 3C). Deletion of the interaction domain in TblncRNA-23 abolished the ability of the lncRNA to increase GPEET or reduce EP proteins. The two substitution mutations increased the level of GPEET 7-fold, with the EP protein at undetectable levels (Fig. 3C). These data suggested that the interaction domain identified by bioinformatics is valid and sufficient to control GPEET expression. Note that deleting the interaction domain of TblncRNA-23 with the GPEET mRNA drastically increased the accumulation of EP protein, suggesting that this domain might also be important for regulating EP by an as yet unknown mechanism.

A Potential base-pair interaction between TblncRNA-23 and GPEET mRNA. (i). The partial sequence and folding of GPEET mRNA from Vassella et al. 12 (in blue), and its proposed interaction with TblncRNA-23 (in red). (ii) pHD1700 plasmid carrying TblncRNA-23 with the stem-loop designed to bind the λ peptide fused to BirA. The positions of the inserted stem-loop tag and the interaction domain with GPEET are marked. The sequence of the interaction between TblncRNA-23 and GPEET is shown, and the mutations introduced are indicated in red. B Northern analysis of cells overexpressing TblncRNA-23. RNA from parental strain (PS), and cells carrying the tagged construct and its mutants were subjected to Northern analysis with the indicated anti-sense probes. The data from (n = 3) experiments are presented as mean ± S.E.M., and p value was determined by Student’s t-test one tailed distribution. Source data are provided as a Source Data file. C Western analysis of GPEET and EP in cells overexpressing TblncRNA-23. Lysates from parental cells and cells carrying the tagged construct and mutants were subjected to western analysis. The expression levels from (n = 3) experiments are presented as mean ± S.E.M. The p values were determined by Student’s t-test one tailed distribution. Methyltransferase-associated protein (MTAP) was used as a loading control. Source data are provided as a Source Data file. D Expression of the reporter gene GARP fused to GPEET/EP 3’ UTR. (i) Whole cell extract from PS and cells overexpressing TblncRNA-23 carrying GARP ORF fused to GPEET 3’ UTR or to 3’ UTR lacking LI and LII domains12, and ii) whole cell lysate from PS and cells overexpressing TblncRNA-23 carrying GARP ORF fused to EP 3’ UTR or to the 3’ UTR lacking the LII domain as in Roditi et al.10 were subjected to western analysis. The protein level is presented as mean ± S.E.M. The p values are indicated and were determined by Student’s t-test one tailed distribution, based on (n = 3) experiments. The level of ZC3H41 was used as a loading control. Source data are provided as a Source Data file.
To examine if the regulation by TblncRNA-23 is mediated by the 3’ UTR of both interacting mRNAs, we used a reporter construct in which the coding sequence of glutamic acid alanine rich protein (GARP) was ligated to different versions of the EP or GPEET 3’ UTR2,10. Expressing the entire GPEET 3’ UTR led to a 2-fold increase in the level of GARP when TblncRNA-23 was overexpressed (Fig. 3D), like the effect on the GPEET gene (Fig. 2). However, the LII domain alone was not sufficient to increase the expression of GARP, suggesting that additional 3’ UTR sequences are necessary. Expressing GARP linked to the 3’ UTR of EP led to a marked decrease in GARP when TblncRNA-23 was overexpressed (Fig. 3D), similar to the regulation pattern of the native EP (Fig. 2). This phenotype could not be achieved when LII was absent from the 3’ UTR. In fact, the level of GARP increased slightly, suggesting that, as in the case of GPEET, LII is important for its regulation by TblncRNA-23 (Fig. 3D). However, it is not sufficient to repress the expression of GARP, suggesting that the regulation involves additional domains of the 3’ UTR.
TblncRNA-23 overexpression and downregulation regulate additional mRNAs that are differentially expressed in early and late PCF
The regulation of EP and GPEET observed upon overexpression of TblncRNA-23 resembles the expression of these proteins in early and late PCF3,26. To examine if TblncRNA-23 regulates additional genes involved in this transition, we determined the transcriptome of cells overexpressing TblncRNA-23. We identified 830 transcripts (out of 10,586) whose levels were significantly changed (p value ranging from 10-160-10-2) by 1.5- to 12- fold between parental strain and overexpressing cells (Supplementary Data 1). Among the dysregulated transcripts are 74 that are differentially expressed in early and late PCF26 (Supplementary Data 1), and 283 are transcripts whose levels changed during the RBP6-induced transition from PCF to metacyclic forms (marked in yellow)32,33. A volcano plot illustrates the changes between PS and the cells OE TblncRNA-23, highlighting transcripts that were dramatically increased (Fig. 4A). Gene ontology (GO) of these transcripts showed a connection to the process of social motility (SoMo), such as cAMP signaling34,35,36 (Supplementary Fig. 5). Among the 74 transcripts that were previously shown to change from early to late PCF3,26, 46 transcripts that were increased in early PCF were elevated in the TblncRNA-23 OE cell line and among 26 transcripts that were shown to be up-regulated in late PCF3,26 17 were downregulated in TblncRNA-23- OE cells (Fig. 4B). We examined the significance of these changes using Chi-squared test and the significant values of 490 (p value = 9.4 e-109) was obtained for genes elevated in early PCF and 114 (p value = 1.04e-26) for genes that were elevated in late PCF. To further validate these findings, we examined the expression of thymidine-7-hydroxylase (TLP7) and found that TblncRNA-23 has the potential to interact with its 5’ UTR (Fig. 4C i). This interaction is supported by the presence of a chimeric RNA species identified in the interactome (Fig. 4C i). Indeed, the level of TLP7 mRNA was increased ~three-fold upon TblncRNA-23 overexpression (Fig. 4C ii). Another example of TblncRNA-23 interaction with 3’ UTRs are with putative pteridine transporter genes encoded on chromosome 1 (exemplified by Tb927.1.2880). The interaction between TblncRNA-23 and the pteridine transporter is also supported by a chimeric molecule between these RNA molecules (Fig. 4D i). Overexpression of TblncRNA-23 reduced the mRNA level, while a mutation introduced into the interaction domain abolished this response (Fig. 4D ii). Taken together, these data suggested that TblncRNA-23 is likely to regulate gene expression associated with the transition from early to late PCF (see below).

A Volcano plot of transcriptome of cells overexpressing TblncRNA-23. The volcano plot is based on DESeq257 using two independent replicates (Supplementary Data 1). The X-axis represents the log2 fold change in gene expression, and Y axis represents the −log10 adjusted p-value (p adj). Genes with differential expression (p adj ≤ 0.05) are highlighted in red, and non-significant genes are in black. B Changes in the levels of mRNA differentially expressed by early and late PCF in cells overexpressing TblncRNA-23. The transcriptome data used to prepare the graph are presented in Supplementary Data 1. Significant mRNAs (p value <=0.05) that are elevated in cells OE TblncRNA-23 and are up-regulated in early PCF are depicted in green, and those that are up-regulated in late PCF are depicted in orange. C Thymidine-7-hydroxylase (TLP7) is regulated by TblncRNA-23. (i) The proposed interaction domain and the chimeric molecules are presented. (ii) Northern analysis of TLP7 under TblncRNA-23 overexpression. RNA from PS and cells overexpressing TblncRNA-23 were subjected to Northern analysis. The data from n = 3 experiments are presented as mean ± S.E.M., and p values were determined by Student’s t-test one-tailed distribution. Source data are provided as a Source Data file. D The pteridine transporter is regulated by TblncRNA-23. (i) The proposed interaction domain and the chimeric molecule are presented. (ii) Pteridine transporter in transgenic cells overexpressing TblncRNA-23. RNA from PS and cells overexpressing TblncRNA-23 and TblncRNA-23 mutated in the putative interaction domain with the pteridine transporter were subjected to Northern analysis. The data from n = 3 experiments is presented as mean ± S.E.M., and p value was determined by Student’s t-test one tailed distribution. Source data are provided as a Source Data file.
Since lncRNA are known to regulate expression at the level of both mRNA stability and translation, we examined the proteome of cells expressing TblncRNA-23 by isotope dimethyl labeling. This method is quantitative, does not require the in vivo incorporation of the isotope, and is fast and efficient37. Analysis of the proteome shows that the levels of 36 proteins were significantly changed (Supplementary Data 2). For 17 proteins, the change in the protein level was consistent with a change in the mRNA level. Among these, 9 are developmentally regulated in early or late PCF (Fig. 5A). Nineteen proteins were affected only at the protein level, and these may be encoded by mRNAs whose translation is regulated by TblncRNA-23. To gain additional support for the effect of TblncRNA-23 on gene expression, the transcriptome of TblncRNA-23-silenced cells were determined after 2.5 days of silencing by RNA-seq of polyA RNA. We identified 675 and 698 transcripts that were up-regulated or down-regulated, respectively, by at least 1.5-fold (p value ≤ 10-2). The transcripts that were shown to be inversely regulated when comparing OE to silenced cells are presented in a heat map (Fig. 5B, Supplementary Data 3). This demonstrates that among 248 transcripts detected in both transcriptomes, 185 transcripts were inversely affected as expected, whereas only 65 transcripts were affected in the same way. In summary, at least ~200 transcripts are directly affected by the level of TblncRNA-23.

A Proteome analysis in comparison to transcriptome analysis from cells overexpressing TblncRNA-23. The proteome was determined by dimethyl labeling. One of two experiments, which gave very similar results (r = 0.9), is presented. The ratio between the level of the protein in the cells overexpressing TblncRNA-23 and PS is presented (Supplementary Data 2). Peptide-level false discovery rates (FDRs) were filtered to 1% using the target-decoy strategy. Abundances were normalized according to the Total Peptide Amount. Protein ratios were calculated based on pairwise ratio. The fold-change (FC) was compared to the parental cells, and the individual p-values shown represent confidence scores calculated by the software based on the consistency and reliability of peptide quantification within each replicate. The changes in the level of the coding mRNA is based on the experiment presented in Supplementary Data 1. Fold Change (FC) and the p-value is provided and reflect the significance of the change relative to the control. The statistical analysis is described in greater detail in the Methods section. The transcripts whose levels changed during the transition from early to late PCF are marked in green and orange. The proteins whose levels were changed, with no effect on their cognate mRNAs, are marked in gray. Proteins whose levels change in metacyclics are marked in yellow. B Heat map comparing the transcriptome of cells overexpressing or silenced for TblncRNA-23. The heatmaps were generated in R (v4.3.2) using the pheatmap package (v1.0.12). Fold-change values derived from DESeq2 analyses were used as input.
Identification of proteins associated with TblncRNA-23 using RNA-protein interaction (RaPID) assays
To identify protein factors that bind to TblncRNA-23, we used the RaPID methodology. The boxB stem-loop was placed at position 133 of TblncRNA-23, and this addition did not interfere with the ability to overexpress the RNA (Fig. 3B). Proteins that associated with the tagged TblncRNA-23 become biotinylated and the purified proteins were analyzed by LC-MS (Supplementary Data 4). To identify proteins that are specifically associated with the RNA, the enrichment of each protein was calculated38; 392 proteins were significantly enriched (Supplementary Data 4). The variability among experiments reflects variation in the efficiency of purification and/or MS analysis. We performed the purification seven times using whole cell extracts and selected to present four of the purifications that enriched the same proteins at levels of ten- to hundred-fold in at least three out of four experiments. Among these 51 proteins were selected in four purifications. The enriched proteins either directly bind to TblncRNA-23 or are present in complexes bound by it. (Supplementary Data 4). The selected proteins were enriched for ribosomal proteins and translation factors as well as with pre-mRNA processing proteins and rRNA processing factors. Many metabolic enzymes were also identified. Such enzymes were shown previously to associate with active ribosomes in mammals39 as well as in trypanosomes40. We identified 38 proteins that are encoded by mRNAs regulated by TblncRNA-23 (highlighted in Supplementary Data 4). Thus, RaPID appears to identify ribosomes that translate TblncRNA-23 substrates. The TblncRNA-23 purification also identified well-defined RNA-binding proteins, known to regulate mRNA expression41 that either bind directly to TblncRNA-23 or to its mRNA substrates (Fig. 6A). Since TblncRNA-23 was localized to the nucleolus, high salt nuclear extracts were prepared and used for the purification. Such purifications should exclusively identify the TblncRNA-23 nuclear complexes. Among the proteins identified by LC-MS in the two biological replicates (r = 0.75) 150 proteins were nuclear (Supplementary Data 5) and ~100 were specific only to the nuclear extract, most likely because of the high salt extraction. These include proteins involved in rRNA processing, chromatin remodeling and nucleopore factors (Fig. 6B, Supplementary Data 5).

A Enrichment of RNA binding proteins and translation factors in RaPID of TblncRNA-23 purified from whole cell extracts. RaPID was performed from whole cell extracts using 150 mM KCl. The table is based on the data presented in Supplementary Data 4. The fold change reflects the number of peptides normalized to the size of the protein and compared to its purification from control cells expressing BirA. The table presents four replicates. B Comparison between RaPID of TblncRNA-23 purified from the nuclear versus whole cell extracts. RaPID purification was performed on nuclear extracts prepared using 400 mM KCl. The data for this table was obtained from Supplementary Data 5. The genomic identity with its annotation and fold enrichment compared to the control (BirA alone) are indicated. One of the two experiments shown in Supplementary Data 4 and 5 is presented. C Fractionation of PUF7 in RNPs separated by sucrose gradient centrifugation. Whole cell extracts expressing of the PTP-tagged PUF7 protein (2 × \({10}^{9}\) cells) were fractionated on a 10-30% sucrose gradient at 70,000 rpm for 3 h using a Beckman SW41 rotor. The fractions were subjected to western analysis with anti-IgG antibodies, and antibodies to NRG142. The optical density of the fractions is presented. Source data are provided as a Source Data file. D PUF7 binds to TblncRNA-23. Whole cell extract from cells expressing PUF7-PTP was subjected to affinity selection on IgG beads. To prepare the cDNA for determining the level of TblncRNA-23, the following amounts were used: 1/400 of the total extract, 1/400 of the supernatant, and 1/40 of the selected RNA. TblncRNA-23 and α-tubulin mRNA was determined by real-time PCR with specific primers. The value (y-axis) represents the normalized expression of TblncRNA-23 with tubulin. The data from n = 3 experiments are presented as mean ± S.E.M., and p values were determined by Student’s t-test one-tailed distribution. Source data are provided as a Source Data file.
PUF7 binds to TblncRNA-23 and regulates many of its mRNA substrates
One of the proteins that was enriched in the RaPID purification was PUF7, which was shown to form complexes with NRG1 (nucleolar regulator of GPEET), PUF10 and BOP142. The silencing of these transcripts prolonged or increased GPEET expression42. Using sucrose gradient fractionation, we detected tagged PUF7, but not NRG1 protein in fractions containing TblncRNA-23 (see Figs. 1E and 6C), suggesting that NRG1 is part of a complex that binds to TblncRNA-23 only in the nucleolus, as opposed to PUF7, which is present in both complexes (Fig. 6C). Direct binding of PUF7 was examined using PTP-tagged protein. The RNA from each sample (total, IgG-bound and supernatant) was examined by real-time PCR using TblncRNA-23 primers. The ratio presented reflects the normalized level of TblncRNA-23 with respect to tubulin. The results showed a 15-fold enrichment of TblncRNA-23 on the beads (Fig. 6D). TblncRNA-23 binds PUF7 and is likely to donate or remove it from its mRNA substrates. Such substrates should be co-regulated by both PUF7 and TblncRNA-23. To identify these substrates the transcriptome of the PUF7-silenced cells was determined. We detected 9,673 transcripts (R = 0.9), of which about 300 transcripts were dysregulated (p < 0.01), with 80 affected both by silencing of PUF7 or by overexpressing TblncRNA-23 (Supplementary Data 6). In many cases, overexpression of TblncRNA-23 increased the levels of these mRNAs, and the same mRNAs were also increased by silencing of PUF7, suggesting TblncRNA-23 removes PUF7 from its mRNA target leading to their stabilization like under PUF7 silencing. In addition, 28 transcripts were inversely affected by TblncRNA-23 silencing i.e. reductions in their levels, suggesting that in the absence of TblncRNA-23 PUF7 is not removed from the mRNA targets and these are therefore destabilized. In other cases, the opposite effect was found, i.e. silencing of PUF7 reduced the mRNA level, suggesting that in these cases PUF7 may stabilize these mRNAs. Thus, TblncRNA-23 functions in modulating the binding of PUF7 to many of its targets and by that controls the fate of these mRNAs.
Overexpression or knockout of TblncRNA-23 affects social motility in early PCF parasites
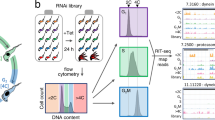
It was previously shown that early PCF exhibit social motility when plated on a semi-solid surface43. To ensure that the overexpression of GPEET and reduction in EP is shared by individual cells in the community, flow cytometry was performed using EP and GPEET antibodies. The results showed that many of cells overexpressing TblncRNA-23 highly expressed GPEET, but showed a reduction in EP (Fig. 7A). The PS and an isogenic strain OE TblncRNA-23 were spotted (2 x 106 cells) on agarose plates. Both cell populations migrated and formed projections (Fig. 7B, Supplementary Fig. 7 i). Cells OE TblncRNA-23 exhibited hyperSoMo (Fig. 7B, Supplementary Fig. 7 i). The communities on the plates were then transferred to nitrocellulose and reacted with antibodies to GPEET and EP. In PS, the late PCF were located in the center of the colony, and cells expressing GPEET were detected at the edges of the projections. In contrast, many more cells in the TblncRNA-23 OE population expressed GPEET, with the exception of a small population in the center that expressed mainly EP (Fig. 7B, Supplementary Fig. 7 i). Moreover, the projections at the edges of colonies were bifurcated. These results suggested that OE of TblncRNA-23 induced the cells to remain at the early PCF stage with active SoMo. The effect on SoMo was examined in the KO strain. The results (Fig. 7C, Supplementary Fig. 7 ii) demonstrated that the KO cells are severely compromised in performing SoMo, compared to PS, but not completely negative. The KO population expressed less GPEET as demonstrated in (Fig. 2E), suggesting that TblncRNA-23 regulates gene expression, which is essential for the early to late PCF transition.

A Flow cytometry analysis of the parental strain and cells overexpressing TblncRNA-23. Cells (5 x \({10}^{5}\)) were stained with anti-GPEET and EP antibodies, as described in Materials and Methods. Flow cytometry analysis was performed. Flow cytometry sequential gating strategy is presented at Supplementary Fig. 6. B Migration of the parental strain and cells overexpressing TblncRNA-23. Cells (2x\({10}^{6}\)) were plated on semi-solid agarose at a distance of 2.5 cm between each colony. Pattern formation was analyzed for 4 days. The parasites from the plates on day 4 were blotted, and the blot was reacted with anti-GPEET and anti-EP. Two more biological replicates are presented in Supplementary Fig. 7 i. Source data are provided as a Source Data file. C Migration of the parental strain and KO cells for TblncRNA-23. Cells (\({10}^{6}\)) were plated on semi-solid agarose at a distance of 2.5 cm between each colony. Pattern formation was analyzed in day 2 and 3 The parasites from day 3 were blotted and the blot was reacted with anti-GPEET and anti-EP. Two more biological replicates are presented in Supplementary Fig. 7 ii. Source data are provided as a Source Data file.
TblncRNA-23 is required for differentiation to metacyclic forms
The OE and silencing (Supplementary Data 1, 3) showed effects on mRNAs involved in the transition to the metacyclic form, based on the transcriptome of cells OE RBP66,7. Indeed, significant enrichment using the Chi-squared test (356 p value 7.910591e-81) of transcripts involved in metacyclic transformation was found. To examine if TblncRNA-23 also participates in controlling this transition, we OE RBP6 in cells silenced for TblncRNA-23 and examined if silencing affected the ability of the parasites to transform to epimastigote and metacyclic forms. Epimastigotes reposition the kinetoplast anterior to the nucleus and express brucei alanine-rich protein (BARP)44. Upon their transformation to metacyclic forms, the kinetoplast moves to the posterior end of the cell6. Microscopic examination of the kinetoplast position in cells either OE RBP6 or OE RBP6 and silenced for TblncRNA-23 for 5 days is presented in (Fig. 8A) and illustrates the difference in localization of the kinetoplast upon induction. Epimastigotes were found upon RBP6 OE and also in cells OE RBP6 that were silenced for TblncRNA-23. To confirm that the cells transformed to epimastigotes, we examined the expression of BARP44. The results (Fig. 8B) confirmed BARP induction in cells OE RBP6 and silenced for TblncRNA-23. Quantification of microscopic experiments recorded after 4 days of induction indicated that 14% of cells transformed to epimastigotes upon RBP6 OE, but more than 80% were transformed to this stage in OE RBP6 cells and silenced for TblncRNA-23. Eight days after induction ~30% of the OE RBP6 cells were metacyclics, whereas in cells that were also silenced for TblncRNA-23 the transition to the metacyclic form was halted (Fig. 8C). Comparing the growth of cultures (Fig. 8D) demonstrated that the OE RBP6 cells grew almost normally but OE RBP6 cells silenced for TblncRNA-23 stopped growing. Next, we determined the transcriptome of the OE RBP6 cells silenced for TblncRNA-23 for 5 days and compared it to the transcriptome of OE RBP6 cells7,8. The transcriptome of RBP6 (OE) revealed changes in 434 transcripts compared to uninduced cells6,7. The transcriptome analysis of cells OE RBP6 and silencing for TblncRNA-23 showed effect on 62 transcripts that their level was inversely expressed when comparing their level in RBP6 OE cells (Supplementary Data 7). Note that it was sufficient to alter the expression of a single mRNA to stop developmental progression9. Thus, TblncRNA-23 also controls the expression of genes that are associated with and/or necessary for the transition from epimastigote to the metacyclic form.

A Microscopic examination of parasites OE RBP6 to cells OE RBP6 and silenced for TblncRNA-23. Cells were treated with tetracycline for 5 days and were stained with DAPI and visualized in Leica SP8 confocal microscope. The cells were stained with DAPI and was subjected to differential interference contrast (DIC). The identity of the cells are indicated. N, nucleus and K, Kinetoplast. Additional images from independent experiments are presented in Supplementary Fig. 8. B Western analysis with BARP antibody. Cells OE RBP6 and silenced for TblncRNA-23 were treated with tetracycline for 5 days and subjected to Western analysis with the indicated antibodies, using ZC3H41 as loading control. The expression levels from (n = 3) experiments are presented as mean ± S.E.M. The p values were determined by Student’s t-test one tailed distribution. Source data are provided as a Source Data file. C Quantitation of the different parasite forms post tetracycline induction. The number of cells imaged are indicated and the percentage of each form is given after 4 and 8 days for cells OE RBP6 and cells OE RBP6 and silenced for TblncRNA-23. D Growth curve of cells. The growth of cells OE RBP6, OE RBP6 and silenced for TblncRNA-23 along with parental cells and cells silenced for TblncRNA-23 was monitored for 6 days. Data are presented as mean ± S.E.M., calculated from three technical replicates. Additional biological replicates of growth curves are presented in Supplementary Fig. 9. Source data are provided as a Source Data file.
Discussion
Most lncRNA species described in eukaryotes to date are transcribed by pol II. This study identified a lncRNA, TblncRNA-23, that is derived from a polycistronic pol I transcript. TblncRNA-23 is produced in the nucleolus and migrates to the cytoplasm where it associates with polyribosomes, translating its targets, affecting their mRNA stability and translation. RaPID of TblncRNA-23 identified hundreds of proteins, including pre-rRNA processing factors, ribosome-associated proteins, and dozens of RNA-binding proteins that likely regulate TblncRNA-23 substrates. The levels of hundreds of mRNAs were affected upon TblncRNA-23 silencing or overexpression, and peptides encoded by tens of these mRNAs were detected, suggesting that TblncRNA-23 regulates these mRNAs during their translation. PCF cells OE or silenced for TblncRNA-23 differentially express mRNAs that are changed during the transition from early to late PCF and upon development to the metacyclic form, and thus, TblncRNA-23 regulates gene expression associated with transition from one life-cycle stage to the next.
Previous studies showed that the clusters of genes containing GPEET and EP, are transcribed by polymerase I45, but the lncRNA regulating their expression had not been reported to date. The transcription of EP was also shown to take place in the nucleolus29,45, supporting the localization we observed for TblncRNA-23 (Fig. 1D). GPEET and EP were shown to be regulated at the mRNA level mostly by the LII domain located in the 3’ UTR of both mRNAs10,11,12,13. The interaction of TblncRNA-23 and GPEET was mapped to the region partially overlapping with LII in the 26-mer domain (Fig. 3a i). In contrast to the GPEET-TblncRNA-23 interaction, we could not identify by bioinformatics any potential interaction with EP mRNA, which might be discontinuous.
In this study, we show the involvement of PUF7 in regulating mRNA levels. Notably, PUF7 was shown to bind TblncRNA-23 (Fig. 6) and to regulate GPEET, and may compete with TblncRNA-23 for GPEET binding. PUF7 binding to GPEET results in its destabilization42. Overexpression of TblncRNA-23 may sequester PUF7, thereby preventing its binding to GPEET mRNA. Indeed, overexpression of TblncRNA-23 enhanced the expression of GPEET, similar to the effect on GPEET upon PUF7 silencing (Fig. 6). PUF7 is likely not the only protein that directly binds to TblncRNA-23. The other mRNA-binding proteins identified might be enriched due to their binding to mRNA targets of TblncRNA-23.
TblncRNA-23 may function at two regulatory levels, mRNA stability discussed above, and translation. Several translation factors were detected in the RaPID purification. Based on proteome analysis, 19 mRNAs were shown to be regulated solely at the level of translation (Supplementary Data 2), suggesting that TblncRNA-23 regulates translation, as well.
TblncRNA-23 was shown to interact with the 5’ and 3’ UTRs of mRNAs but via different domains (Fig. 3C and Fig. 4D). The interaction with the different substrates may deliver RNA binding protein(s) to these mRNA substrates. Alternatively, the binding of TblncRNA-23 to its substrate may create landing pads for RNA binding proteins that do not recognize either RNA individually. Studies from mammalian cells revealed a similar phenomenon. A lncRNA abundant in human skeletal myoblasts, associates via base-pairing with MEF2C mRNA, and stabilizes it by recruiting HuR to the MEF2C 3’ UTR, increasing MEF2C production and affecting myogenesis46. An example that resembles the regulation of GPEET by PUF7 in sequestering decay-promoting RBP is the abundant lncRNA, NORAD. This lncRNA contains many binding sites for Pumilio (PUM1/2), an RNA binding protein, like PUF7 that typically reduces the stability and or translation of its target mRNAs47. The sequestration of Pumilio by NORAD enables the production of several proteins involved in genomic stability47. Specific effects on translation, similar to those observed for TblncRNA-23 were reported with LincRNAp21, which was shown to interact via base-pairing with JUNB and CTNNB1 mRNA, and to repress translation due to recruitment of the RACK repressor to the mRNA-lncRNA complex48. The unique finding presented in this study, which was not reported before, is that RaPID purification co-precipitated the lncRNA substrates (Supplementary Data 4).
The finding that TblncRNA-23 regulates gene expression and is involved in the transition both from early to late PCF and from PCF to metacyclic forms suggests that ncRNAs play a major role in regulating the parasite developmental cycle within the insect host. Until the current study and a recent analysis of GRUMPY snoRNA23 only RNA binding proteins were known to regulate transformation from one stage to the other6,49. Interestingly, despite the high expression of metacyclic-specific genes as a result of TblncRNA-23 overexpression, the parasites did not transform to the metacyclic stage, most likely because they did not express RBP6 (Supplementary Fig. 10). It was previously shown that procyclin knockouts (which were generated by sequentially deleting pairs of procyclin genes and the intervening region) became progressively less efficient at colonizing the salivary glands and generating metacyclic forms50. This was surprising at the time, because neither EP nor GPEET is expressed in the glands. These results can now be understood since each round of gene deletion removed a copy of TblncRNA-23 and the procyclin null mutant was also a TblncRNA-23 knockout. In conclusion, this study highlights the central roles of a lncRNA in the developmental cycle of T. brucei.
Methods
Cell growth and transfection
PCF T. brucei, strain 29-1351, which carries integrated genes for T7 polymerase and the tetracycline repressor, was grown in SDM-79 medium supplemented with 10% fetal calf serum, in the presence of 50 μg/ml hygromycin and 15 μg/ml G418 to generate the RNAi silenced cell lines. The TblncRNA-23 wild-type and mutants were overexpressed from plasmid pHD1700 (https://www.zmbh.uni-heidelberg.de/Clayton/vectors.html).
RNAi construct preparation
Stem-loop constructs were generated to silence TblncRNA-23 and PUF7 using primers listed in Supplementary Table 1, as described51. Stem-loop constructs were linearized by EcorV digestion and integrated into the non-transcribed rRNA spacer.
CRISPR-Cas9 knockout
For the inducible expression of SpCas9 in T. brucei, a plasmid (Cas9-2NLS-eYFP-BSD) was generated by cloning Streptococcus pyogenes Cas9 from plasmid Cas9-NLS-PPOTv4-PURO52 into plasmid 4296-SCD6-2NLS-eYFP-BSD (a gift from Dr Susanne Kramer, University of Wuerzburg, Germany). This plasmid was transfected into T. brucei procyclic 29/13 strain. TblncRNA-23 specific guide RNAs (gRNAs) targeting 5’ and 3’ regions of the gene were designed using the EuPaGDT database (http://grna.ctegd.uga.edu/). PCR product carrying T7 promoter and gRNA was prepared. To prepare a homologous DNA repair (HDR) template, PPOTv4 plasmid carrying coding sequences for tagRFPt and puromycin resistance was used as a template for PCR amplification. To increase the efficiency of HDR integration, long primers carrying 100nt complementarity sequences in both 5’ and 3’ directions were used. The primers used for gRNA synthesis and HDR template are listed in (Supplementary Table 1). SpCas9 expression was induced 12 h prior to transfection with PCR products of gRNAs and HDR. Transfected cells were selected using puromycin, single cells were cloned using Fluorescence-activated cell sorting (FACS), and KO cells were screened by Northern analysis.
Preparation of nuclear and whole cell extracts
T. brucei PCF (108) were harvested and washed with phosphate-buffered saline. The cell pellet was resuspended in hypotonic buffer (10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.5 mm dithiothreitol, 5 μg/ml leupeptin). For whole cell extract, 150 mM KCl salt was used; the cells were subjected to 20 strokes in a Dounce homogenizer in the presence of 0.1% Nonidet P-40. For nuclear preparation, the nuclei were pelleted at 10,000 × g, and proteins were extracted in 400 mM KCl.
Fractionation of RNPs on sucrose gradient
Whole cell extracts were prepared from 5 × 109 T. brucei PCF in a buffer containing 150 mM KCl, 20 mM Tris-pH 7.6, 10 mM MgCl2, 0.5 M DTT and 0.1% NP-40. In addition, 1 μl leupeptin (10 μg/ml) protease inhibitor and 1 μl of RNasin (Thermo Scientific) were added to the lysate. Extracts were fractionated on a 10–30% sucrose gradient by centrifugation for 3 h at 210,000 g in a Beckman SW41 rotor at 4oC. Gradient fractions were used for Western or Northern analyses.
Western analysis
Whole-cell lysates (107 cells) were fractionated by SDS-PAGE, transferred to Protran membranes (Whatman), and reacted with the indicated antibodies. The bound antibodies were detected with goat anti-rabbit immunoglobulin G coupled to horseradish peroxidase and were visualized by ECL-enhanced chemiluminescence (Amersham Biosciences). The dilutions used for the antibodies are EP [Cedarlane (USA) 1:1000], GPEET (1:1000)4, PTB2 (1:10,000)53, ZC3H41 (1:10,000)54, GARP (1:10,000)55, NRG1 (2:10,000)42, and BARP (2:10,000)44. Quantitation of the signals was determined using ImageJ (https://imagej.nih.gov/ij/) software.
Northern analysis
Total RNA was prepared with TRIzol reagent (Sigma), and 20 μg/lane was fractionated on a 1.2% agarose, 2.2 m formaldehyde gel. After transfer, membranes were probed with anti-sense RNA probes. For analyzing small RNAs, total RNA was fractionated on a 6% polyacrylamide gel containing 7 m urea. The RNA was transferred to a nylon membrane (Hybond; Amersham Biosciences) and hybridized with anti-sense RNA probes. The antisense RNA probes were transcribed in vitro by T7 polymerase using a PCR product carrying the T7 promoter as a template. Quantitation of the signals was determined using ImageJ (https://imagej.nih.gov/ij/) software.
In situ hybridization
For high-resolution microscopy, T. brucei PCF were fixed on coverslips using 1.6% formaldehyde and permeabilized using Triton/Tween 20 (1:0.1%) in PBS. In situ hybridization with specific DIG-labeled RNA probes was performed as described54. The nucleolus was localized using anti-NHP2 antibody. Images were acquired using a Leica SP8 confocal microscope equipped with a white light laser and gating.
RapID of TblncRNA-23
To identify proteins that associate with TblncRNA-23, a boxB sequence that binds the λ peptide56 was introduced at position 133 with respect to the SL addition site using PCR with primers carrying the boxB sequence (listed in Supplementary Table 1). The TblncRNA-23 sequence tagged with boxB was cloned into the pHD1700 vector between the ApaI and MluI restriction sites, linearized with NotI, and integrated into the rRNA spacer in a cell line expressing the BirA-λ (biotin ligating enzyme-fused to the λ peptide) from the pPPOTv4 vector57. The proteins biotinylated by BirA-λ were purified on streptavidin beads, as previously described56 and subjected to tandem mass spectrometry, LC-MSMS, at the Smoler Proteomics Center, Technion, Israel. The mass spectrometry proteomics data was analyzed using the MaxQuant software version 2.1.1.038, and have been deposited to the ProteomeXchange Consortium via the PRIDE58 partner repository with the dataset identifier PXD049995.
Poly-A RNA-seq analysis
The RNA-sequencing reads were aligned to the T. brucei genome (v5) using Smalt v_0.7.5 (http://www.sanger.ac.uk/resources/software/ SMALT/) with default parameters. To quantify read counts per gene, multiBamCov from bedtools (v2.30.0) was used to count aligned reads from BAM files. The gene count tables were used as input for the R-Bioconductor package, DESeq2 (v1.38.3)59. The differential expression was assessed using the Wald test (default in DESeq2), and p-values were adjusted using the Benjamini-Hochberg (FDR) correction. mRNAs with an absolute log2 fold-change of >/=1 and FDR < 0.05 were considered differentially expressed. The volcano plot was generated using ggplot2 in R. To evaluate the significance of gene enrichment within list of genes a chi-square test was performed in R using the chisq.test function from https://www.R-project.org/. All sequencing data related to this study have been deposited in the NCBI BioProject database under the accession number PRJNA1066091.
Proteolysis and dimethyl labeling
Cells lysed using SDS buffer (5% SDS, 10 mM DTT, 100 mM Tris pH 8), sonicated twice (90%, 10-10 cycles, 5’), and centrifuged. The proteins in the solution were reduced (60 °C for 30 min), modified with 35.2 mM iodoacetamide in 100 mM ammonium bicarbonate (in dark, at room temperature for 30 min), and digested in 1.5 M Urea, 70 mM ABC with modified trypsin (Promega) at a 1:50 enzyme-to-substrate ratio, overnight at 37oC. A second digestion was performed for 4 h. The resulting peptides were desalted using C18 Top tip columns (Glygen), dried, and re-suspended in 50 mM Hepes (pH 7.9). Labeling by dimethylation was performed as recently described (Rajan et al.52) at the Smoler Proteomics Center, Technion, Israel, and raw data have been deposited to the ProteomeXchange Consortium via the PRIDE58 partner repository with the dataset identifier PXD049995.
Mass spectrometry analysis of proteins
The peptides were resolved by reversed-phase chromatography on 0.075 × 180-mm fused silica capillaries (J&W) packed with Reprosil reversed phase material (Dr Maisch GmbH, Germany). The peptides were eluted with a linear 180 min gradient of 5 to 28%; a 15 min gradient of 28 to 95%; and 25 min with 95% acetonitrile and 0.1% formic acid in water, at a flow rate of 0.15 μl/min. Mass spectrometry was performed by Q-Exactive HFX mass spectrometer (Thermo) in positive mode using repetitively full MS scan followed by collision to induce dissociation (HCD) of the 30 most dominant ions selected from the first MS scan. The mass spectrometry data were analyzed using the Proteome Discoverer 1.4 (Thermo) software, searching against the T. brucei from the TriTrypDB v.35 database (https://tritrypdb.org). Results were filtered with rank 1 peptides and 1% false discovery rate. The ratios were normalized according to the protein’s median ratio. Perseus software (https://maxquant.net/perseus/) was used for statistical analysis of the data. Significant outliers relative to a given population were calculated using intensity-dependent calculation. The truncation was based on the Benjamini-Hochberg correction for multiple hypothesis testing (Significance B). For the combined analysis of different replicates, one-sample t-test was used to determine if the mean was significantly different from a fixed value (0).
RNA immunoprecipitation and qPCR for TblancRNA-23
Extracts from cells (109) that express PUF7 N’PTP42 were prepared as previously described54. RNA was extracted from the total cell extract, and the supernatant and the beads were subjected to RT-qPCR reactions in a Bio-Rad CFX96 thermal cycler. Relative normalized expression of target TBlncRNA-23 relative to total RNA and supernatant RNA were calculated using alpha tubulin mRNA as a reference gene according to the formula of ΔΔCq in CFX MaestroTM Software (https://www.bio-rad.com/en-il/product/cfx-maestro-software-for-cfx-real-time-pcr-instruments).
Cells Flow cytometry analysis of cells expressing EP and GPEET
Cells (5 × 105) were washed and blocked with 2% FBS, and incubated with anti-GPEET (2:1000) and anti-EP (1:10000) antibodies. After washing, cells were further incubated with Alexa Fluor 488-conjugated anti-rabbit IgG secondary antibody (1:1000; 2% FBS/PBS) for GPEET and with Cy5-conjugated anti-mouse IgG secondary antibody (1:1000; 2% FBS/PBS) for EP. The cells were washed and subjected to flow cytometry analysis in a BD LSR Fortessa instrument (BD Biosciences, San Jose, CA, United States). Forward scatter (FSC) and side scatter (SSC) were measured using a 488 nm laser. The 640 nm laser – 670/14 nm detector pair was used to determine the auto-fluorescence of cells. Post-acquisition analysis was done using FlowJo software (ver. 10.9) (Ashland, OR, United States).
Motility assay
Plates were prepared as described4. To inoculate the parasites, 5 μl of cell suspension (106 or 2 × 106 cells/ml) was spotted onto the agarose surface, and the plates were sealed with parafilm and incubated at 27 °C. Colony lift was performed as described4. The blots were reacted with antibodies to EP [Cedarlane (USA) 1:1000)] or GPEET (1:1000), and antibody binding was detected with IgG coupled to horseradish peroxidase and visualized by ECL.
Statistics and reproducibility
All experiments were repeated at least three times with biological replicates unless otherwise indicated. Poly(A) transcriptome, dimethyl labelled proteome, and RaPID (400 mM) were repeated two times. RaPID (150 mM) was repeated seven times, but only four experiments are presented. Statistical analysis and bar-graphs were prepared using Microsoft Excel and DESeq2 (v1.38.3).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available from the corresponding authors upon request. Source data are provided with this paper. The RNA sequencing data generated in this study have been deposited in the NCBI BioProject database under the accession number PRJNA1066091. The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE58 partner repository with the dataset identifier PXD049995. For all main figures and supplemental figures Source data are provided with this paper.
References
Matthews, K. R. Controlling and coordinating development in vector-transmitted parasites. Science 331, 1149–1153 (2011).
Vassella, E. et al. A major surface glycoprotein of trypanosoma brucei is expressed transiently during development and can be regulated post-transcriptionally by glycerol or hypoxia. Genes Dev. 14, 615 (2000).
Naguleswaran, A. et al. Developmental changes and metabolic reprogramming during establishment of infection and progression of trypanosoma brucei brucei through its insect host. PLoS Negl. Trop. Dis. 15, e0009504 (2021).
Imhof, S., Knüsel, S., Gunasekera, K., Vu, X. L. & Roditi, I. Social motility of African trypanosomes is a property of a distinct life-cycle stage that occurs early in tsetse fly transmission. PLoS Pathog. 10, 1–10 (2014).
Shaw, S. et al. Cyclic AMP signalling and glucose metabolism mediate pH taxis by African trypanosomes. Nat. Commun. 13, 1–13 (2022).
Kolev, N., Ramey-Butler, K., Cross, G., Ullu, E. & Tschudi, C. Developmental progression to infectivity in trypanosoma brucei triggered by an RNA-binding protein. Science 338, 1352–1353 (2012).
Christiano, R. et al. The proteome and transcriptome of the infectious metacyclic form of trypanosoma brucei define quiescent cells primed for mammalian invasion. Mol. Microbiol. 106, 74–92 (2017).
Shi, H., Butler, K. & Tschudi, C. A single-point mutation in the RNA-binding protein 6 generates Trypanosoma brucei metacyclics that are able to progress to bloodstream forms in vitro. Mol. Biochem. Parasitol. 224, 50–56 (2018).
Toh, J. Y. et al. Identification of positive and negative regulators in the stepwise developmental progression towards infectivity in trypanosoma brucei. Sci. Rep. 11, 5755 (2021).
Roditi, I., Furger, A., Ruepp, S., Schürch, N. & Bütikofer, P. Unravelling the procyclin coat of trypanosoma brucei. Mol. Biochem. Parasitol. 91, 117–130 (1998).
Vassella, E. et al. Multiple procyclin isoforms are expressed differentially during the development of insect forms of trypanosoma brucei. J. Mol. Biol. 312, 597–607 (2001).
Vassella, E. et al. Expression of a major surface protein of trypanosoma brucei insect forms is controlled by the activity of mitochondrial enzymes. Mol. Biol. Cell 15, 3986 (2004).
Hotz, H. R., Biebinger, S., Flaspohler, J. & Clayton, C. PARP gene expression: control at many levels. Mol. Biochem. Parasitol. 91, 131–143 (1998).
Walrad, P., Paterou, A., Acosta-Serrano, A. & Matthews, K. R. Differential trypanosome surface coat regulation by a CCCH protein that co-associates with procyclin mRNA cis-elements. PLoS Pathog. 5, 1–10 (2009).
Janz, L. & Clayton, C. The PARP and rRNA promoters of trypanosoma brucei are composed of dissimilar sequence elements that are functionally interchangeable. Mol. Cell. Biol. 14, 5804–5811 (1994).
Rudenko, G., Chung, H. M., Pham, V. P. & Van der Ploeg, L. H. RNA polymerase I can mediate expression of CAT and neo protein-coding genes in Trypanosoma brucei. EMBO J. 10, 3387–3397 (1991).
Berberof, M., Pays, A. & Pays, E. A similar gene is shared by both the variant surface glycoprotein and procyclin gene transcription units of Trypanosoma brucei. Mol. Cell. Biol. 11, 1473–1479 (1991).
Michaeli, S. Trans-splicing in trypanosomes: Machinery and its impact on the parasite transcriptome. Future Microbiol. 6, 459–474 (2011).
Clayton, C. The regulation of trypanosome gene expression by RNA-binding proteins. PLoS Pathog. 9, e1003680 (2013).
Michaeli, S. Non-coding RNA and the complex regulation of the trypanosome life cycle. Curr. Opin. Microbiol. 20, 146–152 (2014).
Atayde, VanessaD., Tschudi, C. & Ullu, E. The emerging world of small silencing RNAs in protozoan parasites. Trends Parasitol. 27, 321–327 (2011).
Rajan, K. S. et al. Developmentally regulated novel non-coding anti-sense regulators of mrna translation in trypanosoma brucei. iScience 23, 101780 (2020).
Guegan, F. et al. A long noncoding RNA promotes parasite differentiation in African trypanosomes. Sci. Adv. 8, eabn2706 (2022).
Tinti, M., Kelner-Mirôn, A., Marriott, L. J. & Ferguson, M. A. J. Polysomal mRNA association and gene expression in trypanosoma brucei. Wellcome open Res. 6, 36 (2021).
Liniger, M., Bodenmüller, K., Pays, E., Gallati, S. & Roditi, I. Overlapping sense and antisense transcription units in trypanosoma brucei. Mol. Microbiol. 40, 869–878 (2001).
Naguleswaran, A., Doiron, N. & Roditi, I. RNA-Seq analysis validates the use of culture-derived trypanosoma brucei and provides new markers for mammalian and insect life-cycle stages. BMC Genomics 19, 227 (2018).
Chikne, V. et al. The canonical poly (A) polymerase PAP1 polyadenylates non-coding rnas and is essential for snoRNA biogenesis in trypanosoma brucei. J. Mol. Biol. 429, 3301–3318 (2017).
Barth, S., Hury, A., Liang, X.-H. & Michaeli, S. Elucidating the role of H/ACA-like RNAs in trans-splicing and rRNA processing via RNA interference silencing of the Trypanosoma brucei CBF5 pseudouridine synthase. J. Biol. Chem. 280, 34558–34568 (2005).
Navarro, M. & Gull, K. A pol I transcriptional body associated with VSG mono-allelic expression in Trypanosoma brucei. Nature 414, 759–763 (2001).
Gupta, S. K. et al. Small nucleolar RNA interference in trypanosoma brucei: Mechanism and utilization for elucidating the function of snoRNAs. Nucleic Acids Res. 38, 7236–7247 (2010).
Knüsel, S. & Roditi, I. Insights into the regulation of GPEET procyclin during differentiation from early to late procyclic forms of trypanosoma brucei. Mol. Biochem. Parasitol. 191, 66–74 (2013).
Shi, H., Butler, K. & Tschudi, C. Differential expression analysis of transcriptome data of trypanosoma brucei RBP6 induction in procyclics leading to infectious metacyclics and bloodstream forms in vitro. Data Br. 20, 978–980 (2018).
Doleželová, E. et al. Cell-based and multi-omics profiling reveals dynamic metabolic repurposing of mitochondria to drive developmental progression of Trypanosoma brucei. PLoS Biol. 18, e3000741 (2020).
Imhof, S. & Roditi, I. The social life of african trypanosomes. Trends Parasitol. 31, 490–498 (2015).
Saada, E. A., DeMarco, S. F., Shimogawa, M. M. & Hill, K. L. With a little help from my friends’-social motility in trypanosoma brucei. PLoS Pathog. 11, e1005272 (2015).
McWilliam, K. R. Cell-cell communication in african trypanosomes. Microbiology 169, 001388 (2023).
Nakayama, H., Nobe, Y., Koike, M. & Taoka, M. Liquid chromatography-mass spectrometry-based qualitative profiling of mRNA therapeutic reagents using stable isotope-labeled standards followed by the automatic quantitation software ariadne. Anal. Chem. 95, 1366–1375 (2023).
Hein, M. Y., Luber, C. A., Paron, I., Nagaraj, N. & Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed maxLFQ *. Mol. Cell Proteom. 13, 2513–2526 (2014).
Simsek, D. et al. The mammalian ribo-interactome reveals ribosome functional diversity and heterogeneity. Cell 169, 1051–1065.e18 (2017).
Alves, L. R. et al. Proteomic analysis reveals the dynamic association of proteins with translated mRNAs in Trypanosoma cruzi. Gene 452, 72–78 (2010).
Erben, E. D., Fadda, A., Lueong, S., Hoheisel, J. D. & Clayton, C. A genome-wide tethering screen reveals novel potential post-transcriptional regulators in Trypanosoma brucei. PLoS Pathog. 10, e1004178 (2014).
Schumann Burkard, G. et al. Nucleolar proteins regulate stage-specific gene expression and ribosomal RNA maturation in Trypanosoma brucei. Mol. Microbiol. 88, 827–840 (2013).
Oberholzer, M., Lopez, M. A., McLelland, B. T. & Hill, K. L. Social motility in african trypanosomes. PLoS Pathog. 6, e1000739 (2010).
Urwyler, S., Studer, E., Renggli, C. K. & Roditi, I. A family of stage-specific alanine-rich proteins on the surface of epimastigote forms of trypanosoma brucei. Mol. Microbiol. 63, 218–228 (2007).
Lee, M. G. & Van der Ploeg, L. H. Transcription of protein-coding genes in trypanosomes by RNA polymerase I. Annu. Rev. Microbiol. 51, 463–489 (1997).
Yang, J.-H. et al. Interaction of OIP5-AS1 with MEF2C mRNA promotes myogenic gene expression. Nucleic Acids Res. 48, 12943–12956 (2020).
Lee, S. et al. Noncoding RNA NORAD regulates genomic stability by sequestering PUMILIO Proteins. Cell 164, 69–80 (2016).
Yoon, J. H. et al. LincRNA-p21 suppresses target mRNA translation. Mol. Cell 47, 648–655 (2012).
Mugo, E. & Clayton, C. Expression of the RNA-binding protein RBP10 promotes the bloodstream-form differentiation state in trypanosoma brucei. PLoS Pathog. 13, e1006560 (2017).
Vassella, E. et al. Major surface glycoproteins of insect forms of trypanosoma brucei are not essential for cyclical transmission by tsetse. PLoS One 4, e4493 (2009).
Wang, Z., Morris, J. C., Drew, M. E. & Englund, P. T. Inhibition of trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J. Biol. Chem. 275, 40174–40179 (2000).
Rajan, K. S. et al. A single pseudouridine on rRNA regulates ribosome structure and function in the mammalian parasite trypanosoma brucei. Nat. Commun. 14, 1–16 (2023).
Stern, M. Z. et al. Multiple roles for polypyrimidine tract binding (PTB) proteins in trypanosome RNA metabolism. Rna 15, 648–665 (2009).
Eliaz, D. et al. Exosome secretion affects social motility in trypanosoma brucei. PLOS Pathog. 13, e1006245 (2017).
Schürch, N., Furger, A., Kurath, U. & Roditi, I. Contributions of the procyclin 3’ untranslated region and coding region to the regulation of expression in bloodstream forms of Trypanosoma brucei. Mol. Biochem. Parasitol. 89, 109–121 (1997).
Ramanathan, M. et al. RNA-protein interaction detection in living cells. Nat. Methods 15, 207–212 (2018).
Dean, S. et al. A toolkit enabling efficient, scalable and reproducible gene tagging in trypanosomatids. Open Biol. 5, 140197 (2015).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 50, D543–D552 (2022).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Acknowledgements
This work was supported by a grant from the Israel USA Binational Fund to S.M and C.T 20211103. S.M. holds the David and Inez Myers Chair in RNA silencing of diseases. We wish to thank the Smoler Proteomics Center, Technion, Israel, and Dr. Tamar Ziv for conducting and analyzing our proteome data.
Author information
Authors and Affiliations
Contributions
S.M., I.R., C.T., conceptualization; B.K.G., K.S.R., Y.I.A., A.F., T.D., S.C.C., H.W.B. methodology; B.K.G., T.D., S.C.C., H.W.B. formal analysis; S.M., B.K.G., I.R., C.T., writing - original draft; S.M., I.R., C.T., R.U. supervision and project administration; S.M., C.T., R.U. funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Keith Matthews, Lysangela Alves and Wei Hu, for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Galili-Kostin, B., Rajan, K.S., Ida Ashkenazi, Y. et al. TblncRNA-23, a long non-coding RNA transcribed by RNA polymerase I, regulates developmental changes in Trypanosoma brucei. Nat Commun 16, 3697 (2025). https://doi.org/10.1038/s41467-025-58979-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58979-w
This article is cited by
-
DRBD3 regulates long non-coding RNA abundance and cryptic splice site selection in trypanosomes
Cellular and Molecular Life Sciences (2025)
