
Abstract
Calcium mineralization in biological and geological systems is often regulated by (macro)molecules enriched with anionic functional moieties. Relatively few studies have examined the effects of phosphate-based modifiers that are integral in calcification underlying human bone formation and pathological diseases. Here we mimic posttranslational phosphorylated moieties of a biologically-active inhibitor protein and demonstrate that polyphosphates and phosphonates suppress calcium oxalate nucleation, tailor solvate crystal structure, and irreversibly inhibit crystal growth in ways that significantly deviate from commonly investigated carboxylate-rich modulators of biomineralization. The most potent modifiers exhibit an uncommon dual mode of action, wherein nucleation is suppressed by altering prenucleation clusters and crystal surface growth is impeded irreversibly by inducing lattice strain. Once crystal surfaces are exposed to modifiers, recrystallization is severely restricted. This exemplifies the uniqueness and efficiency of phosphates wherein their multiple modes of action are promising characteristics for designing de novo biologically-inspired molecules as mineralization regulators.
Similar content being viewed by others
Introduction
Regulatory pathways of synthetic, biological, and natural crystallization often rely on foreign species (modifiers) ranging from ions and small molecules to macromolecules, which interact with crystal surfaces to either inhibit or promote solute incorporation1,2. Modifiers that operate by classical mechanisms bind to growth sites on crystal surfaces (Fig. 1: kinks, steps, or terraces) and impede solute attachment or alter local solvent ordering3. Emerging evidence of nonclassical crystallization by species more complex than monomers has led to questions pertaining to the mechanism(s) by which modifiers disrupt the formation of metastable precursors that comprise dense liquids or amorphous particles. These processes are pervasive in calcification (e.g., amorphous calcium carbonate4,5, phosphate6,7, and oxalate8,9) where calcium mineralization is essential to structural elements of coral and shells (e.g., mollusks), the formation of bone and teeth of vertebrates, and pernicious diseases. One of the most frequently studied pathological crystals is calcium oxalate, which is a common constituent of human kidney stones that forms three crystal forms: monohydrate (COM), the most thermodynamically favorable10, dihydrate (COD)9, and trihydrate (COT)11. This study focuses on the two most clinically relevant hydrates: COM, which exhibits hexagonal crystal habit and belongs to the P21/C space group with parameters a = 6.290, b = 14.583, c = 10.116 Å and β = 109.46°; and COD, which exhibits a bipyramidal crystal habit and belongs to the I4/m space group with parameters a = b = 12.371 and c = 7.357 Å12.

Illustration of the three processes influenced by phosphate-based modifiers: I, nucleation from classical or nonclassical (two-step) pathways; II, polymorph (solvate) selection; and III, surface growth of crystals.
Detailed investigations of calcification modifiers have predominantly focused on carboxylates13,14,15 with relatively fewer mechanistic studies examining phosphate-based molecules. Here we demonstrate the unique ability of phosphates to impact three critical processes of crystallization: (i) fully suppress nucleation (Fig. 1, path I), (ii) steer crystal form selection (Fig. 1, path II), and (iii) irreversibly inhibit crystal surface growth (Fig. 1, path III). It is challenging to fully suppress nucleation that follows the pathway envisioned by classical theory, wherein stochastic local fluctuations in composition promote the formation of particles with a critical size16,17. Nucleation, however, may follow a nonclassical (two-step) pathway within precursors that are either fluid-like clusters18,19,20 or amorphous solid/gel particles21,22. Here, we address whether a nonclassical pathway offers a means to suppress nucleation, using calcium oxalate as a model system in combination with a library of molecular modifiers that mimic proteins identified as natural inhibitors of pathological crystallization22. One of the most widely studied proteins is osteopontin (OPN, Fig. 2)23,24, which regulates calcium phosphate bone formation25, vascular calcification, and calcium oxalate renal stones26. The efficacy of OPN is attributed to its high percentage of anionic functional moieties, which are common among modifiers of calcium minerals. A unique characteristic of OPN is its abundance of serine and threonine residues susceptible to posttranslational phosphorylation. Prior studies have established general correlations between the number of phosphate groups in OPN peptide sequences and their efficacy as COM crystal growth inhibitors; however, few studies have exploited phosphates or phosphonates27. One recent example is a study by Kletzmayr et al28. showing that modified myo-inositol hexakisphosphate analogues are highly effective modifiers capable of inhibiting COM nucleation and growth both in situ and in vivo.

The OPN peptide sequence (amino acids, AA, 62 to 85) with serine residues (S, cyan) as sites for phosphorylation. Molecular structures of LTPP, CTMP, HMP, and PA, modifiers of COM crystallization. Background image: photograph of a human kidney stone.
Results and discussion
Impact of phosphates on COM crystallization
Polyphosphates regulate multiple aspects of biocrystallization, ranging from the formation of exoskeletons in living organisms29 to medical treatments in dentistry30. Key advantages of polyphosphates over carboxylate-based modifiers are their enhanced biocompatibility and hydrophilicity31. In general, modifiers can operate by two modes of action: thermodynamic, for instance, by complexing free calcium ions, which lowers their concentration and thus the driving force for crystal growth14; and kinetic, related to modifier interactions with crystal surfaces, which alter the rate of solute incorporation. Here we examine four modifiers of calcium oxalate (Fig. 2): cyclic trimetaphosphate (CTMP), linear tripolyphosphate (LTPP), hexametaphosphate (HMP), and phytic acid (PA). The three polyphosphates exhibit distinct linear and cyclic structures while the organophosphate is a reported growth inhibitor of COM and other minerals13,31.
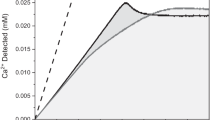
The efficacy of these four molecules was evaluated using a combination of in situ techniques to elucidate modifier – COM crystal interactions at both macroscopic and molecular length scales. The impact of modifiers on the kinetics of bulk crystallization is assessed by ion-selective electrode (ISE) measurements to track the temporal removal of Ca2+ ions from supersaturated growth solutions without any crystal seeds (Fig. 3a) over a short 1-h period. Two modifiers known to be efficient COM growth inhibitors were selected as benchmarks: OPN, from bovine milk, and hydroxycitrate (HCA)1. Prior studies have shown that ISE profiles exhibit a Langmuir-like behavior wherein the percent inhibition of crystallization plateaus at a certain modifier concentration14,15,32. It is widely reported that macromolecules tend to be more effective inhibitors of crystallization compared to small molecules owing to a higher percentage of proximal anionic functional moieties that cooperatively bind to crystal surfaces33. Small molecules rarely exceed 80 – 90% inhibition of crystal growth. This is evident in this study when comparing OPN ( ~70% inhibition, Fig. 3a) to HCA and CTMP which both exhibit around 60% maximum inhibition; however, HMP and PA achieve approximately 100% inhibition of calcium oxalate crystallization at relatively low modifier concentrations.

a Percent inhibition of calcium oxalate crystallization from ISE measurements in the presence of modifiers conducted over a 1-h period (see raw data in Supplementary Fig. 1). Dashed lines are interpolations to guide the eye; data points are an average of at least three separate measurements; and error bars span two standard deviations (some are smaller than the symbol size). b Statistical distribution of cluster cross-sectional areas measured from over 250 LPTEM images. Inset: effective diameters calculated from cross-sectional areas assuming a spherical cluster morphology with error bars showing 95% confidence intervals for population means (see Supplementary Fig. 8 for detailed statistical analysis). c – e LPTEM images of clusters observed in undersaturated calcium oxalate solutions (0.07 mM) in the (c) absence of modifier (inset: image of a blank liquid cell containing only DI water: scale bar = 500 nm) and in the presence of 7 μg mL−1 (d) PA and (e) HMP. Clusters formed in the presence of LTPP are similar to the control (see Supplementary Fig. 9).
Bulk crystallization over a longer period (72 h) reveals that PA is the only modifier that completely suppresses COM nucleation (Supplementary Figs. 2–4). The complete suppression of crystallization by a small molecule at low concentration (0.4 nM) is unusual and highly suggestive of a nonclassical nucleation pathway. To test this hypothesis, we used five characterization techniques to search for calcium oxalate (CaOx) clusters, or precursors, in physiologically-relevant solutions at undersaturated, saturated, and mildly supersaturated conditions (0.10, 0.13, and 0.15 mM CaOx). Oblique illumination microscopy (OIM)34,35, dynamic light scattering (DLS), and small-angle X-ray scattering (SAXS) all failed to detect clusters (Supplementary Table 1), which can be attributed to either poor cluster-solvent contrast or a low number density of clusters that falls below scattering detection limits. Prior studies have reported the presence of calcium oxalate precursors35,36,37,38 under non-physiological conditions where CaOx concentrations are orders of magnitude higher than those used in this study. Examples include cluster formation with disproportionate molar ratios of calcium and oxalate35,38 or when substituting water with an alternative solvent (e.g. ethanol)39. Putnis and coworkers40 reported small clusters ( <10 nm) using atomic force microscopy (AFM) to visualize their attachment on mica substrates. We used a similar protocol for our samples but did not observe clusters (Supplementary Table 2).
The only technique that successfully identified clusters was in situ liquid-phase transmission electron microscopy (LPTEM), which revealed precursors of ~150 nm in size (Fig. 3b and Supplementary Figs. 5–9). The presence of prenucleation clusters is strong evidence for two-step nucleation. These measurements were first carried out in an undersaturated solution (0.07 mM CaOx) without modifier, revealing a small population of clusters (Fig. 3c and Supplementary Fig. 5) that were on average spherical/ellipsoidal, with a significant presence of irregular (non-spheroidal) morphologies. As a result, cross-sectional area measured from LPTEM images was used as a metric for size comparison and an effective cluster diameter was calculated for each population based on a spherical geometry (Fig. 3b, inset). Clusters were also observed upon addition of 7.0 μg mL−1 PA (Fig. 3d and Supplementary Fig. 6) and HMP (Fig. 3e and Supplementary Fig. 7) to a solution of 0.07 mM CaOx. Control clusters formed in the absence of modifier have an average size of 150 ± 5 nm (mean ± 95% confidence interval, 19200 ± 1100 nm2). Clusters formed in the presence of PA and HMP have smaller sizes of 130 ± 5 nm and 140 ± 4 nm (14600 ± 1000 nm2 and 16700 ± 900 nm2), respectively (Fig. 3b, Supplementary Fig. 8). An ANOVA statistical test (p-value <0.0001) was performed to confirm the reported differences in group means are real, and a Tukey multiple comparisons test revealed that each of the three samples resulted in statistically significant differences in cluster size with the trend PA < HMP <control (see Methods, Supplementary Fig. 8). These observations indicate that modifiers alter cluster properties, with PA and HMP decreasing the size of prenucleation calcium oxalate clusters, an effect that could be sufficient to impact nucleation. It is important to note that the LPTEM experiments were carried out at low electron doses, up to a maximum of 1.5 e− Å−2 s−1, thereby minimizing electron beam effects. Measurements of a background sample containing only water (Fig. 3c, inset) showed no evidence of clusters, suggesting minimal potential for radiolytic effects to contribute to the objects observed in LPTEM. Furthermore, cryogenic transmission electron microscopy (Supplementary Fig. 10) of undersaturated calcium oxalate solutions (0.07 mM) in the absence of modifier indicates the presence of clusters of a similar size and shape as those observed in LPTEM (Fig. 3b and Supplementary Fig. 8), further confirming that the observed features are not generated by electron beam irradiation.
The exact mechanism by which modifiers alter cluster formation and suppress nucleation is not well understood. Guo et al41. proposed that rapid adsorption of polymeric additives can reduce the number of active growth sites on subcritical nuclei of COM, creating a thermodynamic driving force for particle dissolution and a concomitant extension of the induction period. It was alternatively proposed that modifiers influence CaOx intermediates. For example, Ruiz-Argudo et al35. reported the formation of small clusters (ca. 2 nm) and amorphous calcium oxalate (ACO) particles (30 – 100 nm) in media containing high concentrations of citrate (1.9 g L−1 or 10 mM) and CaOx (2 mM). Banner et al36. reported a counter observation wherein amorphous clusters form only in the absence of citrate. It has also been reported that certain modifiers can switch the preferred solvate from COM to COD22,42. The pathway(s) favoring metastable solvates, such as COD, may involve the stabilization of high supersaturation43, templating effects35, or alteration of cluster properties36,44,45.
Phosphates select the form of crystal solvates
A second unique action of phosphate modifiers is their facile ability to switch crystal structure from COM to COD at remarkably low concentration (Fig. 4a) – an outcome with pathological relevance based on studies reporting the metastable COD solvate is less prone to aggregation and potentially less likely to adhere to cellular membranes in biological tissues46. Methods to crystallize COD generally require high concentrations of modifiers, such as citrate (10 – 100 mM), or the use of macromolecules, such as polyacrylic acid (≤230 μg mL−1; ca. 5 kDa)9,28,47. Here we observe that LTPP and HMP induce a switch in crystal structure from COM (Fig. 4b) to COD (Fig. 4c) at significantly lower concentrations: 20–30 μg mL−1 LTPP (or 0.8–1.2 mM) and 30 μg mL−1 HMP (or 0.6 mM). Some of the modifiers tested in this study (e.g., CTMP) were unable to generate COD, whereas the most potent modifier (PA) completely suppresses nucleation at concentrations less than 5 μg mL−1 (Fig. 4a). Optical micrographs of glass slides placed at the bottom of bulk crystallization vials show only trace amount of amorphous precipitate in the presence of PA (Supplementary Fig. 2), consistent with ISE measurements (Fig. 3a). The amorphous nature was confirmed by polarized filters in the optical microscope; however, attempts to isolate the precipitate for further characterization was unsuccessful owing to its ultrasmall quantity. It has been reported that PA is an inhibitor of COM crystallization in human urine32 as well as a potent crystal growth modifier of struvite (MgNH4PO4\(\cdot\)6H2O), another pathological crystal48.

a Products from calcium oxalate growth solutions in the presence of modifiers at different concentrations. White corresponds to minute amorphous precipitate observed by optical microscopy (but a quantity too small for extraction and analysis). Gray and red correspond to COM and COD, respectively, confirmed by optical microscopy based on their distinct crystal habit. b, c Representative SEM images of (b) COM and (c) COD crystal habits.
The preferential interactions between modifiers and COM crystal surfaces were assessed by a combination of bulk crystallization, AFM, and microfluidics. Scanning electron microscopy (SEM) images of crystals grown in the presence of modifiers revealed significant differences in crystal habit compared to those prepared in the absence of modifier (Fig. 4b). The least effective modifier, CTMP, produced crystals with slightly elongated hexagonal shapes (Supplementary Fig. 11a). Analysis of growth solutions containing two analogues of CTMP, its linear chain LTPP (Fig. 5a and Supplementary Fig. 11b,c) and the larger ring HMP (Fig. 5b and Supplementary Fig. 11d, f), revealed an ovoid-like morphology similar to crystals in biological environments (e.g., urine sediments)49 where proteins are known to modulate crystal formation by binding to specific crystal surfaces or incorporating into the urinary stone matrix50. Minute concentrations of PA result in ill-defined morphologies (Fig. 5c and Supplementary Fig. 11g,h), suggesting a strong interaction between PA and multiple surfaces of COM crystals. The generation of crystals with curved surfaces is often attributed to the incorporation of additives within crystals51, which is observed by elemental analysis (vide infra). These interactions also change the topography of COM surfaces, which is observed during in situ AFM measurements of unfinished layer growth in the presence of modifiers. LTPP significantly reduces layer advancement in the c-direction with the generation of corrugated step edges (Fig. 5d) characteristic of a step pinning mode of action (Supplementary Movie 1 and Supplementary Fig. 12). Introduction of HMP arrests step advancement (Fig. 5e, Supplementary Movie 2, and Supplementary Fig. 13) whereas PA has the same effect but via surface roughening that obscures the presence of underlying layers (Fig. 5f and Supplementary Fig. 14).

SEM images of crystals prepared with (a) LTPP (5 μg mL−1), (b) HMP (20 μg mL−1), and (c) PA (0.25 μg mL−1). AFM images during in situ growth of COM (100) surfaces after ca. 50 min under flow of supersaturated solutions (supersaturation ratio S = 3.9) containing 1 μg mL−1 of (d) LTPP, (e) HMP, and (f) PA. Scale bars in AFM images equal 500 nm. g Percent inhibition of COM crystal growth in the [100], [010], and [001] directions from microfluidics in supersaturated solutions (S = 3.9) containing 1 μg mL−1 LTPP and 0.1 μg mL−1 of HMP and PA. Error bars span two standard deviations.
The effects of modifiers on COM growth at macroscopic and microscopic scales are determined by in situ microfluidics and AFM, respectively. A microfluidics device prepared using protocols reported by Sosa et al.51. was used to monitor time-resolved changes in COM crystal morphology under conditions where growth solutions with or without modifiers are continuously flowed over seed crystals in a microchannel. Random orientations of seed crystals on either the basal (100) or side (010) faces enable the evaluation of growth along all three principal directions: [100], [010], and [001]. Measurements in the presence of LTPP reveal its preferential binding to COM surfaces in the order of (100) > (121) > (010) based on changes in crystal morphology, which results in decreased thickness in the [100] direction and reduced length-to-width (c/b) ratio (Fig. 5g). Analogous to ISE measurements, low concentrations of HMP and PA (0.1 μg mL−1) suppress COM growth in all crystallographic directions (Fig. 5g). To our knowledge, these two modifiers are the most potent molecular inhibitors of calcium oxalate crystallization with a maximum efficacy achieved at concentrations nearly three orders of magnitude less than those of carboxylates. The ability of a small molecular modifier to fully suppress crystal growth is uncommon, and even more so is the dual capability of modifiers, such as PA, to suppress both nucleation and crystal growth, which we observe here for COM and reported previously for struvite52.
Irreversible mechanism of growth inhibition
The third unique action of phosphates is a mechanism by which they irreversibly inhibit COM surface growth. COM crystals nominally grow by screw dislocations (Fig. 6a) where layers on basal (100) surfaces advance in the [001] direction at a velocity approximately 10-times faster than <121> directions. After COM crystals are exposed to HMP at concentration as low as 1 μg mL−1, surface growth is completely arrested (Fig. 6b). To test whether layer advancement resumes to its original velocity under supply of a fresh (modifier-free) growth solution, we conducted regeneration experiments. Continuous AFM imaging over 10 h shows that step edges remain frozen, indicating irreversible inhibition; however, we observe two-dimensional nucleation and growth of islands on terraces between steps. Although 2D birth and spreading is a general mechanism of crystal growth53, it is uncommon for COM crystal surfaces54. The islands observed during surface regeneration are initially rounded (Fig. 6c) with a height of about 0.6 nm, equal to a single step on (100) faces (Fig. 6d). Under normal growth conditions step advancement can be difficult to track owing to the fast kinetics of surface growth as steps advance out of the frame within 1 min of imaging. Surfaces exposed to HMP, however, could be tracked for much longer times ( >5 h) wherein unfinished layers remain frozen, and 2D nucleation and growth of islands can be captured in real time (Fig. 6e). These islands only advance in the direction opposite that of layers emanating from screw dislocations; and as the islands advance, their shape gradually shifts from rounded to hexagonal (Fig. 6f). The orientation of oxalate ions in the crystal structure causes steps to form acute angles with the underlying terrace in one direction, and obtuse angles in the opposite direction (Fig. 6g). In the absence of modifiers, solute attaches to acute steps on advancing [001] layers; however, growth of 2D islands occurs by solute attachment to obtuse steps. The mechanism for this irregular growth pattern is elusive, but we posit that residual HMP on (or within) COM crystal surfaces is responsible for the observed effects (vide infra).

Two-dimensional nucleation on the terraces between unfinished layers. a In situ AFM image of a COM (100) surface growing under a continuous flow of growth solution with concentration 0.18 mM CaOx (S = 3.9). b Snapshot of the crystal surface after 149 min exposure to HMP. Scale bars equal 1 μm. c High-magnification image of the region within the yellow dashed box in panel (b) showing an arrested step edge (I) and 2D islands (II). d Height profiles across the two dashed lines in panel (c). e AFM images of a COM (100) surface previously exposed to HMP that is subjected to a continuous flow of modifier-free growth solution (Supplementary Movie 3 and Supplementary Fig. 15). f Time-resolved contours of a growing 2D island highlighted in panel e (arrow) where the dashed line is a reference point of growth suppression in the [001] direction. g Schematic of COM crystal habit with indexed facets. Callouts: (left) growth hillock with labeled steps and (right) molecular structure of (100) layers showing the acute [001] and obtuse [00\(\bar{1}\)] step edges projected down the b-axis. The COM crystal is indexed using the Tazzoli notation41 where the [001] direction of an advancing layer is consistent with previous literature76, which is identical to the [101] direction95 based on the Deganello notation96.
The advancement of unfinished layers (Fig. 7a, open symbols) and 2D islands (Fig. 7a, closed symbols) were assessed by AFM in the [001] and [00\(\bar{1}\)] directions, respectively. Islands that form on crystal surfaces after exposure to HMP advance with a step velocity of 0.05 nm/s, which is nearly two orders of magnitude less than the step velocity of pristine crystal surfaces (3 nm/s). We posit the irreversible effect of HMP is due to its incorporation within COM (100) surfaces, thereby imposing strain that hinders growth in the [001] direction while allowing 2D islands to slowly advance in the opposite direction. Irreversible inhibition of COM recrystallization is unexpected since growth typically resumes once the modifier is removed from the surface55,56. For instance, prior studies have demonstrated growth reversibility for COM and other crystalline materials (e.g., struvite and calcite) whereby regeneration with fresh growth solutions returns step advancement to rates achieved prior to their exposure to a modifier. In some instances, an initial delay is observed due to the time required for modifier desorption from crystal surfaces and/or growth to reduce strain imposed by modifier occlusion (as defects) within crystals. It has been shown that growth may resume via the generation of macrosteps while single steps remain immobilized57. We recently reported a different mode of irreversible inhibition for an organic crystal (β-hematin) where nanocrystals in solution deposit on crystal surfaces to reduce the rate of layer advancement, such that attempts to regenerate surface growth failed to restore step velocity to its original value58.

a Step advancement as a function of time monitored by in situ AFM, as schematically illustrated in the insets with orange indicating growth and grey indicating growth suppression. Left axis: steps advancing in the [001] direction in the absence of modifier. Right axis: islands advancing in the [00\(\bar{1}\)] direction during regeneration after exposure to HMP. b ToF-SIMS intensity profiles of phosphate (POx) as a function of sputtering time. Samples were prepared by subjecting COM crystals to a sequence of treatments: (1) growth in a supersaturated solution without modifier for 2 h (control, gray); (2) growth in the same solution containing 1 μg mL−1 modifier for 1 h; and (3) regeneration for 2 h in a supersaturated solution without modifier. The control is the baseline for comparing samples exposed to LTPP (yellow), HMP (red), and PA (orange). The blue shaded transient region (0–6 sec) represents the first monolayers that do not reflect POx absorption. c Temporal evolution in the cross-sectional area of COM crystals within microchannels during a period of growth in the absence of modifier (grey circles) following by regeneration after an initial exposure to HMP (blue squares and triangles) and PA (red diamonds). Optical micrographs of COM crystals during (d) control growth and e, f two separate regeneration tests after crystals are first exposed to HMP (see also Supplementary Movie 4 and Supplementary Fig. 20). Scale bars equal 20 μm. g, h SEM images of crystals regenerated for 12 h after exposure to 1 μg mL−1 HMP. Recovered growth produces distinct morphological features that include protrusions, new crystals nucleating at apical tips and basal surfaces, and overlayers that partially encompass the original crystal. Scale bars equal 5 μm.
Time-of-flight secondary ion mass spectrometry (ToF-SIMS) was used to analyze crystals after a 2-h regeneration period (Fig. 7b). Control crystals that were never exposed to modifiers were used as a zero baseline for comparison. COM crystals grown in the presence of HMP and PA retain a higher amount of phosphate compared to the control, whereas the ToF-SIMS intensity for LTPP is only slightly higher than the control, indicating a small quantity of modifier irreversibly accumulates on COM crystal surfaces. It is difficult to extract information from ToF-SIMS regarding the location of phosphates on either external surface sites or within the outermost layer of COM given that the crystals prepared on substrates do not lay flat, leading to a monotonic reduction in intensity with sputtering time (Fig. 7b). ToF-SIMS data provide evidence that phosphates irreversibly adsorb on (or incorporate within) the outermost layer of COM crystals. These results are consistent with elemental analysis of COM crystals recovered from bulk crystallization assays showing evidence of modifier accumulation ( ≤ 0.07 P/Ca ratio, Supplementary Fig. 16). The residual modifier is likely responsible for irreversible growth observed during in situ AFM and microfluidics experiments. Likewise, modifiers retained by COM crystals seemingly facilitate 2D island nucleation, while also acting as stoppers or inducing strain on the crystal lattice to hinder island advancement in the [001] direction via preferential interactions with acute steps on COM (100) surfaces. For less potent modifiers, such as LTPP, microfluidics studies (Supplementary Fig. 17) confirm a reversible mode of growth inhibition; however, in situ AFM images during regeneration reveal that layered growth resumes through the propagation of macrosteps that form shortly after introduction of modifier-free growth solution (Supplementary Fig. 18). Pinned steps advance slowly and are eventually overtaken by advancing macrosteps (Supplementary Fig. 19), which have step heights nearly two orders of magnitude greater than those of COM (100) single layers.
An alternative thermodynamic pathway of COM crystal growth inhibition was evaluated based on previous studies suggesting HMP and PA inhibit mineralization59 via their ability to chelate metals, thereby reducing supersaturation59. This mechanism, however, is inconsistent with the low concentrations of HMP and PA needed to suppress crystallization, but may become more relevant under conditions where the modifier is used in much higher concentration (i.e., comparable to or exceeding the concentration of Ca2+). To confirm that complexation plays a minor role under the growth conditions selected for our study, ISE bulk crystallization assays were performed over a range of supersaturation in modifier-free growth solutions with fixed oxalate content and a progressively decreasing calcium concentration to simulate Ca2+:modifier complexation. Under these conditions, we would expect a maximum 10% reduction in the rate of COM crystallization (Supplementary Fig. 21), which is well below experimental observations (Fig. 3b). From this analysis, we conclude the primary mechanism of COM crystal growth inhibition is a kinetic pathway involving modifier interactions with crystal surfaces to reduce the rate of solute incorporation.
Evidence of strain imposed by phosphates retained within COM crystals is further gleaned from in situ microfluidics experiments where we measure temporal changes in the cross-sectional area (Fig. 7c). Measurements in the absence of modifier (Fig. 7d and Supplementary Fig. 22) reveal a linear increase in crystal size; however, for crystals first exposed to HMP or PA and then subjected to fresh growth solution, there is an induction period of around 6 h (Fig. 7c) where no change in crystal size is observed (Supplementary Fig. 23 and Movie 5). After this period there is evidence of growth, but the strain imposed by residual modifiers causes anisotropic growth wherein features emerge randomly from the sides and/or basal surfaces (Fig. 7e,f). This haphazard nature of regrowth is why the cross-sectional area was selected to track the kinetics of growth regeneration; however, irregular nucleation and growth of features on either the sides or basal surfaces leads to different rates of regrowth (e.g., HMP data in Fig. 7c). Scanning electron micrographs of COM crystals after a 12-h period of regrowth clearly show the abnormal features that form on original seed crystals as either protrusions (Fig. 7g) or overlayers that partially encompass the original crystal (Fig. 7h) and appear to be in crystallographic registry with the original seed crystal.
In summary, the results presented here show that biologically-inspired molecules rich in phosphate moieties are among the most highly efficient inhibitors of calcium oxalate crystallization. Their multiple modes of action impacting nucleation, crystal structure selection, and growth motivate use of the term “disruptor” to describe their diverse effects, which are counter to mechanisms reported for common inhibitors of biomineralization. We show that potent polyphosphates selected for this study suppress the nonclassical pathway of calcium oxalate nucleation, direct the formation of the more metastable COD hydrate at relatively low modifier concentration, and uniquely inhibit COM growth by embedding in the crystal and putatively inducing strain, leading to irreversible effects that frustrate attempts to regenerate crystal growth. The phosphates impact COM crystal growth via a kinetic mechanism wherein modifier interaction with crystal surfaces leads to their occlusion within the crystal – a mode of action that diverges from previously suggested thermodynamic pathways involving ion complexation. The resulting strain imposed by this mechanism may be utilized in biological systems as a way of regulating mineralization and may also be an advantageous feature for designing therapeutics based on the ability to sustain growth inhibition for a period of time after removal of the modifier from crystallization environments. While this study focuses on clinically relevant growth conditions, the observations reported here require more extensive analysis over a broader range of growth environments (e.g., supersaturation, pH, etc.) with parallel in vivo tests to verify their true potential as treatments of calcium oxalate kidney stone disease. Future studies may also determine whether the effects reported here for phosphate inhibition of crystal nucleation and growth could potentially extend to crystal aggregation, which is not well understood, but has been proposed as a critical process of pathological stone formation60. We also anticipate that the observations reported here may have widespread implications for phosphate-based molecule regulation of biomineralization in other natural or biological systems.
Methods
Materials
The following reagents were obtained from Sigma Aldrich (St. Louis, MO) and used without further purification: calcium chloride dihydrate (ACS Reagent, 99 + %), sodium oxalate (Na2C2O4, >99%), sodium citrate dihydrate ( >99%), sodium chloride ( >99.5%), sodium triphosphate pentabasic ( >98%, linear tripolyphosphate or LTPP), trisodium trimetaphosphate ( >95%, cyclic trimetaphosphate or CTMP), and sodium hexametaphosphate (96%, hexametaphosphate or HMP). Phytic acid dipotassium salt (98%, phytate or PA) was purchased from Boc Sciences. Bovine milk osteopontin for these studies was isolated and donated by E. Sorensen (Aarhus University, Denmark). All experiments used deionized (DI) water (18.2 MΩ) purified by an Aqua Solutions RODI-C-12A system.
The degree of phosphate dissociation is different for each modifier under fixed experimental conditions of pH 6.2 for COM growth solutions based on the pKa values reported in literature. For instance, PA has 12 ionizable H atoms comprised of 3 weak acids (pKa 10.0–12.0), 3 moderately-weak acids (pKa 5.7–7.6), and six strong acids (pKa 1.1–2.1)61. This indicates that 7 to 8 sites per PA molecule will be dissociated. LTPP has 5 ionizable H atoms with pKa values62 ranging from 0.8 to 2, indicating all 5 sites per LTPP molecule will be dissociated. CTMP has 3 ionizable H atoms with an average pKa value63 of around 0.4, indicating all 3 sites per CTMP molecule will be dissociated. HMP has 6 ionizable H atoms with theoretical pKa values59 predicted in the range of -1.2 to 12.0, which suggests 4 sites per HMP molecule will be dissociated.
Calcium oxalate monohydrate (COM) crystallization
COM crystals were prepared using a previously reported protocol64. Stock solutions of calcium chloride and sodium oxalate were prepared at 10 mM concentration by dissolving corresponding quantities of each reagent in deionized water. Stock solutions of 1000 μg mL−1 modifier (LTPP, HMP, CTMP, and PA) and 1.2 M sodium chloride were prepared. Batch crystallization was performed in a 20-mL glass vial by preparing a 10 mL solution of composition 0.7 mM CaCl2:0.7 mM Na2C2O4: 150 mM NaCl: X μg mL−1 modifier (pH = 6.1 ± 0.2). The pH of each growth solution was measured with an Orion 3-Star Plus benchtop meter equipped with a ROSS Ultra electrode (8102BNUWP). All samples were incubated for 1 h at 60 °C prior to the dropwise addition of 10 mM Na2C2O4, 1000 μg mL−1 modifier, and an appropriate amount of NaOH(aq) to adjust the pH (if necessary). All solutions were placed in the incubator at 60 °C for three days. The final crystals were observed in the solution by optical microscopy using a Leica DMi8 instrument. The morphology of crystals was used to determine if modifiers added to growth mixtures caused a switch in structure from COM to COD22,23,42,65,66,67,68,69.
Scanning electron microscopy (SEM)
Electron microscopy was performed using a Phenom Pharos Desktop SEM and a JEOL JEM-6330F field emission scanning electron microscope. As-synthesized crystals were gently rinsed with DI water, and dried in air prior to analysis. SEM samples were prepared by attaching the glass slides to the SEM studs (Ted Pella) using carbon tape and were coated with a layer of gold (about 20 nm thick) to reduce the effects of electron beam charging.
Liquid-phase transmission electron microscopy (LPTEM)
LPTEM was performed on a FEI Tecnai F20 Cryo TEM operating at 200 kV in TEM mode. Before imaging, the liquid cell was prepared by washing the liquid cell chips (Protochips Poseidon, 50 nm flow spacer, 50 nm thick silicon nitride window) with acetone and methanol, followed by plasma cleaning the chips for five minutes to imbue hydrophilicity.
Samples were prepared by mixing an equimolar concentration of Ca and Ox stock solutions (0.07 mM) with 150 mM NaCl. For solutions containing modifiers, the additives were first added to oxalate prior to mixing by inverting the vial three times and allowing the solution to sit undisturbed for two hours. The liquid cell was then assembled in a Protochips Poseidon LPTEM holder by sandwiching 1 μL of the sample between the two chips, arranged with their viewing windows perpendicularly to give a 50 × 50 μm2 effective viewing area. Images were collected with a Gatan K2 Summit detector in counted mode, with a 2 s exposure time and at low dose rates up to 1.5 e−Å−2s−1, as read from Gatan Digital Micrograph software. After imaging each sample, the liquid cell was disassembled to verify that the liquid layer was still present and the observed feature(s) was not formed via sample drying. Negative control experiments with DI water in the liquid cell showed no precipitate, verifying that the observed feature(s) in other samples was not due to impurities in the water or the silicon nitride viewing windows.
LPTEM image analysis
For the analysis of LPTEM images we used ImageJ to evaluate cluster size based on cross-sectional area. Due to variation in cluster contrast, images are not binarized (so as to preserve relevant differences in contrast). Automatic functions in ImageJ to distinguish objects like “detect edges” were not effective due to undefined boundaries in many cases; therefore, all images were analyzed by hand. To aid in cluster identification, images underwent a minimal processing pipeline consisting of a 3 × 3 pixel smoothing filter followed by normalizing their contrast. We used general rules for identifying and distinguishing clusters. Two overlapping areas were considered as separate clusters if their intersection had a higher contrast or if they had a high circularity with a relatively small intersection.
Cross-sectional area was measured by one of two methods in ImageJ. For ellipsoid clusters, the shape was traced using the elliptical tool and the areas were extracted. Areas of non-ellipsoid clusters were measured using the polygon tool to define a cluster shape, which was then fitted to a spline and adjusted to the exact morphology of the cluster area. To translate cross-sectional area into a more practical parameter, effective cluster diameters were calculated from area data assuming circularity of all clusters. A histogram of cross-sectional area as measured from LPTEM is presented in Fig. 3b, and that of effective diameter in Supplementary Fig. 8.
LPTEM statistical data analysis
Population means with 95% Confidence Intervals (CI) were calculated based on each set of cluster sizes, resulting in statistical significance from non-overlapping CI ranges for PA-modified, HMP-modified, and control clusters. To verify these differences in population means, one-way ANOVA testing was performed, giving an F-statistic of 17.8 and p-value < 0.0001 for cross-sectional area comparisons. A follow-up Tukey multiple comparisons test was used to evaluate the significance of each pairwise comparison of group means. The 95% CIs of mean difference for each of the three comparisons were non-overlapping with zero, confirming statistical significance between all three treatments. The same was done for effective cluster diameter (F-statistic 18.1, p-value < 0.0001). See Supplementary Fig. 8 for all statistical analysis.
Cryo-transmission electron microscopy
Control samples of undersaturated calcium oxalate solutions (0.07 mM) were mixed thoroughly and allowed to sit for 2 h. To prepare the CryoEM sample, 3 μL of the mixed solution was deposited onto a Lacey Carbon grid (Lacey Carbon Films with no formvar backing, Ted Pella Inc.). A Vitrobot Mark IV System (Thermo Fisher) was used with a blotting force of 2 and blotting time of 2. To rule out the possibility of imaging ethane (which was used for sample preparation), the electron beam was focused on a particle to induce local heating. If the contrast remained after sustained imaging, we concluded that the region was not composed of volatile ethane.
Kinetic studies of COM crystallization
Time-resolved analysis of COM crystallization was performed in situ using a calcium ion-selective electrode (ISE) from Orion (model 9720BNWP). COM growth was analyzed at 21 ± 2 °C under constant stirring at 1200 rpm using solutions with a supersaturation ratio S of 4.1 prepared with compositions 0.5 mM CaCl2, 0.5 mM Na2C2O4, 150 mM NaCl, and X μg mL−1 modifier. Solutions were prepared using the same procedure outlined above, with modifiers added prior to the addition of Na2C2O4 stock solution. The supersaturation ratio was calculated using the relationship IAP/Ksp, where IAP is the ion activity product (\({a}_{{{Ca}}^{2+}}\cdot {a}_{{C}_{2}{O}_{4}^{2-}}\)) and Ksp is the solubility product for COM at 25 °C reported in literature11. As a benchmark for these kinetic studies we selected osteopontin (OPN), which is one of many proteins22,70,71,72,73 that function as a modifier of calcification13,23,26,56,74,75,76,77,78. Polyphosphates in many native proteins and their molecular analogues regulate multiple aspects of crystallization31,79,80 based on kinetic effects associated with their interactions with crystal surfaces14,81,82,83,84,85. ISE measurements were also used to assess thermodynamic pathways of modifiers to inhibit COM crystallization, which have been reported for many cases of mineralization58,59,86,87,88,89,90 based on the ability of modifiers to chelate metals80,91,92, thereby reducing supersaturation59,93,94.
Oblique illumination microscopy (OIM)
Brownian microscopy was employed to investigate the presence of precursors (clusters) in COM solutions. For these experiments, solutions were prepared according to the aforementioned procedure for COM crystallization with the calcium oxalate concentrations adjusted to achieve conditions ranging from undersaturation to slight supersaturation. OIM measurements were performed using a Nanosight LM10-HS device where calcium oxalate solutions with or without modifier were illuminated using a specifically designed laser beam. Scattered light was collected from a liquid cell of dimensions 120 ×80 x 5 μm3 (length x width x height) containing the sample.
Microfluidics
A microfluidics device was used to examine COM growth at macroscopic length scale. The device, which comprised poly(dimethylsiloxane) (PDMS) on glass, was fabricated and used according to a reported protocol51. This system was monitored under a continuous supply of growth solution using a semiautomatic inverted light microscope (Leica DMi8 equipped with PL Fluotar 5X, 10X, 20X, and N plan L 50X objectives). For in situ crystallization studies, the microchannels were first flushed thoroughly with DI water. The conditions for preparing COM seed crystals were optimized to obtain at least 50 COM crystals that were well dispersed along the channel. First, a 0.85 mM Ca2+ solution was mixed through a y-connector with a second solution containing 0.85 mM C2O42- and 300 mM NaCl. This combined solution was delivered into the channels using a dual syringe pump (CHEMYX Fusion 200) for 100 min at a combined flow rate of 12 mL h−1. Subsequently, these solutions were replaced by one with slightly lower supersaturation (0.8 mM) to further grow seed crystals over a 600-min period to a size (20 – 30 μm) that allows for seeded growth experiments in the absence or presence of modifiers.
Time-resolved imaging of COM crystal growth and inhibition using an inverted optical microscope was performed to quantify the kinetics of COM crystallization. Two solutions were prepared in individual syringes: one containing 0.36 mM CaCl2,(aq) and the second containing 0.36 mM Na2C2O4(aq). The two solutions were mixed using an inline flow configuration that produced a final composition of 0.18 mM CaCl2 and 0.18 mM Na2C2O4. Growth solution was introduced into the microchannels with COM seeds at a rate of 12 mL h−1 (total combined solutions) using a dual syringe pump. For measurements in the presence of a growth modifier, an appropriate amount of each modifier was added to the syringe containing Na2C2O4.
Atomic force microscopy (AFM)
COM crystals were collected on AFM specimen disks (Ted Pella, Inc.) by placing the specimen disks in the synthesis vial prior to the addition of reagents used for COM crystallization. After synthesis, the solution was cooled down, and disks containing COM crystals were retrieved, rinsed thoroughly in DI water, and air-dried prior to AFM analysis. COM crystals were anchored to the specimen disk with either the (100) or (010) surface oriented in the plane of imaging. AFM measurements were performed using an Asylum Cypher ES instrument (Santa Barbara, CA) in contact mode (256 lines/scan) with scanning rates of 2 Hz and 11–25 Hz for ex-situ and in situ studies, respectively. Topographical images were collected using Asylum TR800PSA cantilevers. Solutions used for in situ measurements of COM surface were the same as those used for microfluidics experiments. The growth solution was delivered to the liquid cell using an in-line mixing configuration, where the two solutions were combined immediately before introduction into the AFM liquid cell (similar to microfluidics experiments).
Time-of-flight secondary ion mass spectrometry (ToF-SIMS)
Negative high mass resolution depth profiles were performed using a ToF-SIMS NCS instrument, which combines a TOF.SIMS5 instrument (ION-TOF GmbH, Münster, Germany) and an in situ scanning probe microscope (NanoScan, Switzerland) in the Shared Equipment Authority at Rice University. Samples were prepared by subjecting COM crystals to a sequence of treatments: (i) growth in a supersaturated solution without modifier for 2 h; (ii) growth in the same solution containing 1 μg mL−1 of modifiers for 1 h; and (iii) all samples exposed to modifiers were subjected to a regeneration step for 2 h using a supersaturated solution without modifier. Samples were dried in air before analysis. A bunched 30 keV Bi3+ ion source (with a measured current of 0.2 pA) was used as primary probe for analysis (scanning area of 80 × 80 µm2), and sputtering was performed using Cs+ ions at 1 keV with a typical current around 25 nA and a rastered area of 500 × 500 µm2. The beams were operated in non-interlaced mode, alternating 4 analysis cycles and 0.5 s of a sputter cycle followed by a pause of 3 s for the charge compensation with an electron flood gun. An adjustment of the charge effects was operated using a surface potential. During depth profiling, the cycle time was fixed to 90 µs (corresponding to a mass-to-charge ratio of m/z = 0 – 737 a.m.u.). Ion intensities were normalized by point-to-point normalization using the total ion intensity for a standardization of data to be able to compare depth profiles. Data was binned using 15 points adjacent averaging for improving the signal-to-noise ratio.
References
Farmanesh, S. et al. Specificity of growth inhibitors and their cooperative effects in calcium oxalate monohydrate crystallization. J. Am. Chem. Soc. 136, 367–376 (2014).
Rimer, J. D. et al. Crystal Growth Inhibitors for the Prevention of L-Cystine Kidney Stones Through Molecular Design. Science 330, 337–341 (2010).
Mass, T. et al. Amorphous calcium carbonate particles form coral skeletons. Proc. Natl Acad. Sci. USA 114, 670–678 (2017).
Wallace, A. F. et al. Microscopic evidence for liquid-liquid separation in supersaturated CaCO3 solutions. Science 341, 885–889 (2013).
Gebauer, D., Kellermeier, M., Gale, J. D., Bergström, L. & Cölfen, H. Pre-nucleation clusters as solute precursors in crystallisation. Chem. Soc. Rev. 43, 2348–2371 (2014).
Habraken, W. et al. Ion-association complexes unite classical and non-classical theories for the biomimetic nucleation of calcium phosphate. Nat. Commun. 4, 1507 (2013).
Reznikov, N., Bilton, M., Lari, L., Stevens, M. M. & Kroger, R. Fractal-like hierarchical organization of bone begins at the nanoscale. Science 360, eaao2189 (2018).
Sheng, X., Ward, M. D. & Wesson, J. A. Crystal surface adhesion explains the pathological activity of calcium oxalate hydrates in kidney stone formation. J. Am. Soc. Nephrol. 16, 1904–1908 (2005).
Chauhan, V. P., Yazbek Grobman, G., Vidavsky, N. & Rimer, J. D. Time-resolved dynamics of calcium oxalate dihydrate crystallization. Cryst. Growth Des. 24, 10273–10283 (2024).
Izatulina, A. R. et al. Hydrated calcium oxalates: crystal structures, thermal stability, and phase evolution. Cryst. Growth Des. 18, 5465–5478 (2018).
Tomazic, B. & Nancollas, G. A study of the phase transformation of calcium oxalate trihydrate-monohydrate. Invest. Urol. 16, 329–335 (1979).
Wesson, J. A. & Ward, M. D. Pathological biomineralization of kidney stones. Elements 3, 415–421 (2007).
Chung, J., Sosa, R. & Rimer, J. D. Elucidating the effects of polyprotic acid speciation in calcium oxalate crystallization. Cryst. Growth Des. 17, 4280–4288 (2017).
Chung, J. et al. Molecular modifiers reveal a mechanism of pathological crystal growth inhibition. Nature 536, 446–450 (2016).
Su, Y. et al. Tartronic Acid as a Potential Inhibitor of Pathological Calcium Oxalate Crystallization. Adv. Sci.11, e2400642 (2024).
De Yoreo, J. J. & Vekilov, P. G. Principles of crystal nucleation and growth. Rev. Mineral. Geochem. 54, 57–93 (2003).
Vekilov, P. G. in Crystallization via Nonclassical Pathways Volume 1: Nucleation, Assembly, Observation & Application Vol. 1358 ACS Symposium Series Ch. 2, 19-46 (American Chemical Society, 2020).
Galkin, O. & Vekilov, P. G. Are nucleation kinetics of protein crystals similar to those of liquid droplets?. J. Am. Chem. Soc. 122, 156–163 (2000).
Vekilov, P. G. Dense liquid precursor for the nucleation of ordered solid phases from solution. Cryst. Growth Des. 4, 671–685 (2004).
Nielsen, M. H., Aloni, S. & De Yoreo, J. J. In situ TEM imaging of CaCO3 nucleation reveals coexistence of direct and indirect pathways. Science 345, 1158–1162 (2014).
Mallette, A. J., Shilpa, K. & Rimer, J. D. The current understanding of mechanistic pathways in zeolite crystallization. Chem. Rev. 124, 3416–3493 (2024).
Wesson, J. A., Worcester, E. M. & Kleinman, J. G. Role of anionic proteins in kidney stone formation: interaction between model anionic polypeptides and calcium oxalate crystals. J. Urol. 163, 1343–1348 (2000).
Wesson, J. A. et al. Osteopontin is a critical inhibitor of calcium oxalate crystal formation and retention in renal tubules. J. Am. Chem. Soc. 14, 139–147 (2003).
Steitz, S. A. et al. Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am. J. Pathol. 161, 2035–2046 (2002).
Khan, S. R., Canales, B. K. & Dominguez-Gutierrez, P. R. Randall’s plaque and calcium oxalate stone formation: role for immunity and inflammation. Nat. Rev. Nephrol. 17, 417–433 (2021).
Grohe, B. et al. Control of calcium oxalate crystal growth by face-specific adsorption of an osteopontin phosphopeptide. J. Am. Chem. Soc. 129, 14946–14951 (2007).
Rimer Jeffrey, D. & Alamani Bryan, G. Polyphosphates As Inhibitors Of Calcium Crystallization. US patent US 2020/0009163 A1 (2020).
Kletzmayr, A. et al. Inhibitors of calcium oxalate crystallization for the treatment of oxalate nephropathies. Adv. Sci. 7, 1903337 (2020).
Addison, W. N., Azari, F., Sorensen, E. S., Kaartinen, M. T. & McKee, M. D. Pyrophosphate inhibits mineralization of osteoblast cultures by binding to mineral, up-regulating osteopontin, and inhibiting alkaline phosphatase activity. J. Biol. Chem. 282, 15872–15883 (2007).
Liu, J., Huang, W., Pang, Y. & Yan, D. Hyperbranched polyphosphates: synthesis, functionalization and biomedical applications. Chem. Soc. Rev. 44, 3942–3953 (2015).
Grases, F., Ramis, M. & Costa-Bauza, A. Effects of phytate and pyrophosphate on brushite and hydroxyapatite crystallization. Urol. Res. 28, 136–140 (2000).
Kim, D., Moore, J., McCoy, C. P., Irwin, N. J. & Rimer, J. D. Engaging a battle on two fronts: dual role of polyphosphates as potent inhibitors of struvite nucleation and crystal growth. Chem. Mater. 32, 8672–8682 (2020).
Li, Y., Lubchenko, V. & Vekilov, P. G. The use of dynamic light scattering and brownian microscopy to characterize protein aggregation. Rev. Sci. Instrum. 82, 053106 (2011).
Warzecha, M., Safari, M. S., Florence, A. J. & Vekilov, P. G. Mesoscopic Solute-Rich Clusters in Olanzapine Solutions. Cryst. Growth Des. 17, 6668–6676 (2017).
Ruiz-Agudo, E. et al. A non-classical view on calcium oxalate precipitation and the role of citrate. Nat. Commun. 8, 1–10 (2017).
Banner, D. J. et al. In Situ Liquid-Cell TEM Observation of multiphase classical and nonclassical nucleation of calcium oxalate. Adv. Funct. Mater. 31, 2007736 (2021).
Ihli, J. et al. Precipitation of Amorphous Calcium Oxalate in Aqueous Solution. Chem. Mater. 27, 3999–4007 (2015).
Hajir, M., Graf, R. & Tremel, W. Stable amorphous calcium oxalate: synthesis and potential intermediate in biomineralization. Chem. Commun. 50, 6534–6536 (2014).
Zhang, J., Wang, L., Zhang, W. & Putnis, C. V. Role of Hyperoxaluria/Hypercalciuria in Controlling the Hydrate Phase Selection of Pathological Calcium Oxalate Mineralization. Cryst. Growth Des. 21, 683–691 (2020).
Putnis, C. V., Wang, L., Ruiz-Agudo, E., Ruiz-Agudo, C. & Renard, F. in Crystallization via Nonclassical Pathways Volume 2: Aggregation, Biomineralization, Imaging & Application, Vol. 1383 ACS Symposium Series Ch. 1, 1-35 (American Chemical Society, 2021).
Guo, S., Ward, M. D. & Wesson, J. A. Direct visualization of calcium oxalate monohydrate crystallization and dissolution with atomic force microscopy and the role of polymeric additives. Langmuir 18, 4284–4291 (2002).
Grohe, B. et al. Cooperation of phosphates and carboxylates controls calcium oxalate crystallization in ultrafiltered urine. Urol. Res. 39, 327–338 (2011).
Jung, T., Kim, W.-S. & Choi, C. K. Crystal structure and morphology control of calcium oxalate using biopolymeric additives in crystallization. J. Cryst. Growth 279, 154–162 (2005).
Li, S. et al. Understanding the role of citric acid on the crystallization pathways of calcium oxalate hydrates. Cryst. Growth Des. 19, 3139–3147 (2019).
Dyer, R. & Nordin, B. Urinary crystals and their relation to stone formation. Nature 215, 751–752 (1967).
Doherty, W. O. S., Crees, O. L. & Senogles, E. The Preparation of Calcium Oxalate Dihydrate Crystals. Cryst. Res. Technol. 29, 517–524 (1994).
Sun, X. Y., Ouyang, J. M. & Xu, M. Synthesis, characterization, and cytotoxicity assay of calcium oxalate dihydrate crystals in various shapes. CrystEngComm 18, 5463–5473 (2016).
Williams, J. C. Jr, Zarse, C. A., Jackson, M. E., Witzmann, F. A. & McAteer, J. A. Variability of protein content in calcium oxalate monohydrate stones. J. Endourol. 20, 560–564 (2006).
Daudon, M., Frochot, V., Bazin, D. & Jungers, P. Crystalluria analysis improves significantly etiologic diagnosis and therapeutic monitoring of nephrolithiasis. Comptes Rendus. Chim. 19, 1514–1526 (2016).
Mijowska, S. et al. High Amino Acid Lattice Loading at Nonambient Conditions Causes Changes in Structure and Expansion Coefficient of Calcite. Chem. Mater. 32, 4205–4212 (2020).
Sosa, R. D., Geng, X., Reynolds, M. A., Rimer, J. D. & Conrad, J. C. A microfluidic approach for probing hydrodynamic effects in barite scale formation. Lab Chip 19, 1534–1544 (2019).
Olafson, K. N., Ketchum, M. A., Rimer, J. D. & Vekilov, P. G. Mechanisms of hematin crystallization and inhibition by the antimalarial drug chloroquine. Proc. Natl. Acad. Sci. USA 112, 4946–4951 (2015).
Li, S., Kang, X., He, Q., Tang, W. W. & Gong, J. B. Dual Mechanism of Natural Polyphenols on crystal whiskers formation on calcium oxalate monohydrate crystal surface. Appl. Surf. Sci. 592, 153355 (2022).
Tazzoli, V. & Domeneghetti, C. The crystal structures of whewellite and weddellite: re-examination and comparison. Am. Mineral. 65, 327–334 (1980).
Chakrabarti, R. & Vekilov, P. G. Dual Mode of Action of Organic Crystal Growth Inhibitors. Cryst. Growth Des. 21, 7053–7064 (2021).
Nene, S. S., Hunter, G. K., Goldberg, H. A. & Hutter, J. L. Reversible inhibition of calcium oxalate monohydrate growth by an osteopontin phosphopeptide. Langmuir 29, 6287–6295 (2013).
Ma, W. et al. A second mechanism employed by artemisinins to suppress Plasmodium falciparum hinges on inhibition of hematin crystallization. J. Biol. Chem. 296, 100123 (2021).
Grases, F., Rodriguez, A. & Costa-Bauza, A. Efficacy of mixtures of magnesium, citrate and phytate as calcium oxalate crystallization inhibitors in urine. J. Urol. 194, 812–819 (2015).
Robinson, T. E. et al. Hexametaphosphate as a potential therapy for the dissolution and prevention of kidney stones. J. Mater. Chem. B 8, 5215–5224 (2020).
Alamani, B. G. & Rimer, J. D. Molecular modifiers of kidney stones. Curr. Opin. Nephrol. Hypertens. 26, 256–265 (2017).
Bloot, A. P. M., Kalschne, D. L., Amaral, J. A. S., Baraldi, I. J. & Canan, C. A Review of Phytic Acid Sources, Obtention, and Applications. Food Rev. Int. 39, 73–92 (2023).
Decker, E. A., Chen, B., Panya, A. & Elias, R. J. in Oxidation in Foods and Beverages and Antioxidant Applications (ed Eric A. Decker) 225-248 (Woodhead Publishing, 2010).
Bezold, D., Dürr, T., Singh, J. & Jessen, H. J. Cyclotriphosphate: A Brief History, Recent Developments, and Perspectives in Synthesis. Chem. A Eur. J. 26, 2298–2308 (2020).
Farmanesh, S. et al. High-throughput platform for design and screening of peptides as inhibitors of calcium oxalate monohydrate crystallization. J. Cryst. Growth 373, 13–19 (2013).
Kok, D. J., Blomen, L. J., Westbroek, P. & Bijveot, O. L. Polysaccharide from coccoliths (CaCO3 biomineral) Influence on crystallization of calcium oxalate monohydrate. Eur. J. Biochem. 158, 167–172 (1986).
Chan, B. P. et al. On the catalysis of calcium oxalate dihydrate formation by osteopontin peptides. Colloids Surf. B. 96, 22–28 (2012).
Liu, J., Jiang, H. & Liu, X.-Y. How does bovine serum albumin prevent the formation of kidney stone? A kinetics study. J. Phys. Chem. B 110, 9085–9089 (2006).
Cerini, C. et al. Nucleation of calcium oxalate crystals by albumin: involvement in the prevention of stone formation. Kidney Int. 55, 1776–1786 (1999).
Zhang, C.-Y., Wu, W.-H., Wang, J. & Lan, M.-B. Antioxidant properties of polysaccharide from the brown seaweed Sargassum graminifolium (Turn.), and its effects on calcium oxalate crystallization. Mar. Drugs 10, 119–130 (2012).
Worcester, E. M. Inhibitors of stone formation. Semin. Nephrol. 16, 474–486 (1996).
Hess, B., Nakagawa, Y., Parks, J. H. & Coe, F. L. Molecular abnormality of Tamm-Horsfall glycoprotein in calcium oxalate nephrolithiasis. Am. J. Physiol. Ren. Physiol. 260, F569–F578 (1991).
Scurr, D. & Robertson, W. Modifiers of calcium oxalate crystallization found in urine. III. Studies on the role of Tamm-Horsfall mucoprotein and of ionic strength. J. Urol. 136, 505–507 (1986).
Mo, L. et al. Renal calcinosis and stone formation in mice lacking osteopontin, Tamm-Horsfall protein, or both. Am. J. Physiol. Ren. Physiol. 293, 1935–1943 (2007).
Millan, A., Sohnel, O. & Grases, F. The influence of crystal morphology on the kinetics of growth of calcium oxalate monohydrate. J. Cryst. Growth 179, 231–239 (1997).
Asplin, J., DeGanello, S., Nakagawa, Y. N. & Coe, F. L. Evidence that nephrocalcin and urine inhibit nucleation of calcium oxalate monohydrate crystals. Am. J. Physiol. Ren. Physiol. 261, F824–F830 (1991).
Qiu, S. R. et al. Molecular modulation of calcium oxalate crystallization by osteopontin and citrate. Proc. Natl Acad. Sci. USA 101, 1811–1815 (2004).
Gericke, A. et al. Importance of phosphorylation for osteopontin regulation of biomineralization. Calcif. Tissue Int. 77, 45–54 (2005).
Hoyer, J. R., Asplin, J. R. & Otvos, L. Jr Phosphorylated osteopontin peptides suppress crystallization by inhibiting the growth of calcium oxalate crystals. Kidney Int. 60, 77–82 (2001).
Aiello, D. et al. Sequestering ability of oligophosphate ligands toward Al3+ in aqueous solution. J. Chem. Eng. Data. 62, 3981–3990 (2017).
Bollyn, J. et al. Polyphosphates and fulvates enhance environmental stability of PO4-bearing colloidal iron oxyhydroxides. J. Agric. Food Chem. 64, 8465–8473 (2016).
Lin, Y.-P. & Singer, P. C. Inhibition of calcite crystal growth by polyphosphates. Water Res. 39, 4835–4843 (2005).
Olszynski, M., Prywer, J. & Mielniczek-Brzóska, E. Inhibition of struvite crystallization by tetrasodium pyrophosphate in artificial urine: chemical and physical aspects of nucleation and growth. Cryst. Growth Des. 16, 3519–3529 (2016).
Fleisch, H., Straumann, F., Schenk, R., Bisaz, S. & Allgower, M. Effect of condensed phosphates on calcification of chick embryo femurs in tissue culture. Am. J. Physiol. 211, 821–825 (1966).
De Yoreo, J. J., Qiu, S. R. & Hoyer, J. R. Molecular modulation of calcium oxalate crystallization. Am. J. Physiol. Ren. Physiol. 291, F1123–F1132 (2006).
DeOliveira, D. B. & Laursen, R. A. Control of calcite crystal morphology by a peptide designed to bind to a specific surface. J. Am. Chem. Soc. 119, 10627–10631 (1997).
Zhu, T. et al. Corrosion-Induced Performance Degradation of Phosphorus-Containing Scale Inhibitors at Carbon Steel–Water Interface. Ind. Eng. Chem. Res. 57, 5183–5189 (2018).
Pinto, A. et al. Sodium tripolyphosphate and sodium hexametaphosphate in preventing dental calculus accumulation in dogs. Arq. Bras. Med. Vet. Zoo. 60, 1426–1431 (2008).
Brady, A. G., Williams, L. E., Haught, D. & Abee, C. R. Use of the feed additive sodium hexametaphosphate to prevent dental calculus in squirrel monkeys (Saimiri spp.). J. Am. Assoc. Lab. Anim. Sci. 39, 27–29 (2000).
Grases, F., Perelló, J., Isern, B. & Costa-Bauzá, A. Myo-inositol hexakisphosphate (phytate) inhibits calcium carbonate crystallisation in hard water. Water SA 33 (2007).
Fakier, S., Rodgers, A. & Jackson, G. Potential thermodynamic and kinetic roles of phytate as an inhibitor of kidney stone formation: theoretical modelling and crystallization experiments. Urolithiasis 47, 493–502 (2019).
Abd-El-Khalek, D. E. & Abd-El-Nabey, B. A. Evaluation of sodium hexametaphosphate as scale and corrosion inhibitor in cooling water using electrochemical techniques. Desalination 311, 227–233 (2013).
Konietzny, U. & Greiner, R. in Encyclopedia of Food Sciences and Nutrition (Second Edition) (ed Benjamin Caballero) 4546-4555 (Academic Press, 2003).
Wang, F., Devine, C. L. & Edwards, M. A. Effect of corrosion inhibitors on in situ leak repair by precipitation of calcium carbonate in potable water pipelines. Environ. Sci. Technol. 51, 8561–8568 (2017).
Trueman, B. F., Krkošek, W. H. & Gagnon, G. A. Effects of ortho-and polyphosphates on lead speciation in drinking water. Environ. Sci. Water Res. Technol. 4, 505–512 (2018).
Deganello, S. & OE, P. The crystal structure of calcium oxalate monohydrate (whewellite). Neues Jahrb. für Mineral. Abhandlungen 81, 81–88 (1981).
Olafson, K. N., Nguyen, T. Q., Rimer, J. D. & Vekilov, P. G. Antimalarials inhibit hematin crystallization by unique drug–surface site interactions. Proc. Natl. Acad. Sci. USA 114, 7531–7536 (2017).
Acknowledgements
JDR and PGV received funding primarily from the Welch Foundation (Awards E-1794 and E-2170) and the Welch Foundation Catalyst for Discovery Center for Advanced Bioactive Materials Crystallization (Award V-E-0001) and NSF (Award DMR-2128121). JDR has received additional support from the National Institutes of Health (R01 DK136684). MRJ received funding from the Packard Foundation (2018-68049) and the Welch Foundation (C-2146). We also wish to acknowledge Dr. Sungmin Han for assistance with SEM and Mangalaa Dinivahi and Sowrav Barua for assistance gathering ISE data. ToF-SIMS analysis was carried out with support provided by the National Science Foundation (Award CBET-1626418) using resources of the Shared Equipment Authority at Rice University. We acknowledge the use of the Electron Microscopy Center (EMC) at Rice University. We wish to thank J.A. Wesson (Nephrology Division of the Medical College of Wisconsin, Department of Veterans Affairs Medical Center, Milwaukee, WI) for generously providing osteopontin, which was donated by Esben Sorensen (Aarhus University, Denmark).
Author information
Authors and Affiliations
Contributions
J.D.R. and B.G.A. conceived the project. B.G.A. and V.P.C. performed data collection and analysis for ISE experiments. D.K. performed data collection and analysis for in situ AFM and OIM experiments. D.K. and V.P.C. contributed to data collection and analysis for microfluidics experiments. V.P.C. performed data collection for bulk crystallization and SEM imaging. J.D.R., B.G.A., D.K., V.P.C. and P.G.V. contributed to the design and analysis of experiments. T.T. collected and analyzed ToF-SIMS data. Z.Y., S.D.F and M.R.J. performed LPTEM experiments with help from V.P.C. for sample preparation. V.P.C. and S.D.F. analyzed the LPTEM experiments. J.D.R. and D.K. wrote the original draft of the manuscript. All authors discussed the results and helped revise the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, D., Chauhan, V.P., Alamani, B.G. et al. Bio-inspired multifunctional disruptors of calcium oxalate crystallization. Nat Commun 16, 5229 (2025). https://doi.org/10.1038/s41467-025-60320-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60320-4
