
Abstract
The development of pure organic photosensitizers remains challenging due to the low intersystem crossing efficiency and the instability of triplet excitons. Herein, fused-ring phosphorescent molecules enhance visible-light absorption, with heteroatom-rich structures breaking the restriction of low triplet excitons. A derivative, 2,3,5,6,9,10-hexabutoxy-8-phenyldithieno-tribenzo-pyridine (TPy), exhibits high ISC efficiency and efficiently sensitizes Fe-catalysts for CO2 photoreduction to CO. We further developed a self-assembly method to stabilize triplet excitons by embedding TPy within the rigid core of amphiphilic polymer nanoparticles. The hydrophobic core of the nanoparticles significantly prolongs the excited-state lifetime, while the hydrophilic shell ensures excellent dispersibility and stability. This system achieves a turnover number of 2041 and retains 93.5% of its initial activity after three cycles. Our work provides a general strategy for designing stable and highly efficient organic photosensitizers, paving the way for sustainable photoredox catalysis.
Similar content being viewed by others
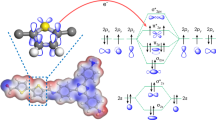

Introduction
Artificial photosynthesis has attracted much academic and industrial interest for utilizing solar energy, avoiding harsh reaction conditions, and reducing the formation of side reactions1,2,3,4. As a key mediator in photocatalytic systems, the development of photosensitizer is a hot research topic. The use of organometallic complexes containing noble metals (such as Ru(bpy)32+ and Ir(ppy)2(bpy)+) has been critical to photocatalysis advancements5,6,7,8,9. These complexes are capable of absorbing visible light to initiate electron or energy transfer reactions from their reactive excited states5,6. The impressive performance of these photosensitizers arises from their photophysical properties, including excellent visible-light absorption and long-lived excited states that can engage in electron transfers6,10,11. However, the development of organometallic complexes is limited by their high cost and complex preparation12,13,14,15. As a result, the development of pure organic photosensitizers would be highly desirable to drive various photocatalytic reactions16,17. As a rule, spin-forbidden relaxation to the ground state prolongs the lifetime of a triplet state compared to the spin-allowed singlet state18,19,20. This provides enough time to mediate intermolecular electron transfer processes. However, most organic molecules only exhibit the short lifetime transitions from the lowest singlet state to the ground state due to the forbidden intersystem crossing (ISC) process20,21. Rational functionalization of boron dipyrromethene and aminoanthraquinone chromophores22,23, as well as the design of molecular donor-accepter structures to break the short-lived excited state limitation24,25,26, have been achieved for photocatalytic reactions (Supplementary Fig. 1 and Supplementary Table 1). However, it is still a significant challenge to develop new, efficient, and stable organic photosensitizers with long-lived triplet excited state.
Organic room temperature phosphorescence (RTP) is a recently reported fascinating phenomenon in which an afterglow emission lasts for several seconds or even minutes after the removal of the irradiation owing to the presence of an ultralong-lived excited state20,27,28,29. The similarity in design between phosphorescent molecules and organic photosensitizers demonstrates that phosphorescent molecules are potential and strong candidates for organic photosensitizers. The abundance and longevity of the triplet excitons in these RTP materials allow pure organic compounds to serve as metal-free redox organic photosensitizers for photoreactions. This is because the photoreaction rate constant may be faster than the phosphorescence decay constant after sufficient interaction between the reaction substrate and RTP materials30,31,32. However, the utilization of RTP materials for photoredox reactions (e.g., CO2 to CO conversion) has not been achieved yet. In addition, understanding the relationship between RTP characteristics and photosensitizing properties remains a challenge. Studying this relationship may provide a great opportunity for the high-throughput pre-estimation of photosensitizing properties in organic systems using spectroscopy. Our work in the field of organocatalytic photoreactions began with our interest in the modulation of triplet exciton decay in organic RTP materials33,34. Host-guest doping strategies achieve high-performance RTP by effectively facilitating the ISC processes and stabilizing triplet excited state27,35,36,37. In combination with nanonization preparation, the stable triplet excited state in the formed doped-RTP nanoparticles (NPs) are not dissipated through radiative decay, but instead can transfer electrons or energy to the surrounding medium38,39, such as 3O2, for photodynamic antibacterial treatment. Therefore, we proposed a strategy of doping the phosphorescent molecules into the core of NPs to construct advanced photosensitizers. The phosphorescent molecules can generate abundant triplet excitons, which can be stabilized by the rigid core of NPs. Under irradiation conditions, electron transfer can occur in NPs, thus improving photocatalytic efficiency.
In this work, we designed and prepared host-guest doped NPs for visible-light-driven reduction of CO2 to CO (Fig. 1). A series of polycyclic aromatic hydrocarbons (PAHs) with buckybowl-based structure was selected as guest candidates. Heteroatoms with abundant long-pair electrons, including oxygen, nitrogen, and sulfur, were introduced into the PAHs, leading to the S1 (n, π*) to Tn (π, π*) spin flip transitions (EI-Sayed rules) and facilitating the ISC process to generate triplet excitons20,29,40. The dissymmetry electrostatic potential distribution on the concave and convex surfaces of the buckybowl structures enables π-π interaction with the aromatic-containing polymer matrix34,41. Therefore, polystyrene (PS) was used as a host to stabilize the triplet excited state, achieving RTP emission (Fig. 1a). Among the selected PAHs, 2,3,5,6,9,10-Hexabutoxy-8-phenyldithieno-triphenyleno-pyridine (TPy) showed the highest triplet exciton generation and the best RTP performance in the PS matrix. Further, to utilize and manipulate the triplet excitons stabilized by PS, self-assembled TPy NPs were prepared by using PS-block-poly(ethylene glycol) (PS-b-PEG) polymer (Fig. 1b). TPy was dispersed into the hydrophobic PS segment to avoid molecular aggregation and motion, which further stabilized the triplet excited state and prolonged their lifetime. The hydrophilic PEG corona made NPs disperse uniformly in the catalytic system, avoiding triplet annihilation caused by aggregation. Thus, the catalytic efficiency of NPs-containing catalytic system was improved by 50% compared to TPy. An impressive turnover number (TON vs. catalyst) of the optimized photocatalytic CO2-to-CO system was 2041 with 51.02 μmol CO yield under 18 h visible light illumination. Remarkably, after three catalytic cycles, the sensitization capacity was effectively restored to 93.5%, realizing a highly sustainable CO2 photoreduction process via a reductive route. Our research clarifies the relationship between the characteristic of RTP and the sensitization ability of photosensitizers, and provides a idea for the development of high-efficiency and stable organic photosensitizers.

a Chemical structures of PS (host) and TPy (guest) and design strategy of PS-based organic RTP materials. b Schematic diagram of photophysical process of self-assembled NPs for CO2 photoreduction.
Results
Photophysical properties and theoretical analysis
A series of PAHs with buckybowl-based structure was prepared in accordance with our prior research42,43,44,45 and denoted TP, TPy, TH, and TQ (Fig. 2a). All the PAHs demonstrate visible-light absorption (1 × 10−5 M in THF, Supplementary Fig. 2), indicating their potential ability to perform photochemical reactions under natural light. The absorption and fluorescence characteristics of these PAHs exhibit a bathochromic shift from TP to TQ, which is attributed to the extension of π-conjugation (Fig. 2b, Supplementary Figs. 3, 4, Supplementary Table 2)46. This indicates the tunable energy level of the designed phosphors. Delayed emission spectra were recorded in 2-MeTHF (1 × 10−5 M) at 77 K (Fig. 2c and Supplementary Fig. 3). Notably, TP, TPy, and TH display distinct phosphorescence emission characteristics at 77 K, whereas TQ demonstrates negligible phosphorescent emission under identical experimental conditions. In this context, assessing the triplet exciton yield of PAHs is pivotal because this parameter is related to the sensitization efficiency of photosensitizers.

a Molecular structures of PAHs. b Normalized UV-vis absorption and fluorescence spectra (at room temperature) of PAHs in THF (1 × 10−5 M). c Normalized prompt and delayed emission spectra (at 77 K) of PAHs in 2-MeTHF (1 × 10−5 M), delayed time: 50 μs, under excitation at 405, 405, 410, 465 nm, respectively. d Diagrams of the TD-DFT calculated energy levels and SOC constants of PAHs.
Theoretical calculations facilitate an understanding the photophysical properties of the PAHs. Highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of PAHs are distributed throughout the molecule, indicating no obvious intramolecular charge transfer (Supplementary Fig. 5). With the increase of conjugation, the energy gap (Eg) between HOMO and LUMO decreases continuously, which is consistent with the UV-vis absorption spectra (Fig. 2b). To gain further insight into electronic transition process of the molecule, the excited state PAHs were calculated based on time-dependent density functional theory (TD-DFT). The hole-electron population of each state was evaluated using the Multiwfn 3.8 package47.
The evaluation of the ISC ability of a molecule can be conducted through a joint analysis of the spin-orbit coupling (SOC) values and the energy difference (ΔEST) between the lowest singlet states (S1) and low-lying triplet states48,49,50. As shown in Tables S3–S6, the dominant orbital transition contributions of the S1 in PAHs exhibit remarkable consistency with those of the T1, indicating the presence of efficient ISC channels. Additionally, the electron-hole distribution of S1 and T1 of PAHs is highly similar (Supplementary Fig. 6), which further ensures that the singlet excitons are easily spin-flipped into triplet excitons51. The calculated SOC constants <S1 | HSO | T1> of TP, TPy, TH, and TQ are 0.16, 1.38, 0.71, and 0.74, respectively. Notably, TP, TPy, and TH exhibit small ΔEST values of 0.15 eV, 0.20 eV, and 0.17 eV, respectively (Fig. 2d), which fall within the energy range conducive to ISC. In contrast, TQ exhibits a large ΔEST (0.78 eV) and a lower T1 energy level (1.24 eV). Although its SOC value is not negligible, thermodynamic considerations strongly favour non-radiative deactivation pathways over phosphorescent emission. This theoretical prediction is evidenced by the absence of detectable phosphorescence in the 77 K delayed emission spectrum of TQ (Supplementary Fig. 7). Consequently, TPy exhibits the highest SOC constants and generates the most triplet excitons, suggesting that TPy possesses the best photosensitizing ability among the four PAHs.
PAHs photosensitizers for photocatalytic CO2 reduction
To reveal the effects of the photophysical processes in the four PAHs on CO2 photoreduction, photocatalytic activities of these molecules were investigated in 5 mL CO2-saturated CH3CN/H2O (v/v = 4/1) solution containing [Fe(qpy)(OH2)2]2+ catalyst (referred to as CAT) and electron donors (EDs): triethylamine (TEA) and 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH)52,53. TEA is used as a deprotonated Brønsted base to ensure the irreversibility of the more oxidizing sacrificial BIH54. The TONCAT of TPy-containing system is as high as 896 (22.41 μmol CO, 18 h irradiation time, 400 nm filter), followed by TH, while the TP and TQ molecules are almost inactive (Fig. 3a). These results reveal that the efficient ISC processes of TPy results in active photosensitizing properties for CO2 photoreduction. Importantly, the organic fused-ring molecule with the high structural stability and a large π-conjugation for charge delocalization42 after the photoredox showed great advantage for CO2 photoreduction.

a Photocatalytic CO2 reduction with PAHs under the illumination of a 300 W Xenon lamp (50 mw cm−2, λ > 400 nm, irradiation time: 18 h) in the presence of PAHs (10 μM), CAT (5 μM), BIH (18 mM) and TEA (0.3 mM) in 5 mL CO2-saturated CH3CN/H2O (v/v = 4/1) solution. b Nanosecond transient absorption spectra of kinetic decay trace of TP, TPy, and TH at 500 nm. These spectra were recorded in CH3CN/H2O (v/v = 4/1) solution after pulsed excitation at 430 nm under argon atmosphere at room temperature. c Delayed emission spectra of TPy@PS at mass ratio of 1:75 in air and vacuum. d Phosphorescence efficiency of PAHs@PS at mass ratio of 1:75.
Further, nanosecond transient absorption spectra of kinetic decay were employed to understand the photosensitizing process of the organic phosphors in CO2 photoreduction system. The TP, TPy, and TH molecules show long-lived triplet excited state capable of sufficient intermolecular electron transfer between the photosensitizers and photoreaction substrates, with triplet excited lifetimes of 74.56 μs, 48.74 μs and 102.58 μs, respectively (Fig. 3b). These findings establish the close relationship between the capacity of PAHs to generate triplet excitons and photosensitization efficiency, particularly in the presence of long-lived triplet excited state. Moreover, the stability of the triplet excited state is a vital factor in ensuring sufficient time for intermolecular electron transfer to boost photosensitization. TPy with the highest ISC efficiency also leads to a relatively short triplet excited state lifetime, potentially impairing the sensitization ability. In addition, TPy shows poor solubility in the photocatalysis solution and gradually aggregates (Supplementary Figs. 8, 9), leading to a decline in photocatalytic efficiency. Therefore, stabilizing both TPy molecules in solution and its triplet excited state may further promote photosensitization efficiency.
RTP phenomenone of the doped system
A polymer host can effectively inhibit molecular motion and stabilize triplet excited state27,28,55,56,57. Herein, inspired by the preparation of organic RTP materials in our previous work56, PS was selected as a polymer matrix, and a series of TPy@PS doped films with different mass ratios were prepared. In prompt spectra, the highest luminescence peak is observed at a mass ratio of 1:75 (Supplementary Fig. 10), attributed to the phosphorescence peak of molecularly dispersed TPy at 620 nm (Fig. 2c). Of note, TPy molecule shows no phosphorescence at room temperature (Supplementary Fig. 11). This indicates that PS is capable of acting as a rigid matrix to inhibit the non-radiative transition of TPy, thereby stabilizing the triplet excited state and facilitating RTP. The robust interaction between the phenyl ring in PS and PAHs can avoid strong π-π stacking interaction between photosensitizers, enhancing stability of triplet excited state34. Therefore, a guest@host doping mass ratio of 1:75 was used for subsequent experiments. In vacuum delayed spectra, TPy@PS shows a strong phosphorescence emission (Fig. 3c, Supplementary Fig. 12), with a phosphorescence quantum yield (QY) is up to 14.84% (Fig. 3d). In comparison, TP@PS and TH@PS have very weak phosphorescence (Supplementary Fig. 13), which further indicates their weak triplet exciton generation ability. TPy@PS shows the highest phosphorescence efficiency, followed by TH@PS and TP@PS (Fig. 3d), which is consistent with theoretical calculations of PAHs (Fig. 2d). These results suggest that utilizing PS as a polymer matrix can stabilize the triplet excites state, leading to improved RTP emission. Importantly, this straightforward doping strategy enables the rapid screening and evaluation of RTP materials as efficient photosensitizers using spectroscopy.
Photophysical properties of self-assembled NPs
To further utilize the triplet excited state stabilized by the PS matrix for CO2 photoreduction, TPy and the amphiphilic block copolymer PS-b-PEG were assembled via microfluidic technology (Fig. 4a)38,58. The properties of NPs are highly related to their size and morphology17,59,60. Large NPs and aggregates may prevent the electron transfer of triplet excited state to the reaction substrate, while triplet excited state may not be stable enough in small NPs39. It is vital for the preparation of monodisperse NPs with controllable aggregates and high uniformity by microfluidic technology. During the self-assembly process, the hydrophobic PS segment within PS-b-PEG acts as the host, wrapping TPy molecules and stabilizing the triplet excited state. Meanwhile, the hydrophilic PEG segment enables the stable dispersion of the NPs in water. TPy do not emit phosphorescence in either oxygenated or deoxygenated water (Fig. 4b). Notably, the NPs also show no phosphorescence in oxygenated water, but emit in argon-deoxygenated water. This phenomenon shows that the hydrophobic PS segment is capable of stabilizing the triplet excited state without hindering the interaction of TPy with various components in the solution, thus manipulating energy decay of triplet excitons. Consequently, the capacity of NPs to generate triplet excitons is evaluated by their phosphorescence emission intensity in deoxygenated water.

a Schematic depiction of microfluidic setup for the assembly of NPs-xk. In deoxidized water, b delayed (200 μs) spectra, c total and phosphorescence QYs of NPs-xk (x = 2, 5, 10, 23, 35). Total QY represents the sum of fluorescence and phosphorescence quantum yields, characterizing the overall photoluminescence efficiency of the RTP material. d DLS measurement of NPs-23k. Insert: TEM images of NPs-23k. cNPs-xk = 3.75 × 10−5 M.
Modifying the hydrophilic-hydrophobic ratio of the amphiphilic polymer can tune the particle size of the self-assembled NPs60,61. Therefore, a series of diblock polymer was prepared with different PS15k-b-PEGxk hydrophilic-hydrophobic ratio (x = 2, 5, 10, 23, 35). With increasing hydrophilic ratio, the size of the NPs becomes smaller (Supplementary Fig. 14). However, when x = 2, insufficient hydrophilic segments result in the formation of unstable NPs that are prone to agglomeration and precipitation in the solution and during the catalytic process. Changing the hydrophilic PEG segment from 2k to 23k in deoxygenated water leads to a significant enhancement of phosphorescence intensity and a prolonged phosphorescence lifetime (Fig. 4b, Supplementary Fig. 15). However, an excessive hydrophilic ratio diminishes the hydrophobicity of the PS segment, causing loose wrapping and a subsequent decrease in phosphorescence intensity and lifetime. This trend is further validated by total and phosphorescence QYs, confirming that NPs-23k generate the highest triplet excitons (Fig. 4c). In addition, time-dependent UV-vis spectral analysis reveals that the resistance to photochemical degradation of NPs-23k is significantly higher than that of TPy under long-term irradiation (Supplementary Fig. 16), which clearly indicates the protection and stability of TPy structure by self-assembled nanomicelles62. Finally, the hydrodynamic diameter of NPs-23k (~ 160 nm, polydispersity index: 0.19) is determined by dynamic light scattering (DLS). The insert transmission electron microscope (TEM) image shows the spherical morphology of NPs-23k (~ 110 nm) in dry state (Fig. 4d, Supplementary Fig. 17). These experiments confirm that NPs-23k exhibit excellent photobleaching resistance, uniform particle size, good dispersion, large surface area, and the highest yield of triplet excitons. Consequently, it can be reasonably inferred that NPs-23k may exhibit a superior sensitization effect on the reactants in the catalytic system.
Self-assembled NPs for photocatalytic CO2 reduction
Finally, the performance of NPs-23k for photocatalytic CO2 reduction was investigated (Fig. 5). NPs-23k generates long-lived triplet excited state of 96.19 μs, which is twice that of TPy molecule (Figs. 3b, 5d). Under irradiation of xenon lamp equipped with 400 nm filter for 18 h, the catalytic efficiency of NPs-23k-containing system is increased by nearly 1.5-fold compared to TPy system, the CO yield can reach 33.40 μmol with a TONCAT value of 1336. (Supplementary Fig. 18). However, the catalytic effect of the system without a filter is inferior. The CO yield of the NPs-23k-containing system is only 7.72 μmol (Supplementary Fig. 19), a value comparable to that observed under irradiation with a 395 nm monochromatic light source (Supplementary Fig. 20). This observation was attributed to the disruption of the organic structure caused by prolonged exposure to high-energy ultraviolet radiation. Furthermore, the NPs-23k-containing catalytic system exhibited superior apparent quantum efficiency (AQE) compared to the TPy system at 395 nm (both 20 and 50 mW·cm−2 light intensities) (Supplementary Table 7). Notably, the AQE of NPs-23k-containing catalytic system at 450 nm demonstrated nearly 2-fold enhancement over the TPy system, highlighting the enhanced sensitization ability of self-assembled NPs. The above findings demonstrates that utilizing PS-b-PEG with host function effectively stabilizes the triplet excited state of TPy. Moreover, in comparison with hydrophobic TPy aggregates (~1.5 μm, Supplementary Figs. 8, 9), the substantial specific surface area and extended PEG segments of spherical NPs-23k enhance the electron diffusion capability22,63,64 and accelerate the electron transfer rate of the excited triplet state (Supplementary Figs. 21, 22, and Supplementary Note 1). Additionally, monodisperse NPs-23k can mitigate the exciton loss induced by triplet exciton annihilation in TPy aggregates65, thus improving the sensitization efficiency. Next, the concentrations of the photosensitizer and EDs in the photocatalytic system were optimized (Table 1). The quantification of the gaseous products shows that the C/H selectivity of photoreduction CO2 to CO is about 95% under all conditions. The TONCAT of NPs-23k-containing system reaches 2041 (51.02 μmol CO) within 18 h illumination (100 mw·cm−2, λ > 400 nm) under the optimal conditions (Table 1, Group 2). Clearly, the simple self-assembly method employed in this work endows TPy with more stable and longer-lived triplet excited state. This enables the facile transfer of electrons to surrounding compounds, thus favoring the photosensitization process.

a Recycle CO2 reduction experiments of TPy and NPs-23k. b Energy diagram depicting the redox potentials of TPy, CAT, and BIH. *Ox and *Red represent excited state oxidation and reduction potentials, respectively. c Phosphorescence quenching of NPs-23k in simulated catalytic system, cNPs-23k = 10 μM. d The decay of NPs-23k at 500 nm, cNPs-23k = 10 μM. e Kinetic traces of reduced NPs-23k and reduced NPs-23k with different concentration of CAT at 500 nm. cNPs-23k = 10 μM, the numbers preceding BIH and CAT represent their relative equivalent to cNPs-23k. These spectra were recorded in CH3CN/H2O (v/v = 4/1) solution after pulsed excitation at 430 nm under argon atmosphere at room temperature. f Reduction mechanism in photocatalytic process.
Subsequently, we also conducted photocatalytic CO2 reduction experiments using NPs-xk (x = 2, 5, 10, 35) as photosensitizers under optimized conditions. The result demonstrates a strong correlation between CO yield and phosphorescence QY, with NPs-23k-containing catalytic system showing optimal photocatalytic activity (Supplementary Fig. 23). This quantitative agreement between photocatalytic performance and phosphorescence characteristics provides compelling experimental validation of phosphorescence QY as a reliable predictor for photosensitizer efficiency.
To investigate the stability of catalytic system under irradiation, recycle experiments were performed by re-adding NPs-23k, CAT, and EDs after 6 h irradiation (Supplementary Fig. 24). Following the addition of CAT and EDs in the 2nd cycle, the photocatalytic activity of the reaction system is almost entirely restored. This indicates that the inactivation of the photocatalytic system is primarily caused by the decomposition or consumption of CAT and EDs. To compare the stability of TPy and NPs-23k, consumed CAT and EDs were re-added after each 6 h cycle. As shown in Fig. 5a, after three cycles (18 h), the sensitization ability of NPs-23k remains at 93.5%, while that of TPy is only 74.9%. Remarkably, the total CO yield of the NPs-23k-containing system reaches 113.6 μmol. This indicates that the stability of NPs-23k photosensitizers is higher than that of TPy, because the vibrational relaxation of TPy is not suppressed in the absence of the PS coating, which leads to declining stability under long-term irradiation. These results further emphasize the significance of assembling TPy with PS-b-PEG. The self-assembly strategy prolongs the lifetime of the triplet excited state, enhances the electron transfer capability, and prevents the loss of triplet excitons, thus effectively improving the photosensitization cyclic ability of the catalytic system.
Mechanism analysis of photocatalytic system
In the absence of photosensitizer (TPy/NPs-23k), CAT, EDs, light, or CO2, trace or no CO was detected (Supplementary Table 8), indicating all of these factors are indispensable for CO2 photoreduction. Moreover, to unequivocally confirm the CO origin, we conducted isotope-labeling experiments using 13CO2, with mass spectrometry analysis demonstrating 13CO as the primary product (Supplementary Fig. 25). These results prove that the CO product is indeed generated from CO2 instead of the decomposition of organic photosensitizers. To elucidate the photocatalytic mechanism, the thermodynamic feasibility of intermolecular electron transfer was evaluated, and cyclic voltammetry (CV) was conducted to study the key components of the reaction system (TPy, CAT, and BIH) in a degassed acetonitrile solution (Supplementary Fig. 26, Supplementary Table 9). The oxidation potential of BIH is far more negative than the excited state reduction potential of TPy (*Red) (0.52 V ≪ 1.95 V vs. SCE), and the excited state oxidation potential of TPy (*Ox) is more negative than the reduction potential of CAT (−1.21 V < −1.05 V vs. SCE) (Fig. 5b). The results indicate that both the reduction and oxidation quenching mechanisms are thermodynamically feasible in the studied photocatalytic system. However, the potential difference for the reduction quenching mechanism is greater than that for the oxidation mechanism (−1.43 V vs. −0.16 V). In addition, the absorption spectra of TPy remained unchanged before and after adding the BIH or CAT (Supplementary Fig. 27), revealing the absence of intermolecular electronic interaction between TPy and CAT or BIH under ground state. Phosphorescence quenching experiments were performed with NPs-23k using CAT or BIH as quenchers. At the concentration scale of CAT and BIH in the photocatalytic system, the phosphorescence intensity of NPs-23k is more efficiently quenched by BIH compared to CAT (Fig. 5c). These findings suggest that the photocatalytic process is primarily governed by the reduction mechanism, which was further supported by nanosecond transient absorption studies (Supplementary Fig. 28, Fig. 5d, e).
Nanosecond transient absorption spectra were utilized to probe the excited state properties and intermolecular electron transfer of NPs-23k and TPy. As shown in Supplementary Fig. 28a, b, a strong bleaching peak below 350 nm is observed upon pulsed laser excitation, while positive peaks at 360 nm and beyond 450 nm correspond to the excited states of NPs-23k and TPy6,22. The excited state map of NPs-23k is similar to that of TPy, indicating that the excited state type of TPy remains unchanged after self-assembly. When BIH is added to the NPs-23k solution, the peak at 360 nm shows a lower intensity and narrower peak width, and the positive absorption after 530 nm almost disappears, indicating the consumption of excited states species (Supplementary Fig. 28b, c). In addition, a negative bleach peak at 410 nm and a new sharper positive signal above 485 nm appears. This new peak is assigned to the reduced NPs-23k with a long lifetime of 191.48 μs (Supplementary Fig. 28c, Fig. 5e). To support this assignment, spectroelectrochemistry experiments were performed at −0.5 V vs. SCE (more negative than the reduction potential) to obtain the absorption spectra of electroreduced species (Supplementary Fig. 29a). As evident in Supplementary Fig. 29b, the differential spectrum obtained through spectroelectrochemical measurements exhibits new absorption peaks at 410 nm and 485 nm, which correspond well with the characteristic features of the reduced transient absorption spectrum (Supplementary Fig. 28c). The subsequent introduction of CAT reduces the lifetime of NPs-23k to 13.27 μs, indicating rapid electron transfer from the reduced NPs-23k to CAT, and the excited state map of the entire system is restored (Fig. 5e, Supplementary Fig. 28b–d). Analogous observations are made in TPy-containing systems (Supplementary Figs. 30, 31). Consequently, the electron transfer route of the NPs-23k-containing and TPy-containing photocatalytic systems are all classified as reduction mechanism (Fig. 5f).
Dicussion
We propose a strategy for self-assembly of phosphorescent molecules with the amphiphilic block copolymer PS-b-PEG to construct stable and efficient photosensitizers. Specifically, at the core of doped NPs-23k, TPy showed the highest ISC efficiency owing to extended conjugation structure and abundance of heteroatoms, as proven by systematic experiments and theoretical calculations, and can sensitize the photoreduction CO2-to-CO catalytic system. Meanwhile, the hydrophobic PS segment with host function stabilizes the triplet excited state of TPy, doubling their lifetime to 96.19 μs and improving the catalytic efficiency by 1.5-fold. Additionally, the hydrophilic PEG segment as corona of self-assembled NPs-23k stably disperses the NPs to prevent aggregation-induced triplet annihilation, ensuring the maximum utilization of triplet excitons of TPy. This self-assembly method endows NPs-23k with better photosensitivity, achieving a TONCAT of 2041 (51.02 μmol) with 18 h illumination, and improves the stability of NPs-23k, maintaining 93.5% photosensitivity after three cycles, and obtaining a CO yield of 113.6 μmol. Finally, NP-23k/TPy-containing photocatalytic systems are determined as reduction mechanism. The strategy employed in this work provides insights for transforming RTP materials with high-phosphorescence QY into efficient photosensitizers, greatly expands the number of organic photosensitizers in photocatalytic systems, and provides the possibility for organic photosensitizers to be used in homogeneous pure water catalysis
Methods
Photocatalytic CO2 reduction
Photocatalytic CO2 reduction was conducted under the illumination of a 300 W Xenon lamp with a 400 nm filter (50 mw cm−2, λ > 400 nm) in the presence of PAHs (TP: 847.06 g·mol−1, TPy: 822.13 gmol−1, TH: 819.96 gmol−1, TQ: 737.00 gmol−1), CAT (601.13 gmol−1), BIH (224.31 gmol−1), and TEA (101.19 gmol−1) in 5 mL CO2-saturated CH3CN/H2O (v/v = 4/1) solution. The following components (in the order indicated), including BIH, photosensitizers, H2O, CAT, CH3CN, and TEA were added to the catalytic system. The photocatalytic system was bubbled with CO2 for 20 min.
Preparation of doped films
Preparation of PS matrix: PS powder (1 g) was dissolved in THF (10 mL) at room temperature with magnetic stirring for 24 h to obtain a homogeneous solution (100 g·L−1), followed by filtering with 0.45 μm syringe filter. Preparation of PAH@PS doped films: According to the different mass ratio of host to guest, PAH (guest) was dispersed in 1 mL PS (100 g·L−1 THF solution). A homogeneous solution was obtained after 10 h magnetic stirring. Then, the films were fabricated with 500 μL solution on each quartz plate (1.5 × 1.5 cm) by drop-coasting method. Finally, solid films with different mass ratio were obtained after heating at 40 °C for 5 h.
Preparation of nanoparticles
Amphiphilic block copolymers (PS15k-b-PEGxk, x = the molecular weight of PEG / 1000 = 2, 5, 10, 23, 35) were purchased from Xi’an Rui xi Biological Technology Co., Ltd. See the supplementary information for more details. TPy and PS15k-b-PEGxk (x = 2, 5, 10, 23, 35) were dissolved in THF solution. The mass ratio of TPy to hydrophobic PS15k segment is 1: 5. Self-assembled NPs-xk were prepared in microfluidic chips. Basically, the organic solution and aqueous solution (DI water) were introduced separately into the microfluidic chip through two inlets, and the syringe pump drove two solutions to mix at the microscale with a controlled velocity, leading to the formation of nanoparticles. The microfluidic chip is composed of a glass substrate and a polydimethylsiloxane (PDMS) membrane, and the structure of the microchannel was designed with two inlets (width: 150 mm, height: 150 mm, length: 1.5 cm and 1.0 cm) and one outlet (width: 300 mm, height: 300 mm and linear length: 3 cm).
Data availability
The data supporting the findings of this study are available within the article and the supplementary file. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Gamache, M. T. et al. Elucidating electron transfer kinetics and optimizing system performance for Escherichia coli-based semi-artificial H2 production. ACS Catal. 13, 9476–9486 (2023).
Chan, A. Y. et al. Exploiting the Marcus inverted region for first-row transition metal-based photoredox catalysis. Science 382, 191–197 (2023).
Onneken, C. et al. Light-enabled deracemization of cyclopropanes by Al-salen photocatalysis. Nature 621, 753–759 (2023).
Liu, R. et al. Linkage-engineered donor-acceptor covalent organic frameworks for optimal photosynthesis of hydrogen peroxide from water and air. Nat. Catal. 7, 195–206 (2024).
Saito, D., Tamaki, Y. & Ishitani, O. Photocatalysis of CO2 reduction by a Ru(II)-Ru(II) supramolecular catalyst adsorbed on Al2O3. ACS Catal. 13, 4376–4383 (2023).
Wang, P. et al. Identification of crucial photosensitizing factors to promote CO2-to-CO conversion. Angew. Chem. Int. Ed. 63, e202312450 (2024).
Zheng, H. et al. Site isolation in metal-organic layers enhances photoredox gold catalysis. J. Am. Chem. Soc. 144, 10694–10699 (2022).
Kumagai, H., Tamaki, Y. & Ishitani, O. Photocatalytic systems for CO2 reduction: metal-complex photocatalysts and their hybrids with photofunctional solid materials. Acc. Chem. Res. 55, 978–990 (2022).
Guo, Z. et al. Selectivity control of CO versus HCOO− production in the visible-light-driven catalytic reduction of CO2 with two cooperative metal sites. Nat. Catal. 2, 801–808 (2019).
Cheung, K. P. S., Sarkar, S. & Gevorgyan, V. Visible light-induced transition metal catalysis. Chem. Rev. 122, 1543–1625 (2022).
Ren, F.-Y. et al. Amphiphilic polycarbonate micellar rhenium catalysts for efficient photocatalytic CO2 reduction in aqueous media. Angew. Chem. Int. Ed. 61, e202200751 (2022).
Nikolaou, V. et al. Antenna effect in noble metal-free dye-sensitized photocatalytic systems enhances CO2-to-CO conversion. Angew. Chem. Int. Ed. 136, e202318299 (2024).
Tang, Q. et al. Bioinspired self-assembly of metalloporphyrins and polyelectrolytes into hierarchical supramolecular nanostructures for enhanced photocatalytic H2 production in water. Angew. Chem. Int. Ed. 63, e202315599 (2024).
Xie, W. et al. Metal-free reduction of CO2 to formate using a photochemical organohydride-catalyst recycling strategy. Nat. Chem. 15, 794–802 (2023).
Yuan, H., Cheng, B., Lei, J., Jiang, L. & Han, Z. Promoting photocatalytic CO2 reduction with a molecular copper purpurin chromophore. Nat. Commun. 12, 1835 (2021).
Zou, W. et al. Metal-free photocatalytic CO2 reduction to CH4 and H2O2 under non-sacrificial ambient conditions. Angew. Chem. Int. Ed. 62, e202313392 (2023).
Yang, H. et al. Packing-induced selectivity switching in molecular nanoparticle photocatalysts for hydrogen and hydrogen peroxide production. Nat. Nanotechnol. 18, 307–315 (2023).
Zhang, X. et al. Ultralong phosphorescence cellulose with excellent anti-bacterial, water-resistant and easy-to-process performance. Nat. Commun. 13, 1117 (2022).
Guo, J., Yang, C. & Zhao, Y. Long-lived organic room-temperature phosphorescence from amorphous polymer systems. Acc. Chem. Res. 55, 1160–1170 (2022).
Sun, H. et al. Engineering tunable ratiometric dual emission in single emitter-based amorphous systems. Angew. Chem. Int. Ed. 63, e202318159 (2024).
Tian, R., Gao, S., Li, K. & Lu, C. Design of mechanical-robust phosphorescence materials through covalent click reaction. Nat. Commun. 14, 4720 (2023).
Chen, K.-K., Guo, S., Ding, M.-J., Lu, T.-B. & Zhang, Z.-M. Heavy-atom-free photosensitizers for high-yield CO2-to-CO conversion. CCS. Chem. 5, 2650–2662 (2023).
Lei, Q. et al. Photocatalytic CO2 reduction with aminoanthraquinone organic dyes. Nat. Commun. 14, 1087 (2023).
Rao, H., Lim, C.-H., Bonin, J., Miyake, G. M. & Robert, M. Visible-light-driven conversion of CO2 to CH4 with an organic sensitizer and an iron porphyrin catalyst. J. Am. Chem. Soc. 140, 17830–17834 (2018).
Bassan, E. et al. Visible-light driven photocatalytic CO2 reduction promoted by organic photosensitizers and a Mn(I) catalyst. Sustain. Energy Fuels 7, 3454–3463 (2023).
Wang, Y., Gao, X.-W., Li, J. & Chao, D. Merging an organic tadf photosensitizer and a simple terpyridine-Fe(III) complex for photocatalytic CO2 reduction. Chem. Commun. 56, 12170–12173 (2020).
Chen, K. et al. A facile strategy for achieving polymeric afterglow materials with wide color-tunability and persistent near-infrared luminescence. Adv. Funct. Mater. 34, 2312883 (2023).
Qian, C. et al. More than carbazole derivatives activate room temperature ultralong organic phosphorescence of benzoindole derivatives. Adv. Mater. 34, 2200544 (2022).
Yu, J. et al. Efficient visible-light-activated ultra-long room-temperature phosphorescence triggered by multi-esterification. Angew. Chem. Int. Ed. 62, e202316647 (2023).
Zeng, Y. et al. Transformation of organic afterglow mechanism from room-temperature phosphorescence to thermally-activated delayed fluorescence in intramolecular charge transfer systems. Dyes Pigm. 219, 111643 (2023).
Herzog, W. et al. Electron transfer between hydrogen-bonded pyridylphenols and a photoexcited rhenium(I) complex. ChemPhysChem 14, 1168–1176 (2013).
Liu, J. et al. Organic afterglow emulsions exhibiting 2.4 s phosphorescence lifetimes and specific protein binding property. Adv. Opt. Mater. 10, 2201502 (2022).
Ren, Y. et al. Clusterization-triggered color-tunable room-temperature phosphorescence from 1,4-dihydropyridine-based polymers. J. Am. Chem. Soc. 144, 1361–1369 (2022).
Zhang, Y. et al. Wide-range color-tunable afterglow emission by the modulation of triplet exciton transition processes based on buckybowl structure. Aggregate 4, e310 (2023).
Dai, W. et al. Halogen bonding: a new platform for achieving multi-stimuli-responsive persistent phosphorescence. Angew. Chem. Int. Ed. 61, e202200236 (2022).
Yao, X. et al. Ultralong organic phosphorescence from isolated molecules with repulsive interactions for multifunctional applications. Nat. Commun. 13, 4890 (2022).
Wang, J. Q. et al. Management of triplet excitons transition: Fine regulation of förster and Dexter energy transfer simultaneously. Light Sci. Appl. 13, 35 (2024).
Chao, C. et al. Microfluidic-based modulation of triplet exciton decay in organic phosphorescent nanoparticles for size-assisted photodynamic antibacterial therapy. J. Mater. Chem. B 11, 3106–3112 (2023).
Kang, L. et al. Host-guest strategy for organic phosphorescence to generate oxygen radical over singlet oxygen. Chem. Mater. 36, 7332–7342 (2024).
Yuan, J. et al. Direct population of triplet excited states through singlet-triplet transition for visible-light excitable organic afterglow. Chem. Sci. 10, 5031–5038 (2019).
Jiang, W., Li, Y. & Wang, Z. Heteroarenes as high performance organic semiconductors. Chem. Soc. Rev. 42, 6113–6127 (2013).
Geng, R. et al. Driving π-plane to π-bowl through lateral coordination at room temperature. Mater. Chem. Front. 2, 1456–1461 (2018).
Hou, X. et al. Tris(S, S-dioxide)-trithiasumanene: strong fluorescence and cocrystal with 1,2,6,7,10,11-hexabutoxytriphenylene. Chem. Commun. 53, 1546–1549 (2017).
Sun, Y. et al. Trichalcogenasumanene ortho-quinones: synthesis, properties, and transformation into various heteropolycycles. Angew. Chem. Int. Ed. 56, 13470–13474 (2017).
Wang, W., Feng, L., Hua, X., Yuan, C. & Shao, X. Stimuli-responsive polycycles based on hetero-buckybowl trithiasumanene. Chin. J. Chem. 39, 3413–3420 (2021).
Yang, Y. et al. Tunable photoresponsive behaviors based on triphenylamine derivatives: the pivotal role of π-conjugated structure and corresponding application. Adv. Mater. 33, 2104002 (2021).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Zhu, T., Yang, T., Zhang, Q. & Yuan, W. Z. Clustering and halogen effects enabled red/near-infrared room temperature phosphorescence from aliphatic cyclic imides. Nat. Commun. 13, 2658 (2022).
Chen, J. et al. Synergistic generation and accumulation of triplet excitons for efficient ultralong organic phosphorescence. Angew. Chem. Int. Ed. 61, e202200343 (2022).
Yang, B. et al. Double-model decay strategy integrating persistent photogenic radicaloids with dynamic circularly polarized doublet radiance and triplet afterglow. J. Am. Chem. Soc. 146, 7668–7678 (2024).
Zhang, X. et al. Highly efficient and persistent room temperature phosphorescence from cluster exciton enables ultrasensitive off-on VOC sensing. Matter 5, 3499–3512 (2022).
Guo, Z. et al. Highly efficient and selective photocatalytic CO2 reduction by iron and cobalt quaterpyridine complexes. J. Am. Chem. Soc. 138, 9413–9416 (2016).
Wei, Y. et al. Highly efficient photocatalytic reduction of CO2 to CO by in situ formation of a hybrid catalytic system based on molecular iron quaterpyridine covalently linked to carbon nitride. Angew. Chem. Int. Ed. 61, e202116832 (2022).
Wang, J.-W., Jiang, L., Huang, H.-H., Han, Z. & Ouyang, G. Rapid electron transfer via dynamic coordinative interaction boosts quantum efficiency for photocatalytic CO2 reduction. Nat. Commun. 12, 4276 (2021).
Li, D. et al. Completely aqueous processable stimulus-responsive organic room temperature phosphorescence materials with tunable afterglow color. Nat. Commun. 13, 347 (2022).
Zhang, Y. et al. Microwave-responsive flexible room-temperature phosphorescence materials based on Poly(vinylidene fluoride) polymer. Angew. Chem. Int. Ed. 62, e202314273 (2023).
Gu, F., Jiang, T. & Ma, X. Visually monitoring the compactness of polymer matrixes coded by disparate luminescence. ACS Appl. Mater. Interfaces 13, 43473–43479 (2021).
Tolabi, H. et al. Progress of microfluidic hydrogel-based scaffolds and organ-on-chips for the cartilage tissue engineering. Adv. Mater. 35, e2208852 (2023).
Burda, C., Chen, X., Narayanan, R. & El-Sayed, M. A. Chemistry and properties of nanocrystals of different shapes. Chem. Rev. 105, 1025–1102 (2005).
Zhao, Z. et al. General synthesis of ultrafine monodispersed hybrid nanoparticles from highly stable monomicelles. Adv. Mater. 33, 2100820 (2021).
Xiao, F. et al. Light-harvesting fluorescent spherical nucleic acids self-assembled from a DNA-grafted conjugated polymer for amplified detection of nucleic acids. Angew. Chem. Int. Ed. 61, e202115812 (2022).
Yu, J. et al. Artificial spherical chromatophore nanomicelles for selective CO2 reduction in water. Nat. Catal. 6, 464–475 (2023).
Steentjes, T., Jonkheijm, P. & Huskens, J. Electron transfer processes in ferrocene-modified poly(ethylene glycol) monolayers on electrodes. Langmuir 33, 11878–11883 (2017).
Zhou, T. et al. Peg-stabilized coaxial stacking of two-dimensional covalent organic frameworks for enhanced photocatalytic hydrogen evolution. Nat. Commun. 12, 3934 (2021).
Hirata, S. Recent advances in materials with room-temperature phosphorescence: photophysics for triplet exciton stabilization. Adv. Opt. Mater. 5, 1700116 (2017).
Acknowledgments
The authors acknowledge the National Natural Scientific Foundation of China (Grant Number: 52473290 (Z.X.C), 22222501 (Z.X.C), 22175023 (Y.P.D) and 22401017 (P.W)), the Natural Science Foundation of Beijing Municipality (2232022 (Z.X.C) and 2242060 (P.S)). This work was also supported by BIT Research and Innovation Promoting Project (2023YCXZ016 (Z.X.C)) and China Postdoctoral Science Foundation (2023M740242 (P.W)).
Author information
Authors and Affiliations
Contributions
Z.X.C., Z.M.Z., and X.F.S. conceived and designed this project. C.C.X., P.W., and Y.M. performed the experiments, C.C.X. and Y.F.Z. carried out the DFT calculation, C.C.X., P.W, C.C., X.C., L.L.K., and G.C.L. analyzed the data, C.C.X. and P.W wrote the article, P.S., J.B.S., B.T., X.F.S., Z.M.Z., Z.X.C., and Y.P.D. revised the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Elena Bassan and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiong, C., Wang, P., Ma, Y. et al. Stabilizing the excited states of organic phosphorescent photosensitizers via self-assembly for CO2 photoreduction. Nat Commun 16, 6140 (2025). https://doi.org/10.1038/s41467-025-61451-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61451-4
