
Abstract
The activation of C—C bonds enables rapid construction of new organic frameworks owing to facile structural reorganization. Nevertheless, enantioselective C—C activation remains heavily underexplored and has been predominantly limited to intramolecular reactions. We herein report two categories of asymmetric intermolecular [4 + 2] annulations between benzocyclobutenones (BCBs) and unsaturated reagents, namely, alkynes and cyclic dicarbonyl compounds. The atroposelective coupling of BCB with several classes of sterically hindered alkynes afford C—N and C—C axially chiral 2-naphthols. The [4 + 2] annulation of BCBs and α-dicarbonyls afford spirocyclic products. Both coupling systems proceed efficiently with excellent regio-, chemo- and enantioselectivity via substrate activation and judicious choice of chiral bidentate phosphine ligands. Synthetic transformations of selected products are demonstrated, and the derived chiral products are shown to be useful additives in C—H bond activation or as ligands in Pd-catalyzed C—C coupling.
Similar content being viewed by others

Introduction
Ever since the seminal reports by Liebeskind1, Jun2,3, and Murakami4,5,6,7, catalytic activation of the C—C bond has been increasingly explored to allow for the development of new synthetic methods owing to facile reorganization of the structural framework8,9,10,11,12,13,14,15,16,17. These coupling systems typically proceed via initial C—C oxidative addition, followed by migratory insertion into an unsaturated bond, and reductive elimination releases the final product. Two strategies are typically employed to ensure the catalytic reactivity. The installation of a proximal directing group facilitates the C—C cleavage facilitated by the entropic effect, which echoes chelation-assisted C—H activation that has been heavily explored18. Alternatively, the employment of a strained ring also activates the C—C bond toward oxidative addition4,5,6,7,11,19. Among strained rings, benzocyclobutenones (BCBs) are readily available and undergo diverse [4 + 2] and, occasionally, [4 + 1]20 annulation with alkynes, alkenes, allenes, dienes, and carbonyls, affording fused and bridged cycles13,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41.
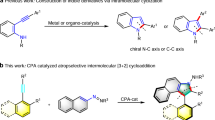
While this type of ‘cut-and-sew’ chemistry, dubbed by Dong21,42, a leading researcher in the field, has witnessed tremendous progress, the asymmetric cut-and-sew of BCBs remains underexplored. Existing reports are predominantly limited to intramolecular reactions (Fig. 1a)43. In 2012, Dong and coworkers reported the first intramolecular [4 + 2] annulation with olefins (Fig. 1a)22. The Xu group later extended the coupling system to other tethered olefins44,45. The Dong group further extended the π-partner to oximes, affording fused lactams43. Subsequently, the Xu and Lu group took advantage of the low aromaticity of furans and realized [4 + 2] annulation via dearomatization46. Alternatively, intramolecular [4 + 2] annulation of cyclobutanones with a different pattern of tethered coupling partner afforded bridged cycles (Fig. 1a)47,48,49. Thus, in 2014, the Cramer group capitalized on the reactivity of an aldehyde-tethered cyclobutanone and realized the Rh(I)-catalyzed intramolecular coupling to give bridged oxacycles (Fig. 1a)47. The Dong group developed intramolecular [4 + 2] coupling with allenes48. Very recently, the Dong group judiciously developed Rh(I)-catalyzed intramolecular annulation of 1,5-enyne-tethered cyclobutanones, which occurred divergently to deliver bis-bicyclic scaffolds and tetrahydroazapinones under catalyst control49. So far, only two intermolecular asymmetric systems have been disclosed (Fig. 1b)50,51. The Krische group utilized ketols as the coupling partner and realized Ru-catalyzed enantioselective [4 + 2] annulation with benzocyclobutenones50. Recently, the group of Feng and Liu succeeded in Sc(III)-catalyzed asymmetric [4 + 1] homologation of benzocyclobutenones with diazo compounds51. Despite the progress, only point-chiral products have been delivered, and it is necessary to develop intermolecular coupling systems to accommodate diverse chirality patterns by taking full advantage of the reactivity of BCBs. On the other hand, structurally related biphenylenes are also strain-activated, and Rh and Pt-catalyzed atroposelective coupling with alkynes has been reported52,53,54. However, these reactions are also limited to intramolecular systems.

a Prior intramolecular asymmetric [4 + 2] reactions of benzocyclobutenones/Cyclobutanones. b Prior intermolecular asymmetric [4 + 2]/[4 + 1] reactions of benzocyclobutenones (only Two Examples, limited to central chirality). c Asymmetric intermolecular [4 + 2] coupling of benzocyclobutenone and alkynes/diketones in this work.
Axially chiral biaryls are an important class of three-dimensional platforms, and atroposelective synthesis of axially chiral compounds has evolved into a dynamic research field55,56,57,58. Axial chirality has been constructed via difunctionalization of alkynes through two approaches, namely, radical and ionic pathways59,60,61,62,63,64,65,66,67.Metal-catalyzed non-radical dicarbonfunctionalization of alkynes is challenging in terms of reactivity and regioselectivity59,60,61,62,63,64. Metal-catalyzed radical difunctionalization of alkynes offered an alternative approach, but existing systems are limited to employment of reactive alkynes and/or stable radical species65,66,67. Alternatively, as a special class of dicarbonfunctionalization of alkynes, Tanaka pioneered in atroposelective synthesis of biaryls via [2 + 2 + 2] cycloaddition between diynes and alkynes via a metallacyclopentadiene intermediate68,69,70,71. On the other hand, the closely related five-membered metalacyclic intermediates are obtainable from oxidative addition of benzocyclobutenones as in the synthesis of 2-naphthols1,31,72,73,74. In this regard, although such naphthol synthesis has been realized by Liebeskind1, Shi31, Harrity72, Martin73, and Wang74, no atroposelective systems have been developed. The reactivity and ready availability of BCBs inspired us to address axial chirality via enantioselective intermolecular [4 + 2] reactions between BCBs and alkynes. In addition, the reactivity of the BCBs also accommodates ketones as a coupling partner75. Here, we report Rh(I)-catalyzed enantioselective annulative coupling of BCBs with alkynes and ketones via C—C bond cleavage, affording C—N axially chiral indolylnaphthols and spirocyclic products, respectively (Fig. 1c).
Results and disccusion
Optimization of the [4 + 2] Annulation Reaction
We conducted our studies with initial identification of a sterically hindered alkyne that could couple with benzocyclobutenone (1a) using a Rh(I) catalyst stabilized by a BINAP ligand. It was found that naphthalene-based alkynes exhibited no reactivity in our initial attempts (see Supplementary Information). These outcomes indicated the intrinsic challenge of this intermolecular C—C activation-[4 + 2] annulation. We reasoned that the challenge in atroposelective [4 + 2] annulation with alkynes may reside in the alkyne insertion process due to the steric bulkiness of the alkyne. Therefore, an electronically biased alkyne such as ynamine60,76,77 was employed, leading to C—N axially chiral biaryls or anilides. Indeed, good reactivity was observed for a 1-alkynyl-2-sulfonylindole and for a related ynamide (see Supplementary Information).
Further optimization studies were conducted using 1-alkynyl-2-sulfonylindole 2 as a substrate in the presence of a cationic Rh(I) catalyst (Table 1). Closely comparable outcomes were obtained when the (S)-BINAP or L2 was used as a ligand (entries 1 and 2). Moving to a Segphos (L3) resulted in improved enantioselectivity (entry 3). The reaction efficiency and selectivity were heavily dependent on the backbone of the chiral ligand, and employment of a bipyridyl-based ligand L5 ((R)-P-Phos) afforded both good activity and enantioselectivity (85% ee, entry 5). A series of solvents were then investigated (entries 6–9), and the highest enantioselectivity was realized in THF (90% ee, entry 7). The counter anion also had noticeable effects, and [Rh(COD)]2OTf outperformed others in terms of enantioselectivity (entry 10, 94% ee), while a neutral dimeric catalyst failed to show any catalytic activity (entry 11). Lowering the catalyst loading to 4 mol% led to a sluggish reaction, and the same trend was also observed when the reaction was conducted at 90 °C. Therefore, the conditions outlined in entry 10 were retained for further studies (Conditions A).
Scope of the Atroposelective [4 + 2] Annulation Reactions
After the establishment of the optimal reaction conditions, we next evaluated the generality of this atroposelective coupling system (Fig. 2, top). Introduction of a 3-methyl group, halogen, or an EDG into the 4-position of the indole ring had a negligible impact on the reactivity and enantioselectivity (4–6, 90–92% ee). The same trend is followed for substrates with an electron-donating or -withdrawing group at the 5- or 6- position (7–11, 93–95% ee). The scope of the alkyne with respect to the aryl terminus was then investigated. Alkynes bearing both electron-donating and -withdrawing groups at the para and meta positions were tolerated (12–20, 89–94% ee). The absolute configuration of the product (R)−16 has been established by X-ray crystallography (CCDC 2360303). The aryl terminus was then smoothly extended to a 2-thienyl group with excellent enantioselectivity (21, 96% ee). The 2-EWG group in the indole ring was also expanded to an ethanesulfonyl (22). Variations of the substituents in the BCB ring was also briefly performed, and alkyl, trifluoromethyl, alkoxyl, halogen, and a fused benzene ring at different positions were all generally compatible (23–31, 84–96% ee). Of note, the 2-substituent of the indole ring is not limited to a sulfonyl group, and a dialkyl phosphate ester also performed well (32, 99% ee). In all cases, the axially chiral products were obtained with exclusive regioselectivity with respect to both the BCB and the alkyne. Extension of the alkyne terminus to an alkyl group, however, met with difficulty. The coupling of an n-propyl based alkyne proceeded in poor enantioselectivity, albeit with excellent catalytic reactivity (product 33). This may indicate the participation of the alkyne terminus during the chiral induction.
![Fig. 2: Scope of the Atroposelective [4 + 2] Annulation Reactionsa.](http://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs41467-025-60109-5/MediaObjects/41467_2025_60109_Fig2_HTML.png?as=webp)
aReaction conditions A: benzocyclobutenone (0.1 mmol), alkyne (0.12 mmol), [Rh(COD)]2OTf (8 mol%), and L5 (8 mol%) in THF (1 mL), 100 °C for 16 h. Isolated yield. Reaction conditions B: benzocyclobutenone (0.1 mmol), alkyne (0.12 mmol), [Rh(COD)]2NTf2 (8 mol%), and L6 (8 mol%) in THF (1 mL) at 80 °C for 16 h. Isolated yield.
In contrast to the compatibility of the 2-sulfonyl/phosphate group in the indole ring, extension to a 2-ester group met with difficulty under the original reaction conditions. Further optimization revealed that an EDG-functionalized Segphos ligand L6 sufficed in combination with a slightly different catalyst [Rh(COD)2]NTf2 (Conditions B, see Supplementary Information for details). The scope of this coupling system was briefly explored (Fig. 2, bottom). While the reaction efficiency is generally lower than that of the 3-sulfonylindole-based alkynes, moderate yields have been realized for the products 35–40. Tolerance of alkyl or halogen groups in the indole ring was observed, and the enantioselectivity varied within a narrow range of 91–94% ee for the substrates examined. To our delight, a structurally related ynamide also reacted smoothly under these conditions to afford product 41 in excellent enantioselectivity.
To further expand the scope of the alkyne, we utilized 1-alkynyl-2-naphthol (42) as another class of electronically biased alkyne, which has been explored as a substrate in organocatalysis78. Extensive screening of the chiral ligand revealed that the (R)-BTFM-Garphos L7 performed well (Conditions C, see the Supplementary Information for optimization studies), and the coupling of benzocyclobutenone and 42 afforded a (S)-configured 2,4’-binaphthol 43 in excellent enantioselectivity and in moderate yield (Fig. 3). Brief studies of the scope of this class of alkyne indicated that electron-donating and -withdrawing groups in the alkyne terminus were compatible, with the enantioselectivity ranging from 93–95% ee (44–49). The absolute configuration of product 46 has been determined by ECD spectroscopy (see Supplementary Data 2 and 3 for details) together with the crystallographic structure of a racemic sample of 45.
![Fig. 3: Asymmetric [4 + 2] Annulation with 1-Alkynyl-2-Naphtholsa.](http://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs41467-025-60109-5/MediaObjects/41467_2025_60109_Fig3_HTML.png?as=webp)
aReaction conditions C: benzocyclobutenone (0.1 mmol), alkyne (0.12 mmol), [Rh(COD)]2BF4 (8 mol%), and L7 (8 mol%) in 2-Me-THF (1 mL), 100 °C for 16 h. Isolated yield.
We found that the four-membered ring was not limited to a BCB, and biphenylene52,53,54 also underwent the same [4 + 2] annulation with alkyne 2 to give 50 in high enantioselectivity and with moderate yield under the reaction Conditions A using a P-Phos ligand in Equation (1). This reaction likely also proceeded through a related rhodacyclic intermediate.

Optimization of the Spirocyclization Reaction
Having established the scope of the atroposelective [4 + 2] annulation, we next explored the intermolecular [4 + 2] spirocyclization between BCB and an activated carbonyl compound. Again, although asymmetric coupling of BCB with ketone has been reported, it is mostly limited to intramolecular reactions79,80. It should be noted that the uncatalyzed reaction between BCB and isatin has been reported to proceed through a ketene methide intermediate81, which may pose an undesired racemic background reaction during the development of our asymmetric system. The Rh-catalyzed reaction of N-methylisatin and a simple BCB substrate using an electron-rich bidentate bisphosphine ligand gave two regioisomeric mixtures in a poor ratio (r.r. = 1.6:1) as a result of competitive cleavage of the C(O)—C(sp2) versus C(O)—C(sp3) bond (Table 2, entry 1), and the major product was isolated in 90% ee. Then, a gem-dimethyl BCB was employed as a substrate to block the competing C(O)—alkyl reaction site. Optimization studies indicated that the enantioselectivity was generally high when different BIPHEP ligands were used, and an electron-rich ligand (L8 and L9) tended to give enhanced reactivity, likely due to a higher tendency of oxidative addition (Table 2). Finally, both excellent yield and enantioselectivity (99% ee) were realized when the L8 ligand was used (Conditions D).
Scope of Enantioselective [4 + 2] Spirocyclization Reactions
The scope of this asymmetric spirocyclization system was then explored (Fig. 4). Tolerance of N-alkyl, -aryl, -benzyl, and -allyl groups in the isatin was verified (52–56, 97–99% ee). In the case of an N-allyl isatin (product 55), the reaction proceeded with isomerization of the allyl to an N-alkenyl group. A diverse spectrum of electron-donating, -withdrawing, and halogen groups at the 4-, 5-, 6-, and 7- positions of the isatin was fully accommodated (56–72), and excellent enantioselectivity was obtained in all cases (96–99% ee). The high reactivity of 4-substituted isatins (products 69 and 70) indicated tolerance of steric effects around the carbonyl group. The presence of nitro (68) and a Weinreb amide (67) in the product leaves room for product transformations. In addition, an aza-isatin was also amenable to the reaction conditions (73, 99% ee). Moreover, unprotected isatins were also fully compatible, as in the isolation of products 74–78 in good to excellent yield and in excellent enantioselectivity (98-99% ee). The dicarbonyl reagents were not limited to isatins, and several other five-membered dicarbonyls also participated well (79 and 80, 98% ee for each). The scope of the BCB substrate was next investigated. Halogen, alkyl, and CF3 groups at different positions of the benzene ring were compatible, and the products 81–86 were obtained in 93-99% ee. The dialkyl substituents at the benzylic position were also extended to several symmetric cyclic ones, affording the bis-spirocyclic product with no loss of reactivity or enantioselectivity (87–90, 97–99% ee). In addition, an unsymmetrically dialkyl-substituted BCB also underwent coupling with N-methylisatin, affording two diastereomeric products in poor d.r. but with excellent enantioselectivity for each product (91 and 91’). The absolute configuration of products 89 (CCDC 2360308), 90 (CCDC 2360232), and 91 (CCDC 2360307) has been secured by X-ray crystallographic analyses. In contrast to the reactivity of these five-membered α-dicarbonyls, no reactivity was detected for acyclic ones such as benzil and α-ketoesters, possibly due to their steric effects. We also attempted to extend the four-membered ring substrate to a biphenylene, but its coupling with 51 only afforded the [4 + 2] target product 92 with poor yield and enantioselectivity (see Equation 2).

![Fig. 4: Scope of Enantioselective [4 + 2] Spirocyclization Reactionsa.](http://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs41467-025-60109-5/MediaObjects/41467_2025_60109_Fig4_HTML.png?as=webp)
aReaction Conditions D: disubstituted benzocyclobutenone (0.1 mmol), dicarbonyl compound (0.12 mmol), [Rh(COD)]2OTf (8 mol%), and L8 ligand (8 mol%) in THF (1 mL), 100 °C for 16 h. Isolated yield. [b] The N-allylisatin was used.
Synthetic Applications of Selected Chiral Products
Extensive synthetic applications of selected chiral products were next demonstrated (Fig. 5). The product 3 was synthesized in high yield at a 2 mmol scale (94% ee, Fig. 5a). The synthesis of naphthol 35 was also demonstrated at the same scale with no loss of enantioselectivity. The spirocyclic product 52’-H was synthesized in high enantioselectivity in overall three steps starting from a readily available reagent (or one step when counted from the simple BCB), while it took five steps even in a racemic synthesis based on a literature report (Fig. 5b)82. The OH group in 3 provides a synthetic handle for diverse transformations (Fig. 5c). Pd-catalyzed oxidative aromatization of 3 a benzofuran ring via C—O cyclization (93). Triflation of the OH group gave product 94, which then underwent Pd-catalyzed C—P coupling to produce a phosphine oxide 95 in decent yield. Standard reduction of 95 then yielded a phosphine 96. In all cases, no erosion of enantioselectivity was observed. A 2-ester-substituted indolylnaphthol (37) also proved to be a useful reagent toward synthetic transformations (Fig. 5d). Methylative protection of the OH followed by saponification gave a carboxylic acid 98. Activation of 98 with oxalyl chloride, followed by treatment with a protected hydroxyamine, afforded an axially chiral amide 99 in 92% ee. O-Methlyation of 35 followed by 3-iodination of the indole ring afforded 10160. Copper-catalyzed phosphination produced 102 as a potential chiral ligand (vide infra). A selected spirocyclic product was also investigated toward synthetic transformations (Fig. 5e). The scale-up synthesis of product 52-Me was achieved with no loss of reactivity or enantioselectivity. LiAlH4 reduction of 52-Me triggered a nucleophilic cyclization-acetalization, affording an oxygen-bridged acetal (103) in excellent enantioselectivity as a single diastereomer. Treatment of 52-Me with an excess of MeLi afforded a related acetal (104) with the introduction of two methyl groups in excellent diastereoselectivity, whereas the enantiopurity was only slightly affected.

a Scale-up synthesis of two axially chiral naphthols. b Comparisons of the synthesis of a spirocyclic product with a reported method. c Synthetic transformations of product 3. d Synthetic transformations of products 35 and 37. e Synthetic transformations of a spirocyclic product. f Catalytic applications of several chiral acids, amides, and phosphines.
The axially chiral acid and carboxamide displayed in the Fig. 5d offered a good chiral additive for asymmetric catalysis (Fig. 5f). The 3-methyl-substituted chiral carboxylic acid (98) was applied as a chiral additive in Rh-catalyzed sp3 C—H desymmetrization-amidation of 8-ethylquinoline, affording amide 105 in 82% ee, a promising initial outcome for further studies. In another desymmetrization reaction, the enantioselective alkynylation reaction of diphenylsulfoxide catalyzed by a chiral Rh(III) catalyst with the assistance of a chiral amide 99 afforded product 106 with a moderate yield and 82% ee. In addition, chiral phosphine 102 (94% ee) was applied as a chiral ligand in Pd-catalyzed allylic substitution, affording 107 in excellent enantioselectivity.
In addition to the synthetic applications, we also preliminarily explored the biological activity of selected spirocyclic lactams against the C. violaceum 12472 and clinical pathogenic bacterium B. cenocepacia 162638 (see Supplementary Information). It was found that compounds 76 and 77-H exhibited quorum sensing activity against both the C. violaceum 12472 and B. cenocepacia 162638, and the quorum sensing activity of compound 77-H was higher than that of 76.
Mechanistic studies
A series of experiments have been conducted to cast light on the mechanism of the atroposelective [4 + 2] annulation (Fig. 6). Ligand substitution between the cationic rhodium(I) COD complex and ligand L5 afforded complex 108 [Rh(L5)(COD)]OTf that is catalytically active (Fig. 6a, left). A stoichiometric interaction between the BCB 1a and 108 in THF-d8 at 100 °C revealed the formation of several new rhodium species with the replacement of the COD ligand (see Supplementary Information). In another experiment, the gem-dimethyl-substituted BCB 1b, Rh(I) catalyst, and ligand L8 reacted to give a mixture, HRMS analysis of which suggested possible formation of an oxidative addition intermediate (Fig. 6a, right). These observations may collectively suggest initial participation of the BCB substrate. Correlation of the ee of ligand L5 with that of the product revealed a linear relation (see Supplementary Information), indicating a 1:1 ratio of the Rh and the L5 ligand. A Hammett study was then performed using a series of alkynes bearing a different para substituents in the benzene ring. The small positive slope of ρ = 0.41 suggests buildup of negative change in the transition state (Fig. 6b), and the fact that a more electronically biased alkyne is more reactive suggests that the alkyne insertion is rate-limiting or occurs prior to the rate-limiting step. To further explore the mechanistic details, kinetic studies using the initial rate method were carried out (Fig. 6c). An inverse first-order dependence of the BCB 1a substrate was found. Our kinetic studies also established the first-order dependence for both the 1-indolylalkyne (2) and the Rh(I) catalyst (108). These kinetic data indicated that both the alkyne and the Rh(I) contributed to the transition state of the rate-limiting step, while the BCB needs to undergo dissociation at a certain stage to give an active intermediate in the catalytic cycle to allow generation of an alkyne-bound active species.

a Possible intermediates. b The Hammett plot of the alkyne substrate. c Kinetic studies (inverse first order dependence for the BCB 1a and first order for both the alkyne 2 and the catalyst 108).
Proposed mechanism
A plausible mechanism was proposed based on these experimental data and literature reports (Fig. 7). The BCB substrate 1 may undergo inhibitive binding to the Rh(I) catalyst prior to its oxidative addition (A). Following dissociation of a benzocyclobutenone 1a and selective C(O)—C(Ar) oxidative addition, a five-membered rhodacyclic intermediate B is generated. Subsequently, coordination of alkyne 2 produces intermediate C. Migratory insertion of the Rh(III)—C(aryl) bond is proposed to give a Rh(III) alkenyl species D, and this migratory insertion is also regioselective with respective to the alkyne ligand; the electronic and steric effect of the alkyne worked in the same direction during the insertion, and the rhodium metal ends up proximal to the less hindered phenyl group in the alkyne unit (D)83,84. Subsequent C—C reductive elimination of this rhodium(III) alkenyl intermediate then furnishes the final product. The migratory insertion is likely stereo-determining. This is because a C—N chiral axis is already constructed upon migratory insertion. In addition, the C—C reductive elimination may proceed with a lower kinetic barrier owing to aromatization as a driving force.

Proposed catalytic cycle for the atroposelective 2-naphthol synthesis.
DFT calculations
To elucidate the detailed mechanism and origins of the selectivities of the coupling of isatins, density functional theory (DFT) calculations were performed using benzocyclobutenone 1b and isatin 51 as the model substrates with L9 as the chiral ligand, which was selected to save the time cost of our computational studies. The calculated energy profile of the most favorable pathway is depicted in Fig. 8a. At the outset, different possible complexations between the Rh catalyst and substrates were evaluated (see Supplementary Data 1 for details). Our computations showed that intermediate IM1, formed by the coordination of two molecules of 1b to the Rh(I) center, is among the most stable species (Supplementary Fig. S29). Therefore, the sum of the free energies of IM1 and substrates were chosen to be the zero on the relative free energy scale (Fig. 8a).

a Calculated energy profile of the most favorable pathway leading to product 52-Me. b Optimized geometries of key migratory insertion transition states. Gibbs free energies and bond distances are given in kcal/mol and Å, respectively.
The reaction is initiated by the dissociation of one molecule of 1b, leading to the formation of intermediate IM2, which was calculated to be endergonic by 5.8 kcal/mol. The ensuing C(O)—C(sp2) bond cleavage occurs via transition state TS1, with an energy barrier of 18.3 kcal/mol relative to IM1, affording the five-membered rhodacycle IM3 irreversibly with a ΔG of − 8.7 kcal/mol. The alternative selectivity of C(O)—C(sp3) oxidative addition was also examined and was found to be energetically disfavored by 2.7 kcal/mol compared to TS1, consistent with the experimentally observed regioselectivity (Supplementary Fig. S30). The preference for the C(O)—C(sp2) bond cleavage is likely attributed to steric repulsion between 1b and the ligand in the C(O)—C(sp3) bond cleavage pathway (see Supplementary Fig. S30 in the Supplementary Information).
Subsequently, the coordination of an incoming isatin 51 to the Rh center of IM3 gives intermediate IM5, which was calculated to be exergonic by 3.7 kcal/mol. Notably, intermediate IM4, generated by the coordination of 1b to the Rh center of IM3, is more stable than IM5 by 3.0 kcal/mol, making it the off-cycle resting state of the catalyst. Indeed, our kinetic studies revealed inverse first-order dependence for the substrate 1b in the coupling with 51 (see Supplementary Information). The migratory insertion of the C(3)=O double bond of the isatin into the Rh—C(sp2) bond then takes place via transition state TS2, leading to the formation of a seven-membered rhodacycle IM6. It is important to note that insertion of the amide C=O double bond into the Rh—C(sp2) bond was safely ruled out, given its drastically high kinetic barrier (Supplementary Fig. S31). Finally, the catalytic cycle is completed upon the C—C reductive elimination via transition state TS3, giving rise to the product-ligated intermediate IM7, from which the ligand exchange with 1b would regenerate intermediate IM1 and release the final product 52-Me.
The computations show that the migratory insertion via transition state TS2 is the rate- as well as the enantioselectivity-determining step, with an overall energy barrier of 28.6 kcal/mol relative to IM4. As shown in Fig. 8b, transition state TS2 is 1.5 kcal/mol lower in energy than TS2’, which is in qualitative agreement with our experimentally observed enantioselectivity. Scrutiny of the optimized geometries reveals the presence of the C—H---π and C—H---O hydrogen-bonding interactions between the chiral ligand and substrates in both transition states. However, these interactions are stronger in TS2 than in TS2’, resulting in the lower energy of TS2, thus accounting for the observed enantioselectivity. It should be noted that the key migratory insertion step employing benzocyclobutenone 1a and 1-alkynyl-2-sulfonylindole 2 as model substrates with L5 as the chiral ligand was also explored (Supplementary Fig. S32). These computations are in line with the enantioselectivity observed in reactions involving alkynes.
In summary, we have realized two classes of rhodium-catalyzed asymmetric intermolecular [4 + 2] annulation reactions between BCBs and unsaturated reagents. The employment of sterically hindered alkynes allowed atroposelective construction of C—N axially chiral indoles. In this system, three sub-classes of alkynes varying at the 3-substituent have been identified. In the coupling of alpha-dicarbonyl compounds such as isatins, the cut-and-sew [4 + 2] annulation afforded spirocyclic products with 100% atom-economy and excellent chemo- and enantioselectivity. Both coupling systems proceeded efficiently with excellent enantioselectivity. Synthetic transformations of selected products have been demonstrated, and the derived chiral acids, carboxamides, and phosphines have been proven to be useful chiral additives in C—H activation-desymmetrization reactions or asymmetric C—C coupling. The ready availability of the strained ring substrates, diverse chiral platforms, intermolecularity of the coupling, and three-dimensionality of the products may inspire further development of new asymmetric C—C activation reactions.
Methods
Synthesis of products 3–33 and 50
Under N2 atmosphere, a screw-cap vial (8 mL) was charged with benzocyclobutenone (0.1 mmol, 1.0 equiv), 1-alkynylindole (0.12 mmol, 1.2 equiv), [Rh(COD)2]OTf (0.008 mmol, 8 mol%), L5 (0.008 mmol, 8 mol%) and THF (anhydrous, 1 mL). After stirred at 100 °C for 16 h, the reaction mixture was filtered through celite and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel (petroleum ether/ ethyl acetate = 3/1) to afford the axially chiral products 3–33 and 50.
Synthesis of products 35–41
Under N2 atmosphere, a screw-cap vial (8 mL) was charged with benzocyclobutenone (0.1 mmol, 1.0 equiv), 1-alkynylindole (0.12 mmol, 1.2 equiv), [Rh(COD)2]NTf2 (0.008 mmol, 8 mol%), L6 (0.008 mmol, 8 mol%) and THF (anhydrous, 1 mL). After stirred at 80 °C for 16 h, the reaction mixture was filtered, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel (petroleum ether/ ethyl acetate = 5/1) to afford the axially product 35–41.
Synthesis of products 43–49
Under N2 atmosphere, a screw-cap vial (8 mL) was charged with benzocyclobutenone (0.1 mmol, 1.0 equiv), 1-alkynyl-2-naphthol (0.12 mmol, 1.2 equiv), [Rh(COD)2]BF4 (0.008 mmol, 8 mol%), L7 (0.008 mmol, 8 mol%) and 2-Me-THF (anhydrous, 1 mL). After stirred at 100 °C for 16 h, the reaction mixture was filtered, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel (petroleum ether/ ethyl acetate = 3/1 or 4/1) to afford the axially product 43–49.
Synthesis of products 52–92
Under N2 atmosphere, a screw-cap vial (8 mL) was charged with disubstituted benzocyclobutenone (0.1 mmol, 1.0 equiv), dicarbonyl compound (0.12 mmol, 1.2 equiv), [Rh(COD)2]OTf (0.008 mmol, 8 mol%), L8 (0.008 mmol, 8 mol%) and THF (anhydrous, 1 mL). After stirred at 100 °C for 16 h, the reaction mixture was filtered, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel (petroleum ether/ethyl acetate = 3/1) to afford the chiral products 52–92.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data generated in this study are provided in the Supplementary Information file. The experimental procedures, data of NMR, HRMS, and HPLC have been deposited in the Supplementary Information file. The crystallographic data for the structures reported in this study are available in the Cambridge Crystallographic Data Center under deposition numbers CCDC 2360303 (16), 2360305 (36), 2360304 (37), 2384769 ((rac)−45), 2371858 (52’-H), 2360308 (89), 2360232 (90), 2360307 (91) and 2360306 (95). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/.
References
Huffman, M. A. & Liebeskind, L. S. Nickel(0)-catalyzed synthesis of substituted phenols from cyclobutenones and alkynes. J. Am. Chem. Soc. 113, 2771–2772 (1991).
Jun, C.-H. & Lee, H. Catalytic carbon−carbon bond activation of unstrained ketone by soluble transition-metal complex. J. Am. Chem. Soc. 121, 880–881 (1999).
Jun, C.-H., Lee, H. & Lim, S.-G. The C−C bond activation and skeletal rearrangement of cycloalkanone imine by Rh(I) catalysts. J. Am. Chem. Soc. 123, 751–752 (2001).
Murakami, M., Itahashi, T. & Ito, Y. Catalyzed intramolecular Olefin insertion into a carbon−carbon single bond. J. Am. Chem. Soc. 124, 13976–13977 (2002).
Matsuda, T., Fujimoto, A., Ishibashi, M. & Murakami, M. Eight-membered ring formation via Olefin insertion into a carbon–carbon single bond. Chem. Lett. 33, 876–877 (2004).
Murakami, M., Ashida, S. & Matsuda, T. Nickel-catalyzed intermolecular alkyne insertion into cyclobutanones. J. Am. Chem. Soc. 127, 6932–6933 (2005).
Murakami, M., Ashida, S. & Matsuda, T. Eight-membered ring construction by [4 + 2 + 2] annulationinvolving β-carbon elimination. J. Am. Chem. Soc. 128, 2166–2167 (2006).
Bishop, K. C. I. Transition metal catalyzed rearrangements of small ring organic molecules. Chem. Rev. 76, 461–486 (1976).
Mitsudo, T. K. Teruyuki. Ruthenium complex-catalyzed formation and cleavage of carbon-carbon σ-bonds. On the requirement of highly qualified tuning of the reaction conditions. Synlett 2001, 0309–0321 (2001).
Kondo, T. & Mitsudo, T. Ruthenium-catalyzed Reconstructive Synthesis of Functional Organic Molecules via Cleavage of Carbon–Carbon Bonds. Chem. Lett. 34, 1462–1467 (2005).
Huang, C.-Y. & Doyle, A. G. The chemistry of transition metals with three-membered ring heterocycles. Chem. Rev. 114, 8153–8198 (2014).
Souillart, L. & Cramer, N. Catalytic C–C Bond activations via oxidative addition to transition metals. Chem. Rev. 115, 9410–9464 (2015).
Chen, P. & Dong, G. Cyclobutenones and Benzocyclobutenones: Versatile Synthons in Organic Synthesis. Chem. Eur. J. 22, 18290–18315 (2016).
Murakami, M. & Ishida, N. Potential of metal-catalyzed C–C single bond cleavage for organic synthesis. J. Am. Chem. Soc. 138, 13759–13769 (2016).
Chen, P., Billett, B. A., Tsukamoto, T. & Dong, G. Cut and Sew” transformations via transition-metal-catalyzed carbon–carbon bond activation. ACS Catal. 7, 1340–1360 (2017).
Fumagalli, G., Stanton, S. & Bower, J. F. Recent methodologies that exploit C–C single-bond cleavage of strained ring systems by transition metal complexes. Chem. Rev. 117, 9404–9432 (2017).
Liu, Q.-S. et al. Ni–Al Bimetallic catalyzed enantioselective cycloaddition of cyclopropyl carboxamide with alkyne. J. Am. Chem. Soc. 139, 18150–18153 (2017).
Onodera, S., Togashi, R., Ishikawa, S., Kochi, T. & Kakiuchi, F. Catalytic, directed C–C bond functionalization of styrenes. J. Am. Chem. Soc. 142, 7345–7349 (2020).
Murakami, M. & Ishida, N. Cleavage of carbon–carbon σ-bonds of four-membered rings. Chem. Rev. 121, 264–299 (2021).
Ochi, S., Zhang, Z., Xia, Y. & Dong, G. Rhodium-catalyzed (4+1) cycloaddition between benzocyclobutenones and styrene-type alkenes. Angew. Chem. Int. Ed. 61, e202202703 (2022).
Xu, T. & Dong, G. Rhodium-catalyzed regioselective carboacylation of Olefins: A C–C bond activation approach for accessing fused-ring systems. Angew. Chem. Int. Ed. 51, 7567–7571 (2012).
Xu, T., Ko, H. M., Savage, N. A. & Dong, G. Highly enantioselective Rh-catalyzed carboacylation of Olefins: Efficient syntheses of chiral poly-fused rings. J. Am. Chem. Soc. 134, 20005–20008 (2012).
Chen, P., Xu, T. & Dong, G. Divergent syntheses of fused β-naphthol and indene scaffolds by rhodium-catalyzed direct and decarbonylative alkyne–benzocyclobutenone couplings. Angew. Chem. Int. Ed. 53, 1674–1678 (2014).
Xu, T. & Dong, G. Coupling of sterically hindered trisubstituted Olefins and benzocyclobutenones by C–C activation: Total synthesis and structural revision of cycloinumakiol. Angew. Chem. Int. Ed. 53, 10733–10736 (2014).
Xu, T., Savage, N. A. & Dong, G. Rhodium(I)-catalyzed decarbonylative spirocyclization through C–C bond cleavage of benzocyclobutenones: An efficient approach to functionalized spirocycles. Angew. Chem. Int. Ed. 53, 1891–1895 (2014).
Chen, P., Sieber, J., Senanayake, C. H. & Dong, G. Rh-catalyzed reagent-free ring expansion of cyclobutenones and benzocyclobutenones. Chem. Sci. 6, 5440–5445 (2015).
Lu, G., Fang, C., Xu, T., Dong, G. & Liu, P. Computational study of Rh-catalyzed carboacylation of olefins: Ligand-promoted rhodacyclen isomerization enables regioselective C–C bond functionalization of benzocyclobutenones. J. Am. Chem. Soc. 137, 8274–8283 (2015).
Lu, Q., Wang, B., Yu, H. & Fu, Y. Mechanistic study on ligand-controlled Rh(I)-catalyzed coupling reaction of alkene-benzocyclobutenone. ACS Catal. 5, 4881–4889 (2015).
Shaw, M. H. & Bower, J. F. Synthesis and applications of rhodacyclopentanones derived from C–C bond activation. Chem. Commun. 52, 10817–10829 (2016).
Deng, L., Chen, M. & Dong, G. Concise synthesis of (−)-cycloclavine and (−)−5-epi-cycloclavine via asymmetric C–C activation. J. Am. Chem. Soc. 140, 9652–9658 (2018).
Guo, J.-H. et al. Site-selective C–C cleavage of benzocyclobutenones enabled by a blocking strategy using nickel catalysis. Angew. Chem. Int. Ed. 60, 19079–19084 (2021).
Hou, S.-H., Prichina, A. Y. & Dong, G. Deconstructive asymmetric total synthesis of morphine-family alkaloid (−)-Thebainone A. Angew. Chem. Int. Ed. 60, 13057–13064 (2021).
Huynh, N. O., Hodík, T. & Krische, M. J. Enantioselective transfer hydrogenative cycloaddition unlocks the total synthesis of SF2446 B3: An aglycone of arenimycin and SF2446 type II polyketide antibiotics. J. Am. Chem. Soc. 145, 17461–17467 (2023).
Jiang, C. et al. Type I [4σ+4π] versus [4σ+4π−1] cycloaddition to access medium-sized carbocycles and discovery of a liver X receptor β-selective ligand. Angew. Chem. Int. Ed. 63, e202405838 (2024).
Okumura, S., Sun, F., Ishida, N. & Murakami, M. Palladium-catalyzed intermolecular exchange between C–C and C–Si σ-bonds. J. Am. Chem. Soc. 139, 12414–12417 (2017).
Zhu, Z. et al. Cobalt-catalyzed intramolecular alkyne/benzocyclobutenone coupling: C–C Bond cleavage via a tetrahedral dicobalt intermediate. ACS Catal. 8, 845–849 (2018).
Lu, H. et al. Divergent coupling of benzocyclobutenones with indoles via C−H and C−C activations. Angew. Chem. Int. Ed. 59, 23537–23543 (2020).
Li, R. et al. A ring expansion strategy towards diverse azaheterocycles. Nat. Chem. 13, 1006–1016 (2021).
Zhang, J., Wang, X. & Xu, T. Regioselective activation of benzocyclobutenones and dienamides lead to anti-Bredt bridged-ring systems by a [4+4] cycloaddition. Nat. Commun. 12, 3022 (2021).
Ano, Y., Takahashi, D., Yamada, Y. & Chatani, N. Palladium-catalyzed skeletal rearrangement of cyclobutanones via C–H and C–C bond cleavage. ACS Catal. 13, 2234–2239 (2023).
Li, R., Shi, X. & Zhao, D. Palladium-catalyzed skeletal reorganization of cyclobutanones invoving successive C—C bond/C—H bond cleavage. Chin. J. Chem. 41, 1679–1683 (2023).
Xue, Y. & Dong, G. Deconstructive synthesis of bridged and fused rings via transition-metal-catalyzed “Cut-and-Sew” reactions of benzocyclobutenones and cyclobutanones. Acc. Chem. Res. 55, 2341–2354 (2022).
Deng, L., Xu, T., Li, H. & Dong, G. Enantioselective Rh-catalyzed carboacylation of C═N bonds via C–C activation of benzocyclobutenones. J. Am. Chem. Soc. 138, 369–374 (2016).
Qiu, B. et al. Catalytic enantioselective synthesis of 3,4-polyfused oxindoles with quaternary all-carbon stereocenters: A Rh-catalyzed C–C activation approach. Org. Lett. 20, 7689–7693 (2018).
Zhang, Y., Shen, S., Fang, H. & Xu, T. Total synthesis of galanthamine and lycoramine featuring an early-stage C–C and a late-stage dehydrogenation via C–H activation. Org. Lett. 22, 1244–1248 (2020).
Li, X. et al. Divergent Rh catalysis: Asymmetric dearomatization versus C–H activation initiated by C–C activation. ACS Catal. 13, 4873–4881 (2023).
Souillart, L. & Cramer, N. Highly enantioselective rhodium(I)-catalyzed carbonyl carboacylations initiated by C–C bond activation. Angew. Chem. Int. Ed. 53, 9640–9644 (2014).
Zhou, X. & Dong, G. Nickel-catalyzed chemo- and enantioselective coupling between cyclobutanones and allenes: Rapid synthesis of [3.2.2] bicycles. Angew. Chem. Int. Ed. 55, 15091–15095 (2016).
Hou, S.-H., Yu, X., Zhang, R., Wagner, C. & Dong, G. Rhodium-catalyzed diastereo- and enantioselective divergent annulations between cyclobutanones and 1,5-enynes: Rapid construction of complex C(sp3)-rich scaffolds. J. Am. Chem. Soc. 144, 22159–22169 (2022).
Ambler, B. R. et al. Enantioselective ruthenium-catalyzed benzocyclobutenone–ketol cycloaddition: Merging C–C bond activation and transfer hydrogenative coupling for type II polyketide construction. J. Am. Chem. Soc. 140, 9091–9094 (2018).
Tan, F. et al. Catalytic asymmetric homologation of ketones with α-alkyl α-diazo esters. J. Am. Chem. Soc. 143, 2394–2402 (2021).
Takano, H., Shiozawa, N., Imai, Y., Kanyiva, K. S. & Shibata, T. Catalytic enantioselective synthesis of axially chiral polycyclic aromatic hydrocarbons (PAHs) via regioselective C–C bond activation of biphenylenes. J. Am. Chem. Soc. 142, 4714–4722 (2020).
Shibata, T., Shiozawa, N., Nishibe, S., Takano, H. & Maeda, S. Pt(ii)-Chiral diene-catalyzed enantioselective formal [4 + 2] cycloaddition initiated by C–C bond cleavage and elucidation of a Pt(ii)/(iv) cycle by DFT calculations. Org. Chem. Front. 8, 6985–6991 (2021).
Ju, C.-W. et al. Evolution of organic phosphor through precision regulation of nonradiative decay. Proc. Natl. Acad. Sci. USA 120, e2310883120 (2023).
Cheng, J. K., Xiang, S.-H., Li, S., Ye, L. & Tan, B. Recent advances in catalytic asymmetric construction of atropisomers. Chem. Rev. 121, 4805–4902 (2021).
Zhang, H.-H. & Shi, F. Organocatalytic atroposelective synthesis of indole derivatives bearing axial chirality: Strategies and applications. Acc. Chem. Res. 55, 2562–2580 (2022).
Qian, P.-F., Zhou, T. & Shi, B.-F. Transition-metal-catalyzed atroposelective synthesis of axially chiral styrenes. Chem. Commun. 59, 12669–12684 (2023).
Zhu, X.-Q., Li, Y.-C. & Ye, L.-W. Enantioselective reaction of diynes and multiynes for the synthesis of axially chiral compounds. Asian J. Org. Chem. 13, e202300554 (2024).
Zou, J.-Y., Wan-Yi, X., Wang, J., Liu, Q. & He, Y. Atroposelective construction of tetrasubstituted axially chiral alkene frameworks. Synthesis 56, 1862–1872 (2024).
Yang, J. et al. Ir/Zn-cocatalyzed chemo- and atroposelective [2+2+2] cycloaddition for construction of C─N axially chiral indoles and pyrroles. Sci. Adv. 9, eadk1704 (2023).
Ma, X. et al. Modular assembly of versatile tetrasubstituted alkenyl monohalides from alkynyl tetracoordinate borons. Chem 9, 1164–1181 (2023).
Li, W. et al. Synthesis of axially chiral alkenylboronates through combined copper- and palladium-catalysed atroposelective arylboration of alkynes. Nat. Synth. 2, 140–151 (2023).
Li, Z.-H., Li, Q.-Z., Bai, H.-Y. & Zhang, S.-Y. Synthetic strategies and mechanistic studies of axially chiral styrenes. Chem. Catal. 3, 100594 (2023).
Mi, R. et al. Rhodium-catalyzed atroposelective access to axially chiral olefins via C−H bond activation and directing group migration. Angew. Chem. Int. Ed. 61, e202111860 (2022).
Li, Q.-Z., Li, Z.-H., Kang, J.-C., Ding, T.-M. & Zhang, S.-Y. Ni-catalyzed, enantioselective three-component radical relayed reductive coupling of alkynes: Synthesis of axially chiral styrenes. Chem. Catal. 2, 3185–3195 (2022).
Lin, Z., Hu, W., Zhang, L. & Wang, C. Nickel-catalyzed asymmetric cross-electrophile trans-aryl-benzylation of α-naphthyl propargylic alcohols. ACS Catal. 13, 6795–6803 (2023).
Fu, L., Chen, X., Fan, W., Chen, P. & Liu, G. Copper-catalyzed asymmetric functionalization of vinyl radicals for the access to vinylarene atropisomers. J. Am. Chem. Soc. 145, 13476–13483 (2023).
Nishida, G., Noguchi, K., Hirano, M. & Tanaka, K. Asymmetric assembly of aromatic rings to produce tetra-ortho-substituted axially chiral biaryl phosphorus Compounds. Angew. Chem. Int. Ed. 46, 3951–3954 (2007).
Nishida, G. et al. Practical enantioselective synthesis of axially chiral biaryl diphosphonates and Dicarboxylates by cationic rhodium(I)/Segphos-catalyzed double [2 + 2 + 2] cycloaddition. Org. Lett. 10, 2849–2852 (2008).
Yokose, D., Nagashima, Y., Kinoshita, S., Nogami, J. & Tanaka, K. Enantioselective synthesis of axially chiral styrene-carboxylic esters by rhodium-catalyzed chelation-controlled [2+2+2] cycloaddition. Angew. Chem. Int. Ed. 61, e202202542 (2022).
Tanaka, K. Transition-metal-catalyzed enantioselective [2+2+2] cycloadditions for the synthesis of axially chiral biaryls. Chem. Asian J. 4, 508–518 (2009).
Auvinet, A.-L. & Harrity, J. P. A. A nickel-catalyzed benzannulation approach to aromatic boronic esters. Angew. Chem. Int. Ed. 50, 2769–2772 (2011).
Juliá-Hernández, F., Ziadi, A., Nishimura, A. & Martin, R. Nickel-catalyzed chemo-, regio- and diastereoselective bond formation through proximal C–C cleavage of benzocyclobutenones. Angew. Chem. Int. Ed. 54, 9537–9541 (2015).
Wang, Y., Ma, P., Ma, N. & Wang, J. Ligand-controlled nickel-catalyzed reactions of benzocyclobutenones with alkynyltrifluoroborates: Diverse construction of polysubstituted naphthols. Org. Lett. 25, 3527–3532 (2023).
Tang, X. et al. Ligand-controlled regiodivergent ring expansion of benzosilacyclobutenes with alkynes en route to axially chiral silacyclohexenyl arenes. J. Am. Chem. Soc. 146, 26639–26648 (2024).
Hong, F.-L. & Ye, L.-W. Transition metal-catalyzed tandem reactions of Ynamides for divergent N-heterocycle synthesis. Acc. Chem. Res. 53, 2003–2019 (2020).
Tanaka, K., Takeishi, K. & Noguchi, K. Enantioselective synthesis of axially chiral anilides through rhodium-catalyzed [2+2+2] cycloaddition of 1,6-diynes with trimethylsilylynamides. J. Am. Chem. Soc. 128, 4586–4587 (2006).
Jia, S. et al. Organocatalytic enantioselective construction of axially chiral sulfone-containing styrenes. J. Am. Chem. Soc. 140, 7056–7060 (2018).
Sun, T. et al. Rhodium(I)-catalyzed carboacylation/aromatization cascade initiated by regioselective C−C activation of benzocyclobutenones. Angew. Chem. Int. Ed. 57, 2859–2863 (2018).
Yang, L. K. et al. Catalytic asymmetric photocycloaddition of triplet aldehydes with benzocyclobutenones. CCS Chem. 7, 573–581 (2024).
Wurm, T., Turnbull, B. W. H., Ambler, B. R. & Krische, M. J. Thermal hetero-diels–alder reaction of benzocyclobutenones with isatins to form 2-oxindole spirolactones. J. Org. Chem. 82, 13751–13755 (2017).
Zhou, F., Ding, M. & Zhou, J. A catalytic metal-free Ritter reaction to 3-substituted 3-aminooxindoles. Org. Biomol. Chem. 10, 3178–3181 (2012).
Ji, D. et al. Palladium-catalyzed asymmetric hydrophosphination of internal alkynes: Atroposelective access to phosphine-functionalized Olefins. Chem 8, 3346–3362 (2022).
Zhan, L.-W., Lu, C.-J., Feng, J. & Liu, R.-R. Atroposelective synthesis of C−N vinylindole atropisomers by palladium-catalyzed asymmetric hydroarylation of 1-alkynylindoles. Angew. Chem. Int. Ed. 62, e202312930 (2023).
Acknowledgements
Financial support from the National Key R&D Program of China (no. 2022YFA1503104), the National Natural Science Foundation of China (Nos. 22371175, 22101167, and 22073066) and the SNNU. The authors thank Dr. Xiao-Xi Li for calculations of the ECD spectrum of the compound 46.
Author information
Authors and Affiliations
Contributions
H.L., Z.Q., W.W., and F.W. conducted the experimental studies. Z.-W.D. and A.-Q.J. performed the biological studies. Y.S. conducted the computational studies under the supervision of G.H. X.L. conceived and directed the project, and acquired the research funding. All authors participated in the manuscript writing and discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, H., Qi, Z., Shi, Y. et al. Catalytic asymmetric intermolecular [4 + 2] annulation of benzocyclobutenones with Alkynes and activated carbonyls via C—C activation. Nat Commun 16, 5741 (2025). https://doi.org/10.1038/s41467-025-60109-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60109-5
