
Abstract
Mitophagy, the selective autophagic elimination of mitochondria, is essential for maintaining mitochondrial quality and cell homeostasis. Impairment of mitophagy flux, a process involving multiple sequential intermediates, is implicated in the onset of numerous neurodegenerative diseases. Screening mitophagy inducers, particularly understanding their impact on mitophagic intermediates, is crucial for neurodegenerative disease treatment. However, existing techniques do not allow simultaneous visualization of distinct mitophagic intermediates in live cells. Here, we introduce an artificial intelligence-assisted fluorescence microscopic system (AI-FM) that enables the uninterrupted recognition and quantification of key mitophagic intermediates by extracting mitochondrial pH and morphological features. Using AI-FM, we identify a potential mitophagy modulator, Y040-7904, which enhances mitophagy by promoting mitochondria transport to autophagosomes and the fusion of autophagosomes with autolysosomes. Y040-7904 also reduces amyloid-β pathologies in both in vitro and in vivo models of Alzheimer’s disease. This work offers an approach for visualizing the entire mitophagy flux, advancing the understanding of mitophagy-related mechanisms and enabling the discovery of mitophagy inducers for neurodegenerative diseases.
Similar content being viewed by others

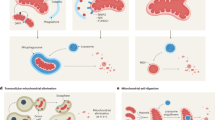

Introduction
Mitochondria, the cellular powerhouses, play crucial roles in energy production, calcium homeostasis, and redox potential modulation1. Neurons, with an estimated pool of two million mitochondria per cell, hundreds of times more than other somatic cells, rely heavily on these organelles to meet local energy demands2. This distinctive feature makes neurons particularly susceptible to mitochondrial defects, contributing to various neurological disorders characterized by varying degrees of mitochondrial dysfunction. Mitophagy is a process of continuous maintenance of mitochondrial quality and cellular homeostasis by selective removal of damaged or aged mitochondria3,4. Impaired mitophagy, leading to the progressive accumulation of dysfunctional mitochondria within the cytoplasm, is a key determinant in the pathogenesis of numerous brain disorders5,6. For example, in Alzheimer’s disease (AD), dysfunctional mitochondria contribute to the excessive production of pathological β amyloid, a hallmark of the disease7,8. Conversely, restoring mitophagy has shown the potential in mitigating symptoms of AD9,10,11. Therefore, techniques that enable dynamic and precise assessment of mitophagic activity are imperative for advancing both research and therapeutic strategies addressing mitochondrial dysfunction in neurological diseases.
Mitophagy flux denotes the process of mitochondrial engulfment by autophagosomes and fusion with autolysosomes for degradation. Throughout this procedure, mitochondria exist in different mitophagic intermediates, including damaged mitochondria, mitophagosomes, and mitolysosomes. The continuous visualization and quantification of mitophagy flux in live cells, particularly focusing on mitophagic intermediates, are crucial for advancing neurodegenerative disease research. Any interruption at any stage of mitophagy, such as inhibiting autophagosome formation or preventing the fusion between autophagosomes and lysosomes, disrupts the process. Modulators restore mitophagy by influencing different mitophagy intermediates, but an increase in one does not always signify mitophagy induction, necessitating assessment for accurate identification of mitophagy inducers. Moreover, mitochondria are unevenly distributed in neurons, with mitophagy occurring in distinct compartments like soma, axons, and synapses. This spatial preference is crucial for neuronal function and the prevention of neurodegenerative diseases12. Therefore, dynamically visualizing the quantity and spatiotemporal distribution of mitophagic intermediates not only enhances mechanistic insights into brain disorders but also accelerates the high-throughput screening of potential mitophagy inducers13,14,15.
The progression of mitophagy is marked by a continuous pH reduction within the mitochondrial matrix, which is normally maintained at a slightly alkaline pH of 7.5–8.016. Upon engulfment by autophagosomes, the mitochondrial pH decreases to a range between 5.8 and 7.017, and further drops to a range between 5.0 and 5.9 after fusion with autolysosomes18,19. Therefore, monitoring pH fluctuations provides an opportunity to dynamically track the mitophagy process. Fluorescence reporters have been explored to track mitochondrial intermediates by sensing their acidities (Supplementary Table 1). For example, the fluorescent protein RFP-GFP-LC3 was engineered to monitor autophagosome dynamics20 but lacks specificity in distinguishing non-selective autophagy from mitophagy. Mitochondria-specific fluorescent proteins such as mt-Keima21 and small molecular fluorescent probes22 are used to track mitochondrial acidities during mitophagy. Even though the above strategies detect the activation state of mitophagy, they fail to simultaneously identify the distinct mitophagic intermediates due to the challenges including multiple staining, spectral overlap, and potential cytotoxicity. Therefore, it is imperative to develop strategies for uninterrupted and simultaneous assessment of the distinct mitophagic intermediates during the mitophagy flux.
Here, we developed an artificial intelligence-assisted fluorescence microscopic system (AI-FM) for the intelligent evaluation of mitophagic flux in live cells (Fig. 1). AI-FM integrated a fluorescent probe, Mcy3, with a deep learning model. Mcy3, designed with mitochondria-targeting specificity and pH-ratiometric responsiveness, was synthesized to report mitochondrial pH during mitophagy. Concurrently, a dual-branch Multi-scale Attention ResNet (DMAN) was developed to extract mitochondrial pH and morphological features, facilitating the recognition and quantification of different mitophagic intermediates throughout the mitophagy flux. AI-FM spatiotemporally and quantitatively monitored the mitophagic intermediates under both mitophagy induction and inhibition conditions, achieving an average test accuracy of 86%. In conjunction with in silico screening targeting the sirtuin 1 (SIRT1) protein, AI-FM identified a mitophagy inducer, Y040–7904, after screening two million compounds. Moreover, we found that Y040–7904 significantly increased the transport of mitochondria to autophagosomes. Notably, Y040–7904 demonstrated potential therapeutic effects in ameliorating disease pathologies both in vivo and in vitro via the SIRT1/FoxO3 pathway. Compared with existing screening methods that rely on manual counting of pixel fluorescence intensity signals of the fluorescent probe9,23, AI-FM not only facilitates high-throughput drug screening but also provides spatiotemporal information on mitophagic intermediates, directly elucidating the efficacy of mitophagy modulators. Overall, the AI-FM system enables precise evaluation of mitophagic intermediates, providing a reliable approach for elucidating the mitophagy-associated pathology and uncovering therapeutic strategies.

a Ratiometric pH fluorescent probes Mcy1–3 are synthesized and Mcy3 is selected for visualizing mitophagic intermediates. Fluorescence images obtained in live cells are analyzed with a homemade deep learning model to quantify mitophagy levels. b Through an extensive in silico screening of a library containing two million compounds, nineteen promising hits have been identified. Further screening by AI-FM has successfully identified a mitophagy inducer candidate, named Y040–7904. c Acting as an agonist of SIRT1, Y040–7904 demonstrates its ability to ameliorate the AD pathological manifestations both in vivo and in vitro. Figure 1 was created in BioRender (Wang, Y. (2025) https://BioRender.com/2x0xwc2) and Adobe Illustrator.
Results
Design, synthesis, and photophysical properties of fluorescent probes
We developed pH-responsive probes, Mcy1–3, for monitoring mitophagy, which are derived from a widely used Förster resonance energy transfer (FRET) fluorophore pair Cy3 and Cy524 (Fig. 2A, Supplementary Fig. 1). Three moieties with varying hydrophobicity were introduced to Cy3 to regulate its mitochondrial targeting ability. A pH-sensitive unsymmetrical Cy5 was chosen as the FRET acceptor to achieve a ratiometric pH response, which is validated by time-dependent density functional theory (TDDFT) calculation (Fig. 2B)25. Representative pH-dependent UV-vis absorption and fluorescence spectra were shown in Fig. 2C, D. With the pH increase, the absorption at 630–650 nm diminished, indicating a conversion between the protonated state and the deprotonated state of Cy5, while the absorption band at 540 nm remained unchanged. When Mcy1–3 was excited at 530 nm at low pH, an intense fluorescence emission at 660 nm was observed, indicating an energy transfer between Cy3 and Cy5. Sequential increases in the fluorescence intensity ratio of I660/I560 were observed with decreasing pH. The pKa values of Mcy1–3 were calculated to be 5.5, 5.0, and 4.6 respectively. Notably, the pKa values of Mcy1–3 were all consistent with the pH ranges in autolysosomes, indicating the suitability of these probes for monitoring mitophagic intermediates (pH 5.0–8.0). The overall fluorescence quantum yield in PBS (pH 4.0) was notably higher than that in PBS at pH 7.0 (Supplementary Table 2). Mcy3 demonstrated the highest quantum yield among the three fluorescent probes.

A Chemical structures of the pH-responsive fluorescence probes Mcy1‒Mcy3 in their protonated and deprotonated forms. B The HOMO and LUMO energy levels, excitation energies, and oscillator strengths for unsymmetrical Cy5, based on TDDFT calculations at the B3LYP/6–311 G(d,p) level. The absorption (C) and fluorescence spectra (D) of Mcy1‒Mcy3 (1 × 10−5 M in PBS, containing 1% DMSO) as a function of pH. The inserts show the ratiometric calibration curve of the fluorescence intensity ratio (I660 nm/I560 nm). Source data are provided as a Source Data file. E Confocal fluorescence images of mouse hippocampal neuronal cell line (HT22 cells) co-incubated with Mitotracker Green (1.0 μM) and Mcy1, Mcy2 or Mcy3 (2.0 μM). PCC: Pearson’s colocalization correlations. Mitotracker Green: Ex=488 nm, Em=500‒530 nm. Mcy1‒Mcy3: Ex=561 nm, Em=580‒700 nm. Scale bar =10 μm. F Fluorescence intensity of Mcy3 (5 μM) upon the treatment of physiological ions and amino acids (200 μM for NH4+, Cu2+, Co2+, Ca2+, Mg2+, Fe2+, Fe3+, H2O2 and 5 mM for glucose, glutathione and cysteine). Data are presented as mean values ± SD. n = 3 samples. Source data are provided as a Source Data file.
To study the mitochondrial targeting specificity of the probes, mouse hippocampal neuronal (HT22) cell line was co-stained with Mcy1–3 and Mitotracker Green (Fig. 2E, Supplementary Fig. 2). Among the three compounds, Mcy3 demonstrated the highest mitochondrial targeting ability (Pearson’s colocalization correlations = 0.92), probably attributed to its positive charge and hydrophobicity enhanced by the lipophilic aliphatic chains26. The outstanding selectivity of Mcy3 was confirmed by incubating the probe with diverse biologically relevant analytes (Fig. 2F). Additionally, Mcy3 showed high photostability under continuous laser scanning for 25 min (Supplementary Fig. 3). Consequently, owing to its mitochondrial targeting ability and favorable photophysical properties, Mcy3 was selected as the probe for mitophagy evaluation under confocal laser scanning microscopy.
Monitoring mitochondrial pH values during mitophagy in live cells using Mcy3
Next, we employed Mcy3 to visualize the mitophagy process (Fig. 3A). The emission band centered at 660 nm (red channel) of Mcy3 is relatively weak due to the alkaline environment of the mitochondrial matrix. Upon encapsulation of mitochondria by autophagosomes, mitochondrial pH decreases, leading to a gradual increase in the red channel emission of Mcy3. During the degradation of mitochondria within lysosomes, the highly acidic lumen of lysosomes further amplifies the intensity of the red channel. The intracellular pH response ability of Mcy3 was first validated in HT22 cells. As shown in Supplementary Fig. 4, the red channel increased proportionally with a decrease in pH values from 7.5 to 4.5. A calibration curve correlating pH with fluorescence intensity ratios (red channel/blue channel) was established. Subsequently, Mcy3 was employed for real-time monitoring of mitophagy, with carbonyl cyanide 3-chlorophenylhydrazone (CCCP) serving as a mitophagy inducer4. Upon CCCP treatment, the red channel fluorescence intensity of Mcy3 increased, while the blue channel remained stable, indicating mitochondrial acidification (Supplementary Fig. 5). Next, we explored the specificity of Mcy3 for mitophagy. Bafilomycin A1, a vacuolar ATPase inhibitor, impairs lysosomal acidification and autophagosome-lysosome fusion27. Treatment with CCCP (10 μM) alongside bafilomycin-A1 (100 nM) for a duration of four hours did not alter the fluorescence ratio of Mcy3 in HT22 cells28,29 (Supplementary Fig. 6). Notably, Mcy3 colocalized well with Mitotracker Green post CCCP treatment, indicating Mcy3 anchored on mitochondria tightly during mitophagy (Supplementary Fig. 7). In addition, Mcy3 exhibited a comparable increase in red fluorescence relative to mt-Keima when detecting mitophagy induced by Earle’s Balanced Salt Solution (EBSS) and Deferiprone (DFP), two well-known mitophagy inducers (Supplementary Fig. 8). These findings underscore the pH responsiveness of Mcy3 in enabling real-time imaging of the mitophagy process.

A Mcy3 monitors mitophagy procedures by determining intracellular pH values. Mcy3 first localizes to mitochondria with neutral pH, characterized by a low intensity ratio between the red and blue channels. Upon autophagosome formation, Mcy3 exhibits an increased fluorescence intensity ratio due to the reduced mitochondrial pH. As mitochondria are further degraded within autolysosomes, Mcy3 shows the highest intensity ratio. Figure 3A was created using Adobe Illustrator. B Representative confocal images depicting mitochondria-autophagosome fusion at indicated timepoints. The top row illustrates GFP-LC3, a marker of autophagosomes. Ex: 488 nm, Em: 500‒530 nm. The second row illustrates the fluorescence of Mcy3 (Ex: 561 nm, Em: 580‒700 nm). The third row illustrates the merge of GFP-LC3 and Mcy3. Scale bar = 10 μm. Arrows indicate the regions of mitophagosomes, where mitochondria and autophagosomes fuse. C Representative confocal images depicting mitochondria-autolysosomes fusion at indicated timepoints. The first row illustrates the fluorescence images of Lysotracker, a marker of lysosomes (Ex: 405 nm, Em: 415‒430 nm) in live HT22 cells at selected time-points post treatment. The second row illustrates the fluorescence of Mcy3 (Ex: 561 nm, Em: 580‒700 nm). The third row illustrates the merge of Lysotracker and Mcy3. Scale bar = 10 μm. D Mean counts of mitophagosomes, mitolysosomes, and automitolysosomes per cell at selected time points following CCCP-induced mitophagy. n = 9 cells. Data are presented as mean values ± SD. Source data are provided as a Source Data file. E Mitophagosome counts after CCCP treatment for 2 h and 100 nM Bafilomycin A1 for 2 h. Data are presented as mean values ± SD. Source data are provided as a Source Data file. F pH values of mitochondria, mitophagosomes, and mitolysosomes measured by Mcy3. Data are presented as mean values ± SD. Statistical significance was determined using One-way ANOVA, followed by Tukey’s multiple comparisons test. Source data are provided as a Source Data file.
Subsequently, we monitored the mitochondrial pH values and transfer during CCCP-induced mitophagy. Real-time imaging of GFP-LC3-expressing HT22 cells was conducted to visualize the fusion between mitochondria and autophagosomes. As shown in Fig. 3B, mitochondria indicated by Mcy3 signals (red channel) gradually overlapped with GFP-LC3 puncta (green channel) at 2–3 h post CCCP treatment, revealing the engulfment of mitochondria by autophagosomes. Similarly, the fusion between mitochondria and autolysosomes was studied in HT22 cells through co-incubation of Mcy3 and Lysotracker (Fig. 3C). Moreover, mitochondrial pH during CCCP-induced mitophagy reduced from 7.46 to 5.45 (Supplementary Fig. 9). Additionally, mitochondria underwent a morphological transition from tubular to punctate patterns at the first hour post CCCP induction. To delve deeper into the dynamics of mitophagic intermediates, fusion events and the number of mitophagosomes (colocalization of Mcy3 with GFP-LC3) and mitolysosomes (colocalization of Mcy3 with LysoTracker) were quantified (Fig. 3D). The number of mitophagosomes gradually increased after 2 h of CCCP treatment (from 6.5 to 43.6 per cell) but decreased at 3 h post-treatment, suggesting that a portion of them may have undergone degradation in lysosomes. Similarly, a noticeable increase in mitochondria-autolysosome fusion was observed after 2 h of CCCP incubation with a gradual increase in the number of mitolysosomes (rising from 3.87 to 33 per cell) and automitolysosomes (rising from 3.1 to 24.7 per cell). The mitophagy flux was also quantified according to the previously reported method30,31. Specifically, the basal flux of mitophagosomes was calculated to be 6.7 mitophagosomes/hour/cell (Supplementary Fig. 10), while the induced flux of mitophagosomes was determined to be 61.33 mitophagosomes/hour/cell (Fig. 3E). Next, the pH values of each intermediate were measured. The pH values of intact mitochondria ranged from 7.4 to 7.9, in line with previous reports32. Mitophagosomes and mitolysosomes exhibited varying pH levels, ranging from 5.0 to 7.0 and 4.5 to 6.2, respectively, suggesting a gradual progression of fusion states rather than a distinct biphasic pattern (Fig. 3F). These findings verify that Mcy3 holds promise in monitoring the mitophagy flux by determining the pH levels of mitophagic intermediates.
Deep learning model for identifying mitophagic intermediates by analyzing fluorescence images
To accurately quantify mitophagy flux, it is essential to determine the number, proportion, and spatiotemporal distribution of mitophagic intermediates within the cytoplasm. A deep learning model named dual-branch Multi-scale Attention ResNet (DMAN)33 was designed to classify three distinct mitophagic intermediates by analyzing microscopic images of HT22 cells treated with Mcy3 during mitophagy induction. DMAN was trained using fluorescence images of live cells labeled with Mcy3 and an additional fluorophore targeting the mitophagic intermediate of interest (Fig. 4A, left). The reliability of the training set was validated through transmission electron microscopy (TEM) analysis of mitophagy events (Supplementary Fig. 11). The model effectively distinguished among mitophagic intermediates by analyzing their morphological characteristics and mitochondrial pH variations indicated by Mcy3. The output of DMAN provides both quantitative proportions of intracellular mitophagic intermediates and pseudocolored distribution maps, illustrating insights into their spatial organization within the cytoplasm (Fig. 4A, right).

A Schematic diagram illustrates the operation of the deep learning model DMAN. First, the training sets were established (left). Images of Mcy3-treated HT22 cells with induced mitophagy were collected. Image patches containing colocalized Mcy3 and GFP-LC3 were used as the training set for mitophagosomes. Image patches with colocalized Mcy3 and Mitotracker served as the training set for mitochondria, while image patches containing colocalized Mcy3 and Lysotracker were used as the training set for mitolysosomes. Mcy3 fluorescence signals from these fluorescence image patches were input into the model. During the testing phase, the outputs include the proportions of mitochondria within different mitophagic intermediates and their pseudo-colored distribution maps in live cells. B Illustration of the dual-branch Multi-scale Attention ResNet (DMAN) model. DMAN is built upon the incorporation of the Residual Network (ResNet) and the Convolutional Block Attention Module (CBAM). During the training phase, DMAN employs a multi-scale approach to extract both fine-grained fluorescence intensity features and coarse-grained morphology features. For fluorescence intensity features, image patches of 32 × 32 pixels are decomposed into three RGB channels. For morphological features, single luminance channels from image patches of 128 × 128 pixels are utilized. The DMAN was trained with the image patches co-labeled with Mcy3, GFP-LC3 and Mitotracker Green as training samples. In the inference phase, a sliding window approach is applied to traverse each pixel in the fluorescence image. The classification of each pixel is determined by the attention scores from both a large window and a small window centered around that pixel. C ROC curves and AUC values in the ten-fold cross-validation. D Confusion matrices for the model’s predictions of the three classes in the ten-fold cross-validation.
In order to simultaneously extract pH information, which is reflected as color intensity at the fine-grained level, and organelle morphology information, which is reflected as structural changes at the coarse-grained level, the DMAN model consists of a dual-channel multi-attention framework shown in Fig. 4B. The color branch in the dual-channel decomposes fine-grained image patches into R, G, B components to extract fluorescence intensity information, while the morphology branch converts coarse-grained image patches into luminance components to extract organelle morphology information. Feature extraction for all four components is based on the Residual Network (ResNet), enhanced by the Convolutional Block Attention Module (CBAM), which can be further decomposed into channel attention and spatial attention. This multiple-attention module allows the network to learn and focus on the features most relevant to the mitophagic intermediates.
In the inference stage of DMAN, we estimate the mitophagic intermediates of each pixel in a fluorescence image by proposing a sliding window strategy. That is, the strategy involves centering on a pixel, intercepting its fine-grained and coarse-grained patches, and then feeding them into the trained DMAN model to output the mitophagic intermediates of the current pixel. By using the sliding window strategy, each pixel of the entire fluorescence map is traversed one by one. By learning typical image features in the training phase, DMAN can generate a normalized map based on the probability that a pixel belongs to one of the three intermediates. Since mitophagy is a continuous process, manual-based methods cannot guarantee a quantitative visualization of the mitophagy process according to a uniform standard, which is the significance of introducing deep learning models.
The performance of DMAN was assessed using ROC (Receiver Operating Characteristic) analysis, depicted in Fig. 4C, to evaluate its class discrimination ability. Additionally, Confusion Matrices, shown in Fig. 4D, were utilized to analyze the agreement between predicted and actual classifications, offering a comprehensive assessment of its classification accuracy across various mitophagic intermediates.
AI-FM dynamically evaluates mitophagy flux in live cells
Upon establishing the DMAN model, we initially validated its accuracy in evaluating the mitophagy levels. The output pseudo-colored distribution map generated by DMAN and the mitophagic intermediates stained with corresponding fluorescence tracers as the golden standard were compared (Fig. 5A). The accuracy (ACC) and area under the curve (AUC) of three mitophagic intermediates predicted by DMAN on the test set are depicted in Fig. 5A and Supplementary Fig. 12A. ACC measures the classification correctness by evaluating the ratio of correctly classified pixels to the total predicted pixels. AUC represents the area under the ROC curve, serving as a pivotal evaluation metric for assessing the performance of classification models.

A Fluorescence images of three mitophagic intermediates identified by fluorescence tracers (left), and pseudo-colored intermediate distribution maps generated by DMAN model (right). In the intermediate maps, the blue areas indicate mitochondria, the green areas indicate mitophagosomes, and the red areas indicate mitolysosomes. The calculated average ACC and AUC values of the model for three mitophagic intermediates in the test set are illustrated under the images. B Upper panel: confocal fluorescence images of HT22 cells pretreated with Mcy3 (2.0 μM) following exposure to the mitophagy inducer CCCP for 0, 60, 120, and 180 min. Blue channel: Ex: 561 nm, Em: 580–600 nm. Red channel: Ex: 561 nm, Em: 620–700 nm. Scale bar = 10 μm. Lower panel: the intermediate maps generated by DMAN model. The predicted mitochondria, mitophagosomes, and mitolysosomes are displayed in blue, green, and red colors, respectively. C The compositions of mitochondria, mitophagosomes, and mitolysosomes at indicated time points post CCCP treatment. n = 15 cells per group. Data are presented as mean values ± SD. Source data are provided as a Source Data file. D Upper panel: Representative fluorescence images of HT22 cells pretreated with Mcy3 followed by co-incubation with CCCP and Bafilomycin A1 (100 nM) for 0, 60, 120, and 180 min. Blue channel: Ex: 561 nm, Em: 580–600 nm. Red channel: Ex: 561 nm, Em: 620–700 nm. Scale bar = 10 μm. Lower panel: the intermediate maps generated by DMAN model. E The compositions of mitochondria, mitophagosomes, and mitolysosomes at indicated time points post CCCP and Bafilomycin A1 treatment. n = 15 cells per group. Data are presented as mean values ± SD. Source data are provided as a Source Data file. F Upper panel: confocal fluorescence images of HT22 cells pretreated with Mcy3 (2.0 μM) following exposure to the mitophagy inducer DFP for 0, 8, 14, and 24 h. Blue channel: Ex: 561 nm, Em: 580–600 nm. Red channel: Ex: 561 nm, Em: 620– 700 nm. Scale bar = 10 μm. Lower panel: the intermediate maps generated by DMAN model. G The compositions of mitochondria, mitophagosomes, and mitolysosomes at indicated time points post DFP treatment. n = 11 cells per group. Data are presented as mean values ± SD. Source data are provided as a Source Data file.
Notably, we developed two single-branch models: the fluorescence intensity branch attention ResNet and the morphology branch attention ResNet, achieving average ACC values of 0.82 and 0.77, respectively (Supplementary Fig. 12B, C). In comparison, DMAN achieved higher ACC and AUC, demonstrating that the combination of pH and morphology features provides a more comprehensive and accurate classification.
In this approach, we not only assessed the proportion of mitochondria enclosed within autolysosomes but also accounted for those sequestered within autophagosomes. Confocal fluorescence imaging of HT22 cells treated with Mcy3 (2.0 μM) followed by CCCP (10 μM) resulted in decreased pH accompanied by morphological alternation of mitochondria(Fig. 5B, upper panel). The lower panel shows the DMAN-generated intermediate distribution maps, indicating an increase in the number of mitochondria fused with autolysosomes. As shown in Fig. 5C, the proportion of mitophagosomes and mitolysosomes increased to 25% and 19%, respectively. This result aligns with mitophagy levels measured by using the mt-Keima reporter, which indicated that approximately 20% of mitochondria underwent mitophagy following 3 h of CCCP treatment (Supplementary Fig. 13). Furthermore, we evaluated the efficacy of DMAN under inhibited mitophagy condition. As depicted in Fig. 5D (upper panel), no significant acidification was observed in the mitochondria of HT22 cells co-treated with CCCP (10 μM) and Bafilomycin A1 (100 nM) at the indicated timepoints (0, 60, 120, and 180 min). However, mitochondria exhibited a noticeable morphological shift from a filamentous to a punctate form. The intermediate distribution map generated illustrated the accumulation of autophagosomes, with up to 39% of mitophagosomes (Fig. 5E). The AI-FM system was also used to analyze DFP-induced mitophagy. We observed that the decreased pH was accompanied by morphological alterations in mitochondria 14 h post DFP treatment, indicating an increase in mitophagy levels (Fig. 5F, G). Specifically, the proportion of intact mitochondria decreased from 99% to 75% after 24 h of DFP treatment, while the proportion of mitochondria within autophagosomes and autolysosomes increased to 9.8% and 14.7%, respectively. Overall, AI-FM effectively quantified the dynamic changes in the proportions and distributions of different mitophagic intermediates.
AI-FM identifies potential mitophagy inducers
After the establishment of AI-FM, we subsequently applied it to identify potential mitophagy inducers. Upon investigating the molecular targets of established mitophagy inducers, it was found that sirtuin 1 protein (SIRT1) controls the formation of autophagic vacuoles, delays multiple aging-related diseases, and extends lifespan34. Thus, we targeted SIRT1 protein and implemented a hierarchical virtual screening strategy to identify candidate molecules for mitophagy induction, as illustrated in Fig. 6, step 1. Initially, we developed a consensus pharmacophore model by aligning the conformations of 8 known SIRT1 activators. Using ROCS from OpenEye (v2022), we performed 3D shape similarity matching to filter approximately two million compounds from the ChemDiv library35. Subsequently, we utilized the DUD-E database36 to generate a set of decoys based on 8 established SIRT1 activators. For molecular docking-based virtual screening, we targeted three potential SIRT protein binding sites (PDB code: 5BTR) using Glide from Schrödinger (v2022)37,38. The results demonstrated that the activation site significantly enriched active molecules, thereby validating our SIRT1 activator screening model. Based on the docking outcomes, we retained molecules with scores lower than -6.0 and strain energies below 6.0 kcal/mol39. Similarity calculations were conducted using the MACCS fingerprint, and clustering was performed with a threshold of 0.740. Finally, integrating protein-ligand key interaction analysis, we identified nineteen candidate compounds.

Step 1: virtual screening schematic model for the discovery of SIRT1 agonists. Nineteen hit compounds were identified through screening a library of two million compounds. Step 2: evaluation of the 19 hits utilizing AI-FM. Live cells are incubated with the 19 hit compounds and Mcy3, and fluorescence images are acquired. Subsequently, the images are analyzed using DMAN to assess mitophagy levels after the treatment of hit compounds. Y040-7904 is finally identified as a potential mitophagy inducer. Figure 6 was created in BioRender (Wang, Y. (2025) https://BioRender.com/25zwqfy) and Adobe Illustrator.
Afterward, we employed AI-FM to evaluate the efficacy of the hit compounds (Fig. 6, Step 2). The mitophagy-inducing efficacies were evaluated in HT22 live cells co-treated with Mcy3 and hit compounds by calculating the proportion and distribution of the mitophagic intermediates (Fig. 7A). The quantification of the proportions of mitophagic intermediates was illustrated in Fig. 7B. It is worth noting that two established mitophagy inducers, Urolithin A (UA)10 and UMI-7711 were tested as the positive controls. In cells treated with UA and UMI-77 for 24 h, the proportions of mitophagosomes were 12.7% and 8.6%, while the proportions of mitolysosomes were 13.0% and 16.5%, respectively. Mitophagy induction levels were categorized using existing modulators as reference points. CCCP, which robustly triggers mitophagy and leads to the formation of 20% mitolysosomes, was used as the benchmark for the highest rank of mitophagy inducers. Modulators inducing more than 20% mitolysosome formation were classified as top-tier mitophagy inducers. Based on the effects of UMI-77 and UA, modulators inducing 13‒20% mitolysosome formation were categorized as second-tier mitophagy inducers. Finally, modulators inducing less than 13% mitolysosome formation were classified as third-tier mitophagy inducers.

A Chemical structures and pseudo-colored intermediate distribution maps processed by DMAN after treatment with two established mitophagy inducers and nineteen test hit compounds. Blue: mitochondria, green: mitophagosomes, red: mitolysosomes. B The calculated proportion of two mitophagic intermediates in live HT22 cells after treatment with mitophagy inducers or the hit compounds under investigation. Source data are provided as a Source Data file. C Surface plasmon resonance (SPR) analysis offers concentration-dependent plots of Y040-7904 to SIRT1 protein. Source data are provided as a Source Data file.
Among the nineteen hit compounds under investigation, D715-0267 and Y040-7904 induced a detectable increase in mitolysosomes (20 μM, 24 h). Y040-7904 notably increased the proportions of mitophagosomes (9%) and mitolysosomes (18%), higher than those induced by the two established mitophagy inducers. Another hit compound, D715-0267, increased the number of mitolysosomes (14%) but had little effect on mitophagosomes (4%). In addition, the formation of autophagosomes and fusion events between mitochondria and autophagosomes were observed in both D715–0267 and Y040-7904-treated cells, indicating that both D715-0267 and Y040-7904 are capable of inducing mitophagy (Supplementary Fig. 14A, B). Analysis using AI-FM revealed that Y040-7904 elicited a higher level of mitophagy compared to D715-0267. Based on these findings, Y040-7904 was selected for further investigation. The mitophagy induction by Y040-7904 in HT22 cells was further confirmed using the mitophagy reporter mt-Keima (Supplementary Fig. 15). Surface plasmon resonance (SPR) analysis was conducted to measure the binding affinity between Y040-7904 and SIRT1 protein and the Kd value was determined to be 191.7 nM (Fig. 7C). To summarize, after the in silico screening and the evaluation through AI-FM, a SIRT1 agonist Y040–7904 with potential mitophagy induction efficacy was identified.
Y040–7904 induces mitophagy through the SIRT1/FoxO3 pathway
Next, the pro-mitophagy effect of Y040-7904 was further investigated. As shown in Fig. 8A–G, the administration of Y040-7904 resulted in a dose- and time-dependent increase in the levels of mitophagy-related proteins, including PINK1, Parkin, phosphorylated Ser65-ubiquitin (pUb), and LC3B-II/LC3B–I. In PINK1 silenced cells, the Y040-7904-induced increase in mitophagy-related proteins, such as pUb and LC3, was abolished (Fig. 8D, E, Supplementary Fig. 16), indicating that Y040-7904 might induce mitophagy via the PINK1-PARKIN pathway. Additionally, we observed increased levels of mitochondrial marker MFN2, TIM23, and TOM20 (Supplementary Fig. 17), along with enhanced nuclear localization of PGC1-1α, a key downstream effector of SIRT1 that regulates mitochondrial biogenesis (Supplementary Fig. 18). These findings suggest that Y040-7904 not only induces mitophagy but also enhances mitochondrial biogenesis. This dual effect mirrors that of Nicotinamide Mononucleotide, a well-known SIRT1-activating mitophagy inducer reported to stimulate both mitophagy and mitochondrial biogenesis41.

A Immunoblotting studies of the levels of mitophagy-associated proteins following treatment with Y040-7904 at concentrations ranging from 5 to 20 μM for 24 h. B Quantification of mitophagy-associated proteins in (A) was performed using Image J software. Data are presented as mean values ± SD. n = 4 independent biological replicates. Statistical significance was determined using two-tailed paired ttest. Source data are provided as a Source Data file. C Western blots of the levels of phosphorylated ubiquitin (p-Ser65-ubiquitin) following treatment with Y040-7904 at concentrations ranging from 5 to 20 μM for 24 h. Representative immunoblot from 3 independent experiments with similar results. D Immunoblotting studies of the levels of LC3II/I and PINK1 following treatment with Y040-7904 in si-NC and si-PINK1-transfected HT22 cells. Representative immunoblot from 2–4 independent experiments with similar results. E Immunoblotting studies of the levels of phosphorylated Ser65-ubiquitin (pUb) following treatment with Y040-7904 in si-NC and si-PINK1-transfected HT22 cells. Representative immunoblot from 3 independent experiments with similar results. F Immunoblotting studies of the levels of mitophagy- associated proteins at various time points (6 h, 10 h, 20 h, and 24 h) post Y040-7904 treatment (20 μM). G Quantification of mitophagy-associated proteins after Y040-7904 treatment in F. Data are presented as mean values ± SD. n = 5–7 independent biological replicates. Statistical significance was determined using two-tailed paired t test. Source data are provided as a Source Data file. H Immunofluorescence images of FoxO3 in control and Y040-7904 (20 μM) treated HT22 cells. Representative fluorescence image from 5 independent captures showing similar data. Scale bar = 10 μm. I Changes in FoxO3 protein in the nucleus and cytoplasm of HT22 cells after Y040-7904 treatment (4 h, 6 h). Representative immunoblot from 3 independent experiments with similar results. J The levels of FoxO3 and LC3 in HT22 cells treated with Y040-7904 or FoxO3 inhibitor AS1842856. K Quantification of indicated proteins in (J). Data are presented as mean values ± SD. n = 3 independent biological replicates. Statistical significance was determined using One-way ANOVA followed by Dunnett’s multiple comparisons test. Source data are provided as a Source Data file. L The illustration describes the mechanism of Y040–7904-induced mitophagy (created using Adobe Illustrator).
Next, we tried to explore the underlying mechanism through which Y040–7904 modulates the PINK1-PARKIN pathway. Forkhead box protein O3a (FoxO3a) is a transcription factor that regulates the expression of PINK1 and Parkin, thereby playing a crucial role in mitophagy42,43. Moreover, the transcription activity of FoxO3a is significantly modulated by SIRT1 in mammalian cells44. Considering the binding of Y040-7904 to SIRT1, we hypothesized that Y040-7904-induced mitophagy may be attributed to FoxO3a. As expected, Y040-7904 significantly promoted the nuclear localization of FoxO3a without affecting the expression of SIRT1 and FoxO3a (Fig. 8H, Supplementary Fig. 19). Consistently, an increase in nuclear FoxO3a levels, accompanied by a concomitant decrease in cytoplasmic FoxO3a levels, was observed in Y040–7904-treated cells (Fig. 8I, Supplementary Fig. 20).
To explore whether the SIRT1-FoxO3a axis is responsible for the pro-mitophagy effect of Y040-7904, the impact of FoxO3 inhibitor AS1842856 on Y040- 7904-induced mitophagy was analyzed. The nuclear translocation of FoxO3a and the up-regulation of LC3II/LC3I induced by Y040-7904 were effectively blocked by AS1842856 (Supplementary Fig. 21, Fig. 8J, K). In addition, Y040-7904 failed to upregulate the nuclear translocation of FoxO3a in SIRT1-silenced cells (Supplementary Fig. 22). These findings demonstrated that Y040-7904 depends on the SIRT1-FoxO3a axis to facilitate the expression of mitophagy-related proteins PINK1 and Parkin, ultimately resulting in an elevation in mitophagy levels (Fig. 8L).
Y040-7904 ameliorates amyloid pathology in both AD cells and Caenorhabditis. elegans models
The crucial regulatory role of mitophagy in AD has sparked our keen interest in the potential therapeutic effect of Y040-7904 on AD-related pathology. The Neuron-2a (N2a) cell line, which have been genetically modified to overexpress the amyloid precursor protein (APP) with Swedish familial double mutations (APP K595N/M596L)45, exhibits an excessive production of APP, APPC-terminal fragment C99 (C99-APP), and β-amyloid proteins along with downregulation of mitophagy-related proteins (Fig. 9A–C, Supplementary Fig. 23), making it a suitable cellular model for AD46. As shown in Fig. 9D, Y040-7904 treatment significantly reduced the levels of APP and its C-terminal fragment C99 in N2a/APPswe cells. Moreover, Y040-7904 effectively decreased intracellular Aβ1-42 levels in a dose-dependent manner (Fig. 9E, Supplementary Fig. 24). These findings underscore the potential of Y040–7904 as a promising agent for mitigating APP and Aβ1-42 accumulation, suggesting its therapeutic relevance in addressing AD pathology.

A A cellular model of AD with APP Swedish mutation was established in Neuron-2a cells (N2a/APPswe cells). Figure 9A was created using Adobe Illustrator. B Western blots demonstrate the expression of APP and C99-APP in N2a/APPswe cells. Data are presented as mean values ± SD. n = 4 independent biological replicates.Statistical significance was determined using two-tailed paired t test. Source data are provided as a Source Data file. C Quantification of Aβ1-42 concentrations through ELISA study in N2a/APPswe cells. Data are presented as mean values ± SD. n = 4 independent biological replicates. Statistical significance was determined using two-tailed unpaired t test. Source data are provided as a Source Data file. D Western blots of amyloid precursor protein (APP) and C99-APP levels in N2a/APPswe cells after Y040-7904 treatment (5 μM‒20 μM). Data are presented as mean values ± SD. n = 3–4 independent biological replicates. Statistical significance was determined using two-tailed paired t test. Source data are provided as a Source Data file. E Levels of Aβ1-42 in N2a/APPswe cells as a function of Y040-7904 concentration. Data are presented as mean values ± SD. n = 2 independent biological replicates. Statistical significance was determined using One-way ANOVA followed by Dunnett’s multiple comparisons test. Source data are provided as a Source Data file. F Western blots depict the levels of β-amyloid 1-42 in the CL4176 worms after Y040-7904 treatment (100‒1000 μM). G Quantification of β-amyloid 1-42 in panel F. Data are presented as mean values ± SD. n = 3 independent biological replicates. Statistical significance was determined using One-way ANOVA followed by Dunnett’s multiple comparisons test. Source data are provided as a Source Data file. H Percentage of paralysis in CL4176 worms treated or non-treated with Y040-7904. Data are presented as mean values ± SD. n = 2 biological replicates, each consisting of ≥100 worms. Statistical significance was determined using two-tailed unpaired t test. Source data are provided as a Source Data file. I Percentage of paralysis in CL2006 worms treated or non-treated with Y040-7904. n = 2 biological replicates, each consisting of ≥100 worms. Data are presented as mean values ± SD. Statistical significance was determined using two-tailed unpaired t test. Source data are provided as a Source Data file. J Western blots depict the levels of mitophagy-associated proteins in N2a/APPswe cells after Y040-7904 treatment (5‒20 μM). Representative immunoblot from 3 independent experiments with similar results. K Western blots depict the levels of APP in N2a/APPswe cells after Y040-7904 treatment with or without PINK1 silencing. Data are presented as mean values ± SD. n = 3 independent biological replicates. Statistical significance was determined using One-way ANOVA followed by Tukey’s multiple comparisons test. Source data are provided as a Source Data file.
Next, we evaluated the in vivo therapeutic potential of Y040-7904 using transgenic C. elegans strains CL417647 and CL200648. The CL4176 strain expresses human Aβ1-42 in muscle cells under a temperature-inducible system. After temperature upshifting (from 16 °C to 25 °C), CL4176 worms overexpress Aβ1-42 (Supplementary Fig. 25) and exhibit paralysis. As shown in Fig. 9F–G, Y040-7904 treatment resulted in a dose-dependent reduction in Aβ1-42 levels in CL4176 worms. At 30 h post temperature upshift, the paralysis rate of CL4176 worms exposed to 1000 μM Y040–7904 was 76.2%, compared to 89.5% in untreated worms (Fig. 9H). In the CL2006 strain, which constitutively expresses Aβ in body wall muscle cells and exhibits age-related progressive paralysis, treatment with 1000 μM Y040-7904 reduced paralysis by 29.3% compared to untreated controls (Fig. 9I). The above findings indicate that Y040-7904 holds therapeutic potential for mitigating Alzheimer’s disease pathology.
Considering that Y040-7904 is a mitophagy inducer, we hypothesized that its impact on N2a/APPswe cells may be attributed to its ability to promote mitophagy. We first assessed the levels of Parkin, PINK1, and LC3II/I in N2a/APPswe cells after Y040-7904 treatment. The results showed that Y040-7904 treatment significantly upregulated the levels of these proteins (Fig. 9J). TEM analysis revealed that Y040–7904–treated N2a/APPswe cells exhibited a greater increase in mitophagosomes/mitolysosomes (4.6 per cell) compared to autophagosomes/autolysosomes (0.8 per cell) (Supplementary Fig. 26). Mitochondrial morphology analysis revealed Y040-7904 treatment increased the number of healthy, tubular-shaped mitochondria with a uniform matrix, indicating restored mitochondrial quality (Supplementary Fig. 27). More importantly, PINK1 silencing reversed the Y040-7904-induced reduction of APP in N2a/APPswe cells (Fig. 9K), supporting the involvement of mitophagy in regulating APP degradation. In addition, silencing of SIRT1 significantly impaired the Y040-7904 induced nuclear translocalization of FoxO3a and attenuated the reduction in Aβ accumulation (Supplementary Figs. 28, 29). These findings collectively demonstrate that Y040-7904 exerts a protective role against amyloid pathologies by enhancing mitophagy via the SIRT1-FoxO3 pathway.
Discussion
Fluorescence reporters have been widely used to track mitophagy both in vivo and in vitro. Several transgenic mouse models, such as the Mt-keima mouse21 and mito-QC mouse49, have been developed to study mitophagy in tissues like the heart. Similarly, Aoyagi et al.50 generated a beta cell-specific mitophagy reporter mouse (CMMR mouse) to assess mitophagy levels in beta cells under metabolic stress. Despite these advances, in vivo quantification of mitophagy still faces challenges in achieving high-resolution, quantitative analysis of mitophagy intermediates, highlighting the need for improved imaging technologies and standardized measurement methods51. In living cells, various fluorescence reporters, including RFP-GFP-LC3, mt-Keima, and small molecular probes, are used to monitor mitophagy (Supplementary Table 1). For example, the fluorescent protein RFP-GFP-LC3 was engineered to monitor autophagosome dynamics based on the pH ratiometric signals between GFP and RFP proteins20. However, RFP-GFP-LC3 lacks specificity and cannot distinguish mitophagy from non-selective macroautophagy22. While mitochondrial-specific probes detect mitophagy by targeting acidic mitochondria or mitolysosomes, they fail to identify mitophagosomes, and factors like mitochondrial acidification may compromise their accuracy. Regarding existing quantitative approaches for measuring mitophagy flux, multiple fluorescence stains for mitochondria, autophagosomes, and lysosomes are commonly used. However, this method has limitations, including fluorescence spectral overlap, potential cytotoxicity and phototoxicity from extensive labeling, and the time-consuming and prone-to-error nature of manual data analysis.
To address these issues, we introduce a deep learning model, DMAN, which allows for the simultaneous monitoring of all mitophagic intermediates using a single fluorescence dye. DMAN extracts and integrates mitochondrial pH values and morphologies at different spatial scales, allowing for a quantitative and intuitive assessment of mitophagy. We applied a multi-scale sliding window approach to analyze the fluorescence images, sliding pixel-by-pixel, and fed the multi-scale image patches within each window into DMAN for recognition. Once the window has traversed the entire image, pixel-level classification is achieved, capturing the dynamic changes of mitophagy. Compared to previous models52, DMAN offers several advantages: 1) DMAN simultaneously captures both mitochondrial pH and multi-scale morphology information to accurately identify mitophagic intermediates; 2) DMAN employs a single-pixel multi-scale sliding window approach to achieve quantitative and visual assessment of the three mitophagic intermediates in one image using a single fluorescent dye. Compared with the single-branch model that solely extracts morphology features, DMAN increased the average accuracy by 12%, underscoring the importance of integrating functional imaging and morphology information. For transitional states in the mitophagy process, DMAN can provide highly informative visual results. Therefore, integrating fluorescent probes with a deep learning model to form AI-FM provides a powerful strategy for simultaneously identifying and visualizing mitophagic intermediates in live cells.
Traditional mitophagy reporters like mt-Keima are valuable tools for compound screening and have identified several modulators such as UMI-7711. However, AI-FM provides enhanced accuracy and efficiency. In AD pathology research, AI-FM provides critical insights into the specific stages at which mitophagy is impaired under different pathological conditions. This is important, as impaired mitophagy typically arises from disruptions at distinct intermediates53. In drug screening, AI-FM assesses how compounds affect mitophagy at different stages, offering deeper insights into their therapeutic potential and mechanisms. This capability is invaluable, as different mitophagy modulators can exert varying impacts on these intermediates18. For instance, AI-FM identified that 24-hour Y040-7904 treatment led to 9% mitophagosomes and 18% mitolysosomes formation. This suggests that mitophagy progression following Y040-7904 treatment is characterized by a marked accumulation of mitolysosomes. Additionally, AI-FM integrates pH and morphology data, improving detection accuracy, reducing false positives, and enabling faster and automated analysis of large chemical libraries.
SIRT1, an NAD + -dependent protein deacetylase, activates PINK1/Parkin-dependent mitophagy both through PGC1α-mediated mitochondrial biogenesis and FoxO3-mediated transcription of mitophagy-related genes54. Small molecular SIRT1 activators, such as nicotinamide, resveratrol, and fisetin, have been reported to promote mitophagy without perturbing mitochondrial function4. Therefore, SIRT1 was selected as the target in the silico screening for mitophagy inducers. In this work, Y040-7904, identified through in silico screening and AI-FM-based screenings, demonstrated superior efficacy in triggering mitophagic flux. Notably, unlike widely used mitophagy modulators such as CCCP, valinomycin, and antimycin A, which collapse mitochondrial membrane potential by disturbing the electrochemical or proton gradient55, Y040-7904 does not induce mitochondrial stress but enhances mitochondrial biogenesis instead. Additionally, Y040-7904 ameliorates amyloid pathologies and restores mitochondrial quality in AD cellular and C. elegans models through activation of the SIRT1-FoxO3 pathway. These findings support Y040-7904 as a promising candidate for mitophagy induction and drug discovery in AD.
We also note several limitations in this work. First, ranking candidate drugs based on their impact on the degree of mitophagy still lacks clear guidelines. Further accurate delineation of the boundary between basal and drug-enhanced mitophagy flux may necessitate analyzing established mitophagy inducers using AI-FM. Second, training and validation datasets comprised only typical images of three mitophagic intermediates. However, test images often encompass transitional states between different intermediates. Although DMAN can output the probability that each image patch belongs to a certain stage of mitophagy based on a uniform criterion by learning from typical images, the accuracy of the test results is still affected by the accuracy of manual labeling. Under the existing technical conditions, accurate manual labeling is difficult to guarantee. Future research could enhance AI-FM by extending its application to quantify mitophagy flux using additional types of mitophagy probes. Incorporating data from probes like mt-Keima alongside Mcy3 would allow for a comparative analysis of their efficiency and robustness. Additionally, AI-FM can improve accuracy and specificity by analyzing super-resolution fluorescence images56, providing detailed insights into organelle morphology and spatial distribution, thus enhancing classification accuracy.
In summary, we proposed an AI-assisted fluorescence microscopic system, AI-FM as a powerful tool for dynamically assessing mitophagic flux in living cells. This system quantitatively analyzes mitophagic flux by utilizing mitochondrial pH values and morphology, aiding in the screening of promising mitophagy inducers. The application of AI-FM holds significant promise for elucidating the mechanisms underlying mitophagy-related pathologies and providing therapeutic strategies for neurodegenerative diseases.
Methods
Absorption spectra
Working solutions (1 × 10−5 M) of Mcy1–3 were prepared by diluting the stock solutions (1 × 10−3 M in DMSO) in PBS with pH ranging from 3.0 to 8.0. Absorption spectra were recorded using a Shimadzu UV-2550 spectrophotometer equipped with a quartz cuvette at room temperature. The scanning wavelength range is 400–900 nm and scanning speed is 1.0 nm/s (slit width: 5 nm). Baseline correction was performed using PBS as a blank.
Emission spectra
Working solutions (1 × 10−5 M) of Mcy1–3 were prepared by diluting the stock solutions (1 × 10−3 M in DMSO) in PBS with pH ranging from 3.0 to 8.0. All emission spectra were performed using a Shimadzu RF-5301PC fluorophotometer with a photomultiplier tube at room temperature. All compounds were excited at maximum absorption wavelength.
Calculations of pKa values
The normalized fluorescent intensities were plotted against pHs and the sigmoidal plots were fitted into sigmoidal curves (Eq. 1) with OriginPro 9.0. \({A}_{1}\) and \({A}_{2}\) represent the maximum and minimum fluorescence intensity respectively. The pKa values were determined as \({x}_{0}\) from this equation.
\(x\): pH; \({{{\rm{y}}}}\): fluorescence intensities at \(x\); \({d}_{x}\): time constant; \({A}_{1}\): initial value of fluorescence intensity; \({A}_{2}\): final value of fluorescence intensity.
Cell culture
HT22 cells and Neuron-2a (N2a)cells were purchased from the American Type Culture Collection (ATCC) and cultured in Dulbecco’s modified Eagle’s medium (MeilunBio) supplemented with 10% fetal Bovine Serum (FBS) (ExCell Bio), 1% penicillin, and streptomycin (MeilunBio) at 37 °C in a humidified atmosphere with 5% CO2/95% air.
Lentiviral transfection of cells
HT22 cells were transduced to overexpress EGFP-LC3 using lentivirus (Hanbio, titer: 1 × 108 TU/mL). The day before transfection, HT22 cells were seeded into 24-well plates and cultured overnight. The next day, transfection was performed at a cell density of between 50–70%. Transfection steps were performed in strict accordance with the manufacturer’s instructions at a multiplicity of infection (MOI) value of 80 in the presence of 5 μg/mL Polybrene (Hanbio). The medium was refreshed 6 h later and the cells were cultured for a further 24 h. Transfected cells were selected with puromycin (2 μg/mL) for 2 days until resistant colonies remained. EGFP-LC3 expression was confirmed by fluorescence microscopy.
HT22 cells were transduced to overexpress mt-Keima using lentivirus (Hanbio, titer: 1 × 108 TU/mL). The day before transfection, HT22 cells were seeded into 24-well plates and cultured overnight. The next day, transfection was performed at a cell density of between 50–70%. Transfection steps were performed in strict accordance with the manufacturer’s instructions at a multiplicity of infection (MOI) value of 50 in the presence of 5 μg/mL Polybrene (Hanbio). The medium was refreshed 6 h later and the cells were cultured for a further 24 h. Transfected cells were selected with puromycin (2 μg/mL) for 2 days until resistant colonies remained. mt-Keima expression was confirmed by fluorescence microscopy.
N2a cells were transduced to overexpress APP Swedish mutation protein using lentivirus (OBiO Technology, titer: 1 × 108 TU/mL). The day before transfection, N2a cells were seeded into 24-well plates and cultured overnight. The next day, transfection was performed at a cell density of between 50–70%. Transfection steps were performed in strict accordance with the manufacturer’s instructions at a multiplicity of infection (MOI) value of 50 in the presence of 5 μg/mL Polybrene (Hanbio). The medium was refreshed 6 h later and the cells were cultured for a further 24 h. Transfected cells were selected with puromycin (2 μg/mL) for 2 days until resistant colonies remained. APP expression was confirmed by fluorescence microscopy.
siRNA-mediated knockdown of SIRT1 and PINK1 in cells
N2a/APPswe cells were transfected with siRNA to knock down SIRT1 expression. SIRT1-targeting siRNA (si-SIRT1) or non-targeting control siRNA (si-NC) were purchased in RiboBio Co., Ltd. Cells were seeded in a 6-well plate to achieve 50–60% confluence. The next day, siRNA (100 nM) and Lipofectamine RNAiMAX (Thermo Fisher, 2 μL/well) were diluted in Opti-MEM (Thermo Fisher) and incubated for 15 min at room temperature before adding to cells. After 4–6 h post-transfection, the media were replaced with fresh DMEM containing 10% FBS and 1% Penicillin-Streptomycin. Cells were incubated for 48–72 h to allow for effective knockdown. Knockdown efficiency was verified by Western blots.
N2a/APPswe cells were transfected with siRNA to knock down PINK1 expression. PINK1-targeting siRNA (si-PINK1) or non-targeting control siRNA (si-NC) was purchased in RiboBio Co., Ltd. Cells were seeded in a 6-well plate to achieve 50–60% confluence. The next day, siRNA (100 nM) and Lipofectamine RNAiMAX (Thermo Fisher, 2 μL/well) were diluted in Opti-MEM (Thermo Fisher) and incubated for 15 min at room temperature before adding to cells. After 4–6 h post-transfection, the media were replaced with fresh DMEM containing 10% FBS and 1% Penicillin-Streptomycin. Cells were incubated for 48–72 h to allow for effective knockdown. Knockdown efficiency was verified by western blot analysis.
shRNA-mediated knockdown of PINK1 in N2A/APP cells
The PINK1-targeting shRNA plasmid (pLKO.1-shPINK1) was kindly provided by Prof. Yunzeng Zou (Zhongshan Hospital, Fudan University). N2a/APP cells were seeded in 6-well plates at a density of 2 × 105 cells per well and allowed to grow to approximately 60–70% confluence. Cells were then transfected with shPINK1 (1 μg/well) using Lipofectamine™ 3000 transfection reagent (Thermo Fisher, 2 μL/well). After 24 h of transfection, the effectiveness of the knockdown was confirmed by western blot analysis.
Live cell microscopy
Cells were cultured in 3.5 mm glass bottom dishes (Cellvis) for confocal microscopic imaging. Fluorescence imaging was performed on a laser scanning confocal microscope Carl Zeiss LSM980 equipped with a 63× oil objective, and Olympus FV3000 equipped with a 60× oil objective. Fluorescence images were analyzed by ImageJ software.
Intracellular pH calibration
HT22 cells were pretreated with DMEM containing Mcy3 (5 μM) for 1 h. Then the media were replaced with high K+ buffer (30 mM NaCl, 120 mM KCl, 1.0 mM CaCl2, 0.50 mM MgSO4, 1.0 mM NaH2PO4, 5.0 mM glucose, 20 mM sodium acetate, 20 mM HEPES, 10 μM nigericin, and 20 mM MES) of various pH values. After 15 min of incubation, the cell imaging was carried out in the confocal fluorescence microscope (Ex = 561 nm).
In silico screening of mitophagy inducers
We generated a decoy set of 8 established SIRT1 activators using the approach provided by the DUD-E website (https://dude.docking.org/generate). Subsequently, we performed virtual screening tests based on molecular docking on the three potential binding sites of the SIRT protein (PDB code: 5BTR). This task was conducted by using Glide module in the software Schrödinger (Schrödinger, Inc., v2022). The test outcomes indicated a significant enrichment effect on the activation site, which affirmed that the model is suitable for virtual screening of SIRT1 activators.
To filter the commercial compound library ChemDiv, we created the consensus pharmacophore model by overlapping the conformations of the 8 established SIRT activators. We used the ROCS module of the software OpenEye (v2022) for 3D shape similarity matching to accomplish this. After filtering, we docked the remaining compounds within the activation site of the SIRT1 protein, retaining only results with a docking score below -6.0, and further filtered for strain energy (total strain energy <6.0 kcal/mol). We calculated similarity using the MACCS molecular fingerprint and clustered the remaining results based on a threshold of 0.7. Finally, we selected 19 candidate compounds by manually inspecting the binding mode of small molecules and SIRT proteins.
Western blot assay
Western blot assay was conducted following established procedures. Samples from diverse cell lines were collected and prepared using 1×RIPA buffer (Beyotime) containing 1% PMSF (Beyotime) and 1% phosphatase inhibitors (Abcam). Protein concentration was determined using the BCA assay (Beyotime) according to the manufacturer’s protocol: protein samples and BSA standards (0–2000 μg/mL) were mixed with BCA working reagent (1:8 ratio) in 96-well plates, incubated at 37 °C for 30 min, and absorbance measured at 562 nm; sample concentrations were calculated from the standard curve. Equal amounts of protein (20 μg per lane) were mixed with 4×loading buffer (Beyotime), boiled at 95 °C for 5 min, and centrifuged briefly before loading onto gels. Proteins were subsequently separated on a SuperPAGE 4–20% Bis-Tris Protein Gel at 120 V running for 1 h and transfer system was set at 300 mA for 0.5 h (for proteins under 30 kDa) on a PVDF membrane (Merck), 1 h (for proteins from 30 kDa to 100 kDa) and 2 h (for proteins between 100 kDa and 180 kDa) on a NC membrane (Cytiva) respectively. Membranes were then probed with a variety of antibodies. Chemiluminescence detection was performed using a ChemiDoc Touch System (Bio-Rad Laboratories). Quantification was performed using ImageJ. Primary antibodies utilized were as follows: APP antibody (PA5-17829, 51-2700, Invitrogen), APP C-Terminal Fragment Antibody (802803, Biolegend), FoxO3a antibody (2497S, Cell Signaling Technology), PINK1 antibody (23274-1-AP, Proteintech), Parkin antibody (66674-1-Ig, Proteintech), LC3B antibody (83506S, Cell Signaling Technology), SIRT1 antibody (8469S, Cell Signaling Technology), Mitofusin-2 antibody (9482S, Cell Signaling Technology), Tom20 antibody (42406S, Cell Signaling Technology), β-Amyloid antibody (8243S, Cell Signaling Technology), Phospho-Ubiquitin (Ser65) antibody (62802S, Cell Signaling Technology), GAPDH antibody (ab9485, Abcam) and β-Tublin antibody (AF2835, Beyotime). The second antibodies used include anti-mouse immunoglobulin G (IgG) and anti-rabbit IgG. Primary antibodies were used at 1:2000 dilution while secondary antibodies at 1:10000 dilution.
Immunofluorescence
For immunofluorescence staining, cells were fixed with 4% paraformaldehyde (Servicebio) for 15 min, then permeabilized with 0.1% Triton X-100 (Beyotime) for 10 min. Non-specific binding sites was blocked by incubating the cells with 1% BSA (Beyotime) for 30 min. Thereafter, cells were incubated overnight with specific primary antibodies (1:200) at 4 °C. Following four washes with PBS, cells were exposed to Alexa Fluor-conjugated secondary antibodies (1:1000) for 1 h at room temperature while shielded from light. Nuclei were stained with DAPI (Servicebio). Primary antibodies used include anti-β-Amyloid 1-42 antibody (44-344, Invitrogen), and FoxO3a antibody (2497S, Cell Signaling Technology).
Enzyme-linked immunosorbent assay (ELISA)
The Aβ1-42(Amyloid Beta 1-42) ELISA Kit (Elabscience) was used for quantifying the Aβ1–42 concentration in N2a/APPswe cells. Cells were washed with pre-cooled PBS and dissociate them using trypsin. The cell suspension was washed for 5 min at 1000×g. The medium was discarded and cells were washed 3 times with pre-cooled PBS. For each 1 × 106 cell, 150–250 μL of pre-cooled PBS was added. Cells were lysed by freeze-thaw cycles or ultrasonic disruption. The lysate was centrifuged for 10 min at 1500 × g at 2–8 °C. Cell fragments was removed and the supernatant was collected for the assay. The method followed the instructions provided by the ELISA kit. Standards or samples were added to a 96-well plate pre-coated with antibodies and incubated for 90 min at 37 °C. After removing the liquid, the biotinylated detection working solution was added to each well and incubated for 60 min at 37 °C. The plate was then washed three times, followed by the addition of the HRP conjugate working solution, and incubated for 30 min at 37 °C. After washing the plate five times, the substrate reagent was added and incubated for 15 min at 37 °C. Finally, the reaction was stopped by adding the stop solution in equal volume, and absorbance was measured at 450 nm.
Surface plasmon resonance technology
Surface plasmon resonance technology (SPR) was employed to determine the binding affinity KD between the molecule Y040-7904 and the receptor SIRT1 using Biacore system (Biacore T200, Sweden). SIRT1 receptors were immobilized on a CM5 sensor chip (GE Healthcare, WI, USA) in HBS-EP+ running buffer (0.01 M HEPES, 0.15 M NaCl, 3 mM EDTA, 0.05% v/v surfactant P20, pH 7.4) at a flow rate of 10 μL/min at 25 °C using the amine coupling kit. Y040-7904 was dissolved in DMSO to a concentration of 20 mM as the stock solution. Working solutions of Y040-7904 were prepared by diluting the stock solution with HBS-EP+ buffer. Each binding cycle between free Y040-7904 and SIRT1 was conducted in HBS-EP+ running buffer at a flow rate of 30 μL/min at 25 °C, with a contact time of 30 s and a dissociation time of 45 s.
C. elegans western blot and paralysis assays
The Caenorhabditis elegans (C. elegans) strains, CL2006 [Punc-54::human A-beta 3–42] and CL4176 [Pmyo-3::human Amyloid beta 1–42], which overexpress human amyloid-beta (Aβ), were employed as the in vivo models. CL4176 worms treated with vehicle or Y040-7904 (100–1000 μM) were maintained at 16 °C for 36 h, followed by a temperature shift to 25 °C for an additional 36 h. For Western blot analysis, 200 worms were collected per group, washed with M9 buffer, mixed with SDS-PAGE loading buffer, boiled at 95 °C for 10 min, and stored at −20 °C until analysis.
For paralysis assays in CL4176 worms, the number of paralyzed worms was recorded 36 h after the temperature shift to 25 °C. More than 100 worms were analyzed per group.
For paralysis assays in CL2006 worms, animals treated with vehicle or Y040–7904 (500–1000 μM) were maintained at 20 °C for 96 h, and the number of paralyzed worms was counted. More than 100 worms were analyzed per group.
DMAN model
We propose a dual-branch Multi-scale Attention ResNet (DMAN) model, which is based on the Residual Network (ResNet) and the Convolutional Block Attention Module (CBAM). This model quantitatively and visually assesses the level of mitophagy from two perspectives: color intensity, reflecting the pH value, and textural information, reflecting organelle morphology and structure. Specifically, the process begins by extracting Mcy3 signals from cell images with gold standards, obtaining original training data with a resolution of 1024 × 1024. Subsequently, the original images are cropped into image patches of 32 × 32 and 128 × 128 pixels, and image patches containing significant organelle information are chosen as the training set.
During the model training and validation phase, the 32 × 32 pixel image patches are mapped to the RGB color space to capture fluorescence intensity information, while the 128 × 128 pixel image patches are mapped to the Lab color space to extract pixel values in the luminance dimension, capturing organelle morphology information. Subsequently, the pH information and organelle morphology information are fed into the color branch and morphology branch of the DMAN, respectively. Within DMAN, the CBAM utilizes channel attention and spatial attention mechanisms, allowing the network to learn and focus on the most critical features in the process of mitophagy. Finally, the features extracted by the dual-branch network are concatenated and processed through average pooling to classify mitochondrial autophagy-related aspects within the images.
During the inference phase of the model, we employ a 32 × 32 sliding window (Window A) and a 128 × 128 sliding window (Window B) of a pixel-centered to slide over the test images pixel by pixel. Window A and Window B are then input into the trained DMAN model to determine the mitophagic intermediates for each pixel in the image. Specifically, the initial step involves setting empirical parameters (pixel threshold α = 13 and darkness threshold β = 0.9) to identify background pixels based on a statistical analysis of background brightness and color. This step helps mitigate the influence of extensive background regions on the classification of mitochondrial autophagy. Subsequently, the pH information extracted by Window A and organelle morphology information extracted by Window B are input into the trained DMAN model, which outputs the classification probabilities for the three mitophagy intermediates for the current pixel. Finally, customized color palettes are applied based on these classification probabilities to label different organelles. This enables the identification and computation of the proportions of mitochondria, autolysosomes, and autophagosomes within the images, thereby elucidating the dynamic changes in mitophagy.
Statistical analysis
Values are presented as mean with SD. Statistical analysis was performed using Origin 2021 software (Microcal Software Inc., Northampton, USA), and GraphPad Prism 8.0. P values were computed using two-tailed t test or one-way analysis of variance. P values < 0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the data generated in this study are provided in the Supplementary Information. Source data are provided with this paper.
Code availability
The code used to develop the model, perform the analyses and generate results in this study is publicly available and has been deposited in Github at https://github.com/sd-spf/DMAN, under Apache-2.0 license. The specific version of the code associated with this publication is archived in Zenodo and is accessible via 10.5281/zenodo.15206014.
References
Shevtsova, E. F., Maltsev, A. V., Vinogradova, D. V., Shevtsov, P. N. & Bachurin, S. O. Mitochondria as a promising target for developing novel agents for treating Alzheimer’s disease. Med. Res. Rev. 41, 803–827 (2021).
Misgeld, T. & Schwarz, T. L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 96, 651–666 (2017).
Youle, R. J. & Narendra, D. P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 (2011).
Georgakopoulos, N. D., Wells, G. & Campanella, M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 13, 136–146 (2017).
Um, J. H. & Yun, J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. 50, 299–307 (2017).
Wang, Y., Wang, P. & Li, C. Fluorescence microscopic platforms imaging mitochondrial abnormalities in neurodegenerative diseases. Adv. Drug Deliv. Rev. 197, 114841 (2023).
Kerr, J. S. et al. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 40, 151–166 (2017).
Gwon, A.-R. et al. Oxidative lipid modification of nicastrin enhances amyloidogenic γ-secretase activity in Alzheimer’s disease. Aging Cell 11, 559–568 (2012).
Xie, C. et al. Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat. Biomed. Eng. 6, 76–93 (2022).
Fang, E. F. et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412 (2019).
Cen, X. et al. Pharmacological targeting of MCL-1 promotes mitophagy and improves disease pathologies in an Alzheimer’s disease mouse model. Nat. Commun. 11, 5731 (2020).
Palikaras, K. & Tavernarakis, N. Regulation and roles of mitophagy at synapses. Mech. Ageing Dev. 187, 111216 (2020).
Lee, J. H. et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Abeta in neurons, yielding senile plaques. Nat. Neurosci. 25, 688–701 (2022).
Goetzl, E. J. et al. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 85, 40–47 (2015).
Onishi, M., Yamano, K., Sato, M., Matsuda, N. & Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 40, e104705 (2021).
Santo-Domingo, J. & Demaurex, N. Perspectives on: SGP symposium on mitochondrial physiology and medicine: the renaissance of mitochondrial pH. J. Gen. Physiol. 139, 415–423 (2012).
Tanida, I. & Waguri, S. Measurement of autophagy in cells and tissues. Methods Mol. Biol. 648, 193–214 (2010).
Maulucci, G. et al. Quantitative analysis of autophagic flux by confocal pH-imaging of autophagic intermediates. Autophagy 11, 1905–1916 (2015).
Saric, A., Grinstein, S. & Freeman, S. A. Measurement of Autolysosomal pH by Dual-Wavelength Ratio Imaging. Methods Enzymol. 588, 15–29 (2017).
Dolman, N. J., Chambers, K. M., Mandavilli, B., Batchelor, R. H. & Janes, M. S. Tools and techniques to measure mitophagy using fluorescence microscopy. Autophagy 9, 1653–1662 (2013).
Sun, N. et al. Measuring In Vivo Mitophagy. Mol. Cell 60, 685–696 (2015).
Li, X., Liang, X., Yin, J. & Lin, W. Organic fluorescent probes for monitoring autophagy in living cells. Chem. Soc. Rev. 50, 102–119 (2021).
Moskal, N. et al. An AI-guided screen identifies probucol as an enhancer of mitophagy through modulation of lipid droplets. PLoS Biol. 21, e3001977 (2023).
Fu, N., Xiong, Y. & Squier, T. C. Synthesis of a targeted biarsenical Cy3-Cy5 affinity probe for super-resolution fluorescence imaging. J. Am. Chem. Soc. 134, 18530–18533 (2012).
Wang, Y. et al. Unsymmetrical pentamethine cyanines for visualizing physiological acidities from the whole-animal to the cellular scale with pH-responsive deep-red fluorescence. RSC Adv. 11, 17871–17879 (2021).
Zhang, R. et al. Reaction-free and MMP-independent fluorescent probes for long-term mitochondria visualization and tracking. Chem. Sci. 10, 1994–2000 (2019).
Mauvezin, C. & Neufeld, T. P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 11, 1437–1438 (2015).
Mauro-Lizcano, M. et al. New method to assess mitophagy flux by flow cytometry. Autophagy 11, 833–843 (2015).
Benischke, A.-S. et al. Activation of mitophagy leads to decline in Mfn2 and loss of mitochondrial mass in Fuchs endothelial corneal dystrophy. Sci. Rep. 7, 6656 (2017).
Du Toit, A., Hofmeyr, J. S., Gniadek, T. J. & Loos, B. Measuring autophagosome flux. Autophagy 14, 1060–1071 (2018).
Loos, B., Du Toit, A. & Hofmeyr, J. H. Defining and measuring autophagosome flux-concept and reality. Autophagy 10, 2087–2096 (2014).
Balut, C. et al. Measurement of cytosolic and mitochondrial pH in living cells during reversible metabolic inhibition. Kidney Int 73, 226–232 (2008).
Yicheng Wang, P. S. et al. In An AI-assisted fluorescence microscopic system for screening mitophagy inducers by simultaneous analysis of mitophagic intermediates, DMAN, https://doi.org/10.5281/zenodo.15206014 (2025).
Hubbard, B. P. & Sinclair, D. A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. 35, 146–154 (2014).
Rush, T. S. 3rd, Grant, J. A., Mosyak, L. & Nicholls, A. A shape-based 3-D scaffold hopping method and its application to a bacterial protein-protein interaction. J. Med. Chem. 48, 1489–1495 (2005).
Mysinger, M. M., Carchia, M., Irwin, J. J. & Shoichet, B. K. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 55, 6582–6594 (2012).
Friesner, R. A. et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 (2004).
Halgren, T. A. et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759 (2004).
Guba, W., Meyder, A., Rarey, M. & Hert, J. Torsion Library Reloaded: A New Version of Expert-Derived SMARTS Rules for Assessing Conformations of Small Molecules. J. Chem. Inf. Model. 56, 1–5 (2016).
Lounkine, E., Hu, Y., Batista, J. & Bajorath, J. Relevance of feature combinations for similarity searching using general or activity class-directed molecular fingerprints. J. Chem. Inf. Model. 49, 561–570 (2009).
Arslan, N. P., Taskin, M. & Keles, O. N. Nicotinamide Mononucleotide and Nicotinamide Riboside Reverse Ovarian Aging in Rats Via Rebalancing Mitochondrial Fission and Fusion Mechanisms. Pharm. Res. 41, 921–935 (2024).
Yu, W. et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: Role of Foxo3A-Parkin-mediated mitophagy. Biochim Biophys. Acta Mol. Basis Dis. 1863, 1973–1983 (2017).
Song, D. et al. FOXO3 promoted mitophagy via nuclear retention induced by manganese chloride in SH-SY5Y cells. Metallomics 9, 1251–1259 (2017).
Giannakou, M. E. & Partridge, L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol. 14, 408–412 (2004).
Zhou, B., Lu, J. G., Siddu, A., Wernig, M. & Sudhof, T. C. Synaptogenic effect of APP-Swedish mutation in familial Alzheimer’s disease. Sci. Transl. Med. 14, eabn9380 (2022).
Xiong, X. et al. Study of mitophagy and ATP-related metabolomics based on beta-amyloid levels in Alzheimer’s disease. Exp. Cell Res. 396, 112266 (2020).
Link, C. D. et al. Gene expression analysis in a transgenic Caenorhabditis elegans Alzheimer’s disease model. Neurobiol. Aging 24, 397–413 (2003).
Link, C. D. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl Acad. Sci. Usa. 92, 9368–9372 (1995).
McWilliams, T. G. et al. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 214, 333–345 (2016).
Aoyagi, K. et al. A new beta cell-specific mitophagy reporter mouse shows that metabolic stress leads to accumulation of dysfunctional mitochondria despite increased mitophagy. Diabetologia 66, 147–162 (2022).
Loos, B., Klionsky, D. J., Du Toit, A. & Hofmeyr, J. S. On the relevance of precision autophagy flux control in vivo - Points of departure for clinical translation. Autophagy 16, 750–762 (2020).
Moen, E. et al. Deep learning for cellular image analysis. Nat. Methods 16, 1233–1246 (2019).
Boland, B. et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 28, 6926–6937 (2008).
Wan, W. et al. Regulation of Mitophagy by Sirtuin Family Proteins: A Vital Role in Aging and Age-Related Diseases. Front. Aging Neurosci. 14, 845330 (2022).
Palikaras, K., Princz, A. & Tavernarakis, N. Mitophagy Modulators. https://doi.org/10.1016/b978-0-12-801238-3.62137-2 (2018).
Chen, Q. et al. A dual-labeling probe to track functional mitochondria-lysosome interactions in live cells. Nat. Commun. 11, 6290 (2020).
Acknowledgements
We thank Yang Li and Lixue Shi for their helpful discussions. We thank Prof. Yunzeng Zou for providing the PINK1 shRNA plasmids. This work was supported by National Key Research and Development Program of China (2023YFA1801200), National Natural Science Foundation of China (92159304, 82394433, 82227806), National Science Fund for Distinguished Young Scholars (82025019), Shanghai Explorer Program (23TS1401100), Cell and Gene Therapy Project of Science and Technology Committee of Shanghai 23J21901700, Natural Science Foundation of Shanghai (22ZR1414700), AI for Science Foundation of Fudan University (FudanX24AI049, FudanX24AI038). Some of the illustrations in Fig. 1 and Fig. 6 were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
Y.W., Z.L., J.Y., Z.S., L.W. and C.L. conceived of and designed the project. P.S., H.Z., and P.W. performed the experiments. Y.L. performed the in Silico screening. Y.W. and Y.Y. wrote the manuscript with the help of others. C.L. supervised the overall project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ben Loos, Jia-Hong Lu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Song, P., Zhou, H. et al. An AI-assisted fluorescence microscopic system for screening mitophagy inducers by simultaneous analysis of mitophagic intermediates. Nat Commun 16, 5179 (2025). https://doi.org/10.1038/s41467-025-60315-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60315-1
