
Abstract
Carbon fibre (CF) is a lightweight next-generation material with numerous applications. CF production is predominantly based on the polyacrylonitrile (PAN) precursor because of the high tensile strength of PAN-based CFs (PAN-CFs). However, expensive and toxic organic solvents are required for PAN-CF production. Moreover, the corresponding thermal stabilisation process is highly energy-intensive, leading to high CO2 emissions. Herein, aqueous polyacrylamide (aqPAM) fibre—prepared via polymerisation and dry spinning using water—is proposed as a water-soluble CF precursor that can be converted into high-performance CF at high carbon yields. The incorporation of small amounts of phosphoric acid into aqPAM considerably reduces the thermal stabilisation time and increases the carbon yield compared with that obtainable using PAN-CF. Moreover, the carbonised aqPAM-based CFs exhibit a high tensile strength and tensile modulus comparable with those of PAN-CFs. The developed process generates less CO2 emissions than PAN-CF production.
Similar content being viewed by others

Introduction
Carbon fibre (CF)1,2,3,4,5,6,7 is a lightweight next-generation material with excellent mechanical properties, good corrosion resistance, and high thermal and electrical conductivities. The precursors used in CF production include polyacrylonitrile (PAN, Fig. 1a) (90%), petroleum pitch (8%), and cellulosic precursor (2%)6. Strong and lightweight PAN-based CF (PAN-CF) can be used in automobiles and aerospace applications instead of steel and aluminium to reduce body weight and, consequently, CO2 emissions2,4. However, the energy consumption and CO2 emissions during PAN-CF production (Fig. 1a) considerably exceed (Fig. 1b)8,9 those during steel and aluminium production10,11. Given that PAN is insoluble in water, expensive, and toxic, high-boiling-point petroleum-derived organic solvents, such as dimethyl sulphoxide (DMSO) or N,N-dimethylformamide, are used during polymerisation of acrylonitrile (AN)12 and wet (or dry-jet wet) spinning13 (Fig. 1a, Supplementary Table 1). Thus, PAN-CF production generates hazardous waste3 and increases the cost (Supplementary Table 2) and the electric energy usage (CO2 emissions) for recycling organic solvent (Supplementary Table 3). Therefore, an alternative precursor without an organic solvent is necessary. For PAN-CF, energy consumption typically accounts for ~37% of the CF production cost from PAN precursor, of which 21% and 14% are related to the stabilisation and carbonisation processes, respectively (Fig. 1c)6. Therefore, the energy consumption of the stabilisation process accounts for ~57% of the total energy consumption (Fig. 1c), which reflects the long heat treatment time (typically 200 − 300 °C, 60 − 90 min)6,7,9. The stabilisation process has also been reported to account for ~63% of the energy consumed during PAN-CF production9. The CO2 emissions associated with the stabilisation process are largely due to electric energy consumption6,14, although small amounts of CO2 are directly exhausted from the fibre during stabilisation6,14. Hence, the energy consumption of the stabilisation process can (and should) be reduced by decreasing its duration. Low-cost CF precursor candidates such as lignin1,2,3,4,15 polyethylene2,16, and polyamide17 have been reported (Supplementary Tables 1 and 2). However, they require prolonged stabilisation times ( ≥1 h, Supplementary Table 1), and hence, the CO2 emissions cannot be reduced; moreover, the mechanical properties of CFs produced from these precursors are inferior to those of PAN-CFs1,2,3 (Supplementary Table 1). Among lignin precursor candidates, ammonium lignosulphonate/poly(vinylpyrrolidone) has been proposed as a water-soluble CF precursor15. However, this precursor requires a long stabilisation time (heating in air up to 250 °C using a heating rate of 1 °C min−1) and exhibits poor mechanical properties (tensile strength = 1.3 GPa, Supplementary Table 1). In addition, lignin itself features a low carbon yield (30 − 40 wt%)15. Herein, we report an ecofriendly water-soluble CF precursor (Fig. 1a, d) that can be converted into high-performance CF with good mechanical properties (tensile strength >3 GPa) and high carbon yields and propose an innovative production process based thereupon with significantly lower CO2 emissions due to rapid stabilisation.

a Innovative CF production process using aqPAM developed as an ecofriendly water-soluble precursor in this study and conventional production process using polyacrylonitrile (PAN) precursor. The polymerisation yield achieved using primarily the acrylamide (AM) monomer is higher (PAM (PAM homopolymer)): ~100%, AM/acrylonitrile (AN) (75/25 mol%): ~100%) than that obtained using primarily the AN monomer (the solution polymerisation yield of AN is usually below ~50% at 70 °C after 8 h12, and a long polymerisation time ( ≥12 h) and high temperature ( >70 °C) were necessary for achieving a yield of ~80%). Polymerisation and spinning of aqPAM in water do not require energy for the recycling and disposal of the organic solvent and residual monomers. b Energy consumption and CO2 emissions during the production of conventional PAN-CF8, steel, and aluminium10,11. c Typical distribution of production cost and energy consumption of PAN-CFs6. d Images and chemical structures of materials used in aqPAM-CF production.
Results
Fabrication and evaluation of a water-soluble CF precursor
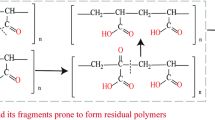
Heat-treating PAN at 250 − 350 °C under air or N2 rapidly generates considerable amounts of heat (exothermic reactions), as determined by differential thermal analysis (DTA) (Fig. 2a (a), 2b (a)) and sealed cell (SC)-differential scanning calorimetry (DSC) (Supplementary Fig. 1a and b, Supplementary Fig. 2e), for the radical cyclisation of the AN monomer units (Supplementary Fig. 3). Therefore, the temperature must be gradually increased from ~240 to ~300 °C and maintained at ≤300 °C6,7,18 using limited amounts of PAN fibre in large ovens9,13 to prevent thermal runaway and consequent fibre breakage. Regarding this range of PAN stabilisation temperatures (240 − 300 °C), a maximum temperature of ~270 °C (e.g. 240 − 270 °C for ≥60 min) was previously used to maximise the CF tensile strength7. Meanwhile, the extent of PAN fibre stabilisation at 300 °C for 60 min was reported to exceed that at 250 °C or 275 °C for 60 min18. In our preliminary studies (Supplementary Table 4), PAN stabilisation at 240 − 300 °C for 60 min resulted in a stabilisation–carbonisation total yield (40.8%) exceeding that obtained at 240 − 270 °C for 60 min (35.4%), and a longer time (240 − 270 °C for 75 min) was therefore necessary for increasing the total yield (to 43.9%) when the maximum temperature was set at 270 °C. In contrast, the precursor proposed herein does not require prolonged heating, is water-soluble, and forms a polycyclic (stabilised) structure after rapid heating (Fig. 1d). Polycyclic structures have high heat resistance and can form polycyclic aromatic structures under N2 at high temperatures. We focused on polyacrylamide (PAM), a low-cost general-purpose polymer19 synthesised via the high (near-quantitative)-yield polymerisation of acrylamide (AM) in water (Fig. 1a, d, Supplementary Fig. 4). Aqueous PAM (aqPAM) can be used for dry spinning20 during fibre production (Fig. 1a, d). The use of water for dry spinning is inexpensive and environmentally friendly, because this spinning method requires only the mild evaporation of non-toxic water from aqPAM–water solutions. The results of density functional theory (DFT) calculations on the AM dimer model (Supplementary Fig. 5) and previous studies on the thermal degradation of PAM19,21 indicate that PAM can undergo cyclisation reactions between neighbouring amide groups (Fig. 1d). The DFT calculations demonstrate that the formation of an imide (glutarimide) ring via NH3 gas elimination is the most favourable reaction when the AM dimer is used (Supplementary Table 5). The AM trimer can form cyclic rings via NH3 and H2O evolution19,21 (Supplementary Fig. 6). As revealed by DTA (Fig. 2a (c)) and SC-DSC (Supplementary Fig. 1c and d), PAM does not rapidly produce heat during cyclisation; instead, endothermic reactions (cyclisation with NH3 and H2O evolution) occur at ~230 °C. The evolution of NH3 and H2O was inferred from the results of thermogravimetry (TG)-mass spectrometry (MS) analysis (Supplementary Fig. 7b). Actually, PAN showed no evolution of NH3 and H2O at ~230 °C (Supplementary Fig. 7d). However, the direct carbonisation yield of PAM under N2 (15.1%, Fig. 2b (b), Supplementary Table 6), attributed to a drastic weight decrease above ~330 °C, is lower than the carbonisation yield of PAN (Fig. 2b (a)). Thus, the formation of glutarimide rings and polycyclic structures—which are highly heat resistant at ≥ ~330 °C—must be accelerated to prevent PAM decomposition. Given that acids can accelerate the formation of glutarimide from diamides22, an acidic catalyst that is heat-resistant at or above the stabilisation temperature ( ≥ ~300 °C) was selected herein (Fig. 1d). The preliminary results of the direct carbonisation of PAMs containing potential catalysts are presented in Supplementary Table 6. Polyphosphoric acid (PPA) is highly heat resistant ( ≥ ~300 °C) and exhibits the highest yield. Phosphoric acid (PA) forms PPA via polycondensation at ≥ ~200 °C (Supplementary Fig. 8a) and is therefore effective for producing carbonised materials. PA acts as a catalyst to form glutarimide rings via NH3 evolution (Supplementary Fig. 8b). The acceleration of the formation of glutarimide rings using PA was also confirmed by experiments using an AM dimer model molecule (glutaramide) (Supplementary Fig. 9a–c). Glutarimide rings were formed even at 200 °C by the addition of PA. The experiments using an AM trimer model molecule (1,3,5-pentanetricarboxylic acid triamide synthesised from 1,3,5-pentanetricarboxylic acid) and PA suggested the existence of polycyclic compounds at 300 °C (Supplementary Fig. 10a and b). In addition, PA was also found to acts as a catalyst to form nitrile groups (Supplementary Fig. 9a–c) due to dehydration reactions of AM units (Supplementary Fig. 8c), although the formation of nitrile groups is much slower than that of glutarimide rings (Supplementary Fig. 9c). PA salts, such as diammonium hydrogen phosphate (DAP), are also effective (Supplementary Table 6, Supplementary Fig. 11). The results of thermogravimetric analysis (TGA) (Fig. 2a (c)) indicate that weight loss due to the cyclisation (Supplementary Figs. 6 and 8b) of PAM containing PA (PAM-PA) is initiated at a temperature lower than that observed for PAM (Fig. 2a (b)). Table 1 and Supplementary Table 7 present the properties of various aqPAM precursors. Stabilisation yields, carbonisation yields, and the overall stabilisation–carbonisation (total) yields of aqPAM and PAN precursors were evaluated by TGA weight losses of them (Supplementary Figs. 12 and 13). When stabilising PAM in short time (10 min), higher temperature ( ≥350 °C) was better for increasing the total yield (Supplementary Table 8). This trend is different from that observed for PAN, which required prolonged stabilisation at lower temperatures to increase the total yield (Supplementary Table 4). Therefore, PAM stabilisation was performed at 350 °C for 10 min (Table 1). PAM-PA (a-2) exhibits higher carbonisation and carbon yields than PAN (b-1); however, the total yield is lower than that of PAN because the stabilisation yield of a-2 (69.7%) is much lower than that of b-1 (90.6%) because of NH3 and H2O elimination. The copolymerisation of AN with AM in water (Supplementary Fig. 4b) improves the total yield to produce water-soluble AM/AN (75/25 mol%) copolymers (a-3 − a-6) (Table 1), although the glass transition temperature (Tg) of AM/AN is minimally lower than that of PAM (Supplementary Table 7). In addition, AM/AN showed a slight exothermic reaction around 240 °C in air as shown in the SC-DSC diagrams (Supplementary Fig. 2a and e). The higher carbonisation yields are partly attributable to the higher theoretical carbon yield (54.1%) of the AM/AN copolymer (Table 1). The 5% decomposition temperature of AM/AN (248 °C) (Supplementary Fig. 14a) is considerably lower than that of PAM (283 °C) (Supplementary Fig. 7a). The residual weight of AM/AN at 600 °C (27.5 wt%) (Supplementary Fig. 14a) is considerably higher than that of PAM (17.8 wt%) (Supplementary Fig. 7a). These results show that AM/AN forms polycyclic structures at lower temperatures than PAM does, which, in turn, prevents thermal decomposition at high temperatures (300 − 600 °C). Figure 2c and Supplementary Fig. 14b show the TG-MS analysis results for AM/AN (75/25 mol%) (a-3). The intensities at m/z = 18 (mass number of H2O) and m/z = 17 (mass number of NH3 and H2O corresponding to ~22%23 of H2O (100%) observed at m/z = 18) during the temperature increase correspond to the concentrations of water vapour and NH3 gas, respectively. When AM/AN (a-3) is used for stabilisation (Supplementary Fig. 15b), radical cyclisation between neighbouring nitrile groups may lead to polycyclisation with amide groups, resulting in higher carbonisation yields. a-5 with 2.9 wt% PA exhibits sharp peaks corresponding to water vapour and NH3 at a temperature ( ~ 238 °C) (Supplementary Fig. 14d) which is lower than that observed for a-3 ( ~ 285 °C) (Supplementary Fig. 14b). These results indicate that glutarimide rings and/or polycyclic rings are formed at a lower temperature in the former case. The incorporation of acrylic acid (AA) units into AM/AN (AM/AN/AA) (a-7–a-10) (Supplementary Fig. 4c and 4d) improves the total and carbon yields. The incorporation of AA or itaconic acid (IA) into PAN24,25,26,27,28 initiates the cyclisation of AN units via an ionic mechanism24,25, and the carbonyl groups further facilitate chain crosslinking via dehydrogenation reactions during subsequent carbonisation25. Therefore, the improved total yields for AM/AN/AA are attributed to the reactions shown in Supplementary Fig. 15c and d. Increasing the AN content from 25 to 33 mol% (a-11) leads to insolubility. However, the incorporation of AA units into AM/AN containing 33 mol% AN (a-12, a-13) and 35 mol% AN (a-14–a-18) leads to good solubility in water ( ≥50 °C), which further improves the carbonisation, total, and carbon yields. The carbon yields of a-15, a-17 and a-18 are very high (94.6%–96.8%, Fig. 2d). The TG-MS results indicate that the water vapour and NH3 gas evolution in a-17 reach their maxima at ~270 and 264 °C, respectively (Supplementary Fig. 14h); these temperatures are marginally lower than those observed for a-16 (Supplementary Fig. 14f).

a Thermogravimetric (TG)–differential thermal analysis (DTA) diagrams (heating rate: 10 °C min−1, in air) of (a) PAN (black line), (b) PAM (PAM homopolymer) (blue line), and (c) PAM containing 2.0 wt% PA (red line). b TG-DTA diagrams (heating rate: 10 °C min−1, under N2) of (a) PAN (black line), (b) PAM (blue line), and (c) PAM containing 2.0 wt% PA (red line). c TG-mass spectrometry (MS) diagrams of aqPAM (AM/AN = 75/25 mol%) under He/O2 (80/20 vol%) atmosphere. d Carbonisation and carbon yields of various precursors measured at 1000 °C via TG-DTA. e Fourier transform infrared (FT-IR) spectra of aqPAM (a–3, AM/AN (75/25 mol%)) fibre, and aqPAM-SFs prepared from a–3, a–5, or a–6 under different stabilisation conditions. IA/IB is the ratio of the intensity (IA) of the absorption peak in the range of 1560–1595 cm−1 to the intensity (IB) of the amide group absorption peak near 1648 cm−1. When phosphoric acid (PA) or diammonium hydrogen phosphate (DAP) was used, the content was 2.9 wt%. The new small band at ~2205 cm–1 was previously observed in the FT-IR spectra of stabilised PAN fibres26 and may be attributed to the α,β-unsaturated nitrile groups resulting from the dehydrogenation or tautomerisation and isomerisation of the ladder polymer. f Relation between the IA/IB of aqPAM-SFs and carbonisation yields. aqPAM-SFs prepared using aqPAM fibres ((●) a–3, AM/AN (75/25 mol%), (■) a–5, AM/AN (75/25 mol%) containing 2.9 wt% PA, and (♦) a–6, AM/AN (75/25 mol%) containing 2.9 wt% DAP) by various stabilisation conditions.
Preparation and evaluation of aqPAM-stabilised fibres (SFs)
The aqPAM fibres obtained after dry spinning (Supplementary Fig. 16) had smooth surfaces and dog bone–shaped cross-sections (Supplementary Fig. 16) typical of dry spinning–prepared fibres3. The aqPAM fibres were drawn at a draw ratio of 4 (Supplementary Table 9), and the drawn fibre bundles were stabilised in a stabilisation oven at 350 °C for 10 min under air to obtain the SFs (Supplementary Table 10, Supplementary Fig. 17). The obtained SFs had smooth surfaces and dog bone–shaped cross-sections (Supplementary Fig. 17). The tensile strengths and tensile moduli of aqPAM fibres were lower than those of the control (modified PAN (mPAN)) fibres (Supplementary Fig. 18), as the employed mPAN had been drawn using a water bath and drying oven in the wet spinning process (for example, 13 times in total). As a result, in the drawn mPAN fibres, the crystallites had a high degree of preferential orientation (DPO) along the fibre axis (0.87), as revealed by wide-angle X-ray scattering (WAXS) measurements (Supplementary Table 11). Given that the aqPAM fibres comprised the amorphous polymer itself, no pronounced orientation preference was observed by WAXS at this stage (DPO = 0.31, Supplementary Table 11). However, the aqPAM molecules can be oriented along the fibre axis during drawing at ≥230 °C and stabilisation because of the formation of rigid glutarimide rings and polycyclic structures along the fibre axis, which leads to a tensile strength (Supplementary Fig. 18) almost identical to that of mPAN-SFs with similar DPOs. The stabilisation of aqPAM was examined by acquiring the Fourier transform infrared (FT-IR) spectra of the SFs obtained after the stabilisation of AM/AN (75/25 mol%) fibres at different temperatures (Fig. 2e). The SFs give rise to bands between ~1560 and 1595 cm–1 (Fig. 2e), which are attributed to a combination of C = C and C = N vibrations and the bending/inclination of the NH group on the stabilised (ladder) structure plane (Supplementary Fig. 19a). These bands are also present in the FT-IR spectra of stabilised PAN structures (Supplementary Fig. 19b)24,26. The ratio of the intensity of the absorption peak in the range of 1560–1595 cm−1 (IA) to the intensity of the amide absorption peak near 1648 cm−1 (IB) was used as the stabilisation index of aqPAM (Fig. 2e, f). In the case of AM/AN, increasing the carbonisation yield to ≥60% requires IA/IB ≥1.5 (Fig. 2f), which is achieved at 350 °C stabilisation with PA or DAP. PAN does not result in a high carbonisation yield (51.5%) after stabilisation at 350 °C for 10 min (Supplementary Table 4), leading to very low total yield (Supplementary Table 4, 39.4%). An extended stabilisation of temperature (e.g. heating from 240 to 270 °C for 75 min) is necessary to increase the total yield of PAN and mPAN (Fig. 2d, Supplementary Table 4). The carbonisation yield of aqPAM-based SFs (aqPAM-SFs) obtained after stabilising a-17 at 300 °C for 10 min is higher than that obtained after stabilising PAN or mPAN at 240–270 °C for 75 min (Fig. 2d). Therefore, the stabilisation time is largely shortened. The stabilisation of a-17 at a marginally higher temperature (320 or 350 °C) further improves the carbonisation and carbon yields (Fig. 2d). The amounts of electric power required for maintaining the interior of the furnace at 300, 320, or 350 °C are similar (Supplementary Table 12). Therefore, shortening the stabilisation time is crucial for reducing CO2 emissions. The high electricity demand during CF production has a major influence on the related CO2 emissions14. The direct CO2 emissions due to the release of gases during PAN fibre stabilisation is small (e.g. ~ 17% of CO2 emissions in CF production14). Therefore, ~83% of the total CO2 emissions can be attributed to electricity consumption (Fig. 3b). The stabilisation from 240 to 270 °C for 75 min is electric energy–intensive (Supplementary Table 13). Hence, the electric energy consumption during the rapid stabilisation of aqPAM at 300 °C for 10 min, 350 °C for 10 min, and 330 °C for 12 min was reduced by ~84%, ~83%, and ~80%, respectively (Supplementary Table 12). This reduction resulted in a decreased electric energy consumption (Fig. 3a) for CF production from precursor fibres by up to ~48%, and the relative index of energy consumption (0.52) was comparable to that of aluminium production (Fig. 1b, 0.48). Given that ~83% of the total CO2 emissions are assumed to be attributed to electricity consumption14, the stabilisation of aqPAM at 300 °C for 10 min reduces the CO2 emissions of the CF production from precursor fibres by ~40% compared with those observed for PAN stabilisation (Fig. 3b). In addition, high-yield polymerisation and dry spinning without organic solvent for producing aqPAM fibres are expected to lead to reduced energy consumption and CO2 emissions for aqPAM fibre production by ≥50%, because recycling organic solvent and AN is not necessary (Supplementary Table 3). The Raman spectra of the sidewalls of the SFs prepared using a-5 or a-17 exhibit G-bands at ~1590 cm−1 (Supplementary Fig. 20), which indicates the formation of graphite structures. For SFs, the intensity ratio between the D-band derived from the defective structure ( ~ 1350 cm−1) and G-band (ID/IG) of SFs derived from a-17 is lower than that observed for a-5, indicating that a-17 more readily forms graphite structures. The solid-state nuclear magnetic resonance (NMR) spectra of the SFs derived from a-17 (Supplementary Fig. 21c, e) resemble those of the mPAN-based SFs (mPAN-SFs, Supplementary Fig. 21d, f)27,29; however, the integrated area of the aromatic C = C peaks from a-17 exceeds that obtained for b-2 (Supplementary Fig. 21g).

a Electric energy consumption-relative index during the production of PAN-CFs6 and aqPAM-CFs from the precursor fibres. b CO2 emissions-relative index during the production of PAN-CFs14 and aqPAM-CFs from the precursor fibres. The difference in the corresponding electric energies of stabilisation between aqPAM fibres and PAN fibres was estimated from the experimental results when our laboratory-sized furnace was used at these temperature conditions (Supplementary Tables 12 and 13). In this study, the energy consumption in the stabilisation operation is solely in the form of electricity. Note that reduced energy consumption and CO2 emissions during high-yield polymerisation and dry spinning without organic solvent for producing aqPAM fibres (with estimated reduction rates of ≥50% for both, compared with producing PAN fibres, as shown in Supplementary Table 3) are not considered here. However, these reductions are expected to lead to further reduction in CO2 emissions for the entire CF production process, potentially by ≥40%.
Preparation and evaluation of aqPAM-CFs
Pre-carbonised fibres (PCFs) were obtained by heating aqPAM-SFs at 800 °C under N2 followed by carbonisation at higher temperature to form the aqPAM-CFs (Fig. 1d, Supplementary Figs. 19 and 22). The cross-sections of the aqPAM-CFs were dog bone-shaped (Fig. 4a, Supplementary Figs. 22 and 23), whereas those of the mPAN-CFs were elliptical (Fig. 4b). Scanning electron microscopy (SEM) images of the cross-sections (the tensile fractured surfaces) of the aqPAM-CFs showed that the fibres had no microvoids in their cross-sections (Fig. 4c, Supplementary Figs. 23a and 23b). The ID/IG value of the centre of the cross-section of aqPAM-SF containing PA prepared after heating a-17 at 330 °C for 12 min minimally exceeded that of the corresponding outer region (Supplementary Fig. 24). However, the ID/IG values of centre of the cross-section of the aqPAM-CF produced after the carbonisation of the aqPAM-SF were comparable with those of the outer region of the cross-section (Fig. 4d and e). Therefore, in the case of aqPAM fibre containing PA, rapid stabilisation worked well for stabilisation (oxidation) to obtain uniformly carbonised CFs. This behaviour was ascribed to abundance of O atoms in aqPAM leading to the formation of glutarimide rings with carbonyl groups (in addition, aqPAM also contains O atoms derived from PA), although this hypothesis requires a detailed evaluation, including the effect of the stabilisation and carbonisation ramp rates on the CF microstructure and properties. The elemental analysis results of the aqPAM-based PCFs (aqPAM-PCFs) show that the carbon content in aqPAM-SFs with PA increases to ~73 wt% (Supplementary Fig. 25a), and phosphorus atoms are detected after stabilisation, pre-carbonisation, and carbonisation (Fig. 5a, Supplementary Fig. 25a, Supplementary Table 14). The TGA results of PA (Supplementary Fig. 26) indicate that PA or PA-derived compounds are still present after stabilisation at 350 °C. For confirming the existence of PA after heating at 350 °C, X-ray photoelectron spectroscopy (XPS) measurements were performed to detect P–O bonding in aqPAM-SFs containing PA (Fig. 5b), which were prepared after stabilisation at 350 °C for 10 min. The P 2p spectrum of aqPAM fibre containing 2.9 wt% PA displayed a peak at 133.3 eV (Fig. 5b) attributable to PA. In contrast, the spectrum of aqPAM-SF obtained after stabilisation at 350 °C for 10 min displayed a peak at ~134.2 eV attributable to the P–O bonds in PPA30,31 formed by the polycondensation of PA, showing that PPA remained on the surface of aqPAM-SFs. The results of SEM–energy-dispersive X-ray spectroscopy (EDS) reveal that the phosphorus atoms are uniformly distributed on the surfaces of the aqPAM fibres, SFs, and CFs (Supplementary Fig. 27). Although the chemical structures of the PA-derived carbonised compounds on the CF surfaces remain unclear, phosphorus atoms are present even after carbonisation at high temperatures, as revealed by the results of XPS measurements (Fig. 5b), elemental composition analyses (Fig. 5a, Supplementary Table 14) and SEM-EDS measurements. The P 2p spectra of aqPAM-CF obtained after carbonisation at 1000–1400 °C displayed signals at ~131.4 and ~134.2 eV, while those of aqPAM-CF obtained after carbonisation at 1100–1500 °C showed peaks at 129.2 and 132.5 eV. The signal of aqPAM-CF (1400 °C) at ~134.2 eV was attributed to PPA. The peak at 132.5 eV was attributed to P bound to O atoms, and those at 129.2 and 131.4 eV were attributed to elemental P and P compounds without O atoms30,31. Owing to the low P content of the samples, no clear evidence of the formation of C–P covalent bonds was obtained. aqPAM fibre containing 2.9 wt% PA (a-5) and aqPAM-CFs obtained after carbonisation at 1000–1400 °C for 3 min were subjected to XPS depth profiling (Supplementary Fig. 28). The P content of the aqPAM fibre with 2.9 wt% PA was 0.7–1.0 at% in the depth range of 0–1000 nm (Supplementary Fig. 28a), which matches the P content (~0.9 wt%) in aqPAM fibre measured by elemental composition analysis (Supplementary Fig. 25a). The P content of the aqPAM-CFs obtained after carbonisation at 1000–1400 °C was determined as 1.7 wt% by elemental composition analysis (Supplementary Fig. 5a), however, the P content measured by XPS depth profiling in the range of 0–1000 nm was very low (0–0.25 at%, Supplementary Fig. 28b). Therefore, most P atoms existed on the surface, as shown in the SEM-EDS P mapping of the sidewalls of aqPAM-CFs (Supplementary Fig. 27). The Raman ID/IG ratio in cross-sections perpendicular to the direction of the fibre axis of a single CF fibre extracted from aqPAM-CFs is 0.9–1.0 at both the outer periphery of the cross-section and the centre of the cross-section of this fibre (Fig. 4d, e). The outer periphery of the cross-section and the centre of the cross-section of mPAN-CFs also showed a similar ID/IG ratio. These results indicated that carbonisation sufficiently progressed in both the outer and inner regions of aqPAM-CFs and mPAN-CFs.

a Optical microscope image of a cross-section of a single fibre extracted from aqPAM-CFs (precursor: a–5, stabilisation condition: 350 °C for 10 min, pre-carbonisation condition: 500–800 °C for 3 min, carbonisation condition: 1000–1400 °C for 3 min). b Optical microscope image of a cross-section of a single fibre extracted from modified PAN (mPAN)-based CFs (mPAN-CFs) (precursor: b–2, stabilisation condition: from 240 to 270 °C for 75 min, pre-carbonisation condition: 500–800 °C for 3 min, carbonisation condition: 1000–1400 °C for 3 min). c SEM images of a cross-section of a single fibre extracted from the aqPAM-CFs (precursor: a–5). d Raman ID/IG mappings of cross-sections perpendicular to the fibre-axis direction of a single CF from aqPAM-CFs (precursor: a–17, stabilisation condition: 330 °C for 12 min, pre-carbonisation condition: 500–800 °C for 3 min, carbonisation condition: 1000–1400 °C for 3 min) and mPAN-CFs (precursor: b–2, stabilisation condition: from 240 to 300 °C for 60 min, pre-carbonisation condition: 500–800 °C for 3 min, carbonisation condition: 1000–1400 °C for 3 min). e Raman spectra of the outer periphery and centre of the cross-sections (a–d) in Fig. 4d of aqPAM-CFs and mPAN-CFs.

a Results of elementary analysis of aqPAM-CFs after the carbonisation of PCFs containing PA at various temperatures, and mPAN-CFs after the carbonisation of PCFs at 1000–1400 °C. The PCFs were prepared by the pre-carbonisation of stabilised fibres at 800 °C. b XPS P 2p spectra of aqPAM fibre, aqPMA-SF, and aqPAM-CFs. Black line indicates P 2p signals of aqPAM (a–5, AM/AN (75/25 mol%) containing 2.9 wt% PA) fibre, red line indicates P 2p signals of aqPAM-SF obtained after stabilisation of a–5 fibre at 350 °C for 10 min, yellow line indicates P 2p signals of aqPAM-CF obtained after the carbonisation at 1000–1400 °C of PCFs derived from the aqPAM-SF, blue line indicates the P 2p signals of aqPAM-CF obtained after the carbonisation at 1100–1500 °C of PCFs derived from the aqPAM-SF.
Properties of aqPAM-CFs
The mechanical properties of single fibres extracted from aqPAM-CF bundles (Supplementary Fig. 22a) were evaluated (Fig. 6a–c). The tensile strength and tensile modulus of aqPAM-CFs derived from SFs (stabilisation of a-5 at 300 °C for 10 min) are 1.46 and 147 GPa, respectively (Fig. 6a, b), while higher values of 1.74 and 176 GPa, respectively, were obtained for aqPAM-CFs derived from SFs (stabilisation of a-5 at 350 °C for 10 min) (Fig. 6a (b), 6b (b)). The aqPAM-CFs derived from a-17 (stabilisation: 350 °C for 10 min) exhibit a higher tensile strength (2.11 GPa) and tensile modulus (210 GPa) (Fig. 6a (g), 6b (g)). The tensile strength (2.10 GPa) and tensile modulus (212 GPa) of the aqPAM-CFs obtained after the 350 °C stabilisation of a-16 without PA are similar (Fig. 6a (h), 6b (h)) to those of the fibres derived from a-17, although the carbonisation yield of a-16 is lower (Table 1, Supplementary Table 15). The tensile strength of the aqPAM-CFs obtained from a-17 stabilised at 320 °C for 12 min (Fig. 6a (c)) is 2.32 GPa; this value is higher than that obtained after 350 °C stabilisation, which may be attributed to the thermal degradation of SFs during their high-temperature (350 °C) stabilisation in air. The elongation of the CFs derived from the 350 °C–stabilised fibres is lower than that of the CFs obtained through 320 °C stabilisation (Fig. 6c), which might be attributed to defects derived from thermal degradation at ≥350 °C in air, as shown in the TGA weight decrease in the temperature range from ~350 to ~450 °C (Supplementary Fig. 14g). The tensile strength increases to 3.19 GPa after 330 °C stabilisation for 12 min (Fig. 6a (d)). The tensile modulus of the CFs derived from 320 °C stabilisation for 12 min is 170 GPa (Fig. 6a (c)) and increases to 229 GPa after 330 °C stabilisation for 12 min (Fig. 6b (d)). Carbonisation at a higher temperature (1500 °C) increases the tensile modulus (263 GPa), although the tensile strength decreases marginally (2.78 GPa) (Fig. 6a (e)). mPAN-based CFs were prepared under identical conditions (1000–1400 °C or 1100–1500 °C) using the same carbonisation furnace (Fig. 6a (i)–(k)). The tensile strength and tensile modulus of mPAN-CFs derived from SFs (stabilisation of b-2 at 240–270 °C for 60 min) are 3.01 and 210 GPa, respectively (Fig. 6a (i), 6b (i)), while the stabilisation of b-2 at 240–270 °C for 75 min results in CFs with tensile strengths of 3.14 or 2.85 GPa and tensile moduli of 205 or 252 GPa (Fig. 6a, b (j) and (k)). For aqPAM and mPAN, carbonisation at 1000–1400 °C yields a higher tensile strength than that at 1100–1500 °C. These results are consistent with previous reports regarding the relationship between the carbonisation temperature and mechanical properties of PAN-CFs32, which may be partly due to an increase in microvoid defects resulting from the elimination of noncarbon (N) elements32. The mechanical properties of aqPAM-CFs (Fig. 6a, b (d) and (e)) are comparable with those of PAN-CFs (Fig. 6a, b (j) and (k)) and significantly superior to those of CFs prepared using other previously reported low-cost CF precursor candidates (Supplementary Table 1). To evaluate the variability in the tensile strength of aqPAM-CFs and mPAN-CFs, we tested 50 single fibre specimens to determine the Weibull distribution33,34,35. The Weibull plots (Fig. 7a and b) of aqPAM-CF (Fig. 6a (d)) and mPAN-CF (Fig. 6a (j)) show that the Weibull moduli (m) of aqPAM-CF and mPAN-CF are 3.5 and 3.9, respectively. CFs are brittle materials that fracture under high tensile stress based on the weakest link theory, which states that fracture occurs at the weakest point of a CF15. Higher m-values indicate a more uniform distribution of defects in the CF, corresponding to a greater homogeneity with narrower strength distribution. The m-values of aqPAM-CF and mPAN-CF were smaller than that of commercial PAN-CF (Toray T300, m = 4.9) (Supplementary Fig. 29) because both aqPAM-CFs and mPAN-CFs were prepared using our laboratory-sized machines (and hence, not perfectly continuous processes), which may have promoted the formation of defects in each process, including winding and unwinding. However, the m-values of aqPAM-CF and mPAN-CF were still in the range of other commercial PAN-CFs (3.1–5.9)33,34,35, showing that the process is stable to some extent and affords a suitable homogeneity. No voids were observed in the SEM images of the tensile fractured surfaces of aqPAM-CFs (Fig. 4c, Supplementary Fig. 23). The CF structures of aqPAM-CFs were also evaluated by WAXS (Supplementary Fig. 30), and the interlayer spacing (d(002)), DPO, and crystallite size (L(002)) were calculated by fitting the (002) reflections. The d(002), DPO, and L(002) values of aqPAM-CFs were similar to those of mPAN-CFs (Supplementary Table 11), probably because aqPAM-SFs and mPAN-SFs featured similar structures (see solid-state NMR spectra, Supplementary Fig. 21). In addition, the in-plane thermal conductivities (TCs) of aqPAM-CFs obtained after carbonisation at 1000–1400 °C ( ~15.4 W m–1 K–1, Supplementary Table 16) were minimally lower than those of mPAN-CFs carbonised at 1000–1400 °C (17.8 W m–1 K–1) but higher than those of commercial PAN-CFs (Toray T300: 10.1 W m–1 K–1). Among these CFs, mPAN-CFs have the highest carbon content (97.0 wt%, Supplementary Table 14) and may therefore exhibit the highest TC. The volume resistivities of aqPAM-CFs (3.2–3.3 × 10–3 Ω cm) were comparable to those of mPAN-CFs (2.1 × 10–3 Ω cm) (Supplementary Table 17). Therefore, overall, the existence of P (PPA, elemental P, or P compounds without O atoms) on the aqPAM-CF surfaces did not adversely affect the CF mechanical properties, TCs, or volume resistivities. The effect of P existence on the affinity of aqPAM-CFs to polymer resins is currently being investigated.

a Tensile strengths, b tensile moduli, and c, elongation at break of single fibres extracted from aqPAM-CF and mPAN-CF bundles. aqPAM-CF and mPAN-CF bundles were produced by continuous stabilisation, continuous pre-carbonisation, and continuous carbonisation. The tensile strengths, tensile moduli, and elongation of aqPAM-CF (d) and mPAN-CF (j) are the averages obtained for 50 single fibres, and the other values refer to the averages obtained for 10 single fibres per sample. The values shown above the bars indicate the mean values. Error bars on the bar plots (a–c) represent standard deviation of the mean values.

a aqPAM-CF derived from aqPAM-SF obtained after stabilisation of a–17 at 330 °C for 12 min (Fig. 6a (d)). b mPAN-CF derived from mPAN-SF obtained after stabilisation of b–2 at 240–270 °C for 75 min (Fig. 6a (j)). P = probability of failure; m = Weibull modulus; σ = tensile strength (MPa); E = tensile modulus (GPa).
Discussion
We presented an innovative CF production process using an ecofriendly, and water-soluble precursor (aqPAM) and water as the only solvent to significantly reduce the associated CO2 emissions. The addition of a small amount of PA or its salts to aqPAM drastically reduced the stabilisation time (from 75 to 10 min) by up to ~87% compared with that of the major conventional PAN precursor. The carbon yield reached ~97% (Fig. 2d), and aqPAM-CFs exhibited a high tensile strength and tensile modulus that were comparable with those of PAN-CFs prepared under identical carbonisation conditions and considerably exceeded those obtained using other previously reported low-cost CF precursor candidates (Supplementary Table 1). This process can reduce the CO2 emissions of the entire CF production by at least ~40% (Fig. 3b) and is scalable, as it does not require the use of large stabilisation ovens to prevent the thermal runway of conventional PAN and achieve its prolonged heating. Large amounts of aqPAM fibres can be handled in stabilisation ovens considerably smaller than those used for PAN processing. We produced aqPAM-CFs (3200 filaments) with a length of ≥15 m (Supplementary Fig. 31), and producing longer aqPAM-CFs ( >1 km) is also possible. As shown in Supplementary Table 12 and 13, we estimated the electric energy consumptions of the stabilisation furnace at various temperatures and their contributions to the total energy consumption. The stabilisation-related energy consumption of aqPAM was calculated to be ~80% smaller than that of PAN (Supplementary Table 12) under the conditions (330 °C, 12 min) yielding the best mechanical properties. This reduction corresponded to a 16.8% decrease in the entire production cost (Supplementary Fig. 32). High-strength PAN-CFs typically have a tensile strength of ≥5 GPa and modulus of ≥300 GPa (e.g. Toray T1100G, Hexcel IM10) while featuring circular cross-sections with diameters of 4.4 − 5.0 μm. Herein, we reported aqPAM-CFs with dog bone–shaped cross-sections and diameters of ~6 μm. The mechanical properties of aqPAM can be further improved by decreasing the diameters, increasing the molecular weight, optimising the manufacturing process to reduce the number of defects36, and improving the crystallite structures (DPO and crystallite sizes) by further increasing the tension in production processes. In addition, aqPAM fibres with circular cross-sections can also be produced by dry spinning by changing spinning conditions, as shown in the case of dry spinning of other polymers15,37, which may lead to further improvement in mechanical properties. Meanwhile, the existence of small amounts of PPA, elemental P, or P compounds without O atoms in aqPAM-CFs (Supplementary Fig. 28b) might act as defects in the carbon structure, potentially reducing the mechanical properties. However, the mechanical properties of aqPAM-CFs derived from AM/AN/AA (60/35/5 mol%) with PA (a-17) and without PA (a-16) were similar (Fig. 6a–c (g) and (h)), although the carbonisation yields of SFs derived from a-16 were lower than those derived from a-17 (Supplementary Table 15). Therefore, in this case, the presence of PPA, elemental P, or P compounds without O atoms does not seem to act as defects reducing the mechanical properties. However, to further improve the mechanical properties, it may be beneficial not to include PA in the fibres. As shown in Supplementary Table 15, our optimised aqPAM, AM/AN/AA (60/35/5 mol%) without PA (a-16), showed high carbonisation yields (72.2% at 1000 °C and 71.3% at 1300 °C, respectively), derived from aqPAM-SFs obtained after a prolonged stabilisation period of 10 to 20 min at 350 °C. The diameters of aqPAM-SFs ( ~ 7.5 μm) (Supplementary Table 10) and aqPAM-CFs ( ~ 6 μm) are larger than those of commercial high-strength CFs with diameters of 4.4 − 5.0 μm. Therefore, decreasing the diameters of aqPAM-SFs without PA may lead to shortened stabilisation times or lower stabilisation temperatures, potentially improving mechanical properties while maintaining high carbonisation yields. In addition, the development of energy-saving carbonisation technologies is important for further reducing CO2 emissions and costs. In particular, the carbonisation temperature can be reduced to obtain PAN/graphene CFs38,39 and PBO-derived CFs40. The molecular dynamics simulations of PAN/graphene CFs suggest that the graphene edges along with the nitrogen and oxygen functional groups play catalytic roles and act as seeds to expedite the alignment of all-carbon rings, which are the starting sites for the growth of graphitic structures39. The incorporation of nanofillers, such as graphenes (e.g. water-soluble graphenes41,42) and boron nitride nanosheets43,44 as one of graphene analogues, into aqPAM is another promising strategy and may lead to energy-saving carbonisation and enhanced physical properties (e.g. mechanical properties and TCs). The use of blending methods45 based on aqPAM and other low-cost precursor candidates may also facilitate cost reduction. We are examining a novel energy-saving carbonisation technology to further reduce CO2 emissions and the cost of aqPAM-CFs by various methods. As a low-CO2-emission high-performance material, aqPAM-CFs can be extensively used for numerous next-generation applications.
Methods
Materials
AM (Kishida Chemical Co., Ltd., code: 020-01225, formula weight (FW): 71.08, purity: 98%), AN (Tokyo Chemical Industry Co., Ltd., code: A0146, FW: 53.06, purity: 99%, density: 0.81 g cm–3), AA (Tokyo Chemical Industry Co., Ltd., code: A0141, FW: 72.06, purity: 99%, density: 1.05 g cm–3), N,N,N’,N’-tetramethylethylenediamine (TMEDA) (Tokyo Chemical Industry Co., Ltd., code: T0147, FW: 116.24, purity: 98%, density: 0.78 g cm–3), ammonium persulphate (APS) (Kishida Chemical Co., Ltd., code: 000-04045, FW: 228.2, purity: 99%), PA (concentration: 85 wt% in aqueous solution, Fujifilm Wako Chemicals Corporation, code: 164-02176), PPA (Fujifilm Wako Chemicals Corporation, code: 162-03032, purity: ≥80%), DAP (Fujifilm Wako Chemicals Corporation, code: 016-03325, FW: 132.06, purity: ≥99%), and PAN (Merck KGaA, code: 181315, Mw: 150000) were purchased and used without further purification.
Synthesis of PAM and PAM containing PA (PAM-PA), and preparation of PAM-PA fibres and films
First, AM (640.0 g, 9.0 mol) and TMEDA (52.7 g, 0.45 mol) were dissolved in 18 L of ion-exchanged water and stirred under a N2 atmosphere. Then, APS (12.6 g, 0.055 mol) was added as a polymerisation initiator for the APS/TMEDA initiator system46. The subsequent radical polymerisation at 60 °C for 3 h yielded an aqueous solution of PAM. For the analysis of the polymer, the obtained aqueous solution was added dropwise to methanol to precipitate PAM to remove some compounds derived from TMEDA. PAM was collected and subjected to vacuum drying at 80 °C for 12 h, and PAM powder (638.8 g, polymerisation yield: 99.8%, density: 1.30 g cm–3; weight-average molecular weight (Mw): 96000; number-average molecular weight (Mn): 37000) was obtained. To prepare PAM containing PA, PA (concentration: 85 wt% in aqueous solution) was added to the aqueous solution of PAM such that the PA content in PAM-PA was 2.0 or 2.9 wt%. The PA-containing PAM aqueous solution was freeze-dried to obtain solid PAM-PA powder. Thereafter, PAM containing 2.0 or 2.9 wt% PA was obtained. The PAM-PA aqueous solution (PAM concentration: ~40 wt%) was used for dry spinning. Precursor multifilament fibres with 100 filaments were produced in a dry-spinning process using a 100-hole spinneret (diameter = 0.07 mm) and wound at 20 m min−1. Prior to winding, the PAM fibres (100 filaments) were coated with silicone oil (1–2 wt%). The aqueous solution that was obtained after polymerisation could also be used for dry spinning without purification because of the high polymerisation yield without any remaining monomers. To prepare the films, the PAM-PA aqueous solution (PAM concentration: 5 wt%) was spread on a plate, and water was naturally volatilised for at least 24 h. PAM-PA films with a thickness of ~40 μm were obtained after further drying at 80 °C for 12 h under vacuum to remove the residual water (film casting method).
Synthesis of AM/AN (75/25 mol%) copolymer and AM/AN containing PA (AM/AN-PA), and preparation of AM/AN-PA fibres and films
First, AM (1551.5 g, 21.8 mol), AN (383.6 g, 7.2 mol), and TMEDA (83.7 g, 0.72 mol) were dissolved in 7.58 L of water and stirred under a N2 atmosphere. A solution of APS (40.3 g, 0.18 mol) in water (100 mL) was then added dropwise to the solution at 50 °C (note that in the case of using large reaction vessels, the initial temperature is better to be set to up to ~50 °C to avoid transient temperature increases to >95 °C under any circumstances), followed by a polymerisation reaction at 79 °C for 3 h. The resultant aqueous solution was heated at 90 °C for 30 min to obtain an AM/AN aqueous solution. For the analysis of the AM/AN copolymer, the aqueous solution was added dropwise into methanol to precipitate the copolymer to remove some compounds derived from TMEDA, which was collected and vacuum-dried at 80 °C for 12 h to obtain a solid powder of AM/AN (75/25 mol%) copolymer (1925.8 g, polymerisation yield: 99.5%, density: 1.26 g cm–3, Mw: 62000, Mn: 27000). For dry spinning, PA (concentration: 85 wt% in aqueous solution) was added to the AM/AN aqueous solution such that the PA content in AM/AN-PA was 2.0 or 2.9 wt%. The solution was stirred and condensed, and an AM/AN-PA aqueous solution (AM/AN concentration: ~45 wt%, PA content in the AM/AN-PA: 2.0 or 2.9 wt%) was obtained. This aqueous solution was used for dry spinning to obtain AM/AN-PA fibres (100 filaments) with silicone oil (1–2 wt%) on the filament surface. The obtained aqueous solution after polymerisation could be directly used for dry spinning without purification because of the high polymerisation yield. Films with ~40 μm thickness were prepared via the casting method using the AM/AN-PA aqueous solution (AM/AN concentration: 5 wt%).
1H NMR (400 MHz, D2O) of AM/AN (Supplementary Fig. 33): δ 3.17–3.23 (m, 1H, CH-CN of AN units), 2.0–2.6 (m, 1H, CH-CONH2 of AM units), 1.3–2.0 (CH2 of the backbone).
13C NMR (100 MHz, D2O) of AM/AN (Supplementary Figs. 34 and 35): δ 177.3–179.5 (carbamoyl group of AM units), 120.8–121.8 (cyano group of AN units), 27–44 (CH and CH2 of the backbone).
IR of AM/AN (Supplementary Fig. 36): ν∼ = 2241 (CN), 1648 (C = O) cm–1.
Synthesis of AM/AN/AA (73/25/2 mol%) terpolymer and AM/AN/AA containing PA (AM/AN/AA-PA), and preparation of AM/AN/AA-PA fibres and films
First, AM (1682.0 g, 23.7 mol), AN (430.0 g, 8.1 mol), AA (46.8 g, 0.65 mol), and TMEDA (94.0 g, 0.81 mol) were dissolved in 11.6 L of water and stirred under a N2 atmosphere. Thereafter, a solution of APS (74.0 g, 0.32 mol) in water (360 mL) was added dropwise to the solution at 50 °C, which was followed by a polymerisation reaction at 79 °C for 3 h. The resulting aqueous solution was heated at 90 °C for 60 min to obtain an AM/AN/AA aqueous solution. The aqueous solution could be directly used for dry spinning, but especially for the analysis, the aqueous solution was added dropwise into methanol to precipitate the terpolymer. The terpolymer was collected and vacuum-dried at 80 °C for 12 h to obtain the solid AM/AN/AA (73/25/2 mol%) terpolymer powder (2152.0 g, polymerisation yield: 99.7%, Mw: 61000, Mn: 27000). PA was added to the AM/AN/AA aqueous solution such that the PA content in AM/AN/AA-PA was 2.0 or 2.9 wt% by mass. The solution was stirred and condensed to obtain an AM/AN/AA terpolymer/PA aqueous solution (polymer concentration: ~45 wt%). This solution was used for dry spinning to obtain AM/AN/AA fibres with silicone oil (1–2 wt%) on the filament surface. Films with ~40 μm thickness were prepared via the casting method using the AM/AN/AA-PA aqueous solution (AM/AN/AA concentration: 5 wt%).
13C NMR (100 MHz, D2O) of AM/AN/AA: δ 179.5–182.0 (carbonyl group of AA units), 177.3–179.5 (carbamoyl group of AM units), 120.8–121.6 (cyano group of AN units), 27–44 (CH and CH2 of the backbone).
IR of AM/AN/AA (Supplementary Fig. 36): ν∼ = 2241 (CN of AN), 1725 (C = O of AA), 1648 (C = O of AM) cm–1.
Synthesis of AM/AN/AA (60/35/5 mol%) terpolymer fibres containing PA (AM/AN/AA-PA) and preparation of AM/AN/AA-PA fibres and films
First, AM (1647.6 g, 23.2 mol), AN (717.5 g, 13.5 mol), AA (139.2 g, 1.9 mol), and TMEDA (112.2 g, 0.97 mol) were dissolved in 9.3 L of water and stirred under a N2 atmosphere. Thereafter, a solution of APS (88.2 g, 0.39 mol) in water (690 mL) was added dropwise to the solution at 50 °C, followed by a polymerisation reaction at 79 °C for 3 h. The resulting aqueous solution was heated at 90 °C for 60 min to obtain an AM/AN/AA aqueous solution. The aqueous solution could be directly used for dry spinning, but especially for the analysis, the aqueous solution was added dropwise into methanol to precipitate the terpolymer. The terpolymer was collected and vacuum-dried at 80 °C for 12 h to obtain an AM/AN/AA (60/35/5 mol%) terpolymer powder (2429.2 g, polymerisation yield: 97.0%, Mw: 66000, Mn: 24000). PA was added to the AM/AN/AA aqueous solution such that the PA content in the AM/AN/AA-PA was 2.0 or 2.9 wt%. The solution was stirred and condensed to obtain an AM/AN/AA terpolymer/PA aqueous solution (polymer concentration: ~45 wt%). This solution was used for dry spinning to obtain AM/AN/AA fibres with silicone oil (1–2 wt%) on the filament surface. Films with a thickness of ~40 μm were prepared via the casting method using the AM/AN/AA-PA aqueous solution (AM/AN/AA concentration: 5 wt%).
Calculations of relative energies of cyclic products using DFT
The DFT calculations were carried out using Gaussian software. The geometry optimisation and energy calculations (Supplementary Table 5) for the molecular models in their closed-shell singlet state were performed at the Becke 3 Parameter Lee-Yang-Parr (B3LYP)/6-31 G(d) level. AM dimer (a) (Supplementary Fig. 5) was used as a molecular model of PAM (PAM homopolymer).
Preparation of aqPAM-SFs from aqPAM fibres
An aqPAM fibre bundle tow (800 filaments, ~3200 dtex), which was spread to increase the tow width for efficient oxidative stabilisation and prevent fusion between fibres, was drawn at a draw ratio of 4 under an air atmosphere in a temperature range of 230–250 °C in an oven. The drawn aqPAM fibre tow (800 filaments, ~400 dtex) was continuously conveyed into a stabilisation oven at a tension (40 cN) and heated at a pre-determined temperature (for example, a temperature gradient of 330 to 350 °C for 12 min using three heating zones (330, 340, and 350 °C)) under a hot air stream. During this stabilisation, the fibres were drawn at a draw ratio of ~2. The SFs were continuously conveyed from the outlet of the stabilisation oven to obtain aqPAM-SFs.
Production of aqPAM-CFs
The obtained SF bundle (800 fibres/bundle) was combined to produce a precursor fibre bundle with 3200 fibres/bundle. This fibre bundle was continuously conveyed into a heat treatment furnace with a temperature gradient of 500 to 800 °C with applied tension of 0.2 cN dtex−1 under a nitrogen atmosphere; the fibres were heated for 3 min (pre-carbonising treatment) to obtain an aqPAM-PCF bundle (3200 fibres/bundle). The aqPAM-PCF bundle was continuously conveyed into a heat treatment furnace with a temperature gradient of 1000 to 1400 °C with applied tension of 0.5 cN dtex−1 under a nitrogen atmosphere for 3 min (carbonising treatment) to obtain a CF bundle (3200 fibres/bundle, density: ~1.76 g cm–3).
Preparation of films of PAN or PAN containing PA (PAN-PA)
PAN was dissolved in DMSO to obtain a PAN/DMSO solution (PAN concentration: 5%). PA (concentration: 85 wt% in aqueous solution) was added into the solution to obtain the PAN-PA/DMSO solution. The obtained PAN or PAN-PA solution was spread on a plate, and each solution was dried under vacuum to produce the corresponding film (thickness: ~40 μm). When mPAN was used instead of PAN, the film of mPAN was prepared using the same method.
Preparation of mPAN-SFs
Stabilisation was performed using 3000-filament mPAN fibres (fineness of single fibre: ~1.2 dtex, average diameter: 11.0 μm). The mPAN fibres were prepared via the radical polymerisation of AN and IA in DMSO12,24 and wet spinning of mPAN/DMSO solutions (mPAN concentration: ~20%)24. mPAN contains AN and IA (2 mol%) units with 1–2 wt% silicone oil on the filament surface. The IA content was confirmed via FT-IR measurements12,24. The Mw and Mn of mPAN were 134,000 and 52,000, respectively (eluent for gel permeation chromatography (GPC) measurement: 1,1,1,3,3,3-hexafluoro-2-propanol). The 3000 filament PAN fibre was continuously conveyed into a stabilisation oven with a temperature gradient of 240 to 270 °C (continuous-type stabilisation condition: gradual heating from 240 to 270 °C for 75 min using five heating zones (240, 248, 255, 263, 270 °C)) under an air stream. Rapid ramping (from 240 to 270 °C in <30 min) and temperatures above 300 °C in the stabilisation oven led to abrupt and intense exothermic reactions, which caused the fibres to overheat, fuse, and break. The mPAN-SFs were continuously conveyed from the outlet of the stabilisation oven.
Production of mPAN-CFs
The mPAN-SFs (3000 filaments) were continuously conveyed into a heat treatment furnace with a temperature gradient of 500 to 800 °C under a nitrogen atmosphere; the fibres were heat-treated for 3 min (pre-carbonising treatment) to obtain a mPAN-PCF bundle. The mPAN-PCF bundle was subsequently conveyed into a heat treatment furnace with a temperature gradient of 1000 to 1400 °C under a nitrogen atmosphere for 3 min (carbonising treatment). Thus, mPAN-CF bundles (3000 fibres/bundle, average diameter: 7.0 μm) were obtained.
Measurements
NMR spectroscopy
1H NMR and 13C NMR measurements were carried out with a JEOL JNM-ECX400P spectrometer at room temperature (rt) (NMR solvent: D2O for solubilising aqPAMs, DMSO-d6 for solubilising mPAN).
FT-IR spectroscopy
FT-IR spectra were obtained using a Nicolet 8700 FT-IR spectrometer (Thermo Fisher Scientific Inc.) via the attenuated total reflection method.
GPC
The Mw and Mn of the obtained aqPAM polymers were measured via GPC (HLC-8220 GPC, Tosoh Corporation) under the following conditions.
Column: TSKgel GMPWXL × 2 + TSKgel G2500PWXL × 1
Eluent: 100 mM sodium nitrate aqueous solution/acetonitrile = 80/20 v/v
Eluent flow rate: 1.0 mL min−1
Column temperature: 40 °C
Molecular weight standard: Polyethylene oxide standard/polyethylene glycol standard
Detector: Differential refractometer
DSC and SC-DSC
DSC analyses (DSC250, TA Instruments Inc.) were performed using Al sample pans. Polymer samples (films, ~5 mg) pre-dried at 100 °C in vacuum were heated from 25 to 200 °C at 10 °C min–1 and cooled at 30 °C min–1 in a flow of N2 (50 mL min–1). Tg values were acquired in the first and second runs. SC-DSC analyses were carried out on a DSC6200R instrument (Hitachi High-Tech Science Corporation) using SUS sealed cell pans in the temperature range of 20–500 °C at a rate of 10 °C min–1 under air or N2.
TG-MS
An alumina pan was filled with the aqPAM ( ~ 2 mg) or PAN ( ~ 2 mg) film and placed on the sample stage of a TG-MS simultaneous measurement apparatus (TG-DTA2020SA/MS9610, Bruker AXS). The aqPAM or PAN film was heated from 30 to 120 °C at a rate of 10 °C min–1 under a flow of a mixture of oxygen (20 vol%) and helium (80 vol%) (gas flow rate: 200 mL min–1) and dried at 120 °C for 1 h in the TG-MS apparatus to remove the remaining water. After cooling on the sample stage, the aqPAM or PAN film was heated from 30 to 600 °C at a rate of 10 °C min–1 under the flow of a mixture of oxygen (20 vol%) and helium (80 vol%) (gas flow rate: 200 mL min–1). Oxygen/helium gas was used instead of air because N2 shows m/z = 28, which is the same as that of CO. An empty alumina pan was placed on the reference sample stage. In conjunction with TG analysis, the MS analysis of the generated gas was conducted in a scanning range of m/z = 2–60 using a quadrupole mass spectrometer. Based on the results of the TG analysis, the ratio of the change in the mass of the aqPAM (wt%) at various temperatures during the temperature increase was calculated with reference to the mass at 100 °C using the following formula.
Here, MT denotes the mass of the AM/AN copolymer at the measurement temperature T (°C) and M100 denotes the mass of the AM/AN copolymer at 100 °C.
Moreover, based on the MS results, the gas generated at m/z = 18 was considered to be water vapour (mass number of H2O), and the intensity at m/z = 17 (mass number of NH3, and H2O corresponding to ~22% of H2O (100%) observed at m/z = 18, measured via mass spectrometry (electron ionisation)23) corresponded to the concentration of NH3 gas.
WAXS
WAXS measurements (Nano Viewer, Rigaku Corporation) were conducted at 40 kV and 30 mA using Cu Kα radiation (λ = 0.15406 nm). All patterns were background-corrected. The reflections were fitted using a bi-Gaussian function. The d-spacing (d(002)) was obtained using the following formula.
where n is the order of diffraction, d is the distance between the (002) planes, and θ is the angle of incidence relative to the planes in question.
The degree of preferred orientation of the crystallites along the fibre axis (DPO) was calculated using the peak width at half-height (w1, w2) according to the following formula.
The crystallite size (L(002)) was evaluated using the Scherrer equation47.
In this formula, K describes the Scherrer factor (K = 0.9), λ equals 0.154 nm, B is the full width at half-maximum of the peak, and θb is the Bragg angle.
Elemental analysis
Elemental analysis of the obtained fibres was conducted to determine the proportion of each element.
Carbon analysis
An elemental analyser (NCH-22F, Sumika Chemical Analysis Service, Ltd.) was used to heat each fibre (SF, PCF, or CF) under an oxygen stream to convert carbon into CO2. The produced CO2 was quantified using a gas chromatograph equipped with a TC detector, and the carbon content in each fibre was calculated.
Hydrogen analysis
An elemental analyser (NCH-22F) was used to heat each fibre (SF, PCF, or CF) under an oxygen stream to convert hydrogen into H2O. The produced H2O was quantified using a gas chromatograph equipped with a TC detector, and the hydrogen content in each fibre was calculated.
Nitrogen analysis
An elemental analyser (NCH-22F) was used to heat each fibre (SF, PCF, or CF) under an oxygen stream to convert nitrogen into N2. The produced N2 was quantified using a gas chromatograph equipped with a TC detector and the nitrogen content in each fibre was calculated.
Oxygen analysis
An elemental analyser (EMGA-920, Horiba, Ltd.) was used to heat each fibre (SF, PCF, or CF) in a graphite crucible under a helium stream to convert oxygen into CO. The produced CO was quantified using a non-dispersive infrared detector and the oxygen content in each fibre was calculated.
Solid-state NMR
Solid-state 13C cross polarisation/total sideband suppression NMR spectra were acquired using a Bruker Avance 400 widebore spectrometer (DSX model) at 100 MHz for 13C with 4 mm sample rotors (5 s pulse repetition, 15000 accumulations). Each sample was packed in a 4 mm–diameter sample rotor and spun at 4 kHz. Solid-state 13C dipolar decoupling magic-angle spinning spectra, wherein the intensities of the resonances are enhanced compared with those in the 13C cross polarisation magic angle spinning spectrum, were measured using the same spectrometer (10 s pulse repetition, 43000 accumulations).
Raman spectroscopy
The Raman spectra of the CF were measured at rt with excitation at 532 nm using a laser Raman spectroscopic analyser (NRS-3300, JASCO Corporation). The peak near 1590 cm−1 indicates the G-band originating from a graphite structure, and the peak near 1350 cm−1 indicates the D-band originating from a defective structure. The intensity ratio between the G- and D-bands (ID/IG) was calculated. Raman ID/IG mappings of cross-sections of fibres were obtained using a Raman microprobe system (inVia Reflex/StreamLine microRaman spectrometer, Renishaw) using a CCD detector.
SEM and SEM–EDS
SEM images of the sidewalls of aqPAM-CFs and the SEM-EDS images of sidewalls of each fibre were obtained using a Phenom ProX instrument. Each fibre was observed after carbon deposition onto the surface. SEM images of aqPAM-CF cross-sections were acquired using a JEOL JSM-IT700 instrument. The cross-sections were obtained via sample fracture. Deposition was not performed for the SEM observation of the cross-sections of aqPAM-CFs because of their sufficient conductivity. SEM-EDS measurements were performed on samples surface-coated with carbon using a Hitachi S-4800 instrument equipped with a Bruker XFlash 5060 EDS detector.
Tensile strength, tensile modulus, and elongation at break
Single fibres were extracted from the obtained CF bundle, and the diameter of each single fibre was measured using an optical microscope (VHX-7000, Keyence Corporation). A micro-strain tester (Micro Autograph MST-I, Shimadzu Corporation) was used for the tensile test (gauge length: 25 mm, and tensile speed: 1 mm/min) at rt in accordance with JIS R7606. The tensile strength, tensile modulus, and elongation at break of the CFs were measured, and the averages of 10 single fibres were calculated. Error bars represent the corresponding standard deviations. For obtaining the Weibull plots of aqPAM-CF, mPAN-CF, and Toray T300 12 K, 50 single fibres were extracted from the bundles for the tensile test. The Weibull modulus (m) was determined by plotting Eq. (5):
where P is the probability of failure, σ is the tensile strength and σ0 is the scale parameter. The Weibull modulus (m) is obtained by plotting ln(ln(1/(1-P))) as a function of lnσ. The Weibull parameter (m) is then determined by linear regression.
XPS
XPS spectra were recorded using a Thermo Fisher Scientific K-Alpha+ spectrometer with a monochromated Al Kα radiation source (1486.6 eV). Experimental energy shifts were corrected relative to the C1s band at 284.6 eV (C–C bond). XPS depth profiling was performed in combination with Ar-ion sputtering.
TC
TC (λ, W m–1 K–1) was calculated from thermal diffusivity (α, m2 s–1) as
where ρ and c are the CF density (kg m–3) and specific heat capacity (J kg–1 K–1).
The in-plane thermal diffusivity of each CF bundle (25 mm × 5 mm; ~0.1 mm thickness) was measured at 27 °C using a thermo-wave analyser (TA3, Bethel Co., Ltd.) under periodic laser heating. The values were reported as the averages of five measurements.
Volume resistivity
The volume resistivity of each single fibre extracted from CF bundles was measured at rt (23 °C) using a digital multimeter (Advantest R6441A digital multimeter) based on JIS R 7609. Copper foil tape was used as the electrode material, and measurements were performed at an electrode distance of 25 mm. Volume resistivity was calculated as
where D is the diameter (μm), Rf is the resistivity (Ω), and Lf is the length of the single fibre (electrode distance, mm). The averages of three measurements were reported.
Data availability
Source data, including coordinates of the optimised structures are available on Figshare (https://doi.org/10.6084/m9.figshare.28390712). All data are available from the corresponding author upon request.
Change history
30 June 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-61365-1
References
Peijs, T., Kirschbaum, R. & Lemstra, P. J. Chapter 5: A critical review of carbon fiber and related products from an industrial perspective. Adv. Ind. Eng. Polym. Res. 5, 90–106 (2022).
Choi, D., Kil, H.-S. & Lee, S. Fabrication of low-cost carbon fibers using economical precursors and advanced processing technologies. Carbon 142, 610–649 (2019).
Brown, K. R. et al. Carbon fibers derived from commodity polymers: A review. Carbon 196, 422–439 (2022).
Mainka, H. et al. Lignin–an alternative precursor for sustainable and cost-effective automotive carbon fiber. J. Mater. Res. Technol. 4, 283–296 (2015).
Morishita, T. & Matsushita, M. Ultra-highly electrically insulating carbon materials and their use for thermally conductive and electrically insulating polymer composites. Carbon 184, 786–798 (2021).
Khayyam, H. et al. Improving energy efficiency of carbon fiber manufacturing through waste heat recovery: A circular economy approach with machine learning. Energy 225, 120113 (2021).
Groetsch, T., Maghe, M., Creighton, C. & Varley, R. J. Environmental, property and cost impact analysis of carbon fibre at increasing rates of production. J. Cleaner Prod. 382, 135292 (2023).
Overview of LCI Data for Carbon Fiber, p. 26 (The Japan Carbon Fiber Manufacturers Association, 2022), https://www.carbonfiber.gr.jp/english/tech/pdf/lci2022_en.pdf.
Guo, Y. et al. Stabilization temperature prediction in carbon fiber production using empirical mode decomposition and long short-term memory network. J. Cleaner Prod. 429, 139345 (2023).
Milford, R. L., Allwood, J. M. & Cullen, J. M. Assessing the potential of yield improvements, through process scrap reduction. Resour. Conserv. Recycl. 55, 1185–1195 (2011).
Seidel, K., Thirunavukkarasu, D., Tjøtta, S. & Vieregge, K. Sustainability assessment of low CO2-emission aluminum materials. ATZ Worldw 122, 62–67 (2020).
Bajaj, P., Sen, K. & Hajir Bahrami, S. Solution polymerization of acrylonitrile with vinyl acids in dimethylformamide. J. Appl. Polym. Sci. 59, 1493–1657 (1996).
Khayyam, H. et al. PAN precursor fabrication, applications and thermal stabilization process in carbon fiber production: experimental and mathematical modelling. Prog. Mater. Sci. 107, 100575 (2020).
Groetsch, T. et al. A modular LCA/LCC-modelling concept for evaluating material and process innovations in carbon fibre manufacturing. Procedia CIRP 98, 529–534 (2021).
Kreis, P. et al. Lignin/poly(vinylpyrrolidone) multifilament fibers dry-spun from water as carbon fiber precursors. Macromol. Mater. Eng. 309, 2300296 (2024).
Brown, K. R., Love-Baker, C., Harrell, T. M. & Li, X. Effect of tension during sulfonation stabilization for UHMWPE-derived carbon fibers. J. Polym. Res. 30, 445 (2023).
Love-Baker, C. et al. Unveiling the microstructural evolution of carbon fibers derived from polyamide-6. J. Polym. Res. 30, 72 (2023).
Dalton, S., Heatley, F. & Budd, P. M. Thermal stabilization of polyacrylonitrile fibres. Polymer 40, 5531–5543 (1999).
Van Dyke, J. D. & Kasperski, K. L. Thermogravimetric study of polyacrylamide with evolved gas analysis. J. Polym. Sci. Part A: Polym. Chem. 31, 1807–1823 (1993).
Imura, Y., Hogan, R. M. C. & Jaffe, M. 10 – Dry spinning of synthetic polymer fibers. In: D. Zhang (Ed.), Advances in Filament Yarn Spinning of Textiles and Polymers, Woodhead Publishing, 187–202 (2014).
Leung, W. M. & Axelson, D. E. Thermal degradation of polyacrylamide and poly(acrylamide-co-acrylate). J. Polym. Sci. Part A: Polym. Chem. 25, 1825–1846 (1987).
Norcross, N. R. et al. Total synthesis of (±)-α-isosparteine, (±)-β-isosparteine, and (±)-sparteine from a common tetraoxobispidine intermediate. J. Org. Chem. 73, 7939–7951 (2008).
Data from NIST Standard Reference Database 69: NIST Chemistry WebBook (accessed 31 August 2024); https://webbook.nist.gov/cgi/cbook.cgi?ID=C7732185&Units=SI&Mask=200#Mass-Spec.
Wangxi, Z., Jie, L. & Gang, W. Evolution of structure and properties of PAN precursors during their conversion to carbon fibers. Carbon 41, 2805–2812 (2003).
Ouyang, Q., Cheng, L., Wang, H. & Li, K. Mechanism and kinetics of the stabilization reactions of itaconic acid-modified polyacrylonitrile. Polym. Degrad. Stab. 93, 1415–1421 (2008).
Terra, B. M., de Andrade, D. A. & de Mesquita, R. N. Characterization of polyacrylonitrile thermal stabilization process for carbon fiber production using intelligent algorithms. Polym. Test. 100, 107238 (2021).
Liu, X. et al. Stabilization of atactic-polyacrylonitrile under nitrogen and air as studied by solid-state NMR. Macromolecules 48, 5300–5309 (2015).
Tan, L., Chen, H., Pan, D. & Pan, N. Investigating the spinnability in the dry-jet wet spinning of PAN precursor fiber. J. Appl. Polym. Sci. 110, 1997–2000 (2008).
Wang, Y., Xu, L., Wang, M., Pang, W. & Ge, X. Structural identification of polyacrylonitrile during thermal treatment by selective 13C labeling and solid-state 13C NMR spectroscopy. Macromolecules 47, 3901–3908 (2014).
Handbook of X-ray Photoelectron Spectroscopy, ULVAC-PHI, Inc. https://www.ulvac-phi.com/en/products/database-books/handbook-of-x-ray-photoelectron-spectroscopy/.
Shimizu, M., Tsushima, Y. & Arai, S. Electrochemical Na-insertion/extraction property of Ni-coated black phosphorus prepared by an electroless deposition method. ACS Omega 2, 4306–4315 (2015).
Qin, X., Lu, Y., Xiao, H., Wen, Y. & Yu, T. A comparison of the effect of graphitization on microstructures and properties of polyacrylonitrile and mesophase pitch-based carbon fibers. Carbon 30, 4459–4469 (2012).
Pardini, L. C. & Manhani, L. G. B. Influence of the testing gage length on the strength, young’s modulus and weibull modulus of carbon fibres and glass fibres. Mater. Res. 5, 411–420 (2002).
Naito, K., Tanaka, Y., Yang, J.-M. & Kagawa, Y. Tensile properties of ultrahigh strength PAN-based, ultrahigh modulus pitch-based and high ductility pitch-based carbon fibers. Carbon 46, 189–195 (2008).
Yang, F. et al. Effect of amorphous carbon on the tensile behavior of polyacrylonitrile (PAN)-based carbon fibers. J. Mater. Sci. 54, 8800–8813 (2019).
Jang, D., Lee, M. E., Choi, J., Cho, S. Y. & Lee, S. Strategies for the production of PAN-based carbon fibers with high tensile strength. Carbon 186, 644–677 (2022).
Andreas, N. Fabrication of large diameter SiC monofilaments by polymer route. J. Eur. Ceram. Soc. 34, 1487–1492 (2014).
Gao, Z. et al. Graphene reinforced carbon fibers. Sci. Adv 6, eaaz4191 (2020).
Rajabpour, S. et al. Low-temperature carbonization of polyacrylonitrile/graphene carbon fibers: A combined ReaxFF molecular dynamics and experimental study. Carbon 174, 345–356 (2021).
Zhang, L. et al. Converting PBO fibers into carbon fibers by ultrafast carbonization. Carbon 159, 432–442 (2020).
Leese, H. S. et al. Reductively PEGylated carbon nanomaterials and their use to nucleate 3D protein crystals: a comparison of dimensionality. Chem. Sci. 7, 2916–2923 (2016).
Zhiyuan Xiong, Z. et al. Ultrahigh concentration exfoliation and aqueous dispersion of few-layer graphene by excluded volume effect. Nat. Commun 15, 10807 (2024).
Morishita, T. & Okamoto, H. Facile exfoliation and noncovalent superacid functionalization of boron nitride nanosheets and their use for highly thermally conductive and electrically insulating polymer nanocomposites. ACS Appl. Mater. Interfaces 8, 27064–27073 (2016).
Elo, T., Parihar, V. S., Nag, R., Bera, A. & Layek, R. Scalable synthesis of highly exfoliated, water-dispersible boron nitride nanosheets for nano-fibrillated cellulose membrane toughening. Chem. Eng. Sci. 276, 118820 (2023).
Scherschel, A. et al. Compatibility of mesophase pitch and linear low-density polyethylene for low-cost carbon fiber. J. Polym. Res. 30, 82 (2023).
Feng, X. D., Guo, X. Q. & Qiu, K. Y. Study of the initiation mechanism of the vinyl polymerization with the system persulfate/N,N,N’,N’-tetramethylethylenediamine. Makromol. Chem. 189, 77–83 (1988).
Scherrer, P. In Kolloidchemie Ein Lehrbuch (Ed.: R. Zsigmondy), Springer, Berlin, 387–409 (1912).
Acknowledgements
We thank Dr. Toshiaki Nakagawa (Toyota Central R&D Labs., Inc. (TCL)), Dr. Makoto Kato (TCL), Dr. Hideoki Fukushima (TCL), Dr. Yoshinori Inoue (TCL), Dr. Tatsuhiro Yamamoto (TCL), Dr. Hiroshi Hohjo (TCL), Dr. Masatoshi Sawamura (TCL), Dr. Noritoshi Iwata (TCL), Dr. Takuro Matsunaga (TCL), Dr. Takashi Iseki (TCL), Dr. Hisaaki Takao (TCL), and Mr. Nobuhiko Koga (TCL and Toyota Motor Corporation (TMC)) for helpful discussions. We thank Ms. Aya Tsuji (TCL), Dr. Yuka Yamada (TCL), Dr. Tomoyuki Koga (TCL), Ms. Megumi Sasaki (TCL), Ms. Haruka Ota, and Ms. Misa Mizuno for performing the related experiments and helpful discussions. We thank Mr. Masao Nakayama (TMC), Mr. Kotaro Harada (TMC), Mr. Hiroyuki Hibino (TMC), Mr. Naoto Kondo (TMC), Mr. Kaito Horikoshi (TMC), Mr. Daisuke Obara (TMC), Mr. Ippei Hamanaka (TMC), Mr. Koji Takao (TMC), and Mr. Nobuo Koyanagi (TMC) for related works and helpful discussions. We thank Mr. Yuki Aoki (TMC), Mr. Takanori Tatematsu (TMC), Mr. Satoshi Aoyama (TMC), Dr. Masayoshi Takami (TMC), Dr. Kazuhiro Kumeta (Nitivy Co., Ltd. (Nitivy)), Mr. Toshiyuki Hayakawa (Nitivy), and Mr. Shohei Sasahara (Nitivy) for helpful discussions.
Author information
Authors and Affiliations
Contributions
T.M. conceived the research concept; designed, synthesised, and evaluated the precursors; and performed the drawing, stabilisation, carbonisation, and evaluation of the SFs and CFs. M.N. synthesised and evaluated the precursors; performed fibre preparations for stabilisation and various measurements of the precursors, SFs, and CFs; and prepared the devices. M.Ma. performed fibre preparations for stabilisation, evaluated the precursor fibres, and performed fibre drawing and heating and SF carbonisation. K.H. conceived the research concept and designed, synthesised, and evaluated the precursors. K.N. and T.M. evaluated the precursor properties by performing measurements, including TG-MS, Raman analysis, and elemental analysis, and performed formal analysis, including DFT calculations. S.T. performed precursor stabilisation and solid-state NMR measurements. T.M., Y.Ki., and M.N. designed and synthesised modified precursors. Y.Ki., M.N., and T.M. performed large-scale precursor polymerisation. N.Sa. performed fibre preparation for stabilisation and the evaluation of precursor and SF properties. H.T. designed and synthesised AM dimer and trimer model molecules and performed experiments using them. T.M., Y.Ka., and M.Ma. designed and prepared the equipment for CF production. A.K. and H.K. evaluated the precursor and CF properties. T.M., M.N., M.Ma., K.N., A.K., H.K., and N.Sh. analysed the data. T.M. and A.O. conceived and started the project. T.M. and A.K. supervised the project. H.M., A.O., M.Mo., and N.Sh. administrated the project. T.M. wrote the paper with input from all authors. All authors discussed the results and contributed to the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Minoo Naebe, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Morishita, T., Narita, M., Matsushita, M. et al. Carbon fibre production using an ecofriendly water-soluble precursor. Nat Commun 16, 4614 (2025). https://doi.org/10.1038/s41467-025-59841-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59841-9
This article is cited by
-
Graphene–amorphous carbon with interwoven networks for enhanced strength
Nature Communications (2025)
