
Abstract
In a standard analysis, pleiotropic variants are identified by running separate genome-wide association studies (GWAS) and combining results across traits. But such statistical approach based on marginal summary statistics may lead to spurious results. We propose a new statistical approach, Debiased-regularized Factor Analysis Regression Model (DrFARM), through a joint regression model for simultaneous analysis of high-dimensional genetic variants and multilevel dependencies. This joint modeling strategy controls overall error to permit universal false discovery rate (FDR) control. DrFARM uses the strengths of the debiasing technique and the Cauchy combination test, both being theoretically justified, to establish a valid post selection inference on pleiotropic variants. Through extensive simulations, we show that DrFARM appropriately controls overall FDR. Applying DrFARM to data on 1031 metabolites measured on 6135 men from the Metabolic Syndrome in Men (METSIM) study, we identify five first-time reported putative causal genes, none of which had been implicated in any prior metabolite GWAS (including the prior METSIM analysis).
Similar content being viewed by others


Introduction
Genetic studies can help identify the contributions of different variants and genes to various processes and pathways. Identifying pleiotropic genes can help us better understand the mechanism of metabolism pathways1,2. Given that technological advances have significantly accelerated the availability of various multi-omics data types (e.g., genomics, epigenomics, transcriptomics, proteomics, metabolomics, glycomics)3, an unprecedented opportunity arises in the characterization and quantification of pleiotropic genes and genetic variants that regulate multiple phenotypes. However, data analytic techniques to detect pleiotropic genes now lag behind the requirements for increasing high-dimensional data; there are few adequate data analytic methods and software tools available to address the complexity and multimodality of biological data in the detection of pleiotropic genes. Valid statistical methods are essential to explore and understand the underlying biology, generate new hypotheses, and design new experiments to deliver potentially better therapeutics as part of the effort to turn data into knowledge that ultimately improves human quality of life.
Our methods development is largely motivated by the objective of identifying pleiotropic genes for various metabolic traits associated with Type 2 diabetes (T2D) in the Metabolic Syndrome in Men (METSIM) cohort4, a longitudinal study of 10,197 middle-aged and older Finnish men that seeks to identify genetic variants that contribute to the risk of metabolic and cardiovascular disease. T2D is a complex trait that largely involves the interplay between multiple genes5,6. Discovering pleiotropic genetic variants is one of the key tasks to understand how multiple genetic variants interact in biochemical pathways, influencing the risk of developing T2D. Currently, most genome-wide association studies (GWAS) do not formally test for pleiotropy. If testing of pleiotropy is performed, they are based on a single-trait, single-variant analysis approach, which tests for the association of each trait with each variant7,8, followed by a second stage of detecting pleiotropic variants using certain GWAS summary statistics9,10,11,12. As evidenced by our investigation in this paper, in comparison to our proposed joint modeling approach, existing approaches based on marginal associations cannot control the false discovery rate (FDR) and hence are susceptible to spurious findings in the study of genetic pleiotropy. This is due largely to the fact that existing marginal methods may over-estimate the variance of individual trait’s residuals, which then affects the calculation of pleiotropy test statistics and ultimately inflates type I error.
We introduce DrFARM as a method to identify pleiotropic variants in which the over-estimation issue is alleviated by adjusting for other genetic variants. DrFARM provides a high-dimensional estimation of the coefficients and inference of pleiotropic variants, as it is developed to handle data with the number of variants exceeding the sample size. Zhou et al.13 proposed a sparse multivariate factor analysis regression model (FARM), a high-dimensional joint modeling approach, to detect the so-called “master regulators” (a.k.a. pleiotropic variants), in which they used sparse group lasso regularization14 to enforce sparsity at both individual-level (entry-level) and group-level (variant-level)13,15. The group sparsity led to the identification of variants being simultaneously associated with multiple traits. The limitation of the sparse multivariate FARM is that it does not quantify uncertainty, and it does not yield FDR control in the discovery of pleiotropic variants. In addition, sparse multivariate FARM ignores relatedness and population structure16,17,18,19,20.
DrFARM is built upon a post-selection debiasing technique to address these limitations, where valid p values are obtained for statistical inference on pleiotropic variants. The debiasing-based post selection (DPS) inference has been studied extensively in the fields of high-dimensional statistics and machine learning21,22,23,24. This method has only limited previous application in genetic data analyses, an area that naturally demands valid DPS inferences25. The critical technical challenge in the utility of DPS inferences lies in the estimation of the precision matrix of the predictors, which is the inverse of the covariance matrix of the predictors. This matrix plays a central role in DPS inference as it is used in desparsifying regularized estimates, which are then known to follow asymptotic distributions, and consequently allows for high-dimensional statistical inference, including valid p values generation. Although several methods for precision matrix estimation exist, such as graphical lasso (Glasso)26, nodewise lasso21, and quadratic optimization23, there is no consensus on which method has the best FDR control, sensitivity of parameter tuning, robustness of numerical performance, and computational efficiency. To the best of our knowledge, this paper is the first to conduct a comprehensive comparison of existing precision matrix estimation methods in DPS inference using large-scale simulations, leading to practical guidelines on the use of DPS inference in the analysis of pleiotropic variants. Such knowledge may be applied to many empirical studies with limited sample sizes encountered by other high-dimensional genetic and omics data analyses.
DrFARM: 1) performs a rigorous, valid statistical test via debiasing to identify potential pleiotropic variants with a proper overall FDR control; 2) accounts for the relatedness and population structure of genetic data in DPS inference; and 3) allows users to choose a precision matrix estimation method in DPS inference. We demonstrate the performance of DrFARM through extensive simulations and make recommendations useful to the application of DrFARM in practical studies. We also reanalyze metabolomics data from the METSIM study to discover new pleiotropic variants and genes.
Results
Motivating example
We begin with a simple but representative simulation example to motivate the proposed method. We illustrate how pleiotropy may lead to complications in statistical inference. Under the setting of two simulated correlated traits, we first illustrate the empirical type I error given by three approaches to identifying pleiotropic variants under the case P < N: I) Fisher combination test approach: p values are first obtained using a single-trait, single-variant analysis (i.e., univariate Yj, j = 1, 2 regressed on single Xi, i = 1, …, P, respectively) and combined for each variant using the Fisher combination test which takes into account the correlation of Y = (Y1, Y2)9,10; II) MANOVA on multivariate marginal model (i.e., multivariate Y regressed on single Xi, i = 1, …, P, respectively); and III) MANOVA on multivariate joint model of P variables (i.e., Y regressed on X = (X1, …, XP)). To further assess the impact of potential over-estimation for the variance of individual trait’s residuals in the marginal analysis, our comparison is extended to several existing methods for identifying pleiotropic variants, including IV) HOPS11, V) PLEIO27, VI) MTAG28 and VII) Primo29. Note that HOPS and PLEIO enable the detection of pleiotropic variants for two traits, MTAG allows the re-estimation of trait-specific effects of individual SNPs under a shared trait model where the Fisher combination test is applied, and Primo permits an integrative analysis across various sources; see more details in “Setup in motivating example” of Methods. Left half of Fig. 1 shows the average empirical type I error of the first three methods I–III. The two methods II (Marginal) and III (Fisher) based on pairwise association testing suffer from severely inflated empirical type I error. In particular, the Fisher combination test gets ~64% average empirical type I error. On the other hand, the empirical type I error of the joint MANOVA model has virtually a constant 5% type I error. This desirable error control is attributed to the fact that the test statistics in the joint modeling correctly estimate each trait’s residual variance. In contrast, without correctly estimating each trait’s residual variance, the same MANOVA modeling, when applied to pairwise marginal models, fails to control the overall type I error (~39% on average). Right half of Fig. 1 unveils similar evidence of poor type I error control by the four existing methods IV–VI (HOPS, PLEIO, MTAG and Primo). Essentially, these marginal analysis approaches overestimate the trait’s residual variance. This simple example implies the need for a joint modeling approach to identifying pleiotropic variants. For illustration, we limited the number of variants to that of a set of genome-wide significant index variants in the original METSIM marginal analysis, as they were the most likely candidates for pleiotropic variants. In practice, it is almost always the case P > N; for example, using 10−6 cutoff instead of 5 × 10−8 can increase the number of variants in the analysis. Thus, our development of DrFARM further extends the joint MANOVA modeling approach for the high-dimensional case with P > N, which is commonly encountered in the study of pleiotropic variants.

The methods under comparison include two possible methods: Joint (joint MANOVA model) and Marginal (marginal MANOVA model), and existing methods: Fisher (Fisher combination test), HOPS, PLEIO, MTAG, and Primo. Each violin shows the distribution of Type I error estimates across 1000 replicates, with a black horizontal crossbar indicating the median. The red horizontal line represents the nominal 5% type I error level.
Overview
We consider a penalized multivariate regression framework that extends the sparse multivariate FARM13 (see “Review of remMap and sparse multivariate FARM” of Methods for more details) to establish valid post-selection statistical inference. Compared to traditional linear mixed models in GWAS, DrFARM enables the adjustment for other variants via the high-dimensional joint modeling between P variants and Q traits and embraces a factor analysis model (FAM) with K latent factors to characterize the between-trait dependence. Additionally, since FAM in DrFARM allows implicitly for missing heritability in GWAS30,31, it is appealing in the analysis of pleiotropic variants. Moreover, a joint analysis of P variants and Q traits can better estimate the loading coefficients in FAM and subsequently improve both estimation and power. DrFARM also extends the sparse multivariate FARM by allowing a certain kinship structure to correlate latent factors in FAM, as opposed to independent latent factors assumed in sparse multivariate FARM. We show that FAM in DrFARM is equivalent to the specification of genetic random effects in the linear mixed model16,17,18,19,20, but the former has parsimonious model constructs and thus is potentially advantageous for model interpretability.
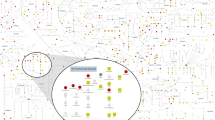
A schematic workflow of DrFARM is given in Fig. 2. To handle simultaneously many variants and traits, in Step 1, DrFARM uses the regularization technique under a sparse group lasso penalty, resulting in both individual (entry-level, i.e., all variant-trait coefficients) level and group (variant-level) level sparsity. Since the sparse estimation does not have the capacity to intentionally control any error rate (e.g., FDR) in the analysis, this method is limited for its use in GWAS when the quantification of sampling uncertainty and discovery rate control is of primary interest. Step 2 of DrFARM implements a rigorous statistical inference through the debiasing technique, leading to valid asymptotic distributions to generate desirable inferential quantities such as p values and confidence intervals for individual association parameters. Step 3 of DrFARM uses the standard FDR control techniques (e.g., Benjamini–Hochberg procedure32) along with the Cauchy combination test (CCT) to calculate combined p values for the detection of pleiotropic variants.

Schematic workflow of the DrFARM method, illustrating the three major steps. The family tree icon symbolizes kinship among related samples. The 3D conformer structure image of the metabolite (hydroxyproline) was obtained from the National Institutes of Health (NIH) PubChem (CID: 5810).
Simulation
We conduct extensive simulation experiments to evaluate the performance of the proposed DrFARM, two of which are reported in detail in this paper. The first compares four methods, including the standard sparse multivariate FARM with no debiasing and three modified sparse multivariate FARM procedures with (i) only inner-debiasing, (ii) only outer-debiasing, and (iii) with double debiasing (i.e., both inner and outer-debiasing) under various choices of precision matrix estimation methods, including Glasso, nodewise lasso, quadratic optimization and naive method (i.e., no use of the precision matrix in inner-debiasing). Inner-debiasing refers to a debiasing step taken within the M-step of the EM algorithm (see Algorithm 1 in Methods); outer-debiasing operates a desparsifying step to ensure the asymptotic normality for individual sparse estimates. The remMap15 model, which does not involve FAM, is also included in the comparison as the most parsimonious joint model. The second simulation investigates the influence of kinship on whether or not to be included in the latent factors of FAM when data are sampled from genetically related subjects. In each simulation setting, we vary the sample size, number of SNPs, number of traits, and number of latent factors. See Supplementary Table 1 in the Supplementary Note 13 for a more detailed description of simulation settings.
In simulation I, we generated data from a standard sparse multivariate FARM assuming independent individuals. As seen in Scenario I in Fig. 3, all methods that do not use outer-debiasing appear to have high FDRs at both individual and group-levels. Similarly, Scenario II in Table 1 suggests that both remMap and the naive method perform poorly in the FDR control without using outer-debiasing. The naive method inflates individual-level and group-level FDRs as high as 27.2% and 65.9%, respectively.

(A) across 100 replicates: A1: remMap.none; A2: remMap.outer; A3: Naive.none; A4: Naive.outer; A5: Glasso.inner; A6: Glasso.double; A7: NL.inner; A8: NL.double; A9: QO.inner; A10: QO.double. NL refers to node-wise lasso, and QO refers to quadratic optimization. In these box plots, the box represents the interquartile range (IQR), the horizontal line inside the box indicates the median, and the whiskers extend to the most extreme data point within 1.5 times the IQR. Data points beyond this range are shown as individual black circle dots.
In regard to the choice of precision matrix estimation, the strategy of the inner-debiasing appears to be very conservative; despite achieving accurate FDR control at 5% for the group-level signals, the FDRs for individual-level signals range from 0.6 to 0.7%. This shows that there is a conservative FDR control by the regularized method. In contrast, for the strategies involving the use of the outer-debiasing, four methods (remMap, naive, Glasso and nodewise lasso) are all able to control their FDRs at levels close to 5% for both individual-level and group-level signals, except the strategy using the quadratic optimization method, the precision matrix estimation yields on average 8.9% FDR for individual signals and 6.8% FDR for group-level signals. In addition to FDR, we compare their performances by MCC (Matthews correlation coefficient), a composite metric of sensitivity and specificity. Supplementary Table 2 in Supplementary Note 13 shows that the naive, Glasso and nodewise lasso with the outer-debiasing show very similar MCCs for the detection of both individual-level and group-level signals. In Scenario I, the MCC values in Supplementary Table 2 indicate that the naive method with the outer-debiasing is slightly more powerful than Glasso and nodewise lasso for the detection of both individual-level and group-level signals. In summary, outer-debiasing is deemed essential to control FDR while not being too conservative.
In simulation II, we simulate data by mimicking GWAS of common variants (≥5% minor allele frequency) in genetically related individuals of on average the third-degree relatedness. Based on our experiences from simulation I, we found that no use of the outer-debiasing leads to an unsatisfactory FDR control, so we here only focus on the results from the methods with the utility of the outer-debiasing. As shown in Fig. 4 (Scenario I), the FDR for individual-level signal for the quadratic optimization method appeared constantly above 5% regardless of accounting for kinship or not, whereas the FDR for group-level signals is controlled under 5%. All the other methods of precision matrix estimation exhibit satisfactory FDR control at levels close to or below 5%. In particular, the FDR for the individual-level signal was uniformly very close to 5%. Furthermore, from the performance results in terms of MCC in Supplementary Tables 3 (Scenario I) and 4 (Scenario II) in Supplementary Note 13, we again observe that the naive method, with or without kinship, is slightly more powerful than both Glasso and nodewise lasso methods for the detection of both individual-level and group-level signals. Incorporating kinship in the analysis does not lead to gains in MCC due largely to the fact that MCC is not a metric of statistical power (or one minus type II error) but a metric of detection accuracy composed of sensitivity and specificity.

In these box plots, the box represents the interquartile range (IQR), the horizontal line inside the box indicates the median, and the whiskers extend to the most extreme data point within 1.5 times the IQR. Data points beyond this range are shown as individual black circle dots.
In conclusion, based on our simulation setup, kinship appears to minimally impact FDR. Thus, one may choose not to use kinship in DrFARM to reduce computational burden. However, given the potential significance of kinship in other contexts, further investigations into its impact on FDR and signal detection are warranted. In addition, among the 3 precision estimation approaches (Glasso, naive method and nodewise lasso) with FDR control, we recommend Glasso as it utilizes the inner-debiasing step, and the computational complexity (or CPU time) is the lowest. Additionally, Fig. 5 shows power curves of DrFARM over effect sizes with different sample sizes. Based on the results, a sample size of 1000 is deemed adequate for DrFARM to achieve desirable power, a sample size requirement akin to GWAS standards (e.g., see Saber and Shapiro33).

The x axis shows the absolute values of the estimated effect sizes arising from debiasing-based post selection (DPS) inference. Such estimates differ in scale from the true effect sizes due to the standardization for both the predictors (X) and outcomes (Y) prior to regularization.
Real data application
Given the high correlation of metabolite abundance for many sets of metabolites across METSIM study participants, we expect to see that many loci exhibit pleiotropy across those metabolite sets. In the original single-metabolite GWAS34, we found at least one significant (p < 7.2 × 10−11) association for 803 of the 1031 tested metabolites. Of the \(322,003=\left(\begin{array}{c}803\\ 2\end{array}\right)\) possible combinations of these metabolites, 334 have a high phenotypic correlation (i.e., ρ ≥ 50%). And of the 334 highly correlated metabolite pairs, 257 (77%) exhibit pleiotropy in at least one locus, where we define pleiotropy as having significant hits for each metabolite within 10 kb of each other (Supplementary Table 3, Yin et al.34). For example, the two medium-chain acylcarnitines hexanoylcarnitine and octanoylcarnitine both have significant lead SNPs at the ACADM locus (encoding the medium-chain acyl-CoA dehydrogenase), which was unsurprising considering this enzyme acts on both metabolites35, and both the metabolites are strongly correlated, ρ = 0.636.
Similarly, 257 (4.5%) of the 5176 unique metabolite pairs sharing a locus (at least one significant hit for each metabolite within 10 kb of each other) in Yin et al.34, have a high phenotype correlation. Thus, at least some of the observed pleiotropy can be explained by the phenotypic correlation of the metabolite concentrations. However, a single locus can also be significantly associated with traits that are not highly correlated at the phenotypic level. For example, hexanoylglycine has a significant association at the ACADM locus even though the phenotypic correlation ρ with hexanoylcarnitine is only 0.185.
Because DrFARM uses the correlation structure across the metabolites to enhance the power to detect genetic associations for individual metabolites, we explored the extent to which the associations identified by DrFARM reflect these phenotypic correlations. Of the 77 = 334−257 highly correlated metabolite pairs with no pleiotropic loci in the original study, DrFARM detected a significant association for an additional 16 of the 77. For example, the caffeine metabolites 1-methylurate and paraxanthine share a phenotypic correlation ρ = 0.578, and yet while paraxanthine was significantly associated with the CYP2A6 locus (p = 2.2 × 10−19 at rs56113850) in the single-metabolite GWAS, 1-methylurate has a p value of only 0.0013 at this same variant in the single-metabolite analysis. In contrast, DrFARM assigns a p value of 3.9 × 10−13 to 1-methylurate at rs56113850. This association is highly plausible given that the CYP2A6 enzyme is responsible for acting on paraxanthine on its way to being converted to 1-methylurate.
In all, DrFARM assigned a p value < 7.2 × 10−11 to 403 (386 pleiotropic + 17 “singleton”) variants (see Supplementary Data 1). These 403 variants collectively yield 2287 significant metabolite associations. While a subset of these 2287 associations involves metabolites that are highly correlated with previously identified metabolites, 70% do not exhibit high correlation to any previously identified metabolite at the same locus. For example, at the GLS2 locus (encoding a glutaminase enzyme), the single-metabolite GWAS identified significant associations for both glutamine and a glutamine derivative, gamma-glutamylglutamine. DrFARM found an additional association for another glutamine derivative, hexanoylglutamine, despite the fact that hexanoylglutamine and glutamine share a phenotypic correlation (ρ) of only 6 × 10−4. Despite the low phenotypic correlation of most of the new metabolite associations from DrFARM compared to the previous single-metabolite results, the vast majority of the new results represent highly plausible biological results. For example, where the previous analysis identified tyrosine as a significant association at the TAT locus (encoding tyrosine aminotransferase), the new analysis identified a significant association for the tyrosine derivative, N-acetyltyrosine. The new analysis also identified a significant association for kynurenine at the KMO locus (encoding kynurenine 3-monooxygenase), for the caffeine derivatives 1-methylurate, 3,7-dimethylurate, 1,7-dimethylurate at the CYP2A6 locus (encoding a caffeine metabolizing enzyme), for the pyrimidine metabolite uracil at the CDA locus (encoding the pyrimidine metabolizing enzyme, cytidine deaminase) and the very long acyl carnitine 5-dodecenoylcarnitine at the ACADVL locus (encoding the very long-chain specific acyl-CoA dehydrogenase).
To further evaluate the DrFARM-identified associations, we performed a colocalization analysis (using HyPrColoc12) comparing DrFARM signals with those from the original METSIM single-metabolite GWAS34. Of the 1748 locus–metabolite associations that DrFARM flagged at p < 7.2 × 10−11 and also retained by colocalization, 1480 were also reported at or below that threshold in the single-metabolite analysis. Among the remaining 268 associations, 229 occurred within 500 kb of a published lead SNP but did not meet the stringent study-wide significance cutoff. Remarkably, 31 of the 39 remaining signals (i.e., those more than 500 kb away from any previously reported association) had already been annotated with a likely causal gene in our earlier genome-wide (but not study-wide) analysis, accounting for 79.5% of the novel locus–metabolite associations highlighted by colocalization. These include three first-time reported metabolite QTL genes (ACER3, AGPAT5, and ELOVL6), each of which plays a key role in lipid or sphingolipid metabolism.
In contrast, HyPrColoc dropped 146 DrFARM associations, including 13 signals with no nearby (±500 kb) published associations. Of these 13, only 7 were previously linked to three putative causal genes (PEMT, SLC7A7, and CETP), each implicated by prior metabolite GWAS, representing 53.8% of the novel locus–metabolite associations from DrFARM that did not pass the colocalization analysis. In addition, there were 393 DrFARM associations in which only a single metabolite achieved study-wide significance (p < 7.2 × 10−11), in which case colocalization analysis was not possible. Notably, two of these signals map to GPD2 and TNFSF11, each representing a first-time reported metabolite QTL gene. We refer the reader to the Supplementary Data for additional details on these loci and for a comprehensive breakdown of the colocalization analysis.
We showed that cross-referencing the DrFARM detected significant associations with biological knowledge gleaned from the rich history of biochemistry provides independent validation of these results. Expanding the current analysis to systematically identify pleiotropic genes for multiple correlated metabolites is a promising future research direction.
Discussion
We developed a new method, DrFARM, to identify potential pleiotropic variants in GWAS. Our methodological contribution centers on post-selection hypothesis testing, adjusting for other genetic variants and confounding factors. DrFARM provides satisfactory FDR control in the detection of both individual-level (entry-level) and group-level (variant-level) signals. In addition, DrFARM incorporates population structure in the latent factors as part of the modeling of between-trait correlations. Being a nontrivial extension from a low-dimensional joint modeling approach, DrFARM overcomes a difficult problem of proper FDR control in the large-P-small-N setting, which has troubled existing pairwise single-variant marginal association testing in the GWAS literature. Our study demonstrates the necessity in including relevant independent variants—as many as possible—in pleiotropy analyses, which has been largely overlooked by existing methodologies. DrFARM is proposed to significantly refine the input to downstream colocalization analyses, such as Moloc36 and HyPrColoc12.
The primary goal of colocalization analysis is twofold: To examine if a certain genomic region is commonly associated with different traits, and to identify which variants are most likely to be responsible for such associations. In contrast, DrFARM enhances the colocalization process by starting with a set of index variants, each being thought of as a statistically independent signal cluster37, which serves as input genetic markers. DrFARM allows for the identification of preliminary pleiotropic variants potentially linked to putative causal gene regions. Those detected candidate variants may be further scrutinized using colocalization techniques tailored for two-trait (Moloc) or multi-trait (HyPrColoc) analyses. This scrutiny step effectively determines the most plausible variant within a signal cluster (now confined within a specific gene region), which leads to the best candidate for true pleiotropy. To illustrate, we provide the HyPrColoc multi-trait colocalization results in Supplementary Data 2. Using 386 pleiotropic variants identified by DrFARM as input, this colocalization analysis yields 368 meaningful clusters of colocalized metabolites. Of these, 63.9% (235/368) of the clusters achieve a posterior probability of colocalization >90%, even when involving a high number of traits (up to 17). Thus, DrFARM not only identifies more reliable and promising candidate gene regions for downstream analysis but also establishes a more robust foundation for colocalization analyses by ensuring that the input consists of potential pleiotropic variants with genuine associations.
A proven advantage of DrFARM is that it can increase power by taking into account the correlation between related traits, enabling identification of associations not identified in single-trait analyses. We identified five unreported candidate genes with DrFARM in the METSIM data analysis. DrFARM is not limited to the association study of metabolites-genetic variants but is applicable to other high-dimensional omics data types such as proteins and glycans. Thus, DrFARM presents an ample opportunity to discover pleiotropic variants in the integrative analysis of multi-trait and multimodal omics data in the modern biology era.
DrFARM has some limitations that deserve further exploration in future research. First, DrFARM is built upon L1 penalty regularization, which is known to suffer from overfitting when predictors are highly correlated. We have seen the sensitivity of FDR on modest or highly correlated SNPs (e.g., correlation ≥0.7), indicating a need to invoke a better regularization method to improve DrFARM with correlated SNPs. Second, DrFARM requires the use of an estimated precision matrix in the outer-debiasing step to calculate p values for inference. Taking our recommended method Glasso (balancing computational efficiency and statistical performance) as an example, the computational complexity is O(P3) to O(P4), depending on the actual sparsity of the precision matrix38. Thus, DrFARM is computationally expensive to handle tens of thousands of variants, which might be improved by feature screening methods39 to reduce dimensionality prior to the application of DrFARM, or by a fast precision matrix estimation method. It is worth noting that DrFARM in its present form may not be scalable to biobank-level datasets. As outlined in Algorithm 1, the computational complexity of DrFARM is O(NPQ). To improve the scalability of DrFARM, a viable future direction is to harness the distributed computational techniques for post-model selection inference, as introduced by Tang et al.40. Through the parallelized computing architecture, the computational burden in the LASSO regularization method can be distributed across multiple CPUs. In this way, DrFARM could significantly increase its scalability, thereby paving the way for its widespread application in large-scale biobank data analysis.
As for future work, one direction is to investigate the latent factors used by DrFARM. Similar to traditional factor analysis, the interpretation of latent factors is a challenging issue. Potentially, geneticists could mine the latent factors to understand the missing heritability in GWAS, similar to how principal component analysis (PCA) has helped to understand population stratification41. Related tasks would include associating these latent factors with different gene regions and elucidating what kind of factor rotation provides a meaningful interpretation for the latent factors. With the ever-increasing size of GWAS cohorts and whole-genome sequencing platforms, another important work is to develop scalable algorithms for estimating ultra-high-dimensional precision matrices, as they play a crucial role in statistical inference with high-dimensional genomics data. Scalability of DrFARM may be further improved by incorporating summary statistics in the proposed analytics useful for the analysis of large-scale biobank data. This task requires a substantial methodological effort on an extension of the EM algorithm for its operation with summary statistics. Finally, another significant direction for future research is the replication of our findings in independent cohorts. While the present study’s results are promising, replicating the newly identified loci in an independent cohort would further validate and strengthen our findings. This limitation may be overcome when we have access to independent datasets in the future. We expect that with the availability of the DrFARM software, researchers in the field may use their own data to replicate our findings, thus reaching broader implications in genetic studies.
Methods
Ethical compliance
This study was approved by the Ethics Committee at the University of Eastern Finland and the Institutional Review Board at the University of Michigan. All participants provided written informed consent.
Setup in motivating example
Consider two correlated traits, Y1 and Y2, constituting a bivariate trait by Y = (Y1, Y2). Suppose that Y is generated from the true model
where X = (X1, ⋯ , XP) is a set of P predictors (e.g., SNPs), β11 and β12 are P-dimensional vector of true coefficients associating X with Y1 and Y2, respectively (notice that some of the coefficients of β11 and β12 can be zero). Since the traits are correlated, we assume and attribute this to an environmental covariance ρ, for Var(ϵ), where ρ ≠ 0.
In practice, it is often assumed that the P SNPs are independent and contribute to the traits independently. However, this assumption may be violated for genetic data due to factors including linkage disequilibrium and population structure42. Nonetheless, it is useful to consider the concept of signal cluster37 and think of SNPs coming from P statistically (roughly) independent sources contributing to the traits.
We set N = 6135, P = 2072 (same as our real data analysis setting), and suppose there are 250 true SNPs that contribute to the two traits. The effect sizes of true SNPs are generated by sampling 500 = 250 × 2 effect sizes from the set of 3443 genome-wide significant associations from prior METSIM single-metabolite GWAS.34. We also set a weak environmental covariance ρ = 0.3. SNPs are generated by sampling 2072 SNPs from a set of 6334 LD-pruned SNPs from chromosome 22 using METSIM data with r2 = 0.01 threshold. The empirical type I error is given by the number of significant discoveries (i.e., p value < 0.05) in the null set divided by 1822 = 2072−250 (the number of null), which is evaluated from 1000 replicates.
In our analysis, we considered various methods to illustrate the validity of the joint modeling approach in type I error control, including I) Fisher combination test9,10; II) MANOVA with two versions of the multivariate marginal models, and III) the multivariate joint model. In addition, we also included four existing methods, including IV) HOPS11, V) PLEIO27, VI) MTAG28 and VII) Primo29. Each of these methods requires different types of inputs, with the details given as follows.
-
HOPS: This method requires a P × Q matrix of Z scores, with Q = 2 being allowed in this method. We used Z scores from pairwise association testing, which are obtained by regressing each Yj, j = 1, 2 on single Xi, i = 1, …, P, respectively, to mimic current GWAS practices, and the magnitude pleiotropy score11p value was used to calculate type I error.
-
PLEIO: Utilizing summary statistics, which are based on standardized phenotype and genotype data, including both effect sizes and standard errors for both traits, PLEIO needs an estimated genetic covariance matrix, and an environmental correlation matrix, with both typically derived from cross-trait LD-score regression (LDSC)43.
-
Our simulation generates a bivariate trait (Y1, Y2) through a linear model with a subset of 6334 LD-pruned SNPs from chromosome 22, in which the subset size varies between simulation replicates. The LDSC is a standard approach for calculating genetic covariance (H) and environmental covariance (E), which was found to be unreliable. This is because LDSC requires LD scores from a reference panel, yet only 50 of the 6334 SNPs had an LD-score available from the European reference panel. Such a low number of SNPs (≤50) can produce unstable estimates for H and E. To address this issue, we employ another commonly used alternative approach to identify null SNPs used in the estimation of E. That is, we selected SNPs with absolute Z scores of magnitude less than 2 for both Y1 and Y2, resulting in an average of 400 SNPs or so over 1000 replicates. Then we estimated E using the covariance matrix of these Z scores. The phenotypic covariance matrix, Σ, was calculated by the sample covariance matrix given by Σ = YTY/(N − 1). Using the decomposition Σ = H + E, we estimated H as Σ − E. Finally, we converted the environmental covariance matrix into a correlation matrix using the R function cov2cor(), and used the PLEIO p value from this output to calculate the type I error.
-
MTAG: This method requires the same inputs as PLEIO, generated in the same manner described above. Since MTAG was used to re-estimate effect size, as opposed to giving an overall p value for pleiotropy, we employed the Fisher combination test to combine two re-estimated p values, providing the final p value for pleiotropy to calculate type I error.
-
Primo: This method requires a P × Q matrix of effect sizes, standard errors and sample sizes. Similar to the procedure used to yield the P × Q matrix of Z scores for PLEIO, we obtained effect sizes and corresponding standard errors through marginal analyses. Since Primo is a Bayesian method, it also requires a vector of length Q for the proportion of test statistics (or a prior) that are non-null for each trait. This proportion was set to (250/2072, 250/2072)T, which is the true proportion used in the simulation. Furthermore, Primo requires the minor allele frequency (MAF) for summary statistics derived from SNP data, such as those from GWAS studies. The MAF was calculated as \(\min \{1-{\overline{X}}_{j}/2,{\overline{X}}_{j}/2\}\), where \({\overline{X}}_{j}\) represents the sample mean of the jth column in the sampled genotype matrix X.
-
The Primo method outputs a P × 2Q posterior probability matrix of association patterns. Given Q = 2, this results in 22 = 4 possible configurations per row (SNP), with the probabilities across these configurations summing to 1. These configurations are (0, 0), (0, 1), (1, 0), and (1, 1), where a value of 0 indicates no association of the SNP with the corresponding trait. For example, (0, 1) signifies an association with only the second trait. Let π1j, π2j, π3j, and π4j denote the posterior probabilities that the jth SNP is associated with the patterns (0, 0), (0, 1), (1, 0), and (1, 1), respectively. For null SNPs, patterns (0, 1), (1, 0), and (1, 1) represent incorrect associations in our simulation. Thus, we compute 1 − π1j = π2j + π3j + π4j and apply a 90% threshold, as per Gleason et al.29, i.e., if 1−π1j > 0.9 or equivalently π1j < 0.1, a SNP is considered a discovery. The type I error rate is calculated as the proportion of null SNPs identified as discoveries over the total number of null SNPs.
Review of remMap and sparse multivariate FARM
Both remMap and sparse multivariate FARM are regularized multivariate regression models that exploit a sparse group lasso penalty to identify “master” predictors (i.e., pleiotropic variants in GWAS). In particular, sparse multivariate FARM extends remMap by modeling residual correlations of traits via a latent factor model13. More specifically, assume P SNPs and Q traits are collected in each individual. Let \({{{{\bf{x}}}}}_{i}={({x}_{i1},\cdots,{x}_{iP})}^{T}\) and \({{{{\bf{y}}}}}_{i}={({y}_{i1},\cdots,{y}_{iQ})}^{T}\) (i = 1, …, N) be normalized SNPs and normalized traits with mean 0 and variance 1, respectively. The multivariate FARM takes the form:
where Θ = {θqp} is a Q × P coefficient matrix, B is a Q × K matrix of factor loadings (K being the number of latent factors). Multivariate FARM assumes the latent factors \({{{{\bf{z}}}}}_{i}={({z}_{i1},\cdots,{z}_{iK})}^{T} \sim {{{\mathrm{MVN}}}}_{K}({{{{\bf{0}}}}}_{K},{{{{\bf{I}}}}}_{K})\) that may be related to either biological systems or environmental exposures. Moreover, ϵi = \({({\epsilon }_{i1},\cdots,{\epsilon }_{iQ})}^{T}\)’s are independent and identically distributed (i.i.d.) errors from MVNQ(0Q, Ψ) with 0Q being a Q-element zero vector and Ψ = diag(ψ1, ⋯ , ψQ) being a Q × Q diagonal matrix. The multivariate FARM further assume ϵi is independent of the latent factors zi.
The multivariate FARM has the following equivalent form:
where \({{{{\bf{Y}}}}}_{N\times Q}={({{{{\bf{y}}}}}_{1},\cdots,{{{{\bf{y}}}}}_{N})}^{T},{{{{\bf{X}}}}}_{N\times P}= {({{{{\bf{x}}}}}_{1},\cdots,{{{{\bf{x}}}}}_{N})}^{T},{{{{\bf{Z}}}}}_{N\times K}= {({{{{\bf{z}}}}}_{1},\cdots,{{{{\bf{z}}}}}_{N})}^{T} \sim {{{\mathrm{MN}}}}_{N\times K}({{{{\bf{O}}}}}_{N\times K}, {{{{\bf{I}}}}}_{N},{{{{\bf{I}}}}}_{K})\) and \({{{{\bf{E}}}}}_{N\times Q}={({{{{\boldsymbol{\epsilon }}}}}_{1},\cdots,{{{{\boldsymbol{\epsilon }}}}}_{N})}^{T} \sim {{{\mathrm{MN}}}}_{N\times Q}({{{{\bf{O}}}}}_{N\times Q},{{{{\bf{I}}}}}_{N}, {{{\mathbf{\Psi }}}})\). Here MNn×m(M, Vr, Vc) denotes the n × m matrix normal distribution with mean matrix M (n × m), row (inter-sample) covariance matrix Vr (n × n) and column (between component) covariance Vc (m × m). The conditional covariance of the response variables given the predictors is Var(yi∣xi) = Σ = BBT + Ψ. To illustrate the role of latent factors, we provided some simulation results (Supplementary Fig. 2) in the Supplementary Note 13 to numerically exhibit the advantage of the FARM to reach parsimonious findings with no sacrifice of FDR. More details can be found in Supplementary Note 6.
The objective function of sparse multivariate FARM is given by
where \({\parallel }{{{\mathbf{\Theta }}}}{\parallel }_{1}={\sum }_{q=1}^{Q}{\sum }_{p=1}^{P}| {\theta }_{qp}|\) and \(\parallel {{{{\mathbf{\Theta }}}}}^{T}{\parallel }_{2,1}=\mathop{\sum }_{p=1}^{P}\sqrt{{\theta }_{1p}^{2}+\cdots+{\theta }_{Qp}^{2}}\), and λ1, λ2 > 0 are tuning parameters controlling the entrywise sparsity and column-wise sparsity in Θ, respectively.
We estimate the parameters (Θ, B, Ψ) in sparse multivariate FARM using the EM-GCD algorithm13, which uses a group-wise coordinate descent (GCD) algorithm for estimating Θ and expectation-maximization (EM) algorithm for estimating both B and Ψ. When there are no latent factors (i.e., K = 0), Model (2) reduces to the remMap model. The objective function of remMap is given by
Here the first term is under the Frobenius norm. Notice that (5) implicitly assumes the variance of the Q trait residuals is equal. The parameter Θ is estimated using a modified version of the active shooting algorithm15,44,45. More details of remMap and sparse multivariate FARM may be found in Peng et al.15 and Zhou et al.13, respectively.
Generalized multivariate FARM
We consider a generalization of the multivariate FARM in DrFARM where the latent factors are allowed to be correlated when study participants are related. That is, we specify Z ~ MNN×K(ON×K, K, IK), where K (N × N) is a prespecified kinship matrix that is scaled to have diagonal elements equal to 1 analogous to a correlation matrix. In GWAS, K is typically estimated separately from available genotype data, e.g., using KING46. To decorrelate samples, we perform an eigendecomposition of K = UDUT17,20,47,48, where U is an N × N orthogonal matrix of eigenvectors and D = diag(δ1, ⋯ , δN) is an N × N diagonal matrix of eigenvalues. Correspondingly, an equivalent form of the generalized multivariate FARM is
where \(\widetilde{{{{\bf{Y}}}}}={{{{\bf{U}}}}}^{T}{{{\bf{Y}}}},\widetilde{{{{\bf{X}}}}}={{{{\bf{U}}}}}^{T}{{{\bf{X}}}},\widetilde{{{{\bf{Z}}}}}={{{{\bf{U}}}}}^{T}{{{\bf{Z}}}} \sim {{{\mathrm{MN}}}}_{N\times K}({{{{\bf{O}}}}}_{N\times K},{{{\bf{D}}}},{{{{\bf{I}}}}}_{K})\) and \(\widetilde{{{{\bf{E}}}}}={{{{\bf{U}}}}}^{T}{{{\bf{E}}}} \sim {{{\mathrm{MN}}}}_{N\times Q}({{{{\bf{O}}}}}_{N\times Q},{{{{\bf{I}}}}}_{N},{{{\mathbf{\Psi }}}})\). That is, for each individual i,
where \({\widetilde{{{{\bf{y}}}}}}_{i},{\widetilde{{{{\bf{x}}}}}}_{i},{\widetilde{{{{\bf{z}}}}}}_{i}\) and \({\widetilde{{{{\boldsymbol{\epsilon }}}}}}_{i}\) are the ith row of \(\widetilde{{{{\bf{Y}}}}},\widetilde{{{{\bf{X}}}}},\widetilde{{{{\bf{Z}}}}}\) and \(\widetilde{{{{\bf{E}}}}}\), respectively. Note that there is an extra δi term in the variance of \({\widetilde{{{{\bf{z}}}}}}_{i}\) compared to zi in (2) due to the presence of kinship dependence among subjects. With the transformation, the likelihood can be obtained as a product of N individual likelihoods, which can be easily evaluated. To deal with latency of \({\widetilde{{{{\bf{z}}}}}}_{i}\)’s, we invoke the EM algorithm by treating the \({\widetilde{{{{\bf{z}}}}}}_{i}\)’s as missing data in the estimation of the model parameters (Θ, B).
The generalized multivariate FARM connects to the multivariate linear mixed model GEMMA given in Zhou and Stephens48: Y = XΘT + G + E, where GN×Q ~ MNN×Q(ON×Q, K, Vg) is genetic random effects, E ~ MNN×Q(ON×Q, IN, Ve), Vg is the Q × Q symmetric matrix of genetic variance component and Ve is the Q × Q symmetric matrix of environmental variance components. In comparison, generalized multivariate FARM is more parsimonious by modeling the random effects G with FAM ZBT ~ MNN×Q(ON×Q, K, BBT) (or equivalently, Vg = BBT). FAM presents simpler covariance structures to both genetic and environmental variance component matrices, and the latent factors may be used to investigate the missing heritability in GWAS (see “Discussion”).
Regularized estimation
The complete data log-likelihood is
where C is a suitable constant.
To identify pleiotropic variants, we employ a regularized estimation method via the sparse group lasso penalty (by predictor/column) λ1∥Θ∥1 + λ2∥ΘT∥2,1 to achieve sparse estimation of Θ, where λ1, λ2 are tuning parameters controlling the entrywise sparsity and column-wise sparsity in Θ, respectively. This penalized estimation is integrated with the EM algorithm that deals with the augmented data log-likelihood with latent factors \(\widetilde{{{{\bf{Z}}}}}\). The penalized log-likelihood function for complete data is given by
where \({g}_{{\lambda }_{1},{\lambda }_{2}}({{{\mathbf{\Theta }}}}):={\lambda }_{1}\parallel {{{\mathbf{\Theta }}}}{\parallel }_{1}+{\lambda }_{2}\parallel {{{{\mathbf{\Theta }}}}}^{T}{\parallel }_{2,1}\) and C is a suitable constant with respect to the parameters (Θ, B, Ψ).
Let t be the iteration number. In the E-step we calculate the first two conditional moments
where \({{{{\bf{W}}}}}_{i}={\delta }_{i}{{{{\bf{B}}}}}^{T}{({\delta }_{i}{{{\bf{B}}}}{{{{\bf{B}}}}}^{T}+{{{\mathbf{\Psi }}}})}^{-1}\) and \({\widetilde{{{{\boldsymbol{\epsilon }}}}}}_{i}^{*}={\widetilde{{{{\bf{y}}}}}}_{i}-{{{\mathbf{\Theta }}}}{\widetilde{{{{\bf{x}}}}}}_{i}\).
In the M-step, we compute \({\theta }_{ij}^{(t+1)}\) (see expression (1) in Supplementary Note 1),
For the detailed derivation, please refer to Supplementary Note 1. Let \(\widehat{{{{\mathbf{\Theta }}}}},\widehat{{{{\bf{B}}}}},\widehat{{{{\mathbf{\Psi }}}}}\) be the regularized estimator for Θ, EM estimator for B and Ψ, respectively. Also, let \(\,{{{\mathrm{E}}}}\,(\widetilde{{{{\bf{Z}}}}}| \widetilde{{{{\bf{Y}}}}})={({{{\mathrm{E}}}}({\widetilde{{{{\bf{z}}}}}}_{1}| {\widetilde{{{{\bf{y}}}}}}_{1}),\cdots,{{{\mathrm{E}}}}({\widetilde{{{{\bf{z}}}}}}_{N}| {\widetilde{{{{\bf{y}}}}}}_{N}))}^{T}.\) Then, we denote the conditional moment based on estimators \(\widehat{{{{\mathbf{\Theta }}}}},\widehat{{{{\bf{B}}}}},\widehat{{{{\mathbf{\Psi }}}}}\) by \(\widehat{\,{{{\mathrm{E}}}}\,}(\widetilde{{{{\bf{Z}}}}}| \widetilde{{{{\bf{Y}}}}})\). Define \({L}^{(t)}=L({\widehat{{{{\mathbf{\Theta }}}}}}^{(t)},{{{{\bf{B}}}}}^{(t)},{{{{\mathbf{\Psi }}}}}^{(t)}),{\widetilde{{{{\bf{Y}}}}}}^{*(t)}=\widetilde{{{{\bf{Y}}}}}-\,{{{\mathrm{E}}}}\,({\widetilde{{{{\bf{Z}}}}}}^{(t)}| \widetilde{{{{\bf{Y}}}}}){{{{\bf{B}}}}}^{(t-1)T}\) and \({\widetilde{{{{\bf{E}}}}}}^{*(t)}=\widetilde{{{{\bf{Y}}}}}-\widetilde{{{{\bf{X}}}}}{{{{\mathbf{\Theta }}}}}_{\,{{\mathrm{db}}}\,}^{(t)}\). The pseudocode of the EM algorithm for parameter estimation is given in Algorithm 1. We highlight two major differences compared to the algorithm implemented in sparse multivariate FARM13: (i) Instead of obtaining an exact minimizer of \(\widehat{{{{\mathbf{\Theta }}}}}\) in M-step 1, we use a one-step update49 to reduce the computational cost. Our numerical studies show that the one-step approximation does not change the final estimate much but greatly improves the overall computational efficiency. (ii) We add a second M-step 2 to calculate a debiased estimate \({{{{\mathbf{\Theta }}}}}_{\,{{\mathrm{db}}}\,}^{(t)}\). This debiasing step helps us to get a more stable estimate of the residual matrix \({\widetilde{{{{\bf{E}}}}}}^{*}\), which subsequently enhances the estimation of the quantities in the FAM (B, Ψ) in M-step 3. We refer to M-step 2 as inner-debiasing. The initial value determination and tuning parameter selection are detailed in the Supplementary Note 3.
Algorithm 1
EM Algorithm for a given pair of tuning parameters (λ1, λ2)
Data: X, Y, K
Result: \(\widehat{{{{\mathbf{\Theta }}}}}=\{{\hat{\theta }}_{ij}\},\widehat{{{{\bf{B}}}}},\widehat{{{{\mathbf{\Psi }}}}},\widehat{\,{{{\mathrm{E}}}}\,}(\widetilde{{{{\bf{Z}}}}}| \widetilde{{{{\bf{Y}}}}})\)
Obtain U and D from eigendecomposition K = UDUT;
Transform \(\widetilde{{{{\bf{X}}}}}={{{{\bf{U}}}}}^{T}{{{\bf{X}}}}\) and \(\widetilde{{{{\bf{Y}}}}}={{{{\bf{U}}}}}^{T}{{{\bf{Y}}}}\);
Fix tolerance ξ;
Initialize Θ(0) and B(0);
Estimate precision matrix \(\widehat{{{{\mathbf{\Omega }}}}}\) from sample covariance matrix
\(\widehat{{{{\bf{C}}}}}=({{{{\bf{X}}}}}^{T}{{{\bf{X}}}})/N\) (except for the nodewise lasso approach);
Set t = 0;
While L(t+1) − L(t) > ξ and L(t+1) < L(t) Set t = t + 1; do
E-step:
Obtain both first and second conditional moments of \(\widetilde{{{{\bf{Z}}}}}\) using (9) and (10);
M-step:
M-Step 1: Update \({\theta }_{ij}^{(t)}\) using (1) in Supplementary Note 3 for all i, j in a coordinate descent search, using the active shooting scheme proposed in Peng et al.15;
M-Step 2: Obtain an inner debiased estimate \({{{{\mathbf{\Theta }}}}}_{\,{{\mathrm{db}}}\,}^{(t)}={{{{\mathbf{\Theta }}}}}^{(t)}+ \frac{1}{N}({\widetilde{{{{\bf{Y}}}}}}^{*(t)T}- {{{{\mathbf{\Theta }}}}}^{(t)}{\widetilde{{{{\bf{X}}}}}}^{T})\,\widetilde{{{{\bf{X}}}}}\,\widehat{{{{\mathbf{\Omega }}}}}\);
M-Step 3: Update B(t) and (t) using (11) and (12) with the residual matrix \({\widetilde{{{{\bf{E}}}}}}^{*(t)}\);
Estimation of variance parameters
The estimates of the trait residual variance (or uniqueness) ψi (for i = 1, …, Q) are part of the parameters output from the EM algorithm. The true ψi’s are typically underestimated in numerical studies. As a remedy, we propose an alternative estimator adjusting for the degrees of freedom given by
where
and \({\widehat{s}}_{i}\) is the number of nonzero in the ith row of \(\widehat{{{{\mathbf{\Theta }}}}}\) (i.e., all the coefficients associated with trait i). Likewise, estimator of variance σ2 is given by
which is suggested by Reid et al.50 (“Overview”), \(\widehat{s}\) is the number of nonzero in the lasso estimator \(\widehat{{{{\boldsymbol{\beta }}}}}\). Note that the diagonal elements of matrix S are extracted to estimate the uniqueness (or the trait-specific variance parameters). This trick has been used in other statistical problems, such as seemingly unrelated regression, to ensure numerical stability; borrowing cross-trait dependence can help remove noise, which avoids aggregating noise from other components into each individual marginal. All off-diagonal elements of S are not used in either inner or outer-debiasing discussed below.
Inference
Single parameter inference
In the univariate regression analysis Y = Xβ + ϵ with ϵ ~ N(0, σ2), a lasso estimator \(\widehat{{{{\boldsymbol{\beta }}}}}\)51 can be desparsified (termed in Van de Geer et al.21) or debiased (termed in Javanmard and Montanari23) by
where
under some regularity conditions, \({\widehat{\sigma }}^{2}\) is an estimator for σ2 when n < p (see “Estimation of variance parameters”). In particular, \({\widehat{{{{\boldsymbol{\beta }}}}}}_{{{{\rm{db}}}}}={({\hat{\beta }}_{{{\mathrm{db}}},1},\ldots,{\hat{\beta }}_{{{\mathrm{db}}},p})}^{T},\widehat{{{{\boldsymbol{\Phi }}}}}=\widehat{{{{\mathbf{\Omega }}}}}\widehat{{{{\bf{C}}}}}{\widehat{{{{\mathbf{\Omega }}}}}}^{T},\widehat{{{{\bf{C}}}}}=({{{{\bf{X}}}}}^{T}{{{\bf{X}}}})/n\), and \(\widehat{{{{\mathbf{\Omega }}}}}\) is the estimated precision matrix which approximates \(n{({{{{\bf{X}}}}}^{T}{{{\bf{X}}}})}^{-1}\) when n < p.
In the same spirit, we propose to debias the regularized estimator \(\widehat{{{{\mathbf{\Theta }}}}}\) in DrFARM by
where \(\widehat{{{{\bf{B}}}}}\) and \(\widehat{\,{{{\mathrm{E}}}}\,}(\widetilde{{{{\bf{Z}}}}}| \widetilde{{{{\bf{Y}}}}})\) are estimators of B and \(\,{{{\mathrm{E}}}}\,(\widetilde{{{{\bf{Z}}}}}| \widetilde{{{{\bf{Y}}}}})\) obtained from the EM algorithm (see Supplementary Note 3). Correspondingly, similar asymptotic properties can be derived for \({\widehat{{{{\mathbf{\Theta }}}}}}_{{{{\rm{db}}}}}=\{{\hat{\theta }}_{{{\mathrm{db}}},ij}\}\) (see Supplementary Note 2). We refer to this as an outer-debiasing step. The outer-debiasing step is different from the inner-debiasing step, which is used inside the EM algorithm. The outer-debiasing step is used outside of the EM algorithm (once the estimation is completed) for statistical inference. Despite the difference in purpose, the outer and inner debiasing steps share a common debiasing expression. It follows that the p value for testing H0: θij = 0 involving the ith trait and jth predictor pij can be calculated by the above estimator with
where \({\widehat{\psi }}_{i}^{*}\) is an estimator for uniqueness (see “Estimation of variance parameters”) and Φ is the cdf of the standard normal distribution.
Hypothesis test for pleiotropy
Let Θj be the jth column of Θ. Testing for pleiotropy (also known as testing the group-level significant association) is equivalent to testing Θj = 0. Of note, the classical MANOVA test statistics, such as Wilk’s Lambda52, Pillai’s Trace53, Hoteling–Lawley Trace54 and Roy’s Greatest Root55 cannot be used when P > N. To use the asymptotic result in Liu and Xie56, we consider the CCT56 for the joint test of Θj = 0. The CCT takes the form
where ωij are nonnegative weights and \(\mathop{\sum }_{j=1}^{d}{\omega }_{ij}=1\). The test statistic follows a Cauchy distribution under the null with an arbitrary dependence structure between pij’s. Liu and Xie demonstrated that CCT can be used for single-trait discovery in GWAS56. For our purpose, we extend the CCT to multi-trait discovery and adjust for multiple testing using the Benjamini–Hochberg procedure32. More specifically, we obtain individual p value pij using (14) and plug it into the CCT test statistic formula (15). The corresponding p value pj is then given by pj = 2Ψ( − ∣Tj∣), where Ψ is the cdf of the standard Cauchy distribution.
Choice of precision matrix estimation
The precision matrix plays a critical role in the debiasing steps. There is a large body of literature on precision matrix estimation. However, to the best of our knowledge, the influence of different estimation methods on the statistical performance of the debiased estimator21,22,23 has not been studied. Here we compare three precision matrix estimation methods: 1) Graphical Lasso (Glasso) maximizes the penalized log-likelihood26 but with unknown theoretical guarantees21; 2) Nodewise lasso (NL), performs row-wise lasso and proved theoretical guarantees in estimation consistency21 and 3) Quadratic optimization (QO) performs a row-wise convex optimization with theoretical guarantees in estimation consistency23.
In our numerical studies, we exploited the precision matrix estimated from Glasso and NL where tuning parameters were selected by the extended Bayesian information criterion (EBIC) with γ = 0.557,58. For Glasso, we used 10 tuning parameters (default setting) using glassopath() of the R package glasso. In the same spirit, for NL, we fitted P regression models Xi regressed on X−i for all i = 1, …, P (where Xi denotes the ith column of X and X−i denotes the matrix after omitting ith column from X) and used 100 tuning parameters (default setting) using R package glmnet. For QO, we used the R code provided on the first authors’ website: https://web.stanford.edu/montanar/sslasso/code.html with the default setting.
Simulation
In each setting, sample size (N), number of predictors (P), number of traits (Q), number of latent factors (K), and number of signals are all varied. We implement the proposed method and use EBIC (γ = 1) for tuning parameter selection. We use 100 replicates for all the methods compared. Details for the implementation of the methods can be found in Supplementary Note 3.
Simulation I
Suppose X = {xnp}, Z = {znk} and E = {ϵnq}. Their entries xnp, znk and ϵnq are independently generated from N(0, 1) for n = 1, …, N, p = 1, …, P, k = 1, …, K and q = 1, …, Q. To generate the Q × P coefficient matrix Θ = {θqp} between the Q traits and P predictors, we specify a sparse indicator matrix Δ = {δqp}. If δqp = 1, then θqp ~ Unif([−1.5, −1] ∪ [1, 1.5]). Otherwise, θqp = 0. Notice that \(\mathop{\sum }_{q=1}^{Q}\mathop{\sum }_{p=1}^{P}{\delta }_{qp}\) is the number of signals fixed in a given scenario. Given a fixed number of pleiotropic variant m (set to be 15% of the number of predictors), the set of pleiotropic variants is randomly drawn from the indices {1, …, P} without replacement. Let M = {q: θpq = 1, for q = 1, …, Q}, i.e., the set of indices corresponding to the pleiotropic variants. The number of traits associated with each j ∈ M follows Multinomial(\(\frac{1}{m}(1,\ldots,1)\)). To specify the factor loading matrix B, we adopt an approach similar to Zhou et al.13. First, we start with an initial matrix \({{{{\bf{B}}}}}^{*}=\{{b}_{qk}^{*}\}\) where \({b}_{qk}^{*}\) are independently generated from Unif(0, τ) where τ >0 is determined empirically and fulfills the signal-to-signal-to-noise ratio (SSNR) = mean(diag(Cov(XΘT))): mean(diag(Cov(ZBT))): mean(diag(Cov(E))) = 1: 3: 5. This SSNR is used to mimic the missing heritability scenario of GWAS and gives the necessity for modeling the latent factors. We perform an eigendecomposition B*B*T = U*Σ*U*T where the column vectors of U* are orthonormal eigenvectors of B*B*T and Σ* is a diagonal matrix with diagonal entries being the eigenvalues of B*B*T. Then we can let V* = sqrt(Σ) and form B = U*V*. Finally, the data are generated using the equation Y = XΘT + ZBT + E.
Simulation II
For this simulation, all settings are kept the same as Simulation I except xni \(\sim\) Bin(2, pi) independently for all n = 1, …, N and Zk ~ MVNN(0, K) independently for k = 1, …, K, where Z = [Z1, …, ZK]. To mimic common variants in GWAS, pi ~ Unif(0.05, 0.95) independently for all i = 1, …, P. We generated kinship K using the standardized X*X*T (i.e., cov2cor() in R) where \({{{{\bf{X}}}}}^{*}=\{{x}_{ni}^{*}\}\) has its entries \({x}_{ni}^{*} \sim \,{{\mathrm{Ber}}}\,(0.25)\) for n = 1, …, N and i = 1, …, P so that the off-diagonal entries of K has a mean of 0.25 to simulate a third-degree relationship (2 × 0.125) between individuals on average46.
Performance metrics
We used true positive rate (TPR), true negative rate (TNR), FDR, and Matthew’s correlation coefficient (MCC)59
to compare the performance of different approaches in simulations I and II, at both the individual-level and group (SNP) level. In particular, for methods that do not provide p values (i.e., without debiasing or with inner-debiasing only), the number of true positive (TP) is the number of nonzero elements in the selected \(\widehat{{{{\mathbf{\Theta }}}}}\) in the signal set for signal-level result and the number of pleiotropic variants with at least one nonzero association for the group (SNP) level result. The number of true negatives (TN) is the number of zeros in the selected \(\widehat{{{{\mathbf{\Theta }}}}}\) in the non-signal set for signal-level result and the number of the non-pleiotropic variant with no association for the group-level result. Then, the number of false positives (FP) and the number of false negatives (FN) are simply given by the number of positive (nonzero coefficients) minus TP, and the number of negatives (zero coefficients) minus TN, respectively. For methods that provide p values (i.e., outer-debiasing or double debiasing), we applied Benjamini–Hochberg procedure32 to both the signal-level and group-level p values at 5% level. To calculate TP, TN, FP and FN, instead of evaluating whether the coefficients are nonzero, we consider whether the adjusted p values are smaller than 0.05.
Power analysis
We use the setting of simulation II to generate some power curves as part of understanding for the performance of DrFARM. Specifically, for each of the 100 simulated datasets, we use the EBIC (γ = 1) to determine sparsity in \(\widehat{{{{\mathbf{\Theta }}}}}\) while using Glasso to estimate the precision matrix. We applied the Benjamini–Hochberg procedure32 to the signal-level p values and declaim significance at 5% FDR level. We recorded the signal detection status, and the empirical percent of correct signal detection was estimated by using the Generalized Additive Model (GAM)60 smoothing technique. The resulting power curves were plotted in Fig. 5 against the corresponding effect size for varying sample sizes of 1000, 2000, and 5000, respectively. The GAMs were fit using the R package mgcv.
METSIM dataset
We use the same METSIM metabolomics GWAS dataset as in Yin et al.34 (N = 6135) to demonstrate the performance of the proposed methods. In the original study, they performed single-variant association tests using a linear mixed-effects model with EPACTS (v3.2.6) https://github.com/statgen/EPACTS on the normalized residual metabolite values, in which they limited their analysis to the 1391 metabolites successfully measured on ≥500 METSIM participants and to the genetic variants with minor allele count (MAC) ≥5. Here, we focused on a subset of P = 2072 nearly-independent index variants identified from this univariate analysis after Bonferroni correction (p < 7.2 × 10−11)34. We chose the set of index variants because they were the most likely candidates for pleiotropic variants. As shown in Yin et al.34, 27.2% of the index variants were associated with more than 2 metabolites using a single-variant association testing approach. Since multivariate regression requires a complete data matrix for traits, we focused on Q = 1031 targeted metabolites that were either complete or imputable using the K-nearest neighbors approach (with 5 neighbors). Examples of non-imputable metabolites include those that were only present ≤3 out of 4 Metabolon panels (data collected at different times). As in Yin et al.34, we regressed the Metabolon-reported metabolite level on covariates (age at sampling, Metabolon batch, and lipid-lowering medication use status for lipid traits only). To obtain covariate-adjusted metabolites with mean 0 and variance 1, we inverse-normalized the residuals from the regression model34. We based the K-nearest neighbor imputation on the inverse-normalized scale. For further details, such as data preprocessing, please refer to Yin et al.34.
METSIM data analysis
We first searched a 10 × 10 tuning parameter grid and picked the optimal tuning parameters using EBIC (γ = 1) for remMap. Then, remMap estimates with the selected tuning parameters were used as the initial value for DrFARM to find the optimal tuning parameters from a refined 5 × 5 grid. As suggested by the simulation, we used DrFARM with double debiasing with Glasso for discovery. We varied K = 1 to 100 (i.e., 5 × 5 × 100 = 2500 grids were searched in total). For a fixed k ∈ {1, …, 100}, the tuning parameter was selected among the 5 × 5 grid. Since we observed EBIC decreases almost monotonically with k, to avoid overfitting, the residual matrix \(\widetilde{{{{\bf{Y}}}}}-\widetilde{{{{\bf{X}}}}}{\widehat{{{{\mathbf{\Theta }}}}}}_{\,{{\mathrm{db}}}\,}^{T}\) was calculated for each k for the selected tuning parameter. The exploratory graph analysis (EGA)61 uses Glasso26 to obtain the sparse inverse covariance matrix for the outcomes of interest and identifies the number of clusters or communities in a graph using a walktrap algorithm62. The number of dense subgraphs (communities or clusters) is declared as the number of latent factors K. Since metabolites are known to be clustered, we used EGA as opposed to common latent factors determination methods such as parallel analysis63,64 or Kaiser-Guttman’s eigenvalue-greater-than-one rule65 for biological interpretability. We performed EGA for each of the 100 residual matrices, and majority voting of the EGA results yielded K = 16. The signal and SNP (group) level results were subject to p < 7.2 × 10−11 (under the Bonferroni correction for 692 metabolite principal components that explained 95% variability in the 1391 correlated metabolite on top of the 5 × 10−8 genome-wide association cutoff, the same cutoff as the original study) for statistical significance. Unlike simulation, in addition to p < 7.2 × 10−11 at the group-level, we also required the significant SNP to have at least two associated metabolites with p < 7.2 × 10−11 to be considered a potential pleiotropic variant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
We used the same METSIM metabolomics GWAS dataset as in Yin et al.34 for the real data analysis. The METSIM metabolomics dataset (n = 6135) used here is a subset of the full METSIM metabolomics data, which is expected to be deposited in dbGaP by the end of 2025. As part of this deposit, we will include ID lists corresponding to the individuals analyzed in this paper. Until the data are available in dbGaP, access can be provided under a Data Use Agreement by request to Dr. Michael Boehnke (boehnke@umich.edu), with responses to requests for data access typically provided within 2 weeks. The simulated datasets used in this paper can be replicated using the R package provided in https://github.com/lapsumchan/drfarm (see the Methods section for details). All association test summary statistics generated from the real data analysis in this manuscript are included in the Supplementary Data that can be fully accessed by readers. All other data supporting simulation experiments and real data analyses are provided in both the main text and Supplementary Information.
Code availability
GATK v3.5 is available at https://gatk.broadinstitute.org/. KING v2.21 is available at https://www.kingrelatedness.com. Beagle v4.1 is available at https://faculty.washington.edu/browning/beagle/b4_1.html. EPACTS v3.2.6 is available at https://github.com/statgen/EPACTS. HOPS v1.0 is available at https://github.com/rondolab/HOPS. PLEIO v2.0 is available at https://github.com/cuelee/pleio. MTAG v1.0.8 is available at https://github.com/JonJala/mtag. Primo v0.2.1 is available at https://github.com/kjgleason/Primo. The R package for DrFARM is available at https://github.com/lapsumchan/drfarm and archived at Zenodo under https://doi.org/10.5281/zenodo.1525215666.
References
Kitano, H. Perspectives on systems biology. New Gener. Comput. 18, 199–216 (2000).
Kitano, H. Systems biology: toward system-level understanding of biological systems. Found. Syst. Biol. 1–36 (2001).
van Karnebeek, C. D. et al. The role of the clinician in the multi-omics era: are you ready? J. Inherit. Metab. Dis. 41, 571–582 (2018).
Laakso, M. et al. The metabolic syndrome in men study: a resource for studies of metabolic and cardiovascular diseases. J. Lipid Res. 58, 481–493 (2017).
Prasad, R. B. & Groop, L. Genetics of type 2 diabetes-pitfalls and possibilities. Genes 6, 87–123 (2015).
Flannick, J. & Florez, J. C. Type 2 diabetes: genetic data sharing to advance complex disease research. Nat. Rev. Genet. 17, 535–549 (2016).
Urrutia, E. et al. Rare variant testing across methods and thresholds using the multi-kernel sequence kernel association test (mk-skat). Stat. Interface 8, 495 (2015).
Sesia, M., Bates, S., Candès, E., Marchini, J. & Sabatti, C. False discovery rate control in genome-wide association studies with population structure. Proc. Natl. Acad. Sci. 118, e2105841118 (2021).
Yang, J. J., Li, J., Williams, L. & Buu, A. An efficient genome-wide association test for multivariate phenotypes based on the fisher combination function. BMC Bioinformatics 17, 1–11 (2016).
Yang, J. J., Williams, L. K. & Buu, A. Identifying pleiotropic genes in genome-wide association studies for multivariate phenotypes with mixed measurement scales. PLoS ONE 12, e0169893 (2017).
Jordan, D. M., Verbanck, M. & Do, R. Hops: a quantitative score reveals pervasive horizontal pleiotropy in human genetic variation is driven by extreme polygenicity of human traits and diseases. Genome Biol. 20, 1–18 (2019).
Foley, C. N. et al. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat. Commun. 12, 1–18 (2021).
Zhou, Y., Wang, P., Wang, X., Zhu, J. & Song, P. X.-K. Sparse multivariate factor analysis regression models and its applications to integrative genomics analysis. Genet. Epidemiol. 41, 70–80 (2017).
Simon, N., Friedman, J., Hastie, T. & Tibshirani, R. A sparse-group lasso. J. Comput. Graph. Stat. 22, 231–245 (2013).
Peng, J. et al. Regularized multivariate regression for identifying master predictors with application to integrative genomics study of breast cancer. Ann. Appl. Stat. 4, 53 (2010).
Yu, J. et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208 (2006).
Kang, H. M. et al. Efficient control of population structure in model organism association mapping. Genetics 178, 1709–1723 (2008).
Kang, H. M. et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010).
Price, A. L., Zaitlen, N. A., Reich, D. & Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 11, 459–463 (2010).
Zhou, X. & Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821–824 (2012).
Van de Geer, S., Bühlmann, P., Ritov, Y. & Dezeure, R. On asymptotically optimal confidence regions and tests for high-dimensional models. Ann. Stat. 42, 1166–1202 (2014).
Zhang, C.-H. & Zhang, S. S. Confidence intervals for low dimensional parameters in high dimensional linear models. J. R. Stat. Soc.: Ser. B 76, 217–242 (2014).
Javanmard, A. & Montanari, A. Confidence intervals and hypothesis testing for high-dimensional regression. J. Mach. Learn. Res. 15, 2869–2909 (2014).
Wang, F., Zhou, L., Tang, L. & Song, P. X. Method of contraction-expansion (moce) for simultaneous inference in linear models. J. Mach. Learn. Res. 22, 192–1 (2021).
Bühlmann, P. High-dimensional statistics, with applications to genome-wide association studies. EMS Surv. Math. Sci. 4, 45–75 (2017).
Friedman, J., Hastie, T. & Tibshirani, R. Sparse inverse covariance estimation with the graphical lasso. Biostatistics 9, 432–441 (2008).
Lee, C. H., Shi, H., Pasaniuc, B., Eskin, E. & Han, B. Pleio: a method to map and interpret pleiotropic loci with GWAS summary statistics. Am. J. Hum. Genet. 108, 36–48 (2021).
Turley, P. et al. Multi-trait analysis of genome-wide association summary statistics using mtag. Nat. Genet. 50, 229–237 (2018).
Gleason, K. J., Yang, F., Pierce, B. L., He, X. & Chen, L. S. Primo: integration of multiple GWAS and omics qtl summary statistics for elucidation of molecular mechanisms of trait-associated snps and detection of pleiotropy in complex traits. Genome Biol. 21, 236 (2020).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Young, A. I. Solving the missing heritability problem. PLoS Genet. 15, e1008222 (2019).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc.: Ser. B 57, 289–300 (1995).
Saber, M. M. & Shapiro, B. J. Benchmarking bacterial genome-wide association study methods using simulated genomes and phenotypes. Microb. Genomics 6, e000337 (2020).
Yin, X. et al. Genome-wide association studies of metabolites in finnish men identify disease-relevant loci. Nat. Commun. 13, 1–14 (2022).
Finocchiaro, G., Ito, M. & Tanaka, K. Purification and properties of short chain acyl-coa, medium chain acyl-coa, and isovaleryl-coa dehydrogenases from human liver. J. Biol. Chem. 262, 7982–7989 (1987).
Giambartolomei, C. et al. A bayesian framework for multiple trait colocalization from summary association statistics. Bioinformatics 34, 2538–2545 (2018).
Lee, Y., Luca, F., Pique-Regi, R. & Wen, X. Bayesian multi-SNP genetic association analysis: control of FDR and use of summary statistics. BioRxiv https://www.biorxiv.org/content/10.1101/316471v1 (2018).
Mazumder, R. & Hastie, T. Exact covariance thresholding into connected components for large-scale graphical lasso. J. Mach. Learn. Res. 13, 781–794 (2012).
Fan, J. & Lv, J. Sure independence screening for ultrahigh dimensional feature space. J. R. Stat. Soc.: Ser. B 70, 849–911 (2008).
Tang, L., Zhou, L. & Song, P. X.-K. Distributed simultaneous inference in generalized linear models via confidence distribution. J. Multivar. Anal. 176, 104567 (2020).
Novembre, J. et al. Genes mirror geography within Europe. Nature 456, 98–101 (2008).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Peng, J., Wang, P., Zhou, N. & Zhu, J. Partial correlation estimation by joint sparse regression models. J. Am. Stat. Assoc. 104, 735–746 (2009).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1 (2010).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Pirinen, M., Donnelly, P. & Spencer, C. C. Efficient computation with a linear mixed model on large-scale data sets with applications to genetic studies. Ann. Appl. Stat. 7, 369–390 (2013).
Zhou, X. & Stephens, M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods 11, 407–409 (2014).
Bickel, P. J. One-step Huber estimates in the linear model. J. Am. Stat. Assoc. 70, 428–434 (1975).
Reid, S. Tibshirani, R. & Friedman, J. A study of error variance estimation in lasso regression. Stat. Sin. 26, 35–67 (2016).
Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B 58, 267–288 (1996).
Wilks, S. S. Certain generalizations in the analysis of variance. Biometrika 24, 471–494 (1932).
Pillai, K. S. Some new test criteria in multivariate analysis. Ann. Math. Statist. 26, 117–121 (1955).
Hotelling, H. A generalized t test and measure of multivariate dispersion. In Proceedings of the second Berkeley symposium on mathematical statistics and probability, 23–41 (University of California Press, 1951).
Roy, S. N. On a heuristic method of test construction and its use in multivariate analysis. Ann. Math. Stat. 24, 220–238 (1953).
Liu, Y. & Xie, J. Cauchy combination test: a powerful test with analytic p value calculation under arbitrary dependency structures. J. Am. Stat. Assoc. 115, 393–402 (2020).
Foygel, R. & Drton, M. Extended Bayesian information criteria for Gaussian graphical models. Adv. Neural Inform. Process. Syst. 1, 604–612 (2010).
Epskamp, S. & Fried, E. I. A tutorial on regularized partial correlation networks. Psychol. Methods 23, 617 (2018).
Matthews, B. W. Comparison of the predicted and observed secondary structure of t4 phage lysozyme. Biochim. Biophys. Acta Protein Struct. 405, 442–451 (1975).
Hastie, T. J. Generalized additive models. In Statistical models in S, 249–307 (Routledge, 2017).
Golino, H. F. & Epskamp, S. Exploratory graph analysis: a new approach for estimating the number of dimensions in psychological research. PloS ONE 12, e0174035 (2017).
Pons, P. & Latapy, M. Computing communities in large networks using random walks. In International symposium on computer and information sciences, 284–293 (Springer, 2005).
Guttman, L. Some necessary conditions for common-factor analysis. Psychometrika 19, 149–161 (1954).
Kaiser, H. F. The application of electronic computers to factor analysis. Educ. Psychol. Meas. 20, 141–151 (1960).
Horn, J. L. A rationale and test for the number of factors in factor analysis. Psychometrika 30, 179–185 (1965).
DrFARM. Software code drfarm: 0.1.0 (0.1.0) for the drfarm method https://doi.org/10.5281/zenodo.15252156 (2025).
Acknowledgements
We thank the participants in the METSIM study. This work was supported by the National Institutes of Health under awards R01 ES033656 (P.X.S.) and R01 HG010731 (G.L.) as well as by the Academy of Finland Grant no. 321428 (M.L.).
Author information
Authors and Affiliations
Contributions
P.X.S., M.B., M.L., and G.L. supervised experiments and analyses. L.S.C., E.B.F., M.L., and P.X.S. designed the study. M.L. enrolled the study participants. M.L. and X.Y.Y. collected, quality-controlled and/or prepared the metabolomics data for association analysis. L.S.C. and E.B.F. analyzed data. M.L. is the principal investigator of the METSIM study. L.S.C. and P.X.S. wrote the manuscript draft. All authors contributed to the interpretation of results and critically reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
E.B.F. is an employee and stockholder of Pfizer. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Cue Hyunkyu Lee and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chan, L.S., Li, G., Fauman, E.B. et al. DrFARM: identification of pleiotropic genetic variants in genome-wide association studies. Nat Commun 16, 5789 (2025). https://doi.org/10.1038/s41467-025-60439-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60439-4
This article is cited by
-
Quantum computing applications in biology
Discover Computing (2025)
