
Abstract
Glioblastoma (GB) is a highly aggressive brain tumor resistant to chemoradiotherapy, largely due to glioma stem-like cells (GSCs) with robust DNA damage repair capabilities. Here we reveal that GSCs enhance their DNA repair capacity by activating non-homologous end-joining (NHEJ) through upregulation of the apoptosis antagonizing transcription factor (AATF), thereby promoting therapeutic resistance in GB. AATF interacts with XRCC4, a core NHEJ subunit, preventing its degradation via ubiquitin-mediated proteasomal processes. Upon DNA damage, AATF undergoes phosphorylation at Ser189 by ATM, leading to its dissociation from XRCC4 and rapid recruitment of XRCC4 to DNA break sites for efficient NHEJ repair. Moreover, AATF depletion or deficient AATF phosphorylation impedes NHEJ in GSCs, sensitizing GB xenografts to chemoradiotherapy. Additionally, elevated levels of AATF inform poor prognosis in GB patients. Collectively, our findings unveil a crucial role of AATF in XRCC4-mediated NHEJ repair, and underscore targeting AATF as a potential strategy to overcome GB resistance to chemoradiotherapy.
Similar content being viewed by others


Introduction
Glioblastoma (GB) is an incurable primary brain tumor with dismal prognosis1,2. While chemoradiotherapy is the most effective non-surgical treatment for GB patients, therapeutic resistance is universal1. Glioma stem-like cells (GSCs) contribute to therapeutic resistance by efficiently repairing DNA damage and maintaining genomic stability, allowing them to survive and grow despite DNA damage3,4,5,6,7,8. However, the molecular mechanism underlying GSCs’ ability to repair DNA damage remains incompletely understood, presenting a major obstacle in improving GB treatment outcomes. A deeper understanding of the DNA damage repair features of GSCs can offer insights into glioma biology and new treatment possibilities for this devastating disease.
Chemoradiotherapy induces various types of DNA damage, with DNA double-strand breaks (DSBs) being particularly detrimental as they can result in chromosomal abnormalities, genomic instability, and cell death if left unrepaired9,10. There are two major pathways for repair of DSBs: homologous recombination (HR) and non-homologous end joining (NHEJ)11,12,13. HR is an error free process that occurs in the S and G2 phases of the cell cycle. In contrast, NHEJ, which is an error-prone and end-to-end ligation process, can occur at any time during the cell cycle12 and considered the primary mechanism for repairing DSBs in human cells13,14. Upon encountering DSBs, NHEJ is initiated by the binding of the Ku70/80 heterodimer to the free DNA ends, facilitating the assembly of essential NHEJ components such as DNA-dependent protein kinase catalytic subunit (DNA-PKcs), X-ray repair cross-complementing protein 4 (XRCC4), and DNA ligase IV (LIG4). The Ku-DNA-PKcs complex plays a crucial role in bringing together the broken DNA ends. Subsequently, XRCC4 forms a complex with and stabilizes LIG4, which is subsequently recruited to DNA ends to seal the broken DNA12,13,14,15. However, the regulation of XRCC4 protein and its fate after translation but before recruitment to DSBs are poorly understood. Although deleterious variants of NHEJ genes increase the risk of GB16,17,18, NHEJ regulation in GB therapeutic resistance warrants additional investigation.
In this work, we demonstrate that GSCs repair DNA damage efficiently by activating the NHEJ pathway through increasing apoptosis antagonizing transcription factor (AATF) levels. We find that AATF binds to XRCC4, the core component of the NHEJ complex, and prevents its proteasomal degradation. In response to DSBs, ATM phosphorylates AATF, causing its dissociation form XRCC4 and allowing the rapid recruitment of XRCC4 to DSB sites to initiate NHEJ repair, thus promoting GSCs’ therapeutic resistance. Therefore, these findings identify AATF as a regulator for XRCC4-mediated NHEJ pathway, suggesting that targeting NHEJ may aid in overcoming GB resistance to chemoradiotherapy.
Results
GSCs exhibit strong capability to repair DNA damage
To investigate the DNA damage repair capability of glioma stem-like cells (GSCs), we utilized preclinical orthotopic GB xenografts. The GSCs were obtained from GB patients and were functionally characterized in previous studies19,20,21,22. We intracranially implanted GSCs into BALB/c nude mice to established orthotopic GB xenografts for twenty days, and treated them with ionizing radiation (IR) (3 Gy) to simulate radiotherapy. Immunohistochemical (IHC) staining demonstrated the typical pathological features of GB, including hyperchromatism, pleomorphism, extensive necrosis, and proliferating blood vessels (Supplementary Fig. 1a). We performed co-immunofluorescence (co-IF) staining with SOX2, a stem cell marker, and histone H2AX phosphorylation (γ-H2AX), a DNA damage marker, in GB xenografts treated with IR for 24 h. The results showed that SOX2+ cells (enriched in GSCs) accumulated less γ-H2AX induced by IR compared to SOX2− cells (Fig. 1a and Supplementary Fig. 1b). Moreover, co-IF staining with SOX2 and cleaved-caspase 3 (CCP3) revealed that less apoptosis was occurred in SOX2+ cells in GB xenografts treated with IR (Supplementary Fig. 1c). In human GB specimens, the SOX2+ cells had lower γ-H2AX accumulation compared to SOX2− cells in tumors (Supplementary Fig. 1d), validating that GSCs are less susceptible to DNA damage than differentiated GB cells (DGCs).

a Co-immunofluorescent (co-IF) staining of γ-H2AX and SOX2 in orthotopic GB xenografts treated with IR (3 Gy) for 24 h. Nuclei were counterstained with Hoechst (blue). Left: representative images; scale bars, 20 μm. Middle: quantifications of γ-H2AX foci, each point represents a single cell (n = 4, biologically independent samples). Right: the percentages of γ-H2AX+ cells, each point represents a biologically independent sample. b IF staining of γ-H2AX in 4121 GSCs and matched DGCs treated with IR (3 Gy). Nuclei were counterstained with Hoechst (blue). Left: representative images; scale bars, 10 μm. Right: quantifications of γ-H2AX foci, each point represents a single cell (n = 3, biologically independent experiments). c, d Schematic model for LC-MS/MS identifying differentially expressed proteins (DEPs) in GSCs and GB cells. Venn diagram (c) and heatmap analysis (d) of DEPs in GB cells vs. NHAs and GSCs vs. DGCs (p < 0.01, fold change > 2.0). e Immunoblot (IB) analysis of γ-H2AX and AATF levels in GSCs and NHA. Cells transduced with shNT or shAATFs (independent sequences). f IF staining of γ-H2AX in 4121 GSCs transduced with shNT or shAATFs. Nuclei were counterstained with Hoechst (blue). Left: representative images; Scale bars, 10 μm. Right: quantifications of γ-H2AX foci, each point represents a single cell (n = 3, biologically independent experiments). g, h IB analysis of expression of indicated proteins in GSCs and their matched DGCs (g), in GSCs, primary GB cells, NPCs, NSCs, and NHAs (h). i Tumor cells isolated from three GB patient samples. CD133+ and CD133- cells sorted using magnetic microbeads (left). IB analysis of AATF and XRCC4 in sorted cells (right). j, k Co-IF staining of AATF and SOX2 (j), or γ-H2AX (k) in 3 human GB specimens. Nuclei were counterstained with Hoechst (blue). Left: representative images; Scale bars, 20 μm. Right: quantifications of γ-H2AX or AATF intensity, each point represents a single cell (n = 3, biologically independent samples). l Immunohistochemical (IHC) analysis of AATF and γ-H2AX in human GB specimens. Left: representative images; Scale bars, 50 μm. Right: Pearson correlation (two-sided) analysis of AATF and γ-H2AX IHC scores in 15 GB samples. Labels hGB#3, hGB#28, hGB#35, and hGB#68 refer to individual GB tumor specimens. For IB, protein levels were normalized to GAPDH or Tubulin. Independent experiments were performed three times with similar results (e, g, h, i). Data represent mean ± SD of at least three independent experiments (a, b, f, j, k). Unpaired two-sided Student’s t-test. Source data are provided as a Source Data file.
We next separated untreated GB xenografts into GSCs and DGCs using magnetic sorting for CD133, a surface marker commonly expressed in GSCs23, and exposed these two cell populations to IR. IF staining showed that IR treatment caused an accumulation of γ-H2AX foci in both cell populations within 30 min (Fig. 1b). However, the γ-H2AX levels returned to the pre-IR basal levels within 8 h in GSCs, while they remained elevated in DGCs (Fig. 1b), suggesting an efficient DNA damage repair in GSCs. This observation was further supported by IB analysis in GSCs and DGCs treated with IR (Supplementary Fig. 1e). These results suggest that GSCs possess strong DNA damage repair capability, which may contribute to their resistance to treatment.
AATF is highly expressed and involved in DNA damage repair in GSCs
To investigate the potential mechanisms responsible for the strong DNA damage repair capability of GSCs, we conducted a screening using liquid chromatography tandem mass spectrometry (LC-MS/MS) on proteins specifically expressed in GSCs and GBs. Two GSCs and their corresponding DGCs, as well as four freshly dissociated human GB cells and non-malignant immortalized normal human astrocytes (NHAs), were included in the analysis (Fig. 1c). We identified 7 proteins consistently elevated, and 11 proteins consistently decreased, in GSCs and GB cells compared to DGCs and NHAs (Fig. 1d and Supplementary data 1). To determine the role of the upregulated proteins in DNA damage repair capability in GSCs, we employed small hairpin RNA sequences (shRNAs) to knock down the expression of 7 elevated proteins in GSCs. IB analysis revealed that knockdown of AATF resulted in a significant accumulation of γ-H2AX in GSCs compared to cells expressing non-targeting shRNA (shNT) (Fig. 1e). However, among the other 6 proteins, only knockdown of PNPH slightly increased the γ-H2AX levels (Supplementary Fig. 1f), indicating that AATF may be involved in DNA damage repair in GSCs.
AATF (apoptosis antagonizing transcription factor) is an evolutionarily conserved RNA polymerase II-binding protein. Studies have indicated that AATF may regulate DNA damage response (DDR), but the underlying molecular mechanism remains elusive24,25. We conducted IF analysis and found that AATF-KD GSCs exhibited an increase in γ-H2AX foci compared to shNT cells (Fig. 1f). Neutral comet assays confirmed that AATF depletion resulted in DNA damage, as evidenced by an increase in the percentage of cells with comet tails observed in AATF-KD GSCs (Supplementary Fig. 1g). Furthermore, to evaluate whether the effects of AATF on DNA damage have broader relevance across different cell types, we depleted AATF expression in U2OS (osteosarcoma), HCT116 (colon cancer), Capan-1 (pancreatic cancer), and HEK293 cells. IB analysis showed that depletion of AATF caused a consistent increase in γ-H2AX levels in these cells (Supplementary Fig. 2a). Additionally, we compared the kinetics of accumulations of γ-H2AX and cleaved caspase-3 in AATF-KD GSCs. Knockdown of AATF led to an accumulation of γ-H2AX at 12 h post-AATF-KD, while an increase in cleaved caspase-3 levels was observed from 36 h post-AATF-KD (Supplementary Fig. 2b), indicating that the increased DNA damage induced by AATF depletion is not an indirect consequence of the elevated apoptosis.
Notably, IB analysis revealed that AATF protein levels were consistently elevated in multiple GSC lines compared to the matched DGCs (Fig. 1g). Additionally, GSCs and primary GB cells, which were isolated from human primary GB specimens then sorted using the surface marker CD133 and cultured in serum-free stem cell medium, exhibited higher AATF protein levels compared to human neural stem cells (NSCs), neural progenitor cells (NPCs), and normal human astrocytes (NHA) (Fig. 1h). However, qPCR analysis showed that AATF mRNA levels were not consistently upregulated in GSCs or GB cells compared to their respective controls (Supplementary Fig. 2c, d), indicating that the increase in AATF protein may be regulated post-transcriptionally. The upregulation of AATF protein in GSCs was further confirmed by IB analysis in CD133+ cells compared to CD133− cells freshly sorted from three human GB specimens (Fig. 1i). Moreover, co-IF staining of human GB specimens revealed that AATF was highly expressed in SOX2+ cells, but was low in GFAP+ (glial fibrillary acidic protein) cells (Fig. 1j and Supplementary Fig. 2e). In contrast, CD45+ (immune cells), CD3+ (T cells), CD68+ (macrophages) and CD31+ (vascular endothelial cells) cells were negative for AATF expression (Supplementary Fig. 2e), suggesting that AATF is primarily expressed in GSCs.
To investigate the correlation between AATF expression and DNA damage in GB tissues in vivo, we performed co-IF staining and found that IR-induced γ-H2AX occurred less frequently in AATF+ cells compared to AATF− cells in mouse GB xenografts (Supplementary Fig. 2f). In human GB tissues, AATF+ cells exhibited lower levels of γ-H2AX than AATF− cells (Fig. 1k). Moreover, IHC staining with the serial sections from 15 human GB specimens revealed an inverse correlation between AATF expression and γ-H2AX levels (Fig. 1l). Together, our findings suggest that AATF is highly expressed in GSCs and may play a role in promoting DNA damage repair in GSCs.
AATF is required for efficient DNA damage repair in GSCs
To investigate the role of AATF in DNA damage repair in GSCs, we treated shNT and AATF-KD GSCs with IR. IB analysis showed that the accumulation of γ-H2AX induced by IR was eliminated in shNT GSCs after 1 h, while AATF-KD GSCs exhibited a continuous increase in γ-H2AX levels (Fig. 2a). IF staining confirmed the significant elevation of γ-H2AX foci in AATF-KD GSCs following IR exposure (Fig. 2b), suggesting that AATF depletion inhibits DNA damage repair. Concordantly, the expression of Flag-tagged AATF, but not a Flag-vector, decreased γ-H2AX levels in GSCs following IR treatment (Fig. 2c, d). Additionally, ectopic expression of Flag-AATF in DGCs, which express low endogenous AATF, also diminished γ-H2AX accumulation in response to IR (Supplementary Fig. 3a, b). These results suggest that high levels of AATF promotes efficient DNA damage repair.

a–d 4121 GSCs transduced with shNT or shAATFs (a, b), or Flag-vector or Flag-AATF (c, d), were treated with IR (3 Gy) for indicated time points. IB (a, c) and IF (b, d) analyses of γ-H2AX are shown. Nuclei counterstained with Hoechst (blue). Left: representative images; Scale bars (b, d), 10 μm. Right: quantifications of γ-H2AX, each point represents a single cell (n = 3, biologically independent experiments). e 4121 GSCs transduced with shNT or shAATF#1 (red), or Flag-vector or Flag-AATF (green), were treated with increasing IR dose for 48 h or 72 h. Cell viability assessed by CellTiter-Glo assay. (Two-way ANOVA). f–j BALB/c nude mice intracranially implanted with GSCs transduced with Luciferase and Tet-on-inducible shNT or shAATF. Mice treated with control, IR (3 Gy, weekly, 3 times), Dox (1 mg/mL in water), or combined treatment from day 9 post-implantation (f, g, top). GB xenografts tracked by bioluminescence; representative images and quantifications are shown (f, g, n = 6, mean ± SD; Unpaired two-sided Student’s t-test). Kaplan–Meier survival plots of mice are shown (h, Log-rank Mantel–Cox test). Co-IF staining of SOX2 with γ-H2AX (i) or cleaved-caspase3 (CCP3) (j) in GB xenografts are shown. Left: representative images; Scale bars, 20 μm. Right: quantifications of γ-H2AX or CCP3 intensity, each point represents a single cell (n = 4, biologically independent samples). k 4121 GSCs transduced with shNT or shAATFs were treated with TMZ (50 μM) for indicated time points. IB analysis of indicated proteins are shown. l, m BALB/c nude mice intracranially implanted with GSCs (Luciferase) transduced with Tet-on-inducible-shNT or shAATF. Mice treated with control, TMZ (40 mg/kg, every 2 days, 5 times), Dox (1 mg/mL in water), or combined treatment from day 9 post-implantation (l, top). GB xenografts tracked by bioluminescence; representative images and quantifications are shown (l, n = 6 mice for each group; mean ± SD; Unpaired two-sided Student’s t-test). Kaplan–Meier survival plots of mice are shown (m, Log-rank Mantel–Cox test). For IB, protein levels were normalized to GAPDH. Independent experiments were performed three times with similar results. The curves represent the relative expression of γ-H2AX protein (a, c, k). Data represent mean ± SD of three independent experiments (a–e, i–k). Unpaired two-sided Student’s t-test. Source data are provided as a Source Data file.
To examine whether AATF is necessary for GSC resistance to IR, we conducted a cell survival analysis. The CellTiter-Glo assay revealed that AATF-KD GSCs showed reduced cell survival upon IR exposure compared to shNT GSCs (Fig. 2e, left). Reciprocally, both GSCs and DGCs expressing Flag-AATF were more resistant to IR treatment than cells expressing Flag-vector (Fig. 2e, right and Supplementary Fig. 3c). Moreover, the combined AATF-KD and IR treatment increased GSC apoptosis compared to each monotherapy, as evaluated by flow cytometry. Conversely, GSCs expressing Flag-AATF showed a decrease in GSC apoptosis induced by IR (Supplementary Fig. 3d). Together, these results suggest that high levels of AATF promote radioresistance of GSCs.
Targeting AATF sensitizes GB to chemoradiotherapy
To investigate whether targeting AATF enhances GB sensitivity to radiotherapy, we established orthotopic GB tumors by transplanting GSCs expressing luciferase and doxycycline (Dox)-inducible shAATF into the brains of nude mice. On day 9 after transplantation, we randomly divided the tumor-bearing mice into groups and treated them with Dox (daily in drinking water), IR (3 Gy, once a week, three times), or a combination treatment (Fig. 2f, g, left) Bioluminescence imaging (BLI) revealed that inducible-KD of AATF inhibited tumor growth compared to the shNT group mice (Fig. 2f, g and Supplementary Fig. 3e) and extended survival in mice bearing either of two GB patient-derived xenografts (Fig. 2h). IR monotherapy also inhibited GB tumor growth (Fig. 2f, g). Notably, the combination therapy showed superior antitumor benefit over either monotherapy and conferred the largest improvement in survival in both GB xenografts (Fig. 2f–h).
To confirm the role of AATF in promoting GSC resistance to DNA damage, we knocked out (KO) AATF expression in GSCs using CRISPR-Cas9 technology. Consistent with our previous findings, AATF-KO using two different sgRNA sequences led to increased γ-H2AX levels in GSCs and sensitized them to IR treatment (Supplementary Fig. 3f, g). Notably, AATF-KO significantly inhibited GB xenograft growth, enhanced the tumor response to IR treatment, and prolonged the survival period of tumor-bearing mice (Supplementary Fig. 3h–j). These results suggest that targeting AATF sensitizes GB to radiotherapy.
To test whether losing AATF increased IR-induced DNA damage in GSCs in vivo, we performed co-IF staining in the GB xenografts tested above. Compared to control group, both inducible-KD of AATF and AATF-KO increased γ-H2AX staining in SOX2+ tumor cells, and the combination therapy led to a further elevation of γ-H2AX levels in SOX2+ tumor cells (Fig. 2i; Supplementary Fig. 3k). Moreover, co-IF staining with antibodies against cleaved-caspase3 (CCP3) and SOX2 revealed that the combination therapy induced a higher rate of GSC apoptosis compared to all other experimental groups (Fig. 2j). These data suggest that targeting AATF inhibits DNA damage repair in GSCs and improves radiotherapy efficacy in GB.
Temozolomide (TMZ) is a critical chemotherapeutic agent induces DNA damage in tumor cells and used to treat GB1,2. To further investigate the role of AATF in cellular response to TMZ, we treated AATF-KD GSCs with TMZ. IB analysis revealed that TMZ treatment increased γ-H2AX levels in shNT GSCs, with a more increase observed in the GSCs with AATF knockdown (Fig. 2k). In addition, in orthotopic GB tumor models, BLI analysis demonstrated that TMZ treatment inhibited tumor growth compared to the control group (Fig. 2l). Notably, combining AATF inducible-KD with TMZ treatment resulted in enhanced antitumor effects compared to TMZ monotherapy and led to the most significant improvement in survival (Fig. 2l, m).
Recurrence of GB is nearly universal, and patients with recurrent GB fare even worse1. To investigate the long-term impact of AATF absence on tumor recurrence after radiotherapy, we established a GB xenograft recurrence model. GSCs were intracranially implanted in nude mice for 12 days, and then the mice were treated with IR (3 Gy, every three days, four times). BLI revealed that tumors were significantly decreased or undetectable after IR treatment at day 22, followed by tumor regrowth starting from day 29, indicating a tumor recurrence (Supplementary Fig. 3l, m). Notably, inducible knockdown of AATF sensitized tumors to IR treatment and delayed tumor recurrence up to day 43 (Supplementary Fig. 3l, m). These results suggest that depletion of AATF enhances GB xenograft sensitivity to radiotherapy and delays long-term tumor recurrence.
AATF interacts with XRCC4 and promotes NHEJ repair
To investigate how AATF promotes DNA damage repair, we conducted an immunoprecipitation (IP) of endogenous AATF and LC-MS analysis to identify potential binding partners of AATF in GSCs. XRCC4 (X-Ray Repair Cross Complementing 4), an essential core component of the non-homologous end-joining (NHEJ) pathway26, was identified as a putative interaction partner of AATF (Supplementary Fig. 4a and Supplementary data 2). Mammalian cells use two major DNA double-strand break (DSB) repair pathways: NHEJ and homologous recombination (HR)12. We first experimentally confirmed AATF-XRCC4 interaction using co-IP experiments with Flag-AATF or Flag-XRCC4 in 293 T cells (Supplementary Fig. 4b). Moreover, an in vitro GST pull-down assay using purified GST-tagged XRCC4 protein revealed that AATF bound to XRCC4 directly (Supplementary Fig. 4c). IP assays with anti-AATF or anti-XRCC4 antibody showed a strong interaction between endogenous AATF and XRCC4 in both GSCs and 293 T cells (Fig. 3a and Supplementary Fig. 4d, e).

a AATF-XRCC4 interaction was analyzed by IP with IgG or anti-AATF antibody in GSCs. IB for indicated proteins are shown. b Schematic of AATF domains (left). Co-IP analysis of AATF-XRCC4 interaction in 293 T cells expressing Flag-AATF mutants and mCherry-XRCC4 (right). c AATF-XRCC4 interaction analyzed by IP with anti-Flag M2 beads in 293 T cells expressing Flag-AATF mutants. d NHEJ repair activity in U2OS cells expressing shNT or shAATFs, transfected with linearized I-Scel NHEJ reporter (EJ5-GFP) (left). HR repair activity assessed using DR-GFP reporter (right). GFP-positive cells, indicating NHEJ and HR repair respectively, analyzed by flow cytometry. e Flag-AATF or Flag-AATF△331–380 mutant was expressed in U2OS cells transduced with shAATF#1. NHEJ activity was assessed as described in (d). f IB showing that full-length AATF, but not △331–380 mutant, decreases γ-H2AX accumulation in GSCs expressing shAATF. g IB showing expression of indicated proteins in GSCs transduced with shNT or shAATFs. h CHX chase analysis of protein degradation in 4121 GSCs transduced with shNT or shAATF#1. XRCC4 levels quantified and normalized to GAPDH. Data presented as mean ± SEM (n = 3, biologically independent experiments). i IB analysis of protein expression in shNT or shAATF#1 GSCs treated with MG132 (10 μM) for 12 h. j IP with anti-XRCC4 antibody and IB with anti-Ub-k48 and anti-Ub antibodies in GSCs (shNT or shAATF#1) treated with MG132. The graph represents the relative levels of Ub (bottom). k Flag-vector or Flag-XRCC4 was expressed in 4121 GSCs transduced with shNT or shAATF#1. The expression of indicated proteins was assessed by IB. l Flag-vector or Flag-XRCC4 was expressed in U2OS transduced with shNT or shAATF#1. The NHEJ activity was assessed as described in (d). For IB, protein levels were normalized to immunoprecipitated AATF (a–c), GAPDH/Tubulin (f, g, i, k), or immunoprecipitated XRCC4 (j). Independent experiments were performed twice (b) or three times (a, c, f–k) with similar results. Data represent the mean ± SD of at least three independent experiments (d, e, l, j). Unpaired two-sided Student’s t-test (d, e, h, j, l). Source data are provided as a Source Data file.
Additionally, we observed a modest interaction between AATF and DNA ligase IV (LIG4), but not with other NHEJ components like Ku80 or DNA-PKcs12,13 (Fig. 3a and Supplementary Fig. 4d). Previous studies have demonstrated that XRCC4 and LIG4 form a complex12,13. To assess whether XRCC4 mediates the interaction between AATF and LIG4, we knocked down XRCC4 expression in GSCs. The IP assay showed that knockdown of XRCC4 disrupted the interaction between AATF and LIG4 (Supplementary Fig. 4f).
AATF comprises an acidic domain at its N-terminus followed by an AATF/Che-1/Traube superfamily-specific domain (AATFD), which consists of two conserved subdomains (AATFD-N and AATFD-C)27 (Fig. 3b, left). To interrogate the structural AATF interactions with XRCC4, we generated and expressed several Flag-tagged truncations constructs of AATF and the full length (FL) mCherry-XRCC4 in 293 T cells. Co-IP experiments showed that deletion of D-N subdomain in AATF disrupted the interaction of AATF-XRCC4 (Fig. 3b, right). Moreover, we found that 50 residues in the AATFD-N subdomain (331–380 aa) were required for AATF-XRCC4 interaction (Fig. 3c). Additionally, we also identified that both the Head and CTR domains of XRCC4 interact with AATF by performing GST-pull down assays (Supplementary Fig. 4g).
To determine whether NHEJ is involved in the observed AATF-mediated DNA damage repair, we utilized two commonly used GFP-based reporters: EJ5-GFP and DR-GFP (Supplementary Fig. 4h), which provide readouts for NHEJ and HR activity, respectively28. Flow cytometry analysis revealed that AATF-KD inhibited NHEJ repair activity (Fig. 3d, left), but had no apparent effect on HR efficiency (Fig. 3d, right). To assess whether the promotion of AATF on NHEJ repair is mediated by its interaction with XRCC4, we expressed Flag-tagged full-length AATF or the XRCC4-binding deficient AATFΔ331–380 mutant in GSCs with endogenous AATF knockdown. The EJ5-GFP reporter assay revealed that expression of AATFFL, but not the AATFΔ331–380 mutant, restored the NHEJ activity of AATF-KD cells (Fig. 3e). IB analysis showed that AATFFL rescued the induction of γ-H2AX in AATF-KD GSCs, whereas no rescue was observed with the AATFΔ331–380 mutant (Fig. 3f). Furthermore, compared to AATFFL, the AATFΔ331–380 mutant failed to reduce the γ-H2AX accumulation in DGCs treated with IR (Supplementary Fig. 3a–c). These results suggest that AATF promotes efficient NHEJ repair through binding to XRCC4.
The previous studies have indicated that AATF acts as a transcriptional regulator, including its role in enhancing p53 transcription29,30. To determine whether AATF promotes NHEJ repair through a transcriptional-dependent pathway, GSCs were treated with Actinomycin D, a transcriptional inhibitor. The results showed that Actinomycin D treatment led to an increase in γ-H2AX in shNT GSCs, while knockdown of AATF caused a further increase in γ-H2AX levels in cells treated with Actinomycin D (Supplementary Fig. 5a). Moreover, we observed that both full-length AATF and the truncated AATFΔ331–380 mutant increased the transcriptional activity of p53 reporter and p53 target genes’ expression in cells with or without IR treatment (Supplementary Fig. 5b–e). XRCC4 knockdown did not significantly alter the expression of these p53 target genes (Supplementary Fig. 5e). These results suggest that AATF promotes NHEJ repair independently of its transcriptional regulation function.
AATF stabilizes XRCC4 by inhibiting its ubiquitin-mediated proteasomal degradation
To investigate the impact of AATF on XRCC4 protein, we conducted IB analysis and found that both AATF-KD and AATF-KO decreased XRCC4 protein levels in GSCs (Fig. 3f, g and Supplementary Fig. 3f). However, AATF-KD did not alter the mRNA expression of XRCC4 (Supplementary Fig. 5f), indicating that AATF may stabilize XRCC4 protein. To test this hypothesis, we performed a cycloheximide (CHX) chase assay and found that the half-life of XRCC4 protein was reduced in AATF-KD GSCs compared to shNT GSCs (Fig. 3h). We also observed a decrease in LIG4 protein levels in AATF-KD GSCs (Fig. 3g), which was consistent with previous reports that XRCC4 is associated with and promotes LIG4 stability26,31. Moreover, AATF depletion caused decreased XRCC4 levels across different types of tumor cells (Supplementary Fig. 2a). Notably, the reduction of XRCC4 protein in AATF-KD GSCs was restored by expressing AATFFL, but not the AATFΔ331–380 mutant (Fig. 3f), suggesting that AATF-XRCC4 interaction is required for stabilizing XRCC4 protein.
We next investigated how AATF increases XRCC4 protein stability. IB analysis showed that treatment with MG132, but not chloroquine, increased XRCC4 protein level in a time-dependent manner in GSCs (Supplementary Fig. 5g). Moreover, MG132 treatment effectively rescued the decreased XRCC4 protein phenotype in AATF-KD GSCs (Fig. 3i and Supplementary Fig. 5h), implicating ubiquitin–dependent proteolysis. Indeed, IP assays showed that both Lys 48-linked poly-ubiquitination and total poly-ubiquitination of XRCC4 were increased in AATF-KD GSCs compared to shNT GSCs (Fig. 3j). These findings suggest that AATF increases XRCC4 protein stability by inhibiting its ubiquitin-mediated degradation.
To evaluate whether AATF promotes NHEJ repair by inhibiting XRCC4 degradation, we expressed Flag-XRCC4 in AATF-KD GSCs. Flag-XRCC4 decreased the accumulation of γ-H2AX in AATF-KD GSCs (Fig. 3k and Supplementary Fig. 5i). The EJ5-GFP reporter assay revealed that Flag-XRCC4 expression restored the NHEJ activity in AATF-KD GSCs (Fig. 3l). Furthermore, expression of Flag-XRCC4 decreased the accumulation of γ-H2AX in DGCs treated with IR, suggesting that elevated levels of XRCC4 enhance DNA damage repair in glioma cells (Supplementary Fig. 5j). To further explore the broader gene networks regulated by AATF in GSCs, we performed RNA-sequencing analysis with control and AATF knockdown GSCs. Our analysis revealed that AATF depletion significantly altered the expression of a large number of genes, with a predominance of upregulated genes (Supplementary Fig. 5k). Gene ontology (GO) analysis of the differentially expressed genes revealed that AATF depletion resulted in a significant downregulation of genes involved in the cell division, DNA replication and DNA repair pathways (Supplementary Fig. 5l), indicating that AATF depletion leads to DNA damage and subsequently cell division defects in GSCs. Together, these results suggest that AATF, by binding to XRCC4, inhibits its ubiquitin-mediated degradation.
DNA damage induces AATF phosphorylation and causes dissociation of the AATF-XRCC4 complex
To assess whether AATF is co-recruited with XRCC4 to DSB sites, we transduced GSCs with mCherry-XRCC4 or GFP-AATF and tracked the localization kinetics of these proteins upon exposure to laser micro-irradiation-induced DNA damage. Interestingly, IF analysis showed that XRCC4 was efficiently recruited to DSB sites, while the localization of AATF was unaffected in micro-irradiation-treated GSCs (Fig. 4a). To investigate whether DNA damage has impact on the interaction between AATF and XRCC4, we performed IP assays at different time points after IR treatment. The strength of the AATF-XRCC4 interaction decreased at 15- and 30-min post-IR treatment, then returned to levels comparable to non-treated cells within 1 h (Fig. 4b, c and Supplementary Fig. 6a, b). The findings suggest that DNA damage disrupts the AATF-XRCC4 interaction, leading to the dissociation of AATF from XRCC4.

a Live-cell imaging of the recruitment of mCherry-XRCC4 and GFP-AATF to laser damage tracks generated by laser micro-irradiation in 4121 GSCs. Left: representative images. Blue arrows indicate the irradiated regions. Right: quantifications of the fluorescence intensity of XRCC4 and AATF on the DNA damage tracks (n = 4, biologically independent experiments; data are presented as mean ± SD; Two-way ANOVA). Scale bars, 5 μm. b, c 4121 GSCs (b), or 4121 GSCs expressing Flag-XRCC4 (c), were treated with IR (7.5 Gy) at indicated time points. The XRCC4-AATF interaction was analyzed by IP assay. d 293 T cells expressing Flag-AATF were pretreated with KU-55933 (ATM inhibitor) or NU7441 (DNA-PKcs inhibitor) for 1 h, and were then treated with IR (7.5 Gy, 30 min) as indicated. The XRCC4-AATF interaction was analyzed by IP with anti-Flag M2 beads. e 4121 GSCs were treated with IR (7.5 Gy) at the indicated time points. ATM-AATF interaction was analyzed by IP with IgG or anti-ATM antibody. f 4121 GSCs expressing Flag-AATF were treated with IR (7.5 Gy) at the indicated time points and collected for IP with anti-Flag M2 beads, and IB with p-SQ antibody. g 4121 GSCs were pretreated with KU-55933 for 1 h, and were then treated with IR (7.5 Gy, 15 min) as indicated. Cell lysates were collected for IP with anti-AATF antibody, and IB with p-SQ antibody. h 4121 GSCs expressing Flag-AATF or Flag-AATFS189A mutants were treated with IR (7.5 Gy, 15 min) and collected for IP with anti-Flag M2 beads, and IB with p-SQ antibody. i 4121 GSCs expressing Flag-AATFWt, Flag-AATFS189A or Flag-AATFS189D mutants were treated with IR (7.5 Gy) for 30 min. The XRCC4-AATF interaction was analyzed by IP with anti-Flag M2 beads. For IB, protein levels were normalized to immunoprecipitated AATF (b, d, f–i), XRCC4 (c), or ATM (e) respectively. Independent experiments were performed three times with similar results. The graphs represent the relative levels of indicated proteins (b, c, e, f, right, mean ± SD). Unpaired two-sided Student’s t-test. Source data are provided as a Source Data file.
We next investigated how cells detect DNA damage and trigger the dissociation of AATF from XRCC4. AATF protein contains three serine/threonine-glutamine (S/T-Q) motifs at its residues Ser189, Thr394, and Thr410, which are the potential phosphorylation sites for kinases involved in activating DNA damage responses, including ATM, ATR, and DNA-PKcs32,33. Subsequently, we tested whether these kinases are responsible for the DNA damage-induced disassociation of the AATF-XRCC4 complex. Co-IP assays revealed that inhibition of ATM by KU-55933, but not inhibition of ATR by VE-821 or DNA-PKcs by NU7441, restored the strength of AATF-XRCC4 interaction that was reduced by IR treatment (Fig. 4d and Supplementary Fig. 6c), suggesting that ATM plays a role in regulating the AATF-XRCC4 interaction following DNA damage.
A previous study revealed that AATF could be phosphorylated by ATM at Ser-189 site upon DNA damage29. According to the PTMs database (https://www.phosphosite.org/), the Ser189 of AATF was identified to be phosphorylated by proteomics analyses. To identify whether AATF is a substrate of ATM in GSCs, we performed an IP analysis with the anti-ATM antibody. AATF was interacted with ATM, and the strength of this interaction was increased by IR exposure (Fig. 4e). Moreover, we conducted a LC-MS analysis on the immunoprecipitated AATF proteins from 293 T cells treated with IR for 15 min, and found that the Ser-189 site of AATF underwent phosphorylation in response to IR treatment (Supplementary Fig. 6d). The IP-coupled IB analysis with a phospho-serine-glutamine (p-SQ) antibody revealed a basal level of AATF phosphorylation, which was increased at 15 min of IR exposure (Fig. 4f). Inhibiting ATM by KU-55933 treatment reduced this IR-induced AATF phosphorylation (Fig. 4g). Additionally, the AATF phosphorylation induced by IR was eliminated in the AATFS189A mutant, where Ser189 was substituted with alanine (Fig. 4h). These results suggest that AATF is phosphorylated by ATM at Ser189 residue in response to DNA damage.
To determine the impact of AATF phosphorylation on AATF-XRCC4 interaction, we carried out IP assays with Flag-AATFWt, Flag-AATFS189A, or Flag-AATFS189D mutants in GSCs. The interaction between AATFWt and endogenous XRCC4 was reduced by IR treatment (Fig. 4i). However, the phospho-deficient AATFS189A mutant exhibited a robust interaction with XRCC4, and this interaction was unaffected upon IR exposure. Conversely, the phospho-mimetic mutant AATFS189D was barely bound to XRCC4 (Fig. 4i). Collectively, these findings suggest that ATM-mediated phosphorylation of AATF promotes dissociation of XRCC4 from AATF.
ATM-mediated phosphorylation of AATF allows recruitment of XRCC4 to DSB sites
To investigate the role of AATF phosphorylation in DNA damage repair, we knocked down endogenous AATF expression and re-expressed Flag-AATFWt, Flag-AATFS189A, or Flag-AATFS189D mutants in GSCs. IB analysis showed that expression of Flag-AATFWt rescued the induction of γ-H2AX and the degradation of XRCC4 in AATF-KD GSCs, whereas no rescue was observed in cells expressing the AATFS189D mutant (Fig. 5a), suggesting that AATF binding is required for XRCC4 stability. Although expression of the AATFS189A mutant restored XRCC4 protein levels, γ-H2AX levels remained elevated in AATF-KD GSCs (Fig. 5a). Furthermore, IB and IF analyses revealed that the expression of AATFS189A or AATFS189D mutant led to increased basal levels of γ-H2AX in GSCs (Fig. 5b, c). The IR-induced γ-H2AX accumulation was higher at 0.5–1.0 h and persisted for a longer duration in GSCs harboring the AATFS189A or AATFS189D mutant compared to AATFWt (Fig. 5b, c). These results indicate that phosphorylation at Ser189 is crucial for AATF’s function in promoting DNA damage repair.

a 4121 GSCs were transduced with shAATFs in combination with Flag-AATFWt, Flag-AATFS189A, or Flag-AATFS189D. IB analysis of indicated proteins are shown. b, c 4121 GSCs, treated as in (a), were treated with IR (3 Gy) at indicated time points. IB (b) and IF (c) analyses of γ-H2AX. Nuclei were counterstained with Hoechst (blue). Left: representative images; scale bars, 10 μm. Right: quantifications of γ-H2AX (b, n = 3 biologically independent experiments; mean ± SEM) (c, each point represents a single cell; n = 5 biologically independent experiments; mean ± SD). d 4121 GSCs, treated as in (a), were treated with IR (7.5 Gy) at indicated time points. Ku80-XRCC4 interaction was analyzed by IP. Left: representative images; Right: quantifications of XRCC4 (n = 3, biologically independent experiments; mean ± SEM). e Live-cell imaging of XRCC4 recruitment to laser micro-irradiation-induced DNA damage in GSCs treated as in (a). Left: representative images; scale bars, 5 μm. Blue arrows indicate irradiated regions. Right: quantifications of XRCC4 fluorescence intensity on DNA damage tracks (n = 4 biologically independent experiments; mean ± SD). f NHEJ activity was assessed in U2OS cells transduced with shAATFs in combination with Flag-AATF (n = 3 biologically independent experiments; mean ± SD). g 4121 GSCs, treated as in (a), were exposed to increasing IR dose for 48 h. Cell viability was assessed by the CellTiter-Glo assay (n = 4 biologically independent experiments; mean ± SD). h–k, BALB/c nude mice intracranially implanted with 4121 GSCs (Luciferase). Mice were treated with IR (3 Gy, weekly, 3 times) from day 9 post-implantation (h, top). GB xenografts were tracked by bioluminescence; representative images and quantifications are shown (h, i, n = 6 mice for each group; mean ± SEM). Kaplan–Meier survival plots of mice (j, Log-rank Mantel–Cox test). Co-IF staining of γ-H2AX and SOX2 in GB xenografts (k, top). Intensity of γ-H2AX staining in SOX2+ cells (k, bottom; each point represents a single cell, n = 3 biologically independent samples, mean ± SD). Scale bars, 20 μm. For IB, protein levels were normalized to GAPDH (a, b) or immunoprecipitated Ku80 (d). Independent experiments were performed three times with similar results (a, b, d). The curves represent relative levels of indicated proteins (b, d, right). Unpaired two-sided Student’s t-test (b–d, f, i, k), or Two-way ANOVA (e, g). Source data are provided as a Source Data file.
Ku70–Ku80 heterodimers are the first factors to bind DSB sites and can then recruit XRCC4-LIG4 to the DNA ends12,13. To further explore whether the dissociation of AATF-XRCC4 promotes recruitment of XRCC4 to DSB sites, we examined XRCC4-Ku80 interaction by conducting IP assays in GSCs exposed to IR. XRCC4 interacted with Ku80, and the interaction strength increased 15 min after IR exposure in GSCs expressing Flag-AATFWt (Fig. 5d, left), but not in GSCs harboring the AATFS189A mutant (Fig. 5d, right). In addition, XRCC4 localization to DSB sites induced by laser micro-irradiation was decreased in GSCs expressing the AATFS189A mutant compared to AATFWt, measured by IF analysis (Fig. 5e). Thus, AATF-phosphorylation-mediated dissociation of AATF-XRCC4 complex enables the rapid recruitment of XRCC4 to DSBs.
To assess whether the phosphorylation of AATF affects NHEJ repair, we performed the EJ5-GFP reporter assay. The results revealed that expression of AATFWt, but not the AATFS189A or AATFS189D mutant, restored the NHEJ activity in AATF-KD cells (Fig. 5f). We next investigated whether interfering AATF phosphorylation sensitizes GSCs to IR. Compared to AATFWt, expressing the AATFS189A or AATFS189D mutant sensitized GSCs to IR treatment and increased IR-induced cell apoptosis (Fig. 5g and Supplementary Fig. 7a). These findings suggest that phosphorylation of AATF facilitates the efficient NHEJ repair.
Studies suggest that the balance between HR and NHEJ is primarily regulated by BRCA1 and 53BP1. Specifically, 53BP1 initiates NHEJ events and is crucial for repairing DSBs, while BRCA1 counters 53BP1 to promote DSB resection and facilitate HR14. To further evaluate the role of AATF in NHEJ repair, we performed Co-IF staining in GSCs. The results showed that IR treatment significantly increased the formation of foci for BRCA1/RAD51 and 53BP1, which were colocalized with γ-H2AX in GSCs (Supplementary Fig. 7b). Interestingly, we also observed that both BRCA1/RAD51 and 53BP1 foci levels were elevated in GSCs with AATF depletion compared to control cells (Supplementary Fig. 7c). We hypothesize that although our study indicates that AATF depletion leads to an accumulation of DNA damage through the inhibition of XRCC4-mediated NHEJ repair, the cells may compensate by enhancing both NHEJ and HR pathways to address this damage.
Additionally, to assess whether ATM-mediated phosphorylation of AATF affects its transcriptional activity, we ectopically expressed Flag-AATFWt, Flag-AATFS189A, or Flag-AATFS189D mutants in cells and exposed them to IR (Supplementary Fig. 7d). The p53 reporter assay revealed that IR exposure increased the transcriptional activity of p53 reporter compared to control group (Supplementary Fig. 7e). Similar to AATFWt, both the AATFS189A and AATFS189D mutants also enhanced the transcriptional activity of p53 reporter in cells treated with or without of IR (Supplementary Fig. 7e). These findings suggest that phosphorylation at Ser189 does not significantly impact AATF’s transcriptional activity, even in the context of DNA damage.
To evaluate the role of AATF phosphorylation in GB therapeutic resistance, we established orthotopic GB xenografts using AATF-KD GSCs expressing AATFWt, AATFS189A, or AATFS189D mutants and treated tumors with IR. BLI revealed that the combined AATF-KD and IR treatment significantly inhibited tumor growth and extended survival. Re-expression of AATFWt enhanced the resistance of tumors to IR treatment and reduced animal survival (Fig. 5h–j and Supplementary Fig. 7f). However, neither expression of AATFS189A or AATFS189D mutant restore the resistance of AATF-KD tumor to radiotherapy (Fig. 5h–j and Supplementary Fig. 7f). Co-IF staining of tumor tissues indicated that, expression of AATFWt, but not AATFS189A or AATFS189D mutant, rescued the DNA damage in GSCs in AATF-KD tumors treated with IR (Fig. 5k). These results suggest that phosphorylation of AATF at Ser189 is crucial for its role in promoting DNA damage repair in GSCs.
Disrupting AATF and XRCC4 interaction sensitizes GB to chemoradiotherapy
To investigate whether disrupting the interaction between AATF and XRCC4 sensitizes GB to chemoradiotherapy, we synthesized a series of FITC-tagged, cell-permeable polypeptides, including K331-S350, E346-R365, V361-K380, corresponding to the AATF (aa 331–380) binding subdomain, along with a scrambled sequence control. The polypeptides were linked to an N-terminal SV40-derived nuclear localization signal (NLS) (Fig. 6a). We treated GSCs with these three polypeptides and performed IF staining. The results showed that all of the three FITC-tagged polypeptides (green) entered GSCs (Fig. 6b). Co-IP assay indicated that the AATF polypeptides treatment reduced the interaction between AATF and XRCC4 in GSCs (Fig. 6c). Moreover, IB analysis revealed that treatment of AATF polypeptides led to reduced XRCC4 protein levels and increased basal γ-H2AX levels in GSCs (Fig. 6d). Of note, the polypeptides treatment elevated the IR- (Fig. 6d, top) and TMZ-induced (Fig. 6d, bottom) γ-H2AX levels, suggesting that blocking AATF-XRCC4 interaction using polypeptides sensitizes GSCs to DNA damage.

a Schematic drawing of the synthesized polypeptides of AATF. NLS, nuclear localization signal. b 4121 GSCs were treated with the indicated polypeptides for 6 h. Live-cell imaging of FITC-tagged polypeptides was performed in three biologically independent experiments, and representative images are shown. Scale bars, 5 μm. c 4121 GSCs were treated with the indicated polypeptides and MG132 (10 μM) for 6 h. AATF-XRCC4 interaction was analyzed by IP with IgG or anti-AATF antibody. d 4121 GSCs preincubated with the indicated polypeptides were treated with IR (3 Gy) (top), or TMZ (50 μM) (bottom) at the indicated time points. IB analysis of γ-H2AX and XRCC4 levels are shown. The curves represent the relative levels of γ-H2AX (right, mean ± SD; Unpaired two-sided Student’s t-test). e–g BALB/c nude mice were intracranially implanted with 4121 GSCs expressing Luciferase. Mice were treated with control, IR (3 Gy, once a week, 3 times), TMZ (40 mg/kg, every two days, 5 times), the mix of three polypeptides (10 mg/kg, every two days, 5 times), or the combined treatment from day 9 post-implantation. GB xenografts were tracked by bioluminescence and the representative images are shown (e). Bioluminescent quantifications of tumor growth are shown (f, n = 6 mice for each group; mean ± SD; Unpaired two-sided Student’s t-test). Kaplan–Meier survival plots of mice are shown (g, Log-rank Mantel–Cox test). For IB, protein levels were normalized to immunoprecipitated AATF (c), or GAPDH (d). Independent experiments were performed three times with similar results (c, d). Source data are provided as a Source Data file.
To determine whether disrupting the interaction of AATF-XRCC4 by these polypeptides enhances chemoradiotherapy efficacy in GB tumors, we established the orthotopic GB xenografts in nude mice. Starting on day 9 post-transplantation, mice were randomly assigned to receive IR, TMZ, AATF polypeptides (K331-S350, E346-R365, and V361-K380), or the combination treatment (Fig. 6e, left). BLI showed that AATF polypeptides treatment reduced tumor growth and enhanced the response of GB xenografts to IR or TMZ treatment (Fig. 6e, f). Furthermore, the combination treatment of AATF polypeptides and IR/TMZ significantly improved the survival of GB tumor-bearing mice compared to monotherapy (Fig. 6g). Collectively, these results further suggest that disrupting AATF-XRCC4 interaction using polypeptides promotes XRCC4 degradation and sensitizes GSCs and GB tumors to chemoradiotherapy.
XRCC4 mediates the function of AATF in promoting GB therapeutic resistance
We next investigated the role of XRCC4 in GSCs. XRCC4 protein levels, but not mRNA levels, were consistently increased in GSCs and primary GB cells compared to DGCs, NSCs, NPCs, and NHAs measured by IB and q-PCR (Fig. 1e–g and Supplementary Fig. 8a). XRCC4-KD increased γ-H2AX accumulation in GSCs (Supplementary Fig. 8b). As anticipated, XRCC4-KD inhibited NHEJ repair activity but did not affect HR repair, as respectively assessed with EJ5-GFP and DR-GFP reporters (Supplementary Fig. 8c). Additionally, treatment of shNT and XRCC4-KD GSCs with IR or TMZ revealed that XRCC4 depletion elevated IR- and TMZ-induced DNA damage and sensitized GSCs to IR treatment (Supplementary Fig. 8d–f). In the established orthotopic GB xenografts, inducible-knockdown of XRCC4 inhibited tumor growth and enhanced the sensitivity of GB xenografts to radiotherapy (Fig. 7a, b).

a, b BALB/c nude mice intracranially implanted with 4121 GSCs (Luciferase) with indicated treatment. Mice were treated with control, IR (3 Gy, weekly, 3 times), Dox (1 mg/mL in water), or the combined treatment from day 9 post-implantation. GB xenografts were tracked by bioluminescence (a, n = 6 mice for each group; mean ± SD). Kaplan–Meier survival plots of mice are shown (b, Log-rank Mantel–Cox test). c Flag-vector or Flag-XRCC4 was expressed in 4121 GSCs transduced with shAATF#1. Cells were treated with IR (3 Gy) at indicated time points. IB analysis of indicated proteins shown. The curve represents relative levels of γ-H2AX (right, mean ± SD). d GSCs expressing shNT, shAATF#1 or shAATF#1 plus Flag-XRCC4 were treated with increasing IR dose for 48 h (n = 5 biologically independent experiments; mean ± SD). Cell viability was assessed by the CellTiter-Glo assay. (Two-way ANOVA). e–g BALB/c nude mice intracranially implanted with 4121 GSCs (Luciferase). GB xenografts were tracked by bioluminescence (e, n = 6 mice for each group; mean ± SD). Kaplan–Meier survival plots of mice are shown (f, Log-rank Mantel–Cox test). IF staining of γ-H2AX or CCP3 in GB xenografts are shown (g, n = 5 biologically independent samples, mean ± SD). Scale bars, 20 μm. h IB analysis of AATF and XRCC4 expression in resected human GB tissues and paired normal brains. i–m IHC analysis of AATF and XRCC4 expression in serial sections of glioma tissue microarrays. Representative images (i) and histoscore quantification (j) of AATF and XRCC4 are shown. Scale bars, 50 μm. (grade I-II, n = 89 independent tissue samples from patients; grade III-IV, n = 67 independent tissue samples from patients). Pearson correlation analysis (two-sided) between AATF and XRCC4 IHC scores is shown (k). Kaplan–Meier survival analysis of patients in high-grade gliomas (l) and all gliomas (m) are shown. (Log-rank Mantel–Cox test). For IB, protein levels were normalized to GAPDH (c, h). Independent experiments were performed three times with similar results (c, h). Unpaired two-sided Student’s t-test (a, c, e, g, j). Source data are provided as a Source Data file.
To evaluate whether XRCC4 protein mediates the promotion of AATF on DNA damage repair, we expressed Flag-vector or Flag-XRCC4 in GSCs with AATF knockdown. Expression of Flag-XRCC4 resulted in a reduction of IR-induced γ-H2AX levels in AATF-KD GSCs (Fig. 7c) and eliminated the sensitivity of AATF-KD GSCs to IR exposure (Fig. 7d). Moreover, in the short exposure IB image, we observed a decrease of Flag-XRCC4 at 12 h compared to 0-h post-IR treatment in AATF-KD GSCs (Fig. 7c). In orthotopic GB xenografts, AATF-KD combined with IR exposure inhibited GB tumor growth, while expressing Flag-XRCC4 compromised the radiosensitivity effects caused by AATF-KD (Fig. 7e, f and Supplementary Fig. 8g). In addition, IF staining revealed that Flag-XRCC4 expression reduced the accumulated DNA damage and inhibited cell apoptosis in IR-treated AATF-KD tumors (Fig. 7g). These results suggest that AATF’s role in promoting DNA damage repair and radioresistance in GSCs is dependent on stabilization of XRCC4 protein.
To determine the clinical relevance of AATF and XRCC4 expression, we performed IB and q-PCR analyses with seven freshly resected human GB specimens. Both AATF and XRCC4 protein levels were consistently elevated in GB tissues compared to paired adjacent normal brain tissues (Fig. 7h). However, the mRNA levels of AATF and XRCC4 were lower or remained unaltered in GBs compared with normal brain tissues (Supplementary Fig. 8h). IHC staining with human glioma tissue microarrays showed that both the percentage and intensity of AATF and XRCC4 staining were increased in high-grade gliomas (III or IV) compared to low-grade gliomas (I or II) (Fig. 7i, j). Moreover, AATF expression was positively correlated with XRCC4 levels in gliomas (Fig. 7k), which was further confirmed by co-IF staining in human GB specimens (Supplementary Fig. 8i). We then subgrouped all gliomas, or the high-grade gliomas, based on the median expression of AATF and XRCC4 as a cutoff. High levels of AATF or XRCC4 were associated with poor survival in all gliomas and GB patients (Fig. 7l, m). Together, these results demonstrate that the elevation of AATF and XRCC4 is associated with poor prognosis in GB.
Discussion
Therapeutic resistance in glioblastoma (GB) is primarily attributed to the presence of self-renewing glioma stem-like cells (GSCs), which exhibit enhanced DNA damage repair efficiency to sustain genomic stability and survive DNA damage4,5,6,8. Our study has revealed that GSCs acquire a potent DNA damage repair capacity by activating the NHEJ pathway through upregulation of AATF.
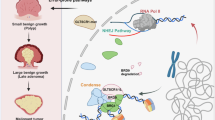
AATF was initially identified as a transcriptional cofactor involved in cell proliferation and survival30,34. Previous studies have demonstrated that in response to DNA damage, AATF can induce p53 transcription, promoting cell survival by favoring cell cycle arrest over apoptosis in breast cancer and colon cancer cells25,29,35. In this study, our findings indicate that the promotion of AATF on DNA damage repair is transcriptionally independent in GSCs. Instead, we have identified AATF as a previously unrecognized regulator of NHEJ pathway, the primary mechanism for DNA damage repair throughout the cell cycle12. We demonstrate that AATF interacts with XRCC4, a core component of NHEJ complex that is crucial for DNA-strand joining12,13. This interaction protects XRCC4 from ubiquitin-mediated proteasomal degradation, thereby maintaining an adequate amount of XRCC4 protein for efficient NHEJ repair (Fig. 8a, left and middle). AATF does not interact with other NHEJ components, such as Ku80 or DNA-PKcs, nor affect the levels of these proteins. Our findings suggest AATF functions as a “binding platform” for XRCC4, increasing XRCC4 protein stability and facilitating its role in DNA damage repair.

Prior to DNA damage, AATF forms a complex with XRCC4; Upon DNA damage, ATM interacts with and phosphorylates AATF at its Ser189 residue, which drives AATF-XRCC4 complex dissociation, thereby allowing rapid recruitment of XRCC4 to DSB sites to drive NHEJ repair (left). Knockdown of AATF results in polyubiquitination and subsequent proteasomal degradation of XRCC4, thus inhibiting NHEJ repair (middle). The non-phosphorylatable AATF mutant variant blocks recruitment of XRCC4 to DSB sites and inhibits NHEJ repair (right).
In response to DNA damage, cells activate DNA damage response (DDR) checkpoints, including ATM, ATR, and DNA-PKcs, leading to cell-cycle arrest and initiation of DNA damage repair10. Our study revealed that ATM interacts with and phosphorylates AATF upon DNA damage, resulting in dissociation of the AATF-XRCC4 complex. This allows free XRCC4 to be rapidly recruited to DNA double strand break (DSB) sites, where it functions as the core component of the NHEJ complex to repair DNA damage (Fig. 8a, left). The phosphorylation of AATF is transient, as it returns to basal levels within one hour after DNA damage (Fig. 4f), coinciding with the restoration of the AATF-XRCC4 interaction (Fig. 4b, c). This suggests that once XRCC4 has sealed the broken DNA, dephosphorylated AATF may re-bind to XRCC4 to stabilize its protein levels. However, the S189 phospho-mimetic mutant (AATFS189D) remains in a constitutively phosphorylated state and fails to efficiently bind XRCC4 (Fig. 4i), which may explain why the phospho-mimetic mutant fails to rescue XRCC4 degradation nor NHEJ repair in AATF-depleted GSCs (Fig. 5a–c, f–k). Moreover, the dissociation of the AATF-XRCC4 complex is crucial for the localization of XRCC4, as inhibiting this dissociation by using an AATF phosphorylation-deficient mutant prevents the recruitment of XRCC4 to DSBs (Fig. 8a, right), suggesting AATF as a link protein that senses DNA damage through ATM-mediated phosphorylation.
Additionally, our findings are consistent with prior research indicating that AATF depletion causes DNA damage36. This study reveals that AATF may suppress RNA:DNA hybrid (R-loop) formation, a known cause of DNA double-strand breaks (DSBs)37, and enable DNA damage repair. Knockdown of AATF results in increased R-loop formation and DSBs36. While we have established that AATF enhances NHEJ repair independent of its role in transcriptional activation, we acknowledge the potential for AATF to regulate genomic homeostasis through additional mechanisms. We hypothesize that AATF has multifaceted roles in promoting DNA damage repair and maintaining genomic stability, which may explain why AATF depletion leads to more severe phenotypes compared to XRCC4 depletion in GSCs. We believe that AATF’s diverse functions in promoting DNA damage repair and genomic stability could contribute to tumor therapeutic resistance.
Consistent with a previous report29, our study also demonstrates that AATF is phosphorylated by ATM at its Ser189 in response to DNA damage, which disrupts the interaction between AATF and XRCC4. Interestingly, Ser189 is not located within the binding region (amino acids 331–380) of AATF for XRCC4. Structural analysis of AATF using AlphaFold 2 reveals that Ser189 is situated within a divergent and intrinsically disordered region, which is spatially close to the binding domain of AATF. Previous research has shown that disordered regions are crucial for protein signaling and regulatory functions38. We hypothesize that phosphorylation of Ser-189 alters the conformation of this disordered region in AATF, thereby affecting its interaction with XRCC4 by interfering with the helical domains of both proteins. Additionally, the Ser189 in AATF is not conserved among mammals. While the conservation of phosphorylated amino acid residues typically indicates their functional significance, it is important to note that phosphorylation events, even if not conserved across species, can still play key roles in protein function. For instance, following UV radiation, p53 is phosphorylated at Ser-33 and Ser-46 residues by p38 kinase and human serine/threonine kinase homeodomain-interacting protein kinase-2 (HIPK2), respectively. This phosphorylation plays a pivotal role in activating p53 in response to DNA damage39,40,41. Interestingly, these two serine residues in p53 are not conserved across human and other animals39. This case supports the notion that phosphorylated amino acid residues, even if not conserved across species, can still be involved in crucial biological functions.
NHEJ repair is the predominant mechanism of DNA damage repair that contributes to resistance of cancer cells to chemoradiotherapy11. Blocking NHEJ repair is thought to be an effective strategy when used in combination with chemoradiotherapy in tumor treatment42. A recent study showed that DNA-PKcs, a NHEJ component, is highly expressed in GSCs. Pharmacological inhibition of DNA-PKcs enhances the sensitivity of GB xenografts to radiotherapy43. Our findings demonstrate that GSCs acquire the enhanced NHEJ repair capability by expressing high levels of AATF, which results in the accumulation of XRCC4 in GSCs. Furthermore, high levels of AATF protein are associated with increased XRCC4 protein and poor clinical outcomes in GB patients, highlighting the potential clinical relevance of these findings regarding AATF modulation. Notably, our study reveals that disrupting the interaction between AATF and XRCC4 using polypeptides effectively sensitizes GSCs to DNA damage induced by IR and TMZ, and improves GB xenograft response to chemoradiotherapy. These findings highlight targeting the AATF-XRCC4 axis as a promising strategy for GB treatment.
In conclusion, our research has uncovered that GSCs acquire a strong ability to repair DNA damage, mainly due to their elevated NHEJ repair efficiency. By targeting AATF to disrupt NHEJ repair, we have shown that GB can be sensitized to chemoradiotherapy, providing a promising strategy to overcome GSC-mediated therapeutic resistance in GB.
Methods
Ethics statement
The experimental protocol was approved by the Ethics Committee of National Center of Biomedical Analysis. All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and with the approval of the Institutional Animal Care and Use Committee of National Center of Biomedical Analysis.
Cell lines, tissues, and cell culture
GSC lines (387, 4121, 3691, 3832, 456, and H2S GSCs) were derived by our laboratory and transferred via a material transfer agreement from Duke University (Durham, NC) and were functionally characterized20,21,44,45. Briefly, cells were isolated from primary GB specimens or patient-derived GB xenografts using the Papain Dissociation System (Worthington Biochemical) according to the manufacturer’s instructions. Isolated cells were recovered in stem cell medium (Neurobasal-A medium with B27 supplement, 10 ng/mL EGF, 10 ng/mL βFGF, 1% penicillin/streptomycin, 1 mM sodium pyruvate, and 2 mM l-glutamine) overnight, then sorted by magnetic cell sorting using the surface marker CD133 (Miltenyi Biotec.) and cultured in stem cell medium as described above. GSCs were derived from the following GB patients: 3691 GSCs, female; 3832 GSCs, female; 4121 GSCs, male (recurrent GB); 387 GSCs, female; 456 GSCs, female; H2S, sex not available. Primary GB cells were obtained from the following GB patients: G#7, male; G#27, male; G#29, male (Anaplastic astrocytoma, Grade III); Primary GB tumor specimens were obtained from the following GB patients: GB#1, male; GB#2, female; GB#3, female; GB#28, female; GB#35, male; GB#37, female; GB#68, sex not available.
Authentication of GSCs was performed with STR analysis and GSCs were regularly tested negative for mycoplasma contamination. GB surgical specimens were collected for this study with the approval of Beijing Fengtai Hospital and PLA General Hospital. Sex was determined by self-reporting, and it was not taken into consideration for the statistical analyses of this study, as it was not the focus of the research. We collected the specimens based on patient availability. Informed consents were received from all the participants whose specimen were used in this study. The glioma tissue microarray (HBraG171Su01) was purchased from Shanghai Outdo Biotechnology Company, Ltd.
The human NPC lines (hNP1 and 15167) derived from fetal brains were cultured and maintained in the stem cell medium described above according to the vendor’s instruction (Lonza). Human NSCs (Meisen CTCC) were cultured and maintained in human neural stem cell medium (Meisen CTCC: CTCC-009-415). NHA cells (Beina Chuanglian Biotechnology Institute) was maintained in glucose-free DMEM supplemented with 5 mM glucose and 10% FBS. All these cells were authenticated by the supplier using STR analysis and were regularly tested negative for mycoplasma contamination.
Mice
Mice used in these studies were 4-week-old female BALB/c nude mice purchased from Beijing Vital River Laboratory Animal Technology. Sex was not considered in the study design and analysis because this study was not designed to detect sex differences. All mice were resided in specific pathogen-free (SPF) conditions. Mice lived in a cycle of 12 h of light and 12 h of darkness. The temperature and humidity of the mouse feeding environment are 18–22 °C and 50–60%, respectively. Animal care was monitored daily by certified veterinary staff and laboratory personnel. A maximum of five mice per cage was allowed. Animals were maintained until manifestation of neurological signs, at which point they were sacrificed by inhalation of carbon dioxide followed by cervical dislocation, and then remove the tissues for in vitro analysis. All surgical procedures were performed under anesthesia by intraperitoneal injection of a ketamine and xylazine cocktail.
Establishment of GSC-derived intracranial GB xenografts and combination treatment
Intracranial transplantation of GSCs to establish GB xenografts was performed19,21. Briefly, GSCs (2 × 104) with indicated treatment were implanted into the right frontal lobe of female BALB/c nude mice. Four-week-old littermates were used for transplantation studies and randomized to be grouped. Mice in each group were treated with doxycycline (Dox) (1 mg/mL, daily in water), irradiation (γ-ray irradiator, 3 Gy, once a week, three times in total), TMZ (40 mg/kg, every two days, five doses in total), or were treated with AATF polypeptides (10 mg/kg, every two days, 5 time in total), starting on the indicated days after tumor implantation. In the recurrent GB model, tumor-bearing mice were treated with irradiation (3 Gy, every three days, four treatments) to eliminate the tumor. To monitor tumor growth, mice transplanted with GSCs, which stably expressed firefly luciferase, were monitored by bioluminescence imaging longitudinally using the Calibar IVIS® Spectrum (PerkinElmer) in vivo imaging system every week. For the survival experiments, animals were maintained until manifestation of neurological signs. The maximum tumor burden permitted by the ethics committee is 1500 mm3, and the tumor weight must not exceed 10% of the mouse’s body weight. The humane endpoint was defined by a combination of clinical signs, including lethargy, hunched posture, and significant tumor volume. In this study, none of the tumor volumes exceeded these limits.
Mice were euthanized and necropsied by exsanguination using cardiac perfusion with PBS under deep anesthesia, when exhibiting signs of declining neurologic status or performance status. Mouse brains were fixed in 4% paraformaldehyde for 24 h at 4 °C, washed with PBS and preserved with 30% sucrose. Mouse brains were placed in OCT compound and stored at −80 °C before sectioning at a thickness of 8 µm until use.
Isolation of CD133 positive and negative glioma cells from human primary GB tissue
Fresh primary glioblastoma tissues were obtained from Beijing Fengtai Hospital and PLA General Hospital and were dissociated into single-cell suspensions using brain tumor dissociation kit (Miltenyi Biotec, Cat# 130-095-942). Then the CD133 positive and negative cells were isolated using CD133 microbead kit (Miltenyi Biotec, Cat# 130-100-857) according to the manufacturer’s protocol.
Immunofluorescence staining, immunohistochemistry, and Immunoblot
For IF staining19,21, cells or tissue sections were fixed in 4% paraformaldehyde (PFA, Sigma-Aldrich) for 15 min at room temperature and then permeabilized in PBS containing 0.3% Triton X-100 (Bio-Rad Laboratories) for 15 min. Samples were blocked with 5% bovine serum albumin and 0.3% Triton X-100 in PBS for 1 h at room temperature, and then incubated with primary antibodies overnight at 4 °C followed by incubation with appropriate secondary fluorescently labeled antibody for 1 h at room temperature. Nuclei were counterstained with Hoechst (Invitrogen). Images were acquired using a laser confocal microscope (Zeiss, LSM880) with Zen 2.1 SP2 software. ImageJ software 1.8.0 (version 1.54 g) was used for image processing after data acquisition.
For IHC staining21,46, a tissue microarray of de-identified formalin-fixed, paraffin-embedded glioma specimens was immunostained with anti-AATF or -XRCC4 antibody. Secondary antibody was labeled with polymer-HRP (horseradish peroxidase) anti-mouse as appropriate. Staining was visualized using DAB (3, 30-diaminobenzidine) chromogen. The staining intensity of AATF or XRCC4 was scored using a modified histochemical scoring (H-score) system to assess both the staining intensity and the percentage of positively stained cells. For the intensity, a score of 1–4 (corresponding to negative, weak, moderate, or strong staining) was recorded and the percentage of positively stained cells at each intensity was estimated. Presence or absence of AATF or XRCC4 staining was scored by at least 2 individuals, one of whom is a pathologist, and consensus scores are reported. Tissue microarrays including low-grade and high-grade gliomas (Shanghai Outdo Biotechnology Company, Ltd). The detailed information of tissue microarrays is listed in Supplementary data 3.
For Western blot analyses19,21, cells were lysed in RIPA buffer supplemented with protease inhibitors and phosphatase inhibitors (Roche), and then incubated on ice for 30 min. Protein concentration was determined by the Bradford assay (Bio-Rad Laboratories). Protein samples were loaded on SDS-PAGE gels and transferred to PVDF membranes (Millipore, Billerica, MA). Blots were incubated with primary antibodies overnight at 4 °C followed by HRP-conjugated species-specific antibodies (Jackson Immuno Research, 1:5000) at room temperature for one hour.
The following antibodies were used: AATF (Proteintech Group for IB, 1:1000, Cat#10282-1-AP; for IF, 1:200, Cat#A301-032a; Santa Cruz for IHC, 1:200, Cat#sc-81225), XRCC4 (Santa Cruz for IB, 1:1000, for IHC 1:200, Cat#sc-365055; Proteintech Group for IF, 1:200,Cat# 15817-1-AP), γ-H2AX (Millipore for IB, 1:1000, Cat#05-636; Cell Signaling for IF 1:200, cat#9718), Flag (Sigma for IB, 1:2000, Cat#F7425), Cleaved-Caspase3 (Cell Signaling for IB, 1:1000; for IF, 1:100, Cat#9664 s), LIG4 (Proteintech Group for IB, 1:1000, Cat#12695-1-AP), DNA-PKcs (Cell Signaling for IB, 1:1000, Cat#4602), Ku80 (Proteintech Group for IB, 1:1000, Cat#16389-1-AP), ATM (Abcam for IB, 1:1000, Cat#ab32420), SOX2 (Santa Cruz for IB, 1:1000, cat#SC-365823; R&D for IF, 1:200,cat#AF2018), CD3 (Thermo Fisher for IF, 1:200, cat#14-0037-82), CD45 (Thermo Fisher for IF, 1:200, cat#58-0459-42), CD68 (Abcam for IF, 1:400, cat# ab955), GFAP (Cell Signaling for IF,1:400,cat#12389). BRCA1 (Proteintech Group for IF, 1;200, cat#22362-1-AP), Rad51 (Cell Signaling for IF, 1:200, cat# 8875), 53BP1 (Novus for IF, 1:200, cat# NB100-304), Ubiquitin (MBL for IB, 1:1000, Cat#D058-3), Ubiquitin, Lys48-specific (Millipore for IB, 1:1000, Cat#05-1307), p-SQ (Cell Signaling for IB, 1:1000, Cat#6966S), Secondary antibody labeled with polymer-HRP anti-rabbit (Jackson for IB, 1:5000, cat#111-035-003), secondary antibody labeled with polymer-HRP anti-mouse (Jackson for IB, 1:5000, cat#115-035-003). Alexa Fluor 488 donkey anti-rabbit IgG (Thermo for IF, 1:400, Cat#A21206), Alexa Fluor 488 donkey anti-mouse IgG (Thermo for IF, 1:400, Cat#A21202), Alexa Fluor 555 donkey anti-mouse IgG (Thermo for IF, 1:400, Cat#A31570), and Alexa Fluor 555 donkey anti-goat IgG (Thermo for IF, 1:400, Cat#A21432).
DNA constructs and lentiviral transfection
Human Flag-AATF, GFP-AATF, Flag-XRCC4, and mCherry-XRCC4 were generated by PCR and were cloned into the pCDH-MCS-T2A-Puro-MSCV lentiviral vector (System Biosciences). Human GST-XRCC4 were generated by PCR and were cloned into the pGEX-4T-1 vector (Addgene). The deletion mutation forms of AATF (Δ181–230aa, Δ231–280aa, Δ281–330aa, Δ331–380aa, Δ381-440aa) were cloned into the pCDH-MCS-T2A-Puro-MSCV vector. The truncated mutant forms of XRCC4 and AATF were cloned into the pGEX-4T-1 vector. Clones expressing AATF or XRCC4 shRNAs were cloned into the pLKO.1 TRC vector (Addgene); Clones expressing AATF or XRCC4 Dox-induced shRNAs were cloned into the Tet-pLKO-puro vector (Addgene).
AATF DOX-induced shRNA: 5′-TTGACTCAGATCGACCATATT-3′; AATF-shRNA#1: 5′-TTGACTCAGATCGACCATATT-3′; AATF-shRNA#2: 5′-GCTCTTGGAAGGAAGG ATCAA-3′; AATF-shRNA#3: 5′-ATGGAGGAAACCAGTGACTTT-3′; XRCC4 DOX-induced shRNA: 5′-CCTCAGGAGAATCAGCTTCAA-3′; XRCC4-shRNA#1: 5′-TATTGCTTGGACACCATTGCA-3′; XRCC4-shRNA#2: 5′-CCTCAGGAGAATCAG CTTCAA-3′.
Lentiviral viral particles were produced in 293 T cells by co-transfection of lentiviral vectors, the packaging vectors pSPAX2 and pVSVG using a standard calcium phosphate transfection method. For rescue experiments, GSCs stably expressing Flag-XRCC4 were transduced with shAATF or non-targeting shRNA via lentiviral infection. After recovering for 48 h, cells were selected by puromycin (2 μg/mL) and plated to assess cell viability, DNA damage or used for in vivo experiments.
Inducible knockdown
For inducible knockdown21, GSCs stably expressing firefly luciferase were infected with lentivirus carrying inducible pLKO-Tet-On-shRNA. GSCs expressing AATF or XRCC4 Dox-induced shRNAs were cultured with 0.1 µg/mL Dox for 6 days before cell lysates were collected to confirm the knockdown efficiency of AATF or XRCC4. After validation of knockdown efficiency in vitro, GSCs expressing Dox-induced shRNAs were intracranially transplanted into nude mice and supplied with water containing 1 mg/mL Dox.
CRISPR-Cas9 gene knockout
The CRISPR design tool from the Broad Institute (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design) was used to design AATF sgRNAs Forward: 5′-CACCGGCTTTAAGTGCCTTGTGACC-3′, Reverse: 5′-AAACGGTCACAAGGCACTTAAAGCC-3′, Forward: 5′-CACCGCCTTTATTTCCCAGGTCACA-3′, Reverse: 5′-AAACTGTGACCTGGGAAATAAAGGC-3′. Oligonucleotides were annealed and cloned into LentiGuide-puro (Addgene) plasmid. For knockout studies, GSCs were transduced with LentiCas9-blast (Addgene) lentiviral construct and selected by blasticidin (10 μg/ml) to obtain GSCs stably expressing Cas9. After that, the GSCs-Cas9 were transduced with a lentiviral plasmid carrying sgRNAs targeting AATF and were selected with puromycin (2 μg/ml) for 48 h. Cell pools were harvested to confirm the knockout efficiency of AATF by IB.
Cell viability assay
Cell viability assays were conducted by plating cells at a density of 2 × 103 cells/well in 96-well plates with 200 μl culture medium in triplicate. Cell titers were determined after the indicated number of days using Cell Titer-Glo Luminescent Cell Viability Assay kit (Promega, Madison). All data were normalized to the initial reading at day 0 and presented as mean ± SD.
Immunoprecipitation and mass spectrometric analysis
Cells were collected and lysed in NP-40 lysis buffer (Boster biological technology, AR0107) supplemented with protease inhibitors and phosphatase inhibitors, incubated on ice for 30 min, and followed by centrifugation at 15,000 × g for 15 min at 4 °C. The supernatant was subjected to immunoprecipitation with the primary antibody (5 μg of antibody as indicated, or isotype control antibodies) overnight at 4 °C. The precipitants were extensively washed six times with IP buffer, boiled with SDS loading buffer and subjected to SDS-PAGE and immunoblot.
For the AATF binding proteins identification, endogenous AATF was immunoprecipitated with anti-AATF antibody or isotype control overnight at 4 °C. The precipitants were extensively washed with wash buffer, boiled with SDS loading buffer and were separated on SDS-PAGE followed by silver staining. Gel fragments were excised, washed, detained in 50% ethanol and 5% acetic acid, dehydrated in acetonitrile, dried in a Speed Vacuum, and digested with trypsin. The peptides were extracted from the polyacrylamide and subjected to LC-MS analysis.
This study utilized an EASY-nLC 1200 liquid chromatography system coupled with a Q Exactive HF mass spectrometer (Thermo Fisher Scientific) to analyze peptides from all samples, including three technical replicates. Samples were first dissolved in 99.9% H2O/0.1% FA and centrifuged at 16,000 × g for 10 min before 500 ng was taken for analysis. Chromatographic conditions included a single-column mode with a separation column of 75 μm × 150 mm (ReproSil-Pur C18-AQ, 1.9 μm particle size), at a temperature of 60 °C. Mobile phases consisted of A: 99.9% H2O + 0.1% FA and B: 80% ACN + 20% H2O + 0.1% FA, with a flow rate of 0.3 μL/min. The elution program gradually increased the concentration of phase B over 75 min. Mass spectrometry settings included a spray voltage of 2.1 KV and a capillary temperature of 320 °C, with Orbitrap resolutions set at 120,000 for precursor ions and 15,000 for fragment ions. Data were qualitatively and quantitatively analyzed using ProteinDiscoverer 2.1.1.21 software, searching against the NCBI human protein database, with precursor and fragment ion tolerances set at 20 ppm and 50 mmu, respectively. Variable modifications considered were N-terminal acetylation and methionine oxidation, employing trypsin digestion allowing up to two missed cleavages, with peptide-level FDR control set at 1%. For experiments analyzing differential protein expression in GSCs and GBMs, DGC and NHA were used as control groups, with three technical replicates performed. For identification AATF binding proteins, IgG was used as a control group, with two biological replicates conducted.
GST-pull down assay
GST-vector and GST-XRCC4 were expressed in BL21 Competent E. coli, purified and incubated with Glutathione Sepharose 4B beads (GE Healthcare, 17-0756-01) according to the manufacturer’s protocol (Amersham Biosciences). For binding assay, Flag-AATF was transfected into 293 T cells. The cells were lysed with NP-40 lysis buffer and then the lysate supernatant was subjected to GST-beads coupled XRCC4 overnight at 4 °C in NP-40 lysis buffer. The precipitants were extensively washed six times with lysis buffer, boiled with SDS loading buffer and subjected to SDS-PAGE.
RNA isolation and real-time PCR
Total RNA was isolated from cultured cells using RNeasy Kit (QIAGEN). Then the RNA was reversely transcribed to cDNA with PrimeScript RT Master Mix (Takara Bio Inc.) according to the manufacturer’s instructions. Real-time PCR was performed with SYBR Green Master Mix (Applied Biosystems) on a cycler (Applied Biosystems QS3). Actin was used for normalization. The primer pairs used to detect the mRNA level are listed below.
AATF, Forward: 5′-CCAGGGTGATTGACAGGTTTG-3′, Reverse: 5′-CCAGTTTT CTAATGCTACCCACT-3′; XRCC4, Forward: 5′-ATGTTGGTGAACTGAGAAAAGCA-3′, Reverse: 5′-GCAATGGTGTCCAAGCAATAAC-3′; TP53, Forward: 5′-CAGCACA TGACGGAGGTTGT-3′, Reverse: 5′-TCATCCAAATACTCCACACGC-3′; P21, Forward: 5′-TGTCCGTCAGAACCCATGC-3′, Reverse: 5′-AAAGTCGAAGTTCCAT CGCTC-3′; PLK2, Forward: 5′- TACGCCGCAAAAATTATTCCTC-3′ Reverse: 5′-TCTTTGTCCTCGAAGTAGTGGT-3′; GAPDH, Forward: 5′-CCAGGTGGTCTCCTCTGACTTC-3’, Reverse: 5′- GTGGTCGTTGAGGGCAATG-3′.
Ubiquitination assay
GSCs were treated with 10 μM MG132 or vehicle control for 6 h before collection. Cells were washed with cold PBS and then lysed with NP-40 lysis buffer (Boster biological technology, AR0107) or denatured protein extracts RIPA supplemented with protease inhibitors (Roche) and N-Ethylmaleimide (Sigma, E3876), incubated on ice for 30 min, and followed by sonication at 75 Hz with a 2-s pause of 3 s for a total of 7 times and centrifugation at 15,000 × g for 15 min at 4 °C. The supernatant was immunoprecipitated with anti-Flag M2 affinity gel (Sigma, A2220) and subjected to western blotting with anti-ubiquitin antibody, anti-ubiquitin Lys48-specific antibody, or anti-Flag antibody to detect ubiquitination of XRCC4.
Flow cytometry analysis
For apoptosis assay, GSCs were harvested and trypsinized as single cell, and then re-suspended in 100 μl AnnexinV-FITC/PI buffer (Promega) and incubated for 15 min at room temperature in dark. Totally 10,000 cells were analyzed per sample and the rate of apoptosis was measured by a flow cytometer (BD Aria III). Flow cytometry assays were performed at least three times per cell line. Data analysis was performed with FlowJo V10 software.
Alkaline comet assay
The alkaline comet assay was performed according to manufacturer protocol (Trevigen #4250-050-K). Briefly, GSCs were harvested and suspended in low-melting point agarose, plated onto provided comet slides, and subjected to electrophoresis and DNA precipitation in neutral electrophoresis buffer. Slides were processed according to manufacturer’s instructions. DNA tails were visualized after SYBR Gold staining using an Invitrogen EvosTM M5000 fluorescence microscope.
HR and NHEJ reporter assays
U2OS cells integrated with DR-GFP or NHEJ-GFP cassettes were used to analysis of HR or NHEJ efficiency, respectively47,48. Cells transduced or transiently transfected with indicated plasmids were then transfected with I-Scel expression plasmid pCBA-Scel (Addgene). Forty-eight hours after transfection, percentages of GFP-positive cells were analyzed by flow cytometry. HR or NHEJ efficiency is presented as the ratio of control cells.
Identification of phosphorylation sites of AATF
For the identification of AATF phosphorylation sites, HEK293T cells were transduced with Flag-tagged AATF. Following collection, cells were lysed in PIP lysis buffer and kept on ice for 30 min, then clarified by centrifugation at 16,000 × g at 4 °C for 10 min. An equal amount of lysate was subsequently incubated with anti-Flag M2 beads at 4 °C overnight. The precipitates were washed thrice with high-salt buffer (PIP lysis buffer adjusted to 300 mM NaCl), boiled with SDS loading buffer, and subjected to SDS-PAGE. The gel was stained with Coomassie brilliant blue, sliced, and subjected to LC-MS/MS analysis at the National Center for Protein Sciences (Beijing). Briefly, gel slices were destained, reduced with DTT, alkylated with iodoacetamide, and digested with trypsin. The resulting peptides were purified and detected using Orbitrap mass spectrometers in positive ionization mode. LC-MS/MS data was analyzed using Proteome Discoverer and the free version of the Mascot Server.
Laser micro-irradiation
GSCs stably expressing indicated plasmids were cultured in glass-bottom dishes and pre-treated with 2 μM Hoechst 33342 Solution (Thermo Scientific) for 10 min. Cells were then subject to a 405-nm localized laser beam (70% laser power, 10 s) on an inverted Nikon A1R microscope. Cells were then imaged using the Nikon TIE microscope and the Nikon NIS software at indicated time. Data analysis was performed with Volocity demo software.
Dual-luciferase report assay
Human p53 promoter (−2000bp–+100 bp) was cloned into pGL3-Basic to construct luciferase reporter vectors. Cells expressing Flag-vector, Flag-AATF, or Flag-AATF△331–380 mutant plasmids were co-transfected with p53 reporter constructs and Renilla luciferase plasmids. After 48 h, luciferase activity was determined by a dual luciferase reporter assay system (Promega) according to the instructions of the manufacturer and measured by a multi-mode microplate reader (SpectraMax iD3).
RNA sequencing
Total RNA was isolated from cells using the RNeasy Mini Kit (QIAGEN). cDNA libraries were generated using the VAHTS Universal V6 RNA-seq Library Prep Kit for Illumina. After library construction, the libraries were first quantified using Qubit 3.0 for preliminary measurement and then diluted to a concentration of 1 ng/μL. Subsequently, the insert size of the libraries was analyzed using an Agilent 2100 Bioanalyzer. Once the insert size met the expected range, the effective concentration of the libraries was accurately determined by qPCR (with a requirement of effective concentration >10 nM), to ensure library quality. Sequencing was then performed on the Illumina platform using a PE150 sequencing strategy, generating paired-end reads of 150 bp in length. Bowtie v1.0.1 was used to build the reference genome index, and the clean data were then aligned to the reference genome using HISAT2 v2.0.5. Fragments per kilobase per millions (FPKMs) of known genes were calculated using eXpress v1.5.1.
Peptides
There sequence encoding AATF (331–380) was: K331-S350 (KQQRRRVPAKRKLEMEDYPS); E346-R365 (EDYPSFMAKRFADFTVYRNR); V361-K380 (VYRNRTLQKWHDKTKLASGK). A control peptide sequence (ALGFTDQKTPKSKRTVNSKQPFKTPSKNLTAY) was designed which had the same amino acid composition and did not bear homology to any known protein in human protein database. The Cell Permeable Peptide (CPP) versions (aa331–380 and control peptides) were tagged with SV40 Large T antigen nuclear localization signal (PKKKRKVEDPYC) in the N-terminus and FITC in the C-terminus. The peptides were dissolved in DMSO to make 100 mM stocks. For in vitro assays in cells, subsequent dilutions were made in 1 × PBS, while for in vivo studies, the compounds were dissolved in 1 × PBS containing 5% DMSO.
Quantification and statistical analysis
All grouped data are presented as mean ± SD or SEM from studies performed at least in triplicate unless otherwise specified. For bar graphs, the unpaired two-sided Student’s t-test was used to compare two groups with equal variances and Welch’s t-test for the comparison of two groups with unequal variances. Multi-group data was compared by Two-way ANOVA. A probability value of 0.05 or less was considered significant. For the survival analysis, Kaplan–Meier survival curves were analyzed by using log-rank statistics comparing the different patient or mouse groups. GraphPad Prism Software 8.3.0 (GraphPad Software, Inc.) was used to examine statistical significance. For IB analysis, protein levels were quantified using ImageJ (version 1.54 g) software.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometric analysis data and RNA-seq data generated for this study have been deposited in the China National Center for Bioinformation under the accession code OMIX004102 and HRA009680, which are publicly available. The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Aldape, K. et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 16, 509–520 (2019).
Stupp, R. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466 (2009).
Bao, S. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 (2006).
Saygin, C., Matei, D., Majeti, R., Reizes, O. & Lathia, J. D. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell 24, 25–40 (2019).
Lathia, J. D., Mack, S. C., Mulkearns-Hubert, E. E., Valentim, C. L. & Rich, J. N. Cancer stem cells in glioblastoma. Genes Dev. 29, 1203–1217 (2015).
Gimple, R. C., Bhargava, S., Dixit, D. & Rich, J. N. Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 33, 591–609 (2019).
Rich, J. N. Cancer stem cells in radiation resistance. Cancer Res. 67, 8980–8984 (2007).
Gimple, R. C., Yang, K., Halbert, M. E., Agnihotri, S. & Rich, J. N. Brain cancer stem cells: resilience through adaptive plasticity and hierarchical heterogeneity. Nat. Rev. Cancer 22, 497–514 (2022).
Pearl, L. H., Schierz, A. C., Ward, S. E., Al-Lazikani, B. & Pearl, F. M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 15, 166–180 (2015).
Lanz, M. C., Dibitetto, D. & Smolka, M. B. DNA damage kinase signaling: checkpoint and repair at 30 years. EMBO J. 38, e101801 (2019).
Trenner, A. & Sartori, A. A. Harnessing DNA double-strand break repair for cancer treatment. Front. Oncol. 9, 1388 (2019).
Scully, R., Panday, A., Elango, R. & Willis, N. A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 20, 698–714 (2019).
Zhao, B., Rothenberg, E., Ramsden, D. A. & Lieber, M. R. The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 21, 765–781 (2020).
Chang, H. H. Y., Pannunzio, N. R., Adachi, N. & Lieber, M. R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 18, 495–506 (2017).
Grawunder, U., Zimmer, D. & Lieber, M. R. DNA ligase IV binds to XRCC4 via a motif located between rather than within its BRCT domains. Curr. Biol.8, 873–876 (1998).
Yan, C. T. et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 449, 478–482 (2007).
Rajaraman, P. et al. DNA repair gene polymorphisms and risk of adult meningioma, glioma, and acoustic neuroma. Neuro-Oncol. 12, 37–48 (2010).
Kang, Y. J. et al. Contribution of classical end-joining to PTEN inactivation in p53-mediated glioblastoma formation and drug-resistant survival. Nat. Commun. 8, 14013 (2017).
Zhan, X. et al. Glioma stem-like cells evade interferon suppression through MBD3/NuRD complex-mediated STAT1 downregulation. J. Exp. Med. 217, e20191340 (2020).
Xie, Q. et al. N(6)-methyladenine DNA modification in glioblastoma. Cell 175, 1228–1243.e1220 (2018).
Huang, H. et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nat. Commun. 12, 3720 (2021).
Chen, L. et al. Phosphocreatine promotes epigenetic reprogramming to facilitate glioblastoma growth through stabilizing BRD2. Cancer Discov. 14, 1547–1565 (2024).
Singh, S. K. et al. Identification of human brain tumour initiating cells. Nature 432, 396–401 (2004).
Kaiser, R. W. J., Erber, J., Hopker, K., Fabretti, F. & Muller, R. U. AATF/Che-1-An RNA binding protein at the nexus of DNA damage response and ribosome biogenesis. Front. Oncol. 10, 919 (2020).
Bruno, T. et al. Che-1 promotes tumor cell survival by sustaining mutant p53 transcription and inhibiting DNA damage response activation. Cancer Cell 18, 122–134 (2010).
Grawunder, U. et al. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature 388, 492–495 (1997).
Bammert, L., Jonas, S., Ungricht, R. & Kutay, U. Human AATF/Che-1 forms a nucleolar protein complex with NGDN and NOL10 required for 40S ribosomal subunit synthesis. Nucleic Acids Res. 44, 9803–9820 (2016).
Bennardo, N., Cheng, A., Huang, N. & Stark, J. M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4, e1000110 (2008).
Bruno, T. et al. Che-1 phosphorylation by ATM/ATR and Chk2 kinases activates p53 transcription and the G2/M checkpoint. Cancer Cell 10, 473–486 (2006).
Iezzi, S. & Fanciulli, M. Discovering Che-1/AATF: a new attractive target for cancer therapy. Front. Genet. 6, 141 (2015).
Bryans, M., Valenzano, M. C. & Stamato, T. D. Absence of DNA ligase IV protein in XR-1 cells: evidence for stabilization by XRCC4. Mutat. Res. 433, 53–58 (1999).
Blackford, A. N. & Jackson, S. P. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA Damage Response. Mol. Cell 66, 801–817 (2017).
Kim, S. T., Lim, D. S., Canman, C. E. & Kastan, M. B. Substrate specificities and identification of putative substrates of ATM kinase family members. J. Biol. Chem. 274, 37538–37543 (1999).
Fanciulli, M. et al. Identification of a novel partner of RNA polymerase II subunit 11, Che-1, which interacts with and affects the growth suppression function of Rb. FASEB J. 14, 904–912 (2000).
Hopker, K. et al. AATF/Che-1 acts as a phosphorylation-dependent molecular modulator to repress p53-driven apoptosis. EMBO J. 31, 3961–3975 (2012).
Jain, M. et al. Inactivation of apoptosis antagonizing transcription factor in tubular epithelial cells induces accumulation of DNA damage and nephronophthisis. Kidney Int. 95, 846–858 (2019).
Hamperl, S. & Cimprich, K. A. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair (Amst.) 19, 84–94 (2014).
Iakoucheva, L. M., Brown, C. J., Lawson, J. D., Obradovic, Z. & Dunker, A. K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 323, 573–584 (2002).
Xu, Y. Regulation of p53 responses by post-translational modifications. Cell Death Differ. 10, 400–403 (2003).
Hofmann, T. G. et al. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 4, 1–10 (2002).
Bulavin, D. V. et al. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 18, 6845–6854 (1999).
Feng, W., Smith, C. M., Simpson, D. A. & Gupta, G. P. Targeting non-homologous and alternative end joining repair to enhance cancer radiosensitivity. Semin. Radiat. Oncol. 32, 29–41 (2022).
Fang, X. et al. Inhibiting DNA-PK induces glioma stem cell differentiation and sensitizes glioblastoma to radiation in mice. Sci. Transl. Med. 13, eabc7275 (2021).
Dixit, D. et al. The RNA m6A reader YTHDF2 maintains oncogene expression and is a targetable dependency in glioblastoma stem cells. Cancer Discov. 11, 480–499 (2021).
Man, J. et al. Hypoxic induction of vasorin regulates notch1 turnover to maintain glioma stem-like cells. Cell Stem Cell 22, 104–118 e106 (2018).
Man, J. et al. Sema3C promotes the survival and tumorigenicity of glioma stem cells through Rac1 activation. Cell Rep. 9, 1812–1826 (2014).
Zhao, W. et al. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 550, 360–365 (2017).
Peng, Y. et al. The deubiquitylating enzyme USP15 regulates homologous recombination repair and cancer cell response to PARP inhibitors. Nat. Commun. 10, 1224 (2019).
Acknowledgements
We thank Dr. Shideng Bao (Cleveland Clinic, USA) for kindly providing NPCs and helpful discussions. We thank Dr. Huadong Pei (The George Washington University School of Medicine and Health Science, USA) for kindly providing the HR and NHEJ reporters. We thank Dr. Xin Jiang (University of Electronic Science and Technology of China, China) for kindly providing the structural prediction and analysis. This research was supported by grants from the National Key Research and Development Program of China (2022YFA1303000 to J.H.M.), and National Natural Science Foundation of China (no. 32270826, 81872153 to J.H.M.).
Author information
Authors and Affiliations
Contributions
J.H.M., L.J.M., and Y.C. conceived the project, designed experiments, and analyzed the data. J.H.M. wrote the paper. J.H.M. and T.Z. supervised the project and acquired funding for this study. J.N.R. provided scientific advices and revised the manuscript. L.J.M. and Y.C. performed most of the experiments with the assistance from L.S.C., Y.Y.L., S.Y.Z., H.W.R., Q.H.Q., C.Z., H.R.W., S.L.C., D.K.X., H.H.H., X.Z.W., and Q.Y.H. J.Q., B.H.L., J.X., and F.Y.L. provided the patient specimens. Q.L.W., T.L., A.L.L., and T.Z. provided scientific advice for the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mi, L., Cai, Y., Qi, J. et al. Elevated nonhomologous end-joining by AATF enables efficient DNA damage repair and therapeutic resistance in glioblastoma. Nat Commun 16, 4941 (2025). https://doi.org/10.1038/s41467-025-60228-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60228-z
