
Abstract
Clinical genetic testing identifies variants causal for hereditary cancer, information that is used for risk assessment and clinical management. Unfortunately, some variants identified are of uncertain clinical significance (VUS), complicating patient management. Case-control data is one evidence type used to classify VUS. As an initiative of the Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) Analytical Working Group we analyze germline sequencing data of BRCA1 and BRCA2 from 96,691 female breast cancer cases and 302,116 controls from three studies: the BRIDGES study of the Breast Cancer Association Consortium, the Cancer Risk Estimates Related to Susceptibility consortium, and the UK Biobank. We observe 11,207 BRCA1 and BRCA2 variants, with 6909 being coding, covering 23.4% of BRCA1 and BRCA2 VUS in ClinVar and 19.2% of ClinVar curated (likely) benign or pathogenic variants. Case-control likelihood ratio (ccLR) evidence is highly consistent with ClinVar assertions for (likely) benign or pathogenic variants; exhibiting 99.1% sensitivity and 95.3% specificity for BRCA1 and 93.3% sensitivity and 86.6% specificity for BRCA2. This approach provides case-control evidence for 787 unclassified variants; these include 579 with strong or moderate benign evidence and 10 with strong pathogenic evidence for which ccLR evidence is sufficient to alter clinical classification.
Similar content being viewed by others
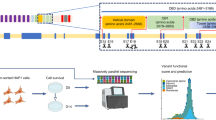
Introduction
Clinical genetic testing of disease-associated susceptibility genes is often complicated by the identification of variants of uncertain clinical significance (VUS)1. These individually rare variants may include missense changes, intronic and small in-frame insertions or deletions, as well as regulatory variants for which the association with disease is uncertain, complicating counselling and clinical management2. This is especially true for BRCA1 and BRCA2, where pathogenic variants (PVs) confer a high and clinically significant risk of breast3,4, ovarian5,6, pancreatic7 and (in the case of BRCA2) prostate cancer8,9; individuals with a VUS are generally not eligible to access well recognized clinical management options offered to individuals with a PV, such as risk-reducing prophylactic surgery to prevent cancer, magnetic resonance imaging (MRI) screening or PARP-inhibitor treatment for cancers that arise10,11,12,13.
To facilitate the classification of variants identified by genetic testing into (likely) benign or (likely) pathogenic, the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) groups14 developed guidelines that incorporate a set of criteria representing different evidence types. In this framework, independent lines of evidence in favor of or against pathogenicity for a variant are weighted according to strength level, where possible based on likelihood ratios (LRs), and following recommendations based on Bayesian modeling of the ACMG/AMP criteria15. Specific guidelines for BRCA1 and BRCA2, based on this framework, have been published by the ClinGen Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) BRCA1 and BRCA2 Variant Curation Expert Panel (VCEP)2. Classification of a variant as (likely) benign or (likely) pathogenic (or remaining as VUS) is determined by joint assessment of the evidence types, either according to the combination of criteria met14,15 or following a points-scoring system recently developed16. Based on previous research by the ENIGMA consortium17, one evidence type adapted for use under the ACMG/AMP framework is that from case-control data. In the ACMG/AMP framework this was included in the PS4 criterion, defined as “the prevalence of the variant in affected individuals is significantly increased over controls, with advice on application that the relative risk (RR) or odds ratio (OR) from case-control studies should be > 5.0, and the confidence interval (CI) around the estimate should not include 1.0”14. ClinGen specifications of the ACMG/AMP framework for BRCA1 and BRCA2 suggest that an OR ≥ 4.0 is assigned at full strength for a statistically significant association with CI not including 2.0[2]. As an initiative of the ENIGMA Analytical Working Group, we have recently proposed a LR-based framework for the analysis of case-control data for variant classification, where derived LRs are applicable under the ACMG/AMP framework for variant classification18. The case-control LR method computes a likelihood ratio, based on the likelihood of the observed data (i.e., the distribution of the variant of interest in cases and controls) under the hypothesis that the variant confers similar age-specific risks as the “average” BRCA1 or BRCA2 PV, relative to the likelihood under hypothesis that the variant is not associated with an increased risk of the disease. By taking into account the known age-specific penetrance of PVs, this approach provides significantly greater power for providing evidence for clinical classification, as compared to traditional “agnostic” case-control analyses. Hence, compared to ORs derived by logistic regression analysis, the LR-based framework has vastly improved performance to provide evidence towards pathogenicity, and more importantly, it can also be used to derive evidence against pathogenicity. For this reason, the BRCA1/2 VCEP specifications also state that ccLR estimates should be used in preference to case-control OR data for application of case-control information (https://cspec.genome.network/cspec/ui/svi/).
In this work, using the case-control LR (ccLR) method and logistic regression analysis, we perform a large-scale assessment of 4291 unique variants in BRCA1 (2248 within the coding sequence (CDS) ± 5 bp) and 6916 unique variants in BRCA2 (4661 CDS ± 5 bp) based on sequencing data from 96,691 female breast cancer cases and 302,116 unaffected female controls from three cohorts: the BRIDGES project of the Breast Cancer Association Consortium (BCAC)4, the Cancer Risk Estimates Related to Susceptibility (CARRIERS) consortium3 and the UK Biobank (UKB)19. In summary, the obtained likelihood ratios strongly align with ClinVar pathogenicity assertions, and we provide case-control evidence towards or against pathogenicity for 787 unclassified variants.
Results
Case-control LRs were aligned to evidence strength levels for or against pathogenicity based on the thresholds recommended under the ACMG/AMG framework15. To evaluate the effect of variant counts on the reliability of the results, we performed calibration analyses considering variants that were present in at least one, two or three individuals in each dataset separately (Supplementary Data 1-2) and in the combined dataset (Supplementary Data 3). Using ClinVar reference sets of (likely) benign and (likely) pathogenic variants, we determined the proportion of benign and pathogenic variants that fell into the different ACMG/AMP evidence strength categories based on variant-specific ccLR estimates15; this allowed us to confirm if variants reaching ccLR evidence at a given evidence strength were enriched in pathogenic variants at the expected rate for that evidence strength level e.g., for variants reaching ccLR pathogenic supporting evidence, we would expect at least 2.08-fold enrichment in the pathogenic reference set compared to the benign reference set.
To ensure low false discovery and false omission rates ( < 0.05) and reach strong evidence for or against pathogenicity when applying ccLR estimates (Supplementary Data 1–3), the analyses indicated that a more conservative approach was necessary. Thus, we restricted results to variants present in at least three individuals in the combined dataset (BRCA1 and BRCA2) and evidence from at least two datasets (only for BRCA2). By applying these criteria, the enrichment ratios for known pathogenic: benign variants within each ccLR evidence category were broadly consistent with the expectation for that category, with the exception of BRCA2 for ccLR benign moderate evidence (Table 1, Supplementary Data 3).
The combined dataset of 96,691 female breast cancer cases and 302,116 unaffected female controls from BRIDGES, CARRIERS, and the UKB contained case-control counts for 11,207 variants. Specifically, 4291 variants were found in BRCA1 and 6,916 in BRCA2 (Supplementary Data 4). The dataset encompasses 21.2% (6495/30,609) of the ClinVar BRCA1 and BRCA2 curated variants, 19.2% (2982/15,565) of the ClinVar variants curated as (likely) benign or pathogenic, and 23.4% (3513/15,044) of the ClinVar variants annotated as VUS. Results for all 11,207 variants are detailed in Supplementary Data 4.
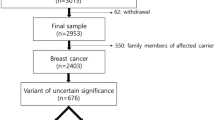
When focusing solely on exonic and proximal intronic sequences (CDS ± 5 bp), covered adequately by all three cohorts, the dataset represented 18.7% (5718/30,609) of the ClinVar database and 21.9% (3297/15,044) of the ClinVar VUS. Of the total CDS ± 5 bp variants, there are 2248 variants in BRCA1 and 4661 in BRCA2 (Supplementary Data 5). Further filtering for maximum credible allele frequency (AF) in the non-founder populations (gnomAD v4.1.0), denoted as filtering allele frequency (FAF) ≤ 0.001 (BRCA1 and BRCA2 VCEP “BA1” benign stand-alone cutoff), present in at least three individuals in the combined dataset, and with evidence from at least two datasets for BRCA2, led to a final set of 1710 variants (681 in BRCA1 and 1029 in BRCA2). These included: 760 variants listed in ClinVar as (likely) pathogenic or (likely) benign; and 950 variants considered to be of uncertain significance because they were not reported in ClinVar or they were listed in ClinVar as VUS or with conflicting classifications of pathogenicity (Fig. 1a). Sequence ontology variant consequence was annotated as follows: 204 presumed loss-of-function (LOF) variants of which 126 were frameshift insertions or deletions, 23 canonical splice site ( ±2 bp), 55 nonsense variants, as well as 31 in-frame insertions or deletions, 367 synonymous, 1082 missense and 26 intronic ( ±3 to ±5 bp) variants (Fig. 1b). Case-control LRs provided evidence in favor of pathogenicity for 264 variants and against pathogenicity for 1185 variants; of these 1449 variants, 787 were considered to be of uncertain significance (Fig. 1c). Case-control results for the final set of 1710 BRCA1 and BRCA2 variants, are summarized and displayed as genomic maps in Fig. 2and Fig. 3.

Donut plots showing the distribution of the a clinical classification (“ClinVar Class”) and b sequence ontology variant consequence (“Consequence”) for the 1717 CDS ± 5 bp variants with filtering allele frequency (FAF) > 0.001 (i.e., variants not meeting the BRCA1 and BRCA2 VCEP “BA1” benign stand-alone criterion), present in at least three individuals in the combined dataset, and with evidence from at least two datasets for BRCA2. The clinical classification status (“ClinVar Class”) of variants was retrieved from the ClinVar database (last accessed on January 7, 2024). c Sankey plot depicting “suggested case-control likelihood ratio (ccLR) ACMG/AMP evidence” (excluding variants with a ccLR of “No evidence”) provided for unclassified variants (i.e., variants not reported in ClinVar or listed in ClinVar as VUS, variants of conflicting classifications of pathogenicity or variants with classification “not provided”), per sequence ontology variant consequence (“Consequence”).

Overlay of the case-control likelihood ratios (LRs) and the logistic regression odds ratio (OR) estimates is represented within each exon (middle panel). Exons are sequentially numbered from 1 to 23 and annotated from right to left to match the MANE Select transcripts. Although BRCA1 was initially described with 24 exons (GenBank Accession ID U14680.1), exon 4 is missing following further assessment of the gene. We implement the most updated version of exon numbering (excluding legacy exon numbering). Case-control LRs (top panel) are represented on a continuous log2-transformed y axis with axis breaks. For the case-control LR analysis, the red color gradient represents LR reaching suggested ACMG/AMP evidence in favor of pathogenicity with strength levels ranging from very strong (dark red) to supporting (yellow). The green color gradient represents LR reaching ACMG/AMP evidence against pathogenicity with strength levels ranging from very strong (dark green) to supporting (light green). Variants with LR of “No evidence” are not plotted. For the logistic regression analysis (bottom panel), orange color represents OR estimates reaching the strong PS4 criterion (OR ≥ 4.0, P value < 0.05, and confidence interval (CI) not including 2.0). Variants with OR estimates not reaching the PS4 criterion are not plotted. Associations were adjusted for age and study country (BCAC dataset), age and ethnic group (CARRIERS dataset), and age and genetic ancestry (UKB dataset). Only variants present in both cases and controls were analyzed. ORs with 95% CIs were estimated for each dataset and combined using a fixed-effects, inverse-variance meta-analysis in the ‘metafor’ R package to derive an overall test of association. A two-sided likelihood ratio test (LRT) was used to calculate P-values. No adjustments were made for multiple comparisons. For visualization purposes, the y-axis for logistic regression is represented in reverse order. LCI, lower confidence interval. Sequence ontology variant consequence (“Consequence”) is represented with different symbols.

Overlay of the case-control likelihood ratios (LRs) and the logistic regression odds ratio (OR) estimates is represented within each exon (middle panel). Exons are sequentially numbered from 1 to 27 and annotated from left to right to match the MANE Select transcripts. Case-control LRs (top panel) are represented on a continuous log2-transformed y axis with axis breaks. For the case-control LR analysis, the red color gradient represents LR reaching suggested ACMG/AMP evidence in favor of pathogenicity with strength levels ranging from very strong (dark red) to supporting (yellow). The green color gradient represents LR reaching ACMG/AMP evidence against pathogenicity with strength levels ranging from very strong (dark green) to supporting (light green). Variants with an LR of “No evidence” are not plotted. For the logistic regression analysis (bottom panel), orange color represents OR estimates reaching the strong PS4 criterion (OR ≥ 4.0, P value < 0.05, and confidence interval (CI) not including 2.0). Variants with OR estimates not reaching the PS4 criterion are not plotted. Associations were adjusted for age and study country (BCAC dataset), age and ethnic group (CARRIERS dataset), and age and genetic ancestry (UKB dataset). Only variants present in both cases and controls were analyzed. ORs with 95% CIs were estimated for each dataset and combined using a fixed-effects, inverse-variance meta-analysis in the ‘metafor’ R package to derive an overall test of association. A two-sided likelihood ratio test (LRT) was used to calculate P-values. No adjustments were made for multiple comparisons. For visualization purposes, the y-axis for logistic regression is represented in reverse order. LCI, lower confidence interval. Sequence ontology variant consequence (“Consequence”) is represented with different symbols.
BRCA1 Likelihood Ratios
Informative case-control LRs were obtained for 604 out of 681 CDS ± 5 bp BRCA1 variants (88.7%) (Fig. 4). Among these, evidence in favor of pathogenicity was observed for 114 variants; of those, 27 were categorized as very strong, 31 as strong, 31 as moderate, and 25 as supporting. Conversely, evidence against pathogenicity was provided for 490 variants; 157 categorized as very strong, 128 as strong, 119 as moderate, and 86 as supporting. In comparison, using logistic regression analysis only 14 variants reached strong evidence in favor of pathogenicity according to the BRCA1/2 ACMG/AMP classification criterion description for OR application (PS4 criterion, OR ≥ 4.0, P value < 0.05 and CI not including 2.0)2, and all of these were also assigned pathogenic evidence using the ccLR method.

Sankey plots for a BRCA1 and b BRCA2. The variants assigned case-control likelihood ratio (LR) evidence in favor of or against pathogenicity (with suggested supporting, moderate, strong, or very strong evidence strength) are simplistically annotated as “Pathogenic”, “Benign”, “Suggested case-control LR (ccLR) ACMG/AMP Evidence”, respectively. Variants with LRs between 0.48 and 2.08 are defined as “No evidence” in the “Suggested ccLR ACMG/AMP Evidence” panel. The clinical classification status (“ClinVar Class”) of variants was retrieved from the ClinVar database (last accessed on January 7, 2024).
Variants having case-control LR evidence in favor of pathogenicity consisted of 64.9% (74/114) LOF (39 frameshift insertions or deletions, 10 canonical splice site and 25 nonsense variants). Missense and synonymous variants accounted for 33.3% (38/114) and 1.8% (2/114), respectively. In contrast, variants with evidence against pathogenicity included 0.8% (4/490) LOF (1 frameshift, and 3 canonical splice site variants), 1.8% (9/490) in-frame, 68.6% (336/490) missense, 2.7% (13/490) intronic and 26.1% (128/490) synonymous variants (Figs. 4 and 5a). Among the 14 variants reaching the PS4 criterion, 78.6% (n = 11) were LOF (8 frameshift and 3 nonsense variants) alongside 21.4% (n = 3) missense variants. For BRCA1 LOF variants, pathogenicity was provided for the majority of the frameshift insertions or deletions (97.5%, 39/40), canonical splice site (76.9%, 10/13) and nonsense (100%, 25/25) variants with informative case-control evidence.

Histograms showing the distribution of case-control likelihood ratios (LRs) categorized by a sequence ontology variant consequence for BRCA1, b ClinVar classification for BRCA1, c sequence ontology variant consequence for BRCA2, d ClinVar classification for BRCA2. Histograms categorized by ClinVar classification are divided into two panels; the top panel represents variants curated in ClinVar as (likely) benign or (likely) pathogenic, while the bottom panel represents unclassified variants (variants not reported in ClinVar or listed in ClinVar as variants of uncertain significance or with conflicting classifications of pathogenicity). For visualization purposes the x axis represents log10(LR) values. Dashed lines represent LRs between 0.48 and 2.08 considered as of “No evidence”.
The proportion of variants with informative case-control evidence towards pathogenicity was 10.2% (38/374) for missense, and 1.5% (2/130) for synonymous variants. All the BRCA1 in-frame (n = 9) and intronic (n = 13) variants with informative case-control evidence were assigned benign evidence (Fig. 4). The majority of the identified CDS ± 5 bp BRCA1 variants were within exon 10 (n = 419), followed by exons 15 (n = 30) and 14 (n = 21). One splice acceptor (c.594-2 A > C) and two splice donor (c.301+1 G > A and c.4096+1 G > A) variants within or proximal to exons 8, 5 and 10 were assigned benign evidence. Furthermore, missense variants with evidence in favor of pathogenicity were enriched in the exons encoding the RING domain (nucleotides 4-294; exons 2-5) (18.4%, 7/38) and the BRCA1-C-Terminal (BRCT) repeats (nucleotides 4987-5577, exons 16-23) (26.3%, 10/38) (Fig. 6). None of the in-frame variants assigned benign case-control evidence (n = 9) fell within the RING or BRCT domains.

Stacked bar plots of the suggested case-control likelihood ratio (LR) ACMG/AMP evidence per exon and sequence ontology variant consequence for a BRCA1 and b BRCA2. Exons are sequentially numbered to match the MANE Select transcripts. Although BRCA1 was initially described with 24 exons (GenBank Accession ID U14680.1), exon 4 is missing following further assessment of the gene; legacy exon numbering for BRCA1 is represented in brackets. Variants assigned case-control LR evidence in favor of or against pathogenicity (with suggested supporting, moderate, strong or very strong evidence strength) are simplistically annotated as “Pathogenic”, and “Benign” in the key, respectively. Variants with LRs between 0.48 and 2.08 are defined as “No evidence”.
Overall, 25% (29/114) of the variants with evidence in favor of pathogenicity and 53.9% (264/490) of the variants with assigned evidence against pathogenicity, were considered to be of uncertain significance prior to evaluation (not reported in ClinVar or listed in ClinVar as VUS, with conflicting classifications of pathogenicity, or with classification “not provided”). For the remaining variants with an established ClinVar clinical classification as (likely) benign or (likely) pathogenic, we observed 99.1% (224/226) consistency for variants assigned benign evidence and 95.3% (81/85) for variants assigned pathogenic evidence (Figs. 4 and 5b). For the 14 variants that reached the PS4 criterion using logistic regression analysis, one was of uncertain significance and the remainder had an established ClinVar clinical classification consistent with PS4 evidence in favor of pathogenicity.
BRCA2 Likelihood Ratios
Informative case-control LRs were obtained for 82.1% of the BRCA2 variants (845/1,029) (Fig. 4). Evidence in favor of pathogenicity was provided for 150 variants, with 24 being very strong, 36 strong, 46 moderate, and 44 supporting. Evidence against pathogenicity was provided for 695 variants, of which 180 reached very strong, 196 strong, 223 moderate, and 96 supporting. Using logistic regression analysis only 17 variants reached strong evidence in favor of pathogenicity following the BRCA1/2 ACMG/AMP PS4 classification criterion (OR ≥ 4.0, P value < 0.05 and CI not including 2.0); all of these were assigned pathogenic evidence using the ccLR method.
Variants assigned ccLR evidence in favor of pathogenicity comprised of 50.7% (76/150) presumed LOF (53 frameshift insertions or deletions, 18 nonsense and 5 canonical splice site variants), 38% (57/150) missense, 8.7% (13/150) synonymous, 1.3% (2/150) intronic and 1.3% (2/150) in-frame variants. In contrast, variants assigned ccLR evidence against pathogenicity comprised of 3% (21/695) presumed LOF (12 frameshift, 8 nonsense and 1 canonical splice site variants), 2.6% (18/695) in-frame, 69.6% (484/695) missense, 1.2% (8/695) intronic and 23.6% (164/695) synonymous variants. The 17 variants reaching the PS4 OR criterion were comprised of 14 presumed LOF (82.4%) (11 frameshift insertions or deletions, 2 nonsense and 1 canonical splice site variants) and 3 (17.6%) missense variants (Figs. 4 and 5c). For BRCA2 LOF variants, we observed that pathogenicity was proposed for the majority of the frameshift insertions or deletions (81.5%, 53/65), canonical splice site (83.3%, 5/6) and nonsense (69.2%, 18/26) variants assigned informative case-control evidence. The proposed pathogenicity rate for non-LOF variants (in-frame, missense, synonymous and intronic variants) with informative case-control evidence, was 10% (2/20) for in-frame, 10.5% (57/541) for missense, 7.3% (13/177) for synonymous and 20% (2/10) for intronic variants (Fig. 4). The majority of the evaluated BRCA2 variants were within exons 11 (n = 479) and 10 (n = 111) (Fig. 6). Two intronic variants (c.425+3 A > G, c.7435+5 T > C) were assigned evidence in favor of pathogenicity, falling within the splice donor sites of exons 4 and 14. Moreover, two in-frame variants within exons 11 and 20 were assigned evidence in favor of pathogenicity. One splice donor variant (c.8331+2 T > C) in the splice donor site of exon 18 was assigned evidence against pathogenicity. Finally, missense variants assigned evidence in favor of pathogenicity were enriched in the exons encoding the DNA binding domain (DBD) (amino acids 2481-3186; exons 15-26) (38.6%, 22/57) (Fig. 6).
Overall, 35.3% (53/150) of the variants with evidence in favor of pathogenicity and 63.5% (441/695) with assigned evidence against pathogenicity, were considered to be of uncertain significance prior to evaluation (not reported in ClinVar or listed in ClinVar as VUS, with conflicting classifications of pathogenicity, or with classification “not provided”). For the remaining variants with an established ClinVar clinical classification as (likely) benign or (likely) pathogenic, we observed 93.3% (237/254) consistency for variants assigned benign evidence and 86.6% (84/97) for variants assigned pathogenic evidence (Figs. 4 and 5b). For the 17 variants that met the PS4 OR criterion, one was of uncertain significance and the remainder had PS4 evidence consistent with an established ClinVar clinical classification.
Concordance with predicted or experimental impact on function
Concordance with evidence in favor or against pathogenicity using the ccLR method was higher for experimental evidence from higher-throughput functional assays compared to in silico prediction methods (Supplementary Data 6). We observed 72.4% sensitivity and 98.7% specificity for functionally evaluated variants located in 13 exons of BRCA1 encoding the RING and BRCT domains (exons 2-5 and 15-23, respectively)20 and 100% sensitivity and 93.3% specificity for functionally evaluated missense BRCA2 variants21. We also observed a strong concordance between the ccLR method and the recent functional evaluation of variants located in the region encoding the DBD of BRCA222,23,24, with maximum sensitivity reaching 66.7% and specificity at 92.1% across three assays. For in silico prediction methods evaluated, including BayesDel25, REVEL26, VEST427, MutPred228 and AlphaMissense29, we observed a moderate concordance with sensitivity ranging from 29.7% to 62.5%, and specificity ranging from 69.1% to 96.3% for BRCA1, and sensitivity ranging from 13.2% to 30.3% and specificity ranging from 88.2% to 95.1% for BRCA2 (Fig. 7 and Supplementary Data 6).

Concordance is shown separately for a BRCA1 and b BRCA2. The top panels for each gene represent case-control likelihood ratios (LRs) compared to variants predicted as benign (“BP4 criterion”, “predicted benign” or “functional”) or pathogenic (“PP3 criterion”, “predicted pathogenic” or “loss-of-function”) by in silico prediction methods (AlphaMissense, BayesDel, MutPred2, VEST4 and REVEL) or through high-throughput functional assays (Findlay et al., 2018, Huang et al., 2025, Sahu et al., 2025, Hu et al., 2024, Mesman et al., 2019). Yellow and green colors represent variants predicted as pathogenic or benign by functional predictors, respectively. For visualization purposes the x axis represents log10(LR) values. Box plots with individual data points display the median value, with whiskers extending to a maximum of 1.5 × interquartile range (IQR) beyond the box. The notch in the box approximates the 95% confidence interval (CI) for the median. Bottom panels for each gene represent sequence-pathogenicity heatmaps demonstrating the concordance between the case-control LR (ccLR) method and functional predictors. For the case-control LR (ccLR) evidence, red color gradient represents LR reaching suggested ACMG/AMP evidence in favor of pathogenicity with strength levels ranging from very strong (dark red) to supporting (yellow). The green color gradient represents LR reaching suggested ACMG/AMP evidence against pathogenicity with strength levels ranging from very strong (dark green) to supporting (light green). Variants with ccLR of “No evidence” are not plotted. For the functional predictors, yellow and green colors represent evidence in favor and against pathogenicity, respectively (expressed as “pathogenic supporting”). The total number of variants included in the concordance analyses is depicted in Supplementary Data 6.
Taken together, high-throughput assay results align with ccLR evidence, offering a robust evaluation framework. Concordance between the ccLR method and predicted or experimental impact is displayed as sequence-pathogenicity heatmaps (Fig. 7), which provide pathogenicity predictions for overlapping variants and functional evidence (binary categories) with ccLR.
Discussion
Significant efforts have been dedicated to the clinical classification of variants in BRCA1 and BRCA2, owing to their elevated risk association with multiple cancer types. To date, only 111 BRCA1 and BRCA2 variants have been assigned case-control LR evidence18,30, and only 20 of these were previously used in clinical classification30. In this work, we present a large-scale multicenter case-control analysis of 11,207 BRCA1 and BRCA2 rare variants, of which 6909 are coding (within CDS ± 5 bp). Using sequencing data from 96,691 female breast cancer cases and 302,116 unaffected controls of the BRIDGES, CARRIERS and the UK Biobank datasets we utilized our recently developed ccLR method18 and logistic regression analysis to leverage case-control data and provide evidence in favor or against variant pathogenicity. Our dataset comprehensively covers all exons, proximal intronic sequences and regulatory regions, accounting for 21.2% of the ClinVar curated variants and 23.4% of the ClinVar VUS, in these genes.
For coding variants, case-control evidence is highly consistent with ClinVar pathogenicity data, exhibiting 99.1% sensitivity and 95.3% specificity in distinguishing (likely) pathogenic and (likely) benign variants for BRCA1 and 93.3% and 86.6% for BRCA2. The somewhat lower sensitivity and specificity for BRCA2 is consistent with the lower penetrance of pathogenic variants in BRCA2 compared to BRCA14, so that the power of the analysis to classify variants, for a given number of observations, is lower for BRCA2. Notably, the majority of presumed LOF variants assigned evidence against pathogenicity are not assigned full or any pathogenic very strong (PVS1) code strength according to the recent ENIGMA classification criteria, since their predicted or known impact on splicing indicates that they are not associated with high risk of cancer2,31.
Using reference sets of (likely) pathogenic and (likely) benign variants drawn from ClinVar; using variants within the CDS ± 2 bp since this captures the regions more regularly tested in a clinical setting, we showed that the ccLR method maintains low false discovery and false omission rates ( <0.05) and can reach strong evidence both in favor and against pathogenicity, following the Bayesian adaptation of the ACMG/AMP framework15,16. Our results provide case-control LR evidence for 1449 variants with suggestive ACMG/AMP code strength levels; of these, 264 have evidence in favor of pathogenicity, and 1185 have evidence against pathogenicity.
This analysis provides evidence to inform clinical classification for 787 variants currently considered of uncertain clinical significance. Specifically, strong or moderate benign evidence (equivalent to −4 or −2 points) observed for 579 unclassified variants should be sufficient to alter clinical classification with at least one other supporting benign evidence type, following standard ACMG/AMP classification protocols for combining evidence types14 and requirement to reach −2 points for a likely benign classification2,15,16. Likewise, strong pathogenic evidence ( +4 points) reached for 10 unclassified variants will provide valuable information towards pathogenicity in combination with other pathogenic evidence types, where +6 points is sufficient to reach a likely pathogenic clinical classification2,14,15,16. This has important implications for clinical management. Individuals with a VUS are generally not eligible to access well-recognized clinical management options offered to individuals with a PV, such as risk-reducing prophylactic surgery to prevent cancer, MRI screening, or PARP-inhibitor treatment for cancers that arise10,11,12,13; thus, delaying effective interventions. Conversely, management of individuals with a VUS as if it were a PV that is actually benign can result in unnecessary anxiety and excessive medical intervention.
While our study provides valuable results that can be used in variant classification assuming the variant follows a high-risk penetrance as observed for “average” BRCA1 and BRCA2 PVs, however, it is important to acknowledge possible sources of bias such as potential sequencing artifacts, variant allele frequency differences between populations, case-control imbalances and possibility of ascertainment bias. We addressed this by applying stringent quality control measures and stratified analyses. It is also important to acknowledge the fact that when assessing numerous variants, particularly those with low counts, some may exhibit outcomes contrary to the expected, by chance. This issue is particularly relevant in the Supporting evidence category, where evidence is relatively weak. While the power to classify variants using case-control data alone can still be limiting, our likelihood-based approach lends itself naturally to combining with other evidence (e.g., co-segregation, tumor pathology, or in silico/functional scores). By integrating ccLR evidence with other lines of evidence, the likelihood of misclassification is minimized.
We further demonstrated that ccLR evidence aligns with functional predictors, offering a robust evaluation framework. These results can serve as a crucial resource for evaluating the consistency of other findings and refining existing methodologies.
Lastly, in order to accommodate the wider use of our case-control results, we provide the “ccLR browser” (https://biostatunitcing.shinyapps.io/ccLRbrowser/) that allows for easier exploration and visualization of our findings.
In summary, we present case-control evidence that strongly aligns with ClinVar pathogenicity assertions for non-VUS. We also provide case-control evidence towards or against pathogenicity for 787 unclassified variants. Our findings will be shared with the ClinGen ENIGMA BRCA1 and BRCA2 Variant Curation Expert Panel (VCEP), for expert panel curation activities that integrate multiple lines of evidence, including bioinformatic, clinical and experimental data, following with FDA-aligned variant classification processes. This can now be used in combination with other evidence for their final clinical classification, which is essential for accurate risk assessment and effective clinical decision-making, providing a larger number of patients and their relatives with clinically informative results.
Methods
This research complies with all relevant ethical regulations and received approval by the Cyprus National Bioethics Committee (EEBK EΠ 2020.01.224). All the studies were approved by the relevant ethics review boards and used appropriate consent procedures.
The BCAC/BRIDGES dataset
The BCAC dataset included 47,201 women with in-situ or invasive breast cancer and 47,316 unaffected controls, from 29 BCAC studies defined as population-based4. All studies were approved by the respective ethics review boards, adhering to appropriate consent procedures. Phenotype data were based on the BCAC database v14. Individuals from a minority ancestry for each study (i.e., non-Asian from the Asian studies and non-European ancestry from the other studies), based on genetic data or self-report, were excluded4. Women of unknown/uncertain ancestry/ethnicity were excluded4. All samples underwent panel sequencing for 34 known or suspected breast cancer susceptibility genes as part of the BRIDGES project4. Details on library preparation, sequencing, and bioinformatics analysis, including variant calling and quality control, are described elsewhere4. The final dataset used consisted of 46,306 women with breast cancer and 43,481 unaffected controls, diagnosed or interviewed at age 21 to 80 years (the age range for which penetrance estimates for BRCA1 and BRCA2 are available) (Fig. 8 and Supplementary Data 7).

Using sequencing data of 96,691 female breast cancer cases and 302,116 unaffected controls from the Breast Cancer Association Consortium (BCAC), the Cancer Risk Estimates Related to Susceptibility (CARRIERS) consortium and the UK Biobank (UKB) we calculated case-control likelihood ratios (LRs) and odds ratios (ORs) for 11,207 BRCA1 and BRCA2 variants, of which 6909 are coding (coding sequence, CDS ± 5 bp). Derived LRs and ORs were further aligned to ACMG/AMP evidence strengths to provide evidence in favor of or against pathogenicity following sensitivity analyses-derived variant exclusion criteria.
The CARRIERS dataset
The CARRIERS consortium dataset included 32,247 women with in-situ or invasive breast cancer and 32,544 unaffected controls from 12 population-based studies3. The CARRIERS study was approved by the institutional review board at the Mayo Clinic, and all participants provided informed consent for research. All samples were subjected to panel sequencing, targeting 37 cancer susceptibility genes. Details on library preparation, sequencing, and bioinformatics analysis, including variant calling and quality control, were previously documented3. Women of unknown/uncertain ethnicity were excluded. The final dataset consisted of 29,832 women with breast cancer and 30,927 unaffected controls diagnosed or interviewed, between the age range of 21 to 80 years (Fig. 8 and Supplementary Data 7).
The UK Biobank dataset
UKB is a prospective cohort of more than 500,000 participants recruited in 22 assessment centers in the United Kingdom between 2006 and 201032. Whole-exome sequencing (WES) data for 454,787 samples were released in October 2021 and were accessed via the UKB DNA Nexus platform19. Genetic ancestry was computed using a genetic principal components analysis from 2318 informative markers33. Women of unknown/uncertain ethnicity were excluded. Cases were defined as individuals with either invasive breast cancer (International Classification of Diseases (ICD)−10 code (C50) or carcinoma in situ (D05) based on linkage to the National Cancer Registration and Analysis Service (CRAS), or self-reported breast cancer incidence. Women diagnosed with ovarian cancer (International Classification of Diseases (ICD)−10 code C56) based on linkage to the National Cancer Registration and Analysis Service (NCRAS), or self-reported, were excluded from the controls. We accounted for both prevalent and incident cases, and only for cancers identified as an individual’s first or second diagnosed cancer. Under these criteria, a total of 20,553 female breast cancer cases and 227,708 female controls were included, diagnosed or interviewed between the age range of 21 to 80 years (Fig. 8 and Supplementary Data 7). Access to the use of the UK Biobank data was granted under application number 102655.
Data preparation
For all datasets, the Ensembl Variant Effect Predictor (VEP) v111 was used to annotate variants34. Annotations include the distance from the upstream or downstream gene, sequence ontology variant consequences, exon/intron number and Human Genome Variation Society (HGVS) nomenclature for the cDNA and protein level; the MANE Select transcript and protein were used (ENST00000357654.3 and ENSP00000350283.3 for BRCA1; ENST00000380152.3 and ENSP00000369497.3 for BRCA2)35. Exons and introns are sequentially numbered to match the MANE Select transcripts. Although BRCA1 was initially described with 24 exons (GenBank Accession ID U14680.1), exon 4 is missing following further assessment of the gene. Herein, we implement the most updated version of exon numbering (excluding legacy exon numbering). Allele frequency was retrieved from the gnomAD v4.1.0 release36. For filtering variants, we used the gnomAD maximum credible AF (the lower bound of the 95% CI) observed across the non-founder populations, including non-Finnish Europeans, African or African Americans, Admixed Americans, East Asians, South Asians, and Middle Easterners. Existing variant class was retrieved from the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/, last accessed on January 7, 2024).
Case-control analysis
The primary analysis involved the calculation of LRs for each individual variant using the ccLR method (https://github.com/BiostatUnitCING/ccLR)18,37. Under a survival analysis framework, the ccLR method compares the likelihood of the distribution of the variant of interest between cases and controls, under the hypothesis that the variant has similar age-specific relative risks as the “average” BRCA1 or BRCA2 PV, compared to the hypothesis that the variant is not associated with increased risk of the disease18. These risks are age-, sex-, and/or country-specific. Hence, the ccLR method requires specification of the age-specific risks in individuals with a PV and in the general population18. These were derived from the age-specific incidence rates for England and Wales (1998-2002) for ages 21-80 years, retrieved by the Cancer Incidence in Five Continents (CI5) Volume IX (https://ci5.iarc.fr/) and age-specific breast cancer odds ratio for individuals with BRCA1 and BRCA2 PVs estimated from the BRIDGES study4.
Case-control LRs were separately calculated for each dataset: BRIDGES, CARRIERS and UKB. To account for possible variant allele frequency differences by country or ethnicity, stratified LR calculations were performed within each dataset (BRIDGES stratified by country, and hence largely also by ethnicity; CARRIERS and UKB stratified by ethnicity) (Supplementary Data 8) and then multiplied across strata to provide a case-control LR for each independent dataset. Dataset-specific LRs were then used to obtain an overall LR for each variant. The LRs were aligned to evidence strength levels for or against pathogenicity based on the thresholds recommended under the ACMG/AMG framework15. Thus, in favor of pathogenicity was classified as very strong, LR ≥ 350; strong, 350 > LR ≥ 18.7; moderate, 18.7 > LR ≥ 4.33; or supporting, 4.33 > LR ≥ 2.08. Likelihood ratios against pathogenicity were classified as very strong, LR ≤ 0.0029; strong, 0.0029 < LR ≤ 0.053; moderate, 0.053 < LR ≤ 0.231; and supporting, 0.231 < LR ≤ 0.48. LRs between 0.48 and 2.08 were considered as “No evidence”.
Associations between variants and breast cancer risk were also assessed by logistic regression, adjusted for age and study country for the BCAC dataset, age and ethnic group for the CARRIERS dataset, and age and genetic ancestry for the UKB dataset. Logistic regression analysis was only performed for variants present in both cases and controls. Odds ratios and standard errors estimated from each dataset were combined in a fixed-effects, inverse-variance meta-analysis using the ‘metafor’ R package to derive an overall test of association. Using the BRCA1/2 VCEP specification based on the PS4 ACMG/AMP classification criterion2, strong evidence in favor of pathogenicity was assigned to variants with OR ≥ 4.0, P value < 0.05, and CI not including 2.0.
Reference sets
To assess the calibration of the ccLR method and perform sensitivity analyses, we selected reference sets of (likely) pathogenic and (likely) benign variants from the list of BRCA1 and BRCA2 variants identified in any of the three datasets. These reference sets included variants located within the CDS or at splice acceptor/donor site dinucleotide positions ( ± 2 bp); since this captures the regions more regularly tested in a clinical setting, classified as such by the ClinGen BRCA1/2 historical expert panel or the ClinGen BRCA1/2 VCEP following ACMG/AMP guidelines2, or by multiple submitters without conflicts in the ClinVar database (last accessed on January 7, 2024). Variants were filtered to exclude any with gnomAD (v4.1.0) maximum credible AF in non-founder populations, denoted as FAF > 0.001, consistent with the BRCA1 and BRCA2 VCEP’s “BA1” benign stand-alone cutoff. Reference sets included 734 BRCA1 variants (448, 369, and 379 with case-control LRs from BRIDGES, CARRIERS, and UKB, respectively) and 1240 BRCA2 variants (762, 554, and 634 with case-control LRs from BRIDGES, CARRIERS, and UKB, respectively).
Calibration of the ccLR method
To evaluate the calibration of the method, we used the case-control LRs for each of the variants in the reference sets and then determined the proportion of benign and pathogenic variants that would be classified in each ACMG/AMP evidence strength category15. We calculated enrichment for variants in each category j to be pathogenic as the ratio of proportions of variants in category j that are pathogenic or benign relative to the overall ratio of pathogenic and benign proportions38, that is:
where \({n}_{{Pj}}\) and \({n}_{{Bj}}\) are the number of pathogenic and benign variants in category j, and \({n}_{P}\) and \({n}_{B}\) are the total number of pathogenic and benign variants.
For calibration purposes, instances where variants were assigned either very strong or strong evidence according to the ACMG/AMP criteria, either in favor or against pathogenicity, were collectively considered as instances of strong evidence in favor of or against pathogenicity, respectively. The Haldane-Anscombe correction was applied for any category where the cell count for the pathogenic or benign variant reference set was zero39.
Concordance with predicted or experimental impact on function
To assess the concordance of the ccLR method with high-throughput functional assays and in silico prediction methods, we compared case-control LR values and suggestive ACMG/AMP code strength levels for variants located within the CDS or at splice acceptor/donor site dinucleotide positions ( ±2 bp); since this captures the regions more regularly tested in a clinical setting, present in at least three individuals in the combined dataset, and with evidence from at least two datasets for BRCA2.
Case-control evidence was compared with high-throughput functional assays performed for functionally critical BRCA1 and BRCA2 domains20,21,22,23,24 and in silico prediction methods. In silico prediction methods included BayesDel25 (without minor allele frequency) which is currently recommended (with optimal score cutpoints) by the ClinGen BRCA1/2 VCEP for the curation of BRCA1 and BRCA2 variants (benign BP4 criterion “Multiple lines of computational evidence suggest no impact on gene or gene product”, ≤ 0.15 for BRCA1 and ≤ 0.18 for BRCA2; pathogenic PP3 criterion “Multiple lines of computational evidence support a deleterious effect on the gene or gene product”, ≥ 0.28 for BRCA1 and ≥ 0.30 for BRCA2)2, as well as other computational tools recommended (with appropriate score thresholds) by the ClinGen SVI Working Group40, including REVEL26 (BP4, ≤ 0.29; PP3, ≥ 0.644), VEST427 (BP4, ≤ 0.449; PP3, ≥ 0.764) and MutPred228 (BP4, ≤ 0.391; PP3, ≥ 0.737). We also investigated the recent proteome-wide variant effect predictor AlphaMissense29.
Sensitivity and specificity metrics were used to assess the concordance of the functional predictors at predicting variants assigned evidence in favor or against pathogenicity using the ccLR method.
Statistics & reproducibility
No statistical method was used to predetermine sample size. The experiments were not randomized and the Investigators were not blinded to allocation during experiments and outcome assessment.
Web resources
Shiny app, https://biostatunitcing.shinyapps.io/ccLRbrowser/
ClinVar database, https://www.ncbi.nlm.nih.gov/clinvar/
gnomAD database, https://gnomad.broadinstitute.org/
ccLR GitHub repository, https://github.com/BiostatUnitCING/ccLR
Cancer Incidence in Five Continents (CI5) Volume IX, https://ci5.iarc.fr/CI5I-X/Default.aspx
LiftOver tool, https://genome.ucsc.edu/cgi-bin/hgLiftOver
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Individual level data for the BCAC data are not publicly available due to ethical review board constraints but are available on request through the BCAC Data Access Co-ordinating Committee (BCAC@medschl.cam.ac.uk). CARRIERS genotype data is available through dbGAP phs002820.v1. Phenotype data are not publicly available due to Institutional Review Board (IRB) constraints but are available on request through the CARRIERS Coordinating Committee (couch.fergus@mayo.edu). Requests for access to UK Biobank data should be made to the UK Biobank Access Management Team (access@ukbiobank.ac.uk). Summary statistics are published in Supplementary Data 4 and in the shiny app (https://biostatunitcing.shinyapps.io/ccLRbrowser/). According to the “UK Biobank best practice approaches on the publication of small numbers, Tables and/or results of any UK Biobank data there should have a minimum number of 5 reported participants within a cell”. Therefore, for variants present in less than 5 individuals, carrier count (Carriers, N) was replaced by “<5”.
Code availability
Source code for the ccLR method is publicly available in GitHub repository: https://github.com/BiostatUnitCING/ccLR37.
References
Federici, G. & Soddu, S. Variants of uncertain significance in the era of high-throughput genome sequencing: a lesson from breast and ovary cancers. J. Exp. Clin. Cancer Res 39, 46 (2020).
Parsons, M. T. et al. Evidence-based recommendations for gene-specific ACMG/AMP variant classification from the ClinGen ENIGMA BRCA1 and BRCA2 Variant Curation Expert Panel. Am. J. Hum. Genet. 111, 2044–2058 (2024).
Hu, C. et al. A population-based study of genes previously implicated in breast cancer. N. Engl. J. Med 384, 440–451 (2021).
Dorling, L. et al. Breast cancer risk genes - association analysis in more than 113,000 women. N. Engl. J. Med 384, 428–439 (2021).
Antoniou, A. et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am. J. Hum. Genet 72, 1117–1130 (2003).
Kuchenbaecker, K. B. et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 317, 2402–2416 (2017).
Hu, C. et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA 319, 2401–2409 (2018).
Li, S. et al. Cancer Risks Associated With BRCA1 and BRCA2 Pathogenic Variants. J. Clin. Oncol. 40, 1529–1541 (2022).
Momozawa, Y. et al. Expansion of cancer risk profile for BRCA1 and BRCA2 pathogenic variants. JAMA Oncol. 8, 871–878 (2022).
Tutt, A. N. J. et al. Adjuvant olaparib for patients with BRCA1- or BRCA2-mutated breast cancer. N. Engl. J. Med 384, 2394–2405 (2021).
Stadler, Z. K. et al. Therapeutic implications of germline testing in patients with advanced cancers. J. Clin. Oncol. 39, 2698–2709 (2021).
Mateo, J. et al. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 30, 1437–1447 (2019).
Daly, M. B. et al. NCCN Guidelines(R) Insights: Genetic/familial high-risk assessment: breast, ovarian, and pancreatic, version 2.2024. J. Natl Compr. Canc Netw. 21, 1000–1010 (2023).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424 (2015).
Tavtigian, S. V. et al. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med 20, 1054–1060 (2018).
Tavtigian, S. V., Harrison, S. M., Boucher, K. M. & Biesecker, L. G. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum. Mutat. 41, 1734–1737 (2020).
Spurdle, A. B. et al. ENIGMA–evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum. Mutat. 33, 2–7 (2012).
Zanti, M. et al. A likelihood ratio approach for utilizing case-control data in the clinical classification of rare sequence variants: application to BRCA1 and BRCA2. Hum. Mutat. 2023, 9961341 (2023).
Backman, J. D. et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 599, 628–634 (2021).
Findlay, G. M. et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222 (2018).
Mesman, R. L. S. et al. The functional impact of variants of uncertain significance in BRCA2. Genet Med 21, 293–302 (2019).
Hu, C. et al. Functional analysis and clinical classification of 462 germline BRCA2 missense variants affecting the DNA binding domain. Am. J. Hum. Genet 111, 584–593 (2024).
Huang, H. et al. Functional evaluation and clinical classification of BRCA2 variants. Nature 638, 528–537 (2025).
Sahu, S. et al. Saturation genome editing-based clinical classification of BRCA2 variants. Nature 638, 538–545 (2025).
Feng, B. J. PERCH: A unified framework for disease gene prioritization. Hum. Mutat. 38, 243–251 (2017).
Ioannidis, N. M. et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet 99, 877–885 (2016).
Carter, H., Douville, C., Stenson, P. D., Cooper, D. N. & Karchin, R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics 14, S3 (2013).
Pejaver, V. et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 11, 5918 (2020).
Cheng, J. et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381, eadg7492 (2023).
Parsons, M. T. et al. Large scale multifactorial likelihood quantitative analysis of BRCA1 and BRCA2 variants: An ENIGMA resource to support clinical variant classification. Hum. Mutat. 40, 1557–1578 (2019).
de la Hoya, M. et al. Combined genetic and splicing analysis of BRCA1 c.[594-2A>C; 641A>G] highlights the relevance of naturally occurring in-frame transcripts for developing disease gene variant classification algorithms. Hum. Mol. Genet 25, 2256–2268 (2016).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12, e1001779 (2015).
Li, Y. et al. FastPop: a rapid principal component derived method to infer intercontinental ancestry using genetic data. BMC Bioinforma. 17, 122 (2016).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Yates, A. D. et al. Ensembl 2020. Nucleic Acids Res 48, D682–D688 (2020).
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625, 92–100 (2024).
Zanti, M., O’Mahony, D. G., Michailidou, K. & Spurdle, A. B. Case-control likelihood ratio (LR). https://github.com/BiostatUnitCING/ccLR, https://doi.org/10.5281/zenodo.15012173 (2023).
Deeks, J. J. & Altman, D. G. Diagnostic tests 4: likelihood ratios. BMJ 329, 168–169 (2004).
Agresti, A. Categorical Data Analysis. 2nd edn, 734 (Wiley, 2003).
Pejaver, V. et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am. J. Hum. Genet 109, 2163–2177 (2022).
Acknowledgements
This research has been conducted using the UK Biobank Resource under application number 102655. Access to the UK Biobank data has been funded by an internal research grant of the Cyprus Institute of Neurology and Genetics. The BRIDGES panel sequencing was supported by the European Union Horizon 2020 research and innovation program BRIDGES (grant number 634935) and the Wellcome Trust (v203477/Z/16/Z). The CARRIERS studies were supported in part by NIH grants R35CA253187, R01CA225662, P50CA116201 and the Breast Cancer Research Foundation (BCRF). This work was supported by Cancer Research UK grant: PPRPGM-Nov20\100002 and by core funding from the NIHR Cambridge Biomedical Research Centre (NIHR203312) [*]. *The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. Additional funding for BCAC is provided by the Confluence project which is funded with intramural funds from the National Cancer Institute Intramural Research Program, National Institutes of Health, the European Union’s Horizon 2020 Research and Innovation Programme (grant numbers 634935 and 633784 for BRIDGES and B-CAST respectively), and the PERSPECTIVE I&I project, funded by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, the Ministère de l’Économie et de l’Innovation du Québec through Genome Québec, the Quebec Breast Cancer Foundation. The EU Horizon 2020 Research and Innovation Programme funding source had no role in study design, data collection, data analysis, data interpretation or writing of the report. ABS was supported by an NHMRC Investigator Fellowship (APP177524). The work of MTP was supported in part by National Institutes of Health grant U24 5U24CA258058-02. SVT’s analytical contribution was funded by NCI grant R01CA264971 (“Upgrading rigor and efficiency of germline cancer gene variant classification for the 2020 s”). PK’s contribution was supported by the Intramural Research Program of the National Cancer Institute. MSC was supported by the National Institute of Health Grants R01CA264971 and U24CA258058. The ABCS study was supported by the Dutch Cancer Society [grants NKI 2007-3839; 2009 4363] and an institutional grant of the Dutch Cancer Society and of the Dutch Ministry of Health, Welfare and Sport. The ACP study is funded by the Breast Cancer Research Trust, UK. KM and AL are supported by the NIHR Manchester Biomedical Research Centre, the Allan Turing Institute under the EPSRC grant EP/N510129/1. The work of the BBCC was partly funded by ELAN-Fond of the University Hospital of Erlangen. For BIGGS, ES is supported by NIHR Comprehensive Biomedical Research Centre, Guy’s & St. Thomas’ NHS Foundation Trust in partnership with King’s College London, United Kingdom. IT is supported by the Oxford Biomedical Research Centre. BOCS is supported by funds from Cancer Research UK (C8620/A8372/A15106) and the Institute of Cancer Research (UK). The BREast Oncology GAlician Network (BREOGAN) is funded by Acción Estratégica de Salud del Instituto de Salud Carlos III FIS PI12/02125/Cofinanciado and FEDER PI17/00918/Cofinanciado FEDER; Acción Estratégica de Salud del Instituto de Salud Carlos III FIS Intrasalud (PI13/01136); Programa Grupos Emergentes, Cancer Genetics Unit, Instituto de Investigación Biomedica Galicia Sur. Xerencia de Xestion Integrada de Vigo-SERGAS, Instituto de Salud Carlos III, Spain; Grant 10CSA012E, Consellería de Industria Programa Sectorial de Investigación Aplicada, PEME I + D e I + D Suma del Plan Gallego de Investigación, Desarrollo e Innovación Tecnológica de la Consellería de Industria de la Xunta de Galicia, Spain; Grant EC11-192. Fomento de la Investigación Clínica Independiente, Ministerio de Sanidad, Servicios Sociales e Igualdad, Spain; and Grant FEDER-Innterconecta. Ministerio de Economia y Competitividad, Xunta de Galicia, Spain. The BSUCH study was supported by the Dietmar-Hopp Foundation, the Helmholtz Society, and the German Cancer Research Center (DKFZ). CCGP is supported by funding from the University of Crete. The CECILE study was supported by Fondation de France, Institut National du Cancer (INCa), Ligue Nationale contre le Cancer, Agence Nationale de Sécurité Sanitaire, de l’Alimentation, de l’Environnement et du Travail (ANSES), Agence Nationale de la Recherche (ANR). The CGPS was supported by the Chief Physician Johan Boserup and Lise Boserup Fund, the Danish Medical Research Council, and Herlev and Gentofte Hospital. The CNIO-BCS was supported by the Instituto de Salud Carlos III, the Red Temática de Investigación Cooperativa en Cáncer and grants from the Asociación Española Contra el Cáncer and the Fondo de Investigación Sanitario (PI11/00923 and PI12/00070). COLBCCC is supported by the German Cancer Research Center (DKFZ), Heidelberg, Germany. Diana Torres was in part supported by a postdoctoral fellowship from the Alexander von Humboldt Foundation. PROCAS is funded from NIHR grant PGfAR 0707-10031. DGE, AH, and WGN are supported by the NIHR Manchester Biomedical Research Centre (IS-BRC-1215-20007). The GENICA was funded by the Federal Ministry of Education and Research (BMBF) Germany grants 01KW9975/5, 01KW9976/8, 01KW9977/0 and 01KW0114, the Robert Bosch Foundation, Stuttgart, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, the Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, as well as the Department of Internal Medicine, Johanniter GmbH Bonn, Johanniter Krankenhaus, Bonn, Germany. Generation Scotland (GENSCOT) received core support from the Chief Scientist Office of the Scottish Government Health Directorates [CZD/16/6] and the Scottish Funding Council [HR03006]. The GESBC was supported by the Deutsche Krebshilfe e. V. [70492] and the German Cancer Research Center (DKFZ). The HABCS study was supported by the German Research Foundation (DFG Do761/15-1), the Claudia von Schilling Foundation for Breast Cancer Research, by the Lower Saxonian Cancer Society, and by the Rudolf Bartling Foundation. The HMBCS was supported by the German Research Foundation (DFG Do761/15-1), a grant from the Friends of Hannover Medical School, and by the Rudolf Bartling Foundation. The HUBCS was supported by German Research Foundation (DFG Do761/15-1), a grant from the German Federal Ministry of Research and Education (RUS08/017), B.M. was supported by grant 17-44-020498, 17-29-06014 of the Russian Foundation for Basic Research, D.P. was supported by grant 18-29-09129 of the Russian Foundation for Basic Research, E.K was supported by the mega grant from the Government of Russian Federation (2020-220-08-2197), and the study was performed as part of the assignment of the Ministry of Science and Higher Education of the Russian Federation (№АААА-А16-116020350032-1). Financial support for KARBAC was provided through the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and Karolinska Institutet, the Swedish Cancer Society, The Gustav V Jubilee foundation and Bert von Kantzows foundation. The KARMA study was supported by Märit and Hans Rausings Initiative Against Breast Cancer. The KBCP was financially supported by the special Government Funding (VTR) of Kuopio University Hospital grants, Cancer Fund of North Savo, the Finnish Cancer Organizations, and by the strategic funding of the University of Eastern Finland. kConFab is supported by a grant from the National Breast Cancer Foundation, and previously by the National Health and Medical Research Council (NHMRC), the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia. Financial support for the AOCS was provided by the United States Army Medical Research and Materiel Command [DAMD17-01-1-0729], Cancer Council Victoria, Queensland Cancer Fund, Cancer Council New South Wales, Cancer Council South Australia, The Cancer Foundation of Western Australia, Cancer Council Tasmania and the National Health and Medical Research Council of Australia (NHMRC; 400413, 400281, 199600). G.C.T. and P.W. are supported by the NHMRC. RB was a Cancer Institute NSW Clinical Research Fellow. The MARIE study was supported by the Deutsche Krebshilfe e.V. [70-2892-BR I, 106332, 108253, 108419, 110826, 110828], the Hamburg Cancer Society, the German Cancer Research Center (DKFZ) and the Federal Ministry of Education and Research (BMBF) Germany [01KH0402]. The MASTOS study was supported by “Cyprus Research Promotion Foundation” grants 0104/13 and 0104/17, and the Cyprus Institute of Neurology and Genetics. The Melbourne Collaborative Cohort Study (MCCS) cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further augmented by Australian National Health and Medical Research Council grants 209057, 396414 and 1074383 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry. MYBRCA is funded by research grants from the Wellcome Trust (v203477/Z/16/Z), the Malaysian Ministry of Higher Education (UM.C/HlR/MOHE/06) and Cancer Research Malaysia. The NBCS has received funding from the K.G. Jebsen Centre for Breast Cancer Research; the Research Council of Norway grant 193387/V50 (to A-L Børresen-Dale and V.N. Kristensen) and grant 193387/H10 (to A-L Børresen-Dale and V.N. Kristensen), South Eastern Norway Health Authority (grant 39346 to A-L Børresen-Dale) and the Norwegian Cancer Society (to A-L Børresen-Dale and V.N. Kristensen). The Ontario Familial Breast Cancer Registry (OFBCR) was supported by grants U01CA164920 and U01CA167551 from the USA National Cancer Institute of the National Institutes of Health. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR) or the Colon Cancer Family Registry (CCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the USA Government or the BCFR or CCFR. The PBCS was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. Genotyping for PLCO was supported by the Intramural Research Program of the National Institutes of Health, NCI, Division of Cancer Epidemiology and Genetics. The PLCO is supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics and supported by contracts from the Division of Cancer Prevention, National Cancer Institute, National Institutes of Health. The POSH study is funded by Cancer Research UK (grants C1275/A11699, C1275/C22524, C1275/A19187, C1275/A15956 and Breast Cancer Campaign 2010PR62, 2013PR044. The SASBAC study was supported by funding from the Agency for Science, Technology and Research of Singapore (A*STAR), the US National Institute of Health (NIH) and the Susan G. Komen Breast Cancer Foundation. The SBCGS was supported primarily by NIH grants R01CA64277, R01CA148667, UMCA182910, and R37CA70867. Biological sample preparation was conducted the Survey and Biospecimen Shared Resource, which is supported by P30 CA68485. The scientific development and funding of this project were, in part, supported by the Genetic Associations and Mechanisms in Oncology (GAME-ON) Network U19 CA148065. SEARCH was funded by Cancer Research UK [C490/A10124, C490/A16561] and supported by the UK National Institute for Health Research Biomedical Research Centre at the University of Cambridge. SGBCC is funded by the National Research Foundation Singapore, NUS start-up Grant, National University Cancer Institute Singapore (NCIS) Centre Grant, Breast Cancer Prevention Programme, Asian Breast Cancer Research Fund, Breast Cancer Screening and Prevention Programme, and the NMRC Clinician Scientist Award (SI Category). Population-based controls were from the Multi-Ethnic Cohort (MEC) funded by grants from the Ministry of Health, Singapore, the National University of Singapore and the National University Health System, Singapore. SKKDKFZS is supported by the DKFZ. The SZBCS was supported by Grant PBZ_KBN_122/P05/2004 and the program of the Minister of Science and Higher Education under the name “Regional Initiative of Excellence” in the 2019-2022 project number 002/RID/2018/19 amount of financing 12 000 000 PLN. UBCS was supported by funding from National Cancer Institute (NCI) grant R01 CA163353 (to N.J. Camp) and the Women’s Cancer Center at the Huntsman Cancer Institute (HCI). Data collection for UBCS was supported by the Utah Population Database (UPDB) and Utah Cancer Registry (UCR). The UPDB is supported by HCI (including Huntsman Cancer Foundation, HCF), the University of Utah, and NCI grant P30 CA042014. The UCR is funded by the NCI’s SEER Program, Contract No. HHSN261201800016I, the US Center for Disease Control and Prevention’s National Program of Cancer Registries (Cooperative Agreement No. NU58DP007131), the University of Utah, and HCF. The BWHS study was supported by NIH grants U01CA164974 and R01CA098663, as well as the Susan G. Komen Foundation grant SAC220228. The NHS was supported by NIH grants P01 CA87969, UM1 CA186107, and U19 CA148065. The NHS2 was supported by NIH grants UM1 CA176726 and U19 CA148065. The CTS and the research reported in this publication were supported by the National Cancer Institute of the National Institutes of Health under award numbers U01-CA199277; P30-CA033572; P30-CA023100; UM1-CA164917; and R01-CA077398. The collection of cancer incidence data used in the California Teachers Study was supported by the California Department of Public Health pursuant to California Health and Safety Code Section 103885; Centers for Disease Control and Prevention’s National Program of Cancer Registries, under cooperative agreement 5NU58DP006344; the National Cancer Institute’s Surveillance, Epidemiology and End Results Program under contract HHSN2612018000321 awarded to the University of California, San Francisco, contract HHSN2612018000151 awarded to the University of Southern California, and contract HHSN2612018000091 awarded to the Public Health Institute. The opinions, findings, and conclusions expressed herein are those of the author(s) and do not necessarily reflect the official views of the State of California, Department of Public Health, the National Cancer Institute, the National Institutes of Health, the Centers for Disease Control and Prevention or their Contractors and Subcontractors, or the Regents of the University of California, or any of its programs. The MEC study was supported by the NIH grant U01-CA164973. The WCHS was supported by the NCI grants R01 CA100598 and R01 CA185623. Wisconsin Women’s Health Study (WWHS) is supported by NCI grant P30CA014520 to the University of Wisconsin Carbone Cancer Center. The American Cancer Society funds the creation, maintenance, and updating of the Cancer Prevention Study-II and Cancer Prevention Study-3 cohorts. CB, JH, AP, and LT’s contribution was supported by American Cancer Society intramural research funding. We thank all the individuals who took part in these studies and all the researchers, clinicians, technicians and administrative staff who have enabled this work to be carried out. ABCS thanks the Blood bank Sanquin, the Netherlands. ABCTB Investigators: Christine Clarke, Deborah Marsh, Rodney Scott, Robert Baxter, Desmond Yip, Jane Carpenter, Alison Davis, Nirmala Pathmanathan, Peter Simpson, J. Dinny Graham, Mythily Sachchithananthan. Samples are made available to researchers on a non-exclusive basis. The ACP study wishes to thank the participants in the Thai Breast Cancer study. Special thanks also go to the Thai Ministry of Public Health (MOPH), doctors and nurses who helped with the data collection process. Finally, the study would like to thank Dr Prat Boonyawongviroj, the former Permanent Secretary of MOPH and Dr Pornthep Siriwanarungsan, the former Department Director-General of Disease Control who have supported the study throughout. BIGGS thanks Niall McInerney, Gabrielle Colleran, Andrew Rowan, Angela Jones. The BREOGAN study would not have been possible without the contributions of the following: Manuela Gago-Dominguez, Jose Esteban Castelao, Angel Carracedo, Victor Muñoz Garzón, Alejandro Novo Domínguez, Maria Elena Martinez, Sara Miranda Ponte, Carmen Redondo Marey, Maite Peña Fernández, Manuel Enguix Castelo, Maria Torres, Manuel Calaza (BREOGAN), José Antúnez, Máximo Fraga and the staff of the Department of Pathology and Biobank of the University Hospital Complex of Santiago-CHUS, Instituto de Investigación Sanitaria de Santiago, IDIS, Xerencia de Xestion Integrada de Santiago-SERGAS; Joaquín González-Carreró and the staff of the Department of Pathology and Biobank of University Hospital Complex of Vigo, Instituto de Investigacion Biomedica Galicia Sur, SERGAS, Vigo, Spain. The BSUCH study acknowledges the Principal Investigator, Barbara Burwinkel, and thanks Peter Bugert, Medical Faculty Mannheim. CCGP thanks Styliani Apostolaki, Anna Margiolaki, Georgios Nintos, Maria Perraki, Georgia Saloustrou, Georgia Sevastaki, Konstantinos Pompodakis. CGPS thanks the staff and participants of the Copenhagen General Population Study. For the excellent technical assistance: Dorthe Uldall Andersen, Maria Birna Arnadottir, Anne Bank, Dorthe Kjeldgård Hansen. CNIO-BCS thanks Guillermo Pita, Charo Alonso, Nuria Álvarez, Pilar Zamora, Primitiva Menendez, the Human Genotyping-CEGEN Unit (CNIO). COLBCCC thanks all patients, the physicians Justo G. Olaya, Mauricio Tawil, Lilian Torregrosa, Elias Quintero, Sebastian Quintero, Claudia Ramírez, José J. Caicedo, and Jose F. Robledo, and the technician Michael Gilbert for their contributions and commitment to this study. PROCAS thanks NIHR for funding. The GENICA Network: Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart, and University of Tübingen, Germany [Hiltrud Brauch, RH, Wing-Yee Lo], Department of Internal Medicine, Johanniter GmbH Bonn, Johanniter Krankenhaus, Bonn, Germany [YDK, Christian Baisch], Institute of Pathology, University of Bonn, Germany [Hans-Peter Fischer], Molecular Genetics of Breast Cancer, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, Germany [UH], Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, Germany [Thomas Brüning, Beate Pesch, Sylvia Rabstein, Anne Lotz]; and Institute of Occupational Medicine and Maritime Medicine, University Medical Center Hamburg-Eppendorf, Germany [Volker Harth]. HABCS thanks Peter Schürmann, Peter Hillemanns, Natalia Bogdanova, Michael Bremer, Johann Karstens, Hans Christiansen and the Breast Cancer Network in Lower Saxony for continuous support. HMBCS thanks Peter Hillemanns, Hans Christiansen and Johann H. Karstens. HUBCS thanks Darya Prokofyeva and Shamil Gantsev. ICICLE thanks Kelly Kohut, Michele Caneppele, Maria Troy. KARMA and SASBAC thank the Swedish Medical Research Counsel. KBCP thanks Eija Myöhänen. kConFab/AOCS wish to thank Heather Thorne, Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow Up Study (which has received funding from the NHMRC, the National Breast Cancer Foundation, Cancer Australia, and the National Institute of Health (USA)) for their contributions to this resource, and the many families who contribute to kConFab. MARIE thanks Petra Seibold, Nadia Obi, Sabine Behrens, Ursula Eilber and Muhabbet Celik. MASTOS thanks all the study participants and express appreciation to the doctors: Yiola Marcou, Eleni Kakouri, Panayiotis Papadopoulos, Simon Malas and Maria Daniel, as well as to all the nurses and volunteers who provided valuable help towards the recruitment of the study participants. The MCCS was made possible by the contribution of many people, including the original investigators, the teams that recruited the participants and continue working on follow-up, and the many thousands of Melbourne residents who continue to participate in the study. MYBRCA thanks study participants and research staff (particularly Patsy Ng, Nurhidayu Hassan, Yoon Sook-Yee, Daphne Lee, Lee Sheau Yee, Phuah Sze Yee and Norhashimah Hassan) for their contributions and commitment to this study. The following are NBCS Collaborators: Kristine K. Sahlberg (PhD), Anne-Lise Børresen-Dale (Prof. Em.), Lars Ottestad (MD), Rolf Kåresen (Prof. Em.) Dr. Ellen Schlichting (MD), Marit Muri Holmen (MD), Toril Sauer (MD), Vilde Haakensen (MD), Olav Engebråten (MD), Bjørn Naume (MD), Alexander Fosså (MD), Cecile E. Kiserud (MD), Kristin V. Reinertsen (MD), Åslaug Helland (MD), Margit Riis (MD), Jürgen Geisler (MD), OSBREAC and Grethe I. Grenaker Alnæs (MSc). NBHS and SBCGS thank study participants and research staff for their contributions and commitment to the studies. The OFBCR thanks Teresa Selander, Nayana Weerasooriya and Steve Gallinger. ORIGO thanks E. Krol-Warmerdam, and J. Blom for patient accrual, administering questionnaires, and managing clinical information. PBCS thanks Louise Brinton, Mark Sherman, Neonila Szeszenia-Dabrowska, Beata Peplonska, Witold Zatonski, Pei Chao, Michael Stagner. We thank the SEARCH team. SGBCC thanks the participants and all research coordinators for their excellent help with recruitment, data and sample collection. SKKDKFZS thanks all study participants, clinicians, family doctors, researchers and technicians for their contributions and commitment to this study. UBCS thanks all study participants as well as the ascertainment, laboratory, analytics and informatics teams at Huntsman Cancer Institute and Intermountain Healthcare for their important contributions to this study. BWHS thanks the contributions of central cancer registries supported through the Centers for Disease Control and Prevention National Program of Cancer Registries (NPCR) and/or the National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) Program. Central registries may also be supported by state agencies, universities, and cancer centers. Participating central cancer registries include the following: A.L., A.R., A.Z., C.A., C.O., C.T., D.E., D.C., F.L., G.A., H.I., I.A., I.L., I.N., K.Y., L.A., M.D., M.A., M.I., M.O., M.S., N.E., N.J., N.M., N.Y., N.C., O.H., O.K., O.R., P.A., S.C., T.N., T.X., V.A., W.A., and W.I. The content is solely the responsibility of the authors and does not necessarily represent the official views of the U.S. Department of Health and Human Services, the National Institutes of Health, the National Cancer Institute, or the state cancer registries. WHI would also like to acknowledge the following short list of WHI investigators: Program Office: (National Heart, Lung, and Blood Institute, Bethesda, Maryland) Jacques Rossouw, Shari Ludlam, Dale Burwen, Joan McGowan, Leslie Ford, and Nancy Geller. WHI Clinical Coordinating Center (Fred Hutchinson Cancer Research Center, Seattle WA) and the WHI investigators (Garnet Anderson, Ross Prentice, Andrea LaCroix, Charles Kooperberg, JoAnn E. Manson, Barbara V. Howard, Marcia L. Stefanick, Rebecca Jackson, Cynthia A. Thomson, Jean Wactawski-Wende, Marian Limacher, Jennifer Robinson, Lewis Kuller, Sally Shumaker, Robert Brunner, Karen L. Margolis) for their dedication, and the study participants for making the program possible. A full list of WHI investigators can be found at: https://www.whi.org/doc/WHI-Investigator-Long-List.pdf. The Women’s Health Initiative (WHI) programme is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, US Department of Health and Human Services through contracts 75N92021D00001, 75N92021D00002, 75N92021D00003, 75N92021D00004, 75N92021D00005. The authors of the CTS would like to thank the CTS Steering Committee that is responsible for the formation and maintenance of the Study within which this research was conducted. A full list of California Teachers Study team members is available at https://www.calteachersstudy.org/team. The CPS3 and CPSII authors express sincere appreciation to all Cancer Prevention Study-II and Cancer Prevention Study-3 participants, and to each member of the study and biospecimen management group. The authors would also like to acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention’s National Program of Cancer Registries and cancer registries supported by the National Cancer Institute’s Surveillance Epidemiology and End Results Program. WWHS thanks Elizabeth Burnside, Irene Ong, John Hampton, and Julie McGregor for study support.
Author information
Authors and Affiliations
Consortia
Contributions
M.Z., D.G.O., M.T.P., K.M., A.B.S. and D.F.E., drafted the manuscript; K.M., A.B.S., D.F.E., F.J.C, D.E.G., M.Z. and D.G.O. designed the study. M.Z., D.O.M., K.Y., K.M., conducted statistical analyses. M.Z., L.D., J.D., M.K.B., Q.W., N.J.B., W.C., C.H., M.N. conducted bioinformatics analyses and data collection. T.U.A., C.B.A., I.L. A., A.C.A., P.L.A., C.B., C.B., N.V.B., S.E.B., M.K.B., K.D.B., N.J.C., A.C., J.E.C., M.H.C., J.C-C., F.C., G.C-T., NBCS Collaborators, D.M.C., K.C., S.M.D., T.D., A.M.D., A.H.E., D.G.E., P.A.F., J.D.F., H.F., M.G-D., M.G-C., G.G., A.G-N., F.G., A.H., C.A.H., U.H., S.N.H., M.B.A.H, W-K.H., J.M.H., R.H., S.J.H., C.H., kConFab Investigators, A.J., E.K.K., Y-D.K., P.K., V.N.K., J.V.L., J.L., G.H.L., S.L., A.L., C.L., A.M., M.E.M., D.M., R.L.M., K.M., K.L.N., R.N-T., N.O., J.E.O., J.R.P., M.I.P., A.V.P., P.D.P.P., E.C.P., M.U.R., K.J.R., E.S., E.J.S., M.K.S., M.C.S., V.K-M.T., S.H.T., L.R.T., D.T., A.T-D., T.T., C.M.V., Q.W., J.N.W., S.Y., S.Y., G.R.Z. provided data. All authors contributed to the review and editing. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The following authors declare conflicts not directly relevant to this work as stated below. VK-MT and GDE have received consultancies from AstraZeneca and Everything Genetic Ltd. ACA and DFE are named creators of the BOADICEA model, which has been licensed by Cambridge Enterprise (University of Cambridge). SJH has received speaker fees from AstraZeneca and Pfizer Ltd. SY has received research funding from AstraZeneca and Repare Therapeutics and participates in the advisory board for AstraZeneca. JWW has received consulting fees from Natera, MyOme, Cancer IQ, equity from Natera, and speaker fees from AstraZeneca. All the other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ken Chen, who co-reviewed with Ziyi Wang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zanti, M., O’Mahony, D.G., Parsons, M.T. et al. Analysis of more than 400,000 women provides case-control evidence for BRCA1 and BRCA2 variant classification. Nat Commun 16, 4852 (2025). https://doi.org/10.1038/s41467-025-59979-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59979-6
