
Abstract
Complex traits exhibit a highly polygenic architecture that complicates gene mapping and molecular characterization. As a model organism for birds, chickens possess high-quality reference panels, functional annotations, and molecular quantitative trait locus maps. However, the genetic mechanisms underlying growth traits have not been systematically analyzed. Here, we develop a 16-generation advanced intercross line of chickens to enhance informative recombination and identify 154 single-gene quantitative trait loci. We use multiple co-localization methods to establish a network landscape of tissue-specific regulatory mutations and functional gene relationships. We leverage gene-clustering and restoration quantitative trait loci within the omnigenic model framework to elucidate the genetic regulation system of growth traits. Cross-species comparisons show the conserved functions of growth-related genes and divergent features of regulatory mechanisms in mammals and birds.
Similar content being viewed by others

Introduction
The sample size of global genomic studies and the number of traits examined have increased rapidly1,2, significantly improving the effectiveness of genome-wide association studies (GWAS) for identifying genetic variants associated with complex human traits. Genetic variations affect biological phenotypes through various molecular mechanisms. Among these, coding variations, which directly affect protein-coding sequences and functions, are the most understood3; however, such variants are always observable at low frequencies owing to purifying selection. In contrast, complex traits are primarily driven by non-coding variants that presumably affect gene regulation4,5. GWAS signals are significantly enriched in regions with regulatory functions, such as chromatin accessibility regions and DNA methylation regions, as well as gene expression quantitative trait locus (eQTLs)6,7,8. Integrating GWAS, molQTLs, and epigenetic feature annotation improves our understanding of how genetic variants influence complex traits by modulating molecular phenotypes and aids in establishing the association between single-nucleotide polymorphisms (SNPs) and target genes, thereby facilitating the identification of true causal genes9. In humans, the v8 freeze of the Genotype-Tissue Expression GTEx data includes 838 post-mortem individuals and 17,382 RNA-seq data10, and the ENCODE Encyclopedia contains 1,063,878 human cis-regulatory elements (CREs), offering a powerful resource for elucidating the mechanisms underlying complex traits11.
Chickens are an important source of protein for human beings, and analyzing the genetic mechanisms of growth traits in chickens is crucial for accelerating the genetic breeding of chickens and increasing the economic value of the chicken meat industry. Chickens are also important model organisms for avian studies and are utilized in research of osteoporosis, obesity, metabolic disorders, and other diseases, providing important models for human medicine12,13. Research on the global chicken reference panel (GCRP), functional annotation (chicken FAANG), and regulatory mapping (chicken GTEx) is advancing14,15,16,17. Despite the availability of extensive gene expression and epigenomic datasets, only a few variant-gene-phenotype links have been established. These observations are generally interpreted within a framework wherein complex traits are driven by the accumulation of minor effects on tissue-specific genes and regulatory pathways. Additionally, in contrast to humans, who have access to large-scale publicly available GWAS data, only the QTL information is publicly available for chickens18. The identification of functional loci is challenging because of the extensive QTL intervals influenced by linkage disequilibrium (LD). This challenge is partially alleviated through fine mapping using artificially designed populations, such as advanced intercross lines (AILs)19,20. AIL populations, the concept of which was first proposed in 1995, can increase recombination and improve the accuracy of QTL localization19. In recent years, AIL populations have been widely established and applied in several species, and the QTLs of AIL populations have been mapped to narrower chromosomal regions and more QTLs have been identified, which helps identify functional genes and causal mutations21. However, these approaches require substantial investments of both time and financial resources.
In this work, we introduced the AIL population that has been maintained for 15 years. The initial F2 population was established through reciprocal crosses between Huiyang Bearded chicken (HB) and the High-Quality Chicken Line A (HQLA), which have significant phenotypic differences in growth traits (Fig. 1a). Subsequent advanced intercross lines (AIL), spanning from F3 to F16 were derived through random mating (detailed in Supplementary Note 1). We collected 4671 samples across different generations for genome sequencing, analyzed changes in this population structure over 16 generations, and conducted gene fine mapping for 75 phenotypes (Fig. 1b). The high accumulation of recombination events facilitated the identification of numerous QTLs at the single-gene level. Through four types of colocalization analysis, we elucidated the intriguing genetic architecture underlying QTLs associated with various growth traits. Additionally, we emphasize the foundational role of regulatory variants in the mechanisms underlying bird growth and developmental traits, as well as the contribution of functional genes to the identification of genes associated with human growth traits.

a A large intercross pedigree was established from two divergent chicken lines, High-Quality chicken Line A (HQLA) and Huiyang Bearded chicken (HB). The average body weight at 7 weeks of age (BW7) is represented in the figure as mean ± standard error. “Cx” and “Hx” denote the number of male and female founders within each population, respectively. Starting from the F2 generation, the figure illustrates mean and standard error of BW7. The horizontal axis indicates the birth time of the samples corresponding to each generation. The red and blue stripes simulate the recombination. b The overall workflow of the data analysis. Step I: Genetic architecture analysis of F0 to F16 using 8M SNPs. The number represents the sample size of each generation. Step II: GWAS analysis based on the genotypes and phenotypes. We collected a total of 75 traits covering five categories, including growth and development (GD), tissue and carcass phenotype (TC), feed intake and efficiency (FE), blood biochemistry (BB), and feather characteristics (FC). Step III: Integration analysis of GWAS with functional annotation of animal genomes (FAANG) and expression quantitative trait locus (eQTL). The numbers represent the count of annotated entries and the number of eQTLs in each tissue, respectively. TssA, Strongly active promoters/transcripts; TssAHet, Flanking active TSS without ATAC; TxFlank, Transcribed at gene; TxFlnkWk, Weak transcribed at gene; TxFlnkHet, Transcribed region without ATAC; EnhA, Strong active enhancer; EnhAMe, Medium enhancer with ATAC; EnhAWk, Weak active enhancer; EnhAHet, Active enhancer no ATAC, hetero; EnhPois, Poised enhancer; ATAC_Is, ATAC island; TssBiv, Bivalent/poised TSS; Repr, Repressed polycomb; ReprWk, Weak repressed polycomb; Qui, Quiescent. GTEx: Genotype-Tissue Expression. Step IV: Cross-breed analyses were conducted using high- and low-body-weight chickens from different breeds. GCRP: Global Chicken Reference Panel (http://farmrefpanel.com/GCRP/#/). Cross-species analyses were performed using the UK Biobank database (https://www.ukbiobank.ac.uk/) and YangLab resources (https://yanglab.westlake.edu.cn/data/ukb_fastgwa/imp/). Additional analyses were performed using the Phenome-Wide Association Study database (https://atlas.ctglab.nl/PheWAS). A total of 20 human traits and 323 human genes (orthologs of chicken growth-related genes) were included. Chicken and human images were obtained from BioRender (https://www.biorender.com/).
Results
Maintenance of AIL population diversity and rapid accumulation of recombination were assessed through genotyping data
We collected 4671 samples from generations F0, F1, F2, F8, F9, F15 and F16 of the AIL population (Supplementary Table 1). The genotypes were obtained using the low coverage sequencing method22, resulting in 8,050,756 SNPs (see “Methods” for details, Supplementary Table 2; Supplementary Fig. 1a, b). The average sequencing depth of these samples was 0.89 ± 0.30. Accuracy assessment of the genotype imputation was conducted using 108 high-depth sequencing samples (Supplementary Data 1), yielding a genotype consistency of 0.99 ± 0.002.
Although the AIL is a reciprocal cross population (high-quality chicken Line A (HQLA) × Huiyang Bearded chicken (HB)), the breeding process of the parent HQLA, which involves initial crossing of two ancestral sources followed by directed selection, imparted a mixed genetic representation from three ancestral sources (Supplementary Fig. 2). Principal component analysis (PCA) indicated that the F16 individuals clustered centrally between the two founders. This population exhibited no population stratification, thereby minimizing the risk of false positive associations (Fig. 2a). To mitigate the bottleneck effect on the genetic diversity of the population, the sample size of each generation was maintained at 1292 ± 407 (minimum: 655 in F3), while that of half-sib families was kept at 94 ± 10 (minimum: 72 in F2). Over successive generations, the nucleotide diversity remained stable following the initial cross between the founders (Fig. 2b), and the inbreeding coefficient remained constant since the F8 generation (Fig. 2c). Additionally, the minor allele frequency spectrum exhibited a similar distribution pattern across generations (Fig. 2d). A mere 0.11% loss of genetic polymorphisms was observed from F0 to F16. These findings suggest that the genetic drift within this population was minimal during the propagation process, with no evident bottleneck effect. Conversely, LD decay analysis indicated that LD declined rapidly with generational transmission (r20.1 = 143 kb in F16 and r20.1 = 259 kb in F2) (Fig. 2e). No change in the frequency of major haplotypes from the F1 generation and an increase in the number of haplotypes suggest that multiple generations of recombination actively fragmented the genomes of this population’s descendants, which significantly enhanced the gene mapping resolution (Fig. 2f, g). In summary, the population is a quality resource for fine mapping.

a Principal component analysis (PCA). b Nucleotide diversity (300 kb windows). The number of windows in HB, HQLA, F1, F2, F8, F9, F15, and F16 was 3170, 3174, 3181, 3189, 3181, 3185, 3184, and 3185, respectively. c Inbreeding coefficient. The sample sizes for HB, HQLA, F2, F8, F9, F15, and F16 were 15, 16, 640, 186, 1131, 360, and 2269, respectively. Violin plots provide a density estimation of the data distribution. Boxplot center lines represent the medians; box limits indicate the upper and lower quartiles; whiskers extend to 1.5 times the interquartile range, and data points beyond this range are considered outliers. d Minor allele frequency (MAF) distribution for the populations of F0 to F16. e Extent of linkage disequilibrium (LD) in different AIL generations. Values are the mean LD r2 values for all pairs of SNPs binned by distance. f Number of haplotypes. g Frequency of major haplotypes. The number of windows in each generation, shown in (f, g), was consistently 3653. For (b, f, g), boxplot center lines represent the medians; box limits indicate the upper and lower quartiles; whiskers extend to 1.5 times the interquartile range, and data points beyond this range are considered outliers. Source data are provided as a Source Data file.
GWAS revealed hundreds of genes responsible for various chicken traits
We collected a series of growth and slaughter traits in the F2 and F9 generations23. In the F16 generation, we expanded our phenotypic records to encompass 75 traits across five categories, including growth and development (GD, n = 36), tissue and carcass phenotypes (TC, n = 23), feed intake and efficiency (FE, n = 9), blood biochemistry (BB, n = 3), and feather characteristics (FC, n = 4). Many of the phenotypic measurements were strongly correlated with each other, suggesting that they may be influenced by pleiotropic genes (Supplementary Fig. 3; Supplementary Data 2 and 3). Additionally, we calculated the SNP heritability for all phenotypes and found that FC exhibited the highest level of heritability, while FE demonstrated the lowest overall heritability. The heritabilities of GD and TC phenotypes were 0.31 ± 0.16 and 0.30 ± 0.13, respectively. These results provided a basis for subsequent phenotypic association analyses (Fig. 3a; Supplementary Data 4).

a Estimation of the heritability of 75 phenotypes from five categories. For (a, c), GD: Growth and development (n = 36); TC: Tissue and carcass (n = 23); FE: Feed intake and efficiency (n = 9); BB: Blood biochemistry (n = 3); FC: Feather characteristics (n = 4). Boxplot center lines represent the medians; box limits indicate the upper and lower quartiles; whiskers extend to 1.5 times the interquartile range, and data points beyond this range are considered outliers. b Summary porcupine plot for 75 phenotypes. Genome-wide representation of all unique QTLs (n = 682, FDR < 5%) identified in this study. The lead SNP at each QTL is marked by a colored dot, with the color corresponding to the type of measure. The y axis shows the −log10 P values and is truncated at −log10 P = 35. The positions of the two strongest QTLs with −log10 P values of 140 and 101 (chromosome 27) are marked with triangles. All SNPs except 682 lead SNPs are shown filtered according to r2 < 0.4, p < 0.05. c Pearson’s correlation between the heritability and number of QTLs. Error bands represent 95% confidence intervals. P value was obtained using two-sided t-test. d Comparison of QTL length of 21 growth and development-related phenotypes in F2, F9, and F16. The number of QTLs identified in F2, F9, and F16 was 131, 348, and 425, respectively. Statistical significance of differences among groups was evaluated using two-sided t-test implemented via the t.test function from the ggsignif package in R (v4.3.2). e Proportion of different QTL types. Trait refers to a QTL detected only in one phenotype. Domain refers to QTL detected in a category (e.g., GD). Multi-domain refers to a QTL detected in more than one category. f Distribution of genes annotated in QTL regions. g MB phenotype Manhattan plot with top SNP (rs14301625) located on the HOXB8 reported causal mutation CNV3. h Manhattan plot of BPCB phenotype, with top SNP (rs14823350) located 5.5 kb downstream of the reported causative mutation in SOX10. Source data are provided as a Source Data file.
We conducted GWAS on 75 phenotypes among which 43 exhibited significant signals (Fig. 3b; Supplementary Data 2). The heritability of a phenotype and the number of QTLs found for this phenotype were significantly correlated (Pearson’s r = 0.64, p = 4.31 × 10−6, Fig. 3c). Fine mapping of these 43 phenotypes resulted in a total of 682 QTLs (Supplementary Data 5). QTL comparisons across different generations revealed that the QTL lengths significantly decreased with transmission through generations (Fig. 3d), as exemplified by the Gallus gallus chromosome 27 (GGA27) QTL (Supplementary Fig. 4a, b; Supplementary Data 6). Considering the impact of sample size, we standardized the sample size of F16 to match that of F2. A comparison of the F16_640 group with all F16 samples revealed no significant difference in QTL length, indicating that increasing the sample size does not influence QTL length. However, a larger sample size facilitated the detection of more QTLs, suggesting that the primary advantage of increasing sample size lies in enhancing the detection of small-effect QTLs rather than altering QTL length. This implies that recombination continues to occur across generations (Supplementary Fig. 4c). The average length of the QTL interval in F16 was 244 ± 343 kb, with 84.2% of the QTL lengths being less than 500 kb (Supplementary Fig. 4d; Supplementary Data 5). Of the 682 QTLs identified, 20.38% were detected in the current Animal QTL database (https://www.animalgenome.org/cgi-bin/QTLdb/GG/index). These QTLs allow us to more comprehensively study growth traits using AILs (Supplementary Fig. 5). By analyzing the relationships between QTLs for different phenotypes, we grouped physically overlapping trait-associated loci, resulting in 396 loci with a total length of 94.7 Mb, covering 9.86% of the genome. Among these, 60.76% were associated with more than one trait, and 25.78% were classified as multi-domain loci (Fig. 3e; Supplementary Data 7).
A total of 624 QTLs contained at least one gene. Of these QTLs, 470 were annotated to at least two genes, and the average number of genes annotated to these QTLs was 9.7 ± 8.9 (Fig. 3f). This includes numerous examples of previously identified causal genes, such as HOXB8, which influences muffs and beards, as well as SOX10, which impacts feather color24,25 (Fig. 3g, h). Additionally, it encompasses previously reported major QTLs on chromosomes 1, 4, and 27 that are associated with chicken growth traits23,26,27,28. Among these, 154 were single-gene QTLs (Supplementary Data 8); 64 of these involved 52 genes that have been validated in mice, indicating that we could directly locate these functionally validated genes (Supplementary Data 9).
Integration of GWAS with eQTL and FAANG identified the relationship of regulatory variants, target genes, and phenotypes
This population has achieved significant advancements in gene fine mapping. However, among the 77,519 significant SNPs identified within QTLs, only 0.33% were missense mutations, while 55.55% and 17.17% were located in the intronic and intergenic regions, respectively (Supplementary Fig. 6). This finding implies that regulatory variants play a dominant role in growth traits. Subsequently, we used the molQTL data from chickenGTEx15 and various functional annotations obtained from ChickenFAANG14 to elucidate the mechanisms underlying these mutations. Among the five molecular QTL and 15 FAANG annotation regions we analyzed, promoters (TxFlnkHet, TxFlnkWk, TssAHet, TssA, TssBiv, and TxFlnk), enhancers (EnhAHet, EnhAWk, EnhAMe, and EnhA), exQTLs, lncQTLs, and eQTLs could explain more heritability than Qui regions without any function (Fig. 4a; Supplementary Table 3). Enrichment analysis indicated that the GWAS signals were significantly enriched in strong promoters (TssA), strong enhancers (EnhA), and eQTL (Fig. 4b; Supplementary Fig. 7). We utilized four complementary approaches to prioritize causal variants and genes, namely summary-data-based Mendelian randomization (SMR, a method used to investigate causal relationships between gene expression and complex traits using summary-level data), fastENLOC-based colocalization, single-tissue transcriptome-wide association study (SPredixcan), and multi-tissue transcriptome-wide association study (SMultixcan). Of the 611 QTL identified for 40 growth phenotypes, 446 contained significant SNPs that affected the expression of at least one gene; we summarized the results of colocalization of these 446 QTLs and found that 245 QTLs could be interpreted by at least one of the methods (Supplementary Fig. 8). We obtained a total of 431 unique functional genes using four methods (Fig. 4c; Supplementary Data 10).

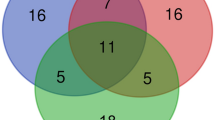
a The heritabilities of molQTLs and functional annotation regions. b GWAS variant enrichment with eQTL in 28 tissues. c Venn diagram of methods for identifying functional genes. d Manhattan plot for serum ALP GWAS conditioned on rs316348444. e eQTL mapping of ST3GAL4 in the small intestine. rs316348444 is a functional SNP derived from SMR. The color indicates the LD of SNPs with the colocalized SNP (rs316348444). f ST3GAL4 promoter and enhancer activity in different genotypes (Wild Type: CC, Mutant Type: TT). Each group consisted of 4 samples. g Differential expression of ST3GAL4 with different genotypes in the small intestine measured by qPCR (CC, n = 9; CT, n = 12, biologically independent samples). Significance of differences among groups in (f, g) were assessed using two-sided t-tests from the ggsignif package in R (v4.3.2). h Loci were classified into 3 categories: no colocalization loci (no GWAS gene loci linked to eGenes in 28 tissues), nearest gene loci (GWAS loci identified through genes closest to the lead SNP) and not-nearest gene loci (GWAS loci identified through genes other than those closest to the lead SNP). eGenes are defined as genes significantly regulated by at least one variant (FDR < 0.05). The number of different loci was identified through SMR. i Shank length at 6 weeks of age (SL6) GWAS Manhattan plot. The color indicates the LD of SNPs with the lead SNP (rs314511712). The red dashed area indicates the QTL range. The gene nearest to rs314511712 is GIP. KCNH6, IGF2BP1, and HOXB7 were functional genes identified via SMR. j Differential expression of KCNH6, IGF2BP1 and HOXB7 across genotypes. Slopes and P values were from tensorqtl. We showed the distance of genes from the lead SNP (rs314511712). For (a, b, f, g, j), boxplot center lines represent the medians; box limits indicate the upper and lower quartiles; whiskers extend to 1.5 times the interquartile range, and data points beyond this range are considered outliers. For (g, j), violin plots provide a density estimation of the data distribution. Source data are provided as a Source Data file.
Alkaline phosphatase (ALP) assay is mainly used to diagnose diseases of the hepatic, biliary, and skeletal systems. ALP in chickens may be related to intestinal function, which in turn affects feed intake29,30. We identified significant signals affecting ALP on chromosome 24 and a conditional control GWAS (rs316348444) further validated this finding (Fig. 4d; Supplementary Fig. 9a). SMR analysis indicated that rs316348444 influenced the ALP phenotype by modulating ST3GAL4 expression in the small intestine (Fig. 4e). A dual-luciferase reporter assay and quantitative polymerase chain reaction (qPCR) confirmed that this mutation enhanced the activities of the promoter and enhancer of ST3GAL4, thereby affecting its expression in the small intestine tissue (Fig. 4f, g; Supplementary Fig. 9b; Supplementary Tables 4 and 5). Additionally, results from human GWAS databases also indicated that ST3GAL4 is associated with serum ALP levels (Supplementary Fig. 9c). These findings confirm that rs316348444 and ST3GAL4 play crucial roles in regulating ALP.
We classified all loci into three categories, namely no colocalization loci (where no GWAS gene loci are linked to eGenes across 28 tissues), nearest gene loci (where GWAS loci are identified through genes closest to the lead SNP) and not-nearest gene loci (where GWAS loci are identified by genes other than those closest to the lead SNP). eGenes are defined as genes significantly regulated by at least one variant (FDR < 0.05). We found that 81.71 ± 3.65% of the identified loci were not-nearest gene loci (SMR = 77.78%, fastENLOC = 86.55%, SPredixcan = 81.82%, and SMultixcan = 80.69%) (Supplementary Fig. 10). For SMR, 108 loci were identified, of which 84 were not-nearest gene loci and 24 were nearest gene loci (Fig. 4h; Supplementary Table 6). For these 84 loci, we identified functional genes located 535 ± 363 kb away from the lead SNP. For example, for shank length at 6 weeks of age (SL6), the lead SNP was rs314511712, and the gene nearest to it was GIP. We did not identify GIP through SMR but identified IGF2BP1 (muscle), HOXB7 (embryo), and KCNH6 (jejunum), which were located 7.5 kb, 119 kb, and 998 kb from the lead SNP, respectively (Fig. 4i, j). The IGF2BP1 gene has been reported in chicken, duck, and goat as a functional gene influencing growth traits such as body size and body weight31,32,33. Similarly, we observed the same phenomenon in shank circumference at 8 weeks of age (SC8) and shank circumference at 12 weeks of age (SC12) (Supplementary Figs. 11 and 12). These results suggest that the eQTL facilitated our identification of the possible functional genes and mutations.
Example of complex QTL analysis: genomic neighbor genes function in different tissues and together contribute to body weight
We identified a significant effect QTL interval at the distal end of chromosome 1 associated with GD and TC phenotypes. The average heritability of this QTL was 7.6 ± 3.67%, contributing to 21.22 ± 7.86% of the total heritability (Fig. 5a). This is a substantial effect QTL previously demonstrated in various populations (Supplementary Table 7). This QTL is unique because it retains ~800 kb after 15 generations of recombination (Fig. 5b). Our previous haplotype association analyses revealed multiple haplotypes with different effects on chicken weight, possibly owing to segregating polymorphisms at multiple, tightly linked mutation loci in the region23. In an omnigenic model, we hypothesized that this genetic process operates through diverse pathways, implying that multiple genes function in different tissues to collectively influence body weight. Utilizing a series of colocalization methods, we identified eight multi-omics relationship pairs, including rs732498434-SETDB2-pituitary, rs735425541-RCBTB1-muscle, rs431853734-ARL11-adipose, rs314814283-KPNA3-retina, rs314099581-TRIM13-spleen, rs315380946-KCNRG-liver, rs316815589-RNASEH2B-muscle, PHF11-muscle (Fig. 5c; Supplementary Figs. 13 and 14). This implies that multiple genes influence body weight in this QTL. They have a neighboring relationship in physical distance, showing a pseudo-main effect GWAS findings.

a Pearson’s correlation between the total heritability and the heritability of GGA1 top QTLs (the most significant QTL on chromosome 1). The total heritability was calculated using the GCTA GRM model, and the heritability of the QTLs was calculated using GCTA bivariate GRM model. Error bands represent 95% confidence intervals. P value was obtained using two-sided t-test. b Body weight at 8 weeks of age (BW8) GWAS Manhattan plot; the red dashed area indicates the QTL range (chr1:170,522,957–171,337,377 bp). The color indicates the LD of SNPs with the lead SNP (rs736348095). c Schematic of the functional modes of different genes in various tissues for the major effect QTL on GGA1. The listed mutations are all within the chr1:170,522,957–171,337,377 bp region. In addition, PHF11 was obtained by TWAS with no SNP level information. Tissue images were obtained from BioRender under an academic license. (https://www.biorender.com/). d q/Q haplotype allele frequency in high- and low-body-weight chickens. High-body-weight breed: HQLA, CBA (commercial Broiler Line A), CBB (commercial Broiler Line B), KB (Cobb), YXB (White Plymouth Rock); Low-body-weight breed: HB, CH (Chahua), DWS (Daweishan), SY (Silkies), ZJ (Tibet); RJF: Red Junglefowl. Source data are provided as a Source Data file.
The proximity of physical distance also facilitated the acceleration of artificial selection in the formation of this QTL. We selected 10 high- and low-weight chickens, as well as red jungle fowl, from the GCRP database17 for outgroup verification. Using the haplotype sharing method34, we identified four Q haplotypes (increased body weight) and one q haplotype (decreased body weight) within this interval (Fig. 5d). Notably, the frequency of the Q haplotype in high-weight chickens was significantly higher than that in low-weight chickens, and vice versa. These Q/q haplotypes were also found in the red jungle fowl. High-weight chickens exhibited an enrichment of standing Q segments to a certain extent, but have not fixed, indicating that there remains potential for further selection in the future.
A discovery caused by the drift of local genetic architecture: NCAPG influences the eviscerated weight
By comparing QTLs across different generations, we identified a QTL on chromosome 4 that affected the eviscerated weight (EW), which was generated in F16 and was not detected in F2 or F9 (Fig. 6a–c; Supplementary Fig. 15). We discovered that the minimum allele frequency of significant SNPs in F16 was notably higher than those in F2 and F9 (Fig. 6d), thereby enhancing the statistical efficiency of GWAS. The sampling GWAS of F16 remained significant (Supplementary Fig. 15), allowing us to eliminate the impact of sample size on the results across different generations. We attribute this intriguing finding to ongoing recombination, suggesting that this population still holds considerable potential for the identification of extra QTLs.

a, b Manhattan plot for eviscerated weight GWAS in F2 and F9. c Manhattan plot for eviscerated weight GWAS in F16. The color indicates the LD of SNPs with the lead SNP in F16 (4_76152106_C_G). d Comparison of the frequency of significant SNPs within the QTLs of F16 EW across generations (For each generation, nsnp = 427). e EW phenotypes of rs14491022 under different genotypes (CC: n = 5, CT: n = 122, TT: n = 994, biologically independent samples). f Functional annotation of features near rs14491022. g Enhancer activity of NCAPG in different genotypes (Wild Type: CC, Mutant Type: TT). Each group consisted of 4 samples (n = 4). h Differential gene expression of NCAPG with different genotypes in duodenum (CC: n = 10, CT: n = 8, biologically independent samples.). Significance of differences among groups in (d, e, g, h) were assessed using two-sided t-tests implemented via the t.test function from the ggsignif package in R (v4.3.2). For (d, e, g, h), boxplot center lines represent the medians; box limits indicate the upper and lower quartiles; whiskers extend to 1.5 times the interquartile range, and data points beyond this range are considered outliers. For (e, h), violin plots provide a density estimation of the data distribution. Source data are provided as a Source Data file.
We confirmed that this was a highly significant QTL in chickens and various species (humans, cattle and sheep), as evidenced by the substantial number of previous studies (Supplementary Table 8). We queried promoter-enhancer annotations and candidate target gene profiles for all significant SNPs within this QTL and found that rs14491022 (4:75,892,495, p = 3.64 × 10−07) was in the NCAPG enhancer region in the jejunum and duodenum, while rs1058217803 (4:76,102,806, p = 5.22 × 10−07) was in the LDB2 promoter region in the hypothalamus (Fig. 6f; Supplementary Fig. 16a, b). A dual-luciferase reporter assay indicated that rs14491022 enhanced the enhancer activity of NCAPG, whereas rs1058217803 enhanced the promoter activity of LDB2. We further collected duodenum and hypothalamus tissue samples of different genotypes for RNA-seq. The results demonstrated that NCAPG expression was elevated in the duodenum for rs14491022, while no change was observed in LDB2 expression in the hypothalamus for rs1058217803 (Fig. 6g, h; Supplementary Fig. 16c, d). Additionally, rs14491022 is also associated with other growth traits, such as LTFW, SC12, LW, and BW10 (Supplementary Table 9). We inferred a genetic model that rs14491022 affected NCAPG enhancer activity and subsequently influenced NCAPG gene expression in the gut, which in turn impacts the EW phenotype in chickens.
Evolutionary patterns of growth trait genetic mechanisms in humans (mammals) and chickens (birds)
To investigate the correlation between growth-related genes identified in chickens and humans, we transformed the GWAS significant genes (uniquely, n = 329) associated with five chicken growth traits into their human orthologous counterparts (Supplementary Data 11). On average, 88.75% of these genes were classified as chicken-human orthologous genes, a proportion that was significantly higher than the overall proportion (50.48%) of orthologous genes in the chicken genome. We also collected GWAS summary statistics for 20 human complex traits from the UK Biobank and Yanglab (Supplementary Data 12 and 13). Linkage disequilibrium score regression (LDSC) analysis showed that the functional genes influencing chicken growth phenotypes also affected the corresponding human traits (Fig. 7a). We further examined the human homologs of chicken growth-related functional genes in the Phenome-Wide Association Study (PheWAS) database. Among the 323 genes analyzed (orthologs of chicken growth-related genes), 80.50% influenced heel bone mineral density, body mass index, sitting height, waist hip ratio, and so on. Using weight as a focal point, notably, significant peaks were observed on chromosomes 13, 4, and 17, which corresponded to chicken chromosomes 1, 4, and 27, respectively (Fig. 7b). The analysis of a series of significant genes revealed a broad spectrum of one-cause pleiotropy (Fig. 7c, Supplementary Fig. 17). Meanwhile, we converted 431 functional genes obtained from chickens to their homologous genes in pigs and further queried the PigGTEx database35 to identify 289 genes affected pigs Production_Traits Meat_and_Carcass_Traits and other growth- and development-related phenotypes (Supplementary Fig. 18). These findings indicate that the functional genes influencing growth traits in humans, pigs, and chickens are highly conserved, suggesting that the growth-related functional genes identified in each species may serve as valuable references for other species.

a Heatmap showing enrichments of five chicken growth traits (indicated at the bottom) associated with selected human traits and diseases (indicated at the left). “*” means 0.01 <p < 0.05, “**” means p < 0.01. b Homologs of functional genes of the chicken body weight (BW) phenotype influence the complex phenotype of humans (from the PheWAS database). c Examples of homologous genes of KCNRG related to chicken body weight phenotypes influencing important growth phenotypes in humans (the human PheWAS was performed using the GWAS atlas, https://atlas.ctglab.nl). d SNP annotation for SNPs converted to human homologs based on the SNPs obtained from SMR affecting chicken growth traits (A total of 173 unique SNPs were obtained, of which 32 were successfully converted to that of humans using the UCSC liftover tool.) e We analyzed genes affecting five phenotypes in chickens and human homologous genes’ intron, exon, and intergenic region lengths. Orange indicates the exon, blue indicates the intron, and gray denotes the intergenic region.
In contrast, we focused on the significant SNPs associated with all growth traits according to the SMR results. Following the conversion to the human genome, results were unattainable for 81.50% of the SNPs. Among the remaining 18.50% of the SNPs, the gene annotation results exhibited considerable variations (Fig. 7d). Notably, growth-related genes in chickens possessed shorter introns and intergenic regions than their human counterparts (Fig. 7e). This result indicates that the regulatory mechanisms governing growth-related gene functions have undergone substantial changes throughout the evolutionary transition from birds to mammals.
Discussion
An AIL is an artificially designed population conducive to fine mapping in many species19,20,36. Overall, similar to the previous QTL interval shortening in the mouse AIL population21, the length of the QTL interval in the F16 generation was ~85% shorter than that in F2, resulting in the identification of 64 single-gene level QTLs, including 52 functional genes previously validated through gene-edited mice. Interestingly, a QTL on chromosome 4 was identified in the F16 generation of this population. This finding is attributed to fluctuations in allele frequency resulting from chromosomal recombination, indicating that this population may also harbor other false negative signals caused by excessive bias in allele frequencies. Our population exhibited higher single-nucleotide and haplotype diversities than the other two chicken AIL populations37,38, which can be attributed to our practice of utilizing more than 72 half-sib families in each generation. However, while we assert that this group possesses genetic diversity from three ancestral sources due to its history, there is a diversity bottleneck in the AIL, resulting from the use of only two parental lines. The multi-parent advanced generation intercross (MAGIC) remarkably contributes to gene mapping39,40; however, the establishment of MAGIC populations in animals remains challenging and infrequent. We also attempted to conduct a meta-GWAS; however, the available data sources did not facilitate its completion. Consequently, the AIL used in this study represents the most effective genetic resource for investigating the growth and development of chickens and potentially other avian species.
Numerous studies have demonstrated that human body weight exhibits a typical polygenic genetic architecture characterized by small effects, with no single large effect QTL across genome41. In contrast, genetic studies of poultry have revealed alterations in the genomic landscape resulting from strong artificial selection. Notably, we identified a significant and unique major QTL associated with growth traits at the distal end of chromosome 1. In a previous F9 report, we concluded that this QTL emerged from a locally highly polygenic genetic architecture rather than being associated with a single major effect locus23,42. Our findings from the F16 generation and the Virginia chicken lines studies further substantiate this notion34. These weight-promoting (Q) or weight-reducing (q) haplotypes were derived from standing variants of the red jungle fowl (Fig. 5d). Under normal conditions, each haplotype harboring weight-related mutations maintains a balance that allows adaptation to natural selection. Following intense artificial selection, genetic selectable variations are released through recombination, resulting in the emergence of popular haplotypes. These functional genes are located in close proximity, increasing the possibility of hitchhiking and facilitating rapid accumulation in high-weight chicken breeds. The compact genome of chickens is particularly conducive to artificial selection, contributing to their status as one of the most widespread species in the world. By integrating chicken FAANG and GTEx data14,15, we discovered that regulatory variants and associated genes were active in distinct target tissues, suggesting that these neighboring genes may not be interconnected within biological pathways. This finding underscores the significant advantages of using eQTL data for linear genomic research. We established multiple relationships among regulatory mutations, genes, target tissues, and phenotypes, providing robust statistical evidence for further functional studies of these genes.
The divergence of birds and mammals can be traced back ~300 million years; however, they exhibit distinctly different evolutionary strategies in their coding and regulatory regions43. Furthermore, chromosome length plays a crucial role in determining overall chromosome architecture in vertebrates. For instance, chickens possess relatively short chromosomes, and their long-range contact frequency is 1.408 times higher than that of humans and mice. This finding suggests that long-range interactions may occur more frequently in chickens. Future studies should explore the impact of long-range contacts on gene regulation and their influence on complex traits via methods such as Hi-C and tensorQTL44. Our results demonstrated the anticipated conservation pattern of orthologous protein-coding genes between chickens and humans45, with this trend being particularly pronounced for functional genes that influence growth-related traits. This suggests that the functional genes identified in chickens contribute to a comprehensive list of those in avian and serve as a reference for research on genes related to human growth and development. This perspective was further supported by the LDSC results (Fig. 7a). Consequently, chickens can be regarded as alternative model animals for studying human growth and developmental phenotypes, particularly given the relatively convenient access to their samples. Conversely, gene regulation may play a significant role in species evolution46. The chicken regulatory region sequences correspond only minimally to those of mammals. Although the number of chicken genes is considered at least comparable to that of humans47, shorter non-coding regions result in a denser appearance of chicken genes. Only 30–40% of the regulatory elements, including functional promoters, predicted promoters, CpG islands, and predicted transcription factor binding sites, are conserved between humans and chickens45. However, over 99% of the avian-specific highly conserved elements are located in non-coding regions and have been under strong selective constraints48. This indicates that the obtained eQTL results represent a paradigm of gene regulation in birds that differs from that in the mammalian model and occupies a crucial position in evolutionary studies. Our findings provide valuable resources for research on the regulatory mechanisms of growth traits across nearly 10,000 bird species.
In summary, we developed a robust AIL population characterized by rich polymorphism and high recombination, which facilitates the fine mapping of genes and the exploration of the complex genetic architecture of QTLs. We identified numerous functional genes associated with growth traits and highlighted their conservation across avian and mammalian species. Multi-omics data are crucial for elucidating the relationship between functional mutations and their corresponding genes as well as for demonstrating the tissue-specific pathways associated with each gene. This resource serves as an excellent systematic foundation for studying chickens and other avian species and provides valuable insights for research on human growth traits.
Methods
Ethics approval
All animals involved in this study were housed and treated in accordance with the guidelines approved by the Animal Welfare Committee of Agrobiotechnology of China Agricultural University (Approval No. SKLAB-2014-06-07). Specifically, all animals were individually housed under controlled conditions (22 ± 2 °C, 50–60% humidity) with exposure to natural daylight and free access to feed and water. During the rearing period, qualified veterinary personnel conducted daily inspections and regularly monitored the chickens’ health and welfare, including assessments of physical appearance, behavior, feed and water intake. Any individuals exhibiting signs of abnormal health were promptly isolated and treated. At the end of the experiment, all chickens were euthanized by cervical dislocation performed by trained personnel. Death was confirmed by the cessation of respiration and heartbeat, as well as the absence of the corneal reflex.
Experimental population and phenotyping
A large distant intercross pedigree was initiated in 2008 from two divergent chicken lines, high-quality chicken Line A (HQLA), a broiler line bred by Guangdong Wiz Agricultural Science and Technology, Co. (Guangzhou, China), and Huiyang Bearded chicken (HB), a native Chinese breed. The body weight of HB chickens at 7 weeks of age was, on average, less than one-third of those of HQLA (p < 2.2 × 10−16). Detailed feeding regimes and F0 to F16 mating schemes have been described in Fig. 1 and earlier by Sheng and Wang et al.23,49.
The data of 75 traits were collected or processed in the F16 generation, including growth and development (GD, n = 36), tissue and carcass (TC, n = 23), feed intake and efficiency (FE, n = 9), blood biochemical (BB, n = 3) and feather characteristics (FC, n = 4) (Supplementary Data 2; Supplementary Note 2).
Similarly, we selected 21 phenotypes shared by F2, F9, and F16, and conducted a comparative analysis with F16 (Supplementary Data 14; Supplementary Note 3).
DNA extraction and Tn5 library construction
Blood samples were collected from the chickens at 5 weeks of age using the Laboratory-made FTA card. DNA was extracted from the blood samples using magnetic bead technology.
Equal amounts of Tn5ME-A/Tn5MErev and Tn5ME-B/Tn5MErev were incubated at 72 °C for 2 min and then placed on ice immediately for 1 min. Tn5 (Karolinska Institutet 171 77 Stockholm, Sweden) was loaded with the Tn5ME-A+rev and Tn5ME-B+rev in 2× Tn5 dialysis buffer at 25 °C for more than 2 h. All linker oligonucleotides were the same as those described in the previous report50.
Tagmentation was conducted at 55 °C for 10 min by mixing 4 μL 5×TAPS-MgCl2, 2 μL dimethylformamide (DMF) (Sigma Aldrich), 1 μL of the Tn5 pre-diluted to 16.5 ng/μL, 10 ng DNA (may be adjusted based on enzyme activity), and nuclease-free water. The total volume of the reaction was 20 μL. Subsequently, 3.5 μL 0.2% SDS was added, and Tn5 was inactivated for another 10 min at 55 °C.
KAPA HiFi HotStart ReadyMix (Roche) was used for PCR amplification by mixing 1 U HotStart DNA Polymerase, 10 μL 5× KAPA HiFi Fidelity Buffer, 2 μL dNTP Mix (10 mM each), 4 + 4 μL i5/i7 primers (5 μM) and 5.5 μL nuclease-free water. The PCR mix was added to the digestion products. The primers were designed for MGI sequencers, with the reverse primers containing different index adapters to distinguish individual libraries. We employed two index systems, one with 96 tags (P_1-96) and the other with 192 tags (C_1-192) to support future expansion. The PCR program was as follows: 9 min at 72 °C, 30 s at 98 °C, and then 9 cycles of 30 s at 98 °C, 30 s at 63 °C, followed by 3 min at 72 °C. The products were quantified using Qubit Fluorometric Quantitation (Invitrogen). The groups of 96 or 192 indexed samples were then pooled with equal amounts.
PCR products were size-selected and purified using the VAHTS DNA Clean Beads (Vazyme). The 0.8× and 1.3× sample volumes of DNA Clean Beads were used to remove most of the short and long fragments, respectively. The fragment sizes obtained via this method were ~300–900 bp, and the fragment size with the highest proportion was 500 bp. Final library quality (concentration and fragment size distribution) was determined using Qubit 4.0 (Qubit® dsDNA BR Assay Kit) and Qsep100, respectively. Sequencing was performed within 48 h after DNA cyclization and rolling circle amplification. All libraries were sequenced in one or two lanes of the MGISEQ-2000 to generate 2 × 100 bp paired-end reads (Supplementary Fig. 19a).
Sequencing and quality control
This study involved two types of sequencing libraries, namely typical whole-genome sequencing libraries (WGS, high coverage for F0 and 75 F16 samples, http://farmrefpanel.com/GCRP/#/, Supplementary Data 1) and Tn5-based low coverage sequencing libraries (LCS). For LCS, 4772 qualified libraries were sequenced using MGISEQ-2000 to generate 2 × 100 bp paired-end reads. The sequencing experiments were performed on the MGISEQ-2000 system at the National Research Facility for Phenotypic and Genotypic Analysis of Model Animals (Beijing) and State Key Laboratory of Animal Biotech Breeding. BCL files as primary sequencing output were converted into FASTQ files using bcl2fastq2 conversion software (version 2.16.0). The sequencing adapter was masked and trimmed during the conversion process. After the trimming step, the PE reads were subjected to a filtering process: SLIDINGWINDOW:4:15 and MINLEN:75 by Trimmomatic-0.36. The average percentage of the clean reads was 95%. The quality control check report of the filtered reads was generated using FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Next, we used a custom script to split each sample index and obtained 4772 samples. Of these, samples with a sequencing depth less than 0.1× of the chicken genome (1.06 Gb) and duplicate samples (IBS > 0.9) were removed. The remaining 4671 samples were retained for subsequent analysis.
Mapping and variant and variant calling
WGS data SNP calling
We analyzed the high-depth sequencing data of 31 F0 founders, F16 samples, and other local chickens and commercial chickens using GTX (Supplementary Data 15). A commercially available FPGA-based hardware accelerator platform was used to map reads to the GRCg6a reference genome (Ensembl version 104) and variant calling. The alignment process was accelerated through the FPGA implementation of a parallel seed-and-extend approach based on the Smith-Waterman algorithm, whereas the variant calling process was accelerated via the FPGA implementation of GATK HaplotypeCaller (PairHMM)51. GATK multi-sample best practice52 was used to call and genotype SNPs for these samples, and the SNPs were hard filtered with a relatively strict option “QD < 2.0||MQ < 40.0||FS > 60.0||SOR > 3.0||MQRankSum < −12.5||ReadPosRankSum < −8.0||QUAL < 30.”
LCS data SNP calling
For LCS data, the clean reads were mapped to the GRCg6a reference genome (Ensembl version 104) using BWA53. This was followed by read group addition, read pair sorting, marking duplicate reads, and building bam index by the Picard tools (http://broadinstitute.github.io/picard, v2.20.4). The indel realignment and base quality recalibration modules in GATK51 were applied to realign the reads around indel candidate loci and to recalibrate the base quality. The above steps are implemented in GTX-align, which is an FPGA-based hardware accelerator system (1–2 min/sample from clean FASTQ to BAM) on the high-performance computing platform of the State Key Laboratory of Agrobiotechnology. Variant calling was performed using the BaseVar and hard filtered with EAF ≥ 0.01 and the depth ≥ 1.5 times the interquartile range. The detailed BaseVar algorithm used to call SNP variants and to estimate allele frequency has been previously described54. We used STITCH55 (v1.1.2) to impute genotype probabilities for all individuals. The key parameter K (number of ancestral haplotypes) was set to 15. Raw SNPs were filtered with an imputation info score > 0.4, call rate > 0.95, and minor allele frequency (MAF) > 0.01. After filtering, 8,050,756 SNPs remained (Supplementary Fig. 19b). We utilized the SNPs from 108 samples, each of which simultaneously had both WGS and LCS data. We converted them to the 0/1/2 format and calculated the concordance between WGS and LCS data, defined as genotypic concordance; a higher concordance indicated a higher imputation accuracy of our LCS data. The SNPEff program56 was used to annotate variants.
RNA-seq library construction and sequencing data analysis
The total RNA of the duodenum and hypothalamus were extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s recommendations. To eliminate DNA contamination, all samples were treated with RNase-free DNase. The concentration and purity of the RNA samples were determined with a Nano Photometer spectrophotometer (Implen, CA, USA), and the RNA integrity number (RIN) of each RNA sample was determined using Agilent 4150 RNA (Agilent Technologies, CA, USA). cDNA synthesis was performed using a PrimerScript™ RT Reagent Kit (Takara, Dalian, China) according to the manufacturer’s instructions. Briefly, 1 μg of total RNA was mixed with DNA Eraser, and the mixture was incubated at 42 °C for 2 min on an EasyCycler® 96-well thermocycler (Analytik, Jena, Germany). Subsequently, oligo-d(T) primer, random hexamers, and dNTPs were added, and the mixture was incubated in a thermocycler at 37 °C for 15 min and 85 °C for 5 s.
The concentration and integrity of the were measured and checked for concentration and integrity using Nano Photometer spectrophotometer (IMPLEN, CA, USA) and an Agilent Bioanalyzer 4150 system (Agilent Technologies, CA, USA). The library was constructed using Hieff NGS Ultima Dual-mode mRNA Library Prep Kit for MGI (Yeasen Biotechnology, Shanghai, China) according to the manufacturer’s protocol. After library construction, the library was converted using the MGIEasy Universal DNA Library Preparation Reagent Kit (BGI, Shenzhen, China) for compatibility and sequenced on the DNBSE-QT7 platform (MGI). For quality control, we used fastp57 default parameters for filtering (v0.22.0). We aligned the clean reads to the GRCg6a reference genome (Ensembl version 104) using hisat258 (v2.2.1) with parameters of “--dta --new-summary -t -p 48 -x $index -1 $fq1 -2 $fq2.” Featurecounts.R of Rsubread (v2.12.0) was used for quantification, and edgeR (v3.40.0) was used for differential analysis.
Population structure analysis
ADMIXTURE59 (v1.3.0) was used for estimating an individual’s ancestry proportions from F0 and F15 genotype data (the F15 individuals were the paternal progenitors of all F16 samples). PCA was performed using Plink60 (v1.90) on the parental HB (n = 15), HQLA (n = 16) and 100 random samples from each of F2, F9, and F16. Vcftools61 (v0.1.17) was used to calculate allele frequency with the parameter “--freq2,” and nucleotide diversity analysis with the parameter “--window-pi 300000.” We used Plink60 (v1.90) with the parameters “--het” to calculate the inbreeding coefficient. We used PopLDdecay62 (v3.41) with parameters of “-MAF 0.01 -MaxDis” to perform LD decay analysis on different generations and use Plot_MultiPop.pl “-bin1 50 -bin2 1500 -break 500 -maxX 2000 -keepR” to draw. For haplotype diversity analysis, we used Plink60 (v1.90) with parameters of “--indep-pairwise 1000 kb 1 0.4” to filter LD on 4671 samples, resulting in 18,325 SNPs. We used five SNPs as windows to perform windowing and calculated each window haplotype type and the main effect haplotype frequency (the sum of the top four main effect haplotype frequencies).
GWAS and fine mapping
The heritability of the 75 phenotypes was calculated using a mixed linear model of GCTA63 (v1.93.3) with parameters “gcta64 --grm test --pheno pheno.txt --reml --out test”, corrected for batch and sex as covariates. We used GCTA63 (v1.93.3) with parameter “--reml-bivar” to calculate the genetic correlation between two phenotypes.
We simultaneously conducted GWAS on 75, 18, and 19 phenotypes of F16, F2, and F9, respectively (Supplementary Data 2, 6 and 14). First, we used GCTA63 (v1.93.3) with parameters “--make-grm” and “--grm-sparse” to construct the kinship and sparse matrices. Subsequently, we used a mixed linear model of fastGWA64 to perform GWAS.
where y is an n × 1 vector of phenotypes; xsnp is a vector of mean-centered genotype values for the variant of interest, with its effect size βsnp; Xc is the incidence matrix of fixed covariates (sex and batch) with their corresponding coefficients βc; g is a vector of the total genetic effects captured by pedigree relatedness; e is a vector of residuals.
For the 75 and 19 phenotypes of the F16 and F9 generations, respectively, we first used the false discovery rate (FDR) for correction and selected SNPs with FDR < 0.05 to determine the initial QTL. SNPs with a distance greater than 500 kb were considered to belong to 2 different QTL. For the 18 phenotypes of F2, owing to the high LD between SNPs, SimpleM.R was first used to determine the number of independent SNPs; further, the initial QTL was determined based on Bonferroni 0.05 (p < 1.26e-7). SNPs with a distance greater than 500 kb were considered to belong to two different QTLs. Next, fine mapping was performed based on the LD r2 < 0.6 of the SNPs in each initial QTL and the top SNP of the QTL.
The heritability of the QTL was calculated using the GCTA63 bivariate GRM model with parameters “gcta64 --reml-bivar --mgrm multi_grm.txt --pheno test.phen”. We assessed the sharing of our detected QTLs with reported QTLs using the QTLdb database (https://www.animalgenome.org/cgi-bin/QTLdb/GG/index, Retrieved 28 December 2023), and if the QTLs of the two collections had overlapped, we considered the two to be shared.
We used Bedtools65 with “bedtools intersect” to annotate all QTLs for the 43 F16 phenotypes. For the QTL where a single gene was identified, we queried its gene function in mice according to the https://genealacart.genecards.org/ database and determined its relationship with growth and their development-related genes and corresponding QTLs.
Integrating eQTL and FAANG with GWAS
We integrated the eQTL data of 28 tissues from the chicken GTEx project and the FAANG annotation of 23 tissues from the chicken FAANG project with 40 F16 growth- and development-related phenotypes with significant GWAS signals to further identify functional genes (Supplementary Table 3). We utilized the GCTA63 bivariate GRM model for the calculation of heritability for each functionally annotated region and molQTL:
where y is the phenotype, Xβ is the fixed effect (sex, batch); g1 is the genetic effect of functional annotation regions or molQTL; g2 is the genetic effect of all SNPs upstream and downstream of the functional region beyond 100 kb; e is a vector of residuals.
For the heritability obtained by “gcta64 --reml-bivar --mgrm multi_grm.txt --pheno test.phen --out test”, considering the different number of SNPs in each interval, we corrected for the number of SNPs and performed a log transformation.
For the growth and development-related phenotypes of the F16 generation that exhibited significant GWAS signals, we utilized all SNPs as a background for calculating enrichment and used a hypergeometric distribution test to calculate P values.
Enrichment factor = (target region significant SNPs/total significant SNPs)/(target region all SNPs/genome-wide total SNPs).
To assess whether eQTLs were significantly enriched among the notable GWAS variants, we used QTLEnrich (v2)10 to measure the enrichment degree between significant eQTLs and GWAS loci.
We performed single- and multi-tissue transcriptome-wide association studies (TWAS) using SPrediXcan66 and SMultiXcan67, which are parts of the MetaXcan (v0.6.11) framework. Briefly, we trained Nested Cross-Validated Elastic Net models with protein-coding genes and their corresponding SNPs within a 1 Mb cis-window across all 28 tissues. Predictive models with cross-validated correlation ρ > 0.1 and prediction performance p < 0.05 were selected for further analysis. Using the SPrediXcan tool and the trained models, we predicted gene-trait associations at the single-tissue level, resulting in single-tissue TWAS outcomes. Subsequently, we employed the SMultiXcan tool to integrate the single-tissue predictions and obtained multiple-tissue TWAS results. After applying multiple testing corrections using the FDR approach, gene-trait associations with corrected p < 0.05 were deemed significant.
To investigate the pleiotropic association between molecular phenotypes and complex traits, we performed a Mendelian Randomization analysis using the SMR software68 (v1.3.1). This software enables the utilization of summary-level data from GWAS and eQTL studies. To configure the SMR software appropriately, the molQTL data generated by tensorQTL69 in this study was first converted into the BESD format using the options “--fastqtl-nominal-format --make-besd.” We then conducted the SMR test and applied multiple testing correction using the FDR approach. Gene-trait pairs with corrected p value < 0.05 were selected and considered significant.
To identify shared genetic variants between GWAS and eQTL, we conducted a colocalization analysis using fastENLOC70 (v2.0). First, we fine-mapped the putative causal variants for each eGene using a Bayesian multi-SNP genetic association analysis algorithm known as the deterministic approximation of posteriors (DAP), with the current version being DAP-G71 (v1.0.0). Using the DAP-G results, we generated a probabilistic annotation of molQTL using the “summarize_dap2enloc.pl” script. Next, we calculated approximate LD blocks with PLINK60 v1.9, using the options: “--blocks no-pheno-req --blocks-max-kb 1000 --make-founders”. The posterior inclusion probability (PIP) for GWAS loci was determined for each LD block using TORUS, with the options: “--load_zval -dump_pip”. By integrating GWAS PIP values, we performed the final colocalization analysis using the fastENLOC70 tool and obtained the gene variant-level colocalization probability (GRCP). GRCP > 0.1 was set as the significance threshold.
Identification of the major effect Q of GGA1 using outgroup data analysis
For the QTL of BW8 (GGA1:170,522,957–171,337,377), we sliding windowed the QTL according to 20 kb and obtained a total of 34 windows, screened the loci with p < 1 × 10−16 in each window, calculated the frequency of each haplotype, and performed haplotype association analysis.
A haplotype-based association analysis was performed in each window using the model:
where Y is a column vector representing the BW8 of the F16 individuals, and X is the design matrix that includes coding for chickens’ sex. For each specific interval, n haplotypes were constructed from m individuals based on several SNPs. Z is the design matrix (m × n) that contains the haplotype counts for each individual, coded as 0, 1, or 2. β is a vector estimating the fixed effect of sex, u is a column vector estimating the allele substitution effects for each haplotype, and e represents the normally distributed residual.
Similarly, we used low-body-weight chickens (HB, CH (Chahua), DWS (Daweishan), SY (Silkies), ZJ (Tibet)) and high-body-weight chickens (HQLA, CBA (commercial Broiler Line A), CBB (commercial Broiler Line B), KB (Cobb), YXB (White Plymouth Rock) and Red Junglefowl (RJF)) from GCRP (http://farmrefpanel.com/GCRP/#/, Supplementary Data 15) to calculate the frequencies of the corresponding haplotypes. Combined with the haplotype association analysis effect values, we determined q/Q; the frequency of high-body-weight chickens minus the low-body-weight chickens of the haplotype was greater than 0.2, and the haplotype was Q for increasing body weight and vice versa for the q haplotype. Combined with the annotations of the candidate functional genes in each window, a total of one q and four Q were identified.
Cross-species analysis
To evaluate the enrichment of genes linked to human traits and diseases in relation to five chicken growth traits (the functional genes identified exceeded 60), we collected GWAS summary statistics for 20 human complex traits from the UK Biobank and Yanglab (Supplementary Data 12 and 13). We mapped the functional genes in chickens to their corresponding human orthologous genes, including 1-1 orthologous, complex orthologous genes, based on the Ensembl database (v102). Subsequently, we conducted LD score regression analysis72 (https://github.com/bulik/ldsc). Heritability enrichment was determined by calculating the proportion of trait heritability attributed to SNPs within the specific annotation relative to the total number of SNPs in that annotation. Additionally, we investigated the impact of homologous human genes, identified from five chicken growth traits, on significant human phenotypes using the PheWAS database (https://atlas.ctglab.nl). Chicken and human SNP conversions were achieved using the UCSC liftover tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver), and SNP annotations were obtained using SNPEff56.
Dual-luciferase reporter assay
For the candidate SNPs identified through GGA4 and GGA24 (Supplementary Table 4), we constructed a promoter vector (pGL3-basic luciferase reporter vector) and an enhancer vector (pGL3-enhancer luciferase reporter vector) to verify the transcriptional activity of the candidate SNPs on functional genes. The target sequence was centered on the candidate SNPs and extended 500 bp up and down. It was synthesized and cloned into the promoter or enhancer vector. DF-1 cells (chicken fibroblast cell line) were cultured in a Dulbecco’s modified Eagle medium (Gibco, USA) supplemented with 10% fetal bovine serum (Gibco, USA), 100 IU/mL penicillin, and 100 μg/mL streptomycin (Gibco, USA). Lipofectamine 3000 reagent (Invitrogen, USA) was used for transient transfection following the manufacturer’s protocols. The recombinant plasmid was transfected into the DF-1 cells together with the PRL-TK plasmid (Promega, USA). The DF-1 cells were then cultured in 24-well culture plates (Thermo Scientific, USA) at 37 °C and 5% CO2 for 48 h. Firefly and Renilla luciferase activities were measured at 48 h post-transfection using a Dual-Luciferase Assay System Kit (Promega, USA) according to the manufacturer’s instructions. Luminescence was detected using a microplate reader (Tecan, Switzerland), and firefly luciferase activities were normalized to Renilla luminescence in each well.
Real-time (RT) PCR experimental verification
Small intestine qRT-PCR was performed by using the SYBR Green Master Mix (Takara, Dalian, China) on Applied BiosystemsTM7300 qRT-PCR system (ABI, CA, USA) according to the manufacturer’s protocol. The data were analyzed by using the 2−ΔΔCT method. The chicken β-actin gene was used as the reference gene for normalization. The primers used for qRT-PCR are listed in Supplementary Table 5.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The DNA and RNA sequencing data generated in this study have been deposited in the NCBI BioProject database with accession numbers PRJNA1197814 and PRJNA1196443. Other breed WGS data are from http://farmrefpanel.com/GCRP/#/. Chicken GTEx data are available at http://chicken.farmgtex.org. FAANG data are available at https://farm.cse.ucdavis.edu/~zhypan/Chicken_FAANG/. GWAS summary data are available on Zenodo (https://zenodo.org/records/15370428)73. Source data are provided with this paper.
Code availability
The processing codes are publicly available on GitHub (https://github.com/xiaoningZhu0506/AIL_GWAS) and Zenodo (https://zenodo.org/records/15370428)73.
References
Visscher, P. M. et al. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22 (2017).
Abdellaoui, A., Yengo, L., Verweij, K. J. H. & Visscher, P. M. 15 years of GWAS discovery: realizing the promise. Am. J. Hum. Genet. 110, 179–194 (2023).
Lappalainen, T., Li, Y. I., Ramachandran, S. & Gusev, A. Genetic and molecular architecture of complex traits. Cell 187, 1059–1075 (2024).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Nicolae, D. L. et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 6, e1000888 (2010).
Banovich, N. E. et al. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet. 10, e1004663 (2014).
Chen, L. et al. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 167, 1398–1414.e24 (2016).
Kumasaka, N., Knights, A. J. & Gaffney, D. J. Fine-mapping cellular QTLs with RASQUAL and ATAC-seq. Nat. Genet. 48, 206–213 (2016).
Qi, T., Song, L., Guo, Y., Chen, C. & Yang, J. From genetic associations to genes: methods, applications, and challenges. Trends Genet. 40, 642–667 (2024).
GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
The ENCODE Project Consortium et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020).
Mellouk, N. et al. Chicken is a useful model to investigate the role of adipokines in metabolic and reproductive diseases. Int. J. Endocrinol. 2018, 1–19 (2018).
Scanes, C. G. Perspectives on the endocrinology of poultry growth and metabolism. Gen. Comp. Endocrinol. 163, 24–32 (2009).
Pan, Z. et al. An atlas of regulatory elements in chicken: A resource for chicken genetics and genomics. Sci. Adv. 9, eade1204 (2023).
Guan, D. et al. Genetic regulation of gene expression across multiple tissues in chickens. Nature Genet. https://doi.org/10.1038/s41588-025-02155-9 (2025).
Zhu, X.-N. et al. Chicken chromatin accessibility atlas accelerates epigenetic annotation of birds and gene fine-mapping associated with growth traits. Zool. Res. 44, 53–62 (2023).
Zhu, D. et al. GCRP: integrated global chicken reference panel from 11,951 chicken genomes. Genom. Proteom. Bioinform. qzaf032. https://doi.org/10.1093/gpbjnl/qzaf032 (2025).
Hu, Z.-L., Park, C. A. & Reecy, J. M. Bringing the animal QTLdb and CorrDB into the future: meeting new challenges and providing updated services. Nucleic Acids Res. 50, D956–D961 (2022).
Darvasi, A. & Soller, M. Advanced intercross lines, an experimental population for fine genetic mapping. Genetics 141, 1199–1207 (1995).
Gonzales, N. M. et al. Genome wide association analysis in a mouse advanced intercross line. Nat. Commun. 9, 5162 (2018).
Parker, C. C., Cheng, R., Sokoloff, G. & Palmer, A. A. Genome-wide association for methamphetamine sensitivity in an advanced intercross mouse line. Genes Brain Behav. 11, 52–61 (2012).
Yang, R. et al. Accelerated deciphering of the genetic architecture of agricultural economic traits in pigs using a low-coverage whole-genome sequencing strategy. GigaScience 10, giab048 (2021).
Wang, Y. et al. Multiple ancestral haplotypes harboring regulatory mutations cumulatively contribute to a QTL affecting chicken growth traits. Commun. Biol. 3, 472 (2020).
Guo, Y. et al. A complex structural variation on chromosome 27 leads to the ectopic expression of HOXB8 and the muffs and beard phenotype in chickens. PLoS Genet. 12, e1006071 (2016).
Gunnarsson, U. et al. The dark brown plumage color in chickens is caused by an 8.3-kb deletion upstream of SOX10. Pigment Cell Melanoma Res. 24, 268–274 (2011).
Yang, R. et al. Genome‑wide association study and genomic prediction for growth traits in yellow-plumage chicken using genotyping-by-sequencing. Genet Sel. Evol. 53, 82 (2021).
Zhong, C. et al. Age-dependent genetic architectures of chicken body weight explored by multidimensional GWAS and molQTL analyses. J. Genet. Genom. 51, 1423–1434 (2024).
Xie, L. et al. Genome-wide association study identified a narrow chromosome 1 region associated with chicken growth traits. PLoS ONE 7, e30910 (2012).
Lallès, J.-P. Recent advances in intestinal alkaline phosphatase, inflammation, and nutrition. Nutr. Rev. 77, 710–724 (2019).
Lallès, J.-P. Intestinal alkaline phosphatase: multiple biological roles in maintenance of intestinal homeostasis and modulation by diet: nutrition Reviews©. Nutr. Rev. 68, 323–332 (2010).
Zhou, Z. et al. An intercross population study reveals genes associated with body size and plumage color in ducks. Nat. Commun. 9, 2648 (2018).
Chen, S. et al. Identification of SNPs in the second intron of IGF2BP1 and their Association with growth traits in Nanjiang Yellow goat. Anim. Biotechnol. 36, 2461176 (2025).
Wang, K. et al. The chicken pan-genome reveals gene content variation and a promoter region deletion in IGF2BP1 affecting body size. Mol. Biol. Evol. 38, 5066–5081 (2021).
Zan, Y. et al. Artificial selection response due to polygenic adaptation from a multilocus, multiallelic genetic architecture. Mol. Biol. Evol. 34, 2678–2689 (2017).
Teng, J. et al. A compendium of genetic regulatory effects across pig tissues. Nat. Genet. 56, 112–123 (2024).
Parker, C. C. et al. Fine-mapping alleles for body weight in LG/J × SM/J F2 and F34 advanced intercross lines. Mamm. Genome 22, 563–571 (2011).
Zan, Y. et al. Genotyping by low-coverage whole-genome sequencing in intercross pedigrees from outbred founders: a cost-efficient approach. Genet. Sel. Evol. 51, 44 (2019).
Van Goor, A. et al. Identification of quantitative trait loci for body temperature, body weight, breast yield, and digestibility in an advanced intercross line of chickens under heat stress. Genet Sel. Evol. 47, 96 (2015).
Yang, H. et al. ABO genotype alters the gut microbiota by regulating GalNAc levels in pigs. Nature 606, 358–367 (2022).
Fourquet, L. et al. An eight-founder wheat MAGIC population allows fine-mapping of flowering time loci and provides novel insights into the genetic control of flowering time. Theor. Appl Genet. 137, 277 (2024).
Yengo, L. et al. A saturated map of common genetic variants associated with human height. Nature 610, 704–712 (2022).
Cao, X. et al. Food intake-related genes in chicken determined through combinatorial genome-wide association study and transcriptome analysis. Anim. Genet. 51, 741–751 (2020).
Zhang, G. et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 346, 1311–1320 (2014).
Li, D. et al. Comparative 3D genome architecture in vertebrates. BMC Biol. 20, 99 (2022).
International Chicken Genome Sequencing Consortium Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432, 695–716 (2004).
King, M.-C. & Wilson, A. C. Evolution at two levels in humans and chimpanzees: their macromolecules are so alike that regulatory mutations may account for their biological differences. Science 188, 107–116 (1975).
Li, M. et al. De novo assembly of 20 chicken genomes reveals the undetectable phenomenon for thousands of core genes on microchromosomes and subtelomeric regions. Mol. Biol. Evol. 39, msac066 (2022).
Seki, R. et al. Functional roles of Aves class-specific cis-regulatory elements on macroevolution of bird-specific features. Nat. Commun. 8, 14229 (2017).
Sheng, Z. et al. Genetic dissection of growth traits in a Chinese indigenous × commercial broiler chicken cross. BMC Genom. 14, 151 (2013).
Picelli, S. et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 24, 2033–2040 (2014).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Liu, S. et al. Genomic analyses from non-invasive prenatal testing reveal genetic associations, patterns of viral infections, and Chinese population History. Cell 175, 347–359.e14 (2018).
Davies, R. W., Flint, J., Myers, S. & Mott, R. Rapid genotype imputation from sequence without reference panels. Nat. Genet. 48, 965–969 (2016).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Zhang, C., Dong, S.-S., Xu, J.-Y., He, W.-M. & Yang, T.-L. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788 (2019).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Jiang, L. et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nat. Genet. 51, 1749–1755 (2019).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825 (2018).
Barbeira, A. N. et al. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet. 15, e1007889 (2019).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Taylor-Weiner, A. et al. Scaling computational genomics to millions of individuals with GPUs. Genome Biol. 20, 228 (2019).
Wen, X., Pique-Regi, R. & Luca, F. Integrating molecular QTL data into genome-wide genetic association analysis: Probabilistic assessment of enrichment and colocalization. PLoS Genet. 13, e1006646 (2017).
Lee, Y., Luca, F., Pique-Regi, R. & Wen, X. Bayesian Multi-SNP Genetic Association Analysis: control of FDR and use of summary statistics. Preprint at https://doi.org/10.1101/316471 (2018).
Schizophrenia Working Group of the Psychiatric Genomics Consortium et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Zhu, X. Chicken AIL population GWAS summary data. Zenodo, https://zenodo.org/records/15370428 (2025).
Acknowledgements
We thank Ning Li, Jian Ji, Xu Liu, Xian Zou, Wei Luo, Chungang Feng, Zheya Sheng, Zhenhui Li, Bolin Cai, Wen Luo, Xiaoli Guo, Jianbo Li, Jingyi He, Xinchun Xu, Hanyu Wu, Chengcheng Zhao, Jiaping Li, Di Lian, Yuzhan Wang, Ruifei Yang, Di Zhu, Xuemin Cao, Haitao Wang, Zhuolin Huang, Ming Li, Juan Wang, Lu Zhang, Jing Tian, Xiaorong Gu, Ying Guo, Jinxiu Li and Dezhi Peng for their valuable contributions during sample collection. We also thank Huaijun Zhou, Zhangyuan Pan, and Dailu Guan for their help in analyzing the data. The analysis was performed on the high-performance computing platform of the State Key Laboratory of Agrobiotechnology and the National Research Facility for Phenotypic and Genotypic Analysis of Model Animals. This study was funded by the Biological Breeding-National Science and Technology Major Project (Grant No. 2023ZD0406805 to X.X.H.), the National Natural Science Foundation of China (Grant No. 32272862 to Y.Z.W.), the National Key R&D Program of China (Grant No. 2024YFF1001500 to Y.Z.W.), the Open project of State Key Laboratory of Animal Biotech Breeding (Grant No. 2025SKLAB6-21 to Y.Z.W.) and the 2115 Talent Development Program of China Agricultural University (Grant to X.X.H.).
Author information
Authors and Affiliations
Contributions
Y.Z.W., X.X.H., and H.Q. conceived and designed the study. X.N.Z. conducted GWAS, fine mapping, and colocalization analysis. C.L. carried out experimental verification. Z.H.B., J.L.R., and R.S. helped generate and analyze the data. C.L.L., D.M.S., and P.C. assisted in sample collection. X.N.Z. and Y.Z.W. wrote the initial manuscript. Y.Z.W., X.X.H., and L.Z.F. were responsible for manuscript revision. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ming-Shan Wang and the other, anonymous, reviewers for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, X., Li, C., Luo, C. et al. Mapping the regulatory genetic landscape of complex traits using a chicken advanced intercross line. Nat Commun 16, 5841 (2025). https://doi.org/10.1038/s41467-025-60834-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60834-x
This article is cited by
-
Genome-wide association studies on body-weight and body-size traits among Zhedong white geese based on a modified genotyping-by-sequencing method
BMC Genomics (2025)
-
Selection signature analysis in chickens divergently selected for growth rate reveals novel candidate genes regulating fat deposition
BMC Genomics (2025)
-
Egg-laying ChickenGTEx resource deciphers context-specific regulatory effects on fertility traits
Nature Communications (2025)
